

Abstract
Background and purpose
Alzheimer's disease (AD) is a common neurodegenerative disease characterized by a neuroinflammatory state, and to date, there is no cure and its treatment represents a large unmet clinical need. The involvement of Th17 cells in the pathogenesis of AD‐related neuroinflammation has been reported in several studies. However, the role of the cytokine, IL‐17 has not been well addressed. Herein, we investigate the effects of IL‐17 neutralizing antibody (IL‐17Ab) injected by i.c.v. or intranasal (IN) routes on amyloid‐β (Aβ)‐induced neuroinflammation and memory impairment in mice.
Experimental approach
Aβ1–42 was injected into cerebral ventricles of adult CD1 mice. These mice received IL‐17Ab via i.c.v. either at 1 h prior to Aβ1–42 injection or IN 5 and 12 days after Aβ1–42 injection. After 7 and 14 days of Aβ1–42 administration, we evaluated olfactory, spatial and working memory and performed biochemical analyses on whole brain and specific brain areas.
Key results
Pretreatment with IL‐17Ab, given, i.c.v., markedly reduced Aβ1–42‐induced neurodegeneration, improved memory function, and prevented the increase of pro‐inflammatory mediators in a dose‐dependent manner at 7 and 14 days. Similarly, the double IN administration of IL‐17Ab after Aβ1–42 injection reduced neurodegeneration, memory decline, and the levels of proinflammatory mediators and cytokines.
Conclusion and implications
These findings suggest that the IL‐17Ab reduced neuroinflammation and behavioural symptoms induced by Aβ. The efficacy of IL‐17Ab IN administration in reducing Aβ1–42 neurodegeneration points to a possible future therapeutic approach in patients with AD.
Linked Articles
This article is part of a themed section on Therapeutics for Dementia and Alzheimer's Disease: New Directions for Precision Medicine. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.18/issuetoc
Abbreviations
- Ab
antibody
- AD
Alzheimer's disease
- ICAM‐1
intercellular adhesion molecule‐1
- IN
intranasal
- MPO
myeloperoxidase
What is already known
Th17 cells are involved in the pathogenesis of AD‐related neuroinflammation.
Th17‐derived proinflammatory cytokine (IL‐17) is expressed and up‐regulated during AD and in other neurological disorders.
What this study adds
In vivo neutralization of IL‐17 rescues amyloid‐β‐induced neuroinflammation and memory impairment
Intranasal administration of IL‐17Ab reduces amyloid‐β‐induced neuroinflammation similar to i.c.v. route.
What is the clinical significance
Our findings pave the way for a future potential clinical use of IL‐17‐site‐directed antibodies in AD and/or CNS-related diseases.
1. INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disorder and one of the most common forms of dementia worldwide (Liu et al., 2017). Pathologically, it is characterized by the deposition of extracellular amyloid‐β (Aβ) in the brain causing neuronal death in the neocortex and hippocampus, leading to irreversible cognitive impairment and behavioural alterations (Taylor, Hardy, & Fischbeck, 2002).
A large body of experimental evidence supports the view that amyloid plaques are (Taylor et al., 2002) key to driving AD pathogenesis through activation of both the innate and the adaptive immune pathways (Hardy & Selkoe, 2002). The ensuing neuroinflammatory response sustains the production and release of neurotoxic and inflammatory mediators (Schwartz & Deczkowska, 2016; Su, Bai, & Zhang, 2016) through the activation of microglia and astrocytes, causing neuronal cell death (Meda et al., 1995; Zuroff, Daley, Black, & Koronyo‐Hamaoui, 2017) and the release of inflammatory neurotransmitters and ROS (Tansey, McCoy, & Frank‐Cannon, 2007).
The release of mediators leads to recruitment of additional monocytes and lymphocytes through the blood–brain barrier, thus promoting their proliferation and resulting in further release of inflammatory factors (Das & Basu, 2008). Inflammatory markers have been detected not only in the brain but also in the body fluids of AD patients, reflecting a systemic neuropathological change (Bagyinszky et al., 2017).
T lymphocytes are particularly involved in the inflammatory response associated with AD and activated T‐cells can easily cross the blood–brain barrier contributing to the ongoing inflammatory repertoire and disease pathogenesis (Togo et al., 2002). In this context, several studies reported the importance of Th17 cells and Th17‐derived proinflammatory cytokines such as IL‐17, IL‐21, IL‐22, IL‐23, GM‐CSF, and IFN‐γ in the pathogenesis of AD (Saresella et al., 2011) and in other neurological disorders such as multiple sclerosis and Parkinson's disease (Miossec & Kolls, 2012; Tahmasebinia & Pourgholaminejad, 2017).
Increased levels of Th17 transcription factor RORγt, in addition to IL‐17 and IL‐22, have been found in the brains of animal models of AD (Zhang, Ke, Liu, Qiu, & Peng, 2013; Zhang, Liu, Sun, & Yin, 2015) and the increase of Th17/IL‐17A axis is sustained by Aβ‐induced oxidative stress (Solleiro‐Villavicencio & Rivas‐Arancibia, 2017). Furthermore, Zenaro et al. (2015) showed a role for neutrophils in AD‐like pathogenesis and cognitive impairment through the release of neutrophil extracellular traps and IL‐17 into the brain, suggesting that the inhibition of neutrophil trafficking and related neuro‐inflammatory onset may be beneficial in this pathology. Therefore, the increased level of IL‐17 and the activation of its signal transduction pathway seem to be involved in neurodegeneration and memory impairment, typical of AD (Diaz, Limon, Chávez, Zenteno, & Guevara, 2012; Yin, Wen, Li, & Wang, 2009). Moreover, a growing number of studies suggest the involvement of IL‐17 in the negative regulation of adult hippocampal neurogenesis (Liu et al., 2014), neuronal differentiation, and injury (Wang et al., 2009).
Given the well‐known contribution of neuroinflammation in AD and the stringent involvement of the proinflammatory cytokine IL‐17, we decided to examined whether a IL‐17 neutralizing antibody (IL‐17Ab) via intracerebroventricular (i.c.v.) or intranasal (IN) routes could ameliorate Aβ‐induced neuroinflammation and memory impairment.
2. METHODS
2.1. Animals
All animal care and experimental procedures were carried out in compliance with the international and national law and policies and approved by Italian Ministry of Health. Animal studies are reported in compliance with the ARRIVE guidelines and with the recommendations made by the British Journal of Pharmacology (EU Directive 2010/63/EU for animal experiments, ARRIVE guidelines, and the Basel declaration including the 3Rs concept; Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015). CD‐1 male mice (10–14 weeks of age, 25–30 g of weight) were purchased from Charles River (Milan, Italy) and kept in an animal care facility under controlled temperature, humidity, and on a 12 hr:12 hr light:dark cycle, with ad libitum access to water and standard laboratory chow diet. All procedures were carried out to minimize the number of animals used (n = 6 per group) and their suffering.
2.2. In vivo animal model and drug administration
Mice were randomly divided into 13 experimental groups, as shown in Table 1, counterbalancing body weight variation across groups. For the in vivo model, we used a well‐established method consisting of a direct Aβ injection as recently described (Maione, Piccolo, et al., 2018). Briefly, before the injection, Aβ1–42 protein was dissolved in PBS (1 μg·μl−1) in tubes that were sealed and incubated for 1 day at 37°C to allow peptide assembly state. Anaesthetized mice (mixture of N2O and O2 70:30 w v−1 containing 2% isoflurane) were injected with aggregated Aβ1–42 peptide (3 μg in 3 μl) or its inactive control peptide Aβ42–1 (3 μg in 3 μl) into cerebral ventricle at a rate of 1 μl·min−1, using a microsyringe (10 μl, Hamilton) according to the procedure previously described (Maione, Piccolo, et al., 2018). The needle was removed after 3 min using three intermediate steps with 1‐min interstep delay to minimize backflow. Another experimental group (Control) included mice that received the surgery procedure and Aβ peptide vehicle injection. After surgery and Ab administration, mice were placed on a thermal pad until they recovered from the anaesthetic. All procedures were performed under strictly aseptic conditions. The Hamilton syringe used for i.c.v. injections was repeatedly washed with distilled water followed by flushing with 1 mg·ml−1 BSA solution, in order to avoid non‐specific binding of peptides to glass. To evaluate the protection profile of the IL‐17Ab against Aβ1–42 peptide‐induced amyloidosis, we used two routes of injection: a single i.c.v. administration of IL‐17Ab (0.01–1 μg in 3 μl) and its related isotype control IgG2A administered at a maximum dose (1 μg in 3 μl), 1 hr prior to the Aβ1–42 injection, or IN administration of IL‐17Ab (0.01–1 μg in 10 μl) and its related isotype control IgG2A administered at its highest dose (1 μg in 10 μl), at 5 and 12 days after injection of Aβ1–42 (Figure 1). The conditions used for the IN administration of IL‐17Ab (volume, body position, and anaesthesia) were based on previously published reports (Pires, Fortuna, Alves, & Falcão, 2009; Southam, Dolovich, O'Byrne, & Inman, 2002).
Table 1.
Schematic representation of in vivo experimental groups and drug administration
| Exp. group | Group name | Aβ42–1 i.c.v. | Aβ1–42 i.c.v. | IgG2A i.c.v. | IgG2A IN | IL‐17Ab i.c.v. | IL‐17Ab IN |
|---|---|---|---|---|---|---|---|
| I | Control | – | – | – | – | – | – |
| II | IL‐17Ab i.c.v. | – | – | – | – | 1 μg in 3 μl | – |
| III | IL‐17Ab IN | – | – | – | – | – | 1 μg in 10 μl |
| IV | Aβ42–1 | 3 μg in 3 μl | – | – | – | – | – |
| V | Aβ1–42 | – | 3 μg in 3 μl | – | – | – | – |
| VI | Aβ1–42 + IgG2A | – | 3 μg in 3 μl | 1 μg in 3 μl | – | – | – |
| VII | Aβ1–42 + IL‐17Ab i.c.v. | – | 3 μg in 3 μl | – | – | 0.01 μg in 3 μl | – |
| VIII | Aβ1–42 + IL‐17Ab i.c.v. | – | 3 μg in 3 μl | – | – | 0.1 μg in 3 μl | – |
| IX | Aβ1–42 + IL‐17Ab i.c.v. | – | 3 μg in 3 μl | – | – | 1 μg in 3 μl | – |
| X | Aβ1–42 + IgG2A | – | 3 μg in 3 μl | – | 1 μg in 10 μl | – | |
| XI | Aβ1–42 + IL‐17Ab IN | – | 3 μg in 3 μl | – | – | – | 0.01 μg in 10 μl |
| XII | Aβ1–42 + IL‐17Ab IN | – | 3 μg in 3 μl | – | – | – | 0.1 μg in 10 μl |
| XIII | Aβ1–42 + IL‐17Ab IN | – | 3 μg in 3 μl | – | – | – | 1 μg in 10 μl |
Figure 1.
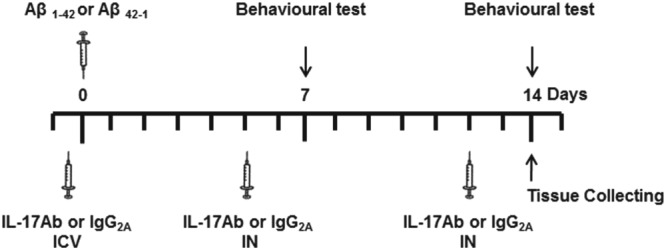
Experimental flow chart for the in vivo model, behavioural studies, and biochemical and molecular analysis with acute and subchronic treatment with IL‐17Ab
2.3. Behavioural studies
At 7 and 14 day post Aβ1–42 administration, mice were tested for novel object recognition (NOR), olfactory discrimination, Y‐maze, and Morris water maze (Figure 1). All tests were performed between 9 a.m. and 2 p.m. in an experimental room with sound isolation and dim light. The animals were carried to the test room for at least 1 hr for acclimation. Behaviour was monitored using a video camera positioned above the apparatus and the videos analysed in a blinded fashion using video tracking software (Any‐maze, Stoelting, Wood Dale, IL, USA).
2.4. Novel object recognition (NOR)
The NOR task exploits a mouse's natural tendency to explore a novel object after previous exposure to two identical objects. Mice were habituated for 10 min into the arena to reduce anxiety associated with the novel arena (plastic arena 30 × 30 × 50 cm). After this habituation stage, mice were ready to perform the task, which was conducted using two trials (familiarization trial [T1] and a test trial [T2]) separated by 30 min. During T1, mice were allowed to explore for 10 min two identical objects (plastic screw‐top tubes) secured to the floor using a small amount of Blu Tack in habituated arenas. For T2, one identical object from T1 was replaced with a novel object (small green flask) and mice were allowed to freely explore for 5 min. T1 and T2 were recorded using a video camera and analysed for the time spent interacting with the novel object. All arenas were cleaned with 80% ethanol prior the test. Novel object exploration was calculated in T2 by (T novel × 100)/(T novel + T identical) with exploration defined as the nose being less than 1 cm from the object when facing the object or actively engaging with the object by sniffing or paw touching. Climbing on the object was not considered exploratory.
2.5. Y‐maze task
Spontaneous alternation is a measure of spatial working memory. Such short‐term working memory was assessed by recording spontaneous alternation behaviour during a single session in the Y‐maze (made with three arms, 40 cm long, 120°C separate) positioned at exactly the same location for all procedures. Each mouse was placed at the end of one arm and allowed to move freely through the maze during a 5 min session. The series of arm entries were visually recorded. An arm choice was considered only when both fore paws and hind paws fully entered the arm. The Y‐maze was cleaned after each test with 80% ethanol to minimize odour cues. Alternation was defined as a successive entrance onto the three different arms. The number of correct entrance sequences (e.g., ABC and BCA) was defined as the number of actual alternations. The number of total possible alternations was therefore the total number of arm entries minus two, and the percentage of alternation was calculated as actual alternations/total alternations × 100 (D'Agostino et al., 2012).
2.6. Morris water maze
To assess spatial memory function, mice were tested by Morris water maze which consisted of a circular pool (diameter 170 cm, height 60 cm) with a transparent platform (10 cm in diameter), submerged under the water surface (1.5 cm). The water temperature of 24 ± 1°C, light intensity, and external visual cues in the room were rigorously reproduced. Swimming was recorded using a camera capture and analysed using video tracking software (Any‐maze, Stoelting, Wood Dale, IL, USA) that divided the pool into four equal quadrants. The platform position remained stable during 4 days in one of the four quadrants. Training phase consisted of four swims per day for 4 days, with about a 15 min intertrial time. Each of the four starting positions was used in randomized order. Each trial was started by placing a mouse into the pool, facing the wall of the tank and terminated as soon as the animal reached the platform with a cut‐off of 60 s. After the test, each mouse was kept warm for an hour and then returned to their home cage. Average of the four trials for each mouse and then the average for each group to give a single path length and escape latency expressed as mean ± SEM, was calculated for each training day. A probe test was also performed 1 hr after the last swim on Day 4. The platform was removed, and each animal was allowed a free 60 s swim. The time spent in the quadrant where the platform was previously placed, was determined. A higher percentage of time spent in the platform quadrant is interpreted as a higher level of memory retention. All tests were conducted in the morning.
2.7. Olfactory discrimination
The task was based on the fact that mice prefer places with their own odour (familiar compartments) instead of places with unfamiliar odors. In this test, mice had access to two adjacent identical chambers separated by an intermediate zone. One chamber contained familiar bedding from its home cage over the last 48 hr (familiar) whereas the other contained fresh bedding (non‐familiar). Mice were placed individually into the intermediate zone and allowed to freely explore each chamber for 10 min. Rodents are capable of discriminating familiar versus nonfamiliar chambers because they prefer their odour to no odour at all (D'Agostino et al., 2012). The time spent in each chamber was recorded and analysed. An olfactory discrimination index was generated according to the following formula: T familiar/(T familiar + T nonfamiliar), where T equal time and/or 0.5 as final value were considered as no preference.
2.8. Cytokine and chemokine protein array
All mice were killed and brains immediately removed. Total brain, prefrontal cortex, and hippocampus (half of the brain was used for cytokine/chemokine analysis and the half for further dissection and related analysis) were collected into a 2.0 ml tube for an immediate preservation in liquid nitrogen and a successive storage at −80°C. The isolated tissues were homogenized in ice‐chilled Tris–HCl buffer (20 mM, pH 7.4) containing 0.32‐M sucrose, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 1 mM sodium orthovanadate, and one protease inhibitor tablet per 50 ml of buffer. Protein concentration was determined by the BioRad protein assay kit (BioRad, Italy). Equal volumes (1.5 ml) of the homogenates were then incubated with the precoated proteome profiler array membranes according to the manufacturer's instructions. Dot plots were detected by using the enhanced chemiluminescence detection kit and Image Quant 400 GE Healthcare software (GE Healthcare, Italy) and successively quantified using GS 800 imaging densitometer software (Biorad, Italy).
2.9. Western blot analysis
Whole brain tissue homogenates (35 μg of protein) were subjected to SDS‐PAGE (10% gel) using standard protocols as previously described (Blaine Stine, Jungbauer, Yu, & Jo LaDu, 2011; Maione, Piccolo, et al., 2018). The proteins were transferred to PVDF membranes in the transfer buffer (25‐mM Tris–HCl [pH 7.4] containing 192‐mM glycine and 20% v/v methanol) at 400 mA for 2 hr. The membranes were saturated by incubation for 2 hr with nonfat dry milk (5% wt/v) in PBS supplemented with 0.1% (v/v) Tween 20 (PBS‐T) for 2 hr at room temperature and then incubated with 1:1000 dilution of anti‐β amyloid or with 1:2000 dilution of anti‐actin (after stripping) overnight at 4°C and then washed three times with PBS‐T. Blots were incubated with a 1:3000 dilution of HRP‐conjugated secondary Ab for 2 hr at room temperature, washed three times with PBS‐T. Protein bands were detected by using the enhanced chemiluminescence detection kit and Image Quant 400 GE Healthcare software (GE Healthcare, Italy). Protein bands were quantified using GS 800 imaging densitometer software (Biorad, Italy) and normalized with respective actin.
2.10. ELISA assay
elisa for Aβ1–42 and S100B was carried out on brain homogenates. Briefly, 100 μl of tissue supernatants, diluted standards, quality controls, and dilution buffer (blank) were added to a precoated plate with monoclonal anti‐Aβ1–42 or S100B for 2 hr. After washing, 100 μl of biotin‐labelled Ab was added for 1 hr. The plate was washed and 100 μl of streptavidin‐HRP conjugate was added and the plate was incubated for a further 30 min period in the dark. The addition of 100 μl of the substrate and stop solution represented the last steps before the reading of absorbance (measured at 450 nm) on a microplate reader. Antigen levels in the samples were determined using a standard curve of Aβ1–42 or S100B and expressed as μg·ml−1.
2.11. Immunofluorescence analysis
Immunofluorescence was performed by the free‐floating method. Briefly, mice were trans‐cardially perfused with PBS pH 7.4 solution followed by 4% paraformaldehyde. Brains were removed from the skull and fixed overnight in the 4% paraformaldehyde solution at 4°C. After being transferred in 30% sucrose solution, brains were cut into 25‐μm sections using a cryostat microtome (Leica, Wetzlar, Germany) throughout the hippocampus. Slices were blocked for 1 hr in 0.1% BSA solution and then incubated overnight at 4°C with primary Ab for the microglial cell marker Iba‐1 (1:1000) or astrocytes cell marker GFAP (1:1000). The day after, sections were washed and incubated for 1 hr with secondary Ab solution (donkey antirabbit IgG Alexa Fluor 488; 1:1000). Slices were washed, mounted onto glass slides, and cover‐slipped with Vectashield mounting medium (Vector Laboratories, Burlingame, CA), and images taken by Leica DM RB microscope using the Leica Application Suite software V.4.1.0 and photographed at 20× magnification. For quantification analysis, from each section containing the hippocampus (bregma level − 1.82 mm), activated astrocytes and microglia were counted from chosen equal area and analysed with Image J software (NIH, Bethesda, MD, USA).
2.12. Myeloperoxidase assay
Leukocyte myeloperoxidase (MPO) activity was assessed by measuring the H2O2‐dependet oxidation of 3,3′,5,5′‐tetramethylbenzidine as previously reported (Maione et al., 2009). Tissues (total brain) were homogenized for 35 s in a solution composed of hexadecyltrimethylammonium bromide (0.5% w v−1) in 50 mM sodium phosphate buffer pH 5.4. After homogenization, samples were centrifuged (4000 g) for 20 min and the supernatant used for the assay. Aliquots of 20 μl were incubated with 160 μl of 3,3′,5,5′‐tetramethylbenzidine and 20 μl of H2O2 (in 80‐mM phosphate buffer, pH 5.4) in 96‐well plates. Plates were incubated for 5 min at room temperature, and OD was read at 620 nm using a plate‐reader (Biorad, Italy). Assay were performed in duplicates and normalized for protein content.
2.13. Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018) and data sharing and presentation in preclinical pharmacology (Alexander et al., 2018; George et al., 2017). The results obtained were expressed as the mean ± SEM. Statistical analysis were performed by using one‐way ANOVA followed by Bonferroni or Dunnett's post‐test, when comparing more than two groups. GraphPad Prism 6.0 software (San Diego, CA, USA) was used for analysis. Data were considered statistically significant when a value of P ≤ 0.05 was achieved.
2.14. Materials
Recombinant mouse IL‐17 (also known as IL‐17A) neutralizing Ab (IL‐17Ab, monoclonal rat IgG2A, clone 50104), its related isotype control (Control; IgG2A, clone 54447) and proteome profiler mouse cytokine Array Kits (cat. ARY006) were purchased from R&D System (Milan, Italy). Aβ1–42 (cat. 1428) and Aβ42–1 (cat. 3391) amyloids peptides were purchased from Tocris (Milan, Italy). For Western blot analysis, the primary antibodies mouse monoclonal anti‐β amyloid were obtained from Novus Biologicals (Milan, Italy). Mouse monoclonal anti‐actin was obtained from Sigma‐Aldrich Co. (Milan, Italy). The HRP‐conjugated IgG secondary antibodies (anti‐mouse) were purchased from Dako (Copenhagen, Denmark). For immunofluorescence analysis, the rabbit polyclonal anti‐glial fibrillary acid protein (GFAP; cat. Z0334) was obtained from Dako (Copenhagen, Denmark) and rabbit anti‐ionized calcium binding adapter molecule (Iba‐1; cat. 019‐19741) was purchased from Wako Chemicals (Neuss, Germany). The secondary Ab donkey antirabbit IgG Alexa Fluor 488 (cat. A21206) was purchased from Invitrogen (USA). The Aβ1–42 and S100B kit were purchased from BioVendor (Rome, Italy). Unless otherwise stated, all the other reagents were from Carlo Erba (Milan, Italy).
2.15. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a; Alexander et al., 2017b; Alexander, Peters et al., 2017).
3. RESULTS
3.1. IL‐17Ab alleviates the Aβ1–42‐induced olfactory and object recognition impairment in a dose‐dependent manner
Consistent with the demonstration that olfactory dysfunction occurs at the early stage of Aβ‐induced pathology, the administration of Aβ1–42 (3 μg in 3 μl, i.c.v.) significantly decreased the capability of mice to discriminate new and familiar odors when tested 7 and 14 days later (Figure 2a–b) compared to Control and Aβ42–1 groups. Interestingly, administration of different doses of IL‐17Ab injected both i.c.v. (0.01–1 μg in 3 μl) and IN (0.01–1 μg in 10 μl) was shown to attenuate the olfactory dysfunction significantly in a dose‐ and time‐dependent manner, both at 7 and 14 days following Aβ1–42 administration.
Figure 2.
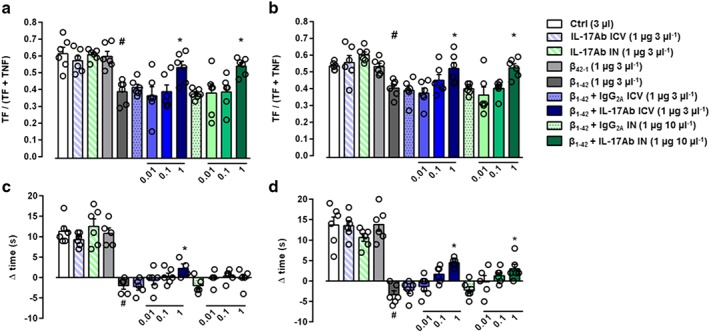
Effect of IL‐17Ab i.c.v. and IN‐treated mice on olfactory discrimination at 7 (a) and 14 (b) days and on novel object recognition at 7 (c) and 14 (d) days. Data were expressed as mean ± SEM (n = 6 per group). # P ≤ 0.05, significantly different from Control‐treated group (Ctrl); *P ≤ 0.05, significantly different from Aβ1–42 peptide‐treated group; one‐way ANOVA with Dunnett's post hoc test
The administration of Aβ1–42 reduced the delta time in the NOR test at Day 7 and at Day 14 (Figure 2c–d). Prior single i.c.v. (0.01–1 μg in 3 μl) injection of IL‐17Ab significantly attenuated the Aβ1–42‐related impairment in the NOR task in a dose‐ and time‐dependent manner compared to Control and Aβ42–1 groups (Figure 2c–d). The injection of IL‐17Ab IN, at different doses (0.01–1 μg in 10 μl) on Day 5, after Aβ1–42‐induced impairment had no effect on Day 7, while after the second IN administration on Day 12, the higher dose was shown to reduce the impairment in the NOR test at Day 14. IL‐17Ab alone and IL‐17Ab isotype control administered via i.c.v. and IN did not alter either olfactory or object recognition impairment (Figure 2).
3.2. IL‐17Ab ameliorates Aβ1–42‐induced spatial and working memory decline
Based on previous results, we selected the most active doses (1 μg for both i.c.v. and IN administration) of IL‐17Ab to test spatial and working memory by the Morris water maze and Y‐maze test. Aβ1–42 (3 μg in 3 μl per mouse, i.c.v.) injection induced an increase of escape latency in Morris water maze, specifically during the second day of the training phase, while the acquisition of the platform position was faster in mice treated with IN injection of IL‐17Ab (Figure 3a). All groups showed the same average speed (Figure 3b). Two different cohorts of mice were subjected to the probe test at 7 or 14 day following Aβ1–42 injection (Figure 3c–d). The time that Aβ1–42‐injected mice spent in the target quadrant was significantly shorter on both Day 7 and 14 compared to Control and Aβ42–1 groups. On Day 7, no significant improvement was observed regarding the time spent in the target quadrant in mice treated with the active doses of IL‐17Ab by i.c.v. while the IN route significantly increased the time spent in the target quadrant, compared to mice that received only Aβ1–42 (Figure 3c). On Day 14, the active doses of IL‐17Ab, administered both i.c.v. and IN, significantly increased the time spent in the target quadrant (Figure 3d). No significant effects were observed in IL‐17Ab administered i.c.v. or IN alone, Aβ42–1 and IL‐17Ab isotype Control groups (Figure 3). These results were also confirmed by the track plots on Day 7 and 14 presented in Figure 3e.
Figure 3.
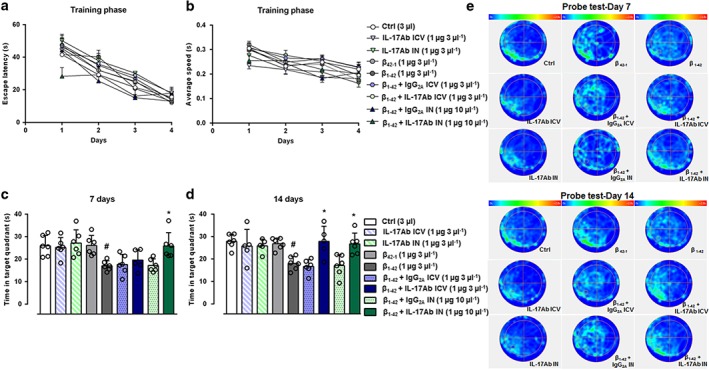
Escape latency (a) and average speed (b) during training phase in the MWM test. Time spent in the target quadrant of the probe test at 7 (c) and 14 (d) days. (e) Schematic plots of probe test at 7 and 14 days for all experimental groups. Data were expressed as mean ± SEM (n = 6 per group). # P ≤ 0.05, significantly different from Control‐treated group (Ctrl); *P ≤ 0.05, significantly different from Aβ1–42 peptide‐treated group; one‐way ANOVA with Bonferroni's post hoc test
Percent of correct alternations in the Y‐maze test was then analysed on Day 7 and 14 days after Aβ1–42 injection (Figure 4). Single prior i.c.v. (1 μg 3 μl−1) administration and double administration of IL‐17Ab IN (1 μg in 10 μl) after the Aβ1–42 significantly attenuated the impairment of spontaneous alternation induced by Aβ1–42 on both Days 7 and 14, in equal manner (Figure 4a–c). Aβ1–42 and IL‐17Ab treatments did not affect the total number of arm entries at these doses (Figure 4b–d). No significant effects were observed in Aβ42–1, IL‐17Ab administered i.c.v. or IN alone and in IL‐17Ab isotype Control groups (Figure 4).
Figure 4.
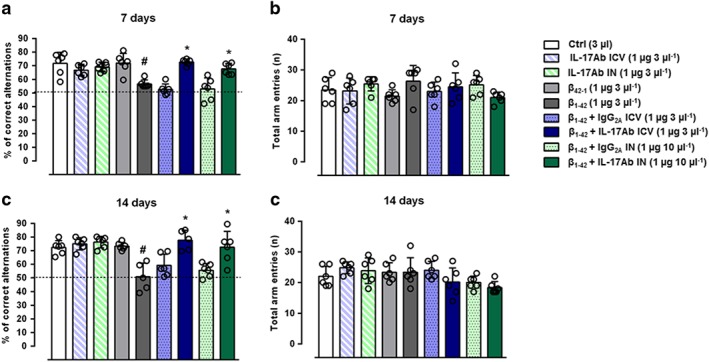
Percent of correct alternations (a–c) and total arm entries (b–d) in the Y‐maze test. Data were expressed as mean ± SEM (n = 6 per group). # P ≤ 0.05, significantly different from Control‐treated group (Ctrl); *P ≤ 0.05, significantly different from Aβ1–42 peptide‐treated group; one‐way ANOVA with Bonferroni's post hoc test
3.3. Effect of IL‐17Ab administration on Aβ1–42, S100B, and MPO levels
We began our biochemical analysis by confirming the behavioural findings with the association of Aβ1–42 level in the brain. As expected, Aβ1–42 was significantly higher in the AD group compared to Control, while its level was significantly lower in IL‐17Ab i.c.v. and IL‐17Ab IN‐treated group (Figure 5a). This correlation failed to reach significance in Aβ42–1, IL‐17Ab administered i.c.v., or IN alone and in IL‐17Ab isotype Control groups. These results were supported by the simultaneously presence of Aβ monomers and large oligomers (most likely dimers and tetramers), at both 7 and 14 day post model induction, in mice injected with fibrillated Aβ peptide, compared with mice injected with not‐fibrillated peptide (Figure S1).
Figure 5.
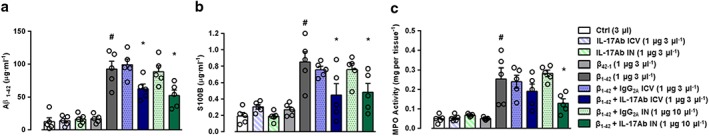
Effect of IL‐17Ab on the production of (a) Aβ1–42 (detected by ELISA assay and expressed as μg·ml−1), release of (b) S100B protein (detected by ELISA assay and expressed as μg·ml−1), and (c) MPO activity (detected by enzymic assay and expressed as mg per tissue) in total brain homogenates. Data were expressed as mean ± SEM (n = 6 per group). # P ≤ 0.05, significantly different from Control‐treated group (Ctrl); *P ≤ 0.05, significantly different from Aβ1–42 peptide‐treated group; one‐way ANOVA with Dunnett's post hoc test
Based on these findings, we decided to examine S100B levels in total brain homogenates by S100B ELISA. As shown in Figure 5b at Day 14, Aβ1–42 (3 μg 3 μl−1) induced a significant increase in S100B levels compared to Control and Aβ42–1 groups. Single i.c.v. administration of IL‐17Ab prior the Aβ1–42 (1 μg in 3 μl), in addition to repeated administration of IL‐17Ab by IN route (1 μg in 10 μl , significantly attenuated S100B levels compared to Aβ1–42‐treated mice. No differences in S100B levels were detected between i.c.v. and IN groups, suggesting that both routes had similar positive effects.
We went on to monitor inflammatory leukocyte infiltration, at Day 14 post Aβ1–42, by measuring MPO activity in brain homogenates. We found a significant increase in MPO activity in Aβ1–42‐injected mice compared to Control and Aβ42–1 groups respectively. The administration of IL‐17Ab by i.c.v. (1 μg in 3 μl ) did not restore MPO activity that was significantly reduced by IL‐17Ab‐injected IN (1 μg in 10 μl; Figure 5c). No significant effects were observed in IL‐17Ab administered i.c.v. or IN alone and in IL‐17Ab isotype Control groups, in terms of S100B levels and MPO activity (Figure 5b–c).
3.4. Effect of IL‐17Ab administration on Aβ1–42‐induced neuroinflammatory activity
Given that Aβ1–42 injection induced neuroinflammation, we investigated the molecular mechanisms behind the neuroprotective effect of IL‐17Ab, by examining both i.c.v. and IN alone (and also their effect prior or after the injection of Aβ1–42 induced impairment) on microglial and astrocyte inflammatory responses. To confirm of activation of neural cells, we measured expression of GFAP (a marker of astrocyte activation) and Iba‐1 (a marker of microglia cell activation) in the hippocampus for different experimental groups 14 days post Aβ1–42 injection. As expected, Aβ1–42‐injected mice showed a significant increase in the expression of both GFAP and Iba‐1 (Figure 6d,j) when compared to Control (Figure 6a,g). Interestingly, both i.c.v. (1 μg 3 μl−1) and IN (1 μg 10 μl−1) injection of IL‐17Ab significantly reduced astrocyte and microglial proliferative responses and activation compared with those in mice that received only Aβ1–42 (Figure 6e–f and Figure 6k–l). GFAP and Iba‐1 staining in animals receiving IL17Ab alone injected i.c.v. or IN was similar to the Control group (Figure 6b–c and Figure 6h–i).
Figure 6.
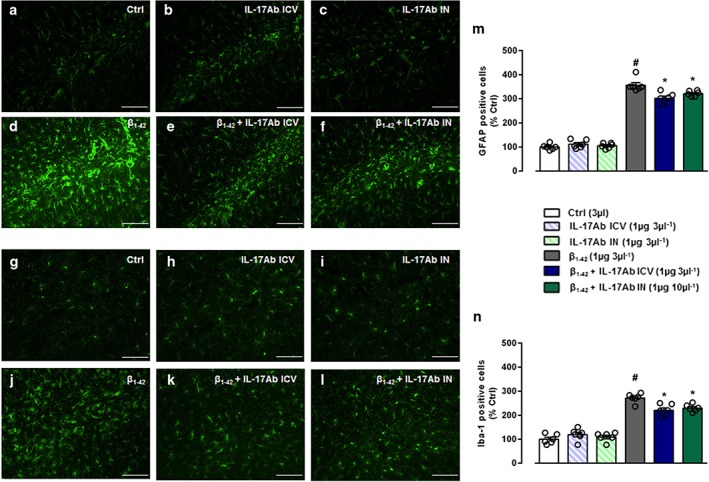
Representative immunofluorescence staining for GFAP and Iba‐1 in the hippocampus of (a–g) Ctrl, (b–h) IL‐17Ab i.c.v., (c–i) IL‐17Ab IN, (d–j) Aβ1–42, (e–k) Aβ1–42 + IL‐17Ab i.c.v., or (f–l) IL‐17Ab IN‐treated mice (scale bar 100 μm). Quantitative analysis of (m) GFAP and (n) Iba‐1 positive cells. Values are expressed as percent mean (±SEM) of positive cells of n = 6 mice in the hippocampus of Ctrl mice. # P ≤ 0.05, significantly different from Ctrl group; *P ≤ 0.05, significantly different from Aβ1–42 peptide‐treated group; one‐way ANOVA with Bonferroni's post hoc test
3.5. Modulation of proinflammatory cyto‐chemokines by IL‐17Ab
Informed by previous results, we also compared the cytokine and chemokine profiles from total brain homogenates of selected groups which were also used for the histological analysis. A similar analysis was performed on homogenates obtained from the hippocampus and prefrontal cortex. As shown in Figure 7, Aβ1–42‐treated mice (Figure 7e) displayed a marked increase in proinflammatory cytokines and chemokines (reference standards shown in Figure 7a) compared to Control group (Figure 7b). In particular, we observed a significant increase in the expression of classical proinflammatory cyto‐chemokines such as IL‐1α, IL‐1β, IL‐1r, IL‐6, IL‐17, CCL3, CCL4 and TNF‐α (Figure 7h–j). In line with our in vivo evidence, we observed a reversal of this proinflammatory onset in IL‐17Ab i.c.v.‐treated mice (Figure 7f) which became more evident after IL‐17Ab IN treatment (Figure 7d). In particular, IN treatment induced a significant decrease in terms of IL‐1β, IL‐17, CXCL1 and CXCL2, compared with IL‐17Ab i.c.v. treatment (Figure 7h–j).
Figure 7.

Inflammatory fluids obtained from total brain homogenates of (b) Ctrl, (c) IL‐17Ab i.c.v., (d) IL‐17Ab IN, (e) Aβ1–42, (f) Aβ1–42 + IL‐17Ab i.c.v., or (g) IL‐17Ab IN‐treated mice were assessed using (a) a proteome profiler cytokine array panel; the modulated cytokines are shown in red. Mean changes (±SEM) of positive spots of three separate experiments with n = 6 mice were expressed as INT per mm2 of densitometric values (h–j). # P ≤ 0.05, significantly different from Ctrl group; *P ≤ 0.05, significantly different from Aβ1–42 peptide‐treated group; one‐way ANOVA with Dunnett's post hoc test
Interestingly, analysing the inflammatory profile from homogenized hippocampus (Figure 8a–g) and prefrontal cortex (Figure 8h–n) tissues, we found a different scenario. In the hippocampus, Aβ1–42‐treated mice presented an increase in the adhesion molecule ICAM‐1, the chemokine CXCL12 and, in particular, in IL‐17 (Figure 8e) compared to Control (Figure 8b) that were reversed after i.c.v. and IN IL‐17Ab administration (Figure 8o). In the prefrontal cortex, we observed a striking difference only in terms of IL‐17 expression in Aβ1–42 group (Figure 8l) that was modulated after IL‐17Ab i.c.v. (Figure 8m) and IN (Figure 8n) treated mice. Cyto‐chemokines expression in animals receiving IL‐17Ab alone injected i.c.v. or IN was similar to Control group in whole brain (Figure 7c–d), hippocampus (Figure 8c–d), and prefrontal cortex (Figure 8j–k).
Figure 8.
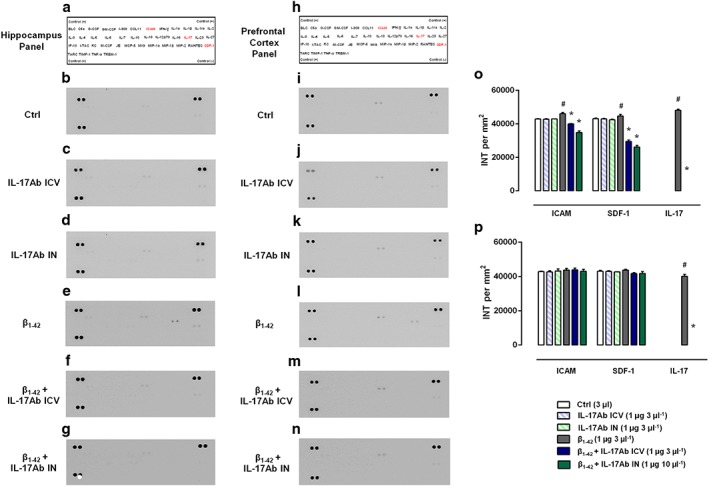
Inflammatory fluids obtained from (a–g) hippocampus and (h–n) prefrontal cortex homogenates of (b and i) Ctrl, (c and j) IL‐17Ab i.c.v., (d and k) IL‐17Ab IN, (e and l) Aβ1–42, (f and m) Aβ1–42 + IL‐17Ab i.c.v., or (g and n) IL‐17Ab IN‐treated mice were assessed using (a and h) a proteome profiler cytokine array; the modulated cytokines are shown in red. Mean changes (±SEM) of positive spots of three separate experiments with n = 6 mice were expressed as INT per mm2 of densitometric values for both hippocampus (o) and prefrontal cortex (p). # P ≤ 0.05, significantly different from Ctrl group; *P ≤ 0.05, significantly different from Aβ1–42 peptide‐treated group; one‐way ANOVA with Dunnett's post hoc test
4. DISCUSSION
Inflammation comprises a large set of diseases that trigger a wide variety of human disorders. Neurological inflammation and autoimmune disorders are an example of immune‐mediated inflammation (Newcombe et al., 2018). In the sites of local inflammation, the concentration of proinflammatory cytokines, for example, IL‐1, IL‐2, IL‐6, IL‐12, IFN‐γ, TNF‐α, IL‐17, and IL‐23 or of the other inflammatory mediators, increases (Kempuraj et al., 2017). Th1 and Th17 cells are the main cellular mediators responsible for immune‐mediated damage that polarize to the site of inflammation in presence of noxious or inflammatory stimuli (D'Acquisto, Maione, & Pederzoli‐Ribeil, 2010). Many studies have suggested a relationship between inflammation severity and Th17 cell‐mediated immune responses (Rostami & Ciric, 2013). The role of Th17 cells has been highlighted in several autoimmune disorders including MS, inflammatory bowel disease, rheumatoid arthritis, and systemic lupus erythematosus as well as in neurological disorders including fronto‐temporal dementia, PD, AD, and schizophrenia (Liu et al., 2014).
AD is the most common and most studied neurodegenerative disorder. It has become a critical issue to human health, especially in ageing societies, and therefore, it is a focus of research in the global scientific community (Newcombe et al., 2018). It is a multifactorial disorder primarily characterized by the deposition of amyloid plaques in the brain, leading to irreversible cognitive impairment and neuroinflammation that mainly involves hippocampus and cortex districts (D'Amelio, Puglisi‐Allegra, & Mercuri, 2018). Although Th17 cells have been acknowledged as crucial mediators of AD, the effector cytokine IL‐17 responsible for their pathogenicity still remains poorly defined and very little is known on its pathophysiological role in the regions of the CNS (hippocampus and prefrontal cortex) usually compromised in this disease.
In this study, we have demonstrated that the neutralization of IL‐17 results in substantial functional recovery of Aβ‐induced neuroinflammation and memory impairment. Moreover, IL‐17Ab administration (both i.c.v. and IN) reversed reactive gliosis and neuroinflammation, as indicated ex vivo by the reduction of Aβ1–42, GFAP, Iba‐1, S100B, and MPO, and by the inhibition of typical proinflammatory cyto‐chemokines in total brain and, more specifically, in the hippocampal and prefrontal cortex regions.
In particular, we show that neutralization of IL‐17 by a single i.c.v. administration ameliorated Aβ‐like symptoms (in terms of olfactory dysfunction, spatial and working memory) both after 7 and 14 days, similar to that observed after a double IL‐17Ab IN administration on days 5 and 12. The difference in terms of time of administration is justified by earlier observations that found a significant increase of Th17 cytokines following 7 days from Aβ injection (Zhang et al., 2013; Zhang et al., 2015). Moreover, very robust effects were observed when the Ab was administered at dose of 1 μg·μl−1, compared with the isotype Control or Ab itself, consistent with previous in vivo reports (Fischer et al., 2015; Mi et al., 2011; Szabo et al., 2017). It is known that a major, yet unmet, objective of neuroprotective or anti‐neurodegenerative treatments is to arrest or to slow down the rapid progression of cognitive impairment. Consistent with this, our results from the behavioural experiments showed that the Aβ‐dependent impairment in terms of spatial learning, reference memory, and olfactory memory was dose‐dependently prevented and reversed by IL‐17Ab treatments.
Due to the particular anatomical features of the nasal cavity, IN administration has been explored as a means of preferential drug delivery to the brain, even though a fraction of the drug may be absorbed by the systemic circulation (Fortuna, Alves, Serralheiro, Sousa, & Falcão, 2014). The route of administration used in our work (IN) ensures high brain concentration levels of IL‐17Ab reaching the olfactory bulb and strongly suggests that the Ab was directly transferred to the brain via the olfactory neuronal pathway, bypassing the blood–brain barrier. To achieve this aim and in the light of earlier work (Pires et al., 2009; Southam et al., 2002), we also used IN administration, paying attention to the effects of volume, time, body position, and anaesthesia.
Next, we started our biochemical investigation by confirming that the well‐replicated behavioural findings were associated with the levels of Aβ1–42 in whole brain, whihcwas used as an index of the pathology. As expected, levels of Aβ1–42 were significantly higher in the AD group compared with the Control group, while its levels were significantly lower in IL‐17Ab i.c.v. and IL‐17Ab IN‐treated group. In addition, our biochemical investigation also revealed that IL‐17Abs (both i.c.v. and IN) attenuated the production of S100B, a glial‐derived protein that is secreted from astrocytes. At low concentrations, this protein is considered a neurotrophic factor and neuronal survival protein during the development of the nervous system (Van Eldik & Wainwright, 2003). Conversely, when overproduced by activated glia, S100B becomes a mediator of pathology, influencing disease progression by acting as proinflammatory cytokine, thus contributing to the exacerbation of neuroinflammation and neuronal dysfunction. There is good clinical evidence that S100B is a component of the neuroinflammatory cycle that drives AD pathogenesis (Mrak & Griffin, 2005). In this context, it is relevant to note that S100B levels are increased in activated astrocytes in the brain regions most severely affected by AD, and its levels correlate with neuritic plaque progression. Additionally, S100B production can be stimulated by mediators associated with AD, including β‐amyloid and proinflammatory cytokines (Van Eldik & Wainwright, 2003). Collectively, this highlights the crucial role of S100B in AD onset and progression and the relevant effects of IL‐17Abs in reducing its expression in mouse whole brain. During the confirmation of the inhibition of the inflammatory state with IL‐17 neutralizing antibodies, we also observed, using immunofluorescence techniques, that expression of GFAP and Iba‐1 (as indices of astrocytes and microglia activation; Gu et al., 2015; Liddelow et al., 2017) was markedly reduced after IL‐17 Abs treatments.
The vasculature of the blood–brain barrier allows only relatively few leukocytes to enter and survey the healthy CNS (Lefkowitz & Lefkowitz, 2008). However, during pathological CNS inflammation (including AD), the number of leukocytes adhering to and penetrating the CNS vasculature increases (Crawford et al., 2001; Russo et al., 2018). Considering that the enzyme MPO is a haem‐protein abundant in circulating phagocytes, tissue neutrophils, and some populations of tissue macrophages but also highly expressed in AD brain, where it colocalized with amyloid plaques (Reynolds et al., 1999), its abnormal expression and modulation could represent another important aspect to be considered in amyloid‐related disorders. Our results showed that the expression levels of the oxidant producing enzyme MPO were modulated by IL‐17Ab, after i.c.v. or IN administration.
In line with previous studies (Xia & Hyman, 1999; Rostasy et al., 2003; Walker et al., 2017), we also found increased expression of different proinflammatory mediators (IL‐1α, IL‐1β, IL‐16, CXCL1, CCL3, IL‐17, and TNF‐α; cytokines and chemokines that are associated with the pathological changes of AD) in the total brain of Aβ‐treated mice, compared with Control mice. Interestingly, we demonstrate, for the first time, a significant reduction of these proinflammatory cascades after both i.c.v. and IN administration of IL‐17Ab. Furthermore, the comparison of the inflammatory profile from hippocampus and prefrontal cortex tissues presented a distinctive induction and modulation of ICAM, SDF‐1, and IL‐17 in Aβ1–42‐treated group compared to Control with a complete absence of IL‐17 in both regions after IL‐17Ab treatment and a significant reduction of ICAM and SDF‐1 only in the hippocampus. This difference could be, in part, correlated with previous observations that have demonstrated that a systemic Th17/IL‐17 response appears before hippocampal neurodegeneration, compared with other CNS regions (Solleiro‐Villavicencio & Rivas‐Arancibia, 2017).
Endothelial ICAM does not only mediate firm adhesion of leukocytes to vascular beds but also triggers signalling cascades within the endothelial cell, which play a crucial role in modulating subsequent leukocyte diapedesis in the CNS, especially during inflammatory conditions (Adamson, Etienne, Couraud, Calder, & Greenwood, 1999). Furthermore, a number of recent reports show that drugs interfering with endothelial ICAM‐1 signalling also efficiently reduce leukocyte migration both in vitro and in animal models of CNS inflammation (Turowski, Adamson, & Greenwood, 2005). In this situation, the chemokine CXCL12 (also known as SDF‐1) could also play a crucial role. While CXCL12 is constitutively expressed in the CNS, its role during neuroinflammation still remains unclear. Several reports have shown that this chemokine moderates remyelination and synaptic plasticity (Merino, Bellver‐Landete, Oset‐Gasque, & Cubelos, 2015) and, in the present context, its pharmacological inhibition (Azizi, Khannazer, & Mirshafiey, 2014) alleviates the release of inflammatory mediators in both sciatic nerve injury and bone cancer models (Shen et al., 2014) as well as NMDA receptor‐mediated neurotoxicity (Sanchez et al., 2016).
Taking our results together, it would appear that neutralization of IL‐17 substantially restrained the release of synergistic cytokines and, consequently, the expression of inflammatory mediators, which sustain the progression of CNS damage. This outcome is in accordance with our previous studies where we have demonstrated and emphasized the role of IL‐17 as a cytokine that sustains, rather than inducing, inflammation (Maione et al., 2009; Maione, Iqbal, et al., 2018).
In conclusion, the experimental findings reported here confirm and extend previous data showing that IL‐17 is a detrimental factor for AD, where this particular cytokine (especially by its selective expression in the hippocampus and prefrontal cortex) could represent a key factor for the “self‐amplifying” neuroinflammatory onset typical of Aβ‐related disease. As a result, future studies will focus on the brain pathophysiology of IL‐17, as a determinant of the developmental neurotoxicity of widely different CNS‐related diseases.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
C.C., F.V., B.B., F.R., P.L., M.P., and F.M. carried out experiments. C.C., A.J.I., C.I., M.C.M., N.M., and F.M. performed data analysis and wrote the paper. C.P.C. and A.C. helped with revision of the paper. All authors read and approved the final version of the paper before submission.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1.
(A) Representative Western blot of Aβ1–42 performed in whole brain lysate at 7‐ and 14‐days post ICV Aβ‐peptide (fibrillated and not fibrillated) administration and related (B) cumulative densiometric analyses of Aβ1–42 protein. Data were expressed as mean ± SEM. Statistical analysis was performed by using one‐way ANOVA followed by Dunnett's post‐test. # P ≤ 0.05 vs 14 days not fibrillated condition, *P ≤ 0.05 vs 7 days not fibrillated condition (N = 5 per group). In C and D are reported the original Western blots related to Aβ1–42 and actin respectively.
Figure S2.
Representative immunofluorescence staining for GFAP in the hippocampus of (A) Control, (B) IL‐17Ab ICV, (C) IL‐17Ab IN, (D) Aβ1–42, (E) Aβ1–42 + IL‐17Ab ICV and (F) IL‐17Ab IN‐treated mice. Original pictures were photographed at 5 X magnification.
Figure S3. Representative immunofluorescence staining for Iba‐1 in the hippocampus of (A) Control, (B) IL‐17Ab ICV, (C) IL‐17Ab IN, (D) Aβ1–42, (E) Aβ1–42 + IL‐17Ab ICV and (F) IL‐17Ab IN‐treated mice. Original pictures were photographed at 5 X magnification.
ACKNOWLEDGEMENTS
We would like to thank Dr Antonio Baiano and also Mr. Giovanni Esposito and Mr. Angelo Russo for animal care and technical assistance.
Cristiano C, Volpicelli F, Lippiello P, et al. Neutralization of IL‐17 rescues amyloid‐β‐induced neuroinflammation and memory impairment. Br J Pharmacol. 2019;176:3544–3557. 10.1111/bph.14586
REFERENCES
- Adamson, P. , Etienne, S. , Couraud, P. O. , Calder, V. , & Greenwood, J. (1999). Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM‐1 via a rho‐dependent pathway. Journal of Immunology, 162, 2964–2973. [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi, G. , Khannazer, N. , & Mirshafiey, A. (2014). The potential role of chemokines in Alzheimer's disease pathogenesis. American Journal of Alzheimer's Disease and Other Dementias, 29, 415–425. 10.1177/1533317513518651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagyinszky, E. , Giau, V. V. , Shim, K. , Suk, K. , An, S. S. A. , & Kim, S. (2017). Role of inflammatory molecules in the Alzheimer's disease progression and diagnosis. Journal of the Neurological Sciences, 376, 242–254. 10.1016/j.jns.2017.03.031 [DOI] [PubMed] [Google Scholar]
- Blaine Stine, W. , Jungbauer, L. , Yu, C. , & Jo LaDu, M. (2011). Preparing synthetic Aβ in different aggregation states. Methods in Molecular Biology, 670, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford, F. C. , Freeman, M. J. , Schinka, J. A. , Morris, M. D. , Abdullah, L. I. , Richards, D. , … Mullan, M. J. (2001). Association between Alzheimer's disease and a functional polymorphism in the myeloperoxidase gene. Experimental Neurology, 167, 456–459. 10.1006/exnr.2000.7560 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers.. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Acquisto, F. , Maione, F. , & Pederzoli‐Ribeil, M. (2010). From IL‐15 to IL‐33: The never‐ending list of new players in inflammation. Is it time to forget the humble aspirin and move ahead? Biochemical Pharmacology, 79, 525–534. 10.1016/j.bcp.2009.09.015 [DOI] [PubMed] [Google Scholar]
- D'Agostino, G. , Russo, R. , Avagliano, C. , Cristiano, C. , Meli, R. , & Calignano, A. (2012). Palmitoylethanolamide protects against the amyloid‐β25‐35‐induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology, 37, 1784–1792. 10.1038/npp.2012.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amelio, M. , Puglisi‐Allegra, S. , & Mercuri, N. (2018). The role of dopaminergic midbrain in Alzheimer's disease: Translating basic science into clinical practice. Pharmacological Research, 130, 414–419. 10.1016/j.phrs.2018.01.016 [DOI] [PubMed] [Google Scholar]
- Das, S. , & Basu, A. (2008). Inflammation: A new candidate in modulating adult neurogenesis. Journal of Neuroscience Research, 86, 1199–1208. 10.1002/jnr.21585 [DOI] [PubMed] [Google Scholar]
- Diaz, A. , Limon, D. , Chávez, R. , Zenteno, E. , & Guevara, J. (2012). Aβ25‐35 injection into the temporal cortex induces chronic inflammation that contributes to neurodegeneration and spatial memory impairment in rats. Journal of Alzheimer's Disease, 30, 505–522. 10.3233/JAD-2012-111979 [DOI] [PubMed] [Google Scholar]
- Fischer, J. A. , Hueber, A. J. , Wilson, S. , Galm, M. , Baum, W. , Kitson, C. , … Schett, G. (2015). Combined inhibition of tumor necrosis factor α and interleukin‐17 as a therapeutic opportunity in rheumatoid arthritis: Development and characterization of a novel bispecific antibody. Arthritis & Rhematology, 67, 51–62. 10.1002/art.38896 [DOI] [PubMed] [Google Scholar]
- Fortuna, A. , Alves, G. , Serralheiro, A. , Sousa, J. , & Falcão, A. (2014). Intranasal delivery of systemic‐acting drugs: Small‐molecules and biomacromolecules. European Journal of Pharmaceutics and Biopharmaceutics, 88, 8–27. 10.1016/j.ejpb.2014.03.004 [DOI] [PubMed] [Google Scholar]
- George, C. H. , Stanford, S. C. , Alexander, S. , Cirino, G. , Docherty, J. R. , Giembycz, M. A. , … Ahluwalia, A. (2017). Updating the guidelines for data transparency in the British Journal of Pharmacology ‐ data sharing and the use of scatter plots instead of bar charts. British Journal of Pharmacology, 174, 2801–2804. 10.1111/bph.13925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, S. M. , Park, M. H. , Hwang, C. J. , Song, H. S. , Lee, U. S. , Han, S. B. , … Hong, J. T. (2015). Bee venom ameliorates lipopolysaccharide‐induced memory loss by preventing NF‐κB pathway. Journal of Neuroinflammation, 12, 124 10.1186/s12974-015-0344-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res., 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, J. , & Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science, 297, 353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Kempuraj, D. , Thangavel, R. , Selvakumar, G. P. , Zaheer, S. , Ahmed, M. E. , Raikwar, S. P. , … Zaheer, A. (2017). Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation and neurodegeneration. Frontiers in Cellular Neuroscience, 11, 216 10.3389/fncel.2017.00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The arrive guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz, D. L. , & Lefkowitz, S. S. (2008). Microglia and myeloperoxidase: A deadly partnership in neurodegenerative disease. Free Radical Biology & Medicine, 45, 726–731. 10.1016/j.freeradbiomed.2008.05.021 [DOI] [PubMed] [Google Scholar]
- Liddelow, S. A. , Guttenplan, K. A. , Clarke, L. E. , Bennett, F. C. , Bohlen, C. J. , Schirmer, L. , … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 2017(541), 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q. , Xin, W. , He, P. , Turner, D. , Yin, J. , Gan, Y. , … Wu, J. (2014). Interleukin‐17 inhibits adult hippocampal neurogenesis. Scientific Reports, 4, 7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Zhang, A. , Sun, H. , Han, Y. , Kong, L. , & Wang, X. (2017). Two decades of new drug discovery and development for Alzheimer's disease. RSC Advances, 7, 6046–6058. 10.1039/C6RA26737H [DOI] [Google Scholar]
- Maione, F. , Iqbal, A. J. , Raucci, F. , Letek, M. , Bauer, M. , & D'Acquisto, F. (2018). Repetitive exposure of IL‐17 into the murine air pouch favors the recruitment of inflammatory monocytes and the release of IL‐16 and TREM‐1 in the inflammatory fluids. Front. Immunol, 9 10.3389/fimmu.2018.02752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione, F. , Paschalidis, N. , Mascolo, N. , Dufton, N. , Perretti, M. , & D'Acquisto, F. (2009). Interleukin 17 sustains rather than induces inflammation. Biochemical Pharmacology, 77, 878–887. 10.1016/j.bcp.2008.11.011 [DOI] [PubMed] [Google Scholar]
- Maione, F. , Piccolo, M. , De Vita, S. , Chini, M. G. , Cristiano, C. , De Caro, C. , … Bifulco, G. (2018). Down regulation of pro‐inflammatory pathways by tanshinone IIA and cryptotanshinone in a non‐genetic mouse model of Alzheimer's disease. Pharmacological Research, 129, 482–490. 10.1016/j.phrs.2017.11.018 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (arrive etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda, L. , Cassatella, M. A. , Szendrei, G. I. , Otvos, L. , Baron, P. , Villalba, M. , … Rossi, F. (1995). Activation of microglial cells by [β]‐amyloid protein and interferon‐[γ]. Nature, 374, 647–650. 10.1038/374647a0 [DOI] [PubMed] [Google Scholar]
- Merino, J. J. , Bellver‐Landete, V. , Oset‐Gasque, M. J. , & Cubelos, B. (2015). CXCR4/CXCR7 molecular involvement in neuronal and neural progenitor migration: Focus in CNS repair. Journal of Cellular Physiology, 230, 27–42. 10.1002/jcp.24695 [DOI] [PubMed] [Google Scholar]
- Mi, S. , Li, Z. , Yang, H. Z. , Liu, H. , Wang, J. P. , Ma, Y. G. , … Hu, Z. W. (2011). Blocking IL‐17A promotes the resolution of pulmonary inflammation and fibrosis via TGF‐β1‐dependent and ‐independent mechanisms. Journal of Immunology, 187, 3003–3014. 10.4049/jimmunol.1004081 [DOI] [PubMed] [Google Scholar]
- Miossec, P. , & Kolls, J. K. (2012). Targeting IL‐17 and TH17 cells in chronic inflammation. Nature Reviews. Drug Discovery, 10, 763–776. [DOI] [PubMed] [Google Scholar]
- Mrak, R. E. , & Griffin, W. S. T. (2005). Glia and their cytokines in progression of neurodegeneration. Neurobiology of Aging, 26, 349–354. 10.1016/j.neurobiolaging.2004.05.010 [DOI] [PubMed] [Google Scholar]
- Newcombe, E. A. , Camats‐Perna, J. , Silva, M. L. , Valmas, N. , Huat, T. J. , & Medeiros, R. (2018). Inflammation: The link between comorbidities, genetics, and Alzheimer's disease. Journal of Neuroinflammation, 1, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires, A. , Fortuna, A. , Alves, G. , & Falcão, A. (2009). Intranasal drug delivery: How, why and what for? Journal of Pharmacy & Pharmaceutical Sciences, 12, 288–311. 10.18433/J3NC79 [DOI] [PubMed] [Google Scholar]
- Reynolds, W. F. , Rhees, J. , Maciejewski, D. , Paladino, T. , Sieburg, H. , Maki, R. A. , & Masliah, E. (1999). Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer's disease. Experimental Neurology, 155, 31–41. 10.1006/exnr.1998.6977 [DOI] [PubMed] [Google Scholar]
- Rostami, A. , & Ciric, B. (2013). Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. Journal of the Neurological Sciences, 333, 76–87. 10.1016/j.jns.2013.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostasy, K. , Egles, C. , Chauhan, A. , Kneissl, M. , Bahrani, P. , Yiannoutsos, C. , … Navia, B. A. (2003). SDF‐1α is expressed in astrocytes and neurons in the AIDS dementia complex: An in vivo and in vitro study. Journal of Neuropathology and Experimental Neurology, 62, 617–626. 10.1093/jnen/62.6.617 [DOI] [PubMed] [Google Scholar]
- Russo, R. , Cattaneo, F. , Lippiello, P. , Cristiano, C. , Zurlo, F. , Castaldo, M. , … Miniaci, M. C. (2018). Motor coordination and synaptic plasticity deficits are associated with increased cerebellar activity of NADPH oxidase, CAMKII, and PKC at preplaque stage in the TgCRND8 mouse model of Alzheimer's disease. Neurobiology of Aging, 68, 123–133. 10.1016/j.neurobiolaging.2018.02.025 [DOI] [PubMed] [Google Scholar]
- Sanchez, A. B. , Medders, K. E. , Maung, R. , Sánchez‐Pavón, R. P. , Ojeda‐Juárez, D. , & Kaul, M. (2016). CXCL12 induced neurotoxicity critically depends on NMDA receptor‐gated and L‐type Ca2+ channels upstream of p38 MAPK. Journal of Neuroinflammation, 13, 252 10.1186/s12974-016-0724-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saresella, M. , Calabrese, E. , Marventano, I. , Piancone, F. , Gatti, A. , Alberoni, M. , … Clerici, M. (2011). Increased activity of Th‐17 and Th‐9 lymphocytes and a skewing of the post‐thymic differentiation pathway are seen in Alzheimer's disease. Brain, Behavior, and Immunity, 25, 539–547. 10.1016/j.bbi.2010.12.004 [DOI] [PubMed] [Google Scholar]
- Schwartz, M. , & Deczkowska, A. (2016). Neurological disease as a failure of brain‐immune crosstalk: The multiple faces of neuroinflammation. Trends in Immunology, 37, 668–679. 10.1016/j.it.2016.08.001 [DOI] [PubMed] [Google Scholar]
- Shen, W. , Hu, X. M. , Liu, Y. N. , Han, Y. , Chen, L. P. , Wang, C. C. , & Song, C. (2014). CXCL12 in astrocytes contributes to bone cancer pain through CXCR4‐mediated neuronal sensitization and glial activation in rat spinal cord. Journal of Neuroinflammation, 11, 75 10.1186/1742-2094-11-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solleiro‐Villavicencio, H. , & Rivas‐Arancibia, S. (2017). Systemic Th17/IL‐17A response appears prior to hippocampal neurodegeneration in rats exposed to low doses of ozone. Neurologia, pii: S0213‐4853(17), 30194–30199. [DOI] [PubMed] [Google Scholar]
- Southam, D. S. , Dolovich, M. , O'Byrne, P. M. , & Inman, M. D. (2002). Distribution of intranasal instillations in mice: Effects of volume, time, body position, and anesthesia. American Journal of Physiology. Lung Cellular and Molecular Physiology, 282, L833–L839. 10.1152/ajplung.00173.2001 [DOI] [PubMed] [Google Scholar]
- Su, F. , Bai, F. , & Zhang, Z. (2016). Inflammatory cytokines and Alzheimer's disease: A review from the perspective of genetic polymorphisms. Neuroscience Bulletin, 32, 469–480. 10.1007/s12264-016-0055-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo, P. A. , Goswami, A. , Mazzuca, D. M. , Kim, K. , O'Gorman, D. B. , Hess, D. A. , … Haeryfar, S. M. (2017). Rapid and rigorous IL‐17A production by a distinct subpopulation of effector memory T lymphocytes constitutes a novel mechanism of toxic shock syndrome immunopathology. Journal of Immunology, 198, 2805–2818. 10.4049/jimmunol.1601366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmasebinia, F. , & Pourgholaminejad, A. (2017). The role of Th17 cells in auto‐inflammatory neurological disorders. Progress in Neuro‐Psychopharmacology & Biological Psychiatry, 79, 408–416. 10.1016/j.pnpbp.2017.07.023 [DOI] [PubMed] [Google Scholar]
- Tansey, M. G. , McCoy, M. K. , & Frank‐Cannon, T. C. (2007). Neuroinflammatory mechanisms in Parkinson's disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Experimental Neurology, 208, 1–25. 10.1016/j.expneurol.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J. P. , Hardy, J. , & Fischbeck, K. H. (2002). Toxic proteins in neurodegenerative disease. Science, 296, 1991–1995. 10.1126/science.1067122 [DOI] [PubMed] [Google Scholar]
- Togo, T. , Akiyama, H. , Iseki, E. , Kondo, H. , Ikeda, K. , Kato, M. , … Kosaka, K. (2002). Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. Journal of Neuroimmunology, 124, 83–92. 10.1016/S0165-5728(01)00496-9 [DOI] [PubMed] [Google Scholar]
- Turowski, P. , Adamson, P. , & Greenwood, J. (2005). Pharmacological targeting of ICAM‐1 signaling in brain endothelial cells: Potential for treating neuroinflammation. Cellular and Molecular Neurobiology, 1, 153–170. [DOI] [PubMed] [Google Scholar]
- Van Eldik, L. J. , & Wainwright, M. S. (2003). The Janus face of glial‐derived S100B: Beneficial and detrimental functions in the brain. Restorative Neurology and Neuroscience, 3‐4, 97–108. [PubMed] [Google Scholar]
- Walker, D. G. , Lue, L. F. , Tang, T. M. , Adler, C. H. , Caviness, J. N. , Sabbagh, M. N. , … Beach, T. G. (2017). Changes in CD200 and intercellular adhesion molecule‐1 (ICAM‐1) levels in brains of Lewy body disorder cases are associated with amounts of Alzheimer's pathology not α‐synuclein pathology. Neurobiology of Aging, 54, 175–186. 10.1016/j.neurobiolaging.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. D. , Zhao, Y. F. , Wang, G. Y. , Sun, B. , Kong, Q. F. , Zhao, K. , … Li, H. L. (2009). IL‐17 potentiates neuronal injury induced by oxygen‐glucose deprivation and affects neuronal IL‐17 receptor expression. Journal of Neuroimmunology, 212, 17–25. 10.1016/j.jneuroim.2009.04.007 [DOI] [PubMed] [Google Scholar]
- Yin, Y. , Wen, S. , Li, G. , & Wang, D. (2009). Hypoxia enhances stimulating effect of amyloid β peptide (25‐35) for interleukin 17 and T helper lymphocyte subtype 17 upregulation in cultured peripheral blood mononuclear cells. Microbiology and Immunology, 53, 281–286. 10.1111/j.1348-0421.2009.00120.x [DOI] [PubMed] [Google Scholar]
- Xia, M. Q. , & Hyman, B. T. (1999). Chemokines/chemokine receptors in the central nervous system and Alzheimer's disease. Journal of Neurovirology, 5, 32–41. 10.3109/13550289909029743 [DOI] [PubMed] [Google Scholar]
- Zenaro, E. , Pietronigro, E. , Della Bianca, V. , Piacentino, G. , Marongiu, L. , Budui, S. , … Constantin, G. (2015). Neutrophils promote Alzheimer's disease‐like pathology and cognitive decline via LFA‐1 integrin. Nature Medicine, 8, 880–886. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Ke, K. F. , Liu, Z. , Qiu, Y. H. , & Peng, Y. P. (2013). Th17 cell‐mediated neuroinflammation is involved in neurodegeneration of aβ1‐42‐induced Alzheimer's disease model rats. PLoS One, 8, e75786 10.1371/journal.pone.0075786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Liu, M. , Sun, H. , & Yin, K. (2015). Matrine improves cognitive impairment and modulates the balance of Th17/Treg cytokines in a rat model of Aβ1‐42‐induced Alzheimer's disease. Cent Eur J Immunol, 40, 411–419. 10.5114/ceji.2015.56961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuroff, L. , Daley, D. , Black, K. L. , & Koronyo‐Hamaoui, M. (2017). Clearance of cerebral Aβ in Alzheimer's disease: Reassessing the role of microglia and monocytes. Cellular and Molecular Life Sciences, 74, 2167–2201. 10.1007/s00018-017-2463-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
(A) Representative Western blot of Aβ1–42 performed in whole brain lysate at 7‐ and 14‐days post ICV Aβ‐peptide (fibrillated and not fibrillated) administration and related (B) cumulative densiometric analyses of Aβ1–42 protein. Data were expressed as mean ± SEM. Statistical analysis was performed by using one‐way ANOVA followed by Dunnett's post‐test. # P ≤ 0.05 vs 14 days not fibrillated condition, *P ≤ 0.05 vs 7 days not fibrillated condition (N = 5 per group). In C and D are reported the original Western blots related to Aβ1–42 and actin respectively.
Figure S2.
Representative immunofluorescence staining for GFAP in the hippocampus of (A) Control, (B) IL‐17Ab ICV, (C) IL‐17Ab IN, (D) Aβ1–42, (E) Aβ1–42 + IL‐17Ab ICV and (F) IL‐17Ab IN‐treated mice. Original pictures were photographed at 5 X magnification.
Figure S3. Representative immunofluorescence staining for Iba‐1 in the hippocampus of (A) Control, (B) IL‐17Ab ICV, (C) IL‐17Ab IN, (D) Aβ1–42, (E) Aβ1–42 + IL‐17Ab ICV and (F) IL‐17Ab IN‐treated mice. Original pictures were photographed at 5 X magnification.