

Abstract
No disease modifying drugs have been approved for Alzheimer's disease despite recent major investments by industry and governments throughout the world. The burden of Alzheimer's disease is becoming increasingly unsustainable, and given the last decade of clinical trial failures, a renewed understanding of the disease mechanism is called for, and trialling of new therapeutic approaches to slow disease progression is warranted. Here, we review the evidence and rational for targeting brain iron in Alzheimer's disease. Although iron elevation in Alzheimer's disease was reported in the 1950s, renewed interest has been stimulated by the advancement of fluid and imaging biomarkers of brain iron that predict disease progression, and the recent discovery of the iron‐dependent cell death pathway termed ferroptosis. We review these emerging clinical and biochemical findings and propose how this pathway may be targeted therapeutically to slow Alzheimer's disease progression.
Linked Articles
This article is part of a themed section on Therapeutics for Dementia and Alzheimer's Disease: New Directions for Precision Medicine. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.18/issuetoc
Abbreviations
- ACSL4
acyl‐CoA synthase 4
- AD
Alzheimer's disease
- APOE
apolipoprotein E
- APP
amyloid precursor protein
- Aβ
β‐amyloid plaque
- BBB
blood brain barrier
- CDK5
cyclin dependent kinase 5
- GPx4
glutathione peroxidase 4
- GSK
glycogen synthase kinase
- MCI
mild cognitive impairment
- NAC
n‐acetylcysteine
- PUFA
polyunsaturated fatty acids
- QSM
quantitative susceptibility mapping
1. INTRODUCTION
A major health care problem worldwide is that current treatments for neurodegenerative diseases such as Alzheimer's disease (AD) are limited to symptomatic benefit, and there are no drugs that affect the underlying disease process. In recent years, there has been a major push by industry and governments throughout the world to develop new disease modifying drugs for AD. Drug development for AD has largely been directed at the amyloid‐β (Aβ) peptide, owing to the dominant hypothesis of the field that posits Aβ as the principal driver of disease, which causes toxicity by inducing a cascade of changes in the brain. However, despite massive public and private investment, and scores of large‐scale trials, there has yet to be a disease modifying drug approved for AD. Indeed, after another recent setback, an editorial from the New England Journal of Medicine (Murphy, 2018) concluded that
We may very well be nearing the end of the amyloid‐hypothesis rope, at which point one or two more failures will cause us to loosen our grip and let go.
And that
… the field is clearly in need of innovative ideas.
Here, we outline the landscape of AD as a backdrop for considering the potential for iron to contribute to disease pathogenesis. Although the connection between iron and AD is not new, indeed it was first described in 1953 by Goodman, there have been several recent advances that have stimulated renewed interest in this topic. The development of fluid and imaging biomarkers for brain iron have provided a new insight into the role of iron in the clinical progression of AD. Additionally, the recent discovery of the iron‐dependent cell death process termed ferroptosis has introduced a new framework to understand mechanistically how iron could drive neurodegeneration in this disease. We review these emerging topics with the view to describing how this pathway could be targeted with drugs to achieve a disease modifying benefit for patients.
2. CANONICAL AD PATHOLOGY
Research into the pathogenesis of AD has focused on abnormal and abundant proteinaceous deposits of extracellular Aβ plaques and intracellular neurofibrillary tangles. Aβ is considered a parent pathology of tangles in AD because tangles are observed in the absence of Aβ in several neurodegenerative diseases including progressive supranuclear palsy, whereas Aβ plaque, once thought to be an AD‐specific pathology and now increasingly observed in a range of neurodegenerative diseases, is always accompanied by the presence of tangles at the symptomatic stage of the disease (Robinson et al., 2018).
Plaque deposition occurs slowly and insidiously in AD. Aβ deposits occur over approximately a 30‐year window in the presymptomatic phase of the disease (Villemagne et al., 2013), resulting in a large number of community dwelling asymptomatic people with accumulating amyloid burden, indeed approximately 30% of cognitively normal individuals over the age of 65 have above‐threshold amyloid deposition in their brain identified by PET imaging (Villemagne et al., 2013). Individuals with elevated Aβ deposition and cognitive performance in the normal range go on to develop cognitive decline and progression towards dementia, but at a rate that is variable between subjects (Lim et al., 2014).
Neurofibrillary tangles, which predominantly consist of phosphorated tau protein, deposit in the brain in a pattern more consistent, both temporally and spatially, with neurodegeneration in AD. Whereas Aβ deposition begins 30 years prior to disease, tangles develop approximately 5 years before diagnosis, which accompanies preclinical hippocampal volume loss (Jack et al., 2013). The extent of tangle burden is more closely associated with cognitive decline than that of the extent of Aβ burden (Nelson et al., 2012). This reflects what occurs at the anatomical level, whereas Aβ pathology begins in precuneus and frontal cortical structures before spreading inward over time, tangle pathology begins in the entorhinal and hippocampus regions before extending to neocortical structures later in the disease—a pattern that is more consistent with the neurodegeneration in AD (Braak & Braak, 1991). Indeed, Aβ plaques only develop in the hippocampus very late in the disease despite this area being among the first and most affected brain structures of AD. If Aβ is the major instigator of AD pathogenesis, it is unclear why the pathology of AD is not generally observed in the major area affected in the disease.
3. CLINICAL TRIAL SETBACKS FOR DRUGS THAT TARGET Aβ
Aβ has been the most pursued drug target for AD; however, after now scores of clinical trials, a drug has not yet been identified that convincingly slows disease progression or modifies the course of the disease. The first approach to remove amyloid was active immunotherapy, using AN‐1792. Immunization against Aβ prevented and rescued the AD behavioural and pathological phenotypes of an AD mouse model (Schenk et al., 1999), which led to future clinical development. In extended analysis of the Phase I clinical trial of AN‐1792 that examined the brain of treated patients at autopsy, in the treatment group plaque was reduced or completely removed, but the cognitive presentation during life was not different between the groups (Holmes et al., 2008). The development of AN‐1792 was haltered when, in a Phase 2 trial of mild to moderate AD, 6% of patients reported severe side effects including four with aseptic meningoencephalitis (Hock et al., 2003). In that Phase 2 trial, subjects that responded to immunization had improved cognition and attenuated brain volume loss compared with placebo patients (Vellas et al., 2009). ACC‐001 was subsequently tested in clinical trials; however, this was suspended in 2008 due to the development of vasculitis in a single patient, and exploratory analysis showed no beneficial effect of the vaccine (Pasquier et al., 2016).
Passive immunotherapy for Aβ has been a more favoured approach, with several antibodies having now undergone advanced testing in multiple Phase 3 studies. Solanezumab and bapineuzumab were the two earliest passive immunotherapy drugs and have progressed furthest in clinical development. Solanezumab was shown to increase Aβ42 in CSF (indicating target engagement) in Phase 2 clinical trials, but this was not accompanied by improvement in cognition (Farlow et al., 2012). In two Phase 3 trials, solanezumab had no overall benefit to cognition; however, secondary analysis of a subgroup of mild AD patients that were pooled from both trials revealed a slowing of decline associated with treatment (Doody et al., 2014). This prompted a further Phase 3 trial of mild AD cases, where the drug had no impact on disease progression (Honig et al., 2018).
Bapineuzumab was also an antibody approach that proved effective in lowering Aβ burden in two Phase 2 studies (Rinne et al., 2010), and, although it had no overall effect on cognition, there appeared to be a treatment effect in people who did not express the APOE ε4 risk allele (Salloway et al., 2009). On the basis of this, four Phase 3 trials commenced; two of the completed trials failed to show a clinical improvement, and the other trials were discontinued (Salloway et al., 2014).
Despite these setbacks of the immunotherapy front‐runners, there are several more antibodies that have been tested or are still currently being tested. Ponezumab targets the C‐terminus of Aβ40 but a Phase 2 trial showed that ponezumab had no effect on cognition (Landen et al., 2017) and the development of ponezumab was discontinued. Gantenerumab, another Aβ immunotherapy, showed evidence of target engagement in a Phase 3 study, but an interim futility analysis halted the study early because of lack of clinical efficacy (Ostrowitzki et al., 2017). Crenezumab (Cummings et al., 2018) and aducanumab (Sevigny et al., 2016) are two additional antibodies that have shown target engagement in early studies and are currently undergoing testing in larger clinical trials.
In addition to immunization approaches, small molecules that aim to inhibit the possessing of the amyloid precursor protein (APP) into the Aβ species have also been developed. Aβ is generated by the proteolytic processing by β‐secretase and γ‐secretase. The γ‐secretase inhibitor, semagacestat, was shown to reduce Aβ in a Phase 2 trial (Fleisher et al., 2008); however, the Phase 3 trial was halted when an interim analysis revealed that the drug worsened cognition and increased the incidence of skin cancer (Doody et al., 2013). The increased risk of cancer may relate to notch protein, a tumour suppressor and also a substrate for γ‐secretase. This prompted the development of γ‐secretase inhibitors that were selective for APP over notch, such as avagacestat. Clinical trials of avagacestat were abandoned because of poor tolerability, including gastrointestinal and dermatological adverse events, and worsening of cognition in mild to moderate AD patients (Coric et al., 2012). The γ‐secretase modulator, tarenflurbil, was subsequently developed, and in a Phase 2 study of people with mild to moderate AD, there was no overall effect on cognition; however, secondary analysis revealed that mild AD subjects on a higher dose appeared to have a small benefit to cognitive performance (Wilcock et al., 2008). This lead to a Phase 3 study; however, no clinical benefit was observed, and further development of this drug was abandoned (Green et al., 2009).
Inhibitors of β‐secretase have more recently undergone development. Verubecestat, a potent β‐secretase inhibitor, was the front‐runner and has undergone the most advanced testing. The drug reduced Aβ concentration in plasma, CSF, and brain in animal models of AD and also lowered CSF Aβ in AD patients (Kennedy et al., 2016). However, a Phase 3 clinical trial showed no benefit to cognition despite lowering amyloid load (Egan et al., 2018). Several other β‐secretase inhibitors are currently in clinical trials, such as elebecestat, CNP520, and LY3202626, or abandoned because of toxicity or lack of efficacy, such as atabecestat and lanabecestat.
4. COPATHOLOGIES OF AD—ALTERNATIVE DRUG TARGETS?
There have now been multiple large‐scale clinical trials using different strategies that lower Aβ but have not been effective in slowing clinical disease progression. There are multiple reasons why this could be the case: that the drugs were treated too late in the disease, that Aβ does not contribute to disease progression, or that other pathologies also contribute to disease progression. In additional to the canonical plaque and tangle pathology, it is increasingly recognized that AD often presents with protein pathology characteristic of other neurodegenerative diseases. Indeed, the α‐synuclein containing Lewy body—most commonly associated with Parkinson's disease, can be found in 30–50% of Alzheimer's cases (Robinson et al., 2018). Similarly, cytosolic deposition of transactive response DNA‐binding protein of 43 kDa (TDP‐43), most commonly associated with amyotrophic lateral sclerosis and frontotemporal dementia, is present in 20% to 30% of AD patients.
Iron is also found to be elevated in AD. Although this was an early identified pathology, first described in 1953 (Goodman, 1953), the contribution of iron to disease progression has only recently been demonstrated. The potential for iron to contribute to neurodegeneration in AD is demonstrated by the class of diseases termed neurodegeneration with brain iron accumulation (NBIA), where excess brain iron caused by genetic mutations is the principal driver of disease (Schneider et al., 2013). If iron can cause neurodegeneration in NBIA, then it is plausible that iron elevation in AD could also contribute to disease progression in AD. Here, we will review the literature describing the iron changes in AD brain, how this occurs, to what degree iron impacts on disease progression, by what mechanisms iron could drive neurodegeneration, and, importantly, how iron can be therapeutically targeted.
5. IRON PATHOLOGY IN AD
Iron is the most abundant transition metal in the brain and is utilized for neurotransmitter synthesis (such as 5‐HT, noradrenaline and dopamine), myelin production, and neuron development among many other cellular functions. Iron levels in the brain accumulate with age (Aquino et al., 2009), which may engender risk of disease. In AD, cortical iron levels are elevated beyond the already high levels that occur during age, as measured by various methods in post‐mortem tissue (Andrasi, Farkas, Scheibler, Reffy, & Bezur, 1995; Bulk et al., 2017; Bulk et al., 2018; Collingwood et al., 2005; Connor, Menzies, St Martin, & Mufson, 1992; De Reuck et al., 2014; Duce et al., 2010; Galazka‐Friedman et al., 2011; Goodman, 1953; Hallgren & Sourander, 1960; Smith et al., 2010; Smith, Harris, Sayre, & Perry, 1997; van Duijn et al., 2017).
The amount of iron (Bulk et al., 2018) and ferritin (the major iron‐binding protein of the cell; Kwiatek‐Majkusiak et al., 2015) in brain tissue is associated with the extent of amyloid deposition. As well as being elevated in neuronal tissue, iron is enriched in the amyloid plaque itself (Meadowcroft, Peters, Dewal, Connor, & Yang, 2014), where the iron is observed to be found in a mineralized magnetite species in humans and mice models (Ayton, James, & Bush, 2017; Everett et al., 2018; Plascencia‐Villa et al., 2016; Telling et al., 2017). Magnetite is not a normal feature of brain tissue, so it is likely that this iron species was deposited by an abnormal pathological process. Possibly, Aβ itself could drive the mineralization of iron, because iron–mineral nanoparticles can be induced in vitro using the Aβ peptide (Tahirbegi, Pardo, Alvira, Mir, & Samitier, 2016).
Advancements in MRI imaging have allowed for new imaging sequences that are sensitive to iron to detect iron levels in living patients. Similar to the post‐mortem findings, MRI measures of iron have been shown to be elevated in living people with clinical AD (Acosta‐Cabronero et al., 2013; Ayton, Fazlollahi, et al., 2017; Hwang et al., 2016; Luo et al., 2013; Qin et al., 2011; Raven, Lu, Tishler, Heydari, & Bartzokis, 2013; Rodrigue, Daugherty, Haacke, & Raz, 2013; van Rooden et al., 2014) and associated with the extent of plaque pathology as measured by PET Aβ imaging (Ayton, Fazlollahi, et al., 2017; Tiepolt et al., 2018; van Bergen et al., 2016; van Bergen et al., 2018; Zhao et al., 2017). Therefore, iron has been shown to be enriched in AD brains and directly associated with amyloid plaque both in vivo and ex vivo.
6. MECHANISMS OF IRON ELEVATION IN AD
How does brain iron increase in AD? It is unlikely that iron is elevated due to factors that affect body iron load such as diet, or peripheral disorders of iron metabolism such as haemochromatosis, because the blood brain barrier (BBB) causes a dissociation between brain and peripheral pools of iron leading to a poor relationship between iron in the body and iron in the brain (Ayton, Faux, et al., 2015; Pirpamer et al., 2016). Although it has been reported that blood biomarkers of iron during midlife are inversely proportional to later life cognitive performance (Andreeva et al., 2013), this association has not yet been directly linked with the development of AD. It is also not yet established whether iron changes in AD are a contributing factor or consequence of other disease processes such as vascular changes and neuroinflammation.
6.1. Vascular pathology in AD
Vascular risk factors and vascular disease are strongly linked with AD pathogenesis; 80% of AD patients have vascular pathology and 32% of AD patients suffer from cerebrovascular disease at death (Toledo et al., 2013). Vascular lesions result in microbleeds that deposit iron in the brain—indeed the MRI method for detecting microbleeds takes advantage of the paramagnetic properties of iron containing hemosiderin to detect the lesion (Liu et al., 2012). It is possible that accumulating vascular burden leads to iron deposition in AD.
6.2. Neuroinflammation
The AD brain is characterized by activated microglia and evidence of neuroinflammation (Heneka, Carson, et al., 2015). Inflammation is a condition that can markedly alter iron homeostasis. Inflammation signals to the cell to take up and retain iron so that pathogens lack access to iron for their growth (Wessling‐Resnick, 2010). Iron elevation in AD could therefore lie downstream of neuroinflammation in AD. But the inappropriate sequestration of iron in a situation without an infection but where the neuroinflammatory apparatus is activated could lead to secondary iron toxicity that propels the neurodegenerative process.
6.3. Iron and AD biochemistry
The neuronal iron pathway has been reviewed extensively previously (Figure 1; Ayton, Lei, & Bush, 2013); here, we focus on how the major proteins involved in AD: tau, the APP (which is cleaved to generate Aβ), and apolipoprotein E (APOE) have recently been shown to impact on brain iron homeostasis. Altered metabolism of this pathway may therefore contribute to iron elevation in AD. For example, tau knockout mice were shown to induce age‐dependent iron accumulation that resulted in neurodegeneration accompanying cognitive and motor impairment that can be rescued by iron chelation (Lei et al., 2012; Lei et al., 2014; Lei et al., 2015; Lei et al., 2016; Tuo et al., 2017). Intoxication of pathogenic species of Aβ in mice also resulted in tau‐dependent iron elevation (Li et al., 2015).
Figure 1.
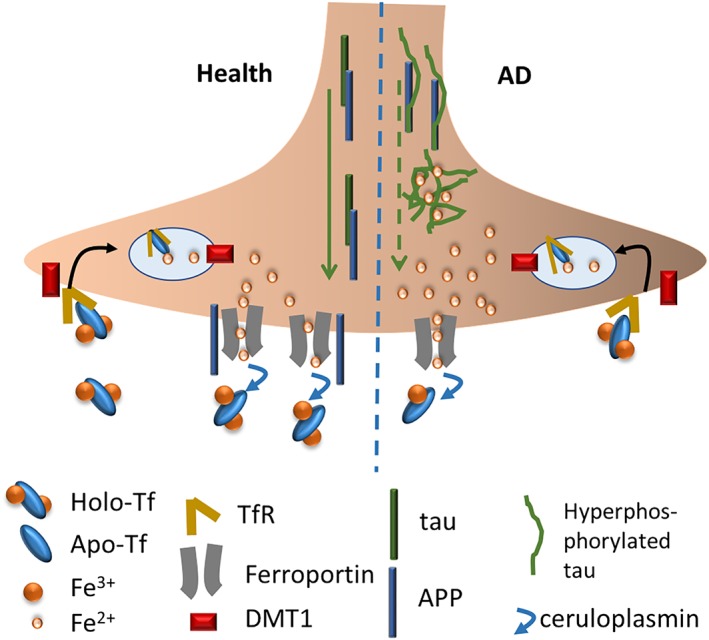
The neuronal iron pathway in health and AD. In health, iron is imported by the transferrin receptor (TfR) and divalent metal iron transporter 1 (DMT1) endocytic pathway. The transferrin protein binds two iron atoms (holo‐TF) and this is endocytosed into the cell via TfR1, and the iron is released into the cytosol for use by the cell. Excess iron is exported via ferroportin, which uses the ferroxidase, ceruloplasmin to oxidize the iron for transferrin loading. APP acts to stabilize the surface presentation of ferroportin to facilitate iron export. Tau protein supports the trafficking of APP to the cell surface. In AD, tau is phosphorylated and aggregated, leading to less tau to function to facilitate APP transport to the surface where it acts to promote ferroportin‐mediated iron efflux, leading to intraneuronal iron accumulation
Loss of tau was shown to prevent the presentation of APP at the cell surface (Lei et al., 2012), where it can act to stabilize the presentation of ferroprotein and promote iron export from the cell (Duce et al., 2010; McCarthy, Park, & Kosman, 2014; Wong et al., 2014). Loss of APP in mice similarly results in iron elevation and neurodegeneration associated with age and other co‐morbidities, which can be rescued by iron chelators (Ayton et al., 2014; Ayton, Lei, et al., 2015; Belaidi et al., 2018).
APOE has also recently been linked to brain iron homeostasis. APOE is a lipoprotein that exists in three variants within the community: ε2, ε3, and ε4. APOE ε4 carriers are at increased risk for AD and these subjects were also shown to have elevated ferritin (major iron‐binding protein and reporter of brain iron load) in their CSF (Ayton, Faux, et al., 2015). CSF APOE levels were also shown to be highly correlated with CSF ferritin levels, but the fact that ferritin levels varied according to APOE genotype suggests that APOE was driving changes in iron, and not the reverse. In agreement with these data, the ε4 allele was shown in people with mild cognitive impairment (MCI) to predict elevated iron as measured by MRI (van Bergen et al., 2016)
6.4. Iron and cognitive decline in AD
Collectively, the above findings describe how iron might be elevated in AD, but just because something is changed in the disease, it does not mean that this is a contributing factor to the disease. The establishment of fluid and imaging reporters of brain iron has opened a new window into the role of iron in disease pathogenesis and has also introduced new biomarkers into the diagnostic toolbox for AD. Biomarkers for amyloid, either using PET or CSF, represent the most advanced diagnostics for AD. Amyloid biomarkers have been used to demonstrate preclinical disease activity occurring in ~30% of people aged over 65 (Villemagne et al., 2013). So, although the role of Aβ in contributing to disease pathogenesis is unclear, the utility of Aβ‐based biomarkers is not disputed. It is clear that Aβ pathology defines the disease, but it is unclear whether the protein deposition contributes to the disease. People who are cognitively normal, yet positive for Aβ pathology, are at increased risk of cognitive decline and progression towards AD, but variability in rates of decline between people precludes biomarkers for Aβ being used in clinical settings to predict cognitive decline in the near term (Lim et al., 2014). The field therefore requires prognostic biomarkers that predict decline upon pathology, and there has been recent progress to achieve this using a biomarker of iron.
As previously described, the development of MRI sequences sensitive to iron levels, and in particular quantitative susceptibility mapping (QSM), has afforded the opportunity to investigate whether iron is associated with disease progression and whether iron biomarkers might have utility in prognostication for AD. Higher QSM values, particularly in the hippocampus, are associated with accelerated cognitive decline over 6 years in people with PET‐confirmed Aβ pathology, and along the clinical severity spectrum of AD (Ayton, Fazlollahi, et al., 2017). In cross‐sectional analysis, the severity of cognitive impairment in people with a clinical diagnosis of AD was associated with QSM (Du et al., 2018), and T2* cortical phase shifts (also sensitive to tissue iron) was associated with cognitive performance in people with subjective memory complaints (van Rooden et al., 2016) and people with AD (Luo et al., 2013).
Similar to the imaging biomarkers for iron, CSF ferritin is a fluid biomarker for brain iron that also demonstrated prognostic utility for AD. Ferritin is the major iron‐binding protein of the body, and serum ferritin has been used routinely as biomarker of body iron stores for decades. CSF is poorly correlated with serum ferritin, revealing a disconnect between brain and body stores of iron (Ayton, Faux, et al., 2015). Unlike the affected cortical brain regions in AD, CSF ferritin is not elevated in AD (Ayton, Faux, et al., 2015). This is likely because CSF ferritin represents iron values in the whole brain, not just the cortical regions affected by AD. But those people who have generally high brain iron load reflected by CSF ferritin have worse cognitive performance throughout the clinical spectrum of AD (Ayton, Faux, et al., 2015). Intriguingly, CSF ferritin levels were shown to be elevated in people carrying the ε4 risk allele of APOE, and CSF ferritin levels were strongly associated with future cognitive loss over 7 years in people carrying the ε4 allele but who were cognitively normal at the beginning of the study (Ayton, Faux, & Bush, 2017).
The fact that brain iron level predicts risk of decline possibly suggests that iron is a mediating factor in the pathogenesis of AD. The reverse could also be true—iron could be elevated as an epiphenomenon in people who are at risk of cognitive decline. This correlation/causation question may be resolved by a clinical trial of an iron lowering drug, to see if lowering iron impacts on disease progression. However, preclinical and clinical findings, reviewed below, have also demonstrated potential mechanisms for how iron could contribute to neurodegeneration in AD that may position iron is a deleterious agent in the AD brain.
7. MECHANISMS OF IRON IN THE PATHOGENESIS OF AD
As previously described, iron deposition within plaques hints at a role of iron in plaque genesis and/or propagation. These ex vivo data are supported by in vitro findings demonstrating that iron can accelerate the aggregation of the Aβ peptide (Huang et al., 1999; Liu et al., 2011; Mantyh et al., 1993; Ott, Dziadulewicz, & Crowther, 2015), which is prevented by iron chelation (Huang et al., 2004). Iron therefore has the ability to cause Aβ aggregation, and the fact that iron is enriched in plaques provides the “smoking gun” evidence that iron is involved in plaque genesis. In vivo evidence that iron might be contributing to plaque deposition is obtained from biomarker studies. Elevated plasma ferritin is associated with PET amyloid load (Goozee et al., 2017), and CSF ferritin has been shown to predict longitudinal loss of Aβ42 levels in CSF (which report elevated amyloid plaque due to sequestration of free Aβ into the protopathic deposit; Ayton, Diouf, & Bush, 2018).
Ferric iron (Fe3+)‐stimulated aggregation of Aβ causes the reduction of the iron to the ferrous species Fe2+ (redox‐active form), thus propagating a continued source of ROS (Everett et al., 2014), which may contribute to the neurotoxicity of Aβ. Iron binding to His6, His13, and His14 of Aβ is suggested to mediate the redox activity of the iron–Aβ interaction (Nakamura et al., 2007). Iron aggregated Aβ is toxic to cultured neurons (Liu et al., 2011; Schubert & Chevion, 1995) possibly by inducing oxidative stress (Rival et al., 2009).
Iron may also increase the production of Aβ by stimulating expression of its parent protein, APP, and altering its processing. APP has a 5`UTR iron responsive element that suppresses translation of the protein in low iron conditions (Rogers et al., 2002). High iron conditions has been shown to accelerate degeneration in a mouse model of AD (Becerril‐Ortega, Bordji, Freret, Rush, & Buisson, 2014).
Iron also colocalizes with tau‐containing neurofibrillary tangles in post‐mortem cases with AD and NBIA (Smith et al., 1997; Tofaris, Revesz, Jacques, Papacostas, & Chataway, 2007), possibly suggesting that iron may bind to tau and affect its toxic potential. Iron has been shown to induce the aggregation of tau (Yamamoto et al., 2002), and neurofibrillary tangles enriched with iron may act as a source of oxidative stress (Sayre et al., 2000; Smith et al., 1997).
Iron has also been shown to enhance hyperphosphorylation of tau by stimulating the cyclin dependent kinase 5 (CDK5) complex and glycogen synthase kinase (GSK)‐3β kinase activity and diminish the activity of protein phosphatase 2A, the major tau phosphatase in the cells (Jin Jung et al., 2013; Xie et al., 2012). Indeed, deferoxamine treatment in an AD mouse model supressed iron‐induced tau hyperphosphorylation through inhibition of CDK5 and GSK3 activity (Guo et al., 2013).
Iron could also contribute to AD pathogenesis by directly causing oxidative stress. Iron is able to cycle between ferrous (Fe2+) and ferric (Fe3+) states within biology. The cell makes use of this chemistry for many redox reactions; however, this same chemistry can allow for deleterious oxidative stress. Ferrous iron catalyses the formation of hydroxyl radicals (OH•), an especially potent ROS, from physiologically available hydrogen peroxide. Neurodegeneration in AD may also be the cause of the newly described cell death process, ferroptosis.
8. FERROPTOSIS
Ferroptosis is a type of regulated necrosis that was first described in 2012 as an iron‐dependent cell death that is genetically, biochemically, and morphologically distinguishable from other types of programmed cell death (Dixon et al., 2012). Iron‐induced lipid peroxidation causes catastrophic membrane rupture resulting in ferroptotic cell death unless this is controlled by glutathione peroxidase 4 (GPx4)—the master regulator of ferroptosis. GPx4 is a lipid repair enzyme that is unique among the glutathione peroxidase family because it is the only enzyme that is able to detoxify lipid hydroperoxides directly in membranes. GPx4 uses GSH as a cofactor, and therefore, GSH availability is required for the activity of the protein. The rate liming factor of GSH synthesis is cysteine levels within the cell. Cysteine is an amino acid that may be taken up by the cell in an oxidized form called cystine by the glutamate/cystine antiporter, system xCT. Within the cell, cystine is reduced to cysteine by cystathionine β‐synthase, and cysteine is available for GSH synthesis. Inhibition of system xCT and cystine uptake using the small molecule erastin was the first induction mechanism of ferroptosis to be demonstrated, which results in GSH depletion and GPx4 inactivation, and accumulating lipid peroxidation (Dixon et al., 2012).
Particular membrane lipids are oxidized during ferroptosis. Polyunsaturated fatty acids (PUFA) specifically containing arachidonic acid or adrenic acid undergo esterification with CoA by acyl‐CoA synthase 4 (ACSL4) to form phosphatidylethanolamine (Doll et al., 2017). Membrane phosphatidylethanolamines are vulnerable to peroxidation by specific iron‐dependent lipoxygenases (Yang et al., 2016): 5‐LOX (ALOX5), 12S‐LOX (ALOX12), 12R‐LOX (ALOX12B), 15‐LOX1 (ALOX15), 15‐LOX‐2 (ALOX15B), and E‐LOX (ALOXE3). This vulnerability is by design, to allow for regulated cell death by ferroptosis.
Iron is a rate‐limiting substrate for lipid peroxidation by lipoxygenases, but labile iron within the cell also causes lipid peroxidation. Iron chelation can completely inhibit ferroptotic cell death, whereas elevation of iron increases vulnerability to ferroptosis (Dixon et al., 2012). During ferroptosis, the activation of “ferritinophagy,” the cellular process that degrades iron‐containing cytosolic ferritin in lysosomes to release “labile” iron into the cell, provides the fuel for ferroptosis. Ferritinophagy is the normal process by which the cell releases iron from ferritin when iron is required to be used for various cell functions. However, ferritinophagy is activated during cysteine deficiency induced ferroptosis despite iron levels being in the normal range. The release of iron from ferritin propergates the ferroptosis signal. Inhibition of autophagy protects against ferroptosis by lowering the amount of iron release by ferritin that can oxidize lipids (Gao et al., 2016; Hou et al., 2016). Therefore, and importantly, ferroptosis is inducible by the iron that resides within the cell—ferroptosis does not require the toxic application of iron. In other words, ferroptosis does not describe the cell death process that occurs when iron is in poisonous excess; rather, it describes the process by which the cell recruits its iron to kill itself. A cell with more iron will have increased susceptibility to ferroptosis, but a cell need not have iron in excess to die by ferroptosis.
Therefore, if ferroptosis were occurring in AD, we may not expect to see every person exhibiting iron elevation; however, we should expect to see that people with elevated iron have a faster disease trajectory. Indeed, this is what is observed with the iron‐related biomarkers (Ayton, Faux, et al., 2015; Ayton, Faux, & Bush, 2017; Ayton, Fazlollahi, et al., 2017). Iron biomarkers thus are not suitable to determine whether ferroptosis is occurring, even if they do predict susceptibility towards ferroptosis. As yet, there are no biomarkers that can detect ferroptosis in humans, but there a number of lines of evidence to support a role for ferroptosis in AD neurodegeneration.
Brain GSH levels can be measured in living patients using magnetic resonance spectroscopy, which may represent one biomarker of ferroptotic stress. Magnetic resonance spectroscopy has been used to demonstrate decreased GSH levels in AD patients (Mandal, Saharan, Tripathi, & Murari, 2015). GSH has also been shown to be associated with amyloid load in cognitively normal older adults (Chiang et al., 2017).
Lipid peroxides, the toxic species of ferroptosis, has been shown to be elevated in AD brain (Bradley, Markesbery, & Lovell, 2010; Hajimohammadreza & Brammer, 1990; Markesbery, Kryscio, Lovell, & Morrow, 2005; Montine et al., 2001; Reed et al., 2008; Williams, Lynn, Markesbery, & Lovell, 2006), and increased levels of serum oxidative stress markers and shortfalls in serum antioxidants are also observed in the blood of MCI and AD subjects (Schrag et al., 2013). In a longitudinal study examining lipid peroxidation levels and cognitive performance in MCI patients, decreased lipid antioxidants were shown to be associated with accelerated cognitive decline (Baldeiras et al., 2010). AD mouse models, supplemented with arachidonic acid (the major lipid substrate for ferroptosis), have been shown to be more prone to oxidation in the hippocampus compared with wild type mice (Furman et al., 2016). Additionally, elevated levels of acrolein‐conjugated protein in AD patients' plasma is negatively correlated with their cognitive performance in Mini–Mental State Examination and clinical dementia rating (Tsou et al., 2018).
9. THERAPEUTICALLY TARGETING IRON AND FERROPTOSIS IN AD
The ferroptosis pathway can be targeted at multiple levels. Increasing cystine or GSH levels will promote GPx4 activity; this can be achieved using n‐acetylcysteine (NAC), a cystine precursor. The expression of GPx4 can also be increased by elevating selenium levels. This will be discussed in more detailed below, but briefly, GPx4 is a selenoprotein that is dependent on selenium within the cell in the form of selenocysteine for its production. Increasing selenium levels with compounds such as selenate or selenomethionine promotes the protein expression of GPx4. Ferroptosis can also be inhibited by interfering with the biochemistry of PUFA. For example, ferroptosis can be prevented by inhibitors of ACSL4 an enzyme that esterifies PUFA, such as arachidonic acid, into phosphatidylethanolamines (the major substrate of ferroptosis). Preventing the oxidation of lipids, by using compounds such as iron chelators or lipoxygenase inhibitors, including vitamin E, also prevents ferroptosis. Here, we review the evidence for agents that impact on the ferroptosis that have been tested in an AD context (Figure 2). Unlike the clinical trials targeting Aβ, many of the aforementioned trials are early, exploratory studies that require dose optimization and replication with larger numbers of subjects. Also, none of these trials were enacted with the view of targeting ferroptosis, a concept that emerged only recently. So, although the following compounds may hinder the ferroptotic cascade, the trialling of specific and potent antiferroptotic compounds would more formally test whether ferroptosis contributes to neurodegeneration in AD.
Figure 2.
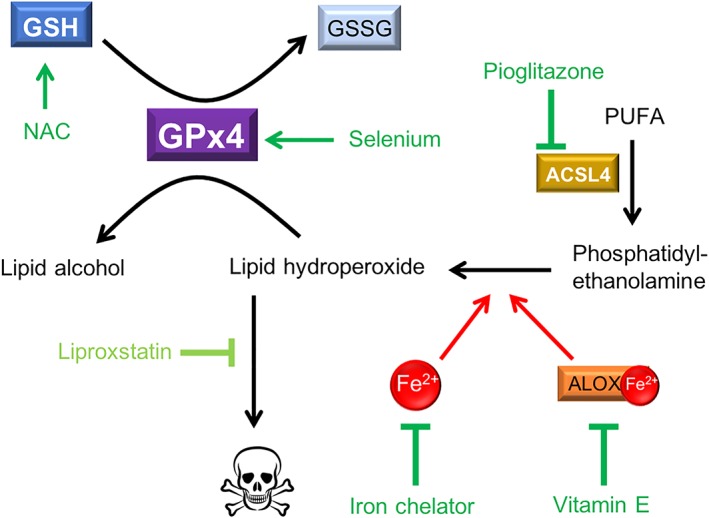
Approaches to pharmacological ferroptosis inhibition in AD. Iron‐induced peroxidation of phosphatidylethanolamines within membranes causes ferroptotic cell death unless this is inhibited by antiferroptotic defences within the cell. There have been several drugs tested in the AD context that bolster the antiferroptotic defences. GPx4 is the master regulator of ferroptosis, and its expression can be increased by selenium supplementation (e.g., sodium selenate and selenomethionine), and its activity can be enhanced by elevating levels of GSH cofactor, which can be achieved using NAC to increase the cystine precursor. Phosphatidylethanolamines are derived from PUFA that undergo esterification by ACSL4, and the diabetes drug, pioglitazone, has recently been identified as an ACSL4 inhibitor. Peroxidation of lipids can be prevented using iron chelators, or lipoxygenase inhibitors such as vitamin E (and related metabolites). Liproxstatin is the most specific and potent antiferroptosis drug currently available, and although drugs that act like liproxstatin may provide the most benefit, they have not yet been investigated in clinical trials
9.1. Iron
Among the ferroptosis‐related targets, iron chelation has been most pursued for AD. In 1991, a single blind Phase 2 clinical trial reported that desferrioxamine slowed cognitive decline in subjects with AD by 50% over a 24‐month period, an effect attributed to binding brain aluminium, even though desferrioxamine was designed as an iron chelator (Crapper McLachlan et al., 1991). The evidence for environmental exposure to aluminium playing a role in AD subsequently weakened (Flaten, 2001), and given that the amount of iron in the brain is substantially higher than the concentration of aluminium, it is reasonable to suspect that the actions of the drug were on iron. Despite this trial showing a positive outcome, this desferrioxamine treatment for AD was not followed up in subsequent clinical trials.
One of the limitations of desferrioxamine is that it has limited BBB penetrance. An internasal formulation of desferrioxamine was developed to overcome this, and it was shown to improve cognition in an animal model of AD (Hanson et al., 2012), while decreasing GSK activity, oxidative stress, and soluble Aβ species (Fine et al., 2015). Similarly, intranasal deferoxamine treatment lowered tau hyperphosphorylation by inhibition of CDK5 and GSK3 (Guo et al., 2013). Another iron chelator, deferasirox, was shown to lower age‐dependent iron accumulation and Aβ levels in the ageing rat brain (Banerjee, Sahoo, Anand, Bir, & Chakrabarti, 2016). An orally bioavailable iron chelator, M30, has also been shown to improve memory performance and lower amyloid and tangle pathology in a mouse model of AD (Kupershmidt, Amit, Bar‐Am, Youdim, & Weinreb, 2012), but has not been tested clinically.
Deferiprone is an orally available iron chelator that is currently in clinical use and has been shown to cross the BBB. In animal models of Parkinson's disease, it has been shown to improve motor performance (Aguirre et al., 2015; Ayton, Lei, Duce, et al., 2013; Ayton, Lei, et al., 2015; Devos et al., 2014; Dexter et al., 2011; Workman et al., 2015). Deferiprone is neuroprotective in neuronal culture models of Aβ toxicity (Molina‐Holgado, Gaeta, Francis, Williams, & Hider, 2008; Part et al., 2015), and it has been shown to reduce Aβ burden and tau phosphorylation in a rabbit model of AD (Prasanthi et al., 2012). Deferiprone was shown to lower brain iron levels (as assessed by MRI and CSF ferritin) and improve motor performance in a Phase 2 study of Parkinson's disease (Devos et al., 2014), and there is now a Phase 2 study underway for AD (clinicaltrials.gov identifier: NCT03234686).
9.2. Selenium
Selenium, in the form of selenocysteine, is an essential amino acid that is required for the synthesis of GPx4, and dietary selenium and selenium supplementation has been explored for its potential antioxidant role in AD. Selenium can be administered as one of several organic (e.g., selenocysteine and selenomethionine) or inorganic (e.g., selenate and selenite) species. Dietary supplementation of selenium (mainly as selenomethionine) was shown to improve cognitive performance in a clinical trial of a cohort of selenium deficient patients with mild cognitive impairment (MCI; Cardoso et al., 2016). Dietary selenium is most bioavailable in the organic selenomethionine form; however, a clinical trial of selenomethionine (200 μg·day−1) did not reduce dementia incidence in a cohort of healthy, nonselenium‐deficient men over 60 (Kryscio et al., 2017). Inorganic selenate has been shown to improve cognitive performance in mouse models of tauopathies (Corcoran et al., 2010; Shultz et al., 2015; van Eersel et al., 2010), which prompted a Phase 2, 24‐week double‐blind, placebo controlled, clinical trial of sodium selenate in Alzheimer's patients (Malpas et al., 2016). Selenate lessened deterioration in brain structures as measured by diffusion tensor MRI, but overall did not impact on cognition in this 24‐week period. However, in secondary analysis, the elevation of selenium in CSF was variable in patients treated with 30 mg of selenate daily, and it was found that there was a relationship between increased CSF selenium and improved cognitive performance (Cardoso et al., 2019).
9.3. Lipid oxidation
Pioglitazone is a drug that has been used for type 2 diabetes and shown to lower blood sugar. It has also recently been characterized as an inhibitor of ACSL4, and long‐term use of pioglitazone has been shown to reduce the risk of dementia in type 2 diabetes patients (Heneka, Fink, & Doblhammer, 2015). Inhibition of the oxidation of PUFA by the 12/15 lipoxygenase inhibitor, PD146176, was shown to protect against cognitive decline in a mouse model of AD (Di Meco et al., 2017). Metabolites of vitamin E and vitamin E itself (more weakly) also inhibit lipoxygenase and protect against ferroptosis (Hinman et al., 2018), and there has been some evidence of a beneficial effect of vitamin E in clinical trials. Vitamin E was shown to slow disease progression in patients with moderately severe AD over a 2‐year period (Sano et al., 1997), which was supported in a more recent clinical trial, which also demonstrated that patients with mild to moderate AD taking vitamin E had slowing of disease progression (Dysken et al., 2014). However, a prior study showed no effect of vitamin E treatment in preventing conversion from mild cognitive imparment to AD over a 3‐year period (Petersen et al., 2005).
NAC is a compound used to treat paracetamol intoxication and can also improve lipid antioxidant defences by elevating cysteine, the rate limiting substrate for GSH, which in turn acts as a necessary cofactor for GPx4. NAC was shown to restore memory function in a mouse model of AD (Hsiao, Kuo, Chen, & Gean, 2012). A small clinical trial of NAC showed no significant effect on the primary outcome cognitive measures, but an improvement on some subanalysis over 6 months of treatment (Adair, Knoefel, & Morgan, 2001).
10. CONCLUSION
It is unclear what causes neurodegeneration in AD. The field has focused on the Aβ peptide, but therapeutically targeting this has not led to improved patient outcomes. The mechanism of toxicity of Aβ is also not established, nor if and how it recruits the tau protein. Ferroptosis is one potential mechanism that could contribute to neurodegeneration in AD. This concept is supported by pathological changes in iron occurring in affected cortical brain region in AD and that these changes correlate with disease outcomes. Importantly, early clinical trial data hint that targeting this pathway may slow disease progression. The cumulative evidence supports the establishment of larger clinical trials with novel and specific ferroptosis inhibitors to determine whether disease modification can be achieved by targeting this pathway.
10.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
Supported by funds from the Australian Research Council, the Australian National Health & Medical Research Council (NHMRC). The Florey Institute of Neuroscience and Mental Health acknowledges support from the Victorian Government, in particular funding from the Operational Infrastructure Support Grant.
Nikseresht S, Bush AI, Ayton S. Treating Alzheimer's disease by targeting iron. Br J Pharmacol. 2019;176:3622–3635. 10.1111/bph.14567
REFERENCES
- Acosta‐Cabronero, J. , Williams, G. B. , Cardenas‐Blanco, A. , Arnold, R. J. , Lupson, V. , & Nestor, P. J. (2013). In vivo quantitative susceptibility mapping (QSM) in Alzheimer's disease. PLoS One, 8, e81093 10.1371/journal.pone.0081093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair, J. C. , Knoefel, J. E. , & Morgan, N. (2001). Controlled trial of N‐acetylcysteine for patients with probable Alzheimer's disease. Neurology, 57, 1515–1517. 10.1212/WNL.57.8.1515 [DOI] [PubMed] [Google Scholar]
- Aguirre, P. , Mena, N. P. , Carrasco, C. M. , Munoz, Y. , Perez‐Henriquez, P. , Morales, R. A. , … Núñez, M. T. (2015). Iron chelators and antioxidants regenerate neuritic tree and nigrostriatal fibers of MPP+/MPTP‐lesioned dopaminergic neurons. PLoS One, 10, e0144848 10.1371/journal.pone.0144848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol, 174, S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol, 174, S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrasi, E. , Farkas, E. , Scheibler, H. , Reffy, A. , & Bezur, L. (1995). Al, Zn, Cu, Mn and Fe levels in brain in Alzheimer's disease. Archives of Gerontology and Geriatrics, 21, 89–97. 10.1016/0167-4943(95)00643-Y [DOI] [PubMed] [Google Scholar]
- Andreeva, V. A. , Galan, P. , Arnaud, J. , Julia, C. , Hercberg, S. , & Kesse‐Guyot, E. (2013). Midlife iron status is inversely associated with subsequent cognitive performance particularly in perimenopausal women. The Journal of Nutrition., 143, 1974–1981. 10.3945/jn.113.177089 [DOI] [PubMed] [Google Scholar]
- Aquino, D. , Bizzi, A. , Grisoli, M. , Garavaglia, B. , Bruzzone, M. G. , Nardocci, N. , … Chiapparini, L. (2009). Age‐related iron deposition in the basal ganglia: Quantitative analysis in healthy subjects. Radiology, 252, 165–172. 10.1148/radiol.2522081399 [DOI] [PubMed] [Google Scholar]
- Ayton, S. , Diouf, I. , Bush, A. I. , & Alzheimer's disease Neuroimaging I (2018). Evidence that iron accelerates Alzheimer's pathology: A CSF biomarker study. Journal of Neurology, Neurosurgery, and Psychiatry, 89, 456–460. 10.1136/jnnp-2017-316551 [DOI] [PubMed] [Google Scholar]
- Ayton, S. , Faux, N. G. , & Bush, A. I. (2017). Association of cerebrospinal fluid ferritin level with preclinical cognitive decline in APOE‐ε4 carriers. JAMA Neurology, 74, 122–125. 10.1001/jamaneurol.2016.4406 [DOI] [PubMed] [Google Scholar]
- Ayton, S. , Faux, N. G. , Bush, A. I. , & Alzheimer's Disease Neuroimaging Initiative I (2015). Ferritin levels in the cerebrospinal fluid predict Alzheimer's disease outcomes and are regulated by APOE. Nature Communications, 6, 6760 10.1038/ncomms7760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton, S. , Fazlollahi, A. , Bourgeat, P. , Raniga, P. , Ng, A. , Lim, Y. Y. , … Bush, A. I. (2017). Cerebral quantitative susceptibility mapping predicts amyloid‐β‐related cognitive decline. Brain: A Journal of Neurology, 140, 2112–2119. 10.1093/brain/awx137 [DOI] [PubMed] [Google Scholar]
- Ayton, S. , James, S. A. , & Bush, A. I. (2017). Nanoscale imaging reveals big role for iron in Alzheimer's disease. Cell Chemical Biology, 24, 1192–1194. [DOI] [PubMed] [Google Scholar]
- Ayton, S. , Lei, P. , & Bush, A. I. (2013). Metallostasis in Alzheimer's disease. Free Radical Biology & Medicine, 62, 76–89. 10.1016/j.freeradbiomed.2012.10.558 [DOI] [PubMed] [Google Scholar]
- Ayton, S. , Lei, P. , Duce, J. A. , Wong, B. X. , Sedjahtera, A. , Adlard, P. A. , … Finkelstein, D. I. (2013). Ceruloplasmin dysfunction and therapeutic potential for parkinson disease. Annals of Neurology, 73, 554–559. 10.1002/ana.23817 [DOI] [PubMed] [Google Scholar]
- Ayton, S. , Lei, P. , Hare, D. J. , Duce, J. A. , George, J. L. , Adlard, P. A. , et al. (2015). Parkinson's Disease Iron Deposition Caused by Nitric Oxide‐Induced Loss of APP. Journal of Neuroscience, 35, 3591–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton, S. , Zhang, M. , Roberts, B. R. , Lam, L. Q. , Lind, M. , McLean, C. , … Duce, J. A. (2014). Ceruloplasmin and β‐amyloid precursor protein confer neuroprotection in traumatic brain injury and lower neuronal iron. Free Radical Biology Medicine, 69, 331–337. 10.1016/j.freeradbiomed.2014.01.041 [DOI] [PubMed] [Google Scholar]
- Baldeiras, I. , Santana, I. , Proenca, M. T. , Garrucho, M. H. , Pascoal, R. , Rodrigues, A. , … Oliveira, C. R. (2010). Oxidative damage and progression to Alzheimer's disease in patients with mild cognitive impairment. Journal of Alzheimer's Disease, 21, 1165–1177. 10.3233/JAD-2010-091723 [DOI] [PubMed] [Google Scholar]
- Banerjee, P. , Sahoo, A. , Anand, S. , Bir, A. , & Chakrabarti, S. (2016). The oral iron chelator, deferasirox, reverses the age‐dependent alterations in iron and amyloid‐β homeostasis in rat brain: Implications in the therapy of Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 49, 681–693. [DOI] [PubMed] [Google Scholar]
- Becerril‐Ortega, J. , Bordji, K. , Freret, T. , Rush, T. , & Buisson, A. (2014). Iron overload accelerates neuronal amyloid‐β production and cognitive impairment in transgenic mice model of Alzheimer's disease. Neurobiology of Aging, 35, 2288–2301. [DOI] [PubMed] [Google Scholar]
- Belaidi, A. A. , Gunn, A. P. , Wong, B. X. , Ayton, S. , Appukuttan, A. T. , Roberts, B. R. , … Bush, A. I. (2018). Marked age‐related changes in brain iron homeostasis in amyloid protein precursor knockout mice. Neurotherapeutics, 15(4), 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1991). Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathologica, 82, 239–259. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Bradley, M. A. , Markesbery, W. R. , & Lovell, M. A. (2010). Increased levels of 4‐hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radical Biology & Medicine, 48, 1570–1576. 10.1016/j.freeradbiomed.2010.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulk, M. , Abdelmoula, W. M. , Nabuurs, R. J. A. , van der Graaf, L. M. , Mulders, C. W. H. , Mulder, A. A. , … van der Weerd, L. (2017). Postmortem MRI and histology demonstrate differential iron accumulation and cortical myelin organization in early‐ and late‐onset Alzheimer's disease. Neurobiology of Aging, 62, 231–242. [DOI] [PubMed] [Google Scholar]
- Bulk, M. , Kenkhuis, B. , Graaf, L. M. V. , Goeman, J. J. , Natte, R. , & Weerd, L. V. (2018). Postmortem T2*‐weighted MRI imaging of cortical iron reflects severity of Alzheimer's disease. Journal of Alzheimer's disease: JAD, 65(4), 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso, B. R. , Apolinario, D. , da Silva Bandeira, V. , Busse, A. L. , Magaldi, R. M. , Jacob‐Filho, W. , & Cozzolino, S. M. (2016). Effects of Brazil nut consumption on selenium status and cognitive performance in older adults with mild cognitive impairment: A randomized controlled pilot trial. European Journal of Nutrition, 55, 107–116. 10.1007/s00394-014-0829-2 [DOI] [PubMed] [Google Scholar]
- Cardoso, B. R. , Roberts, B. R. , Malpas, C. B. , Vivash, L. , Genc, S. , Saling, M. M. , … Bush, A. I. (2019). Supranutritional sodium selenate supplementation delivers selenium to the central nervous system: Results from a randomized controlled pilot trial in Alzheimer's disease. Neurotherapeutics, 16(1), 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, G. C. , Mao, X. , Kang, G. , Chang, E. , Pandya, S. , Vallabhajosula, S. , … Shungu, D. C. (2017). Relationships among cortical glutathione levels, brain amyloidosis, and memory in healthy older adults investigated in vivo with (1)H‐MRS and Pittsburgh compound‐B PET. AJNR American Journal of Neuroradiology, 38, 1130–1137. 10.3174/ajnr.A5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingwood, J. F. , Mikhaylova, A. , Davidson, M. , Batich, C. , Streit, W. J. , Terry, J. , & Dobson, J. (2005). In situ characterization and mapping of iron compounds in Alzheimer's disease tissue. Journal of Alzheimer's Disease: JAD, 7, 267–272. 10.3233/JAD-2005-7401 [DOI] [PubMed] [Google Scholar]
- Connor, J. R. , Menzies, S. L. , St Martin, S. M. , & Mufson, E. J. (1992). A histochemical study of iron, transferrin, and ferritin in Alzheimer's diseased brains. Journal of Neuroscience Research, 31, 75–83. 10.1002/jnr.490310111 [DOI] [PubMed] [Google Scholar]
- Corcoran, N. M. , Martin, D. , Hutter‐Paier, B. , Windisch, M. , Nguyen, T. , Nheu, L. , … Hovens, C. M. (2010). Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer's disease model. Journal of Clinical Neuroscience: Official Journal of the Neurosurgical Society of Australasia, 17, 1025–1033. 10.1016/j.jocn.2010.04.020 [DOI] [PubMed] [Google Scholar]
- Coric, V. , van Dyck, C. H. , Salloway, S. , Andreasen, N. , Brody, M. , Richter, R. W. , … Berman, R. M. (2012). Safety and tolerability of the γ‐secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Archives of Neurology, 69, 1430–1440. 10.1001/archneurol.2012.2194 [DOI] [PubMed] [Google Scholar]
- Crapper McLachlan, D. R. , Dalton, A. J. , Kruck, T. P. , Bell, M. Y. , Smith, W. L. , Kalow, W. , & Andrews, D. F. (1991). Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet, 337, 1304–1308. 10.1016/0140-6736(91)92978-B [DOI] [PubMed] [Google Scholar]
- Cummings, J. L. , Cohen, S. , van Dyck, C. H. , Brody, M. , Curtis, C. , Cho, W. , … Paul, R. (2018). ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology, 90, e1889–e1897. 10.1212/WNL.0000000000005550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Reuck, J. L. , Deramecourt, V. , Auger, F. , Durieux, N. , Cordonnier, C. , Devos, D. , … Bordet, R. (2014). Iron deposits in post‐mortem brains of patients with neurodegenerative and cerebrovascular diseases: A semi‐quantitative 7.0 T magnetic resonance imaging study. European Journal Of Neurology, 21(7), 1026–1031. [DOI] [PubMed] [Google Scholar]
- Devos, D. , Moreau, C. , Devedjian, J. C. , Kluza, J. , Petrault, M. , Laloux, C. , … Bordet, R. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxidants & Redox Signaling, 21, 195–210. 10.1089/ars.2013.5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter, D. T. , Statton, S. A. , Whitmore, C. , Freinbichler, W. , Weinberger, P. , Tipton, K. F. , … Crichton, R. R. (2011). Clinically available iron chelators induce neuroprotection in the 6‐OHDA model of Parkinson's disease after peripheral administration. Journal of Neural Transmission, 118, 223–231. 10.1007/s00702-010-0531-3 [DOI] [PubMed] [Google Scholar]
- Di Meco, A. , Li, J. G. , Blass, B. E. , Abou‐Gharbia, M. , Lauretti, E. , & Pratico, D. (2017). 12/15‐Lipoxygenase inhibition reverses cognitive impairment, brain amyloidosis, and tau pathology by stimulating autophagy in aged triple transgenic mice. Biological Psychiatry, 81, 92–100. 10.1016/j.biopsych.2016.05.023 [DOI] [PubMed] [Google Scholar]
- Dixon, S. J. , Lemberg, K. M. , Lamprecht, M. R. , Skouta, R. , Zaitsev, E. M. , Gleason, C. E. , … Stockwell, B. R. (2012). Ferroptosis: An iron‐dependent form of nonapoptotic cell death. Cell, 149, 1060–1072. 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll, S. , Proneth, B. , Tyurina, Y. Y. , Panzilius, E. , Kobayashi, S. , Ingold, I. , … Conrad, M. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature Chemical Biology, 13, 91–98. 10.1038/nchembio.2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody, R. S. , Raman, R. , Farlow, M. , Iwatsubo, T. , Vellas, B. , Joffe, S. , … Mohs, R. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer's disease. The New England Journal of Medicine, 369, 341–350. 10.1056/NEJMoa1210951 [DOI] [PubMed] [Google Scholar]
- Doody, R. S. , Thomas, R. G. , Farlow, M. , Iwatsubo, T. , Vellas, B. , Joffe, S. , … Solanezumab Study Group (2014). Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. The New England Journal of Medicine, 370, 311–321. 10.1056/NEJMoa1312889 [DOI] [PubMed] [Google Scholar]
- Du, L. , Zhao, Z. , Cui, A. , Zhu, Y. , Zhang, L. , Liu, J. , … Ma, G. (2018). Increased iron deposition on brain quantitative susceptibility mapping correlates with decreased cognitive function in Alzheimer's disease. ACS Chemical Neuroscience, 9(7), 1849–1857. [DOI] [PubMed] [Google Scholar]
- Duce, J. A. , Tsatsanis, A. , Cater, M. A. , James, S. A. , Robb, E. , Wikhe, K. , … Bush, A. I. (2010). Iron‐export ferroxidase activity of β‐amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell, 142, 857–867. 10.1016/j.cell.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dysken, M. W. , Sano, M. , Asthana, S. , Vertrees, J. E. , Pallaki, M. , Llorente, M. , … Guarino, P. D. (2014). Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM‐AD VA cooperative randomized trial. Jama, 311, 33–44. 10.1001/jama.2013.282834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, M. F. , Kost, J. , Tariot, P. N. , Aisen, P. S. , Cummings, J. L. , Vellas, B. , … Michelson, D. (2018). Randomized trial of verubecestat for mild‐to‐moderate Alzheimer's disease. The New England Journal of Medicine, 378, 1691–1703. 10.1056/NEJMoa1706441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett, J. , Cespedes, E. , Shelford, L. R. , Exley, C. , Collingwood, J. F. , Dobson, J. , … Telling, N. D. (2014). Ferrous iron formation following the co‐aggregation of ferric iron and the Alzheimer's disease peptide β‐amyloid (1‐42). J R Soc Interface, 11 20140165, 10.1098/rsif.2014.0165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett, J. , Collingwood, J. F. , Tjendana‐Tjhin, V. , Brooks, J. , Lermyte, F. , Plascencia‐Villa, G. , … Telling, N. D. (2018). Nanoscale synchrotron X‐ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer's disease subjects. Nanoscale, 10(25), 11782–11796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow, M. , Arnold, S. E. , van Dyck, C. H. , Aisen, P. S. , Snider, B. J. , Porsteinsson, A. P. , … Siemers, E. R. (2012). Safety and biomarker effects of solanezumab in patients with Alzheimer's disease. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 8, 261–271. 10.1016/j.jalz.2011.09.224 [DOI] [PubMed] [Google Scholar]
- Fine, J. M. , Renner, D. B. , Forsberg, A. C. , Cameron, R. A. , Galick, B. T. , Le, C. , … Hanson, L. R. (2015). Intranasal deferoxamine engages multiple pathways to decrease memory loss in the APP/PS1 model of amyloid accumulation. Neuroscience Letters, 584, 362–367. 10.1016/j.neulet.2014.11.013 [DOI] [PubMed] [Google Scholar]
- Flaten, T. P. (2001). Aluminium as a risk factor in Alzheimer's disease, with emphasis on drinking water. Brain Research Bulletin, 55, 187–196. 10.1016/S0361-9230(01)00459-2 [DOI] [PubMed] [Google Scholar]
- Fleisher, A. S. , Raman, R. , Siemers, E. R. , Becerra, L. , Clark, C. M. , Dean, R. A. , … Thal, L. J. (2008). Phase 2 safety trial targeting amyloid β production with a γ‐secretase inhibitor in Alzheimer disease. Archives of Neurology, 65, 1031–1038. 10.1001/archneur.65.8.1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman, R. , Murray, I. V. , Schall, H. E. , Liu, Q. , Ghiwot, Y. , & Axelsen, P. H. (2016). The fate of uniformly radiolabeled arachidonic acid in Alzheimer mouse brain: Plaque‐associated oxidative degradation. ACS Chemical Neuroscience, 7(3), 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galazka‐Friedman, J. , Bauminger, E. R. , Szlachta, K. , Koziorowski, D. , Tomasiuk, R. , Jaklewicz, A. , … Friedman, A. (2011). Iron in Alzheimer's and control hippocampi—Mössbauer, atomic absorption and ELISA studies. Acta Physica Polonica a, 119, 81–82. 10.12693/APhysPolA.119.81 [DOI] [Google Scholar]
- Gao, M. , Monian, P. , Pan, Q. , Zhang, W. , Xiang, J. , & Jiang, X. (2016). Ferroptosis is an autophagic cell death process. Cell Research, 26, 1021–1032. 10.1038/cr.2016.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, L. (1953). Alzheimer's disease: A clinico‐pathologic analysis of twenty‐three cases with a theory on pathogenesis. The Journal of Nervous and Mental Disease, 118, 97–130. 10.1097/00005053-195308000-00001 [DOI] [PubMed] [Google Scholar]
- Goozee, K. , Chatterjee, P. , James, I. , Shen, K. , Sohrabi, H. R. , Asih, P. R. , … Martins, R. N. (2017). Elevated plasma ferritin in elderly individuals with high neocortical amyloid‐β load. Molecular Psychiatry. 10.1038/mp.2017.1146 [DOI] [PubMed] [Google Scholar]
- Green, R. C. , Schneider, L. S. , Amato, D. A. , Beelen, A. P. , Wilcock, G. , Swabb, E. A. , … Tarenflurbil Phase 3 Study Group (2009). Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. Jama, 302, 2557–2564. 10.1001/jama.2009.1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, C. , Wang, P. , Zhong, M. L. , Wang, T. , Huang, X. S. , Li, J. Y. , & Wang, Z. Y. (2013). Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochemistry International, 62, 165–172. 10.1016/j.neuint.2012.12.005 [DOI] [PubMed] [Google Scholar]
- Hajimohammadreza, I. , & Brammer, M. (1990). Brain membrane fluidity and lipid peroxidation in Alzheimer's disease. Neuroscience Letters, 112, 333–337. 10.1016/0304-3940(90)90226-Y [DOI] [PubMed] [Google Scholar]
- Hallgren, B. , & Sourander, P. (1960). The non‐haemin iron in the cerebral cortex in Alzheimer's disease. Journal of Neurochemistry, 5, 307–310. 10.1111/j.1471-4159.1960.tb13369.x [DOI] [PubMed] [Google Scholar]
- Hanson, L. R. , Fine, J. M. , Renner, D. B. , Svitak, A. L. , Burns, R. B. , Nguyen, T. M. , … Frey, W. H. (2012). Intranasal delivery of deferoxamine reduces spatial memory loss in APP/PS1 mice. Drug Delivery and Translational Research, 2, 160–168. 10.1007/s13346-011-0050-2 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka, M. T. , Carson, M. J. , El Khoury, J. , Landreth, G. E. , Brosseron, F. , Feinstein, D. L. , … Kummer, M. P. (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurology, 14, 388–405. 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka, M. T. , Fink, A. , & Doblhammer, G. (2015). Effect of pioglitazone medication on the incidence of dementia. Annals of Neurology, 78, 284–294. 10.1002/ana.24439 [DOI] [PubMed] [Google Scholar]
- Hinman, A. , Holst, C. R. , Latham, J. C. , Bruegger, J. J. , Ulas, G. , McCusker, K. P. , … Shrader, W. D. (2018). Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15‐lipoxygenase. PLoS One, 13, e0201369 10.1371/journal.pone.0201369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock, C. , Konietzko, U. , Streffer, J. R. , Tracy, J. , Signorell, A. , Muller‐Tillmanns, B. , … Nitsch, R. M. (2003). Antibodies against β‐amyloid slow cognitive decline in Alzheimer's disease. Neuron, 38, 547–554. 10.1016/S0896-6273(03)00294-0 [DOI] [PubMed] [Google Scholar]
- Holmes, C. , Boche, D. , Wilkinson, D. , Yadegarfar, G. , Hopkins, V. , Bayer, A. , … Nicoll, J. A. R. (2008). Long‐term effects of Aβ42 immunisation in Alzheimer's disease: Follow‐up of a randomised, placebo‐controlled phase I trial. Lancet, 372, 216–223. 10.1016/S0140-6736(08)61075-2 [DOI] [PubMed] [Google Scholar]
- Honig, L. S. , Vellas, B. , Woodward, M. , Boada, M. , Bullock, R. , Borrie, M. , … Siemers, E. (2018). Trial of solanezumab for mild dementia due to Alzheimer's disease. The New England Journal of Medicine, 378, 321–330. 10.1056/NEJMoa1705971 [DOI] [PubMed] [Google Scholar]
- Hou, W. , Xie, Y. , Song, X. , Sun, X. , Lotze, M. T. , Zeh, H. J. 3rd , … Tang, D. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy, 12, 1425–1428. 10.1080/15548627.2016.1187366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao, Y. H. , Kuo, J. R. , Chen, S. H. , & Gean, P. W. (2012). Amelioration of social isolation‐triggered onset of early Alzheimer's disease‐related cognitive deficit by N‐acetylcysteine in a transgenic mouse model. Neurobiology of Disease, 45, 1111–1120. 10.1016/j.nbd.2011.12.031 [DOI] [PubMed] [Google Scholar]
- Huang, X. , Atwood, C. S. , Hartshorn, M. A. , Multhaup, G. , Goldstein, L. E. , Scarpa, R. C. , … Bush, A. I. (1999). The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry, 38, 7609–7616. 10.1021/bi990438f [DOI] [PubMed] [Google Scholar]
- Huang, X. , Atwood, C. S. , Moir, R. D. , Hartshorn, M. A. , Tanzi, R. E. , & Bush, A. I. (2004). Trace metal contamination initiates the apparent auto‐aggregation, amyloidosis, and oligomerization of Alzheimer's Aβ peptides. Journal of Biological Inorganic Chemistry: JBIC: A Publication of the Society of Biological Inorganic Chemistry, 9, 954–960. 10.1007/s00775-004-0602-8 [DOI] [PubMed] [Google Scholar]
- Hwang, E. J. , Kim, H. G. , Kim, D. , Rhee, H. Y. , Ryu, C. W. , Liu, T. , … Jahng, G. H. (2016). Texture analyses of quantitative susceptibility maps to differentiate Alzheimer's disease from cognitive normal and mild cognitive impairment. Medical Physics, 43, 4718–4728. 10.1118/1.4958959 [DOI] [PubMed] [Google Scholar]
- Jack, C. R. Jr. , Knopman, D. S. , Jagust, W. J. , Petersen, R. C. , Weiner, M. W. , Aisen, P. S. , … Trojanowski, J. Q. (2013). Tracking pathophysiological processes in Alzheimer's disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurology, 12, 207–216. 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Jung, K. , Hyun Kim, D. , Kyeong Lee, E. , Woo Song, C. , Pal Yu, B. , & Young Chung, H. (2013). Oxidative stress induces inactivation of protein phosphatase 2A, promoting proinflammatory NF‐κB in aged rat kidney. Free Radical Biology & Medicine, 61, 206–217. 10.1016/j.freeradbiomed.2013.04.005 [DOI] [PubMed] [Google Scholar]
- Kennedy, M. E. , Stamford, A. W. , Chen, X. , Cox, K. , Cumming, J. N. , Dockendorf, M. F. , … Forman, M. S. (2016). The BACE1 inhibitor verubecestat (MK‐8931) reduces CNS β‐amyloid in animal models and in Alzheimer's disease patients. Science Translational Medicine, 8, 363ra150 10.1126/scitranslmed.aad9704 [DOI] [PubMed] [Google Scholar]
- Kryscio, R. J. , Abner, E. L. , Caban‐Holt, A. , Lovell, M. , Goodman, P. , Darke, A. K. , … Schmitt, F. A. (2017). Association of antioxidant supplement use and dementia in the prevention of Alzheimer's disease by vitamin e and selenium trial (preadvise). JAMA Neurology, 74, 567–573. 10.1001/jamaneurol.2016.5778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupershmidt, L. , Amit, T. , Bar‐Am, O. , Youdim, M. B. , & Weinreb, O. (2012). The novel multi‐target iron chelating‐radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer's disease. Antioxidants & Redox Signaling, 17, 860–877. 10.1089/ars.2011.4279 [DOI] [PubMed] [Google Scholar]
- Kwiatek‐Majkusiak, J. , Dickson, D. W. , Tacik, P. , Aoki, N. , Tomasiuk, R. , Koziorowski, D. , & Friedman, A. (2015). Relationships between typical histopathological hallmarks and the ferritin in the hippocampus from patients with Alzheimer's disease. Acta Neurobiologiae Experimentalis, 75, 391–398. [PubMed] [Google Scholar]
- Landen, J. W. , Cohen, S. , Billing, C. B. Jr. , Cronenberger, C. , Styren, S. , Burstein, A. H. , … Binneman, B. (2017). Multiple‐dose ponezumab for mild‐to‐moderate Alzheimer's disease: Safety and efficacy. Alzheimer's & Dementia, 3, 339–347. 10.1016/j.trci.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, P. , Ayton, S. , Appukuttan, A. T. , Moon, S. , Duce, J. A. , Volitakis, I. , … Bush, A. I. (2016). Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Molecular Psychiatry, 22, 369–406. [DOI] [PubMed] [Google Scholar]
- Lei, P. , Ayton, S. , Appukuttan, A. T. , Volitakis, I. , Adlard, P. A. , Finkelstein, D. I. , & Bush, A. I. (2015). Clioquinol rescues Parkinsonism and dementia phenotypes of the tau knockout mouse. Neurobiology of Disease, 81, 168–175. 10.1016/j.nbd.2015.03.015 [DOI] [PubMed] [Google Scholar]
- Lei, P. , Ayton, S. , Finkelstein, D. I. , Spoerri, L. , Ciccotosto, G. D. , Wright, D. K. , … Bush, A. I. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP‐mediated iron export. Nature Medicine, 18, 291–295. 10.1038/nm.2613 [DOI] [PubMed] [Google Scholar]
- Lei, P. , Ayton, S. , Moon, S. , Zhang, Q. , Volitakis, I. , Finkelstein, D. I. , & Bush, A. I. (2014). Motor and cognitive deficits in aged tau knockout mice in two background strains. Molecular Neurodegeneration, 9, 29 10.1186/1750-1326-9-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Lei, P. , Tuo, Q. , Ayton, S. , Li, Q. X. , Moon, S. , … Bush, A. I. (2015). Enduring elevations of hippocampal amyloid precursor protein and iron are features of β‐amyloid toxicity and are mediated by tau. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics, 12, 862–873. 10.1007/s13311-015-0378-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, Y. Y. , Maruff, P. , Pietrzak, R. H. , Ames, D. , Ellis, K. A. , Harrington, K. , … Rowe, C. C. (2014). Effect of amyloid on memory and non‐memory decline from preclinical to clinical Alzheimer's disease. Brain: A Journal of Neurology, 137, 221–231. 10.1093/brain/awt286 [DOI] [PubMed] [Google Scholar]
- Liu, B. , Moloney, A. , Meehan, S. , Morris, K. , Thomas, S. E. , Serpell, L. C. , … Crowther, D. C. (2011). Iron promotes the toxicity of amyloid β peptide by impeding its ordered aggregation. The Journal of Biological Chemistry, 286, 4248–4256. 10.1074/jbc.M110.158980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T. , Surapaneni, K. , Lou, M. , Cheng, L. , Spincemaille, P. , & Wang, Y. (2012). Cerebral microbleeds: Burden assessment by using quantitative susceptibility mapping. Radiology, 262, 269–278. 10.1148/radiol.11110251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Z. , Zhuang, X. , Kumar, D. , Wu, X. , Yue, C. , Han, C. , & Lv, J. (2013). The correlation of hippocampal T2‐mapping with neuropsychology test in patients with Alzheimer's disease. PLoS One, 8, e76203 10.1371/journal.pone.0076203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpas, C. B. , Vivash, L. , Genc, S. , Saling, M. M. , Desmond, P. , Steward, C. , … O'Brien, T. J. (2016). A phase IIa randomized control trial of VEL015 (Sodium Selenate) in mild‐moderate Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 54, 223–232. 10.3233/JAD-160544 [DOI] [PubMed] [Google Scholar]
- Mandal, P. K. , Saharan, S. , Tripathi, M. , & Murari, G. (2015). Brain glutathione levels—A novel biomarker for mild cognitive impairment and Alzheimer's disease. Biological Psychiatry, 78, 702–710. 10.1016/j.biopsych.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Mantyh, P. W. , Ghilardi, J. R. , Rogers, S. , DeMaster, E. , Allen, C. J. , Stimson, E. R. , & Maggio, J. E. (1993). Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of β‐amyloid peptide. Journal of Neurochemistry, 61, 1171–1174. 10.1111/j.1471-4159.1993.tb03639.x [DOI] [PubMed] [Google Scholar]
- Markesbery, W. R. , Kryscio, R. J. , Lovell, M. A. , & Morrow, J. D. (2005). Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Annals of Neurology, 58, 730–735. 10.1002/ana.20629 [DOI] [PubMed] [Google Scholar]
- McCarthy, R. C. , Park, Y. H. , & Kosman, D. J. (2014). sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Reports, 15, 809–815. 10.15252/embr.201338064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadowcroft, M. D. , Peters, D. G. , Dewal, R. P. , Connor, J. R. , & Yang, Q. X. (2014). The effect of iron in MRI and transverse relaxation of amyloid‐β plaques in Alzheimer's disease. NMR in Biomedicine, 28, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina‐Holgado, F. , Gaeta, A. , Francis, P. T. , Williams, R. J. , & Hider, R. C. (2008). Neuroprotective actions of deferiprone in cultured cortical neurones and SHSY‐5Y cells. Journal of Neurochemistry, 105, 2466–2476. 10.1111/j.1471-4159.2008.05332.x [DOI] [PubMed] [Google Scholar]
- Montine, T. J. , Kaye, J. A. , Montine, K. S. , McFarland, L. , Morrow, J. D. , & Quinn, J. F. (2001). Cerebrospinal fluid aβ42, tau, and f2‐isoprostane concentrations in patients with Alzheimer disease, other dementias, and in age‐matched controls. Archives of Pathology & Laboratory Medicine, 125, 510–512. [DOI] [PubMed] [Google Scholar]
- Murphy, M. P. (2018). Amyloid‐β solubility in the treatment of Alzheimer's disease. The New England Journal of Medicine, 378, 391–392. 10.1056/NEJMe1714638 [DOI] [PubMed] [Google Scholar]
- Nakamura, M. , Shishido, N. , Nunomura, A. , Smith, M. A. , Perry, G. , Hayashi, Y. , … Hayashi, T. (2007). Three histidine residues of amyloid‐β peptide control the redox activity of copper and iron. Biochemistry, 46, 12737–12743. 10.1021/bi701079z [DOI] [PubMed] [Google Scholar]
- Nelson, P. T. , Alafuzoff, I. , Bigio, E. H. , Bouras, C. , Braak, H. , Cairns, N. J. , … Beach, T. G. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. Journal of Neuropathology and Experimental Neurology, 71, 362–381. 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowitzki, S. , Lasser, R. A. , Dorflinger, E. , Scheltens, P. , Barkhof, F. , Nikolcheva, T. , … Fontoura, P. (2017). A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alzheimer's Research & Therapy, 9, 95 10.1186/s13195-017-0318-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, S. , Dziadulewicz, N. , & Crowther, D. C. (2015). Iron is a specific cofactor for distinct oxidation‐ and aggregation‐dependent Aβ toxicity mechanisms in a Drosophila model. Disease Models & Mechanisms, 8, 657–667. 10.1242/dmm.019042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Part, K. , Kunnis‐Beres, K. , Poska, H. , Land, T. , Shimmo, R. , & Zetterström Fernaeus, S. (2015). Amyloid β induced ROS‐burst through NADPH oxidase is sensitive to iron chelation in microglial Bv2 cells. Brain Research., 1629, 282–290. 10.1016/j.brainres.2015.09.034 [DOI] [PubMed] [Google Scholar]
- Pasquier, F. , Sadowsky, C. , Holstein, A. , Leterme Gle, P. , Peng, Y. , Jackson, N. , … ACC‐001 (QS‐21) Study Team (2016). Two phase 2 multiple ascending‐dose studies of vanutide cridificar (ACC‐001) and QS‐21 adjuvant in mild‐to‐moderate Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 51, 1131–1143. 10.3233/JAD-150376 [DOI] [PubMed] [Google Scholar]
- Petersen, R. C. , Thomas, R. G. , Grundman, M. , Bennett, D. , Doody, R. , Ferris, S. , … Alzheimer's Disease Cooperative Study Group (2005). Vitamin E and donepezil for the treatment of mild cognitive impairment. The New England Journal of Medicine, 352, 2379–2388. [DOI] [PubMed] [Google Scholar]
- Pirpamer, L. , Hofer, E. , Gesierich, B. , De Guio, F. , Freudenberger, P. , Seiler, S. , … Schmidt, R. (2016). Determinants of iron accumulation in the normal aging brain. Neurobiology of Aging, 43, 149–155. 10.1016/j.neurobiolaging.2016.04.002 [DOI] [PubMed] [Google Scholar]
- Plascencia‐Villa, G. , Ponce, A. , Collingwood, J. F. , Arellano‐Jimenez, M. J. , Zhu, X. , Rogers, J. T. , … Perry, G. (2016). High‐resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer's disease. Scientific Reports, 6, 24873 10.1038/srep24873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanthi, J. R. , Schrag, M. , Dasari, B. , Marwarha, G. , Dickson, A. , Kirsch, W. M. , & Ghribi, O. (2012). Deferiprone reduces amyloid‐β and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol‐enriched diet. Journal of Alzheimer's Disease: JAD, 30, 167–182. 10.3233/JAD-2012-111346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Y. , Zhu, W. , Zhan, C. , Zhao, L. , Wang, J. , Tian, Q. , & Wang, W. (2011). Investigation on positive correlation of increased brain iron deposition with cognitive impairment in Alzheimer disease by using quantitative MR R2' mapping. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban, 31, 578–585. [DOI] [PubMed] [Google Scholar]
- Raven, E. P. , Lu, P. H. , Tishler, T. A. , Heydari, P. , & Bartzokis, G. (2013). Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer's disease detected in vivo with magnetic resonance imaging. Journal of Alzheimer's disease: JAD, 37(1), 127–136. [DOI] [PubMed] [Google Scholar]
- Reed, T. , Perluigi, M. , Sultana, R. , Pierce, W. M. , Klein, J. B. , Turner, D. M. , … Butterfield, D. A. (2008). Redox proteomic identification of 4‐hydroxy‐2‐nonenal‐modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiology of Disease, 30, 107–120. 10.1016/j.nbd.2007.12.007 [DOI] [PubMed] [Google Scholar]
- Rinne, J. O. , Brooks, D. J. , Rossor, M. N. , Fox, N. C. , Bullock, R. , Klunk, W. E. , … Grundman, M. (2010). 11C‐PiB PET assessment of change in fibrillar amyloid‐β load in patients with Alzheimer's disease treated with bapineuzumab: A phase 2, double‐blind, placebo‐controlled, ascending‐dose study. Lancet Neurology, 9, 363–372. 10.1016/S1474-4422(10)70043-0 [DOI] [PubMed] [Google Scholar]
- Rival, T. , Page, R. M. , Chandraratna, D. S. , Sendall, T. J. , Ryder, E. , Liu, B. , … Lomas, D. A. (2009). Fenton chemistry and oxidative stress mediate the toxicity of the β‐amyloid peptide in a Drosophila model of Alzheimer's disease. The European Journal of Neuroscience, 29, 1335–1347. 10.1111/j.1460-9568.2009.06701.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. L. , Lee, E. B. , Xie, S. X. , Rennert, L. , Suh, E. , Bredenberg, C. , … Trojanowski, J. Q. (2018). Neurodegenerative disease concomitant proteinopathies are prevalent, age‐related and APOE4‐associated. Brain: A Journal of Neurology, 141, 2181–2193. 10.1093/brain/awy146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigue, K. M. , Daugherty, A. M. , Haacke, E. M. , & Raz, N. (2013). The role of hippocampal iron concentration and hippocampal volume in age‐related differences in memory. Cerebral Cortex, 23, 1533–1541. 10.1093/cercor/bhs139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, J. T. , Randall, J. D. , Cahill, C. M. , Eder, P. S. , Huang, X. , Gunshin, H. , … Gullans, S. R. (2002). An iron‐responsive element type II in the 5′‐untranslated region of the Alzheimer's amyloid precursor protein transcript. The Journal of Biological Chemistry, 277, 45518–45528. 10.1074/jbc.M207435200 [DOI] [PubMed] [Google Scholar]
- Salloway, S. , Sperling, R. , Fox, N. C. , Blennow, K. , Klunk, W. , Raskind, M. , … Bapineuzumab 301 and 302 Clinical Trial Investigators (2014). Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. The New England Journal of Medicine, 370, 322–333. 10.1056/NEJMoa1304839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway, S. , Sperling, R. , Gilman, S. , Fox, N. C. , Blennow, K. , Raskind, M. , … For the Bapineuzumab 201 Clinical Trial Investigators (2009). A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology, 73, 2061–2070. 10.1212/WNL.0b013e3181c67808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano, M. , Ernesto, C. , Thomas, R. G. , Klauber, M. R. , Schafer, K. , Grundman, M. , … Thal, L. J. (1997). A controlled trial of selegiline, α‐tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's disease cooperative study. The New England Journal of Medicine, 336, 1216–1222. 10.1056/NEJM199704243361704 [DOI] [PubMed] [Google Scholar]
- Sayre, L. M. , Perry, G. , Harris, P. L. , Liu, Y. , Schubert, K. A. , & Smith, M. A. (2000). In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: A central role for bound transition metals. Journal of Neurochemistry, 74, 270–279. [DOI] [PubMed] [Google Scholar]
- Schenk, D. , Barbour, R. , Dunn, W. , Gordon, G. , Grajeda, H. , Guido, T. , … Seubert, P. (1999). Immunization with amyloid‐β attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature, 400, 173–177. 10.1038/22124 [DOI] [PubMed] [Google Scholar]
- Schneider, S. A. , Dusek, P. , Hardy, J. , Westenberger, A. , Jankovic, J. , & Bhatia, K. P. (2013). Genetics and Pathophysiology of neurodegeneration with brain iron accumulation (NBIA). Current Neuropharmacology, 11, 59–79. 10.2174/157015913804999469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag, M. , Mueller, C. , Zabel, M. , Crofton, A. , Kirsch, W. M. , Ghribi, O. , … Perry, G. (2013). Oxidative stress in blood in Alzheimer's disease and mild cognitive impairment: A meta‐analysis. Neurobiology of Disease, 59, 100–110. 10.1016/j.nbd.2013.07.005 [DOI] [PubMed] [Google Scholar]
- Schubert, D. , & Chevion, M. (1995). The role of iron in β amyloid toxicity. Biochemical and Biophysical Research Communications, 216, 702–707. 10.1006/bbrc.1995.2678 [DOI] [PubMed] [Google Scholar]
- Sevigny, J. , Chiao, P. , Bussiere, T. , Weinreb, P. H. , Williams, L. , Maier, M. , … Sandrock, A. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature, 537, 50–56. 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- Shultz, S. R. , Wright, D. K. , Zheng, P. , Stuchbery, R. , Liu, S. J. , Sashindranath, M. , … O'Brien, T. J. (2015). Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain, 138, 1297–1313. 10.1093/brain/awv053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M. A. , Harris, P. L. , Sayre, L. M. , & Perry, G. (1997). Iron accumulation in Alzheimer disease is a source of redox‐generated free radicals. Proceedings of the National Academy of Sciences of the United States of America, 94, 9866–9868. 10.1073/pnas.94.18.9866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M. A. , Zhu, X. , Tabaton, M. , Liu, G. , McKeel, D. W. Jr. , Cohen, M. L. , … Perry, G. (2010). Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. Journal of Alzheimer's Disease: JAD, 19, 363–372. 10.3233/JAD-2010-1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirbegi, I. B. , Pardo, W. A. , Alvira, M. , Mir, M. , & Samitier, J. (2016). Amyloid Aβ42, a promoter of magnetite nanoparticle formation in Alzheimer's disease. Nanotechnology, 27, 465102 10.1088/0957-4484/27/46/465102 [DOI] [PubMed] [Google Scholar]
- Telling, N. D. , Everett, J. , Collingwood, J. F. , Dobson, J. , van der Laan, G. , Gallagher, J. J. , … Hitchcock, A. P. (2017). Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of Alzheimer's disease. Cell Chemical Biology, 24, 1205–1215.e3. 10.1016/j.chembiol.2017.07.014 [DOI] [PubMed] [Google Scholar]
- Tiepolt, S. , Schafer, A. , Rullmann, M. , Roggenhofer, E. , Netherlands Brain B , Gertz, H. J. , … Barthel, H. (2018). Quantitative susceptibility mapping of amyloid‐β aggregates in Alzheimer's disease with 7T MR. Journal of Alzheimer's Disease: JAD, 64, 393–404. 10.3233/JAD-180118 [DOI] [PubMed] [Google Scholar]
- Tofaris, G. K. , Revesz, T. , Jacques, T. S. , Papacostas, S. , & Chataway, J. (2007). Adult‐onset neurodegeneration with brain iron accumulation and cortical α‐synuclein and tau pathology: A distinct clinicopathological entity. Archives of Neurology, 64, 280–282. [DOI] [PubMed] [Google Scholar]
- Toledo, J. B. , Arnold, S. E. , Raible, K. , Brettschneider, J. , Xie, S. X. , Grossman, M. , … Trojanowski, J. Q. (2013). Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain: A Journal of Neurology, 136, 2697–2706. 10.1093/brain/awt188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou, H. H. , Hsu, W. C. , Fuh, J. L. , Chen, S. P. , Liu, T. Y. , & Wang, H. T. (2018). Alterations in acrolein metabolism contribute to Alzheimer's disease. Journal of Alzheimer's Disease, 61, 571–580. [DOI] [PubMed] [Google Scholar]
- Tuo, Q. Z. , Lei, P. , Jackman, K. A. , Li, X. L. , Xiong, H. , Li, X. L. , … Bush, A. I. (2017). Tau‐mediated iron export prevents ferroptotic damage after ischemic stroke. Molecular Psychiatry, 22, 1520–1530. 10.1038/mp.2017.171 [DOI] [PubMed] [Google Scholar]
- van Bergen, J. M. , Li, X. , Hua, J. , Schreiner, S. J. , Steininger, S. C. , Quevenco, F. C. , … Unschuld, P. G. (2016). Colocalization of cerebral iron with amyloid β in mild cognitive impairment. Scientific Reports, 6, 35514 10.1038/srep35514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bergen, J. M. G. , Li, X. , Quevenco, F. C. , Gietl, A. F. , Treyer, V. , Meyer, R. , … Unschuld, P. G. (2018). Simultaneous quantitative susceptibility mapping and Flutemetamol‐PET suggests local correlation of iron and β‐amyloid as an indicator of cognitive performance at high age. NeuroImage, 174, 308–316. 10.1016/j.neuroimage.2018.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duijn, S. , Bulk, M. , van Duinen, S. G. , Nabuurs, R. J. A. , van Buchem, M. A. , van der Weerd, L. , & Natté, R. (2017). Cortical iron reflects severity of Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 60, 1533–1545. 10.3233/JAD-161143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eersel, J. , Ke, Y. D. , Liu, X. , Delerue, F. , Kril, J. J. , Gotz, J. , & Ittner, L. M. (2010). Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer's disease models. Proceedings of the National Academy of Sciences of the United States of America, 107, 13888–13893. 10.1073/pnas.1009038107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooden, S. , Buijs, M. , van Vliet, M. E. , Versluis, M. J. , Webb, A. G. , Oleksik, A. M. , … van der Grond, J. (2016). Cortical phase changes measured using 7‐T MRI in subjects with subjective cognitive impairment, and their association with cognitive function. NMR in Biomedicine, 29, 1289–1294. 10.1002/nbm.3248 [DOI] [PubMed] [Google Scholar]
- van Rooden, S. , Doan, N. T. , Versluis, M. J. , Goos, J. D. , Webb, A. G. , Oleksik, A. M. , … van der Grond, J. (2014). 7T T2*‐weighted magnetic resonance imaging reveals cortical phase differences between early‐ and late‐onset Alzheimer's disease. Neurobiology of Aging, 36, 20–26. [DOI] [PubMed] [Google Scholar]
- Vellas, B. , Black, R. , Thal, L. J. , Fox, N. C. , Daniels, M. , McLennan, G. , … AN1792 (QS‐21)‐251 Study Team . (2009). Long‐term follow‐up of patients immunized with AN1792: Reduced functional decline in antibody responders. Current Alzheimer Research, 6, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne, V. L. , Burnham, S. , Bourgeat, P. , Brown, B. , Ellis, K. A. , Salvado, O. , … Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group (2013). Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: A prospective cohort study. Lancet Neurolology, 12, 357–367. 10.1016/S1474-4422(13)70044-9 [DOI] [PubMed] [Google Scholar]
- Wessling‐Resnick, M. (2010). Iron homeostasis and the inflammatory response. Annual Review of Nutrition, 30, 105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock, G. K. , Black, S. E. , Hendrix, S. B. , Zavitz, K. H. , Swabb, E. A. , Laughlin, M. A. , & Tarenflurbil Phase II Study investigators (2008). Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: A randomised phase II trial. Lancet Neurology, 7, 483–493. 10.1016/S1474-4422(08)70090-5 [DOI] [PubMed] [Google Scholar]
- Williams, T. I. , Lynn, B. C. , Markesbery, W. R. , & Lovell, M. A. (2006). Increased levels of 4‐hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in mild cognitive impairment and early Alzheimer's disease. Neurobiology of Aging, 27, 1094–1099. 10.1016/j.neurobiolaging.2005.06.004 [DOI] [PubMed] [Google Scholar]
- Wong, B. X. , Tsatsanis, A. , Lim, L. Q. , Adlard, P. A. , Bush, A. I. , & Duce, J. A. (2014). β‐Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PloS one, 9, e114174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman, D. G. , Tsatsanis, A. , Lewis, F. W. , Boyle, J. P. , Mousadoust, M. , Hettiarachchi, N. T. , … Duce, J. A. (2015). Protection from neurodegeneration in the 6‐hydroxydopamine (6‐OHDA) model of Parkinson's with novel 1‐hydroxypyridin‐2‐one metal chelators. Metallomics: Integrated Biometal Science, 7, 867–876. 10.1039/C4MT00326H [DOI] [PubMed] [Google Scholar]
- Xie, L. , Zheng, W. , Xin, N. , Xie, J. W. , Wang, T. , & Wang, Z. Y. (2012). Ebselen inhibits iron‐induced tau phosphorylation by attenuating DMT1 up‐regulation and cellular iron uptake. Neurochemistry International, 61, 334–340. 10.1016/j.neuint.2012.05.016 [DOI] [PubMed] [Google Scholar]
- Yamamoto, A. , Shin, R.‐W. , Hasegawa, K. , Naiki, H. , Sato, H. , Yoshimasu, F. , & Kitamoto, T. (2002). Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer's disease. Journal of Neurochemistry, 82, 1137–1147. [DOI] [PubMed] [Google Scholar]
- Yang, W. S. , Kim, K. J. , Gaschler, M. M. , Patel, M. , Shchepinov, M. S. , & Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proceedings of the National Academy of Sciences of the United States of America, 113, E4966–E4975. 10.1073/pnas.1603244113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Raichle, M. E. , Wen, J. , Benzinger, T. L. , Fagan, A. M. , Hassenstab, J. , … Yablonskiy, D. A. (2017). In vivo detection of microstructural correlates of brain pathology in preclinical and early Alzheimer disease with magnetic resonance imaging. NeuroImage, 148, 296–304. 10.1016/j.neuroimage.2016.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]