

Abstract
Astrocytes are essential for CNS health, regulating homeostasis, metabolism, and synaptic transmission. In addition to these and many other physiological roles, the pathological impact of astrocytes (“reactive astrocytes”) in acute trauma and chronic disease like Alzheimer's disease (AD) is well established. Growing evidence supports a fundamental and active role of astrocytes in multiple neurodegenerative diseases. With a growing interest in normal astrocyte biology, and countless studies on changes in astrocyte function in the context of disease, it may be a surprise that no therapies exist incorporating astrocytes as key targets. Here, we examine unintentional effects of current AD therapies on astrocyte function and theorize how astrocytes may be intentionally targeted for more efficacious therapeutic outcomes. Given their integral role in normal neuronal functioning, incorporating astrocytes as key criteria for AD drug development can only lead to more effective therapies for the millions of AD sufferers worldwide.
Linked Articles
This article is part of a themed section on Therapeutics for Dementia and Alzheimer's Disease: New Directions for Precision Medicine. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.18/issuetoc
Abbreviations
- Aβ
amyloid‐β
- AChEI
AChE inhibitor
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- APPswe/PS1
APP Swedish mutation/presenilin 1
- APPswe
APP Swedish mutation
- BBB
blood–brain barrier
- BDNF
brain‐derived neurotrophic factor
- BuChE (Bche)
butyrylcholinesterase
- GDNF
glial cell‐derived neurotrophic factor
- GFAP
glial fibrillary acidic protein
- GLT
glutamate transporter
- NGF
nerve growth factor
1. INTRODUCTION
Alzheimer's disease (AD) is the most common neurodegenerative disorder and cause of dementia worldwide. Characterized by progressive language, memory, and cognitive loss, AD has been historically categorized as a disease of proteopathic stress caused by abnormal amyloid‐β (Aβ) and tau aggregation with primary toxic consequences affecting neuronal health and function (De Strooper & Karran, 2016; Ginsberg, Che, Counts, & Mufson, 2006; Harding et al., 2018; Mattsson, Schott, Hardy, Turner, & Zetterberg, 2016; Yue & Jing, 2015). More recently, a growing interest has focused on other non‐neuronal CNS cell types and their potential roles in AD aetiology and progression. Of particular interest are astrocytes, given their fundamental roles as regulators of homeostasis, metabolism, and clearance as well as modulators of synaptic plasticity and transmission. Recent evidence established a clear link between immune cell‐induced reactive astrocyte function that leads to neuronal and oligodendroglial cell death. Most strikingly, reactive astrocytes are localized to regions of degeneration in human AD patient post‐mortem tissue (Liddelow et al., 2017), mouse models of tauopathy (Shi et al., 2017), and many other neurodegenerative diseases (Liddelow et al., 2017; Rothhammer et al., 2018; Yun et al., 2018). Given this integral and active role of astrocytes in neurodegeneration, this review will evaluate how current AD pharmacological agents affect astrocyte function and will hypothesize how astrocytes may be targeted for future drug development. We also use the term “reactive astrocyte” or “reactivity” to encompass all AD pathological states of astrocytes. It should be noted that it is unknown exactly how many different states of astrocytes are present during different states of AD progression, or if each state represents a bona fide “reactive astrocyte” or an intermediary and transient state change that may be responsive to, rather than inducive of, AD pathology. This review will not encompass discussions on bioavailability or mode of transport of drug candidates; rather, we will focus on astrocyte targets and the functional changes predicted to occur if they are effectively modulated.
1.1. Effect of AD therapeutics on astrocyte function
1.1.1. AChE inhibitors
In the 1970s, initial biochemical analysis of AD patient brains identified substantial cholinergic deficits, including decreased choline acetyltransferase levels, decreased ACh uptake, decreased ACh release, and fewer cholinergic neurons (among other stereotypical AD pathologies; Francis, Palmer, Snape, & Wilcock, 1999). Paired with emerging knowledge that ACh is critical for learning and memory, the “cholinergic hypothesis of AD” was established (Bartus, Dean, Beer, & Lippa, 1982; Pabst et al., 2016; Revathikumar et al., 2016). This model describes degenerating cholinergic neurons directly causing cognitive dysfunction. In response, AChE inhibitors (AChEIs) were introduced as a means to improve cholinergic neuronal function. Currently, three out of the four current FDA‐approved AD treatments are AChEIs: donepezil, rivastigmine, and galantamine (Alzheimer's Association, 2017). The final non‐AChEI FDA‐approved AD therapy is memantine, which targets glutamate overactivity (discussed below). Regardless of varying modes of action (Supporting Information Table S1), these AChEIs function by delaying breakdown of ACh in the synaptic cleft, increasing the time ACh remains at the synapse, elongating potential for cholinergic neurotransmission (Birks, 2006; Tabet, 2006).
Astrocytes have functional nicotinic and muscarinic receptors and (controversially) may express butyrylcholinesterase (Bche/BuChE) and Ache. Levels of expression and catalytic function appear to vary depending on the stage of development/age and species investigated (Anderson et al., 2008; Elhusseiny, Cohen, Olivier, Stanimirovic, & Hamel, 1999; Pabst et al., 2016; Revathikumar et al., 2016; Teaktong et al., 2003; Thullbery, Cox, Schule, Thompson, & George, 2005). In AD pathology, the number of glial fibrillary acidic protein (GFAP) immunopositive astrocytes in the hippocampus and cortex with nicotinic receptor subtype α7 (α7 nAChR)‐immunoreactivity is increased from 40 –100% in sporadic AD and 50–60% in Swedish amyloid precursor protein (APPswe) 670/671 AD cases, compared to less than 50% in non‐symptomatic patient controls (Teaktong et al., 2003; Yu, Guan, Bogdanovic, & Nordberg, 2005). It should be noted however that this co‐immunofluorescence does not provide significant resolution for spatial localization of receptors within GFAP+ astrocytes. Therefore, increased nicotinic receptor presence in GFAP+ astrocytes may be due to astrocyte‐specific up‐regulation of these receptors and/or may be an artefact from astrocyte‐mediated synapse elimination, a well‐defined early hallmark of AD (Chung, Welsh, Barres, & Stevens, 2015; Clarke & Barres, 2013; Eroglu & Barres, 2010; Hong et al., 2016). Additionally, global cholinesterase activity is markedly changed in AD, with decreased AChE and increased BuChE activity (Giacobini, 2004; Perry, Perry, Blessed, & Tomlinson, 1978). Although less commonly expressed in the brain (representing ~20% of all cholinesterase activity), reports of astrocyte BuChE (Giacobini, 2004; Revathikumar et al., 2016) and increased BuChE may be correlated with astrocyte reactivity (Kadir et al., 2011). Such reports are based on in vitro culture experiments from human fetal material in the presence of serum and therefore may not be representative of either physiological or aged pathological state of astrocytes. Given the prevalence and active role of astrocytes in AD pathology, how do AChEIs and the subsequent increase in ACh affect astrocyte function?
Standard morphological characterization using GFAP immunostaining reveals a loss of traditional astrocyte morphology. This cellular hypertrophy is a standard measurement of astrocyte “health” and is often used to measure cellular atrophy and loss of function in a wide range of neurological disorders including AD (see Pekny et al., 2016 and Verkhratsky, Marutle, Rodriguez‐Arellano, & Nordberg, 2015 for comprehensive review of this topic). Often used as a proxy of multiple forms of astrocyte reactivity, GFAP+ hypertrophied astrocytes return to a physiologically “normal” morphology after AChEI treatment (Liu et al., 2015; Mohamed, Keller, & Kaddoumi, 2016; Unger, Svedberg, Yu, Hedberg, & Nordberg, 2006; Wu, Zhao, Chen, Cheng, & Zhang, 2015)—suggesting a return to physiologically astrocyte functions. Though not an effective measure of astrocyte pathology, there is a gross morphological change in reactive astrocytes upon entering a pathological state (such methods of describing reactive astrocytes lack required fidelity to determine level or state of a reactive phenotype). After 2 months of rivastigmine treatment, a 50% decrease in GFAP levels was detected by fluorescence intensity in the hippocampi of APPswe mice (Mohamed et al., 2016). These mice express human APPswe double mutations (K670N and M671L) that result in significant amyloid plaque deposition by 9 months of age (Hsiao et al., 1996). Similarly, APPswe/presenilin 1 (APPswe/PS1) mice—which express both the human Swedish mutation and have a deletion of PS1, causing significant behavioural deficits, accelerated plaque deposition, and increased inflammation (Holcomb et al., 1998; Jankowsky et al., 2004)—display decreased astrocyte responses, as identified by morphological changes (i.e., fewer swollen, hypertrophic processes) and decreased GFAP levels, after long‐term galantamine treatment (Wu et al., 2015). AChEIs also reduce astrocyte production of ROS and inhibit oxidative stress‐related apoptosis (Liu et al., 2015; Makitani, Nakagawa, Izumi, Akaike, & Kume, 2017). Protective mechanisms are postulated to be due to nicotinic receptor interaction and subsequent activation of the PI3K–Akt pathway (Makitani et al., 2017).
Astrocytes are also key modulators of neuroinflammation in the CNS, secreting pro‐inflammatory and/or anti‐inflammatory cytokines depending on the stimulus/mode of injury (Anderson et al., 2016; Zamanian et al., 2012). Choline receptor stimulation via AChEIs or nicotine agonists can reduce many pro‐inflammatory signals (Tabet, 2006)—making them an appealing target. When activated by IL‐1β, cultured fetal human astrocytes up‐regulated many pro‐inflammatory cytokines (Revathikumar et al., 2016). Pretreating cultures with nicotine prior to IL‐1β stimulation leads to a reduction in IL‐6, TNF‐α, IL‐1β, IL‐8, and IL‐13 production. However, this decreased level of cytokine production never returned to non‐stimulated baselines, suggesting ACh stimulation may only modestly attenuate pro‐inflammatory responses. Additionally, no differences were detected for IFNγ, IL‐2, IL‐44, IL‐10, and IL‐12p70 secretion, and therefore, modulation of these inflammatory cytokines may not be universal (Revathikumar et al., 2016). One should also be wary of such investigations completed in serum‐containing culture conditions, as astrocyte cultures maintained in serum‐containing growth media irreversibly change the transcriptome and several functions of astrocytes (Foo et al., 2011). Although these results may indeed be indicative of bona fide astrocyte responses in vivo, they should be reproduced under more physiological conditions that replicate true astrocyte function. Treating aged APPswe/PS1 mice with galantamine for 2 months also decreases astrocyte production of pro‐inflammatory cytokines, TNF‐α and IL‐6 (Wu et al., 2015). In comparison, bulk hippocampal levels of TNF‐α and IL‐6, as assessed by ELISA, do not change with AChEI treatment (Wu et al., 2015). These discrepancies are likely due to differences in cell type‐specific contributions masked by analysing bulk tissue preparations, as multiple cell types (e.g., microglia, astrocytes, neurons, and endothelial cells) secrete these cytokines (Zhang et al., 2014). Follow‐up studies using single‐cell transcriptomics are essential to understand nuances of how different cells may contribute to a phenotype (i.e., heterogeneity between cell types and heterogeneity of response within a specific cell type) as a means to better understand the phenotype of interest and subsequently develop more efficacious drug targets that take this information into consideration.
Stimulation of nicotinic receptors on striatal hydrogen peroxide‐treated murine astrocytes in vitro suppresses caspase‐9 (Casp9) expression and subsequently inhibits mitochondrial‐dependent apoptotic pathways, permitting maintenance of mitochondrial membrane integrity/pressure and BAX/Bcl‐2 ratios (Liu et al., 2015). LPS‐stimulated neonatal murine astrocytes treated with GTS21, a nicotinic agonist, provide additional support for an astrocyte role in modulating neuroinflammation—decreasing pro‐inflammatory cytokine production measured by NF‐κB pathway inhibition (Patel, McIntire, Ryan, Dunah, & Loring, 2017). In addition to primary effects of AChEIs on astrocyte function, one should consider potential secondary effects—for example, how do AChEIs affect microglia, the resident immune cells of the brain? Neurotoxic astrocyte reactivity is dependent on microglia activation (Liddelow et al., 2017). Therefore, if microglia‐related inflammation decreases, so too would astrocyte reactivity (Liddelow et al., 2017; Yun et al., 2018). Given that ACh attenuates microglial cytokine production, anti‐inflammatory and neuroprotective pathways would be promoted (Liu et al., 2015; Revathikumar et al., 2016; Tabet, 2006). Under these parameters, the cholinergic system may be considered a modulator of astrocyte‐associated neuroinflammation both directly and indirectly (Revathikumar et al., 2016).
Astrocytes provide significant neuronal support through secretion of neurotrophic factors, and choline receptor stimulation, via AChEIs or ACh agonists, and may promote secretion of these factors. As an example, treatment with metrifonate, an irreversible AChEI, up‐regulates secretion of neurotropic factors nerve growth factor (NGF) and brain‐derived neurotrophic factor (BDNF) in cultured rat cortical astrocytes through nicotinic receptor interactions (Mele & Juric, 2014). Additionally, stimulation of nicotinic ACh receptors on oxidative‐stressed midbrain murine astrocytes ameliorates glial cell‐derived neurotrophic factor (GDNF) secretion (Liu et al., 2015). Therefore, activation of astrocyte choline receptors under pathological conditions may provide added support for neighbouring output cells.
1.1.2. NMDA receptor antagonists
Glutamate is the primary excitatory neurotransmitter in the CNS and is critical for learning and memory (Dzamba, Honsa, & Anderova, 2013; Verkhratsky & Kirchhoff, 2007). Overstimulation of NMDA receptors by glutamate is well documented in many neurodegenerative diseases including AD (Reisberg et al., 2003). This overstimulation promotes excessive Ca2+ influx, resulting in free radical production, activation of proteolytic processes, and ultimately cell death (Lee et al., 2010; Lipton, 2006). Modulating glutamate‐mediated NMDA receptor stimulation however is a significant challenge, as glutamate receptor activity is imperative for normal neurotransmission (Lipton, 2006; Suhs et al., 2016). Memantine, an uncompetitive NMDA receptor antagonist that is FDA‐approved for treatment of AD, meets these selective binding requirements by preferentially binding to NMDA receptors when glutamate activates the channel for a significant period of time (Lipton, 2004). In this manner, memantine modulates excessive activation by glutamate without interfering with normal levels of glutamate‐based synaptic transmission (Lipton, 2006; Reisberg et al., 2003).
Understanding astrocyte NMDA receptor expression and functionality is complex due to incomplete understanding of variations in expression levels and functionality between species, at different developmental stages, and in different brain regions (Lee et al., 2010; Suhs et al., 2016). For instance, adult rat CA1 hippocampal astrocytes appear not to express NMDA receptors (Krebs, Fernandes, Sheldon, Raymond, & Baimbridge, 2003), while NMDA receptors are abundant in astrocytes from post‐mortem human cortex (Conti, Barbaresi, Melone, & Ducati, 1999). This may be due to species differences in the transcriptome, and presumably function, of CNS cells (e.g., see Zhang et al., 2016 for comparison of rodent and human glial cells) or areas of the brain assessed. Analysis of astrocyte transcriptome data from the aged mouse in different brain regions however suggests that any difference is more likely species‐dependent (Boisvert, Erikson, Shokhirev, & Allen, 2018; Clarke et al., 2018). More recent single‐cell transcriptome analyses show that many NMDA receptor transcripts are indeed highly expressed by individual astrocytes in particular brain regions (Saunders et al., 2018). Grin2c, for instance, is highly localized to astrocytes in the posterior cortex and striatum, while Grin2b is more equally expressed by other cell types in the same brain regions.
NMDA receptors can be composed of varying subunits: GluN1, GluN2, and GluN3. Two GluN1 subunits are essential for membrane trafficking, while GluN2 and/or GluN3 subunits vary and will subsequently determine pharmacological properties of the receptor (e.g., glutamate affinity and cation permeability), as well as regional and developmental specificity (Dzamba et al., 2013; Palygin, Lalo, & Pankratov, 2011). NMDA receptors present on non‐electrically excitable cells, including those on glia, are postulated to be non‐functional at physiological resting potentials due to extracellular Mg2+ acting as a block on cation influx (Palygin et al., 2011). However, due to glial NMDA receptor subunit differences, these receptors are only weakly sensitive to extracellular Mg2+ and therefore may be activated even at negative resting membrane potentials (Dzamba et al., 2013; Palygin et al., 2011; Verkhratsky & Kirchhoff, 2007). As there is limited Mg2+ block on astrocyte NMDA receptors, these cells may be even more vulnerable to NMDA‐associated excitotoxicity because they do not require depolarization for glutamate binding (Verkhratsky & Kirchhoff, 2007). Consequently, astrocytes may be more sensitive to NMDA receptor antagonists (Palygin et al., 2011). It is therefore important to assess how NMDA receptor antagonists affect astrocyte function in the treatment of AD, and much work going forward will be required to determine the effect of such drugs in non‐neuronal cells with grossly different receptor structure.
NMDA receptor antagonists do not significantly alter gene expression profiles of AD‐associated astrocytes. After 5 months of treatment with riluzole, another NMDA receptor antagonist, 5X familial AD mice, which have five familial‐associated AD mutations including amyloid precursor protein (APP; K670M/N671L, 1716V, and V717I) and PS1 (M146L and L286V) mutations and result in a highly robust AD phenotype (Oakley et al., 2006), have significant reductions of Aβ pathology (including decreased APP, Aβ40, Aβ42, and Aβ oligomer production as well as decreases in both the number and size of plaques) and improved memory performance (Okamoto et al., 2018). However, bulk RNA sequencing of brains from treated mice reveals only a modest return towards physiological astrocyte‐specific transcriptomic expression levels. Disease‐associated microglial‐specific expression is reduced towards normal baselines (Keren‐Shaul et al., 2017), which may indirectly reduce astrocyte reactivity through inhibition of microglia‐mediated neuroinflammation. Although cell type specificities were determined post hoc in these analyses, results suggest that NMDA receptor antagonists have limited ability to modulate reactive astrocytes (Okamoto et al., 2018). In agreement with these findings, memantine treatment over 10 days in 10‐month‐old APPswe mice does not decrease intensity of GFAP+ immunoreactivity, a generic metric of astrocyte reactivity, associated with Aβ plaques (Unger et al., 2006).
Although the primary pharmacological function of NMDA receptor antagonists is associated with blocking cation voltage channels and consequent inhibition of glutamate release, treatment may also increase neurotrophic factor secretion (through unknown mechanisms). Conditioned medium from striatal neonate rat astrocyte cultures exposed to riluzole provides trophic support to motor neuron cultures (Peluffo, Estevez, Barbeito, & Stutzmann, 1997). Additionally, NGF, BDNF, and GDNF synthesis and secretion increase by 109‐, 2‐, and 3.1‐fold, respectively, when cultured murine astrocytes are exposed to riluzole (Mizuta et al., 2001). However, these results may not be directly extrapolated to disease‐relevant treatments, as doses provided in this study were 500 times higher than the maximal plasma concentration of a therapeutic dose.
Astrocyte glutamate transporters (GLTs) are key regulators of glutamate concentration in the synaptic cleft (Carbone, Duty, & Rattray, 2012; Yoshizumi, Eisenach, & Hayashida, 2012). Under excitotoxic conditions, excessive Ca2+ influx can induce cell death in cells, including astrocytes (Lee et al., 2010). Application of NMDA receptor antagonists can attenuate this glutamate‐induced toxicity (Lee et al., 2010) and may be due to up‐regulation of GLTs, such GLT‐1 (EAA2; Slc1a2; Carbone et al., 2012). In striatal murine astrocyte cultures, stressed by removal of growth factor components, subsequent application of riluzole produces a doubling in GLT‐1 protein levels and a minor increase (~30%) in glutamate uptake (Carbone et al., 2012). However, this increased glutamate uptake may have paradoxical effects. Riluzole‐induced glutamate uptake by astrocytes may promote subsequent glutamate Ca2+‐dependent release by increasing intracellular stores of Ca2+ via the reverse mode of Na+/Ca2+ transport (Yoshizumi et al., 2012). This effect is likely due to low concentrations of riluzole used, as higher concentrations would inhibit voltage‐dependent Na+ and Ca2+ channels (Yoshizumi et al., 2012). In addition, glutamate release by astrocytes is contentious, and Ca2+‐dependent vesicular release of glutamate may be overrepresented in in vitro models (Nedergaard & Verkhratsky, 2012). Therefore, the relevance of this phenomenon in AD treatment models may be minimal.
1.2. Therapeutics in the pipeline
As of mid‐2018, the Alzforum Therapeutic Database lists ~100 drug candidates in the clinical pipeline, ranging from Phase I to IV. The majority of these drug agents target amyloid pathology, tau pathology, and/or global neuroinflammation. Notably, there is limited, substantiated information known assessing the effect of these drugs on astrocyte function (Figure 1).
Figure 1.
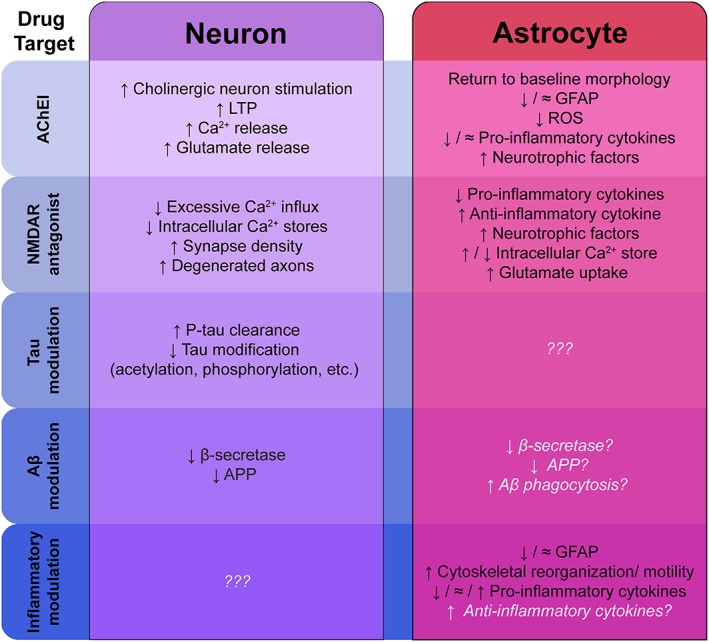
How do Alzheimer's disease therapies affect neurons versus astrocytes? Major drug classes are listed with experimentally validated concepts and mechanisms in black text. White text represents hypothetical effects. Aβ, amyloid‐β; AChEI, AChE inhibitor; APP, amyloid precursor protein; GFAP, glial fibrillary acidic protein; LTP, long‐term potentiation; NMDAR, NMDA receptor; P‐tau, phosphorylated tau; ROS, reactive oxygen species
1.2.1. Amyloid β
Abnormal aggregation of Aβ is a key pathological feature of AD. Neurotoxic astrocytes and pro‐inflammatory disease‐associated microglia are spatially and biochemically associated with this Aβ pathology. As previously discussed, reactive astrocytes localize to areas of degeneration/Aβ plaques in AD (Liddelow et al., 2017; Shi et al., 2017; Wisniewski & Wegiel, 1991). Additionally, astrocytes have tangible levels of APP, β‐secretase (Bace1), and γ‐secretase (Aph1a), and synthesis of these proteins increases in reactive astrocytes under pathological conditions, such as exposure to pro‐inflammatory cytokines (Hong et al., 2003) or Aβ oligomers and fibrils (Zhao, O'Connor, & Vassar, 2011) in astrocyte cultures from APPswe transgenic mice as well as in human AD patient samples (Hartlage‐Rubsamen et al., 2003). More recent evidence shows a strong correlation between Aβ, microglia, astrocytes, and overall neuronal health in a novel 3D culture system (Park et al., 2018). It will be exciting to see, going forward, how modulation of individual cell components of this system alters deposition of Aβ and ultimately neuronal viability and function. Therefore, the effect of Aβ modifying drugs (i.e., enhanced clearance and inhibition of key enzymes) on astrocyte function should be carefully evaluated.
1.2.2. Tau
Another pathological hallmark of AD, the inclusion of neurofibrillary tangles, is characterized by hyperphosphorylated tau (Hampel et al., 2010; Javidnia, Hebron, Xin, Kinney, & Moussa, 2017). Under normal physiological conditions, tau is primarily localized to mature neuronal axons and aids in stabilizing microtubule assembly and regulating axonal transport (Garwood, Cooper, Hanger, & Noble, 2010; Leyns & Holtzman, 2017). Phosphorylation of tau determines its affinity for tubulin (Leyns & Holtzman, 2017). If tau is inappropriately or excessively phosphorylated, microtubule assembly becomes inhibited (Leyns & Holtzman, 2017). Although primarily expressed in neurons, tau is also expressed at low levels in oligodendrocytes (LoPresti, Szuchet, Papasozomenos, Zinkowski, & Binder, 1995) and astrocytes (Buee, Bussiere, Buee‐Scherrer, Delacourte, & Hof, 2000; Chiarini, Armato, Gardenal, Gui, & Dal Pra, 2017; Kahlson & Colodner, 2015; Leyns & Holtzman, 2017). Additionally, astrocytes produce cytokines, such as TNF‐α and IL‐6 (Wu et al., 2015), that subsequently intensify tau hyper‐phosphorylation, exacerbating overall tau pathology (von Bernhardi, Eugenin‐von Bernhardi, & Eugenin, 2015). This presence of tau may inhibit neuroprotective capabilities of astrocytes. In co‐cultures of neurons and astrocytes isolated from either C57BL/6 or mutant human tau P301S transgenic mice, in which tau is expressed under the Thy1.2 (Cd90) promoter and therefore only present in neurons (Allen et al., 2002), P301S astrocytes fail to protect neurons from cell death (Sidoryk‐Wegrzynowicz et al., 2017). Upon transplantation of C57BL/6 astrocytes or addition of C57BL/6 astrocyte‐conditioned medium to neuron cultures, neuronal survival significantly improved, indicating that astrocytes develop pathological changes upon exposure to P301S neurons. This neuroprotective phenomenon was also replicated in vivo by transplanting C57BL/6 astrocytes into P301S mice (Hampton et al., 2010). Astrocytes may also mediate Aβ‐induced tau phosphorylation and cleavage. Mixed rat cortical in vitro cultures have significantly increased tau phosphorylation, compared to neuronal monocultures, suggesting astrocyte presence exacerbates neurotoxicity (Garwood, Pooler, Atherton, Hanger, & Noble, 2011). Therefore, tau may affect glia (“astrogliotauopathy”; Kovacs et al., 2016), and glia may affect tau pathology. The effects of tau modulating treatments (i.e., tau aggregation inhibition and phosphorylated tau clearance) on glial function are yet to be confirmed.
1.2.3. Inflammation
Inflammation is a critical component of AD pathology. Given astrocytes, along with microglia and peripheral immune cells, function as key regulators of inflammation in the CNS (Anderson et al., 2016; Zamanian et al., 2012), modulation of these cells may help prevent pro‐inflammatory responses. As an example, minocycline, an antibiotic with well‐defined anti‐inflammatory properties, would be a key drug candidate for such immunomodulation. Minocycline treatment over 2 weeks reduces the number of activated astrocytes in the cortex of young human tau transgenic mice (Garwood et al., 2010), as identified by intensity of GFAP immunohistochemistry and morphology (i.e., fewer hypertrophied soma and thick processes). Interestingly, no differences in astrocyte morphology were identified in the hippocampus, which may be due to regional‐specific heterogeneity of astrocytes and/or may highlight the limited ability to identify the reactive state of an astrocyte using simplistic GFAP intensity and morphological assessment. As anticipated, minocycline treatment significantly reduced global levels of many pro‐inflammatory factors (GM‐CSF, I‐309, eotaxin, IL‐6, IL‐10, M‐CSF, MCP‐1 (CCL2), MCP‐5 (CCL12), and TREM‐1), while other factors remained similar to untreated baselines (BCL, C5a, G‐CSF, sICAM‐1, IFNγ, IL‐1α, IL‐1ra, IL‐2, IL‐3, IL‐4, IL‐5, IL‐7, IL‐13, IL‐12p70, IL‐16, IL‐23, IL‐27, IP‐10 (CXCL10), iTAC (CXCL11), KC (mouse CXCL1), MIG (CXCL9), MIO‐1α, MIP‐2 (human CXCL1), RANTES (CCL5), SDF‐1 (CXCL‐12β), TARC (CCL17), TIMP‐1, and TNF‐α). Surprisingly, a handful of cytokine factors increased (IL‐17, IL‐1β, and MIP‐1β (CCL4); Garwood et al., 2010). Since levels of cytokines were gathered from bulk tissue preparations, follow‐up assessments of cell type‐specific contributions to these cytokine changes are needed to add nuance to what cell type therapeutics should target.
Another drug class with potential to modulate inflammation in AD and induce intriguing effects on astrocyte function is non‐steroidal anti‐inflammatory drugs. Chronic (9‐month) treatment with CHF5074 (a novel γ‐secretase modulator) in Tg2576 mice increased localization of reactive astrocytes around Aβ plaques (Imbimbo et al., 2010). These changes may be indicative of cytoskeletal reorganization to promote migration to areas of injury (Lichtenstein et al., 2010), potentially allowing for increased astrocyte‐mediated neuroprotection (i.e., phagocytosis of Aβ plaques).
1.3. Future specific astrocyte targets as a potential AD therapy
There are several possible targeting strategies to ameliorate astrocyte dysfunction during AD. One does however need to determine what stage during disease would provide the most effective astrocyte‐specific targets. Many studies report astrocyte “activation” early in the pathogenesis of AD (Carter et al., 2012; Owen et al., 2009; Schipper et al., 2006); however, we still understand very little about their role in the initiation, progression, or possible resolution of disease (Figure 2). Although no astrocyte‐specific therapies currently exist for AD, there are several promising possibilities. We will briefly discuss a small number of these here.
Figure 2.
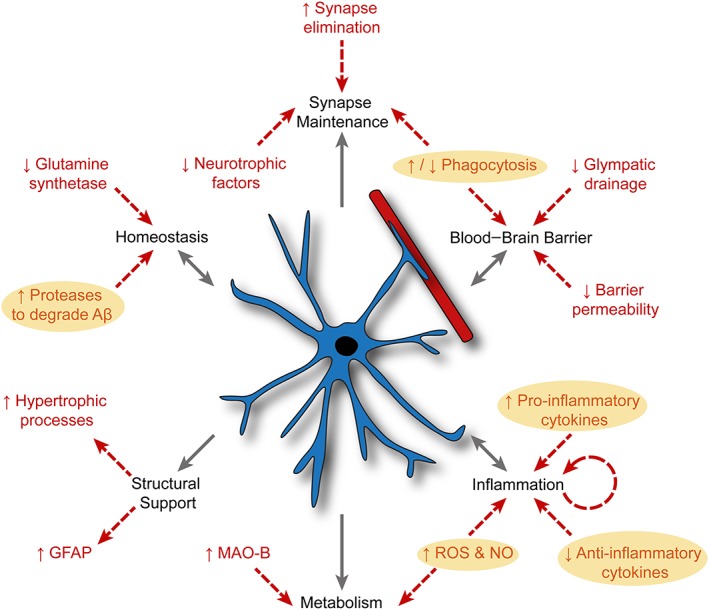
Alzheimer's disease significantly disrupts many major astrocyte functions. Major astrocyte functions are listed in black. Red text and arrows indicate documented examples of Alzheimer's disease‐associated astrocyte perturbation. Yellow circles indicate added influence of activated microglia. Aβ, amyloid‐β; GFAP, glial fibrillary acidic protein; NO, nitric oxide; MAO‐B, monoamine oxidase B; ROS, reactive oxygen species
1.3.1. Homeostasis/trophic support of neurons and synapses
Astrocytes secrete a number of known and unknown neurotrophic factors required for normal neuronal health and extension of neurite outgrowths (Meyer‐Franke, Kaplan, Pfrieger, & Barres, 1995; Verkhratsky, Matteoli, Parpura, Mothet, & Zorec, 2016). These factors also likely play a key role in synaptic plasticity and signal strength (with unhealthy neurons likely unable to maintain such connections). The importance of activity‐dependent plasticity should not be overlooked however, as neuronal activity is known to cause release of many neurotrophic factors, like NGF and BDNF (Ernfors, Bengzon, Kokaia, Persson, & Lindvallt, 1991; Isackson, Huntsman, Murray, & Gall, 1991). There is also strong evidence that neuronal activity can control synthesis, secretion, and signalling mediated by such neurotrophins (Kang & Schuman, 1991; see also Schinder & Poo, 2000; Schipper et al., 2006 for review). But what role would astrocytes play in such a neuron‐dominated hypothesis?
Astrocytes are intimately involved at the synapse and along with the pre‐ and post‐synaptic neuronal terminals form the “tripartite synapse” (Araque, Parpura, Sanzgiri, & Haydon, 1999) or “astrocyte cradle” (Verkhratsky & Nedergaard, 2014). In this location, astrocytes help form structural and functional synapses (Allen et al., 2012; Blanco‐Suarez, Liu, Kopelevich, & Allen, 2018; Christopherson et al., 2005; Singh et al., 2016; Stogsdill et al., 2017), but in the context of disease, the capacity of astrocytes to continue such functions is decreased (Liddelow et al., 2017). Additionally, due to loss of synaptic density early in AD (Hong et al., 2016), it is likely that astrocytes play a major role propagating disease progression. Would targeting astrocytes to produce more synaptogenic factors, like glypicans, thrombospondins, neuroligins, hevin/sparc, and chordin‐like 1, alleviate synaptic and memory loss in patients? Or would such efforts cause an imbalance in neuron/synapse compliments leading to additional complications like epilepsy?
In contrast to neurons in the peripheral nervous system, CNS neurons are not maintained by exogenous delivery of target‐derived neurotrophic factors following axotomy (Ernfors, Lee, Kucera, & Jaenisch, 1994; Farinas, Jones, Backus, Wang, & Reichardt, 1994). Although reactive astrocytes present in regions of neurodegeneration in AD patient post‐mortem tissues are known to secrete a potent neurotoxin (Liddelow et al., 2017), this toxicity is not due to a lack of astrocyte‐derived neurotrophins. As such, it seems unlikely that simply adding more trophic support will be sufficient to maintain neuronal numbers (although this may be different at different stages of disease). As such, future pharmaceuticals targeting astrocyte–neuron viability will likely be more effective if they block secretion of toxic factors from astrocytes or receptor binding/uptake into neurons.
1.3.2. Neuroinflammation
Inflammation is the body's immune response that drives self‐protection. It both removes harmful stimuli and begins the healing process. Infection, traumatic injury, and disease that damage tissue are unable to heal without an inflammatory response. However, we also know that inflammation can be a major contributor to cognitive decline and neurodegeneration. In most cases, it is apparent that the problem with inflammation is not how often it starts but how often it fails to subside (Nathan & Ding, 2010). It is therefore not the presence of inflammation that is a likely initiator/progressor of AD but non‐resolving inflammation that is causing most problems.
AD pathology is overwhelmingly characterized by such detrimental chronic, non‐resolving inflammation. Resident microglial cells and infiltrating peripheral immune cells, like macrophages, primarily drive this inflammation (Heneka et al., 2015; Zenaro et al., 2015). More recently, the involvement of inflammation‐induced reactive astrocytes is also gaining interest (Liddelow et al., 2017; Shi et al., 2017; Zamanian et al., 2012). Multiple lines of communication exist between the nervous and immune system cells, and dysregulation of this communication represents a fundamental principle underlying neuroinflammation. Immune cell‐derived inflammatory molecules are critical for the regulation of host responses to inflammation—an important part of wound healing and repair. In a chronic setting, however, this maintained state of inflammation inside the CNS correlates (perhaps initiates?) with neurodegenerative diseases like AD. So how would one target inflammation with pharmacotherapeutics?
In the field of multiple sclerosis, much effort has been placed in developing anti‐inflammatory agents to help alleviate symptoms and progression for patients. Immunomodulatory drugs have beneficial properties (e.g., fingolimod and laquinimod), which exert effects by targeting peripheral immune cells and reactive astrocytes—indicating that their modulation may be a promising therapeutic option (Bruck et al., 2012; Choi et al., 2011). But care must be taken. TNF‐α blockers have been shown to have a relatively safe profile, but non‐selective inhibitory drugs of TNF‐α in multiple sclerosis are contradictory in their effects. The earliest investigation of anti‐TNF‐α therapies in demyelinating mouse models reported beneficial anti‐inflammatory effects. As a result, clinical trials of lenercept (van Oosten et al., 1996) and infliximab lenercept (Lenercept MS Study Group and University of British Columbia MS/MRI Analysis Group, 1999) were rushed to human clinical trials—and caused a paradoxical increase in disease activity. In addition, dangerous side effects, including increased risk of secondary infection, T‐cell lymphoma, congestive heart failure, lupus‐like syndromes, diabetes mellitus, psoriasis, and myasthenia gravis (muscle weakness), among others, were reported (Bosch, Saiz et al., 2011; British Thoroacic Society Standards of Care Committee, 2005; Bruzzese et al., 2015; Ramos‐Casals et al., 2007; Strangfeld & Listing, 2006). So is anti‐inflammatory therapy a cross too dangerous to bear?
The very first suspicion that anti‐inflammatory drugs may decrease risk of AD was from the observation that people with rheumatoid arthritis on anti‐inflammatory therapy had an unexpectedly low prevalence of dementia (McGeer, McGeer, Rogers, & Sibley, 1990). While extensive laboratory and epidemiological studies have also suggested that anti‐inflammatory medications can defer or prevent AD occurrence, there remain several studies that do not corroborate these findings. On the whole, however, it seems anti‐inflammatory therapies that target chronic, non‐resolving inflammation are beneficial (see Wang et al., 2015 for meta‐analysis). So why this disconnect, and why have no blockbuster anti‐AD drugs hit the market? This may be partially due to low penetrance of most anti‐inflammatory drugs across the blood–brain barrier (BBB) into the CNS. It may also be a consequence of therapeutics unable to discern between new acute (helpful) inflammation compared with old chronic (harmful) inflammation already present in the brain. An ideal anti‐inflammatory treatment would be both a CNS penetrant and sufficiently specific to target only that inflammatory response (be it immune cells or astrocytes) that is driving degeneration and dementia. As immune cells are primed to respond rapidly to insult and inflammation, and resolve almost as quickly upon removal of the initiating pathogen, more successful therapies in the future may instead target chronic astrocyte inflammatory responses. It may be that the slow‐to‐respond and chronically reactive astrocytes prove a better target for specific astrocyte anti‐inflammatory therapeutics.
1.3.3. BBB maintenance
Astrocytes, although not involved in the formation of the BBB (instead, this is initiated by Wnt/β‐catenin signalling and pericytes; Daneman et al., 2009; Daneman et al., 2010; Daneman, Zhou, Kebede, & Barres, 2010), are intimately involved in its normal regulation and functioning. Many studies have implicated cerebrovascular disorders with AD—suggesting an important role of the blood vasculature in the initiation and progression of the disease (Farkas & Luiten, 2001; Viswanathan & Greenberg, 2011; see also de la Torre, 2004 and Jellinger, 2008 for clinical review). In addition, there is the complication of normal age‐related deterioration of the BBB (Montagne et al., 2015), in much the same way that there are ageing changes in the astrocyte transcriptome that drives them to a pro‐inflammatory reactive state (Boisvert et al., 2018; Clarke et al., 2018). This makes it difficult to discern the causative or correlative role of barrier breakdown in the context of AD.
The deposition of Aβ (Carrano et al., 2011; Roher et al., 2003) and tau (Forman et al., 2005; Vidal et al., 2000) in the cerebral vasculature appears to drive a pro‐inflammatory environment, setting up a positive feedback look leading to greater cytotoxic events and barrier permeability. In validation of these hypotheses, depletion of tau in rTg4510 tauopathy mice decreases extravasation of IgG, indicative of a less permeable vascular barrier in the brain (Blair et al., 2015). This suggests barrier integrity can be maintained/revived by mediating levels of extracellular AD pathological proteins. As microglia and astrocytes are the major phagocytes in the brain, it is likely that these cells are clearing such debris during disease. But what happens to their phagocytic ability during disease?
Microglia, along with astrocytes, are known to localize with amyloid plaques in the brains of AD patients, and it has been shown that amyloid can stimulate microglia to phagocytose these protein deposits (Bard et al., 2000; Schenk et al., 1999). Similarly, blocking microglial phagocytosis of amyloid causes an increase in plaque load in brains of amyloidogenic human APP mice (Wyss‐Coray et al., 2001), which have human APP Swedish double mutations as well as the London (V717F) mutation that result in elevated levels of Aβ, plaque deposition, and gliosis (Mucke et al., 2000). Closer inspection of these results shows that experiments that blocked microglial phagocytosis were accompanied by an increase in expression of the complement component C3 (recently shown to be highly expressed by neurotoxic reactive astrocytes; Liddelow et al., 2017). What connections therefore exist between microglial removal of pathogenic proteins, astrocyte activation, and BBB permeability?
As stated above, astrocytes are not involved in the formation of the BBB (Daneman et al., 2009) but are intimately involved in its maintenance and restoration following insult from injury and in disease. The astrocyte endfeet that encapsulate blood vessels form a secondary barrier between the periphery and the CNS termed the “glia limitans” (see Engelhardt & Coisne, 2011; Verkhratsky, Nedergaard, & Hertz, 2015). It is also these endfeet with high levels of the water channel aquaporin 4 (AQP4) that mediate glymphatic drainage of toxic metabolites out of the CNS during sleep (Iliff et al., 2013). As there is impairment of these paravascular clearance pathways in both normal ageing (Kress et al., 2014) and in disease (Peng et al., 2016; Weller, Subash, Preston, Mazanti, & Carare, 2008), the targeting of such structures may indeed prove beneficial in removal of AD pathogenic proteins. In this way, one would not need to target phagocytic function of cells per se but instead could improve passive drainage of such molecules out of the brain through the glymphatic system.
2. CONCLUSIONS
There are currently ~5.5 million Americans living with AD with a corresponding healthcare cost of around 259 billion US dollars. Over 16 million Americans are expected to develop AD by 2050 with a corresponding estimated healthcare cost of over 1 trillion dollars (Alzheimer's Association, 2017). Current FDA‐approved drugs provide modest symptomatic relief for AD patients. Could this be because glial populations are not being accounted for, even though they respond to these drugs? Could taking astrocyte modulation into account enhance pharmacological action in neurons? Systematic review of clinical trials reveals AChEI treatment is modestly effective compared to placebo groups (Lanctôt et al., 2003; Zhang & Neubert, 2011). Additionally, 30–40% of AD patients do not respond to AChEI treatment (McGleenon, Dynan, & Passmore, 1999). The same is true of any current AD drug class under investigation. While headlines in the press bemoan the failure of drugs almost every month, we are learning more and more about this devastating disease.
With increasing age as the primary risk factor for AD development and a steadily growing ageing world population, a greater understanding of AD aetiology and pathogenesis is imperative in order to help alleviate the physical, emotional, and financial burden of this devastating disease on patients and their families. Our understanding of the non‐neuronal contributions to chronic neurodegenerative diseases is leaving its infancy and entering adolescence—there is much upheaval and excitement to come.
2.1. Nomenclature of target and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently achieved in the Concise Guide to PHARMACOLOGY 2017/2018 (Alexander, Christopoulos, et al., 2017; Alexander, Fabbro, et al., 2017; Alexander, Kelly, et al., 2017a, b, c; Alexander, Peters, et al., 2017).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Table S1. Supporting information.
Sadick JS, Liddelow SA. Don't forget astrocytes when targeting Alzheimer's disease. Br J Pharmacol. 2019;176:3585–3598. 10.1111/bph.14568
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Overview. British Journal of Pharmacology, 174, S1–S16. 10.1111/bph.13882/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Other ion channels. British Journal of Pharmacology, 174, S195–S207. 10.1111/bph.13881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017c). The Concise Guide To PHARMACOLOGY 2017/18: Transporters. British Journal of Pharmacology, 174, S360–S446. 10.1111/bph.13883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). The concise guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, B. , Ingram, E. , Takao, M. , Smith, M. J. , Jakes, R. , Virdee, K. , … Goedert, M. (2002). Abundant tau filaments and nonapoptoic neurodegeneration in transgenic mice expressing human P301S tau protein. Journal of Neuroscience, 22(21), 9340–9351. 10.1523/JNEUROSCI.22-21-09340.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, N. J. , Bennett, M. L. , Foo, L. C. , Wang, G. X. , Chakraborty, C. , Smith, S. J. , & Barres, B. A. (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature, 486(7403), 410–414. 10.1038/nature11059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Association (2017). 2017 Alzheimer's disease facts and figures. Alzheimer's & Dementia, 13(4), 325–373. 10.1016/j.jalz.2017.02.001 [DOI] [Google Scholar]
- Anderson, A. A. , Ushakov, D. S. , Ferenczi, M. A. , Mori, R. , Martin, P. , & Saffell, J. L. (2008). Morphoregulation by acetylcholinesterase in fibroblasts and astrocytes. Journal of Cellular Physiology, 215(1), 82–100. 10.1002/jcp.21288 [DOI] [PubMed] [Google Scholar]
- Anderson, M. A. , Burda, J. E. , Ren, Y. , Ao, Y. , O'Shea, T. M. , Kawaguchi, R. , … Sofroniew, M. V. (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature, 532(7598), 195–200. 10.1038/nature17623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque, A. , Parpura, V. , Sanzgiri, R. P. , & Haydon, P. G. (1999). Tripartite synapses: Glia, the unacknowledged partner. Trends in Neuroscience, 22(5), 208–215. [DOI] [PubMed] [Google Scholar]
- Bard, F. , Cannon, C. , Barbour, R. , Burke, R.‐L. , Games, D. , Grajeda, H. , … Yednock, T. (2000). Peripherally administered antibodies against amyloid β‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Medicine, 6(8), 916–919. [DOI] [PubMed] [Google Scholar]
- Bartus, R. T. , Dean, R. L. , Beer, B. , & Lippa, A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science, 217(4558), 408–414. [DOI] [PubMed] [Google Scholar]
- Birks, J. (2006). Cholinesterase inhibitors for Alzheimer's disease (review). Cochrane Database System Review, (1), CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair, L. J. , Frauen, H. D. , Zhang, B. , Nordhues, B. A. , Bijan, S. , Lin, Y. C. , … Dickey, C. A. (2015). Tau depletion prevents progressive blood–brain barrier damage in a mouse model of tauopathy. Acta Neuropathologica Communications, 3, 8 10.1186/s40478-015-0186-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco‐Suarez, E. , Liu, T. F. , Kopelevich, A. , & Allen, N. J. (2018). Astrocyte‐secreted chordin‐like 1 drives synapse maturation and limits plasticity by increasing synaptic GluA2 AMPA receptors. Neuron, 100(5):1116–1132.e13. 10.1016/j.neuron.2018.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert, M. M. , Erikson, G. A. , Shokhirev, M. N. , & Allen, N. J. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Reports, 22(1), 269–285. 10.1016/j.celrep.2017.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch, X. , Saiz, A. , Ramos‐Casals, M. , & BIOGEAS Study Group (2011). Monoclonal antibody therapy‐associated neurological disorders. Nature Reviews Neurology, 7, 165–172. 10.1038/nrneurol.2011.1 [DOI] [PubMed] [Google Scholar]
- British Thoroacic Society Standards of Care Committee (2005). BTS recommendations for assessing risk and for managing Mycobacterium tuberculosis infection and disease in patients due to start anti‐TNF‐alpha treatment. Thorax, 60(10), 800–805. 10.1136/thx.2005.046797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck, W. , Pfortner, R. , Pham, T. , Zhang, J. , Hayardeny, L. , Piryatinsky, V. , … Wegner, C. (2012). Reduced astrocytic NF‐κB activation by laquinimod protects from cuprizone‐induced demyelination. Acta Neuropathologica, 124, 411–424. 10.1007/s00401-012-1009-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzese, V. , Marrese, C. , Scolieri, P. , Hassan, C. , Lorenzetti, R. , & Zullo, A. (2015). Myasthenia gravis onset during rheumatic disease: A new paradoxical effect of anti‐TNF alpha therapy? International Journal of Rheumatic Diseases, 18, 375–376. [DOI] [PubMed] [Google Scholar]
- Buee, L. , Bussiere, T. , Buee‐Scherrer, V. , Delacourte, A. , & Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Research Reviews, 33, 95–130. [DOI] [PubMed] [Google Scholar]
- Carbone, M. , Duty, S. , & Rattray, M. (2012). Riluzole elevates GLT‐1 activity and levels in striatal astrocytes. Neurochemistry International, 60(1), 31–38. 10.1016/j.neuint.2011.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano, A. , Hoozemans, J. J. , van der Vies, S. M. , Rozemuller, A. J. , van Horssen, J. , & de Vries, H. E. (2011). Amyloid beta induces oxidative stress‐mediated blood–brain barrier changes in capillary amyloid angiopathy. Antioxidants & Redox Signaling, 15(5), 1167–1178. 10.1089/ars.2011.3895 [DOI] [PubMed] [Google Scholar]
- Carter, S. F. , Scholl, M. , Almkvist, O. , Wall, A. , Engler, H. , Langstrom, B. , & Nordberg, A. (2012). Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C‐deuterium‐L‐deprenyl: A multitracer PET paradigm combining 11C‐Pittsburgh compound B and 18F‐FDG. Journal of Nuclear Medicine, 53(1), 37–46. 10.2967/jnumed.110.087031 [DOI] [PubMed] [Google Scholar]
- Chiarini, A. , Armato, U. , Gardenal, E. , Gui, L. , & Dal Pra, I. (2017). Amyloid β‐exposed human astrocytes overproduce phospho‐tau and overrelease it within exosomes, effects suppressed by calcilytic NPS 2143‐further implications for Alzheimer's therapy. Frontiers in Neuroscience, 11, 217 10.3389/fnins.2017.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J. W. , Gardell, S. E. , Herr, D. R. , Rivera, R. , Lee, C.‐W. , Noguchi, K. , … Chun, J. (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1‐phosphate receptor 1 (S1P1) modulation. Proceedings of the National Academy of Sciences, 108(2), 751–756. 10.1073/pnas.1014154108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson, K. S. , Ullian, E. M. , Stokes, C. C. , Mullowney, C. E. , Hell, J. W. , Agah, A. , … Barres, B. A. (2005). Thrombospondins are astrocyte‐secreted proteins that promote CNS synaptogenesis. Cell, 120(3), 421–433. 10.1016/j.cell.2004.12.020 [DOI] [PubMed] [Google Scholar]
- Chung, W. S. , Welsh, C. A. , Barres, B. A. , & Stevens, B. (2015). Do glia drive synaptic and cognitive impairment in disease? Nature Neuroscience, 18(11), 1539–1545. 10.1038/nn.4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L. E. , & Barres, B. A. (2013). Emerging roles of astrocytes in neural circuit development. Nature Reviews Neuroscience, 14(5), 311–321. 10.1038/nrn3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L. E. , Liddelow, S. A. , Chakraborty, C. , Munch, A. E. , Heiman, M. , & Barres, B. A. (2018). Normal aging induces A1‐like astrocyte reactivity. Proceedings of the National Academy of Sciences, 115(8), E1896–E1905. 10.1073/pnas.1800165115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti, F. , Barbaresi, P. , Melone, M. , & Ducati, A. (1999). Neuronal and glial localization of NR1 and NR2A/B subunits of the NMDA receptor in the human cerebral cortex. Cerebral Cortex, 9, 110–120. [DOI] [PubMed] [Google Scholar]
- Daneman, R. , Agalliu, D. , Zhou, L. , Kuhnert, F. , Kuo, C. J. , & Barres, B. A. (2009). Wnt/beta‐catenin signaling is required for CNS, but not non‐CNS, angiogenesis. Proceedings of the National Academy of Sciences, 106(2), 641–646. 10.1073/pnas.0805165106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman, R. , Zhou, L. , Agalliu, D. , Cahoy, J. D. , Kaushal, A. , & Barres, B. A. (2010). The mouse blood–brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS One, 5(10), e13741 10.1371/journal.pone.0013741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman, R. , Zhou, L. , Kebede, A. A. , & Barres, B. A. (2010). Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature, 468(7323), 562–566. 10.1038/nature09513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper, B. , & Karran, E. (2016). The cellular phase of Alzheimer's disease. Cell, 164(4), 603–615. 10.1016/j.cell.2015.12.056 [DOI] [PubMed] [Google Scholar]
- de la Torre, J. C. (2004). Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. The Lancet Neurology, 3(3), 184–190. 10.1016/s1474-4422(04)00683-0 [DOI] [PubMed] [Google Scholar]
- Dzamba, D. , Honsa, P. , & Anderova, M. (2013). NMDA receptors in glial cells: Pending questions. Current Neuropharmacology, 11, 250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhusseiny, A. , Cohen, Z. , Olivier, A. , Stanimirovic, D. B. , & Hamel, E. (1999). Functional acetylcholine muscarinic receptor subtypes in human brain microcirculation: Identification and cellular localization. Journal of Cerebral Blood Flow and Metabolism, 19, 794–802. [DOI] [PubMed] [Google Scholar]
- Engelhardt, B. , & Coisne, C. (2011). Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two‐walled castle moat surrounding the CNS castle. Fluids and Barriers of the CNS, 8(1), 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors, P. , Bengzon, J. , Kokaia, Z. , Persson, H. , & Lindvallt, O. (1991). Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron, 7, 165–176. [DOI] [PubMed] [Google Scholar]
- Ernfors, P. , Lee, K.‐F. , Kucera, J. , & Jaenisch, R. (1994). Lack of neurotrophin‐3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell, 77, 503–512. [DOI] [PubMed] [Google Scholar]
- Eroglu, C. , & Barres, B. A. (2010). Regulation of synaptic connectivity by glia. Nature, 468(7321), 223–231. 10.1038/nature09612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinas, I. , Jones, K. R. , Backus, C. , Wang, X.‐Y. , & Reichardt, L. F. (1994). Severe sensory and sympathetic deficits in mice lacking neurotrophin‐3. Nature, 369, 658–661. [DOI] [PubMed] [Google Scholar]
- Farkas, E. , & Luiten, P. G. M. (2001). Cerebral microvascular pathology in aging and Alzheimer's disease. Progress in Neurobiology, 64, 575–611. [DOI] [PubMed] [Google Scholar]
- Foo, L. C. , Allen, N. J. , Bushong, E. A. , Ventura, P. B. , Chung, W. S. , Zhou, L. , … Barres, B. A. (2011). Development of a method for the purification and culture of rodent astrocytes. Neuron, 71(5), 799–811. 10.1016/j.neuron.2011.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman, M. S. , Lal, D. , Zhang, B. , Dabir, D. V. , Swanson, E. , Lee, V. M. , & Trojanowski, J. Q. (2005). Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. Journal of Neuroscience, 25(14), 3539–3550. 10.1523/JNEUROSCI.0081-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, P. T. , Palmer, A. M. , Snape, M. , & Wilcock, G. K. (1999). The cholinergic hypothesis of Alzheimer's disease: A review of progress. Journal of Neurology, Neurosurgery, and Psychiatry, 66, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garwood, C. J. , Cooper, J. D. , Hanger, D. P. , & Noble, W. (2010). Anti‐inflammatory impact of minocycline in a mouse model of tauopathy. Frontiers in Psychiatry, 1, 136 10.3389/fpsyt.2010.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garwood, C. J. , Pooler, A. M. , Atherton, J. , Hanger, D. P. , & Noble, W. (2011). Astrocytes are important mediators of Aβ‐induced neurotoxicity and tau phosphorylation in primary culture. Cell Death & Disease, 2(6), e167–e167. 10.1038/cddis.2011.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini, E. (2004). Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacological Research, 50(4), 433–440. 10.1016/j.phrs.2003.11.017 [DOI] [PubMed] [Google Scholar]
- Ginsberg, S. D. , Che, S. , Counts, S. E. , & Mufson, E. J. (2006). Single cell gene expression profiling Alzheimer's disease. NeuroRx, 3, 302–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel, H. , Blennow, K. , Shaw, L. M. , Hoessler, Y. C. , Zetterberg, H. , & Trojanowski, J. Q. (2010). Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Experimental Gerontology, 45(1), 30–40. 10.1016/j.exger.2009.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton, D. W. , Webber, D. J. , Bilican, B. , Goedert, M. , Spillantini, M. G. , & Chandran, S. (2010). Cell‐mediated neuroprotection in a mouse model of human tauopathy. The Journal of Neuroscience, 30(30), 9973–9983. 10.1523/JNEUROSCI.0834-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlage‐Rubsamen, M. , Zeitschel, U. , Apelt, J. , Gartner, U. , Franke, H. , Stahl, T. , … Rossner, S. (2003). Astrocytic expression of the Alzheimer's disease beta‐secretase (BACE1) is stimulus‐dependent. Glia, 41(2), 169–179. 10.1002/glia.10178 [DOI] [PubMed] [Google Scholar]
- Heneka, M. T. , Carson, M. J. , Khoury, J. E. , Landreth, G. E. , Brosseron, F. , Feinstein, D. L. , … Kummer, M. P. (2015). Neuroinflammation in Alzheimer's disease. The Lancet Neurology, 14(4), 388–405. 10.1016/s1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb, L. , Gordon, M. N. , McGowan, E. , Yu, X. , Benkovic, S. , Jantzen, P. , … Duff, K. (1998). Accelerated Alzheimer‐type phenotype in transgenic mice carrying both mutant amyloid precursor prlotein and presenilin 1 transgenes. Nature Medicine, 4(1), 97–100. [DOI] [PubMed] [Google Scholar]
- Hong, H. S. , Hwang, E. M. , Sim, H. J. , Cho, H. J. , Boo, J. H. , Oh, S. S. , … Mook‐Jung, I. (2003). Interferon γ stimulates β‐secretase expression and sAPPβ production in astrocytes. Biochemical and Biophysical Research Communications, 307(4), 922–927. 10.1016/s0006-291x(03)01270-1 [DOI] [PubMed] [Google Scholar]
- Hong, S. , Beja‐Glasser, V. F. , Nfonoyim, B. M. , Frouin, A. , Li, S. , Ramakrishnan, S. , … Stevens, B. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352(6286), 712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao, K. , Chapman, P. , Nilsen, S. , Eckman, C. , Harigaya, Y. , Younkin, S. , … Cole, G. (1996). Correlative memory deficits, AB elevation, and amyloid plaques in transgenic mice. Science, 274(5284), 99–102. 10.1126/science.274.5284.99 [DOI] [PubMed] [Google Scholar]
- Iliff, J. J. , Lee, H. , Yu, M. , Feng, T. , Logan, J. , Nedergaard, M. , & Benveniste, H. (2013). Brain‐wide pathway for waste clearance captured by contrast‐enhanced MRI. Journal of Clinical Investigation, 123(3), 1299–1309. 10.1172/JCI67677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo, B. P. , Giardino, L. , Sivilia, S. , Giuliani, A. , Gusciglio, M. , Pietrini, V. , … Calza, L. (2010). CHF5074, a novel gamma‐secretase modulator, restores hippocampal neurogenesis potential and reverses contextual memory deficit in a transgenic mouse model of Alzheimer's disease. Journal of Alzheimer's Disease, 20(1), 159–173. 10.3233/JAD-2010-1366 [DOI] [PubMed] [Google Scholar]
- Isackson, P. J. , Huntsman, M. M. , Murray, K. D. , & Gall, C. M. (1991). BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: Temporal patterns of induction distinct from NGF. Neuron, 6, 937–948. [DOI] [PubMed] [Google Scholar]
- Jankowsky, J. L. , Fadale, D. J. , Anderson, J. , Xu, G. M. , Gonzales, V. , Jenkins, N. A. , … Borchelt, D. R. (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta‐amyloid peptide in vivo: Evidence for augmentation of a 42‐specific gamma secretase. Human Molecular Genetics, 13(2), 159–170. 10.1093/hmg/ddh019 [DOI] [PubMed] [Google Scholar]
- Javidnia, M. , Hebron, M. L. , Xin, Y. , Kinney, N. G. , & Moussa, C. E. (2017). Pazopanib reduces phosphorylated tau levels and alters astrocytes in a mouse model of tauopathy. Journal of Alzheimer's Disease, 60(2), 461–481. 10.3233/JAD-170429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger, K. A. (2008). The pathology of “vascular dementia”: A critical update. Journal of Alzheimer's Disease, 14(1), 107–123. [DOI] [PubMed] [Google Scholar]
- Kadir, A. , Marutle, A. , Gonzalez, D. , Scholl, M. , Almkvist, O. , Mousavi, M. , … Nordberg, A. (2011). Positron emission tomography imaging and clinical progression in relation to molecular pathology in the first Pittsburgh compound B positron emission tomography patient with Alzheimer's disease. Brain, 134(Pt 1), 301–317. 10.1093/brain/awq349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlson, M. A. , & Colodner, K. J. (2015). Glial tau pathology in tauopathies: Functional consequences. Journal of Experimental Neuroscience, 9(Suppl 2), 43–50. 10.4137/JEN.S25515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H. , & Schuman, E. M. (1991). Long‐lasting neurotrophin‐induced in the enhancement of synaptic transmission adult hippocampus. Science, 267(5204), 1658–1662. [DOI] [PubMed] [Google Scholar]
- Keren‐Shaul, H. , Spinrad, A. , Weiner, A. , Matcovitch‐Natan, O. , Dvir‐Szternfeld, R. , Ulland, T. K. , … Amit, I. (2017). A unique microglia type associated with restricting development of Alzheimer's disease. Cell, 169(7), 1276–1290 e1217. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Kovacs, G. G. , Ferrer, I. , Grinberg, L. T. , Alafuzoff, I. , Attems, J. , Budka, H. , … Dickson, D. W. (2016). Aging‐related tau astrogliopathy (ARTAG): Harmonized evaluation strategy. Acta Neuropathologica, 131(1), 87–102. 10.1007/s00401-015-1509-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs, C. , Fernandes, H. B. , Sheldon, C. , Raymond, L. A. , & Baimbridge, K. G. (2003). Functional NMDA receptor subtype 2B is expressed in astrocytes after ischemia in vivo and anoxia in vitro. Journal of Neuroscience, 23(8), 3364–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress, B. T. , Iliff, J. J. , Xia, M. , Wang, M. , Wei, H. S. , Zeppenfeld, D. , … Nedergaard, M. (2014). Impairment of paravascular clearance pathways in the aging brain. Annals of Neurology, 76(6), 845–861. 10.1002/ana.24271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanctôt, K. L. , Herrmann, N. , Yau, K. K. , Khan, L. R. , Liu, B. A. , LouLou, M. M. , & Einarson, T. R. (2003). Efficacy and safety of cholinesterase inhibitors in Alzheimer's disease: A meta‐analysis. Canadian Medical Association Journal, 169(6), 557–564. [PMC free article] [PubMed] [Google Scholar]
- Lee, M. C. , Ting, K. K. , Adams, S. , Brew, B. J. , Chung, R. , & Guillemin, G. J. (2010). Characterisation of the expression of NMDA receptors in human astrocytes. PLoS One, 5(11), e14123 10.1371/journal.pone.0014123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenercept MS Study Group and University of British Columbia MS/MRI Analysis Group (1999). TNF neutralization in MS: Results of a randomized, placebo‐controlled multicenter study. Neurology, 53, 457–465. [PubMed] [Google Scholar]
- Leyns, C. E. G. , & Holtzman, D. M. (2017). Glial contributions to neurodegeneration in tauopathies. Molecular Neurodegeneration, 12(1), 50 10.1186/s13024-017-0192-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein, M. P. , Carriba, P. , Baltrons, M. A. , Wojciak‐Stothard, B. , Peterson, J. R. , Garcia, A. , & Galea, E. (2010). Secretase‐independent and RhoGTPase/PAK/ERK‐dependent regulation of cytoskeleton dynamics in astrocytes by NSAIDs and derivatives. Journal of Alzheimer's Disease, 22(4), 1135–1155. 10.3233/JAD-2010-101332 [DOI] [PubMed] [Google Scholar]
- Liddelow, S. A. , Guttenplan, K. A. , Clarke, L. E. , Bennett, F. C. , Bohlen, C. J. , Schirmer, L. , … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton, S. A. (2004). Failures and successes of NMDA receptor antagonists: Molecular basis for the use of open‐channel blocks like memantine in the tretment of acute and chronic neurologic insults. NeuroRx: The Journal of the American Society for Experimental NeuroTherapeutics, 1, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton, S. A. (2006). Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nature Reviews Drug Discovery, 5(2), 160–170. 10.1038/nrd1958 [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Zeng, X. , Hui, Y. , Zhu, C. , Wu, J. , Taylor, D. H. , … Hu, J. (2015). Activation of alpha7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress‐induced apoptosis: Implications for Parkinson's disease. Neuropharmacology, 91, 87–96. 10.1016/j.neuropharm.2014.11.028 [DOI] [PubMed] [Google Scholar]
- LoPresti, P. , Szuchet, S. , Papasozomenos, S. C. , Zinkowski, R. P. , & Binder, L. I. (1995). Functional implications for the microtubule‐associated protein tau: Localization in oligodendrocytes. Proceedings of the National Academy of Sciences of the United States of America, 92, 10369–10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makitani, K. , Nakagawa, S. , Izumi, Y. , Akaike, A. , & Kume, T. (2017). Inhibitory effect of donepezil on bradykinin‐induced increase in the intracellular calcium concentration in cultured cortical astrocytes. Journal of Pharmacological Sciences, 134(1), 37–44. 10.1016/j.jphs.2017.03.008 [DOI] [PubMed] [Google Scholar]
- Mattsson, N. , Schott, J. M. , Hardy, J. , Turner, M. R. , & Zetterberg, H. (2016). Selective vulnerability in neurodegeneration: insights from clinical variants of Alzheimer's disease. Journal of Neurology, Neurosurgery, and Psychiatry, 87(9):1000–1004. 10.1136/jnnp-2015-311321 [DOI] [PubMed] [Google Scholar]
- McGeer, P. L. , McGeer, E. , Rogers, J. , & Sibley, J. (1990). Anti‐inflammatory drugs and Alzheimer's disease. The Lancet Neurology, 335, 1037. [DOI] [PubMed] [Google Scholar]
- McGleenon, B. M. , Dynan, K. B. , & Passmore, A. P. (1999). Acetylcholinesterase inhibitors in Alzheimer's disease. British Journal of Clinical Pharmacology, 48, 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele, T. , & Juric, D. M. (2014). Metrifonate, like acetylcholine, up‐regulates neurotrophic activity of cultured rat astrocytes. Pharmacological Reports, 66(4), 618–623. 10.1016/j.pharep.2014.02.025 [DOI] [PubMed] [Google Scholar]
- Meyer‐Franke, A. , Kaplan, M. R. , Pfrieger, F. W. , & Barres, B. A. (1995). Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron, 15, 805–819. [DOI] [PubMed] [Google Scholar]
- Mizuta, I. , Ohta, M. , Ohta, K. , Nishimura, M. , Mizuta, E. , & Kuno, S. (2001). Riluzole stimulates nerve growth factor, brain‐derived neurotrophic factor and glial cell line‐derived neurotrophic factor synthesis in cultured mouse astrocytes. Neuroscience Letters, 310, 117–120. [DOI] [PubMed] [Google Scholar]
- Mohamed, L. A. , Keller, J. N. , & Kaddoumi, A. (2016). Role of P‐glycoprotein in mediating rivastigmine effect on amyloid‐beta brain load and related pathology in Alzheimer's disease mouse model. Biochimica et Biophysica Acta, 1862(4), 778–787. 10.1016/j.bbadis.2016.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne, A. , Barnes, S. R. , Sweeney, M. D. , Halliday, M. R. , Sagare, A. P. , Zhao, Z. , … Zlokovic, B. V. (2015). Blood–brain barrier breakdown in the aging human hippocampus. Neuron, 85(2), 296–302. 10.1016/j.neuron.2014.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke, L. , Masliah, E. , Yu, G.‐Q. , Mallory, M. , Rockenstein, E. M. , Tatsuno, G. , … McConlogue, L. (2000). High‐level neuronal expression of AB(1‐42) in wild‐type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. Journal of Neuroscience, 20(11), 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan, C. , & Ding, A. (2010). Nonresolving inflammation. Cell, 140(6), 871–882. 10.1016/j.cell.2010.02.029 [DOI] [PubMed] [Google Scholar]
- Nedergaard, M. , & Verkhratsky, A. (2012). Artifact versus reality—How astrocytes contribute to synaptic events. Glia, 60(7), 1013–1023. 10.1002/glia.22288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley, H. , Cole, S. L. , Logan, S. , Maus, E. , Shao, P. , Craft, J. , … Vassar, R. (2006). Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: Potential factors in amyloid plaque formation. Journal of Neuroscience, 26(40), 10129–10140. 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, M. , Gray, J. D. , Larson, C. S. , Kazim, S. F. , Soya, H. , McEwen, B. S. , & Pereira, A. C. (2018). Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early‐onset Alzheimer's disease. Translational Psychiatry, 8(1), 153 10.1038/s41398-018-0201-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, J. B. , Di Domenico, F. , Sultana, R. , Perluigi, M. , Cini, C. , Pierce, W. M. , & Butterfield, D. A. (2009). Proteomics‐determined differences in the concanavalin‐A‐fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer's disease and mild cognitive impairment: implications for progression of AD. Journal of Proteome Research, 8(2), 471–482. 10.1021/pr800667a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst, M. , Braganza, O. , Dannenberg, H. , Hu, W. , Pothmann, L. , Rosen, J. , … Beck, H. (2016). Astrocyte intermediaries of septal cholinergic modulation in the hippocampus. Neuron, 90(4), 853–865. 10.1016/j.neuron.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Palygin, O. , Lalo, U. , & Pankratov, Y. (2011). Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. British Journal of Pharmacology, 163(8), 1755–1766. 10.1111/j.1476-5381.2011.01374.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. , Wetzel, I. , Marriott, I. , Dreau, D. , D'Avanzo, C. , Kim, D. Y. , … Cho, H. (2018). A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer's disease. Nature Neuroscience, 21(7), 941–951. 10.1038/s41593-018-0175-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, H. , McIntire, J. , Ryan, S. , Dunah, A. , & Loring, R. (2017). Anti‐inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF‐κB pathway and activation of the Nrf2 pathway. Journal of Neuroinflammation, 14(1), 192 10.1186/s12974-017-0967-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny, M. , Pekna, M. , Messing, A. , Steinhauser, C. , Lee, J. M. , Parpura, V. , … Verkhratsky, A. (2016). Astrocytes: A central element in neurological diseases. Acta Neuropathologica, 131(3), 323–345. 10.1007/s00401-015-1513-1 [DOI] [PubMed] [Google Scholar]
- Peluffo, H. , Estevez, A. , Barbeito, L. , & Stutzmann, J. M. (1997). Riluzole promotes survival of rat motoneurons in vitro by stimulating trophic activity produced by spinal astrocyte monolayers. Neuroscience Letters, 228, 207–211. [DOI] [PubMed] [Google Scholar]
- Peng, W. , Achariyar, T. M. , Li, B. , Liao, Y. , Mestre, H. , Hitomi, E. , … Deane, R. (2016). Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiology of Disease, 93, 215–225. 10.1016/j.nbd.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, E. K. , Perry, R. H. , Blessed, G. , & Tomlinson, B. E. (1978). Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathology and Applied Neurobiology, 4(4), 273–277. [DOI] [PubMed] [Google Scholar]
- Ramos‐Casals, M. , Brito‐Zeron, P. , Munoz, S. , Soria, N. , Galiana, D. , Bertolaccini, L. , … Khamashta, M. A. (2007). Autoimmune diseases induced by TNF‐targeted therapies: Analysis of 233 cases. Medicine (Baltimore), 86(4), 242–251. 10.1097/MD.0b013e3181441a68 [DOI] [PubMed] [Google Scholar]
- Reisberg, B. , Doody, R. , Stöffler, A. , Schmitt, F. , Ferris, S. , & Möbius, H. J. (2003). Memantine in moderate‐to‐severe Alzheimer's disease. The New England Journal of Medicine, 248(13), 1333–1341. [DOI] [PubMed] [Google Scholar]
- Revathikumar, P. , Bergqvist, F. , Gopalakrishnan, S. , Korotkova, M. , Jakobsson, P. J. , Lampa, J. , & Le Maitre, E. (2016). Immunomodulatory effects of nicotine on interleukin 1β activated human astrocytes and the role of cyclooxygenase 2 in the underlying mechanism. Journal of Neuroinflammation, 13(1), 256 10.1186/s12974-016-0725-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher, A. E. , Kuo, Y.‐M. , Esh, C. , Knebel, C. , Weiss, N. , Kalback, W. , … Kokjohn, T. A. (2003). Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer's disease. Molecular Medicine, 9(3/4), 112–122. [PMC free article] [PubMed] [Google Scholar]
- Rothhammer, V. , Borucki, D. M. , Tjon, E. C. , Takenaka, M. C. , Chao, C. C. , Ardura‐Fabregat, A. , … Quintana, F. J. (2018). Microglial control of astrocytes in response to microbial metabolites. Nature, 557(7707), 724–728. 10.1038/s41586-018-0119-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, A. , Macosko, E. Z. , Wysoker, A. , Goldman, M. , Krienen, F. M. , de Rivera, H. , … McCarroll, S. A. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell, 174(4), 1015–1030. e1016. 10.1016/j.cell.2018.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk, D. , Barbour, R. , Dunn, W. , Gordon, G. , Grajeda, H. , Guido, T. , … Seubert, P. (1999). Immunization with amyloid‐beta attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature, 400(8), 173–177. [DOI] [PubMed] [Google Scholar]
- Schinder, A. F. , & Poo, M. (2000). The neurotrophin hypothesis for synaptic plasticity. Trends in Neuroscience, 23(12), 639–645. [DOI] [PubMed] [Google Scholar]
- Schipper, H. M. , Bennett, D. A. , Liberman, A. , Bienias, J. L. , Schneider, J. A. , Kelly, J. , & Arvanitakis, Z. (2006). Glial heme oxygenase‐1 expression in Alzheimer disease and mild cognitive impairment. Neurobiology of Aging, 27(2), 252–261. 10.1016/j.neurobiolaging.2005.01.016 [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Yamada, K. , Liddelow, S. A. , Smith, S. T. , Zhao, L. , Luo, W. , … Holtzman, D. M. (2017). ApoE4 markedly exacerbates tau‐mediated neurodegeneration in a mouse model of tauopathy. Nature, 549(7673), 523–527. 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk‐Wegrzynowicz, M. , Gerber, Y. N. , Ries, M. , Sastre, M. , Tolkovsky, A. M. , & Spillantini, M. G. (2017). Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathologica Communications, 5(1), 89 10.1186/s40478-017-0478-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, S. K. , Stogsdill, J. A. , Pulimood, N. S. , Dingsdale, H. , Kim, Y. H. , Pilaz, L. J. , … Eroglu, C. (2016). Astrocytes assemble thalamocortical synapses by bridging NRX1α and NL1 via hevin. Cell, 164(1–2), 183–196. 10.1016/j.cell.2015.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogsdill, J. A. , Ramirez, J. , Liu, D. , Kim, Y. H. , Baldwin, K. T. , Enustun, E. , … Eroglu, C. (2017). Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature, 551(7679), 192–197. 10.1038/nature24638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strangfeld, A. , & Listing, J. (2006). Infection and musculoskeletal conditions: Bacterial and opportunistic infections during anti‐TNF therapy. Best Practice & Research: Clinical Rheumatology, 20(6), 1181–1195. 10.1016/j.berh.2006.08.010 [DOI] [PubMed] [Google Scholar]
- Suhs, K. W. , Gudi, V. , Eckermann, N. , Fairless, R. , Pul, R. , Skripuletz, T. , & Stangel, M. (2016). Cytokine regulation by modulation of the NMDA receptor on astrocytes. Neuroscience Letters, 629, 227–233. 10.1016/j.neulet.2016.07.016 [DOI] [PubMed] [Google Scholar]
- Tabet, N. (2006). Acetylcholinesterase inhibitors for Alzheimer's disease: Anti‐inflammatories in acetylcholine clothing! Age and Ageing, 35(4), 336–338. 10.1093/ageing/afl027 [DOI] [PubMed] [Google Scholar]
- Teaktong, T. , Graham, A. , Court, J. , Perry, R. , Jaros, E. , Johnson, M. , … Perry, E. (2003). Alzheimer's disease is associated with a selective increase in alpha7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia, 41(2), 207–211. 10.1002/glia.10132 [DOI] [PubMed] [Google Scholar]
- Thullbery, M. D. , Cox, H. D. , Schule, T. , Thompson, C. M. , & George, K. M. (2005). Differential localization of acetylcholinesterase in neuronal and non‐neuronal cells. Journal of Cellular Biochemistry, 96(3), 599–610. 10.1002/jcb.20530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger, C. , Svedberg, M. M. , Yu, W. F. , Hedberg, M. M. , & Nordberg, A. (2006). Effect of subchronic treatment of memantine, galantamine, and nicotine in the brain of Tg2576 (APPswe) transgenic mice. Journal of Pharmacology and Experimental Therapeutics, 317(1), 30–36. 10.1124/jpet.105.098566 [DOI] [PubMed] [Google Scholar]
- van Oosten, B. W. , Barkhof, F. , Truyen, L. , Boringa, J. B. , Bertelsmann, F. W. , von Blomberg, B. M. , … Polman, C. H. (1996). Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti‐tumor necrosis factor antibody cA2. Neurology, 47(6), 1531–1534. [DOI] [PubMed] [Google Scholar]
- Verkhratsky, A. , & Kirchhoff, F. (2007). NMDA receptors in glia. The Neuroscientist, 13(1), 28–37. 10.1177/1073858406294270 [DOI] [PubMed] [Google Scholar]
- Verkhratsky, A. , Marutle, A. , Rodriguez‐Arellano, J. J. , & Nordberg, A. (2015). Glial asthenia and functional paralysis: A new perspective on neurodegeneration and Alzheimer's disease. The Neuroscientist, 21(5), 552–568. 10.1177/1073858414547132 [DOI] [PubMed] [Google Scholar]
- Verkhratsky, A. , Matteoli, M. , Parpura, V. , Mothet, J. P. , & Zorec, R. (2016). Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. European Molecular Biology Organization Journal, 35(3), 239–257. 10.15252/embj.201592705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky, A. , & Nedergaard, M. (2014). Astroglial cradle in the life of the synapse. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 369(1654), 20130595 10.1098/rstb.2013.0595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky, A. , Nedergaard, M. , & Hertz, L. (2015). Why are astrocytes important? Neurochemical Research, 40(2), 389–401. 10.1007/s11064-014-1403-2 [DOI] [PubMed] [Google Scholar]
- Vidal, R. , Calero, M. , Piccardo, P. , Farlow, M. R. , Unverzagt, F. W. , Méndez, E. , … Ghetti, B. (2000). Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE‐epsilon4 allele. Acta Neuropathologica, 100(1), 1–12. [DOI] [PubMed] [Google Scholar]
- Viswanathan, A. , & Greenberg, S. M. (2011). Cerebral amyloid angiopathy in the elderly. Annals of Neurology, 70(6), 871–880. 10.1002/ana.22516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernhardi, R. , Eugenin‐von Bernhardi, L. , & Eugenin, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Frontiers in Aging Neuroscience, 7, 124 10.3389/fnagi.2015.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Tan, L. , Wang, H. F. , Tan, C. C. , Meng, X. F. , Wang, C. , … Yu, J. T. (2015). Anti‐inflammatory drugs and risk of Alzheimer's disease: An updated systematic review and meta‐analysis. Journal of Alzheimer's Disease, 44(2), 385–396. [DOI] [PubMed] [Google Scholar]
- Weller, R. O. , Subash, M. , Preston, S. D. , Mazanti, I. , & Carare, R. O. (2008). Perivascular drainage of amyloid‐beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathology, 18(2), 253–266. 10.1111/j.1750-3639.2008.00133.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski, H. M. , & Wegiel, J. (1991). Spatial relationships between astrocytes and classical plaque components. Neurobiology of Aging, 12, 593–600. [DOI] [PubMed] [Google Scholar]
- Wu, Z. , Zhao, L. , Chen, X. , Cheng, X. , & Zhang, Y. (2015). Galantamine attenuates amyloid‐beta deposition and astrocyte activation in APP/PS1 transgenic mice. Experimental Gerontology, 72, 244–250. 10.1016/j.exger.2015.10.015 [DOI] [PubMed] [Google Scholar]
- Wyss‐Coray, T. , Lin, C. , Yan, F. , Yu, G.‐Q. , Rohde, M. , McConlogue, L. , … Mucke, L. (2001). TGF‐β1 promotes microglial amyloid‐β clearance and reduces plaque burden in transgenic mice. Nature Medicine, 7(5), 612–618. [DOI] [PubMed] [Google Scholar]
- Yoshizumi, M. , Eisenach, J. C. , & Hayashida, K. (2012). Riluzole and gabapentinoids activate glutamate transporters to facilitate glutamate‐induced glutamate release from cultured astrocytes. European Journal of Pharmacology, 677(1–3), 87–92. 10.1016/j.ejphar.2011.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, W. F. , Guan, Z. Z. , Bogdanovic, N. , & Nordberg, A. (2005). High selective expression of alpha7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer's disease and patients carrying Swedish APP 670/671 mutation: A possible association with neuritic plaques. Experimental Neurology, 192(1), 215–225. 10.1016/j.expneurol.2004.12.015 [DOI] [PubMed] [Google Scholar]
- Yue, C. , & Jing, N. (2015). The promise of stem cells in the therapy of Alzheimer's disease. Transl Neurodegener, 4, 8 10.1186/s40035-015-0029-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun, S. P. , Kam, T. I. , Panicker, N. , Kim, S. , Oh, Y. , Park, J. S. , … Ko, H. S. (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nature Medicine, 24, 931–938. 10.1038/s41591-018-0051-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian, J. L. , Xu, L. , Foo, L. C. , Nouri, N. , Zhou, L. , Giffard, R. G. , & Barres, B. A. (2012). Genomic analysis of reactive astrogliosis. Journal of Neuroscience, 32(18), 6391–6410. 10.1523/JNEUROSCI.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenaro, E. , Pietronigro, E. , Della Bianca, V. , Piacentino, G. , Marongiu, L. , Budui, S. , … Constantin, G. (2015). Neutrophils promote Alzheimer's disease‐like pathology and cognitive decline via LFA‐1 integrin. Nature Medicine, 21(8), 880–886. 10.1038/nm.3913 [DOI] [PubMed] [Google Scholar]
- Zhang, G. , & Neubert, T. A. (2011). Comparison of three quantitative phosphoproteomic strategies to study receptor tyrosine kinase signaling. Journal of Proteome Research, 10(12), 5454–5462. 10.1021/pr200697x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Chen, K. , Sloan, S. A. , Bennett, M. L. , Scholze, A. R. , O'Keeffe, S. , … Wu, J. Q. (2014). An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. Journal of Neuroscience, 34(36), 11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Sloan, S. A. , Clarke, L. E. , Caneda, C. , Plaza, C. A. , Blumenthal, P. D. , … Barres, B. A. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron, 89(1), 37–53. 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , O'Connor, T. , & Vassar, R. (2011). The contribution of activated astrocytes to AB production: Implications for Alzheimer's disease pathogenesis. Journal of Neuroinflammation, 8(150), 150 10.1186/1742-2094-8-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Supporting information.