

Abstract
Background and Purpose
Doxorubicin is a highly effective anticancer agent but eventually induces cardiotoxicity associated with increased production of ROS. We previously reported that a pathological protein interaction between TRPC3 channels and NADPH oxidase 2 (Nox2) contributed to doxorubicin‐induced cardiac atrophy in mice. Here we have investigated the effects of ibudilast, a drug already approved for clinical use and known to block doxorubicin‐induced cytotoxicity, on the TRPC3‐Nox2 complex. We specifically sought evidence that this drug attenuated doxorubicin‐induced systemic tissue wasting in mice.
Experimental Approach
We used the RAW264.7 macrophage cell line to screen 1,271 clinically approved chemical compounds, evaluating functional interactions between TRPC3 channels and Nox2, by measuring Nox2 protein stability and ROS production, with and without exposure to doxorubicin. In male C57BL/6 mice, samples of cardiac and gastrocnemius muscle were taken and analysed with morphometric, immunohistochemical, RT‐PCR and western blot methods. In the passive smoking model, cells were exposed to DMEM containing cigarette sidestream smoke.
Key Results
Ibudilast, an anti‐asthmatic drug, attenuated ROS‐mediated muscle toxicity induced by doxorubicin treatment or passive smoking, by inhibiting the functional interactions between TRPC3 channels and Nox2, without reducing TRPC3 channel activity.
Conclusions and Implications
These results indicate a common mechanism underlying induction of systemic tissue wasting by doxorubicin. They also suggest that ibudilast could be repurposed to prevent muscle toxicity caused by anticancer drugs or passive smoking.
Abbreviations
- Nox
NADPH oxidase
- NRCMs
neonatal rat cardiomyocytes
- CSM
cigarette sidestream smoke‐containing medium
- PLA
proximity ligation assay
- Br‐cAMP
8‐bromoadenosine 3′,5′‐cyclic monophosphate
- MeHg
methyl mercury
- Top2
DNA topoisomerase II
- MuRF
muscle ring‐finger protein.
What is already known
Formation of TRPC3‐Nox2 protein complex contributes to doxorubicin‐induced cardiotoxicity in rodents.
What this study adds
Ibudilast attenuates muscle toxicity induced by doxorubicin treatment or passive smoking by inhibiting TRPC3‐Nox2 interaction.
What is the clinical significance
Ibudilast could be repurposed to prevent muscle toxicity caused by anticancer drugs or passive smoking.
1. INTRODUCTION
Doxorubicin is a highly effective anthracycline‐based anticancer agent used to treat a variety of haematological and solid malignancies (Yeh & Bickford, 2009). However, it is difficult to use at high doses, because of strong adverse events such as cardiac and skeletal muscle atrophy and impaired immune function (Gilliam et al., 2012; Hassan et al., 2005). Indeed, the frequency of cardiac decline and heart failure occurring within 1 year after the end of the final administration of doxorubicin is 3–26% (Yeh & Bickford, 2009). Therefore, some cancer patients are forced to stop treatment with doxorubicin. Furthermore, although the reduction of the cumulative dose below 450 mg·m−2 diminishes the incidence of cardiac toxicity, cardiac functional abnormalities have been reported even in patients treated with lower doses of doxorubicin (Lipshultz et al., 2005; Vejpongsa & Yeh, 2014).
Besides the adjustment of doxorubicin dose, several additional strategies for primary prevention of cardiotoxicity (i.e., slow infusion, liposomal formulations, and anthracycline analogues) are attempted but not routinely used (Raj, Franco, & Lipshultz, 2014). Only one drug, dexrazoxane, has been approved for clinical use by the Food and Drug Administration to prevent the cardiac toxicity of anthracycline. Despite being able to effectively reduce the occurrence of heart failure in patients, dexrazoxane seems also to have serious adverse effects, including immunosuppression and dysfunction of liver and kidney (Štěrba et al., 2013). Thus, eliminating doxorubicin‐induced adverse events is an urgent challenge to be solved for safe and effective anthracycline‐based clinical chemotherapeutic strategies.
NADPH oxidase (Nox) is one of ROS‐generating enzymes, and two Nox isozymes (Nox2 and Nox4) are expressed in a mammalian heart. Nox2‐mediated ROS production physiologically participates in mechanochemical transduction in the heart during left ventricular diastolic filling and mechanically stretching of cardiomyocytes (Prosser, Ward, & Lederer, 2011). In contrast, up‐regulation of Nox2 protein is reportedly associated with severity of congestive heart failure in humans (Lassègue, San Martín, & Griendling, 2012). Notably, Nox2‐deficient mice exhibit resistance to doxorubicin‐induced cardiotoxicity (Zhao et al., 2010), suggesting the involvement of Nox2 up‐regulation in doxorubicin‐induced oxidative stress in the heart.
The transient receptor potential (TRP) channels have been attracting attention as multimodal cation channels regulated by environmental factors, including mechanical stress, in vertebrates (Kaneko & Szallasi, 2014). In particular, two members of the TRPC subfamily, TRPC3 and TRPC6, have been shown to participate in the development of pathological hypertrophy caused by neurohumoral factors and mechanical stress (Numaga‐Tomita et al., 2018). TRPC3 interacts with Nox2 via TRPC3‐specific C‐terminal sites, thereby protecting Nox2 from proteasome‐dependent degradation and amplifying Ca2+‐dependent Nox2 activation through TRPC3‐mediated background Ca2+ entry (Kitajima et al., 2016). Nox2 also stabilizes TRPC3 proteins to enhance mechanical stress‐induced background Ca2+ entry.
Recently, we reported that the extent of doxorubicin‐induced formation of TRPC3‐Nox2 complexes correlated well with the severity of doxorubicin‐induced cardiac atrophy and that the inhibition of TRPC3‐Nox2 physical interaction suppressed doxorubicin‐induced myocardial atrophy in mice (Shimauchi et al., 2017). Thus, a drug already approved for clinical use which can also inhibit the formation of TRPC3‐Nox2 complexes could be repurposed to prevent doxorubicin‐induced adverse events.
In this study, we carried out a large‐scale screen of 1,271 clinically approved chemical compounds, focusing on the inhibition of doxorubicin‐induced cytotoxicity using RAW264.7 cells, which express high levels of TRPC3 channels and Nox2. We found that ibudilast, an inhibitor of PDE4, strongly suppressed doxorubicin‐induced ROS production and cytotoxicity by destabilizing the TRPC3‐Nox2 complex. Ibudilast attenuated doxorubicin‐induced systemic tissue wasting and body weight loss in mice. Furthermore, TRPC3‐Nox2 complexes were also formed in cardiac cells by exposure to cigarette sidestream smoke, suggesting that TRPC3‐Nox2 complexes may regulate cardiac events evoked by a range of environmental risk factors.
2. METHODS
2.1. Cell culture
RAW264.7 (ATCC Cat# TIB‐71, RRID:CVCL_0493), HEK293 (ATCC Cat# CRL‐1573, RRID:CVCL_0045), H9c2 (ATCC Cat# CRL‐1446, RRID:CVCL_0286), C2C12 (ATCC Cat# CRL‐1772, RRID:CVCL_0188), HepG2 (ATCC Cat# HB‐8065, RRID:CVCL_0027), and A549 (ATCC Cat# CCL‐185, RRID:CVCL_0023) cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin. 4T1 (ATCC Cat# CRL‐2539, RRID:CVCL_0125) cells were cultured in RPMI1640 supplemented with 10% FBS and 1% penicillin and streptomycin. Plasmid DNAs were transfected into HEK293 cells with X‐tremeGENE9 (Roche, Switzerland) according to manufacturer's instruction. Isolation of neonatal rat cardiomyocytes (NRCMs) were performed as described previously (Kitajima et al., 2016).
2.2. Screening assay
RAW264.7 cells (1 × 104 cells per well) were plated onto 96‐well plates 1 day before the treatment. Compounds of the library were transferred to the plates to yield a final concentration of 10 μM. Each compound was tested on two separate microplates (the corresponding concentration of DMSO was prepared as a control [vehicle]). After 1‐hr incubation at 37°C, cells were treated with doxorubicin (3 μM) for 12 hr at 37°C and then fixed in 4% paraformaldehyde. Cells were washed twice with PBS, permeabilized using 0.1% Triton X‐100 in PBS for 5 min, and treated with 3% BSA in PBS for 1 hr at room temperature. Then, nuclei were stained with DAPI for 1 hr; cell images were captured; and the number of cells was counted using IN Cell Analyzer 2000 (GE Healthcare Life Sciences). Cytoprotection was measured by the number of cells, with or without compounds. Cell viability was expressed as the percentage of control. To examine the effect of post‐treatment with ibudilast, cells were treated with ibudilast (10 μM) or DMSO (vehicle) from 1 hr after cisplatin treatment (20 μM for 24 hr).
2.3. Cell viability assay in H9c2 cells and NRCMs
H9c2 cells (1 × 105 cells per well) or NRCMs (1 × 105 cells per well) were plated onto 96‐well plates 1 day before the treatment. Cells were treated with or without PDE inhibitors (10 μM) for 1 hr and then doxorubicin (3 μM) was added for 24 hr. Samples were fixed in 4% paraformaldehyde. After washing twice with PBS, cells were permeabilized using 0.1% Triton X‐100 in PBS for 5 min then blocked using 3% BSA in PBS for 1 hr at room temperature. After incubation with nuclear staining using DAPI for 1 hr, images were captured, and the number of cells was counted by using IN Cell Analyzer 2000 (GE Healthcare Life Sciences). Cell viability was represented as the percentage of control.
2.4. Morphological analysis of C2C12 cells
C2C12 myoblasts were grown for 4 days. On reaching 80% confluence, culture medium was changed to serum‐restricted differentiation medium (DMEM supplemented with 2% heat‐inactivated horse serum [Gibco]) and grown for five additional days. On the fifth day following serum restriction, matured myotubes were pretreated with ibudilast (10 μM) for 1 hr followed by doxorubicin (3 μM) for 24 hr. Cells were visualized by Giemsa staining and imaged with a fluorescence microscope. Surface areas of at least 40 cells were analysed in each experiment using ImageJ software. To examine the effect of post‐treatment with ibudilast, cells were treated with ibudilast (10 μM) or DMSO (vehicle) from 1 hr after doxorubicin treatment (3 μM for 24 hr).
2.5. Intracellular Ca2+ imaging
Measurement of intracellular Ca2+ concentrations were performed with Fura 2‐AM (Dojindo, Japan) as previously described (Nishimura et al., 2016). After aspirating the culture medium from the dishes and washing the cells with DMEM, freshly prepared 1‐μM Fura 2‐AM diluted in DMEM was added to the dishes and incubated for 30 min at room temperature. The dye solution was then replaced with HEPES‐buffered saline solution containing 140‐mM NaCl, 5.6‐mM KCl, 10‐mM glucose, 10‐mM HEPES (pH 7.4), 1‐mM MgCl2, and 1.8‐mM CaCl2. Fluorescence images were recorded and analysed using a video image analysis system (Aquacosmos, Hamamatsu photonics, Japan).
2.6. Whole cell patch clamp
TRPC3 channel currents were measured using the whole‐cell patch‐clamp technique with an EPC‐10 patch‐clamp amplifier (Heka Elektronik). Patch electrodes with a resistance of 3–4 MΩ (when filled with internal solution) were made from 1.5‐mm borosilicate glass capillaries (Sutter Instrument). Voltage ramps (−100 to +100 mV) of 250 ms were recorded every 2 s from a holding potential of −60 mV, and the currents were normalized based on cell capacitance. Cells were allowed to settle in the perfusion chamber in the external solution containing 140‐mM NaCl, 5.6‐mM KCl, 0.8‐mM MgCl2, 1.8‐mM CaCl2, 10‐mM HEPES, and 10‐mM glucose (pH 7.4). The pipette solution contained 120‐mM CsOH, 120‐mM aspartate, 20‐mM CsCl, 2‐mM MgCl2, 5‐mM EGTA, 1.5‐mM CaCl2, 10‐mM HEPES, and 10‐mM glucose (pH 7.2, adjusted with Tris base). Cells were superfused with standard external solution in the presence or absence of carbachol focally using a Y‐tube perfusion system.
2.7. Animals
All animal care and experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health (NIH Publication No. 85–23, revised 1996) and were approved by the ethics committees at National Institute for Physiological Sciences or the Animal Care and Use Committee, Kyushu University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. Male C57BL/6 mice (19–23 g, 8–10 weeks old, RRID:IMSR_JAX:000664) were obtained from CLEA Japan, Inc. (Tokyo Japan). Male and female 1‐ to 3‐day‐old Sprague–Dawley rat pups (RRID:RGD_70508) for the isolation of NRCMs were purchased from Japan SLC. All mice were housed in individually ventilated cages, with aspen wood chip bedding, in groups of three mice per cage and kept under controlled environmental conditions (12‐hr light/12‐hr dark cycle, room temperature 21–23°C, and humidity 50–60%) with free access to standard laboratory food pellets and water.
2.8. Animal model
Male mice were randomly divided into four groups (n = 7 per group). All mice were anaesthetized with medetomidine (0.3 mg·kg−1), midazolam (4 mg·kg−1), and butorphanol (5 mg·kg−1) by intraperitoneal injection, then osmotic pumps (ALZET) for sustained administration of ibudilast (10 mg·kg−1·day−1) or vehicle were implanted intraperitoneally 3 days before doxorubicin administration. After the operation, the surgical wound was sutured, and cefalexin was given to prevent infection. And then mice were given doxorubicin (15 mg·kg−1, i.p.) or saline and killed 2 weeks later. The doses of doxorubicin and ibudilast were determined with reference to previous studies (Kagitani‐Shimono et al., 2005; Poland, Hahn, Knapp, Beardsley, & Bowers, 2016; Shimauchi et al., 2017). Mice were killed by cervical dislocation, and tissues were removed and processed for histological examination or frozen for further analysis. No behavioural changes were observed during the protocols. Neither piloerection nor hunched posture was apparent during the experimental procedures.
2.9. Blinding and randomization
Laboratory animals were randomly assigned to experimental groups, and treatments were assessed blindly. The order of treatment administration was also randomized. All animal samples were studied, and analysis was carried out in a blinded manner.
2.10. Measurements of ROS production in gastrocnemius muscle
ROS production in gastrocnemius muscle was measured using dihydroethidium (DHE) staining. Frozen tissue was cut into 16‐μm‐thick sections and incubated with DHE (10 μM) for 30 min at 37°C. The sections were fixed in 4% paraformaldehyde for 10 min to stop the reaction. The DHE fluorescence intensity was analysed using ImageJ software.
2.11. NADPH oxidase activity
NADPH‐dependent superoxide production was measured in gastrocnemius muscle homogenates using lucigenin (5 μM)‐enhanced chemiluminescence (NADPH 300 μM; 100‐μg protein; 37°C; Bendall, Cave, Heymes, Gall, & Shah, 2002).
2.12. Western blotting assay
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Cells and tissues were homogenized in RIPA buffer containing 0.1% SDS, 0.5% sodium deoxycholate, 1% NP‐40, 150‐mM NaCl, 50‐mM Tris–HCl (pH 7.4), and protease inhibitor cocktail (Nacalai, Japan). Samples (10 μg) were then separated by SDS‐PAGE and transferred onto PVDF membranes (Millipore, USA). The membranes were blocked with Tris‐buffered saline plus 0.05% Tween‐20 (TBST) containing 1% BSA and incubated with primary antibodies overnight at 4°C. After this incubation, the membranes were washed with TBST and incubated with HRP‐conjugated secondary antibodies. The blots were visualized using Western Lightning Plus ECL (PerkinElmer, USA). Blots were normalized to those obtained with antibodies against GAPDH or β‐actin for stressed cardiomyocytes and tissues or normal cardiomyocytes, respectively. The expression of GAPDH is relatively stable compared to other loading controls of cytoskeletal proteins in stressed muscle tissue and myocytes (Kitajima et al., 2016; Shimauchi et al., 2017).
Images were captured with an ImageQuant LAS 4000 (GE Healthcare Life Sciences) and quantified using ImageQuant TCL software (GE Healthcare Life Sciences). Primary antibodies against Nox2 (1:1,000, Santa Cruz Biotechnology, Cat# sc‐130543, RRID:AB_2261483), TRPC3 (1:1,000, Alomone Labs, Cat# ACC‐016, RRID:AB_2040236), muscle ring‐finger protein (MuRF; 1:1,000, Santa Cruz Biotechnology, Cat# sc‐398608), FLAG (1:5,000, Sigma‐Aldrich Cat# A8592, RRID:AB_439702), GFP (1:1,000, Cell Signaling Technology Cat# 2037, RRID:AB_1281301), β‐actin (1:2,000, Cell Signaling Technology, Cat# 4967, RRID:AB_330288), and GAPDH (1:2,000, Santa Cruz Biotechnology, Cat# sc‐25778, RRID:AB_10167668) were diluted with TBST containing 1% BSA. To analyse expression of endogenous Nox2 in vitro, the total membrane fraction was isolated as described previously (Numaga et al., 2010).
2.13. Morphometric analyses
Gastrocnemius muscle cells were determined by visual inspection of haematoxylin and eosin‐stained sections. The cross‐sectional area of myocytes was measured using ImageJ software.
2.14. Immunoprecipitation assay
For immunoprecipitation, HEK293 cells expressing FLAG‐tagged Nox2 and TRPC3‐EGFP were lysed in a lysis buffer containing 1% Triton X‐100, 140‐mM NaCl, 1‐mM EDTA, 20‐mM Tris–HCl (pH 7.4), and protease inhibitor cocktail (Nacalai, Japan). Cell lysates were incubated with FLAG M2 agarose (Sigma, USA) for 12 hr to immunoprecipitate FLAG‐Nox2. Immune complexes were washed three times with the lysis buffer and eluted with 0.1 mg·ml−1 FLAG peptide (Sigma, USA) in the lysis buffer. The immunoprecipitated proteins were detected by western blotting, as described above.
2.15. Measurements of cell size and ROS production in H9c2 cells and NRCMs
NRCMs were visualized by fluorescent staining with Alexa Fluor 488‐conjugated Phalloidin (Thermo Fisher Scientific), and cell surface area was observed using a fluorescence microscope and analysed from at least 40 cells in each experiment using ImageJ software. Production of ROS in H9c2 and NRCMs was measured using H2DCFDA as described previously (Kitajima et al., 2011). Cells were loaded with H2DCFDA (10 μM) for 10 min at 37°C. The DCF fluorescence excited by illumination at 488 nm was observed at room temperature using a confocal laser microscopy.
Production of ROS in H9c2 cells was measured using DHE staining. DHE staining was performed by incubation with DHE (2 μM) for 1 hr at 37°C. Cells (2 × 105 cells/35 mm dish) were fixed in 2% paraformaldehyde for 10 min to stop the reaction. The DCF and DHE florescence intensity was analysed from at least 40 cells in each experiment using ImageJ software.
2.16. Immunohistochemistry
For immunohistochemistry, HEK293 cells expressing Myc‐tagged p22phox and TRPC3‐EGFP were fixed in 1% paraformaldehyde in PBS and then washed twice in PBS. The fixed cells were permeabilized using 0.1% Triton X‐100 in PBS for 5 min on ice then blocked with 3% BSA in PBS for 1 hr at room temperature. Antibodies against c‐Myc (1:200, Cell Signaling Technology Cat# 2276, RRID:AB_331783) and GFP (1:200, Cell Signaling Technology Cat# 2956, RRID:AB_1196615) were diluted in incubation solution containing 0.01% Triton X‐100, 3% BSA in PBS over night at 4°C. After incubation with the secondary antibody, images were captured using a confocal laser‐scanning microscope (LSM700, Zeiss, Germany)..
2.17. Preparation of cigarette sidestream smoke‐containing medium
Preparation of cigarette sidestream smoke‐containing medium (CSM) was carried out using the method previously described (Ibuki, Toyooka, Zhao, & Yoshida, 2014). Briefly, cigarette sidestream smoke generated by the spontaneous combustion of five cigarettes (tar: 14 mg, nicotine: 1.2 mg, Japan Tobacco Co.) was trapped in 100 ml of DMEM (100% CSM) by bubbling using a dry vacuum pump (pumping speed: 45 L·min−1; DA‐30D; ULVAC, Kanagawa, Japan).
2.18. Duolink proximity ligation assay
NRCMs were treated with or without 5% CSM for 24 hr. Samples were fixed in 4% paraformaldehyde for 10 min. After washing twice with PBS, cells were permeabilized using 0.1% Triton X‐100 in PBS for 10 min and blocked with PBS containing 0.1% TritonX‐100 and 1% BSA for 1 hr at room temperature. Cells were incubated with antibodies against TRPC3 (1:100, Santa Cruz Biotechnology, Cat# sc‐514670) and Nox2 (1:50, Abcam Cat# ab80508, RRID:AB_1603890) at 4°C overnight. Incubation with proximity ligation assay (PLA) probes and detection were carried out according to the manufacturer's instructions (Duolink® In Situ Red Starter Kit Mouse/Rabbit, Sigma). Cells were counterstained with Alexa Fluor 488‐conjugated Phalloidin or CF488‐conjugated wheat germ agglutinin for 30 min at room temperature. Images were acquired using fluorescence microscope.
2.19. RNA‐isolation and real‐time PCR
Tissues were homogenized, and total RNA was extracted using a TRI Reagent (Sigma, USA). cDNA was synthesized with a ReverTra Ace qPCR RT Master Mix (Toyobo, Japan) according to the manufacturer's instructions. Quantitative real‐time PCR was performed using an ABI StepOnePlus Real Time System (Applied Biosystems) and KAPA SYBR FAST qPCR kits (Roche, Switzerland) according to the manufacturer's instructions. Primer sequences used for Nox4 amplification were forward 5′‐GAAGGGGTTAAACACCTCTGC‐3′ and reverse 5′‐ATGCTCTGCTTAAACACAATCCT‐3′ and for 18s rRNA amplification were forward 5′‐ATTAATCAAGAACGAAAGTCGCAGGT‐3′ and reverse 5′‐TTTAAGTTTCAGCTTTGCAACCATACT‐3′. The cycling conditions were one cycle at 95°C during 10 min, followed by 40 cycles of 30 s at 94°C, 30 s at 60°C, and 10 s at 72°C. 18s rRNA expression was used to normalize cDNA levels. The relative fold gene expressions of samples were calculated with the 2−∆∆Ct method.
2.20. Plasmid DNA
Detailed information of FLAG‐tagged Nox2, Myc‐tagged p22phox, and TRPC3‐EGFP were described in the previous study (Kitajima et al., 2011).
2.21. Data and statistical analysis
G*Power3.1.9.2 software was used to calculate the sample size for each group (RRID:SCR_013726). All results are presented as the mean ± SEM from at least five independent experiments and were considered significant if P < .05. Statistical comparisons were made using unpaired t test for two group comparisons or using one‐way ANOVA with Tukey's post hoc test or two‐way ANOVA with Sidak's post hoc test for comparisons among three or more groups when F achieved was P < .05, and there was no significant variance in homogeneity. Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software, LaJolla, CA). Some results were normalized to control to avoid unwanted sources of variation. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018) and data sharing and presentation in preclinical pharmacology (Alexander et al., 2018; George et al., 2017).
2.22. Materials
ATP, 8‐bromoadenosine 3′,5′‐cyclic monophosphate (Br‐cAMP), dibutyryl‐cAMP, LPS from Escherichia coli [0111: B4], and enoximone were purchased from Sigma (St. Louis, MO, USA). Pyr3 was purchased from R&D Systems (Minneapolis, USA). Ibudilast, cilostazol, sildenafil, tadalafil, amrinone, mitomycin C, carbachol and d‐glucose were purchased from Wako Pure Chemical (Osaka, Japan). Milrinone, minoxidil, methyl mercury (MeHg), methotrexate, and doxorubicin were purchased from Tokyo Chemical Industry (Tokyo, Japan). The approved drugs library of 1,271 compounds were purchased from Prestwick Chemical (Illkirch, France).
2.23. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017a, b; Alexander, Striessnig et al., 2017).
3. RESULTS
3.1. Ibudilast inhibits doxorubicin‐induced cell death
Doxorubicin induces cell death in macrophages (Hassan et al., 2005) in which TRPC3 and Nox2 are functionally expressed (Noubade et al., 2014; Solanki, Dube, Tano, Birnbaumer, & Vazquez, 2014; Tano et al., 2014). We investigated doxorubicin‐induced cytotoxicity using a mouse macrophage cell line RAW264.7 (Figure 1a). We confirmed that treatment with Pyr3, a TRPC3 inhibitor, significantly attenuated doxorubicin‐induced cell death. Using 1,271 clinically approved drugs, we found that ibudilast, a PDE4 inhibitor, was the most potent inhibitor of doxorubicin‐induced cytotoxicity (Figure 1b,c and Table S1). To test the involvement of PDE4 enzymis activity in doxorubicin‐induced cell death, cells were treated with PDE‐resistant cell‐permeable cAMP analogues, Br‐cAMP, and dibutyryl‐cAMP (Dastidar, Rajagopal, & Ray, 2007). Neither Br‐cAMP nor dibutyryl‐cAMP suppressed doxorubicin‐induced cell death (Figure 1d), suggesting that the inhibition of doxorubicin‐induced cell death by ibudilast was independent of PDE4 inhibitor activity. Next, we examined whether ibudilast also attenuates cell death caused by other anticancer drugs. Ibudilast attenuated cisplatin‐induced cell death (Figure 1e) but had no effect on mitomycin‐ and methotrexate‐induced cell death (Figure 1f,g). These results suggest that ibudilast attenuates the cytotoxicity, particularly that caused by doxorubicin and cisplatin, via an action different from the inhibition of PDE4.
Figure 1.
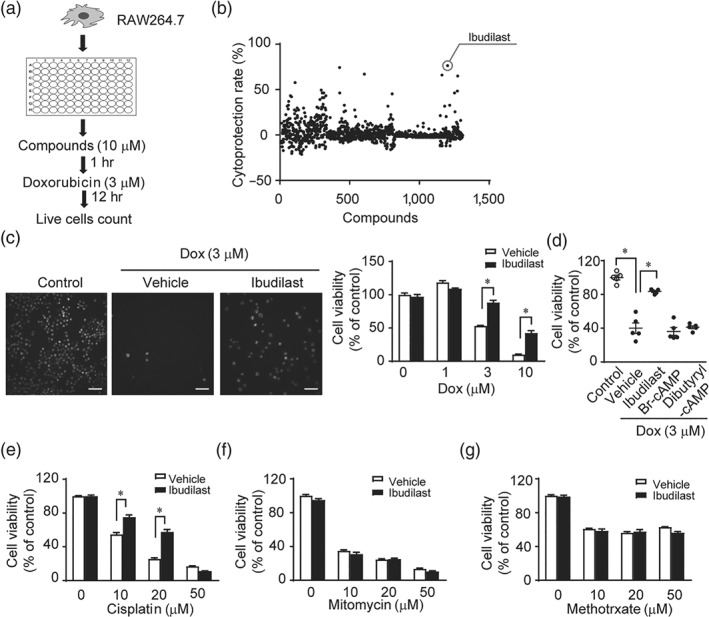
Ibudilast inhibits doxorubicin‐induced cytotoxicity in RAW264.7 macrophage cell line. (a) Schematic flow chart of the high‐throughput screening to identify compounds that can attenuate cytotoxicity caused by doxorubicin treatment. (b) Results of cytoprotection screen. (c) Effect of ibudilast on doxorubicin‐induced cytotoxicity. RAW 264.7 cells were pretreated with or without ibudilast (10 μM) for 30 min before the addition of doxorubicin at 1, 3, 10, and 30 μM. The number of living cells was counted 12 hr after doxorubicin treatment (n = 5). Scale bar: 50 μm. (d) Effect of cAMP analogues (10 μM) on doxorubicin‐induced cytotoxicity in RAW 264.7 cells (n = 5). (e–g) Effect of ibudilast on cisplatin‐induced cytotoxicity (e, n = 6), mitomycin‐induced cytotoxicity (f, n = 6), and methotrexate‐induced cytotoxicity (g, n = 6) in RAW 264.7 cells. Data are shown as the mean ± SEM. *P < .05, significantly different as indicated; one‐way ANOVA with Tukey's comparison test (d) or two‐way ANOVA with Sidak's comparison test (c, e, f, and g)
3.2. Ibudilast attenuates doxorubicin‐induced toxicity in H9c2, NRCMs, and C2C12 myoblasts
The most important adverse event caused by doxorubicin is cardiotoxicity (Spallarossa et al., 2004; Yeh & Bickford, 2009) but doxorubicin also causes skeletal muscle atrophy (Gilliam et al., 2012). Therefore, we examined the effect of ibudilast on doxorubicin‐induced cytotoxicity in H9c2 rat cardiac myoblasts, NRCMs, and C2C12 skeletal muscle myoblasts. Among the eight PDE inhibitors, only ibudilast attenuated doxorubicin‐induced cytotoxicity in H9c2 cells and NRCMs (Figure 2a,b). Pre‐ and post‐treatment with ibudilast also attenuated C2C12 cell atrophy by doxorubicin treatment (Figure 2c and Figure S1A). We further examined whether ibudilast has affected the anticancer effects of doxorubicin in a number of cancer cell lines (4T1 cells, HepG2 cells, and A549 cells). Ibudilast showed little suppression of doxorubicin‐induced cytotoxicity in all the tested cell lines (Figure 2d). Thus, ibudilast attenuated doxorubicin‐induced cytotoxicity in cardiac and skeletal myoblasts, without compromising the anticancer effects of doxorubicin.
Figure 2.
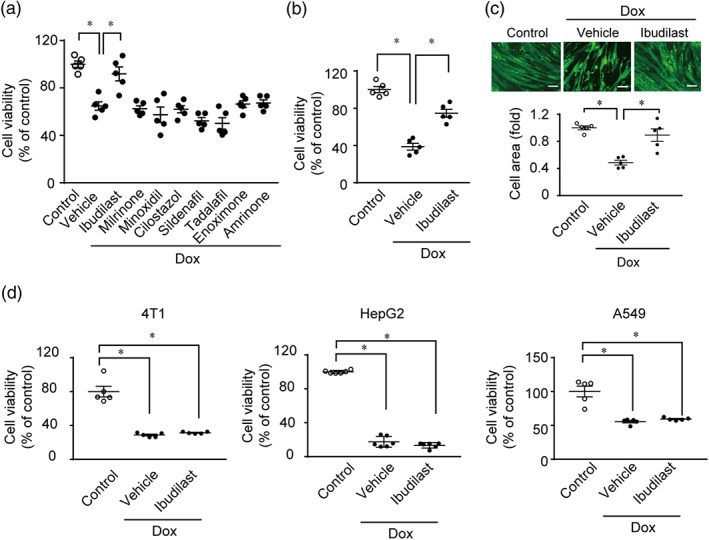
Ibudilast attenuates doxorubicin‐induced cytotoxicity in NRCMs, and H9c2 and C2C12 myoblasts but does not suppress the anticancer effects of doxorubicin in cancer cell lines. (a) Effect of PDE inhibitors on doxorubicin‐induced cytotoxicity in H9c2 cells (n = 5). Cells were treated with or without 10 μM of ibudilast, milrinone, minoxidil, cilostazol, sildenafil, tadalafil, enoximone, and amrinone for 1 hr and then doxorubicin (3 μM) was added for 24 hr. (b) Effect of ibudilast on doxorubicin‐induced atrophy in NRCMs (n = 5). Cells were treated with ibudilast (10 μM) or DMSO (vehicle) for 1 hr and then with doxorubicin (3 μM) for 24 hr. (c) Effects of ibudilast on doxorubicin‐induced atrophy in C2C12 cells (n = 5). Cells were treated with ibudilast (10 μM) or DMSO (vehicle) for 1 hr and then treated with doxorubicin (3 μM) for 24 hr. Scale bar: 100 μm. (d) Effects of ibudilast on the viability of cancer cell lines (4T1, HepG2, and A549 cells) treated with doxorubicin (n = 5). Cells were treated with or without ibudilast (10 μM) for 1 hr and then treated with doxorubicin (3 μM) for 24 hr. Data are shown as the mean ± SEM. *P < .05, significantly different as indicated; one‐way ANOVA with Tukey's comparison test
3.3. Ibudilast inhibits the physical interaction between TRPC3 channels and Nox2
Doxorubicin increases the number of TRPC3‐Nox2 complexes and causes excessive ROS production in NRCMs and Pyr3 suppressed doxorubicin‐induced cell atrophy and ROS production by disrupting TRPC3‐Nox2 physical interaction (Shimauchi et al., 2017). Next, we assessed whether ibudilast was as effective as Pyr3, in suppressing doxorubicin‐induced cardiotoxicity. A shown in Figure 3a,b, ibudilast suppressed doxorubicin‐induced cardiomyocyte atrophy and ROS production.
Figure 3.
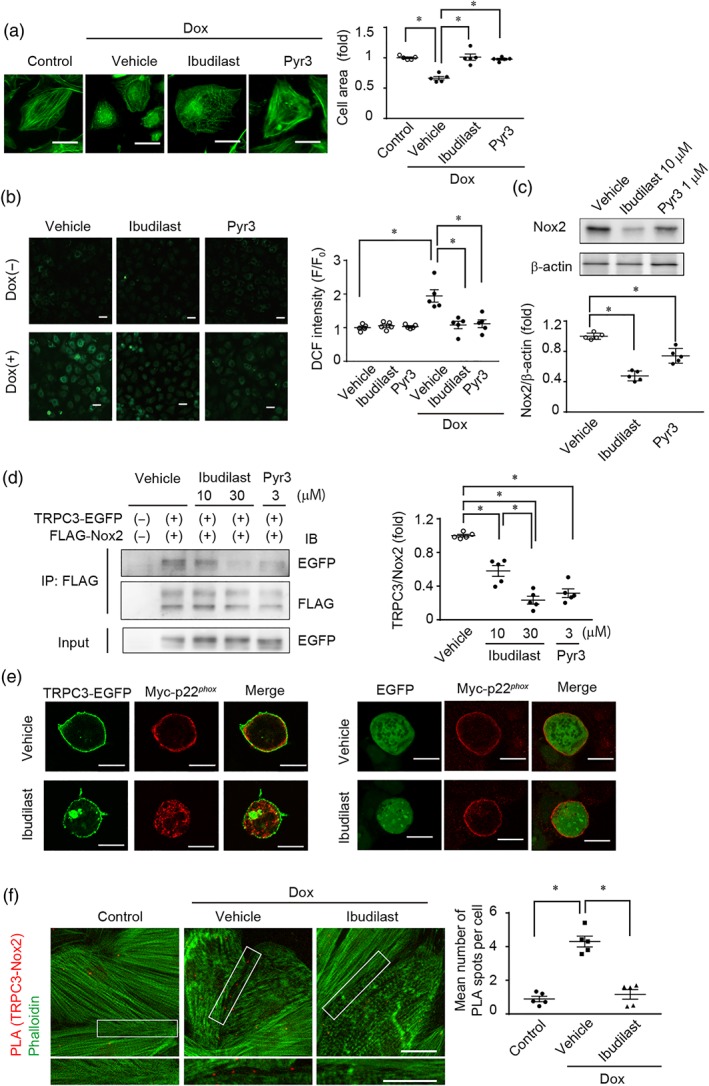
Ibudilast inhibits TRPC3‐Nox2 protein signal complex. (a, b) Effects of ibudilast and Pyr3 on doxorubicin‐induced atrophy (a) and ROS production (b) in NRCMs. NRCMs were treated with doxorubicin (3 μM) for 12 hr (n = 5). Scale bars: 20 μm (a) and 40 μm (b). (c) Effect of ibudilast and Pyr3 on the basal protein abundances of Nox2 in NRCMs (n = 5). (d) Effect of ibudilast and Pyr3 on the interaction between FLAG‐Nox2 and TRPC3‐EGFP in HEK293 cells (n = 5). (e) Effect of ibudilast on the interaction between Myc‐p22phox and TRPC3‐EGFP. Scale bars: 10 μm. (f) Co‐localization of TRPC3 with Nox2 in NRCMs visualized by using Duolink PLA. Scale bars; 20 μm. Data are shown as the mean ± SEM. *P < .05, significantly different as indicated; one‐way ANOVA with Tukey's comparison test
Doxorubicin and cisplatin markedly increased ROS production in H9c2 cells among four anticancer drugs (Figure S2A). Treatment with ibudilast significantly suppressed also cisplatin‐induced ROS production (Figure S2B).
In NRCMs, the basal amount of Nox2 was decreased by ibudilast, as effectively as by Pyr3 (Figure 3c), suggesting that ibudilast can reduce Nox2 protein stability by disrupting the TRPC3‐Nox2 physical interaction. Therefore, we examined the effect of ibudilast on TRPC3‐Nox2 complex formation. Immunoprecipitation of FLAG‐tagged Nox2 and TRPC3‐EGFP demonstrated that ibudilast suppressed interaction between Nox2 and TRPC3 channels (Figure 3d). Nox2 forms a huge complex with p22phox and p47phox (Sumimoto, Miyano, & Takeya, 2005) and TRPC3 channels also interact with p22phox (Kitajima et al., 2016). Therefore, we examined the effect of ibudilast on localization of p22phox and TRPC3 channels by immunostaining, in HEK293 cells expressing TRPC3‐EGFP and Myc‐tagged p22phox. Ibudilast did not alter the membrane localization of p22phox without TRPC3‐EGFP. In vehicle‐treated cells, TRPC3 channels and p22phox were co‐localized at the plasma membrane. In ibudilast‐treated cells, TRPC3 channels were localized at the plasma membrane, while p22phox was localized to intracellular endomembranes (Figure 3e). Finally, we analysed the doxorubicin‐induced formation of native TRPC3‐Nox2 complex and its subcellular localization in NRCMs by PLA. Treatment with doxorubicin significantly increased PLA signal especially in the plasma membrane and ibudilast suppressed the doxorubicin‐induced co‐localization of TRPC3 channels with Nox2 at the plasma membrane in NRCMs (Figure 3f and Figure S4). These data suggest that ibudilast suppresses doxorubicin‐induced cardiotoxicity by interfering with the formation of TRPC3‐Nox2 complexes at the plasma membrane.
3.4. Ibudilast does not inhibit TRPC3 channel activity
We have previously reported that cilostazol (PDE3 inhibitor) and sildenafil (PDE5 inhibitor) attenuate TRPC3 and TRPC6 channel activities through PKA‐ or PKG‐dependent phosphorylation of TRPC proteins (Nishida et al., 2010; Nishioka et al., 2011). Therefore, we examined whether ibudilast suppresses TRPC3 channel activity by PDE4 inhibition. TRPC3‐mediated Ca2+ influx, which was completely suppressed by Pyr3, was scarcely attenuated by ibudilast (Figure 4a,b) or diphenylene iodonium chloride (DPI: an inhibitor of NADPH oxidase; Figure S3). Treatment with cilostazol or sildenafil did significantly attenuate TRPC3‐mediated Ca2+ influx (Figure 4b), while these two compounds failed to suppress doxorubicin‐induced cytotoxicity (Figure 2a). TRPC3‐mediated currents were not suppressed by the pretreatment with ibudilast (Figure 4c–e). These results suggest that ibudilast inhibits the TRPC3‐Nox2 complex without compromising TRPC3 channel activity.
Figure 4.
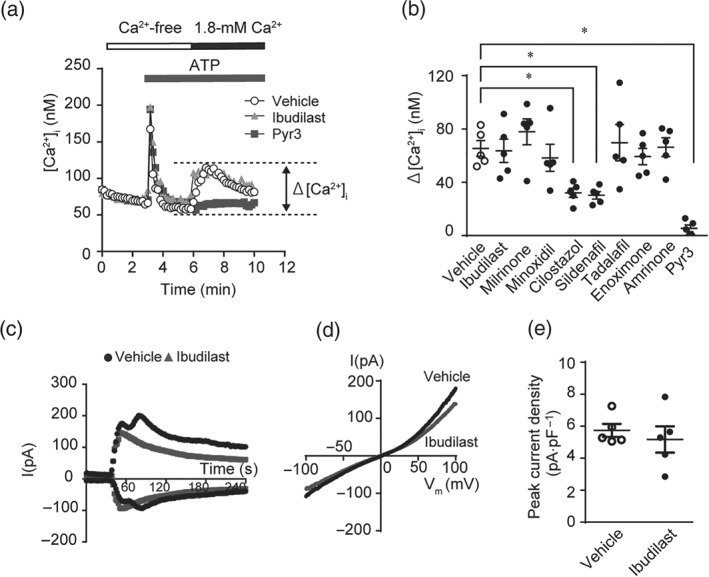
Ibudilast does not inhibit TRPC3 channel activity. (a) Average time courses of ATP‐stimulated changes in intracellular Ca2+ concentration ([Ca2+]i) in TRPC3‐overexpressing HEK293 cells. Ca2+ release‐mediated [Ca2+]i increase was first induced by ATP (100 μM) in Ca2+‐free external solution, and then Ca2+ influx‐mediated [Ca2+]i increase was induced by the addition of Ca2+ (1.8 mM). (b) Peak changes in [Ca2+]i induced by ATP in the presence of extracellular Ca2+ (n = 5). Cells were pretreated with or without 10 μM of ibudilast, milrinone, minoxidil, cilostazol, sildenafil, tadalafil, enoximone, and amrinone, and 3 μM of Pyr3 for 30 min before ATP stimulation. (c–e) Representative time courses (c), representative leak‐subtracted I–V relationships (d), and averaged peak current density (e, n = 5 cells per condition) of carbachol (CCh)‐induced TRPC3 currents recorded in TRPC3‐EGFP‐expressing HEK293 cells treated with or without ibudilast. Currents recorded at 100 or −100 mV of membrane potential every 2 s were plotted. Cells were treated with 100‐μM CCh 1 min after the start of recording. Ibudilast (10 μM) was applied 30 min prior to the start of and throughout the current recording. Data are shown as the mean ± SEM. *P < .05, significantly different as indicated; one‐way ANOVA with Tukey's comparison test
3.5. Ibudilast attenuates tissue wasting by doxorubicin treatment in vivo
We next investigated whether ibudilast attenuates doxorubicin‐induced cytotoxicity in vivo. Doxorubicin administration to mice resulted in a reduction of spontaneous activity and food intake, causing marked decrease in body weight (Figure 5a). Co‐treatment with doxorubicin and ibudilast prevented the doxorubicin‐induced weight loss, as well as the wasting of the heart, spleen, and gastrocnemius muscle tissues (Figure 5a–e). Doxorubicin treatment had no effect on the weights of soleus and extensor digitorum longus muscles (Table 1). In addition, treatment with ibudilast significantly suppressed doxorubicin‐induced up‐regulation of TRPC3, Nox2, and MuRF proteins in gastrocnemius muscle (Figure 5f). Nox4 is expressed in the heart, skeletal muscle, kidney, and lung tissues (Teixeira et al., 2017) and Nox2 altered Nox4 expression in the heart or lung endothelial cells (Pendyala et al., 2009; Zhao et al., 2010). In our experiments, treatment with doxorubicin or ibudilast had no effect on Nox4 mRNA expression in gastrocnemius muscles (Figure 5g). ROS production and NADPH oxidase activity in gastrocnemius muscle of doxorubicin ‐treated mice were markedly increased compared to those of control mice. The doxorubicin‐induced ROS production and NADPH oxidase activation were substantially attenuated by ibudilast co‐treatment (Figure 5h,i).
Figure 5.
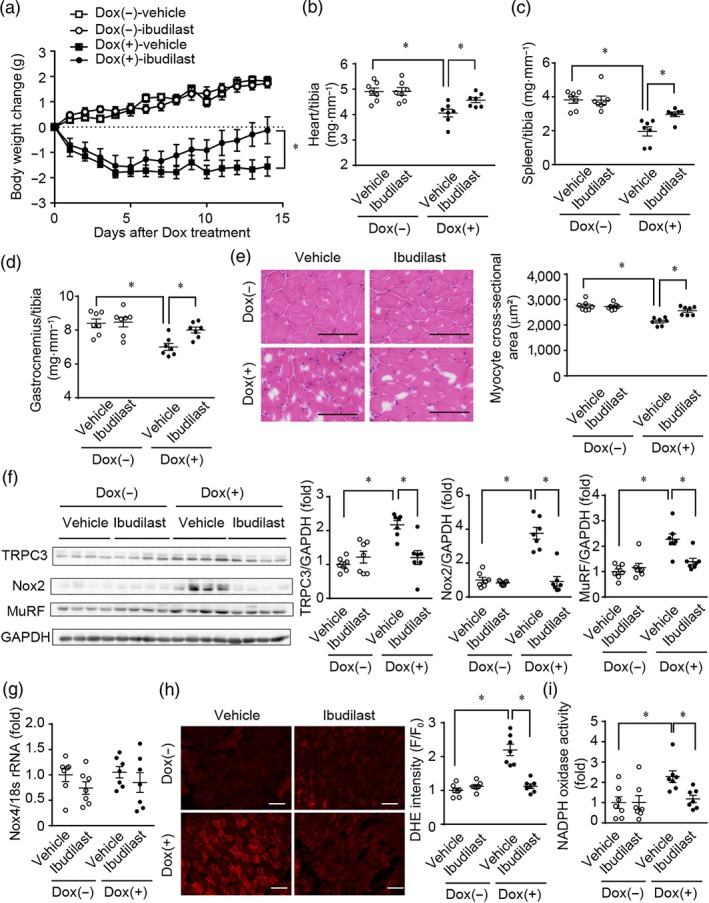
Treatment of mice with ibudilast attenuates doxorubicin‐induced systemic tissue wasting. (a) Reduction of mouse body weight by doxorubicin treatment. C57BL/6J mice were administered doxorubicin (15 mg·kg−1, i.p.). Osmotic pumps including ibudilast (10 mg·kg−1·day−1) or vehicle were implanted intraperitoneally 3 days before doxorubicin administration. (b–d) Tissue weights of the gastrocnemius muscle (b), spleen (c), and heart (d). (e) Representative haematoxylin and eosin staining images (left) and quantitative results of average myocyte cross‐sectional area in mouse gastrocnemius muscles (right). Scale bar: 100 μm. (f) Protein expression of TRPC3, Nox2, and MuRF in mouse gastrocnemius muscles. (g) Expression levels of Nox4 mRNAs in mouse gastrocnemius. (h) Representative dihydroethidium (DHE) fluorescence images (left) and semiquantitative results of average DHE fluorescence intensity in mouse gastrocnemius muscles (right). Scale bar: 100 μm. (i) Quantitative results of average NADPH oxidase activity in mouse gastrocnemius muscles. Data are shown as the mean ± SEM (n = 7). *P < .05, significantly different as indicated; two‐way ANOVA with Tukey's comparison test in (a) and Sidak's comparison test in (b–h)
Table 1.
Effects of doxorubicin (Dox), with or without ibudilast on skeletal muscles in C57BL/6 mice
| Tissue ratio | Dox(−) vehicle (N = 7) | Dox(−) ibudilast (N = 7) | Dox(+) vehicle (N = 7) | Dox(+) ibudilast (N = 7) |
|---|---|---|---|---|
| GM/TL (mg·mm−1) | 8.4 ± 0.3 | 8.5 ± 0.3 | 7.0 ± 0.2* | 8.0 ± 0.2# |
| GM/BW (mg·g−1) | 6.1 ± 0.2 | 6.6 ± 0.2 | 6.0 ± 0.2 | 6.5 ± 0.2 |
| SOL/TL (mg·mm−1) | 0.64 ± 0.03 | 0.68 ± 0.04 | 0.52 ± 0.04* | 0.62 ± 0.03 |
| SOL/BW (mg·g−1) | 0.47 ± 0.02 | 0.53 ± 0.03 | 0.45 ± 0.03 | 0.50 ± 0.02 |
| EDL/TL (mg·mm−1) | 0.78 ± 0.04 | 0.76 ± 0.03 | 0.61 ± 0.01* | 0.71 ± 0.04 |
| EDL/BW (mg·g−1) | 0.57 ± 0.03 | 0.59 ± 0.03 | 0.52 ± 0.02 | 0.57 ± 0.03 |
Tissue ratios are as follows: GM/TL; gastrocnemius weight/tibial length, GM/BW; gastrocnemius weight / body weight, SOL/TL; soleus weight / tibial length, SOL/BW; soleus weight / body weight, EDL/TL; extensor digitorum longus weight / tibial length, EDL/BW; extensor digitorum longus weight / body weight. Data are shown as the mean ± SEM.
P < .05, significantly different from Dox(−) vehicle.
P < .05, significantly different from Dox(+) vehicle; two‐way ANOVA followed Sidak's comparison test.
3.6. Cigarette sidestream smoke up‐regulates ROS production via formation of TRPC3‐Nox2 complex in cardiomyocytes
Various environmental factors, including cigarette smoking, pathogen infection, hyperglycaemia, and heavy metals contaminants in foods, are known to increase the risk of cardiac events (Held et al., 2007; Meurrens et al., 2007; Nemoto et al., 2002). We thus investigated whether environmental stress factors could also induce cardiac vulnerability via the same mechanism as used by doxorubicin treatment. H9c2 cells were treated with 5% CSM, LPS (1 μg·ml−1), high glucose (40 mM), or MeHg (1 μM) for 24 hr. We found that only CSM, out of the four environmental stresses, increased ROS production in H9c2 cell (Figure 6a). Treatment with ibudilast or Pyr3 significantly suppressed CSM‐induced ROS production and up‐regulation of Nox2 proteins (Figure 6b,c). Post‐treatment with ibudilast also attenuated CSM‐induced ROS production (Figure S1C). These results suggest that formation of TRPC3‐Nox2 complex also participates in oxidative stress‐mediated cardiac fragility caused by exposure to CSM and that ibudilast can prevent cardiac tissue wasting caused by cigarette smoke exposure, as well as that caused by doxorubicin treatment.
Figure 6.
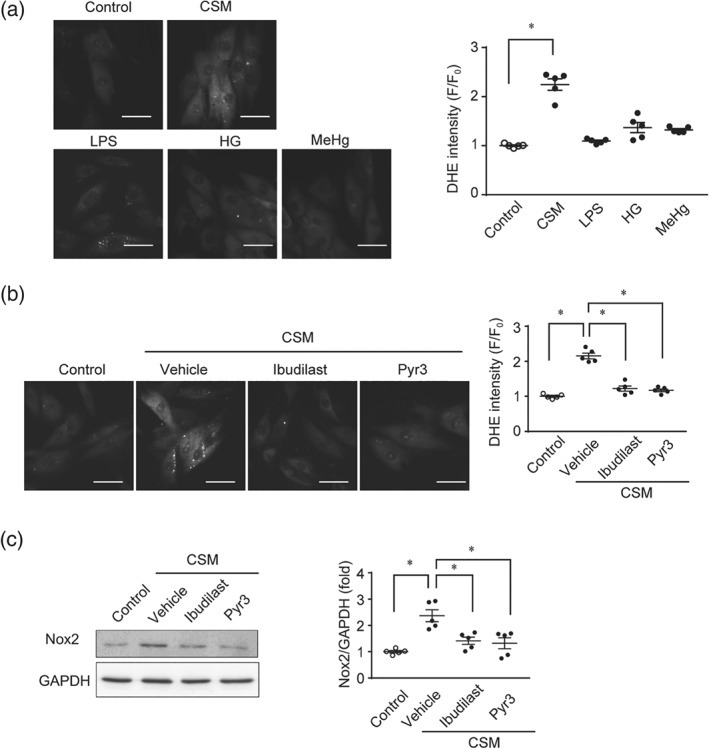
Cigarette sidestream smoke enhances ROS production via formation of TRPC3‐Nox2 protein complex in cardiomyocytes. (a) DHE‐positive ROS production in H9c2 cells. H9c2 cells were treated with cigarette sidestream smoke‐containing medium (CSM, 5%), LPS (1 μg·ml−1), high glucose (HG, 40 mM), and methyl mercury (MeHg, 1 μM) for 24 hr (n = 5). Scale bar: 50 μm. (b, c) Effects of ibudilast and Pyr3 on CSM‐induced ROS production (b) and Nox2 up‐regulation (c) in H9c2 cells (n = 5). Scale bar: 50 μm. Cells were treated with the ibudilast (10 μM) and Pyr3 (3 μM) 30 min prior to CSM treatment (5% for 24 hr, n = 5). Data are shown as the mean ± SEM. *P < .05, significantly different as indicated; one‐way ANOVA with Tukey's comparison test
4. DISCUSSION
We demonstrated that ibudilast attenuated doxorubicin‐induced cytotoxicity via a mechanism other than PDE4 inhibition. Ibudilast suppressed the doxorubicin‐induced formation of TRPC3‐Nox2 complexes without inhibiting TRPC3 channel activity. Furthermore, ibudilast attenuated the doxorubicin‐induced ROS production and in vitro cytotoxicity in cardiomyocytes, skeletal muscle cells, and immune cells, and in a model of tissue wasting and body weight loss in mice, in vivo.
Ibudilast is used for the treatment of bronchial asthma and post‐ischaemic sequelae (Houslay, Schafer, & Zhang, 2005; Kawasaki, Hoshino, Osaki, Mizushima, & Yano, 1992; Lee et al., 2012). PDE inhibitors increase intracellular cAMP levels and exhibit bronchodilation and cytoprotective effects (Dastidar et al., 2007; Houslay et al., 2005). However, addition of cAMP analogues or other PDE inhibitors failed to prevent doxorubicin‐induced cytotoxicity (Figures 1d and 2a), suggesting that ibudilast specifically attenuates this cytotoxicity, independently of PDE4 inhibition. Additionally, ibudilast has beneficial effects in alcohol dependence and multiple sclerosis (Bell et al., 2015; Fox et al., 2018). Ibudilast is also reported to inhibit macrophage migration inhibitory factor and toll‐like receptor 4 (Cho et al., 2010; Ruiz‐Pérez et al., 2016). We demonstrated that ibudilast attenuated the formation of pathological TRPC3‐Nox2 protein complexes induced by doxorubicin treatment or exposure to cigarette sidestream smoke. We previously reported that overexpression of the cytosolic C‐terminal 56 peptides of TRPC3 proteins required for the interaction with Nox2 suppressed doxorubicin‐induced Nox2 protein stabilization and cardiotoxicity by inhibiting TRPC3‐Nox2 complex formation. Ibudilast failed to attenuate TRPC3 channel activity, as did cilostazol and sildenafil (Figure 4), but TRPC3 channel activity per se is not required for the formation of TRPC3‐Nox2 protein complexes and Nox2 stabilization in the heart (Kitajima et al., 2016). Why ibudilast and Pyr3 can suppress the TRPC3‐Nox2 interaction is still unclear, but one possibility is that these compounds may directly bind to the C‐terminal Nox2‐associating region of TRPC3. Further structure‐biological action studies are required to understand the molecular mechanism underlying inhibition of TRPC3‐Nox2 complex by ibudilast. In contrast, cilostazol and sildenafil attenuated TRPC3‐mediated Ca2+ influx activity but failed to inhibit doxorubicin‐induced H9c2 cell death (Figures 2a and 4b). These results suggest that inhibition of Nox2 protein stability through disrupting TRPC3‐Nox2 complex, rather than inhibiting TRPC3 channel activity, is essential to suppress Nox2‐mediated cytotoxicity caused by doxorubicin or cisplatin treatment.
Dexrazoxane is a therapeutic agent that reduces the risk of adverse events induced by anthracycline anticancer drugs and is used to improve the necrosis associated with the extravasation of doxorubicin (Kreidieh, Moukadem, & El Saghir, 2016). The anticancer activity of doxorubicin has been attributed to its targeting of DNA topoisomerase II (Top2) by stabilizing a Top2‐doxorubicin‐DNA covalent complex, referred to as cleavable or cleavage complex (Tewey, Rowe, Yang, Halligan, & Liu, 1984). Dexrazoxane reduces the cardiotoxicity of doxorubicin by either preventing binding of Top2 to chromosomal DNA or inducing proteasomal degradation of Top2 (Yi et al., 2007). However, dexrazoxane did not protect against doxorubicin‐induced myelosuppression and weight loss (Hofland, Thougaard, Sehested, & Jensen, 2005) and increased incidence of secondary malignant neoplasms, particularly acute myeloid leukaemia, in children with Hodgkin's lymphoma (Tebbi et al., 2007). On the other hand, ibudilast suppressed cell death following cisplatin treatment as well (Figure 1e) and doxorubicin‐induced weight loss (Figure 5). As ibudilast is an approved medicine and its safety is known, our findings would suggest a new therapeutic strategy to prevent the systemic toxicity caused by treatment with anticancer drugs.
ROS are also involved in the systemic toxicity caused by cumulative exposure to environmental stresses, including smoking (Kim, Han, & Lee, 2014). Smoking increases not only direct damage to the lung but also the risk of cardiac events (Baba et al., 2006). Ibudilast and Pyr3 suppressed ROS production and Nox2 up‐regulation caused by cigarette sidestream smoke exposure (Figure 6). These results suggest that formation of TRPC3‐Nox2 protein complex underlies CSM‐induced oxidative stress and cardiotoxicity. Cigarette smoke contains more than 5,000 substances, many of which are carcinogenic aldehydes, and complex exposure to multiple environmental electrophiles will increase the risk of cardiovascular diseases and premature death (Fehily, Elwood, & Yarnell, 1989; Talhout et al., 2011).
We found that ibudilast has potency to inhibited doxorubicin‐induced wasting in several tissues in vivo. Our findings also suggest that the pathology‐specific interaction between TRPC3 and Nox2 could be a general mechanism underlying wasting in a range of tissues. Indeed, ibudilast has pleiotropic effects on amyotrophic lateral sclerosis and multiple sclerosis (Crisafulli, Brajkovic, Cipolat Mis, Parente, & Corti, 2018; Fox et al., 2018). Further studies will elucidate the involvement of TRPC3‐Nox2 complexes in these intractable diseases and allow us to propose the inhibition of TRPC3‐Nox2 complex as an innovative therapeutic strategy for their prevention and treatment.
In summary, we revealed that ibudilast inhibited doxorubicin‐induced cytotoxicity both in vivo and in vitro. Our findings suggest that the pathology‐specific interaction between TRPC3 channels and Nox2 will be a general mechanism underlying the tissue wasting caused by anticancer drug treatment.
AUTHOR CONTRIBUTIONS
M.N. and K.N. designed the research and wrote the paper. K.N., T.N‐T., Y.F., T.T., and C.T. performed the experiments. T.Y., N.M., S.K., S.O., Y.I., Y‐T.A., and K.U. contributed new reagents/analytic tools and provide critical suggestions. K.N., T.N‐T., Y.F., T.T., C.T., and A.N. analysed and interpreted the data. M.N. edited the paper.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1.
Post‐treatment with ibudilast attenuates cytotoxicity induced by Dox, cisplatin and CSM. (A) Effect of ibudilast on Dox‐induced atrophy in C2C12 cells (n=5). Cells were treated with ibudilast (10 μM) or DMSO (vehicle) from 1h after Dox treatment (3 μM for 24 h). Scale bar: 100 μm. (B) Effect of ibudilast on cisplatin‐induced cell death in RAW264.7 cells (n=5). Cells were treated with ibudilast (10 μM) or DMSO (vehicle) from 1h after cisplatin treatment (20 μM for 24 h). (C) Effect of ibudilast on CSM‐induced ROS production in H9c2 (n=5). Cells were treated with ibudilast (10 μM) or DMSO (vehicle) from 1h after CSM treatment (5% for 24 h). Scale bar: 50 μm. Data are shown as the mean ± SEM. Significance was imparted using one‐way ANOVA followed by Tukey's comparison test. *P<0.05.
Figure S2.
Ibudilast attenuates cisplatin‐induced ROS production in H9c2. (A) DCF‐positive ROS production in H9c2 cells. Cells were treated with Dox (3 μM), cisplatin (20 μM), mitomycin, (20 μM) and methotrexate (20 μM) for 24 h (n=5). Scale bar: 50 μm. (B) Effect of ibudilast on cisplatin‐induced ROS production in H9c2 cells (n=5). Scale bar: 50 μm. H9c2 cells were treated with the ibudilast (10 μM) 30 min prior to cisplatin treatment (20 μM for 24 h, n=5). Data are shown as the mean ± SEM. Significance was imparted using one‐way ANOVA followed by Tukey's comparison test. *P<0.05.
Figure S3.
NADPH oxidase inhibition has no impact on TRPC3 channel activity. (A) Effect of DPI on TRPC3 channel activity. Average time courses of ATP‐stimulated changes in intracellular Ca2+ concentration ([Ca2+]i) in TRPC3‐overexpressing HEK293 cells. (B) Peak changes in [Ca2+]i induced by ATP (100 μM) in the presence of extracellular Ca2+ (n=5). Cells were pretreated with or without 1 μM of DPI for 30 min before ATP stimulation. Data are shown as the mean ± SEM.
Figure S4.
Formation of TRPC3‐Nox2 protein complex at the plasma membrane. Co‐localization of TRPC3 with Nox2 in NRCMs visualized by using Duolink PLA with WGA. Scale bar: 20 μm.
Table S1.
Result of Cytoprotection rates by the treatment with chemical compounds.
ACKNOWLEDGEMENTS
We thank Dr. Soichiro Akashi for supporting drug screening. We appreciate the technical assistance from The Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences. This work was supported by grants from Japan Society for the Promotion of Science (JSPS KAKENHI; 18K14921 to K.N., 19K16363 to T.T., and 16H05092 and 19H03383 to M.N.) and Innovative Areas (Research in a Proposed Research Area “Oxygen Biology” (26111011 to K.U.) from the Ministry of Education, Culture, Sports, Science and Technology. This work was also supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research [BINDS]) from Japan Agency for Medical Research and Development (AMED; JP18am0101091 to S.O.) and Smoking Research Foundation and Ono Medical Research Foundation (to M.N.).
Nishiyama K, Numaga‐Tomita T, Fujimoto Y, et al. Ibudilast attenuates doxorubicin‐induced cytotoxicity by suppressing formation of TRPC3 channel and NADPH oxidase 2 protein complexes. Br J Pharmacol. 2019;176:3723–3738. 10.1111/bph.14777
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Striessnig, J. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Voltage‐gated ion channels. British Journal of Pharmacology, 174, S160–S194. 10.1111/bph.13884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology . British Journal of Pharmacology, 175, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba, S. , Iso, H. , Mannami, T. , Sasaki, S. , Okada, K. , Konishi, M. , & Tsugane, S. (2006). Cigarette smoking and risk of coronary heart disease incidence among middle‐aged Japanese men and women: The JPHC Study Cohort I. European Journal of Preventive Cardiology, 13, 207–213. 10.1097/01.hjr.0000194417.16638.3d [DOI] [PubMed] [Google Scholar]
- Bell, R. L. , Lopez, M. F. , Cui, C. , Egli, M. , Johnson, K. W. , Franklin, K. M. , & Becker, H. C. (2015). Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addiction Biology, 20, 38–42. 10.1111/adb.12106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall, J. K. , Cave, A. C. , Heymes, C. , Gall, N. , & Shah, A. M. (2002). Pivotal role of a gp91phox‐containing NADPH oxidase in angiotensin II‐induced cardiac hypertrophy in mice. Circulation, 105, 293–296. 10.1161/hc0302.103712 [DOI] [PubMed] [Google Scholar]
- Cho, Y. , Crichlow, G. V. , Vermeire, J. J. , Leng, L. , Du, X. , Hodsdon, M. E. , … Lolis, E. J. (2010). Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proceedings of the National Academy of Sciences of the United States of America, 107, 11313–11318. 10.1073/pnas.1002716107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisafulli, S. G. , Brajkovic, S. , Cipolat Mis, M. S. , Parente, V. , & Corti, S. (2018). Therapeutic strategies under development targeting inflammatory mechanisms in amyotrophic lateral sclerosis. Molecular Neurobiology, 55, 2789–2813. 10.1007/s12035-017-0532-4 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastidar, S. G. , Rajagopal, D. , & Ray, A. (2007). Therapeutic benefit of PDE4 inhibitors in inflammatory diseases. Current Opinion in Investigational Drugs, 8, 364–372. [PubMed] [Google Scholar]
- Fehily, A. M. , Elwood, P. C. , & Yarnell, J. W. G. (1989). Cigarettes and heart disease. The Lancet, 334, 114–115. 10.1016/S0140-6736(89)90364-4 [DOI] [PubMed] [Google Scholar]
- Fox, R. J. , Coffey, C. S. , Conwit, R. , Cudkowicz, M. E. , Gleason, T. , Goodman, A. , … Zabeti, A. (2018). Phase 2 trial of ibudilast in progressive multiple sclerosis. New England Journal of Medicine, 379, 846–855. 10.1056/NEJMoa1803583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, C. H. , Stanford, S. C. , Alexander, S. , Cirino, G. , Docherty, J. R. , Giembycz, M. A. , … Ahluwalia, A. (2017). Updating the guidelines for data transparency in the British Journal of Pharmacology—Data sharing and the use of scatter plots instead of bar charts. British Journal of Pharmacology, 174, 2801–2804. 10.1111/bph.13925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliam, L. A. , Moylan, J. S. , Patterson, E. W. , Smith, J. D. , Wilson, A. S. , Rabbani, Z. , & Reid, M. B. (2012). Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. American Journal of Physiology. Cell Physiology, 302, C195–C202. 10.1152/ajpcell.00217.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan, F. , Islam, S. , Mu, M. M. , Ito, H. , Koide, N. , Mori, I. , … Yokochi, T. (2005). Lipopolysaccharide prevents doxorubicin‐induced apoptosis in RAW 264.7 macrophage cells by inhibiting p53 activation. Molecular Cancer Research, 3, 373–379. 10.1158/1541-7786.MCR-05-0046 [DOI] [PubMed] [Google Scholar]
- Held, C. , Gerstein, H. C. , Yusuf, S. , Zhao, F. , Hilbrich, L. , Anderson, C. , … ONTARGET/TRANSCEND Investigators (2007). Glucose levels predict hospitalization for congestive heart failure in patients at high cardiovascular risk. Circulation, 115, 1371–1375. 10.1161/CIRCULATIONAHA.106.661405 [DOI] [PubMed] [Google Scholar]
- Hofland, K. F. , Thougaard, A. V. , Sehested, M. , & Jensen, P. B. (2005). Dexrazoxane protects against myelosuppression from the DNA cleavage‐enhancing drugs etoposide and daunorubicin but not doxorubicin. Clinical Cancer Research, 11, 3915–3924. 10.1158/1078-0432.CCR-04-2343 [DOI] [PubMed] [Google Scholar]
- Houslay, M. D. , Schafer, P. , & Zhang, K. Y. J. (2005). Keynote review: Phosphodiesterase‐4 as a therapeutic target. Drug Discovery Today, 10, 1503–1519. 10.1016/S1359-6446(05)03622-6 [DOI] [PubMed] [Google Scholar]
- Ibuki, Y. , Toyooka, T. , Zhao, X. , & Yoshida, I. (2014). Cigarette sidestream smoke induces histone H3 phosphorylation via JNK and PI3K/Akt pathways, leading to the expression of proto‐oncogenes. Carcinogenesis, 35, 1228–1237. 10.1093/carcin/bgt492 [DOI] [PubMed] [Google Scholar]
- Kagitani‐Shimono, K. , Mohri, I. , Fujitani, Y. , Suzuki, K. , Ozono, K. , Urade, Y. , & Taniike, M. (2005). Anti‐inflammatory therapy by ibudilast, a phosphodiesterase inhibitor, in demyelination of twitcher, a genetic demyelination model. Journal of Neuroinflammation, 2, 10 10.1186/1742-2094-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko, Y. , & Szallasi, A. (2014). Transient receptor potential (TRP) channels: A clinical perspective. British Journal of Pharmacology, 171, 2474–2507. 10.1111/bph.12414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki, A. , Hoshino, K. , Osaki, R. , Mizushima, Y. , & Yano, S. (1992). Effect of ibudilast: A novel antiasthmatic agent, on airway hypersensitivity in bronchial asthma. Journal of Asthma, 29, 245–252. 10.3109/02770909209048938 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. J. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biology, 8, e1000412 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. , Han, C. H. , & Lee, M. Y. (2014). NADPH oxidase and the cardiovascular toxicity associated with smoking. Toxicology Research, 30, 149–157. 10.5487/TR.2014.30.3.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima, N. , Numaga‐Tomita, T. , Watanabe, M. , Kuroda, T. , Nishimura, A. , Miyano, K. , … Nishida, M. (2016). TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Scientific Reports, 6, 37001 10.1038/srep37001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima, N. , Watanabe, K. , Morimoto, S. , Sato, Y. , Kiyonaka, S. , Hoshijima, M. , … Nishida, M. (2011). TRPC3‐mediated Ca2+ influx contributes to Rac1‐mediated production of reactive oxygen species in MLP‐deficient mouse hearts. Biochemical and Biophysical Research Communications, 409, 108–113. 10.1016/j.bbrc.2011.04.124 [DOI] [PubMed] [Google Scholar]
- Kreidieh, F. Y. , Moukadem, H. A. , & El Saghir, N. S. (2016). Overview, prevention and management of chemotherapy extravasation. World Journal of Clinical Oncology, 7, 87–97. 10.5306/wjco.v7.i1.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassègue, B. , San Martín, A. , & Griendling, K. K. (2012). Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circulation Research, 110, 1364–1390. 10.1161/CIRCRESAHA.111.243972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. Y. , Cho, E. , Ko, Y. E. , Kim, I. , Lee, K. J. , Kwon, S. U. , … Kim, J. S. (2012). Ibudilast, a phosphodiesterase inhibitor with anti‐inflammatory activity, protects against ischemic brain injury in rats. Brain Research, 1431, 97–106. 10.1016/j.brainres.2011.11.007 [DOI] [PubMed] [Google Scholar]
- Lipshultz, S. E. , Lipsitz, S. R. , Sallan, S. E. , Dalton, V. M. , Mone, S. M. , Gelber, R. D. , & Colan, S. D. (2005). Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. Journal of Clinical Oncology, 23, 2629–2636. 10.1200/JCO.2005.12.121 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP . British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurrens, K. , Ruf, S. , Ross, G. , Schleef, R. , von Holt, K. , & Schlüter, K. D. (2007). Smoking accelerates the progression of hypertension‐induced myocardial hypertrophy to heart failure in spontaneously hypertensive rats. Cardiovascular Research, 76, 311–322. 10.1016/j.cardiores.2007.06.033 [DOI] [PubMed] [Google Scholar]
- Nemoto, S. , Vallejo, J. G. , Knuefermann, P. , Misra, A. , Defreitas, G. , Carabello, B. A. , & Mann, D. L. (2002). Escherichia coli LPS‐induced LV dysfunction: Role of toll‐like receptor‐4 in the adult heart. American Journal of Physiology ‐ Heart and Circulatory Physiology, 282, H2316–H2323. 10.1152/ajpheart.00763.2001 [DOI] [PubMed] [Google Scholar]
- Nishida, M. , Watanabe, K. , Sato, Y. , Nakaya, M. , Kitajima, N. , Ide, T. , … Kurose, H. (2010). Phosphorylation of TRPC6 channels at Thr69 is required for anti‐hypertrophic effects of phosphodiesterase 5 inhibition. Journal of Biological Chemistry, 285, 13244–13253. 10.1074/jbc.M109.074104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, A. , Sunggip, C. , Tozaki‐Saitoh, H. , Shimauchi, T. , Numaga‐Tomita, T. , Hirano, K. , … Nishida, M. (2016). Purinergic P2Y6 receptors heterodimerize with angiotensin AT1 receptors to promote angiotensin II‐induced hypertension. Science Signaling, 9, ra7 10.1126/scisignal.aac9187 [DOI] [PubMed] [Google Scholar]
- Nishioka, K. , Nishida, M. , Ariyoshi, M. , Jian, Z. , Saiki, S. , Hirano, M. , … Kurose, H. (2011). Cilostazol suppresses angiotensin II‐induced vasoconstriction via protein kinase A‐mediated phosphorylation of the transient receptor potential canonical 6 channel. Arteriosclerosis, Thrombosis, and Vascular Biology, 31, 2278–2286. 10.1161/ATVBAHA.110.221010 [DOI] [PubMed] [Google Scholar]
- Noubade, R. , Wong, K. , Ota, N. , Rutz, S. , Eidenschenk, C. , Valdez, P. A. , … Ouyang, W. (2014). NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature, 509, 235–239. 10.1038/nature13152 [DOI] [PubMed] [Google Scholar]
- Numaga, T. , Nishida, M. , Kiyonaka, S. , Kato, K. , Katano, M. , Mori, E. , … Mori, Y. (2010). Ca2+ influx and protein scaffolding via TRPC3 sustain PKCβ and ERK activation in B cells. Journal of Cell Science, 123, 927–938. 10.1242/jcs.061051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numaga‐Tomita, T. , Oda, S. , Nishiyama, K. , Tanaka, T. , Nishimura, A. , & Nishida, M. (2018). TRPC channels in exercise‐mimetic therapy. Pflügers Archiv – European Journal of Physiology, 471(3), 507–517. 10.1007/s00424-018-2211-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendyala, S. , Gorshkova, I. A. , Usatyuk, P. V. , He, D. , Pennathur, A. , Lambeth, J. D. , … Natarajan, V. (2009). Role of Nox4 and Nox2 in hyperoxia‐induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxidants and Redox Signaling, 11, 747–764. 10.1089/ars.2008.2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland, R. S. , Hahn, Y. K. , Knapp, P. E. , Beardsley, P. M. , & Bowers, M. S. (2016). Ibudilast attenuates expression of behavioral sensitization to cocaine in male and female rats. Neuropharmacology, 109, 281–292. 10.1016/j.neuropharm.2016.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser, B. L. , Ward, C. W. , & Lederer, W. J. (2011). X‐ROS signaling: Rapid mechano‐chemo transduction in heart. Science, 333, 1440–1445. 10.1126/science.1202768 [DOI] [PubMed] [Google Scholar]
- Raj, S. , Franco, V. I. , & Lipshultz, S. E. (2014). Anthracycline‐induced cardiotoxicity: A review of pathophysiology, diagnosis, and treatment. Current Treatment Options in Cardiovascular Medicine, 16, 315 10.1007/s11936-014-0315-4 [DOI] [PubMed] [Google Scholar]
- Ruiz‐Pérez, D. , Benito, J. , Polo, G. , Largo, C. , Aguado, D. , Sanz, L. , & Gómez de Segura, I. A. (2016). The effects of the toll‐like receptor 4 antagonist, ibudilast, on sevoflurane's minimum alveolar concentration and the delayed remifentanil‐induced increase in the minimum alveolar concentration in rats. Anesthesia and Analgesia, 122, 1370–1376. 10.1213/ANE.0000000000001171 [DOI] [PubMed] [Google Scholar]
- Shimauchi, T. , Numaga‐Tomita, T. , Ito, T. , Nishimura, A. , Matsukane, R. , Oda, S. , … Nishida, M. (2017). TRPC3‐Nox2 complex mediates doxorubicin‐induced myocardial atrophy. JCI Insight, 2 10.1172/jci.insight.93358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanki, S. , Dube, P. R. , Tano, J. Y. , Birnbaumer, L. , & Vazquez, G. (2014). Reduced endoplasmic reticulum stress‐induced apoptosis and impaired unfolded protein response in TRPC3‐deficient M1 macrophages. American Journal of Physiology ‐ Cell Physiology, 307, C521–C531. 10.1152/ajpcell.00369.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spallarossa, P. , Garibaldi, S. , Altieri, P. , Fabbi, P. , Manca, V. , Nasti, S. , … Patrone, F. (2004). Carvedilol prevents doxorubicin‐induced free radical release and apoptosis in cardiomyocytes in vitro. Journal of Molecular and Cellular Cardiology, 37, 837–846. 10.1016/j.yjmcc.2004.05.024 [DOI] [PubMed] [Google Scholar]
- Štěrba, M. , Popelová, O. , Vávrová, A. , Jirkovský, E. , Kovaříková, P. , Geršl, V. , & Šimůnek, T. (2013). Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxidants and Redox Signaling, 18, 899–929. 10.1089/ars.2012.4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto, H. , Miyano, K. , & Takeya, R. (2005). Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochemical and Biophysical Research Communications, 338, 677–686. 10.1016/j.bbrc.2005.08.210 [DOI] [PubMed] [Google Scholar]
- Talhout, R. , Schulz, T. , Florek, E. , van Benthem, J. , Wester, P. , & Opperhuizen, A. (2011). Hazardous compounds in tobacco smoke. International Journal of Environmental Research and Public Health, 8, 613–628. 10.3390/ijerph8020613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tano, J. Y. , Solanki, S. , Lee, R. H. , Smedlund, K. , Birnbaumer, L. , & Vazquez, G. (2014). Bone marrow deficiency of TRPC3 channel reduces early lesion burden and necrotic core of advanced plaques in a mouse model of atherosclerosis. Cardiovascular Research, 101, 138–144. 10.1093/cvr/cvt231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbi, C. K. , London, W. B. , Friedman, D. , Villaluna, D. , De Alarcon, P. A. , Constine, L. S. , … Schwartz, C. L. (2007). Dexrazoxane‐associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. Journal of Clinical Oncology, 25, 493–500. 10.1200/JCO.2005.02.3879 [DOI] [PubMed] [Google Scholar]
- Teixeira, G. , Szyndralewiez, C. , Molango, S. , Carnesecchi, S. , Heitz, F. , Wiesel, P. , & Wood, J. M. (2017). Therapeutic potential of NADPH oxidase 1/4 inhibitors. British Journal of Pharmacology, 174, 1647–1669. 10.1111/bph.13532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewey, K. M. , Rowe, T. C. , Yang, L. , Halligan, B. D. , & Liu, L. F. (1984). Adriamycin‐induced DNA damage mediated by mammalian DNA topoisomerase II. Science, 226, 466–468. 10.1126/science.6093249 [DOI] [PubMed] [Google Scholar]
- Vejpongsa, P. , & Yeh, E. T. H. (2014). Prevention of anthracycline‐induced cardiotoxicity: Challenges and opportunities. Journal of the American College of Cardiology, 64, 938–945. 10.1016/j.jacc.2014.06.1167 [DOI] [PubMed] [Google Scholar]
- Yeh, E. T. , & Bickford, C. L. (2009). Cardiovascular complications of cancer therapy: Incidence, pathogenesis, diagnosis, and management. Journal of the American College of Cardiology, 53, 2231–2247. 10.1016/j.jacc.2009.02.050 [DOI] [PubMed] [Google Scholar]
- Yi, L. L. , Kerrigan, J. E. , Lin, C. P. , Azarova, A. M. , Tsai, Y. C. , Ban, Y. , & Liu, L. F. (2007). Topoisomerase IIβ‐mediated DNA double‐strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Research, 67, 8839–8846. [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , McLaughlin, D. , Robinson, E. , Harvey, A. P. , Hookham, M. B. , Shah, A. M. , … Grieve, D. J. (2010). Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with doxorubicin chemotherapy. Cancer Research, 70, 9287–9297. 10.1158/0008-5472.CAN-10-2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Post‐treatment with ibudilast attenuates cytotoxicity induced by Dox, cisplatin and CSM. (A) Effect of ibudilast on Dox‐induced atrophy in C2C12 cells (n=5). Cells were treated with ibudilast (10 μM) or DMSO (vehicle) from 1h after Dox treatment (3 μM for 24 h). Scale bar: 100 μm. (B) Effect of ibudilast on cisplatin‐induced cell death in RAW264.7 cells (n=5). Cells were treated with ibudilast (10 μM) or DMSO (vehicle) from 1h after cisplatin treatment (20 μM for 24 h). (C) Effect of ibudilast on CSM‐induced ROS production in H9c2 (n=5). Cells were treated with ibudilast (10 μM) or DMSO (vehicle) from 1h after CSM treatment (5% for 24 h). Scale bar: 50 μm. Data are shown as the mean ± SEM. Significance was imparted using one‐way ANOVA followed by Tukey's comparison test. *P<0.05.
Figure S2.
Ibudilast attenuates cisplatin‐induced ROS production in H9c2. (A) DCF‐positive ROS production in H9c2 cells. Cells were treated with Dox (3 μM), cisplatin (20 μM), mitomycin, (20 μM) and methotrexate (20 μM) for 24 h (n=5). Scale bar: 50 μm. (B) Effect of ibudilast on cisplatin‐induced ROS production in H9c2 cells (n=5). Scale bar: 50 μm. H9c2 cells were treated with the ibudilast (10 μM) 30 min prior to cisplatin treatment (20 μM for 24 h, n=5). Data are shown as the mean ± SEM. Significance was imparted using one‐way ANOVA followed by Tukey's comparison test. *P<0.05.
Figure S3.
NADPH oxidase inhibition has no impact on TRPC3 channel activity. (A) Effect of DPI on TRPC3 channel activity. Average time courses of ATP‐stimulated changes in intracellular Ca2+ concentration ([Ca2+]i) in TRPC3‐overexpressing HEK293 cells. (B) Peak changes in [Ca2+]i induced by ATP (100 μM) in the presence of extracellular Ca2+ (n=5). Cells were pretreated with or without 1 μM of DPI for 30 min before ATP stimulation. Data are shown as the mean ± SEM.
Figure S4.
Formation of TRPC3‐Nox2 protein complex at the plasma membrane. Co‐localization of TRPC3 with Nox2 in NRCMs visualized by using Duolink PLA with WGA. Scale bar: 20 μm.
Table S1.
Result of Cytoprotection rates by the treatment with chemical compounds.