

Abstract
Dysfunction of cell bioenergetics is a common feature of neurodegenerative diseases, the most common of which is Alzheimer's disease (AD). Disrupted energy utilization implicates mitochondria at its nexus. This review summarizes some of the evidence that points to faulty mitochondrial function in AD and highlights past and current therapeutic development efforts. Classical neuropathological hallmarks of disease (β‐amyloid and τ) and sporadic AD risk genes (APOE) may trigger mitochondrial disturbance, yet mitochondrial dysfunction may incite pathology. Preclinical and clinical efforts have overwhelmingly centred on the amyloid pathway, but clinical trials have yet to reveal clear‐cut benefits. AD therapies aimed at mitochondrial dysfunction are few and concentrate on reversing oxidative stress and cell death pathways. Novel research efforts aimed at boosting mitochondrial and bioenergetic function offer an alternative treatment strategy. Enhancing cell bioenergetics in preclinical models may yield widespread favourable effects that could benefit persons with AD.
Linked Articles
This article is part of a themed section on Therapeutics for Dementia and Alzheimer's Disease: New Directions for Precision Medicine. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.18/issuetoc
Abbreviations
- Aβ
β‐amyloid
- AD
Alzheimer's disease
- APOE
apolipoprotein E
- APP
amyloid precursor protein
- BDNF
brain derived neurotrophic factor
- BHB
β‐hydroxybutyrate
- COX
cytochrome c oxidase
- ETC
electron transport chain
- FDG
fluorodeoxyglucose
- LCR
low‐capacity runner
- mitoQ
mitoquinone mesylate
- mtDNA
mitochondrial DNA
- mPTP
mitochondrial permeability transition pore
- NR
Nicotinamide riboside
- OOA
oxaloacetate
- PD
Parkinson's disease
1. MITOCHONDRIAL AND CELL BIOENERGETIC DYSFUNCTION IN ALZHEIMER'S DISEASE
Mitochondria are maternally inherited, intracellular organelles with key roles covering energy metabolism and second messenger signalling to programmed cell death. Oxygen‐dependent ATP synthesis is mediated by enzyme complexes that make up the mitochondrial electron transport chain (ETC) in a process called oxidative phosphorylation. Mitochondriopathies are diseases classically recognized for mitochondria dysfunction as the inciting insult. These may be genetically inherited (mitochondrial proteins encoded by mitochondrial DNA [mtDNA] or nuclear DNA) or acquired (toxins or other secondary insults). Mitochondriopathies are typically heterogeneous in their presentation, with multiorgan involvement in common. Characterization of the various mitochondriopathies in the past identified skeletal muscle and brain to be the primary target organs, with variable effects on liver, heart, kidneys, and endocrine glands (Scholte, 1988). Although genetically determined mitochondriopathies are best recognized as disorders of childhood and managed by paediatric specialists, mitochondrial impairment is increasingly recognized in adult‐onset neurodegenerative disorders, including Alzheimer's disease (AD), Parkinson's disease (PD), and progressive supranuclear palsy (Parker, Filley, & Parks, 1990; Swerdlow, 2009; Swerdlow et al., 2000, 1996). Neuronal susceptibility to mitochondrial dysfunction might be explained by the fact that neurons are highly dependent on oxidative phosphorylation (Cardoso, Carvalho, Correia, Seiça, & Moreira, 2016).
AD is a progressive neurodegenerative disorder characterized by memory deficits and cognitive impairment. There is no cure. The classical neuropathological hallmarks of AD are abnormal aggregation of amyloid plaques and neurofibrillary tangles, containing β‐amyloid (Aβ) and hyperphosphorylated tau (τ) respectively. Definitive AD diagnosis is classically made post mortem, once neuropathology has confirmed the characteristic plaque and tangle deposition in an individual with clinical diagnostic criteria during life. As such, studies on AD‐related pathogenesis and therapeutics have focused largely on amyloid precursor protein (APP) metabolism, Aβ toxicity, and τ phosphorylation. Autosomal dominant mutations in APP processing genes lead to the uncommon, early‐onset, Mendelian AD cases. These mutations can be located in the APP gene or presenilin genes (a γ secretase subunit necessary for APP processing) and have been used extensively in transgenic mice to induce brain amyloidosis and to model that aspect of AD pathology. Interest in better understanding other pathological features has been slowly gaining traction, including inflammation pathways, insulin resistance, mitochondrial dysfunction, and oxidative stress (Toledo et al., 2018). Evidence‐based disease staging generated by the Alzheimer's Disease Neuroimaging Initiative and other longitudinal studies supports the notion that AD is not a linear downstream consequence of Aβ deposition alone but rather a multifactorial disease (Iturria‐Medina, Carbonell, Sotero, Chouinard‐Decorte, & Evans, 2017; Veitch et al., 2018). Some argue that mitochondrial dysfunction is a primary insult driving the more prevalent, sporadic late‐onset AD pathophysiology and refer to this as a “mitochondrial cascade hypothesis” (Swerdlow et al., 2000; Swerdlow, Burns, & Khan, 2014; Swerdlow & Khan, 2004, 2009).
2. EVIDENCE FOR MITOCHONDRIAL DYSFUNCTION IN AD
Some of the first clues hinting at mitochondria dysfunction in AD developed from observations of regional hypometabolism in AD subjects on brain imaging. Altered brain glucose metabolism in AD subjects is detectable in living subjects using fluorodeoxyglucose PET (FDG‐PET). Low glucose consumption (FDG uptake on imaging) is thought to be due to reduced glycolysis, synaptic dysfunction, and neuron loss (Foster et al., 1983; Friedland et al., 1983; Fukuyama et al., 1994; Mosconi et al., 2010; Silverman et al., 2001; Veitch et al., 2018). AD brains seem to compensate for the glucose hypometabolism by shifting to amino acids and fatty acids as alternative energy sources (Toledo et al., 2018). These observations have undoubtedly implicated alterations in energy metabolic pathways in AD. Although the primary insult in AD is still a subject of debate, the observed brain metabolic changes are thought to reflect or include impaired mitochondrial function.
Increased oxidative stress and ROS damage in AD brains provide other clues implicating mitochondria in AD pathogenesis, as mitochondria are the major generators of ROS (Bonda et al., 2014; Wang et al., 2014). ROS generated by a faulty ETC may result in damage to DNA, lipids, and proteins. Anatomical regional susceptibility to oxidative damage seems to correlate with brain regions most affected in the disease, namely, the frontal, parietal, and temporal lobes (Wang, Markesbery, & Lovell, 2006). That mtDNA exhibits higher levels of oxidized nucleotide bases compared to nuclear DNA suggests differential ROS‐induced damage (Wang et al., 2006). Finally, cell lines containing mtDNA from AD subjects demonstrate increased ROS and free radical production, compared with controls (Swerdlow et al., 1997).
Decreased regional blood flow and oxygen utilization in AD brains provide perhaps more direct evidence implicating impaired mitochondrial respiration in disease (Frackowiak et al., 1981; Fukuyama et al., 1994; Sims, Finegan, Blass, Bowen, & Neary, 1987). While, on the one hand, AD brains exhibit reductions in numbers of intact mitochondria (Hirai et al., 2001; Wang et al., 2009), impaired mitochondrial function is attributed to loss or dysfunction of specific ETC enzymes. These enzymes include (a) cytochrome c oxidase (COX; i.e., complex IV; Maurer, Zierz, & Möller, 2000; Mutisya, Bowling, & Beal, 1994; Parker et al., 1990), (b) pyruvate dehydrogenase complex (Gibson, Starkov, Blass, Rata, & Beal, 2010), and (c) the Krebs cycle α‐ketoglutarate dehydrogenase complex (Gibson et al., 2010).
One mechanism by which mitochondrial enzyme expression can be compromised is through the depletion of the mtDNA that encodes subunits of these enzymes. For example, studies using AD cybrid models exhibit reduced COX activity. Because cybrids are generated by fusing anuclear cells (platelets) derived from AD patients with cell lines depleted of endogenous mtDNA, they directly implicate mtDNA changes in the enzyme deficits observed (Swerdlow et al., 1997). Work by others has shown that mtDNA from AD patients host more lesions than control subjects (Krishnan, Ratnaike, De Gruyter, Jaros, & Turnbull, 2012; Wang et al., 2006). These mtDNA lesions could either represent maternally inherited mutations or mutagenesis acquired in life, secondary to nucleotide damage accumulated during aging (Wallace, 2005). Samples from asymptomatic adult children whose mothers carry an AD diagnosis exhibit reduced COX activities, hinting at a heritable component (Mosconi et al., 2010). Nevertheless, aging‐related cellular mechanisms are also likely to be involved. Studies investigating a role for DNA repair enzyme activity in AD brains have pointed to deficits in this process (Canugovi, Shamanna, Croteau, & Bohr, 2014; Lovell, Xie, & Markesbery, 2000; Weissman et al., 2007). From a mechanistic standpoint, there is evidence to link impaired base excision repair capacity in AD brains with lower mtDNA copy number (Soltys et al., 2019). Others speculate that oxidative stress (and not DNA repair capacity) is the main cause of aging‐dependent mtDNA damage (Wang et al., 2014).
3. BRAIN METABOLIC/MITOCHONDRIAL DYSFUNCTION DURING NORMAL AGING
Age is the strongest risk factor for developing AD. This underscores the need to factor in aging‐related biological changes in the study of AD. Mitochondrial dysfunction may play a role in the increased susceptibility to AD that accompanies aging. The aging brain features decreased energy utilization, including decreased glucose utilization (Chételat et al., 2013; De Santi et al., 1995; Kalpouzos et al., 2009; Marano et al., 2013; Willis et al., 2002) and decreased respiratory capacity (Navarro & Boveris, 2007). On closer examination, mitochondrial ETC Complex I and IV functions decline with age (Navarro & Boveris, 2007). In rodents, the OXYS rat model of accelerated aging exhibits brain mitochondrial disruption. These alterations comprise mitochondrial gene expression changes, age‐dependent ultrastructural differences, and decreased activity of several ETC enzymes (Complexes I, IV, and V; Stefanova et al., 2018). A key pathogenic finding is that mitochondrial dysfunction precedes Aβ aggregation in the OXYS rat model (Stefanova et al., 2018; Tyumentsev et al., 2018). A more direct link between mitochondrial function and aging was demonstrated in a series of experiments, whereby dampening mitochondrial ROS production prolonged lifespan in mice (Schriner et al., 2003).
4. CLASSICAL AD TARGETS AND THEIR LINK TO MITOCHONDRIAL DYSFUNCTION
For decades, Aβ has been the primary target for disease‐modifying AD therapies, but to date, Aβ‐focused approaches have produced disappointing results in clinical trials. Meanwhile, efforts to understand the multifactorial processes driving AD pathology have shed light on interactions between mitochondrial dysfunction and more “classical” triggers of AD (i.e., Aβ, τ, or apolipoprotein E [APOE]). While there are data to support a model whereby these protein species incite mitochondrial insult, there is also evidence to support a process through which dysfunctional mitochondria may trigger abnormal processing of amyloid and τ proteins. That APP, Aβ, and τ directly interact with mitochondria hints at possible mechanisms by which dysfunction of one might affect the other. This review summarizes these data and argues that irrespective of the primary pathogenic insult, targeting mitochondria and cell bioenergetics for the treatment of AD is reasonable.
Amyloid‐based transgenic mouse models of AD display age‐related mitochondrial impairments (Fu, Xiong, Lovell, & Lynn, 2009; Hauptmann et al., 2009). The most studied mechanism by which APP/Aβ might affect mitochondrial function is by direct interaction and inhibiting oxidative phosphorylation. In AD human brains, APP (full length or C‐terminal fragment) and Aβ are associated with the mitochondrial membrane in brain regions affected in disease (Caspersen et al., 2005; Devi, Prabhu, Galanti, Avadhani, & Anandatheerthavarada, 2006). Similar observations have been described in APP transgenic mouse models, whereby APP appears trapped in the mitochondrial membrane (Caspersen et al., 2005). This is thought to affect mitochondrial function via APP/Aβ accumulation in mitochondrial import channels, which in turn block entry and function of ETC enzymes and incite abnormal accumulation of toxic hydrogen peroxide species (Caspersen et al., 2005). APP overexpression in a human‐derived cortical cell line or in mouse transgenic brains leads to reductions in ATP levels, impaired COX activity, and disrupted mitochondrial transmembrane potential (Anandatheerthavarada, Biswas, Robin, & Avadhani, 2003). Specific mitochondrial‐binding partners for Aβ have been described and are thought to influence its role in pathology. Enhanced interaction between Aβ and the mitochondrial enzyme Aβ‐binding alcohol dehydrogenase in the mitochondria exacerbates Aβ toxicity, free radical generation, and learning deficits (Lustbader et al., 2004). At the outer mitochondrial membrane, Aβ interferes with the mitochondrial permeability transition pore (mPTP), a critical step in apoptosis (Du et al., 2008).
While these examples emphasize APP/Aβ‐triggered mitochondrial insult, mitochondrial dysfunction itself may promote aberrant Aβ production. Chemical inhibition of mitochondrial ETC function increases proteolytic APP processing (Gabuzda, Busciglio, Chen, Matsudaira, & Yankner, 1994). Moreover, AD‐derived cybrids (which contain mtDNA from human AD subjects) display increased intracellular and secreted Aβ, along with increased oxidative stress and apoptotic markers (Khan et al., 2000). In efforts to better understand how age‐related mtDNA mutations contribute to AD pathology in vivo, some groups have employed genetic approaches to augment mtDNA lesions during the lifespan of the mouse. The results have unveiled a relationship between Aβ processing and mtDNA defects.
To investigate a link between mtDNA damage and AD, one group induced double strand breaks in mtDNA using a modified bacterial endonuclease PstI directed to the mitochondria of CaMK2α‐positive neurons in mouse brains (Mito‐PstI). This approach results in mtDNA deletions, reduced levels of mtDNA‐encoded COXI, and reduced COX activity (Fukui & Moraes, 2009; Pinto, Pickrell, Fukui, & Moraes, 2013). When crossed to APPswe;PSEN1(A246E) mice, mitochondrial lesions provoked by Mito‐PstI yielded reductions in Aβ plaques and Aβ42 levels. Unexpectedly, mitochondrial lesions in APPswe;PSEN1(A246E);Mito‐PstI mice did not exacerbate oxidative stress. The authors proposed a model in which Aβ production is necessary to generate toxic ROS species. Whatever the case, the data also suggest that mitochondria contribute to Aβ processing and its abnormal aggregation. These observations are certainly confounded by the fact that Mito‐PstI expression was suppressed until age 6 months, when Aβ processing changes had already begun (Pinto et al., 2013). Whether the Mito‐PstI model mimicked the mitochondrial dysfunction profile seen in AD is unclear (Maurer et al., 2000; Swerdlow, 2011).
An alternative approach to experimentally disrupt mtDNA was achieved by inactivating native mtDNA repair mechanism in mice. The inactivating mutation (D257A) of the mtDNA polymerase PolgA accelerates the accumulation of mtDNA mutations with age, and this results in premature aging and bioenergetic deficits linked to mitochondria (Trifunovic et al., 2004). In contrast to the above studies, when PolgA D257A knockin mice were crossed to the APP (Ld) mouse model, Aβ42 levels and Aβ plaque burden were exacerbated (Lokesh, Kujoth, Prolla, Van Leuven, & Vassar, 2014). In this case, increased Aβ protein burden was attributed to impaired clearance mechanisms. Although oxidative stress in the brain was not examined in this study, others have shown that PolgA D257A mice exhibit increased markers of apoptosis systemically, without signs of oxidative stress (Kujoth et al., 2005). Albeit with disparate directional effects on Aβ, all these studies suggest that mtDNA lesions play an important role in APP processing, and this may be due to mtDNA‐encoded proteins, such as ETC enzymes, and possibly drive effects independently of oxidative damage.
Pathological forms of τ are similarly associated with AD neuropathology and mitochondrial dysfunction. Studies using platelet‐derived mitochondria from patients point to enzymic mitochondrial dysfunction in tauopathies that include AD and progressive supranuclear palsy (Swerdlow et al., 2000). Further, altered mitochondrial trafficking in AD brains correlates with abnormal τ aggregation. Neurons positive for neurofibrillary tangles (Alz50+) demonstrate more severe mitochondrial distribution defects than neurons without τ tangles (Kopeikina et al., 2011). This suggests either there is a threshold of mitochondrial dysfunction that determines tangle formation or that tangle formation exacerbates mitochondrial dysfunction. Data from transgenic mice engineered to accumulate mutant human τ do not support the first possibility but are at least consistent with the latter possibility (Kopeikina et al., 2011). Transgenic mice expressing the human τ MAPT(P301L) mutation exhibit oxidative stress, alterations in antioxidant enzymes, and deficits in mitochondrial ETC enzymes (Complexes I and V; David et al., 2005). Various τ species have been shown to incite mitochondrial insult. For example, either phosphorylated τ or truncated τ forms disrupt mitochondrial membrane potential, cause oxidative stress, and hinder the calcium buffering capacity of the cell (Pérez, Jara, & Quintanilla, 2018). Moreover, truncated τ provokes mitochondrial dysfunction in vitro (Quintanilla, Matthews‐Roberson, Dolan, & Johnson, 2009).
τ‐related mitochondrial dysfunction is likely to be caused by a toxic gain‐of‐function mechanism rather than loss‐of‐function. Recent work in mice shows that complete genetic ablation of endogenous τ (mapt−/−) displays neuroprotective effects and improves mitochondrial function. Indeed, mapt−/− mice exhibit reduced oxidative damage, decreased mPTP formation, enhanced markers of mitochondrial biogenesis, enhanced ATP production, and improved cognition (Jara, Aránguiz, Cerpa, Tapia‐Rojas, & Quintanilla, 2018). For a more comprehensive review on the relationship between τ and mitochondrial deficits, please refer to Eckert, Schulz, Rhein, and Götz (2010) and Eckert, Nisbet, Grimm, and Götz (2014).
The mouse models described above individually address either APP‐ or τ‐related features of disease and consequently either APP‐ or τ‐related mitochondrial disturbances. The 3xTg AD mouse model expresses three mutations linked to familial AD (APPswe, MAPT P301L, and PSEN1 M146V) that simultaneously address Aβ amyloid and τ pathological features. Some of the mitochondrial deficits described above are reproduced in the 3xTg triple‐transgenic model. Oxidative stress is marked, with more lipid peroxidation, elevated antioxidant enzyme activities, and augmented hydrogen peroxide production (Resende et al., 2008; Yao et al., 2009). The 3xTg mice also display diminished mitochondrial enzyme activities (COX and PDH) and decreased mitochondrial respiration (Yao et al., 2009). A remarkable finding in 3xTg females is that mitochondrial dysfunction occurs even at a young age and before detectable evidence of Aβ plaques or tangle pathology. In fact, embryonically derived hippocampal neurons displayed alterations in bioenergetic pathways, involving decreased mitochondrial respiration and increased glycolysis (Yao et al., 2009). That these mice were genetically modified to express three mutant genes cannot preclude the influence of soluble or undetectable APP or τ proteins on the bioenergetic changes described. Therefore, while these data cannot argue that dysfunctional mitochondria drive AD pathology, it suggests that mitochondrial dysfunction is a very early step in AD pathophysiology.
A major limitation of the APP/Aβ‐ or τ‐induced mitochondrial toxicity studies is that they experimentally overexpress mutant protein, unavoidably biasing them towards a model in which Aβ and/or τ is the primary insult. The low‐capacity runner (LCR) rat offers a less proteotoxic aging model, by comparison. The LCR and high‐capacity runner rats were selected over generations for their divergent intrinsic aerobic exercise capacity (VO2). LCR rats harbour genetic elements acquired through artificial selection that determine aging‐related metabolic impairments and, in this way, provide a model of disease with native, non‐transgenic, protein expression profiles. LCR rats display cellular and systemic metabolic dysfunction, in which mitochondrial impairments are considered early (if not primary) features of pathology. Hippocampi of LCR rats demonstrate age‐related atrophy and neurodegeneration, coupled to mitochondrial functional abnormalities and alterations in proteins that assist in mitochondrial biogenesis and oxidative phosphorylation (Choi et al., 2014). Presumably as a consequence of bioenergetic impairments, aged LCR rat hippocampi exhibit greater native phospho‐τ build up in the mitochondria when compared to high‐capacity runner rats. LCR rats are likely to model a pathway in which abnormal phospho‐τ accumulation is a consequence of mitochondrial dysfunction.
The APOE allele type is the main genetic risk factor for sporadic AD. Individuals with APOE ε4 carrier status have a higher risk for AD, while APOE ε2 carriers demonstrate decreased risk (Corder et al., 1993; Corder et al., 1994). Although this link is incompletely understood, one proposed mechanism is by APOE allele effects on Aβ levels. There is clear evidence that APOE influences Aβ protein burden, and there is promising preclinical evidence for APOE ε2 gene therapy to promote Aβ clearance in the APP Tg2576 model (Hudry et al., 2013). Whereas APOE ε4 exacerbates Aβ accumulation and synaptic toxicity, APOE carrier status might also influence susceptibility by its effects at the level of the mitochondria. Akin to a potential amyloid clearance mechanism, APOE ε4/3 carriers with more severe cognitive status display a higher degree of brain mitochondrial APP accumulation, compared with APOE ε4 non‐carriers or APOE ε4 carriers with less severe cognitive status (Devi et al., 2006). Additionally, mitochondrial APP burden is more widespread in APOE ε4/3 brains.
Beyond Aβ, evidence suggests that APOE status may determine mitochondrial function. Brains from young, non‐demented APOE ε4 individuals exhibit lower brain mitochondrial COX activity than controls (Perkins et al., 2016; Valla et al., 2010). In addition to alterations in mitochondrial ETC enzymes, expression of proteins involved in ketone and glucose metabolism is also affected (Valla et al., 2010). That APOE ε4 brains do not already harbour high levels of toxic Aβ or τ species at a young age suggests that (a) observed metabolic aberrations precede the predicted Aβ or τ dysregulation and that (b) metabolic abnormalities play a critical, upstream role in pathology. Impaired mitochondrial enzyme activities in APOE ε4 carriers are also evident after onset if cognitive impairment and clinical AD diagnosis have been established. A small clinical study from our group examined platelet mitochondrial enzyme activities in AD subjects. Having age, sex, and AD diagnosis in common, APOE ε4 carriers demonstrated lower mitochondrial enzyme activities than ε4 non‐carriers (Wilkins et al., 2017). In vitro, APOE ε4 neurotoxic protein fragments interact with mitochondria, cause mitochondrial dysfunction, and hinder cell survival (Chang et al., 2005).
5. IN SEARCH OF THERAPIES FOR AD
The standard of care for clinically diagnosed AD patients includes cholinesterase inhibitors (donepezil, rivastigmine, and galantamine) and memantine (Birks, 2006; Birks, Chong, & Grimley Evans, 2015; Birks & Harvey, 2018; Loy & Schneider, 2006; McShane, Areosa Sastre, & Minakaran, 2006). The former blocks degradation of ACh, the neurotransmitter of the cholinergic neurons affected in AD. As disease progresses, the cholinergic neurons are lost in the basal forebrain, and partly for that reason, the therapeutic potential is limited. Memantine is an NMDA receptor blocker, and this mechanism is thought to attenuate excess glutamate neurotransmitter activity. Both pharmacological approaches address cognitive/memory symptoms, but neither halts disease progression. Non‐pharmacological tools for AD include lifestyle modifications (i.e., diet and exercise), and these are being evaluated at the preclinical level and in clinical trials (Ströhle et al., 2015).
The amyloid cascade hypothesis has driven the development of AD therapeutics over the past several decades and continues to guide the development of disease‐modifying interventions (Cummings, Aisen, et al., 2016; Cummings, Morstorf, & Lee, 2016). Some of the disappointments in human trials targeting Aβ reflect an inability to reliably alter the course of the disease, and in some cases, interventions appear to have accelerated cognitive decline or produced toxic side effects. Based on these clinical trial disappointments, and based on the observation that neurofibrillary tangle deposition more closely tracks with the regional and symptomatic progression of disease, τ‐targeted therapies have received increasing attention in recent years (Cummings, Lee, Ritter, & Zhong, 2017, 2018).
As imaging tools and biomarkers help us to study the onset and progression of AD, systematic and comprehensive efforts to apply these tools offer new insights into disease processes. Indeed, Alzheimer's Disease Neuroimaging Initiative analyses increasingly consider AD as a multifactorial condition (Iturria‐Medina et al., 2017; Veitch et al., 2018). Accordingly, investigators predict that combinatorial therapeutic approaches for AD are more likely to yield positive results than Aβ‐directed therapies alone (Iturria‐Medina et al., 2017). The fact that bioenergetic and mitochondrial dysfunction contribute to AD justifies harnessing mitochondrial and bioenergetic medicine approaches to treat AD.
6. MITOCHONDRIAL AND BIOENERGETIC MEDICINE
Cell energy metabolism involves mitochondrial and non‐mitochondrial biochemical pathways that either depend or not on oxygen utilization. In brief, mitochondrial bioenergetic pathways consist of (a) the Krebs cycle, (b) fatty acid β‐oxidation (to generate reducing equivalents for oxidative phosphorylation or to make ketone bodies that can serve as direct energy sources), and (c) oxidative phosphorylation (oxygen‐dependent ATP generation in the mitochondrial ETC). Glycolysis anaerobically breaks down glucose molecules to make ATP. While glycolysis takes place in the cytosol, it generates products that in turn feed mitochondrial carbon fluxes.
Mitochondrial medicine seeks to compensate for or correct disease‐associated mitochondrial dysfunction (Luft, 1994). Examples of mitochondrial medicine agents typically include vitamins, cofactors, electron acceptors, and redox molecule precursors, some of which have been tested in AD clinical trials but with limited success. Cellular energy producing pathways extend beyond the mitochondrial compartments (Swerdlow, 2014). The term “bioenergetic medicine” was proposed recently to describe the strategic restoration of energy metabolism‐related fluxes in AD and other diseases which show impairments of those fluxes. For an in‐depth review, please refer to Swerdlow (2014). Interventions designed to improve bioenergetics in the brain represent important therapeutic strategies to delay, prevent, or treat age‐related neurodegenerative diseases, the most common of which is AD (Swerdlow, 2009, 2014, 2016).
7. TREATMENT STRATEGIES TARGETING MITOCHONDRIA IN AD: TRADITIONAL STRATEGIES
To date, treatment strategies targeting aspects of mitochondrial dysfunction have focused on forestalling mitochondrial processes linked to terminal cell fates, namely, oxidative stress and apoptosis. Brains from AD patients and AD mouse models exhibit oxidative stress early on. (Nunomura et al., 2001; Praticò, Uryu, Leight, Trojanoswki, & Lee, 2001; Rhein et al., 2009; Wang et al., 2006; Yao et al., 2009). Therefore, it is reasonable to hypothesize that antioxidant treatment should prove beneficial for AD. However, clinical trials testing antioxidants in AD patients have not yielded clear‐cut benefits and may have detrimental effects (Lloret et al., 2009; Miller et al., 2005). In the case of vitamin E, promising clinical trials initially demonstrated an attenuation in cognitive decline when compared with the placebo group (Sano et al., 1997), but other studies later argued that the findings might not generalize to larger groups (Lloret et al., 2009).
It is worth recognizing that while antioxidant supplementation has fallen short in clinical trials, preclinical studies continue to show promise in their therapeutic potential, especially when targeted directly to mitochondria and when therapy is started early. A compelling story highlighting the role of mitochondrial oxidative stress in AD‐related pathology comes from APP transgenic mice (Tg2576) crossed to mice that overexpress catalase in the mitochondria (i.e., MCAT). The enzyme catalase is normally localized to the peroxisome, where it catalyses the removal of hydrogen peroxide. The resulting AD‐MCAT mice demonstrated extended lifespan and decreased AD neuropathology markers (i.e., APP, oligomeric Aβ, Aβ42, and Aβ40) compared to appropriate controls (Mao et al., 2012). These results point specifically to mitochondrial oxidative stress as a contributing factor that favours formation of toxic APP derivatives. Observations such as these suggest targeting antioxidants specifically to the mitochondria might be a suitable approach for antioxidant‐based treatment success in the context of aging and AD.
Mitoquinone mesylate (mitoQ) is a mitochondrial‐targeted antioxidant. MitoQ prevented Aβ‐induced cell death and oxidative stress in mouse cortical neurons in vivo (McManus, Murphy, & Franklin, 2011). MitoQ treatment also favourably altered the development of AD‐like pathology in vivo. Starting at 2 months of age, 3xTg female mice were treated with mitoQ for 5 months, and, not surprisingly, the treatment prevented oxidative stress damage. Beyond antioxidant effects, mitoQ administration successfully reduced Aβ accumulation, astrogliosis, synaptic loss, brain caspase activation, and improved cognitive performance (McManus et al., 2011). These studies emphasize the role of mitochondrial oxidative stress in driving a variety of disease features. Additionally, these findings imply that early intervention might be a key to therapeutic effectiveness. At present, mitoQ is being tested in a small clinical trial (Table 1), albeit for its role in endothelial NO production, and whether it benefits cerebrovascular blood flow in AD subjects (NCT03514875).
Table 1.
Interventional clinical trials for “Alzheimer's disease” and “mitochondria”
| Agent name | Effect | Mitochondrial effect | clinicaltrias.gov ID | Phase | Subjects/enrolment | Study design | Study arms | Outcome measures (biomarkers, cognitive, imaging, other) | Completion date |
|---|---|---|---|---|---|---|---|---|---|
| MitoQ | Antioxidant | Mitochondria‐targeting antioxidant | NCT03514875 | Not applicable | AD, early onset AD. 12 subjects, 50–85 years old. Gender: all | Randomized. Crossover assignment, Masking: double (participant, investigator), Primary purpose: treatment | Arm 1: baseline and mitoQ intake, 2‐week washout period, placebo intake Arm 2: baseline and placebo intake, 2‐week washout period, mitoQ intake | Carotid artery blood flow, Oxidative stress (serum markers), Cerebrovascular oxygenation, Brain electrical activity, Endothelial function | May 2019 |
| R + pramipexole | Antioxidant | Free‐radical scavenger that accumulates into the brain and mitochondria | NCT01388478 | Phase 2 | Mild‐to‐moderate AD. 20 participants, 55 years and older. Gender: all | Single group assignment. Open label. Primary purpose: treatment | 50 mg BID for 4 weeks, then 100 mg BID for 4 weeks, then 150 mg BID for 16 weeks. Dose will only increase if the participant is not having side effects. Study period: 24 weeks (~6 months) | Reduction of oxidative stress, Cognitive performance, Changes in cerebral glucose metabolism, Adverse events | January 2014 |
| EGCg (Sunphenon EGCg) | Antioxidant Modulates cell survival Induces α‐secretase and endothelin‐converting enzymes Prevents aggregation of Aβ to toxic oligomers | Modulates mitochondrial function | NCT00951834 | Phase 2/3 | Early stage AD. 21 participants, age 60+ years. Gender: all | Randomized. Parallel assignment. Masking: quadruple (participant, care provider, investigator, outcomes assessor). Primary purpose: treatment | Arm 1: home donepezil, plus: Months 1–3: 200 mg EGCg/day (200–0‐0 mg), Months 4–6: 400 mg EGCg/day (200–0‐200 mg), Months 7–9: 600 mg EGCg/day (400–0‐200 mg), Months 10–18: 800 mg EGCg/day (400–0‐400 mg). Arm 2: home donepezil, plus Placebo. Study period: 18 months | ADAS‐Cog, MMSE, CIBIC‐plus, WHO‐QOL‐Bref, Trail Making Test, MVGT, Brain atrophy (brain MRI), Safety and tolerability, Time to hospitalization, Time to death related to AD | February 2015 |
| DL‐3‐n‐butylphthalide | Antioxidant. Improves microcirculation | Alleviates oxidative damage and mitochondria dysfunction | NCT02711683 | Mild‐to‐moderate AD. 120 participants, age 50–85 years. Gender: all | Observational model: cohort; time perspective: prospective | Group 1: DL‐3‐n‐butylphthalide in patients already receiving donepezil. Group 2: No NMP in patients already receiving donepezil, Study period: 48 weeks | ADAS‐Cog, CIBIC‐plus, ADCS‐ADL, NPI | January 2018 | |
| Resveratrol with glucose, and malate | Resveratrol: antioxidant found in red wine | Resveratrol: antioxidant; Glucose: precursor for oxidative metabolism; Malate: primer of the Krebs cycle. | NCT00678431 | Phase 3 | 27 participants, age 50–90 years. Gender: all | Randomized. Parallel assignment. Masking: quadruple (participant, care provider, investigator, outcomes assessor). Primary purpose: treatment | Arm 1: placebo. Arm 2: resveratrol with glucose, and malate. Study period: 1 year | ADAS‐Cog, CGIC | June 2011 |
| S‐equol | Nonhormonal oestrogen receptor; β agonist | Potentiates mitochondrial function in rat hippocampal neurons | NCT02142777 | Phase 1 | Very mild or mild AD. 15 participants, age 60 to 90 years. Gender: female only | Single group assignment. Masking: single (participant). Primary purpose: treatment | Placebo (2 weeks), then 10 mg S‐equol (2 weeks), then placebo (2 weeks) | Platelet mitochondria cytochrome oxidase (COX) activity, Safety of S‐equol | April 2016 |
| Dimebon (latrepirdine) | Antihistamine, Neuroprotective Antiapoptotic NMDA receptor antagonist | May benefit AD patients, possibly by affecting mPTP | NCT00675623 | Phase 3 | Mild‐to‐moderate AD. 598 participants, Age 50+ years. Gender: all | Randomized parallel assignment. Masking: quadruple (participant, care provider, investigator, outcomes assessor). Primary purpose: treatment | Arm A: 5‐mg Dimebon TID, Arm B: 20‐mg Dimebon TID, Arm C: placebo TID. Study period: 6 months | ADAS‐Cog, CIBIC‐plus, ADCS‐ADL, NPI | December 2009 |
Note. AD: Alzheimer's disease; ADAS‐Cog: Alzheimer's disease assessment scale‐cognitive subscale; CIBIC‐plus: Clinician's Interview‐Based Impression of Change Plus Caregiver Input; ADCS‐ADL: Alzheimer's Disease Cooperative Study‐Activities of Daily Living; NPI: Neuropsychiatric Inventory; MMSE: Mini Mental State Examination; WHO‐QOL‐Bref: World Health Organization Quality of Life (abbreviated form); MVGT: Munich verbal memory test; CGIC: Clinical Global Impression of Change; BID: twice a day; TID: three times a day; mPTP: mitochondrial permeability transition pore; EGCg: epigallocatechin gallate.
Cell death is the ultimate irreversible fate for degenerating neurons. Mitochondria play a critical role in programmed cell death, specifically in the formation of the mPTP. Recent efforts screened low MW inhibitors of mPTP for their potential to attenuate Aβ‐induced cell toxicity and mitochondrial dysfunction and identified compound candidates (Elkamhawy et al., 2018).
8. TREATMENT STRATEGIES TARGETING MITOCHONDRIA: AD DRUG DEVELOPMENT PIPELINE
The AD drug development pipeline continues to emphasize the amyloid cascade hypothesis, with Aβ as the main target. Recent reviews have evaluated AD therapies and identified inflammatory mediators and τ as emerging targets of interest (Cummings, Aisen, et al., 2016; Cummings, Morstorf, et al., 2016). This section will focus on current clinical trials with special attention to interventions targeting mitochondria or related pathways in the context of AD. To this end, we resorted to data available at the clinicaltrials.gov database (accessed October 15–20, 2018), using a systematic search method described below. We found 927 clinical trials that presumably affect the mitochondria for any disease or condition, of which 670 are interventional studies. Of these, 640 target adults 18 and older.
First, we were interested in identifying the clinical trials targeting mitochondria in AD relative to neurological diseases. As an initial approach, we conducted three separate searches for clinical trials for either “brain diseases,” “dementia,” or “Alzheimer's disease” as the condition or disease under investigation. For each of these, we further selected for search results with “mitochondria” in the other terms search bar. Because AD is an adult‐onset neurodegenerative disease, and mitochondrial diseases afflict the paediatric population in distinct clinical profiles, we narrowed down the results further to only include “adult (18–64)” and “older adult (65+)” in the age eligibility criteria. We allowed the results to include all possible status (recruiting, completed, etc.), to include “all” sexes, to include all Phases (1–4), to include with or without results, and to include all funder types.
We downloaded the tabulated information for each of the search results and used the clinical trial numbers (NCT) to generate area‐proportional Venn diagrams using BioVenn (Hulsen, de Vlieg, & Alkema, 2008). There are 132 clinical trials for “brain diseases” that involve mitochondria in adults (Figure 1a). Of these, 18 studies target “dementia” specifically. Eleven of the 18 dementia trials target AD, while five target Huntington's disease, one targets diabetes, and the last targets a spectrum of rare neurological diseases. When we narrowed down the studies to include only “interventional” study types, we found most of the clinical trials are interventional, and 10 involve mitochondria (Figure 1b).
Figure 1.
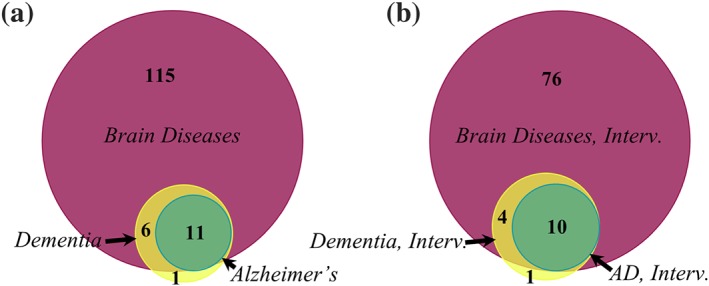
Clinical trials with the search term “Mitochondria” for neurological conditions classified as “brain diseases,” “dementia,” or “Alzheimer's disease” (AD). (a) Includes all results; (b) includes only the interventional trials
The clinical trials studying “AD and mitochondria” are tabulated in Table 1. The trials span various phases, with Phase 1 (n = 2), Phase 2 (n = 3), and Phase 3 (n = 4). Of the agents being tested, five are considered antioxidant therapies (R+pramipexole, Sunphenon epigallocatechin gallate (EGCg), DL‐3‐n‐butylphthalide (NBP), mitoQ, resveratrol), one is described as a mitochondrial enhancer molecule (S‐equol), and one is an antihistimine/antiapoptotic agent (Dimebon). Three of the clinical trials are ongoing. We examined the characteristics of these trials with emphasis on the drug target and what results were linked to these trials.
R+pramipexole is described as a free‐radical scavenger that accumulates within brain mitochondria and has neuroprotective effects. The Phase 2 clinical trial (NCT01388478) involved escalating the dose of R+pramipexole as tolerated for 24 weeks total in mild‐to‐moderate AD subjects and was completed in 2014. We found one report (PMID 26682692) with results related to this study (Bennett, Burns, Welch, & Bothwell, 2016). The authors reported mild to moderate adverse effects, with 4 out of 19 subjects withdrawing. There was 84.8–93.3% mean compliance. CSF protein biomarkers linked to AD (Aβ42, τ, and phospho‐τ) were examined and found no change 6 months after treatment when compared to baseline. FDG‐PET imaging revealed increased glucose uptake in cerebellum. We found no AD Phase 3 R+pramipexole studies.
The green tea extract Sunphenon EGCg was tested in Phase 2/3 clinical trial in 21 participants with early stage AD (MMSE 20–26, on donepezil; NCT00951834). Preclinical efforts with Sunphenon EGCg were promising for antioxidant, anti‐apoptotic, and anti‐Aβ accumulation effects and are reviewed in Cascella, Bimonte, Muzio, Schiavone, and Cuomo (2017). Additionally, Sunphenon EGCg is described as a modulator of cell survival and mitochondrial function. The study is reported as being completed in 2015. We did not find publications linked to this study.
NBP is a synthetic compound with antioxidant effects that also attenuates mitochondrial dysfunction. It is being evaluated in an observational clinical trial (NCT02711683) in 120 subjects with mild‐to‐moderate AD already receiving donepezil (to be compared to AD subjects on donepezil and not on NBP). NBP is approved in China for treatment of ischaemic stroke and is hypothesized to improve cognitive outcomes by its beneficial effects on the microcirculation. The data acquisition phase of the study is noted as completed in January 2018, but we found no related publications reported at this time.
S‐equol is a non‐hormonal agonist at the oestrogen receptor β, which potentiates mitochondrial function in rat hippocampal neurons (Yao et al., 2013). A Phase 1 study on S‐equol (NCT02142777) focused on safety and tolerability of the agent in 15 female subjects with very mild or mild AD and was completed in 2016. Treatment consisted of 2 weeks drug on, 2 weeks drug off, and finally, a 2‐week drug washout period. We found two articles linked to this study. The first (PMID28598847) reported mean compliance of 96.4% to S‐equol with no serious adverse events (Wilkins et al., 2017). Platelet mitochondrial enzyme activities were evaluated. By the COX biomarker, 11 of 15 subjects showed a positive drug response pattern, as defined by comparing the slope of the measurement change when going from no treatment to treatment to the slope when going from treatment to no treatment. A trend pointed to a more pronounced effect in APOE ε4 non‐carriers, although comparisons between the carrier and non‐carrier groups were not statistically different. The second report linked to this study (PMC5406545) analysed pretreatment samples alone to determine APOE ε4 carrier status effects on mitochondrial enzyme activities. The authors found that platelet‐derived mitochondrial COX and CS activity were lower in APOE ε4 carriers than non‐carriers at baseline. S‐equol is currently being evaluated in an active phase 2 clinical trial (NCT03101085). This study consists of APOE ε4 non‐carrier male and female AD subjects treated with S‐equol or placebo for 1 month. The study will again ascertain effects on platelet mitochondrial enzyme activity (COX and CS) and additionally examine changes in cognitive performance.
9. AD DRUG DEVELOPMENT PIPELINE: BIOENERGETICS BEYOND MITOCHONDRIA
As discussed above, energy metabolism pathways extend beyond the mitochondria. We therefore widened the scope of our search to better assess the number of AD studies that indirectly target mitochondria or other energy metabolism pathways. When searching for “bioenergetic medicine” alone, we found 20 clinical trials, but none focused on AD (not shown). Thus, we broadened the search using either “metabolism” or “energy metabolism” in other terms. There are 235 clinical trials studying metabolism and AD (Figure 2a). Surprisingly, of the 11 AD studies involving “mitochondria,” only two overlap with “metabolism” studies, and none overlap with “energy metabolism.” All but one of the “energy metabolism” trials are also categorized as “metabolism” trials. Energy metabolism studies in AD include physical exercise (n = 3), the metabolic intermediate oxaloacetate (OAA; n = 1), and a transcranial electromagnetic device (n = 1) as therapeutic interventions. Table 2 lists some characteristics of these trials. The metabolic intermediate OAA listed in this table is being tested by our group in a Phase 1b trial (NCT02593318) and will acquire data on several parameters, including safety, cognitive measures, brain glucose metabolic rate by FDG‐PET, changes in brain neurochemicals by magnetic resonance spectroscopy, and drug plasma levels.
Figure 2.
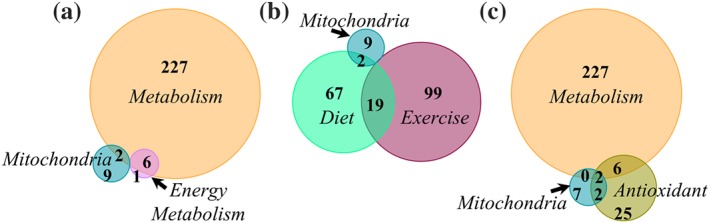
Clinical trials involving metabolism pathways or lifestyle modification interventions for Alzheimer's disease (AD). (a) AD “Metabolism” is the major focus in clinical trials targeting cell energy pathways and does not meaningfully overlap with AD “Mitochondria” trials. (b) Lifestyle interventions that may affect biological energy pathways in AD include diet and exercise; only two AD “mitochondria” trials overlap with AD “diet” trials. (c) Most clinical trials testing antioxidants for AD are registered in a category distinct from interventions involving the mitochondria or other metabolism pathways
Table 2.
Clinical trials for “Alzheimer's disease” and “energy metabolism”
| Intervention | clinicaltrias.gov ID | Phase | Enrolment | Study design | Outcome measures (biomarkers, cognitive, imaging, other) | Completion date |
|---|---|---|---|---|---|---|
| Physical exercise | NCT02708485 | Not applicable | 15 |
Randomized, parallel assignment. Open label Primary purpose: treatment |
Brain glucose consumption, brain acetoacetate consumption | July 2017 |
| 3‐month exercise intervention programme | NCT02253732 | Not applicable | 30 |
Single group assignment. Open label. Primary purpose: prevention |
muscle microRNAs and myokinescognitive function, motoric function, muscle functional test, swhole body energy metabolism | March 2018 |
|
Combination of aerobic and strength exercises Stretching and balance exercise programme |
NCT02520232 | Not Applicable | 360 |
Randomized, parallel assignment. Masking: single (outcomes assessor). Primary purpose: prevention |
BMI, heart variability at rest, BP, Executive functions, Depression score, Self‐efficacy score, Quality of life score, Score at Verbal working‐memory span, Logic memory score, Brain glucose metabolism, Grey and white matter volumes in regions of interest, Cerebral perfusion in the same regions of interest, Resting State Networks Activity; Senior Fitness Test score, Actimetry, Stage of change score related to physical activity, Reaction time in a two‐choice reaction time task, Error rate in a two‐choice reaction time task | September 2018 |
| Drug: oxaloacetate | NCT02593318 | Phase 1 | 30 |
Non‐randomized, parallel assignment. Open label. Primary purpose: treatment |
Correlation between oral oxaloacetate intake and plasma levels, Change in safety assessments (safety labs, physical and neurological exams, vital signs, cognitive measures, signs and symptoms), FDG‐PET, lactate by magnetic resonance spectroscopy | September 2018 |
| Ecological environmental therapy | NCT02462291 | Not applicable | 163 |
Randomized, parallel assignment Masking: triple (care provider, investigator, outcomes assessor) Primary purpose: treatment |
BMI, BP, serum markers (glucose, cholesterol), salivary cortisol, Evaluations of behavioural disorders, cognitive status, Daily energy expenditure, activity of daily life; number of medications, number of patients treated with each of quetiapine, citalopram, donepezil, memantine, ticlopidine | December 2015 |
| Transcranial electromagnetic device: MemorEM 1000 | NCT02958930 | Phase 1 | 12 |
Intervention, single group assignment. Open label. Primary purpose: treatment |
Blood and CSF markers (β‐amyloid, τ, markers of immune function, markers of oxidative stress), ADAS‐Cog, Adverse Event Assessment, ADCS‐ADL score, MMSE score, global deterioration score, Hachinski score, Rey AVLT score, Trails A & B score, Digit span score, Clock draw score, FDG‐PET, resting state fMRI, diffusion tensor imaging, susceptibility‐weighted imaging | August 2018 |
| Drug: 15 O water | NCT00001972 | 123 | August 2002 |
Note. BMI: body mass index; FDG‐PET: fluorodeoxyglucose PET; ADAS‐Cog: Alzheimer's disease assessment scale‐cognitive subscale; ADCS‐ADL: Alzheimer's Disease Cooperative Study‐Activities of Daily Living; MMSE: Mini Mental State Examination; fMRI: functional MRI.
To include lifestyle modification interventions that may biologically influence bioenergetics, we found 118 studies on AD and exercise (103 categorized as interventional) and 88 on AD and diet (77 categorized as interventional; Figure 2b). Nineteen AD studies overlap between “diet” and “exercise.” Only two AD “mitochondria” studies overlap with “diet” (mitoQ or resveratrol with glucose and malate). Because oxidative stress is thought to be at least in part provoked by mitochondrial dysfunction, we searched for “antioxidant” treatments for AD. Using the search definitions “Alzheimer's disease” and “antioxidant” yielded 35 clinical trials; four of these (Sunphenon EGCg, mitoQ, resveratrol with glucose and malate, and R+pramipexole) are also categorized as AD “mitochondria” studies (Figure 2c). The trials testing resveratrol with glucose and malate or R+pramipexole are the two in common for all three search terms (AD & Metabolism, AD & Mitochondria, and AD & Antioxidant).
Finally, we asked how mitochondria studies for AD compared to clinical trials focused on amyloid, τ, or APOE. We searched for “Alzheimer's disease” in disease or condition and either “amyloid,” “tau,” or “APOE” in other terms. Consistent with Cummings et al. (2018), we found the majority of AD clinical trials focus on amyloid (436 trials), τ second (204 trials), and APOE third (81 trials; Figure 3a). Twenty studies include all three search terms. For unclear reasons, narrowing the search term to “interventional” studies augmented the search results of clinical trials involving AD and amyloid (n = 363), AD and τ (n = 243), or AD and APOE (n = 123).
Figure 3.
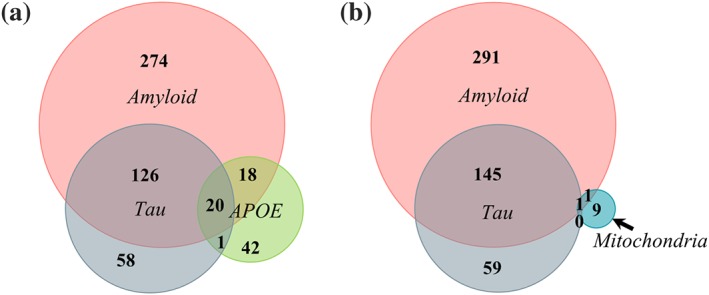
Amyloid is the major focus in clinical trials for Alzheimer's disease (AD). (a) Relationship between clinical trials for AD with search terms “amyloid,” “tau,” or “APOE.” (b) Trials investigating mitochondria in AD relative to trials investigating amyloid or tau
AD studies on mitochondria scarcely overlap with those involving amyloid (n = 2), τ (n = 1), or APOE (n = 0; not shown; Figure 3b). This is surprising considering evidence suggests an interplay between mitochondrial dysfunction and amyloid, τ, and APOE. One trial (NCT02062099) overlaps with “amyloid,” “tau,” and “mitochondria” search terms for AD. This is a pilot study using radioligands in PET imaging to correlate neuroinflammation ([18F]‐DPA‐714) and amyloid load ([18F]‐AV‐45) in subjects with memory complaint, mild cognitive impairment, or AD. The link to mitochondria is that the radioligand [18F]‐DPA‐714 binds receptors in the outer mitochondrial membrane that are up‐regulated with neuroinflammation. Because this is a diagnostic study evaluating radioligands as biomarkers of disease (not testing therapeutic agents), we did not include it in Table 1. We did not find published results from this study. Finally, NCT00951834 is linked to both “amyloid” and “mitochondria” in AD studies. This study was discussed above and tests the green tea extract Sunphenon EGCg in AD participants and is listed in Table 1.
10. TREATMENT STRATEGIES TARGETING MITOCHONDRIA: NOVEL APPROACHES IN PRECLINICAL STUDIES
As discussed above, mitochondria‐based treatment strategies have largely centred on oxidative stress and apoptosis pathways. There is evidence that enhancing mitochondrial function itself may ameliorate AD pathology (Mattson & Arumugam, 2018). Novel approaches seek to bolster mitochondrial function using intermediates of cell bioenergetic pathways for their therapeutic potential as they seem to show benefits in AD models (Murray et al., 2016).
Nicotinamide riboside (NR), an NAD+ precursor, extends lifespan in mice (Zhang et al., 2016). A 3xTg model with polymerase b haploinsufficiency (3xTg;polb+/−) was designed to render 3xTg mice unable to repair DNA defects (Hou et al., 2018). When both 3xTg and 3xTg;polb+/− mice were treated with NR for several months, the results pointed to favourable reductions in DNA damage, neuroinflammation, and apoptosis of hippocampal neurons. NR supplementation was biologically meaningful in hampering adverse AD features. AD‐related neuropathology indicated a preference for biochemical improvement in phospho‐τ protein burden, as there was no reversal in Aβ accumulation or Aβ‐related neuropathology. This is consistent with previous reports (Green et al., 2008). NR treatment failed to modulate Aβ production or viability in a cell line overexpressing mutant APPswe (Hou et al., 2018). Mechanistically, these findings seem to uncouple Aβ and mitochondrial dysfunction or at least suggest that targeting cell bioenergetics by enhancing NAD+/NADH preferentially improves aspects of τ pathology. However, NR treatment does seem to lower Aβ production in the Tg2576 model in vivo (which overexpresses the APPswe mutation alone). The discrepancy in Aβ biology might be attributable to the existing protein burden at treatment onset. The 3xTg and 3xTg;polb+/− mice received NR after 16 months of age, while the Tg2576 mice initiated treatment much earlier at 7 months of age. The notion of overdue treatment onset is supported by the observation that if NR administration is delayed in much older 3xTg mice (20 months), it no longer lowers phospho‐τ species (Green et al., 2008). Regardless of the differences on Aβ effects, NR administration in these studies invariably improved memory/cognitive function tests in vivo and improved synaptic function in brain slices. Together, these findings suggest that enhancing cell bioenergetics can improve brain function and may alter AD‐related protein biology but also underscore the importance of timing of treatment in relation to the onset of Aβ or τ pathologies. We did not find any ongoing AD clinical trials using “nicotinamide riboside” and “Alzheimer's Disease” as clinicaltrials.gov search terms.
OAA is an intermediate in several biochemical reactions and metabolic pathways, including the Krebs cycle and gluconeogenesis. In AD models, Aβ‐mediated inhibition of synaptic activity in the hippocampus is alleviated by OAA treatment (Zhang, Mably, Walsh, & Rowan, 2017). Moreover, OAA administration in pre‐symptomatic amyotrophic lateral sclerosis transgenic (SOD1 G93A mutant) mice preserved strength (Nishimune et al., 2017). Our group observed that i.p. OAA administration for 2 weeks in young wild type mice was sufficient to boost markers of mitochondrial biogenesis and promote expression of mtDNA‐encoded genes. Further, OAA treatment achieved broader beneficial effects in the brain, including insulin pathway activation, neurogenesis, and reduced neuroinflammatory markers (Wilkins et al., 2014). When tested on SH‐SY5Y neuroblastoma cells, OAA enhanced glycolysis and cell respiration, and this correlated with an increased NAD+/NADH ratio (Wilkins et al., 2016). As mentioned above, OAA is being tested in a Phase 1b clinical trial in AD subjects (NCT02593318; Table 2).
Both aging and AD are associated with decreased brain glucose utilization, while ketone utilization is maintained (Cunnane et al., 2016; Mattson & Arumugam, 2018). Efforts to exploit ketones as an energy fuel source are being investigated. Ketone bodies are generated from fatty acid oxidation during fasting or extended periods of exercise and may be utilized in the brain by conversion to acetyl‐CoA to make ATP in the mitochondria (Mattson, Moehl, Ghena, Schmaedick, & Cheng, 2018). A recent pilot study in AD participants showed cognitive improvement in those subjects that were compliant with a ketogenic diet (Taylor, Sullivan, Mahnken, Burns, & Swerdlow, 2018). Administration of a ketone ester that elevated levels of the ketone body β‐hydroxybutyrate (BHB) led to improvement in behavioural deficits in 3xTg‐AD transgenic mice (Kashiwaya et al., 2013). It appears that BHB has the ability to induce brain derived neurotrophic factor (BDNF) expression in mouse brain, and this is linked to mitochondrial biogenesis and metabolic benefits (Marosi et al., 2017; Sleiman et al., 2016). An earlier clinical trial testing a caprylic triglyceride, which undergoes hepatic metabolism to BHB, showed cognitive benefits 45 days after initiating treatment (Henderson et al., 2009). We found 5 studies for “ketone diet and AD,” 1 study for “ketone ester and AD,” and 18 studies for “ketone and AD.”
The search for drugs that can alter energy metabolism in AD brains also includes the “repurposing” of drugs designed for diabetes. A recent report advocates that antidiabetic medications may help by reversing dysregulated transcriptional changes in AD (Katsel et al., 2018). The authors identified transcriptional changes in the insulin receptor pathway and microvasculature‐related genes in AD brains. These gene changes were reversed in the brains of AD patients who had received antidiabetic medications in life. The conclusions are limited by the patient groups analysed. The AD group on antidiabetic medication also carried a diabetes diagnosis, while the no‐treatment AD group did not have diabetes. Likewise, the study did not include a group with AD and diabetes but without antidiabetic treatment (as these would be harder to encounter). As such, the potentially favourable transcriptional effects by antidiabetic medication could be attributed to the effects of diabetes itself on the brain. Still, the report contributes by sharing insight into a subset of gene expression pathways that are altered in AD brains and indicates that metabolic changes (by medications to treat diabetes and/or diabetes itself) modulate the brain in potentially beneficial ways. A previous report by the same group hinted at this possibility with the findings that brains of diabetic patients who had been on both insulin and oral antidiabetic agents exhibited lower amyloid plaque burden compared to the other groups examined (including non‐diabetics, diabetics on insulin, or diabetics on oral agent monotherapy; Beeri et al., 2008).
Oral antidiabetic agents have yielded promising results when tested in disease models. The gluconeogenesis inhibitor molecule metformin extends lifespan in mice, lowers hyperphosphorylated τ in a diabetes mouse model, and reverses AD features in APP/PS1 mice (Chen et al., 2019; Martin‐Montalvo et al., 2013; Ou et al., 2018). Glucagon‐like peptide 1 (GLP1) receptor agonists act as an insulin sensitizing hormone and show neuroprotective effects (Li et al., 2009; Li et al., 2010). The thiazolidinediones (rosiglitazone and pioglitazone) work by activating PPARs and thereby enhance glucose utilization. Preclinical studies have demonstrated encouraging effects with rosiglitazone treatment. Rodent data show rosiglitazone induces mitochondrial biogenesis, improves memory, lowers Aβ burden, and lowers phospho‐τ (O'Reilly & Lynch, 2012; Pedersen et al., 2006; Strum et al., 2007; Yoon et al., 2010). In a Phase 2 human study, AD subjects (APOE ε4 non‐carriers) exhibited cognitive improvement on rosiglitazone (Risner et al., 2006), but benefits were not observed in a subsequent Phase 3 trial (Gold et al., 2010; Harrington et al., 2011).
Additional approaches to protect mitochondrial function in AD include the glial‐derived octadecaneuropeptide, which blocks chemically induced oxidative stress, apoptosis, and mitochondrial dysfunction. This effect benefits both neurons and astrocytes in culture (Kaddour et al., 2018).
11. CONCLUSIONS
Regardless of whether it is accepted as an inciting insult or a consequence of Aβ or τ pathologies, mitochondrial dysfunction in AD is a well‐accepted finding. Such dysfunction includes mtDNA lesions and reduced ETC enzyme function in the brains of AD subjects and AD mouse models alike. AD‐related neuropathological markers (APP, Aβ, and τ) and genetic risk factors (APOE) have been shown to interact with and alter mitochondrial function. There is also evidence to support the notion that abnormal mitochondrial function itself may trigger neurodegeneration and aberrant APP or τ processing.
We used the clinicaltrials.gov database as a resource to identify research efforts that have advanced into clinical trials. We found that therapies targeting conventional AD neuropathology (amyloid, τ, and APOE) continue to predominate, while studies involving energy metabolism pathways (mitochondria, metabolism, energy metabolism, antioxidants, exercise, and diet) lag behind by comparison. Metabolism, exercise, and dietary interventions are emphasized in the latter category. Antioxidant and antiapoptotic agents continue to represent the main pharmacological agents for addressing mitochondrial‐related dysfunction in AD models. Metabolic intermediates, such as NR, OAA, and ketones, seem to address a wide array of biological changes and, in some preclinical studies, have produced modifying outcomes.
As more advanced research tools allow us to better understand the pathological changes of AD, it becomes increasingly apparent that AD should be recognized as a multifactorial disease. Combining interventions that target several different processes may, therefore, make sense. With that in mind, this review emphasizes the rationale for testing bioenergetic approaches in AD.
11.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017a; Alexander, Cidlowski et al., 2017; Alexander, Peters et al., 2017; Alexander, Fabbro et al., 2017b; Alexander, Christopoulos et al., 2017).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
The authors are supported by the University of Kansas Alzheimer's Disease Center (P30 AG035982). J.M.P.O. is supported by a Mabel A. Woodyard Fellowship in Neurodegenerative Disorders.
Perez Ortiz JM, Swerdlow RH. Mitochondrial dysfunction in Alzheimer's disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol. 2019;176:3489–3507. 10.1111/bph.14585
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174, S208–S224. 10.1111/bph.13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandatheerthavarada, H. K. , Biswas, G. , Robin, M. A. , & Avadhani, N. G. (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. Journal of Cell Biology, 161(1), 41–54. 10.1083/jcb.200207030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeri, M. S. , Schmeidler, J. , Silverman, J. M. , Gandy, S. , Wysocki, M. , Hannigan, C. M. , … Haroutunian, V. (2008). Insulin in combination with other diabetes medication is associated with less Alzheimer neuropathology. Neurology, 71(10), 750–757. 10.1212/01.wnl.0000324925.95210.6d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, J. , Burns, J. , Welch, P. , & Bothwell, R. (2016). Safety and tolerability of R(+) pramipexole in mild‐to‐moderate Alzheimer's disease. Journal of Alzheimer's Disease, 49(4), 1179–1187. 10.3233/JAD-150788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birks, J. (2006). Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database of Systematic Reviews, (1). Art. No.: CD005593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birks, J. , Chong, L. , & Grimley Evans, J. (2015). Rivastigmine for Alzheimer's disease. Cochrane Database of Systematic Reviews. 10.1002/14651858.CD001191.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birks, J. , & Harvey, R. (2018). Donepezil for dementia due to Alzheimer's disease. Database of Systematic Reviews. 10.1002/14651858.CD001190.pub3 [DOI] [Google Scholar]
- Bonda, D. J. , Wang, X. , Lee, H. G. , Smith, M. A. , Perry, G. , & Zhu, X. (2014). Neuronal failure in Alzheimer's disease: A view through the oxidative stress looking‐glass. Neuroscience Bulletin, 30(2), 243–252. 10.1007/s12264-013-1424-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canugovi, C. , Shamanna, R. , Croteau, D. , & Bohr, V. (2014). Base excision DNA repair levels in mitochondrial lysates of Alzheimer's disease. Neurobiology of Aging, 35(6), 1293–1300. 10.1016/j.neurobiolaging.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso, S. , Carvalho, C. , Correia, S. C. , Seiça, R. M. , & Moreira, P. I. (2016). Alzheimer's disease: From mitochondrial perturbations to mitochondrial medicine. Brain Pathology, 26(5), 632–647. 10.1111/bpa.12402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella, M. , Bimonte, S. , Muzio, M. R. , Schiavone, V. , & Cuomo, A. (2017). The efficacy of epigallocatechin‐3‐gallate (green tea) in the treatment of Alzheimer's disease: An overview of pre‐clinical studies and translational perspectives in clinical practice. Infectious Agents and Cancer, 12(1), 1–7. 10.1186/s13027-017-0145-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspersen, C. , Wang, N. , Yao, J. , Sosunov, A. , Chen, X. , Lustbader, J. W. , … Yan, S. D. (2005). Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB Journal, 19(14), 2040–2041. 10.1096/fj.05-3735fje [DOI] [PubMed] [Google Scholar]
- Chang, S. , Ma, T. r. , Miranda, R. D. , Balestra, M. E. , Mahley, R. W. , & Huang, Y. (2005). Lipid‐ and receptor‐binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proceedings of the National Academy of Sciences, 102(51), 18694–18699. 10.1073/pnas.0508254102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. L. , Luo, C. , Pu, D. , Zhang, G. Q. , Zhao, Y. X. , Sun, Y. , … Xiao, Q. (2019). Metformin attenuates diabetes‐induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagic clearance. Experimental Neurology, 311, 44–56. 10.1016/j.expneurol.2018.09.008 [DOI] [PubMed] [Google Scholar]
- Chételat, G. , Landeau, B. , Salmon, E. , Yakushev, I. , Bahri, M. A. , Mézenge, F. , … Fellgiebel, A. (2013). Relationships between brain metabolism decrease in normal aging and changes in structural and functional connectivity. NeuroImage, 76, 167–177. 10.1016/j.neuroimage.2013.03.009 [DOI] [PubMed] [Google Scholar]
- Choi, J. , Chandrasekaran, K. , Demarest, T. G. , Kristian, T. , Xu, S. , Vijaykumar, K. , … Russell, J. W. (2014). Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Annals of Clinical and Translational Neurology, 1(8), 589–604. 10.1002/acn3.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder, A. E. H. , Saunders, A. M. , Strittmatter, W. J. , Schmechel, D. E. , Gaskell, P. C. , Small, W. , Roses AD, Haines JL, Pericak‐Vance MA. (1993). Gene dose of Apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science, 261(5123), 921–923. [DOI] [PubMed] [Google Scholar]
- Corder, E. , Saunders, A. , Risch, N. , Strittmatter, W. , Schmechel, D. , Gaskell, P. , … Pericak‐Vance, M. A. (1994). Protective effect APOE type 2 allele for late onset Alzheimer disease. Nature Genetics, 7, 180–184. [DOI] [PubMed] [Google Scholar]
- Cummings, J. , Aisen, P. S. , Dubois, B. , Frölich, L. , Jack, C. R. , Jones, R. W. , … Scheltens, P. (2016). Drug development in Alzheimer's disease: The path to 2025. Alzheimer's Research and Therapy, 8(1), 1–12. 10.1186/s13195-016-0207-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, J. , Lee, G. , Ritter, A. , & Zhong, K. (2017). Alzheimer's disease drug development pipeline: 2017. Alzheimer's and Dementia: Translational Research and Clinical Interventions, 3, 195–214. 10.1016/j.trci.2018.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, J. , Lee, G. , Ritter, A. , & Zhong, K. (2018). Alzheimer's disease drug development pipeline: 2018. Alzheimer's and Dementia: Translational Research and Clinical Interventions, 4, 195–214. 10.1016/j.trci.2018.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, J. , Morstorf, T. , & Lee, G. (2016). Alzheimer's drug‐development pipeline: 2016. Alzheimer's & Dementia: Translational Research & Clinical Interventions, 2(2016), 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane, S. C. , Courchesne‐Loyer, A. , Vandenberghe, C. , St‐Pierre, V. , Fortier, M. , Hennebelle, M. , … Castellano, C. A. (2016). Can ketones help rescue brain fuel supply in later life? Implications for cognitive health during aging and the treatment of Alzheimer's disease. Frontiers in Molecular Neuroscience, 9, 1–21. 10.3389/fnmol.2016.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, D. C. , Hauptmann, S. , Scherping, I. , Schuessel, K. , Keil, U. , Rizzu, P. , … Götz, J. (2005). Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. Journal of Biological Chemistry, 280(25), 23802–23814. 10.1074/jbc.M500356200 [DOI] [PubMed] [Google Scholar]
- De Santi, S. , de Leon, M. , Convit, A. , Tarshish, C. , Rusinek, H. , Tsui, W. , … Volkow, N. (1995). Age‐related changes in brain: II. Positron emission tomography of frontal and temporal lobe glucose metabolism in normal subjects. The Psychiatric Quarterly, 66, 357–370. 10.1007/BF02238755 [DOI] [PubMed] [Google Scholar]
- Devi, L. , Prabhu, B. , Galanti, D. , Avadhani, N. , & Anandatheerthavarada, H. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. Neurobiology of Disease, 26(35), 9057–9068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, H. , Guo, L. , Fang, F. , Chen, D. , Sosunov, A. A. , McKhann, G. M. , … Yan, S. D. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nature Medicine, 14(10), 1097–1105. 10.1038/nm.1868. 10.1038/nm.1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, A. , Nisbet, R. , Grimm, A. , & Götz, J. (2014). March separate, strike together—Role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer's disease. Biochimica et Biophysica Acta ‐ Molecular Basis of Disease, 1842(8), 1258–1266. 10.1016/j.bbadis.2013.08.013 [DOI] [PubMed] [Google Scholar]
- Eckert, A. , Schulz, K. L. , Rhein, V. , & Götz, J. (2010). Convergence of amyloid‐β and tau pathologies on mitochondria in vivo. Molecular Neurobiology, 41(2–3), 107–114. 10.1007/s12035-010-8109-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkamhawy, A. , Park, J. E. , Hassan, A. H. E. , Pae, A. N. , Lee, J. , Park, B. G. , & Roh, E. J. (2018). Synthesis and evaluation of 2‐(3‐arylureido)pyridines and 2‐(3‐arylureido)pyrazines as potential modulators of Aβ‐induced mitochondrial dysfunction in Alzheimer's disease. European Journal of Medicinal Chemistry, 144, 529–543. 10.1016/j.ejmech.2017.12.045 [DOI] [PubMed] [Google Scholar]
- Foster, N. , Chase, T. , Fedio, P. , Patronas, N. , Brooks, R. , & Di Chiro, G. (1983). Alzheimer's disease: Focal cortical changes shown by positron emission tomography. Neurology, 33(8), 961–965. [DOI] [PubMed] [Google Scholar]
- Frackowiak, R. , Pozzilli, C. , Legg, N. , Du Boulay, G. , Marshall, J. , Lenzi, G. , & Jones, T. (1981). Regional cerebral oxygen supply and utilization in dementia. A clinical and physiological study with oxygen‐15 and positron tomography. Brain, 104, 753–778. [DOI] [PubMed] [Google Scholar]
- Friedland, R. , Budinger, T. , Ganz, E. , Yano, Y. , Mathis, C. , Koss, B. , … Derenzo, S. E. (1983). Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F]fluorodeoxyglucose. Journal of Computer Assisted Tomography, 7(4), 590–598. 10.1097/00004728-198308000-00003 [DOI] [PubMed] [Google Scholar]
- Fu, Y. J. , Xiong, S. , Lovell, M. A. , & Lynn, B. C. (2009). Quantitative proteomic analysis of mitochondria in aging PS‐1 transgenic mice. Cellular and Molecular Neurobiology, 29(5), 649–664. 10.1007/s10571-009-9359-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui, H. , & Moraes, C. T. (2009). Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Human Molecular Genetics, 18(6), 1028–1036. 10.1093/hmg/ddn437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama, H. , Ogawa, M. , Yamauchi, H. , Yamaguchi, S. , Kimura, J. , Yonekura, Y. , & Konishi, J. (1994). Altered cerebral energy metabolism in Alzheimer's disease: A PET study. Journal of Nuclear Medicine: Official Publication, Society of Nuclear Medicine, 35(1), 1–6. [PubMed] [Google Scholar]
- Gabuzda, D. , Busciglio, J. , Chen, L. B. , Matsudaira, P. , & Yankner, B. A. (1994). Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. Journal of Biological Chemistry, 269(18), 13623–13628. [PubMed] [Google Scholar]
- Gibson, G. , Starkov, A. , Blass, J. , Rata, R. , & Beal, M. (2010). Cause and consequence: Mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age‐associated neurodegenerative diseases. Biochimica et Biophysica Acta, 1802(1), 122–134. 10.1016/j.bbadis.2009.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold, M. , Alderton, C. , Zvartau‐Hind, M. , Egginton, S. , Saunders, A. M. , Irizarry, M. , … Sawchak, S. (2010). Rosiglitazone monotherapy in mild‐to‐moderate alzheimer's disease: Results from a randomized, double‐blind, placebo‐controlled phase III study. Dementia and Geriatric Cognitive Disorders, 30(2), 131–146. 10.1159/000318845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, K. N. , Steffan, J. S. , Martinez‐Coria, H. , Sun, X. , Schreiber, S. S. , Thompson, L. M. , & LaFerla, F. M. (2008). Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231‐phosphotau. Journal of Neuroscience. 10.1523/JNEUROSCI.3203-08.2008, 28, 11500–11510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington, C. , Sawchak, S. , Chiang, C. , Davies, J. , Donovan, C. , Saunders, A. , … Gold, M. (2011). Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild‐to‐moderate Alzheimer's disease: Two phase 3 studies. Current Alzheimer Research, 8(5), 592–606. 10.2174/156720511796391935 [DOI] [PubMed] [Google Scholar]
- Hauptmann, S. , Scherping, I. , Dröse, S. , Brandt, U. , Schulz, K. L. , Jendrach, M. , … Müller, W. E. (2009). Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiology of Aging, 30(10), 1574–1586. 10.1016/j.neurobiolaging.2007.12.005 [DOI] [PubMed] [Google Scholar]
- Henderson, S. , Vogel, J. , Barr, L. , Garvin, F. , Jones, J. , & Constantini, L. (2009). Study of the ketogenic agent AC‐1202 in mild to moderate Alzheimer's disease: A randomized, double‐blind, placebo‐controlled, multicenter trial. Nutrition and Metabolism, 6, 31 10.1186/1743-7075-6-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, K. , Aliev, G. , Nunomura, A. , Fujioka, H. , Russell, R. L. , Atwood, C. S. , … Smith, M. A. (2001). Mitochondrial abnormalities in Alzheimer's disease. The Journal of Neuroscience, 21(9), 3017–3023. 10.1523/JNEUROSCI.21-09-03017.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, Y. , Lautrup, S. , Cordonnier, S. , Wang, Y. , Croteau, D. L. , Zavala, E. , … Bohr, V. A. (2018). NAD + supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proceedings of the National Academy of Sciences, 115(8), E1876–E1885. 10.1073/pnas.1718819115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry, E. , Dashkoff, J. , Roe, A. D. , Takeda, S. , Koffie, R. M. , Hashimoto, T. , … Hyman, B. T. (2013). Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Science Translational Medicine, 5(212), 1–5. 10.1126/scitranslmed.3007000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsen, T. , de Vlieg, J. , & Alkema, W. (2008). BioVenn—A web application for the comparison and visualization of biological lists using area‐proportional Venn diagrams. BMC Genomics, 9(1), 488 10.1186/1471-2164-9-488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturria‐Medina, Y. , Carbonell, F. M. , Sotero, R. C. , Chouinard‐Decorte, F. , & Evans, A. C. (2017). Multifactorial causal model of brain (dis)organization and therapeutic intervention: Application to Alzheimer's disease. NeuroImage, 152, 60–77. 10.1016/j.neuroimage.2017.02.058 [DOI] [PubMed] [Google Scholar]
- Jara, C. , Aránguiz, A. , Cerpa, W. , Tapia‐Rojas, C. , & Quintanilla, R. A. (2018). Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biology, 18, 279–294. 10.1016/j.redox.2018.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddour, H. , Hamdi, Y. , Amri, F. , Bahdoudi, S. , Bouannee, I. , & Leprince, J. (2018). Antioxidant and anti‐apoptotic activity of octadecaneuropeptide against 6‐OHDA toxicity in cultured rat astrocytes. 10.1007/s12031-018-1181-4 [DOI] [PubMed]
- Kalpouzos, G. , Chételat, G. , Baron, J. C. , Landeau, B. , Mevel, K. , Godeau, C. , … Desgranges, B. (2009). Voxel‐based mapping of brain gray matter volume and glucose metabolism profiles in normal aging. Neurobiology of Aging, 30(1), 112–124. 10.1016/j.neurobiolaging.2007.05.019 [DOI] [PubMed] [Google Scholar]
- Kashiwaya, Y. , Bergman, C. , Lee, J. H. , Wan, R. , King, M. T. , Mughal, M. R. , … Veech, R. L. (2013). A ketone ester diet exhibits anxiolytic and cognition‐sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer's disease. Neurobiology of Aging, 34(6), 1530–1539. 10.1016/j.neurobiolaging.2012.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsel, P. , Roussos, P. , Beeri, M. S. , Gama‐Sosa, M. A. , Gandy, S. , Khan, S. , & Haroutunian, V. (2018). Parahippocampal gyrus expression of endothelial and insulin receptor signaling pathway genes is modulated by Alzheimer's disease and normalized by treatment with anti‐diabetic agents. PLoS One, 13(11), e0206547 10.1371/journal.pone.0206547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. M. , Cassarino, D. S. , Abramova, N. N. , Keeney, P. M. , Borland, M. , Trimmer, P. A. , … Bennett, J. P. Jr. (2000). Alzheimer's disease cybrids replicate beta‐amyloid abnormalities through cell death pathways. Annals of Neurology, 48(2), 148–155. [DOI] [PubMed] [Google Scholar]
- Kopeikina, K. J. , Carlson, G. A. , Pitstick, R. , Ludvigson, A. E. , Peters, A. , Luebke, J. I. , … Spires‐Jones, T. L. (2011). Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. American Journal of Pathology, 179(4), 2071–2082. 10.1016/j.ajpath.2011.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan, K. J. , Ratnaike, T. E. , De Gruyter, H. L. M. , Jaros, E. , & Turnbull, D. M. (2012). Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer's disease. Neurobiology of Aging, 33(9), 2210–2214. 10.1016/j.neurobiolaging.2011.08.009 [DOI] [PubMed] [Google Scholar]
- Kujoth, G. , Hiona, A. , Pugh, T. , Someya, S. , Panzer, K. , Wohlgemuth, S. , … Prolla, T. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science, 309(5733), 481–484. 10.1126/science.1112125 [DOI] [PubMed] [Google Scholar]
- Li, Y. , Duffy, K. B. , Ottinger, M. A. , Ray, B. , Bailey, J. A. , Holloway, H. W. , … Greig, N. H. (2010). GLP‐1 receptor stimulation reduces amyloid‐β peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer's disease. Journal of Alzheimer's Disease, 19(4), 1205–1219. 10.3233/JAD-2010-1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Perry, T. , Kindy, M. S. , Harvey, B. K. , Tweedie, D. , Holloway, H. W. , … Greig, N. H. (2009). GLP‐1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proceedings of the National Academy of Sciences, 106(4), 1285–1290. 10.1073/pnas.0806720106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloret, A. , Badía, M. C. , Mora, N. J. , Pallardó, F. V. , Alonso, M. D. , & Viña, J. (2009). Vitamin e paradox in alzheimer's disease: It does not prevent loss of cognition and may even be detrimental. Journal of Alzheimer's Disease, 17(1), 143–149. 10.3233/JAD-2009-1033 [DOI] [PubMed] [Google Scholar]
- Lokesh, K. , Kujoth, G. , Prolla, T. , Van Leuven, F. , & Vassar, R. (2014). Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer's disease. Molecular Neurodegeneration, 9(1), 1–18. 10.1186/1750-1326-9-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell, M. A. , Xie, C. , & Markesbery, W. R. (2000). Decreased base excision repair and increased helicase activity in Alzheimer's disease brain. Brain Research, 855(1), 116–123. 10.1016/S0006-8993(99)02335-5 [DOI] [PubMed] [Google Scholar]
- Loy, C. , & Schneider, L. (2006). Galantamine for Alzheimer's disease and mild cognitive impairment. Cochrane Database of Systematic Reviews, (1), Art. No.: CD001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft, R. (1994). The development of mitochondrial medicine. Proceedings of the National Academy of Sciences, 91, 8731–8738. 10.1073/pnas.91.19.8731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustbader, J. W. , Cirilli, M. , Lin, C. , Xu, H. W. , Takuma, K. , Wang, N. , … Wu, H. (2004). ABAD directly links Aβ to mitochondrial toxicity in Alzheimer's disease. Science, 304(5669), 448–452. 10.1126/science.1091230 [DOI] [PubMed] [Google Scholar]
- Mao, P. , Manczak, M. , Calkins, M. J. , Truong, Q. , Reddy, T. P. , Reddy, A. P. , … Reddy, P. H. (2012). Mitochondria‐targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer's disease: Implications for neuroprotection and lifespan extension. Human Molecular Genetics, 21(13), 2973–2990. 10.1093/hmg/dds128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marano, C. , Workman, C. , Kramer, E. , Hermann, C. , Ma, Y. , Dhawan, V. , … Smith, G. S. (2013). Longitudinal studies of cerebral glucose metabolism in late‐life depression and normal aging. International Journal of Geriatric Psychiatry, 28(4), 417–423. 10.1002/gps.3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marosi, K. , Kim, S. W. , Moehl, K. , Scheibye‐Knudsen, M. , Cutler, R. , Camandola, S. , & Mattson, M. P. (2017). 3‐Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. Journal of Neurochemistry, 139(5), 769–781. 10.1111/jnc.13868.3-Hydroxybutyrate [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Montalvo, A. , Mercken, E. M. , Mitchell, S. J. , Palacios, H. H. , Mote, P. L. , Scheibye‐Knudsen, M. , … de Cabo, R. (2013). Metformin improves healthspan and lifespan in mice. Nature Communications, 4, 2192 10.1038/ncomms3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson, M. P. , & Arumugam, T. V. (2018). Hallmarks of brain aging: Adaptive and pathological modification by metabolic states. Cell Metabolism, 27(6), 1176–1199. 10.1016/j.cmet.2018.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson, M. P. , Moehl, K. , Ghena, N. , Schmaedick, M. , & Cheng, A. (2018). Intermittent metabolic switching, neuroplasticity and brain health. Nature Reviews Neuroscience, 19(2), 81–94. 10.1038/nrn.2017.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer, I. , Zierz, S. , & Möller, H. (2000). A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiology of Aging, 21(3), 455–462. 10.1016/S0197-4580(00)00112-3 [DOI] [PubMed] [Google Scholar]
- McManus, M. J. , Murphy, M. P. , & Franklin, J. L. (2011). The mitochondria‐targeted antioxidant mitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. Journal of Neuroscience, 31(44), 15703–15715. 10.1523/JNEUROSCI.0552-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane, R. , Areosa Sastre, A. , & Minakaran, N. (2006). Memantine for dementia. Cochrane Database of Systematic Reviews, (2), Art. No.: CD003154. [DOI] [PubMed] [Google Scholar]
- Miller, E. , Pastor‐Barriuso, R. , Dalal, D. , Riemersma, R. A. , Appel, L. , & Guallar, E. (2005). Meta‐analysis: High‐dosage vitamin E supplementation may increase all‐cause mortality. Annals of Internal Medicine, 142(1), 37–46. [DOI] [PubMed] [Google Scholar]
- Mosconi, L. , Berti, V. , Glodzik, L. , Pupi, A. , De Santi, S. , & de Leon, M. (2010). Pre‐clinical detection of Alzheimer's disease using FDG‐PET, with or without amyloid imaging. Journal of Alzheimer's Disease, 20(3), 843–854. 10.3233/JAD-2010-091504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A. J. , Knight, N. S. , Cole, M. A. , Cochlin, L. E. , Carter, E. , Tchabanenko, K. , … Clarke, K. (2016). Novel ketone diet enhances physical and cognitive performance. FASEB Journal, 30(12), 4021–4032. 10.1096/fj.201600773R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutisya, E. , Bowling, A. , & Beal, M. (1994). Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. Journal of Neurochemistry, 63(6), 2179–2184. [DOI] [PubMed] [Google Scholar]
- Navarro, A. , & Boveris, A. (2007). The mitochondrial energy transduction system and the aging process. American Journal of Physiology Cell Physiology, 292(2), C670–C686. 10.1152/ajpcell.00213.2006 [DOI] [PubMed] [Google Scholar]
- Nishimune, H. , Tungtur, S. , Wilkins, H. , Swerdlow, R. , Sage, J. , Agbas, A. , & Barohn, R. (2017). Beneficial effect of oxaloacetate for the neuromuscular function of SOD1G93A mice. Theme 3: In Vivo Experimental Models, Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, 18(suppl 2), 130–151. 10.1080/21678421.2017.1371517 [DOI] [PubMed] [Google Scholar]
- Nunomura, A. , Perry, G. , Aliev, G. , Hirai, K. , Takeda, A. , Balraj, E. , … Smith, M. A. (2001). Oxidative damage is the earliest event in Alzheimer disease. Journal of Nueuropathology and Experimental Neurology, 60(8), 759–767. 10.1093/jnen/60.8.759 [DOI] [PubMed] [Google Scholar]
- O'Reilly, J. A. , & Lynch, M. (2012). Rosiglitazone improves spatial memory and decreases insoluble Aβ 1‐42 in APP/PS1 mice. Journal of Neuroimmune Pharmacology, 7(1), 140–144. 10.1007/s11481-011-9282-7 [DOI] [PubMed] [Google Scholar]
- Ou, Z. , Kong, X. , Sun, X. , He, X. , Zhang, L. , Gong, Z. , … Xuan, A. (2018). Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain, Behavior, and Immunity, 69, 351–363. 10.1016/j.bbi.2017.12.009 [DOI] [PubMed] [Google Scholar]
- Parker, W. Jr. , Filley, C. , & Parks, J. (1990). Cytochrome oxidase deficiency in Alzheimer's disease. Neurology, 40, 1302–1303. [DOI] [PubMed] [Google Scholar]
- Pedersen, W. A. , McMillan, P. J. , Kulstad, J. J. , Leverenz, J. B. , Craft, S. , & Haynatzki, G. R. (2006). Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Experimental Neurology, 199(2), 265–273. 10.1016/j.expneurol.2006.01.018 [DOI] [PubMed] [Google Scholar]
- Pérez, M. J. , Jara, C. , & Quintanilla, R. A. (2018). Contribution of Tau pathology to mitochondrial impairment in neurodegeneration. Frontiers in Neuroscience, 12(JUL), 1–14. 10.3389/fnins.2018.00441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins, M. , Wolf, A. B. , Chavira, B. , Shonebarger, D. , Meckel, J. P. , Leung, L. , … Valla, J. (2016). Altered energy metabolism pathways in the posterior cingulate in young adult Apolipoprotein E ϵ4 carriers. Journal of Alzheimer's Disease, 53(1), 95–106. 10.3233/JAD-151205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, M. , Pickrell, A. M. , Fukui, H. , & Moraes, C. T. (2013). Mitochondrial DNA damage in a mouse model of Alzheimer's disease decreases amyloid beta plaque formation. Neurobiology of Aging, 34(10), 2399–2407. 10.1016/j.neurobiolaging.2013.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praticò, D. , Uryu, K. , Leight, S. , Trojanoswki, J. Q. , & Lee, V. M.‐Y. (2001). Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. The Journal of Neuroscience, 21(12), 4183–4187. 10.1523/JNEUROSCI.21-12-04183.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla, R. A. , Matthews‐Roberson, T. A. , Dolan, P. J. , & Johnson, G. V. W. (2009). Caspase‐cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons. Journal of Biological Chemistry, 284(28), 18754–18766. 10.1074/jbc.M808908200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resende, R. , Moreira, P. I. , Proença, T. , Deshpande, A. , Busciglio, J. , Pereira, C. , & Oliveira, C. R. (2008). Brain oxidative stress in a triple‐transgenic mouse model of Alzheimer disease. Free Radical Biology and Medicine, 44(12), 2051–2057. 10.1016/j.freeradbiomed.2008.03.012 [DOI] [PubMed] [Google Scholar]
- Rhein, V. , Song, X. , Wiesner, A. , Ittner, L. M. , Baysang, G. , Meier, F. , … Eckert, A. (2009). Amyloid‐ and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proceedings of the National Academy of Sciences, 106(47), 20057–20062. 10.1073/pnas.0905529106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risner, M. E. , Saunders, A. M. , Altman, J. F. B. , Ormandy, G. C. , Craft, S. , Foley, I. M. , … Rosiglitazone in Alzheimer's Disease Study Group (2006). Efficacy of rosiglitazone in a genetically defined population with mild‐to‐moderate Alzheimer's disease. Pharmacogenomics Journal, 6(4), 246–254. 10.1038/sj.tpj.65003697 [DOI] [PubMed] [Google Scholar]
- Sano, M. , Ernesto, C. , Thomas, R. G. , Klauber, M. R. , Schafer, K. , Grundman, M. , … for the Members of the Alzheimer's Disease Cooperative Study (1997). A controlled trial of selegiline, alpha‐tocopherol, or both as treatment for Alzheimer's disease. New England Journal of Medicine, 336(17), 1216–1222. 10.1056/NEJM199704243361704 [DOI] [PubMed] [Google Scholar]
- Scholte, H. (1988). The biochemical basis of mitochondrial diseases. Journal of Bioenergetics and Biomembranes, 20, 161–191. 10.1007/BF00768393 [DOI] [PubMed] [Google Scholar]
- Schriner, S. E. , Linford, N. J. , Martin, G. M. , Treuting, P. , Ogburn, C. E. , Emond, M. , … Rabinovitch, P. S. (2003). Extension of murine life span by overexpression of catalase targeted to mitochondria. Science, 299(5611), 1342–1346. 10.1126/science.1077991 [DOI] [PubMed] [Google Scholar]
- Silverman, D. , Small, G. , Chang, C. , Lu, C. , Kung De Aburto, M. , Chen, W. , … Phelps, M. E. (2001). Positron emission tomography in evaluation of dementia: Regional brain metabolism and long‐term outcome. Jama, 286(17), 2120–2127. 10.1001/jama.286.17.2120 [DOI] [PubMed] [Google Scholar]
- Sims, N. , Finegan, J. , Blass, J. , Bowen, D. , & Neary, D. (1987). Mitochondrial function in brain tissue in primary degenerative dementia. Brain Research, 436(1), 30–38. 10.1016/0006-8993(87)91553-8 [DOI] [PubMed] [Google Scholar]
- Sleiman, S. F. , Henry, J. , Al‐Haddad, R. , El Hayek, L. , Haidar, E. A. , Stringer, T. , … Chao, M. V. (2016). Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β‐hydroxybutyrate. eLife, 5, e15092 10.7554/eLife.15092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltys, D. T. , Pereira, C. P. M. , Rowies, F. T. , Farfel, J. M. , Grinberg, L. T. , Suemoto, C. K. , … Souza‐Pinto, N. C. (2019). Lower mitochondrial DNA content but not increased mutagenesis associates with decreased base excision repair activity in brains of AD subjects. Neurobiology of Aging, 73, 161–170. 10.1016/j.neurobiolaging.2018.09.015 [DOI] [PubMed] [Google Scholar]
- Stefanova, N. A. , Ershov, N. , Maksimova, K. , Muraleva, N. , Tyumentsev, M. , & Kolosova, N. (2018). The rat prefrontal‐cortex transcriptome: Effects of aging and sporadic Alzheimer's disease‐like pathology. Journal of Gerontology: Biological Sciences and the Journal of Gerontology: Medical Sciences, 74(1), 33–43. 10.1093/gerona/gly198 [DOI] [PubMed] [Google Scholar]
- Ströhle, A. , Schmidt, D. , Schultz, F. , Fricke, N. , Staden, T. , Hellweg, R. , … Rieckmann, N. (2015). Drug and exercise treatment of Alzheimer disease and mild cognitive impairment: A systematic review and meta‐analysis of effects on cognition in randomized controlled trials. The American Journal of Geriatric Psychiatry, 23(12), 1234–1249. [DOI] [PubMed] [Google Scholar]
- Strum, J. C. , Shehee, R. , Virley, D. , Richardson, J. , Mattie, M. , Selley, P. , … Roses, A. (2007). Rosiglitazone induces mitochondrial biogenesis in mouse brain. Journal of Alzheimer's Disease, 11(1), 45–51. 10.3233/JAD-2007-11108 [DOI] [PubMed] [Google Scholar]
- Swerdlow, R. (2009). Mitochondrial medicine and the neurodegenerative mitochondriopathies. Pharmaceuticals, 2(3), 150–167. 10.3390/ph2030150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow, R. (2011). Role and treatment of mitochondrial DNA‐related mitochondrial dysfunction in sporadic neurodegenerative diseases. Current Pharmaceutical Design, 17(31), 3356–3373. 10.2174/138161211798072535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow, R. (2014). Bioenergetic medicine. British Journal of Pharmacology, 171(8), 1854–1869. 10.1111/bph.12394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow, R. , Burns, J. , & Khan, S. (2014). The Alzheimer's disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim Biophys Acta., 1842(8). 1219031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow, R. , Golbe, L. , Parks, J. , Cassarino, D. , Binder, D. , Grawey, A. , … Parker, W. D. (2000). Mitochondrial dysfunction in cybrid lines expressing mitochondrial genes from patients with progressive supranuclear palsy. Journal of Neurochemistry, 75, 1681–1684. [DOI] [PubMed] [Google Scholar]
- Swerdlow, R. , & Khan, S. (2004). A “mitochondrial cascade hypothesis” for sporadic Alzheimer's disease. Medical Hypotheses, 63, 8–20. [DOI] [PubMed] [Google Scholar]
- Swerdlow, R. , & Khan, S. (2009). The Alzheimer's disease mitochondrial cascade hypothesis: An update. Experimental Neurology, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow, R. , Parks, J. , Cassarino, D. , Maguire, D. , Maguire, R. , Bennett, J. , … Parker, W. D. Jr. (1997). Cybrids in Alzheimer's disease: A cellular model of the disease? Neurology, 49(4), 918–925. 10.1212/WNL.49.4.918 [DOI] [PubMed] [Google Scholar]
- Swerdlow, R. , Parks, J. , Miller, S. , Tuttle, J. , Trimmer, P. , Sheehan, J. , … Parker, W. D. Jr. (1996). Origin and functional consequences of the complex I defect in Parkinson's disease. Annals of Neurology, 40, 663–671. [DOI] [PubMed] [Google Scholar]
- Swerdlow, R. H. (2016). Bioenergetics and metabolism: A bench to bedside perspective. Journal of Neurochemistry, 139, 126–135. 10.1111/jnc.13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, M. K. , Sullivan, D. K. , Mahnken, J. D. , Burns, J. M. , & Swerdlow, R. H. (2018). Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer's disease. Alzheimer's and Dementia: Translational Research and Clinical Interventions, 4, 28–36. 10.1016/j.trci.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo, J. B. , Arnold, M. , Chang, R. , Rebecca, A. , Han, X. , Thambisetty, M. , … Indiana, T. (2018). Metabolic network failures in Alzheimer's disease: A biochemical road map. Alzheimers Dement, 13(9), 965–984. 10.1016/j.jalz.2017.01.020.Metabolic [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic, A. , Wredenberg, A. , Falkenberg, M. , Spelbrink, J. , Rovio, A. , Bruder, C. , … Larsson, N. G. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature, 429(6990), 417–423. 10.1038/nature02517 [DOI] [PubMed] [Google Scholar]
- Tyumentsev, M. , Stefanova, N. , Muraleva, N. , Rumyantseva, Y. , Kiseleva, E. , Vavilin, V. , & Kolosova, N. (2018). Mitochondrial dysfunction as a predictor and driver of Alzheimer's disease‐like pathology in OXYS rats. Journal of Alzheimer's Disease, 63(3), 1075–1088. 10.3233/JAD-180065 [DOI] [PubMed] [Google Scholar]
- Valla, J. , Yaari, R. , Wolf, A. , Kusne, Y. , Beach, T. , Roher, A. , … Reiman, E. M. (2010). Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE ε4 allele, the major late‐onset Alzheimer's susceptibility gene. Journal of Alzheimer's Disease, 22(1), 307–313. 10.3233/JAD-2010-100129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitch, D. P. , Weiner, M. W. , Aisen, P. S. , Beckett, L. A. , Cairns, N. J. , Green, R. C. , … Alzheimer's Disease Neuroimaging Initiative (2018). Understanding disease progression and improving Alzheimer's disease clinical trials: Recent highlights from the Alzheimer's Disease Neuroimaging Initiative. Alzheimer's & Dementia, 1–46. 10.1016/j.jalz.2018.08.005 [DOI] [PubMed] [Google Scholar]
- Wallace, D. C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annual Review of Genetics, 39, 359–407. 10.1146/annurev.genet.39.110304.095751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Markesbery, W. R. , & Lovell, M. A. (2006). Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. Journal of Neurochemistry, 96(3), 825–832. 10.1111/j.1471-4159.2005.03615.x [DOI] [PubMed] [Google Scholar]
- Wang, X. , Su, B. , Lee, H. G. , Li, X. , Perry, G. , Smith, M. A. , & Zhu, X. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. Journal of Neuroscience, 29(28), 9090–9103. 10.1523/JNEUROSCI.1357-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Wang, W. , Li, L. , Perry, G. , Lee, H. , & Zhu, X. (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer's disease ☆. Biochimica et Biophysica Acta, 1842(8), 1240–1247. 10.1016/j.bbadis.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman, L. , Jo, D. G. , Sørensen, M. M. , de Souza‐Pinto, N. C. , Markesbery, W. R. , Mattson, M. P. , & Bohr, V. A. (2007). Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Research, 35(16), 5545–5555. 10.1093/nar/gkm605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins, H. M. , Harris, J. L. , Carl, S. M. , E, L. , Lu, J. , Eva Selfridge, J. , … Swerdlow, R. H. (2014). Oxaloacetate activates brain mitochondrial biogenesis, enhances the insulin pathway, reduces inflammation and stimulates neurogenesis. Human Molecular Genetics, 23(24), 6528–6541. 10.1093/hmg/ddu371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins, H. M. , Koppel, S. , Carl, S. M. , Ramanujan, S. , Weidling, I. , Michaelis, M. L. , … Swerdlow, R. H. (2016). Oxaloacetate enhances neuronal cell bioenergetic fluxes and infrastructure. Journal of Neurochemistry, 137(1), 76–87. 10.1111/jnc.13545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins, H. M. , Koppel, S. J. , Bothwell, R. , Mahnken, J. , Burns, J. M. , & Swerdlow, R. H. (2017). Platelet cytochrome oxidase and citrate synthase activities in APOE ε4 carrier and non‐carrier Alzheimer's disease patients. Redox Biology, 12, 828–832. 10.1016/j.redox.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins, H. M. , Mahnken, J. D. , Welch, P. , Bothwell, R. , Koppel, S. , Jackson, R. L. , … Swerdlow, R. H. (2017). A mitochondrial biomarker‐based study of S‐equol in Alzheimer's disease subjects: Results of a single‐arm, pilot trial. Journal of Alzheimer's Disease, 59(1), 291–300. 10.3233/JAD-170077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, M. W. , Ketter, T. A. , Kimbrell, T. A. , George, M. S. , Herscovitch, P. , Danielson, A. L. , … Post, R. M. (2002). Age, sex and laterality effects on cerebral glucose metabolism in healthy adults. Psychiatry Research ‐ Neuroimaging, 114(1), 23–37. 10.1016/S0925-4927(01)00126-3 [DOI] [PubMed] [Google Scholar]
- Yao, J. , Irwin, R. W. , Zhao, L. , Nilsen, J. , Hamilton, R. T. , & Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer'spathology in female mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America, 106(34), 14670–14675. 10.1073/pnas.0903563106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, J. , Zhao, L. , Mao, Z. , Chen, S. , Wong, K. C. , To, J. , & Brinton, R. D. (2013). Potentiation of brain mitochondrial function by S‐equol and R/S‐equol estrogen receptor β‐selective phytoSERM treatments. Brain Research, 1514, 128–141. 10.1016/j.brainres.2013.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, S. Y. , Park, J. S. , Choi, J. E. , Choi, J. M. , Lee, W. J. , Kim, S. W. , & Kim, D. H. (2010). Rosiglitazone reduces tau phosphorylation via JNK inhibition in the hippocampus of rats with type 2 diabetes and tau transfected SH‐SY5Y cells. Neurobiology of Disease, 40(2), 449–455. 10.1016/j.nbd.2010.07.005 [DOI] [PubMed] [Google Scholar]
- Zhang, D. , Mably, A. J. , Walsh, D. M. , & Rowan, M. J. (2017). Peripheral interventions enhancing brain glutamate homeostasis relieve amyloid β‐ and TNFα‐mediated synaptic plasticity disruption in the rat hippocampus. Cerebral Cortex, 27(7), 3724–3735. 10.1093/cercor/bhw193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Ryu, D. , Wu, Y. , Gariani, K. , Wang, X. , Luan, P. , … Auwerx, J. (2016). NAD repletion enhances life span in mice. Science, 6(6292). 10.1126/science.aaf2693 [DOI] [PubMed] [Google Scholar]