

Summary
Tumor-associated macrophages (TAMs) represent a major component of the tumor microenvironment supporting tumorigenesis. TAMs re-education has been proposed as a strategy to promote tumor inhibition. However, whether this approach may work in prostate cancer is unknown. Here we find that Pten-null prostate tumors are strongly infiltrated by TAMs expressing C-X-C chemokine receptor type 2 (CXCR2), and activation of this receptor through CXCL2 polarizes macrophages toward an anti-inflammatory phenotype. Notably, pharmacological blockade of CXCR2 receptor by a selective antagonist promoted the re-education of TAMs toward a pro-inflammatory phenotype. Strikingly, CXCR2 knockout monocytes infused in Ptenpc−/−; Trp53pc−/− mice differentiated in tumor necrosis factor alpha (TNF-α)-releasing pro-inflammatory macrophages, leading to senescence and tumor inhibition. Mechanistically, PTEN-deficient tumor cells are vulnerable to TNF-α-induced senescence, because of an increase of TNFR1. Our results identify TAMs as targets in prostate cancer and describe a therapeutic strategy based on CXCR2 blockade to harness anti-tumorigenic potential of macrophages against this disease.
Keywords: prostate cancer, tumor immunology, immunomodulation, immune response to cancer, tumor associated macrophages
Graphical Abstract
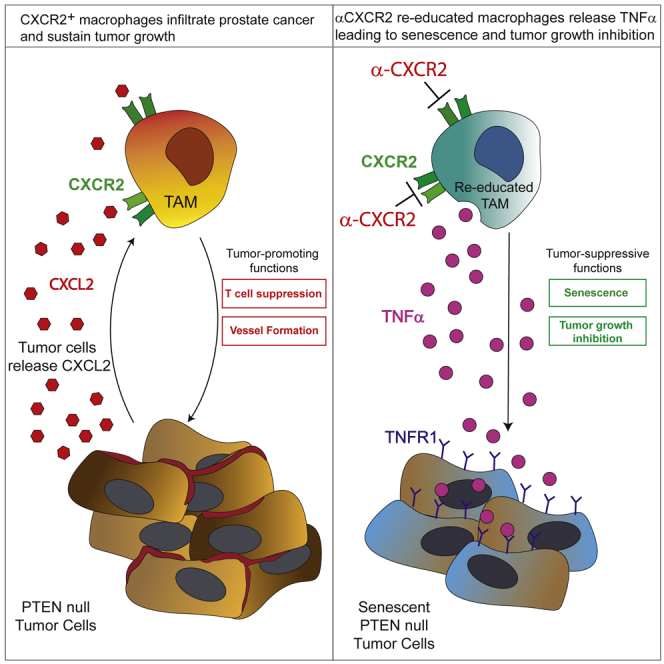
Highlights
-
•
CXCR2 blockade drives re-education of tumor-associated macrophages (TAMs)
-
•
Infusion of CXCR2-KO monocytes in tumor-bearing mice blocks tumor progression
-
•
PTEN deletion sensitizes tumor cells to TNF-α-induced senescence and growth arrest
Di Mitri et al. show that CXCR2 blockade in prostate cancer triggers TAMs re-education, leading to tumor inhibition. CXCR2-KO monocytes infused in Ptenpc−/−; Trp53pc−/− tumor-bearing mice differentiate into TNFα-releasing pro-inflammatory macrophages that induce senescence in tumor cells. PTEN-null tumors display higher sensitivity to TNF-α-induced senescence because of TNFR1 upregulation.
Introduction
Immunotherapy based on reactivation of T cells in the tumor microenvironment is emerging as an effective strategy to treat cancer. However, in several tumors, such as in prostate cancer, T cells constitute only a minor component of the tumor immune response compared with tumor-associated macrophages (TAMs), the most abundant noncancerous cell type (Nava Rodrigues et al., 2018). Macrophages are a plastic immune population that can be polarized in vitro in anti-inflammatory subsets by cytokines, such as IL-4 and IL-13, or into pro-inflammatory immune cells by IFNγ. However, these extreme macrophage phenotypes do not fully recapitulate the plasticity of TAMs in vivo, where macrophages are polarized to different and more complex phenotypes. Indeed cancer cells can influence TAMs polarization by releasing cytokines, glucocorticoids, extracellular vesicles, and extracellular matrix components that give rise to a large spectrum of pro-tumoral macrophages (Cassetta et al., 2019). In prostate cancer, TAMs and other myeloid subsets constitute up to 70% of tumor immune subsets (Calcinotto et al., 2018) and are known to influence tumor growth by controlling adaptive immunity, angiogenesis, tumor cell proliferation, and metastasis formation, thus playing a fundamental role in cancer initiation, progression, and resistance to treatment (Baer et al., 2016, Bingle et al., 2002, Guerriero et al., 2017, Kaneda et al., 2016, Mantovani et al., 2006, Qian and Pollard, 2010). As a consequence, TAMs provide an ideal target for therapy in cancer patients. Although strategies to deplete TAMs have been extensively investigated in the clinical setting in different cancer types (Guerriero et al., 2017, Pienta et al., 2013, Ries et al., 2014), the reported overall benefit for cancer patients has been negligible (Mantovani et al., 2017). The limited success of this approach has been ascribed to the plasticity of TAMs. In the tumor microenvironment, TAMs work as either anti- or pro-tumoral components, and the removal of anti-tumoral TAMs blunts the efficacy of TAM-depleting therapies (Mantovani et al., 2017). Exploiting the tumor-homing ability of TAMs and their plasticity by treatments that can re-educate TAMs toward a pro-inflammatory, anti-tumorigenic functional status may lead to more effective and long-lasting responses in cancer patients. In this regard, re-education of TAMs toward a pro-inflammatory, anti-tumorigenic functional status has been recently proposed as a potential therapeutic approach to treat different types of cancer, including prostate cancer (Guerriero et al., 2017, Hagemann et al., 2009, Pyonteck et al., 2013, Salvagno et al., 2019). In prostate cancer the frequency and activation state of infiltrating macrophages have been associated with disease progression and therapy resistance (Escamilla et al., 2015, Lanciotti et al., 2014, Nonomura et al., 2010). Nevertheless, current knowledge on the interplay between macrophages and prostate cancer is still limited, and further investigation is required. Moreover, it is unknown whether compounds that re-educate TAMs in order to promote their tumor-suppressive function may suppress the proliferation of aggressive prostate cancer. In the present paper we identify the macrophage receptor CXCR2 and CXCR2 signaling as major drivers of TAMs polarization in prostate tumors, and we propose a therapeutic strategy based on blockade of the CXCL-CXCR2 pathway or infusion of CXCR2 knockout (KO) monocytes to harness the anti-tumorigenic potential of macrophages against prostate cancer.
Results
CXCR2-Expressing TAMs Infiltrate Prostate Cancer
To better understand the mechanism by which prostate tumor cells affect the functional polarization of TAMs, we compared two Pten-null prostate conditional (pc−/−) mouse models, in which Trp53 expression in prostate is respectively maintained or depleted (Alimonti et al., 2010, Chen et al., 2005). These two models recapitulate pathological and molecular features of human prostate cancer at different stages of progression, with Ptenpc−/−; Trp53pc−/− tumors more invasive compared with Ptenpc−/−; Trp53pc+/+ (Ptenpc−/−) (Figures 1A–1C). Flow cytometry analyses showed that both Ptenpc−/− and Ptenpc−/−; Trp53pc−/− tumors are strongly infiltrated by CD45+CD11b+LY6G−F4/80+ macrophages and that TAMs frequency slightly increases with tumor progression (Figures 1D and 1E; see Figure S1A for the gating strategy). Immunofluorescence staining confirmed the prominent infiltration of TAMs in both Ptenpc−/− and Ptenpc−/−; Trp53pc−/− tumors (Figure 1F; Figure S1B). Importantly, TAMs are localized mainly in the vimentin+ surrounding stroma of prostatic tumors (Figures 1G and S1C). A prominent increase of CD68+ TAMs was also detected in human prostate cancers compared with PIN sections (Figures S1D–S1F), thus confirming previous evidence (Lanciotti et al., 2014). Strikingly, we found that the majority of tumor-infiltrating CD45+CD11b+LY6G−F4/80+ macrophages expressed the CXCR2 receptor, at levels comparable with the CD45+CD11b+LY6G+F4/80− granulocytic subset, as shown by flow cytometry performed on single-cell suspension obtained from Ptenpc−/−; Trp53pc−/− tumors (Figures 1H and 1I). Accordingly, protein profiling performed through a cytokine array revealed a significant upregulation of the CXCL1, CXCL2, and CXCL5, three ligands of the C-X-C motif chemokine receptor 2 (CXCR2), in both Ptenpc−/− and Ptenpc−/−; Trp53pc−/− tumors (Figure 1J). Analysis of public available gene expression data confirmed the upregulation of ELR+CXCL chemokines in Pten-null tumors (Figure S1G).
Figure 1.

Tumor-Associated Macrophages (TAMs) Infiltrate Prostate Cancer and Express CXCR2
(A) Representative images of H&E and Ki-67 IHC staining. Original magnification, 20×. Scale bar: 100 μm.
(B and C) Anterior prostate lobe volume (mm3) (B) (see STAR Methods) and Ki-67 quantification (C) in 12-week-old Ptenpc+/+, Ptenpc−/−; Trp53pc+/+ (Ptenpc−/−), and Ptenpc−/−; Trp53pc−/−mice (n = 5 mice per group).
(D and E) Representative FACS plots of immunophenotyping (D) and quantification (E) of tumor-infiltrating CD11b+Ly6G−F4/80+ macrophages in 12-week-old Ptenpc−/− and Ptenpc−/−; Trp53pc−/− mice. Events are gated on CD45+CD11b+ cells (n = 5 per group).
(F) Representative confocal immunofluorescence (IF) images and quantification showing the localization of F4/80+ (red) macrophages in Ptenpc−/−; Trp53pc−/−prostatic tumors. Prostatic epithelial tissue is stained with αPan-Cytokeratin antibody (PCK; green). Cells were counterstained with the nuclear marker DAPI (blue). Scale bar, 10 μm (n = 3 mice per group).
(G) Representative confocal immunofluorescence images of F4/80+ (red) tumor-infiltrating macrophages in Ptenpc−/−; Trp53pc−/−prostatic tumors. Stromal cells are stained with anti-vimentin antibody (vimentin; green). Cells were counterstained with the nuclear marker DAPI (blue). Scale bar, 10 μm (n = 3 mice per group).
(H and I) Representative FACS analysis (H) and quantification (I) of the mean fluorescence index (MFI) per cell of CXCR2 expression on TAMs and neutrophils in Ptenpc−/−; Trp53pc−/− prostatic tumors (n = 6 mice). Mean fluorescence intensity was measured on CD11b+CD45+F480+Ly6G− TAMs and CD11b+CD45+F480−LY6G+ neutrophils.
(J) Graph showing analysis of the protein expression profiling from epithelia isolated from Ptenpc−/− and Ptenpc−/−; Trp53pc−/− tumors. One hundred ten proteins were analyzed by mean of the XL mouse protein array kit (R&D). All proteins included in our panel were assessed for their possible involvement in macrophage polarization by applying text-mining algorithms (Agilent Literature Search 3.1.1, in Cytoscape 3.1.1) for a minimum of 40 papers per protein. A node’s color represents mean fold change for Ptenpc−/− versus wild-type and Ptenpc−/−; Trp53pc−/− versus wild-type. Genes consistently yielding a >2-fold increase for both comparisons and showing significant matching trends for their gene expression profiles (Figure S1E) are magnified. Classical and alternatively activating polarizers are annotated with blue as font color and displayed as diamonds.
Error bars are mean ± SEM. ∗∗∗p < 0.001.
CXCR2 Engagement Tilts Macrophages Polarization toward a Pro-tumorigenic Functional State In Vitro
To assess the role of the CXCL-CXCR2 axis in the polarization of macrophages, we isolated and differentiated bone marrow-derived macrophages (BMDMs) from C57BL/6 mice and exposed them to CXCL2 recombinant protein, the most upregulated CXCL chemokine in our tumor models, in a polarization assay in vitro. Gene expression analysis performed on these cells revealed a marked upregulation of arginase and CD206, genes that are generally associated with anti-inflammatory macrophages, in CXCL2-educated cells, while pro-inflammatory genes such as tumor necrosis alpha (TNF-α) and IL-12 were not significantly enriched or were even decreased in these samples (Figure 2A). Subsequently we applied an unbiased gene expression analysis to CXCL2-stimulated macrophages. Pro-inflammatory IFNγ/LPS-polarized macrophages and anti-inflammatory and pro-angiogenic IL-4/IL-13 alternatively activated macrophages were used as controls (Martinez and Gordon, 2014). Interestingly, analysis of pivotal gene sets (GSs) revealed a significant overlap (83.6%) in the transcriptional landscape of CXCL2 and IL-4/IL-13-stimulated macrophages (Figure S2A). In addition, profiling of 110 proteins performed by a proteome profiler cytokine array showed a substantial correspondence in the type of proteins secreted by the two groups (Figure S2B). To therefore test the functional status of CXCL2-stimulated macrophages, we exposed macrophages to CXCL2 in vitro for 24 h, we washed the cells to replace normal DMEM, and after 48 h we collected the conditioned media (c.m.). Strikingly, c.m. from CXCL2-polarized macrophages reduced CD8+ proliferation in a suppression assay (Figures 2B and 2C; see Figure S2C for the gating strategy). These findings were further confirmed on CD8+ T cells sorted from murine lymph nodes and CD8+ cells gated from murine peripheral blood (Figures S2D and S2E). Further functional analysis showed that CXCL2-stimulated macrophages promote the formation of capillary-like structures (tubes) from endothelial murine cells (Figure 2D). Results reported above indicate that CXCL2 promotes macrophage differentiation toward an alternative activation state. In line with this evidence, we detected increased protein levels of pSTAT6 in both canonical (IL-4/IL-13) and CXCL2-polarized macrophages (Takeda et al., 1996) (Figure 2E). Therefore, we checked whether CXCR2 inhibition could affect IL-4/IL-13 polarization. Our flow cytometry analysis showed an increase in the levels of CXCR2 upon IL-4/IL-13 activation of BMDMs (Figure S2F). Finally, treatment with two different CXCR2 antagonists (αCXCR2) reverted the macrophage polarization toward the anti-inflammatory state driven by IL-4 and IL-13, as shown by the decrease expression of prototypic genes and a CD8+T cells suppression assay in vitro (Figures 2F and 2G).
Figure 2.

CXCL2 Administration Induces a Suppressive and Pro-angiogenic Functional State in Macrophages In Vitro
(A) RT-qPCR gene expression analysis of BMDMs polarized in vitro upon administration of CXCL2 recombinant protein (n = 4).
(B and C) FACS analysis (B) and quantification (C) showing a carboxyfluorescein succinimidyl ester (CFSE) proliferation assays performed on isolated splenocytes exposed to macrophage-derived conditioned media (n = 3). Plots show the percentage of CD8+CFSE− proliferating cells. Macrophages were polarized in presence of stimuli for 48 h, then media was washed out and replaced. Conditioned media for the experiment was collected after 24 h.
(D) Representative pictures of immunofluorescence staining (left panel) and quantification (right panel) showing a tube formation assays performed on CECs (cardiac endothelial murine cells) exposed to macrophage-derived conditioned media (n = 3). Macrophages were polarized in presence of stimuli for 48 h, then media was washed out and replaced. Conditioned media for the experiment was collected after 24 h.
(E) Western blot analysis (left panel) showing the levels of total Stat6, phosphorylated Stat6, and HSP90 in IL-4/IL-13 and CXCL2-polarized macrophages. The bar graph (right panel) shows the levels of pStat6 expression. The levels of pStat6 expression were normalized for the levels of total Stat6 in each sample.
(F) RT-qPCR gene expression analysis of alternative macrophages prototypic markers on macrophages polarized in vitro in absence or presence of αCXCR2 1 (1 μΜ, SB265610) and αCXCR2 (1 μΜ, SB225002) (n = 5).
(G) FACS analysis and quantification of a CFSE proliferation assay on isolated splenocytes exposed to macrophage-derived conditioned media in absence or presence of SB265610. Quantification is based on the frequency of CD8+CFSE− proliferating cells (n = 5).
Error bars are mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Pharmacological Disruption of the CXCL2-CXCR2 Pathway Triggers Tumor Inhibition and TAMs Re-education
Altogether, the findings reported above support the importance of the CXCL-CXCR2 axis in the induction of an anti-inflammatory functional state in macrophages. To validate this hypothesis in vivo, we took advantage of the Ptenpc−/−; Trp53pc−/− mouse model, which develops highly aggressive prostate cancers compared with Ptenpc−/− mice. Therefore we treated Ptenpc−/−; Trp53pc−/− mice with AZD5069, a potent and selective antagonist of CXCR2 under clinical development (Steele et al., 2016) (ClinicalTrials.gov: NCT03177187). Gross macroscopic and histological analyses of Ptenpc−/−; Trp53pc−/− tumors treated withAZD5069 showed a strong suppression of tumor size and presence of normalized prostate areas compared with the untreated control (Figure 3A). Remarkably, AZD5069 treatment induced a strong reduction of Ki-67 staining and upregulation of senescence as detected by increased levels of pHP1γ and p16 (Figures 3A–3E; Figures S3A and S3B). Western blot analysis showed increased levels of GATA4, an additional marker of senescence (Kang et al., 2015), in tumors treated with AZD5069 (Figures S3C and S3D). Importantly, αCXCR2 treatment in vitro did not affect prostate epithelial cell proliferation and did not drive senescence per se, demonstrating that the effect of αCXCR2 is indirect (Figure S3E). To evaluate the effect of the CXCR2 blockade on infiltrating macrophages, we investigated the immune infiltrate of tumors by fluorescence-activated cell sorting (FACS) analysis. Our results showed a reduction of F4/80+CD11c+/−CD206+ cells, previously described as pro-angiogenic macrophages in different tumors, in favor of F4/80+CD11c+CD206− pro-inflammatory TAMs (Figures 3F and 3G) (Mazzieri et al., 2011). Accordingly, RT-qPCR analysis showed a significant increase in TNFα mRNA levels (generally associated with pro-inflammatory TAMs) and downregulation of arginase and CD206 levels (generally associated with anti-inflammatory TAMs) in TAMs isolated from mice treated with αCXCR2 (Figure 3H) (Mosser and Edwards, 2008, Rath et al., 2014). These findings were also validated in an isogenic TRAMP-C1 cell line in which Pten was deleted by using a Crispr-Cas9 system thereby generating the Pten-null; Trp53-inactivated prostate epithelial cells (Pten−/−-TRAMP-C1) (Figure S4A). Pten−/−-TRAMP-C1 cells were subcutaneously injected in vivo, and mice were treated with AZD5069. As observed in the transgenic model, AZD5069 treatment resulted in a reduction in tumor size (Figure S4B) and in the re-education of TAMs (Figure S4C). Of note, re-educated macrophages expressed a higher amount of TNFα upon αCXCR2 treatment (Figure S4D). Pten−/−-TRAMP-C1 allografts also showed an increased senescence induction upon aCXCR2 treatment, as demonstrated by the upregulation of p16, pH2AX foci, and GATA4 expression, three well-characterized markers of senescence (Figures S4E–S4H) (Kang et al., 2015).
Figure 3.

αCXCR2-Mediated TAMs Reprogramming Induces Tumor Regression and Modulates T Cell Response and Vessel Size.
Mice were treated with αCXCR2 (AZD5069 100 mg/kg) or vehicle, starting at the age of tumor formation (8 weeks), for 3 weeks.
(A) Representative images of H&E (right panel) and Ki-67 IHC staining (right panel). Original magnification, 20×; scale bar, 50 μm.
(B) pHp1γ IF staining. Original magnification, 20×; scale bar, 25 μm.
(C) Volume of anterior prostate lobes (see STAR Methods); n = 7.
(D) and E) Ki-67 (D) and pHp1γ quantification (E) in Ptenpc−/−; p53pc−/− prostatic tumors upon αCXCR2 treatment (n = 7 and n = 3, respectively).
(F and G) Representative immunophenotyping plot (F) and quantification (G) of CD11c+CD206− pro-inflammatory and CD11c+/−CD206+ tumor-promoting macrophages (Mazzieri et al., 2011) infiltrating the tumor upon administration of αCXCR2. Events are gated on CD45+CD11b+F4/80+ cells (n = 5 mice per group).
(H) RT-qPCR gene expression analysis on CD45+/F4/80+ macrophages sorted from tumors (n = 4).
(I) Unbiased systematic analyses of coordinated alterations following gene expression profiling in TAMs isolated from the prostate of untreated and αCXCR2-treated mice (n = 3 per group). Graph shows significantly altered clusters of immunity-associated effector processes. Bars represent the average log2FDRq values of the gene sets included in each cluster.
(J) Representative images of CD31 IHC staining on anterior lobe prostate of Ptenpc−/−; Trp53pc−/−mice upon αCXCR2 treatment. Original magnification 20×.
(K and L) Quantification of area (K) and perimeter (L) of vessels in the tumor area (n = 3).
(M and N) Quantification of the frequency of infiltrating CD4+CD25− (M) and CD8+ T cells (N) in prostatic tumors before and after αCXCR2 treatment. Events are gated on CD45+ cells (n = 5 mice per group).
(O) Volume of anterior prostate lobes (see STAR Methods) at the end of the treatments.
(P) Quantification of CD45+CD11b+F4/80+CD206+ anti-inflammatory macrophages infiltrating the tumor upon treatments. Ptenpc−/−; Trp53pc−/−mice were treated with αCXCR2 (AZD5069 100 mg/kg), 1A8 (αLy6G antibody), or αCXCR2/1A8 for 3 weeks, starting at the age of tumor formation (8 weeks). Events are gated on CD45+CD11b+ cells (n = 4 mice per group).
Error bars are mean ± SEM. ∗p < 0.05, ∗p < 0.01, and ∗∗∗p < 0.001.
Notably, treatment of Ptenpc−/−; Trp53pc−/− mice with AZD5069 strongly affected the gene expression profile of CD45+CD11b+LY6G−F4/80+ sorted TAMs. Indeed, unbiased systematic analyses of coordinated alterations following gene expression profiling in sorted TAMs, resulted in the over- and under-representation of 312 GSs in AZD5069 treated mice compared with untreated animals (Figure 3I). Among the gene processes that were modulated in sorted macrophages upon AZD5069 administration, we detected a significant activation of pro-inflammatory immunity-associated effector processes and depletion of processes related to pro-tumorigenic functions, IL-10 signaling, and collagen degradation (Figures S4I and S4J). This evidence indicates a reprogramming of TAMs from a pro-tumorigenic to a pro-inflammatory phenotype in αCXCR2-treated mice. In accordance with the results of the GS analysis and our in vitro evidence, AZD5069 treatment resulted in a strong inhibition of vessel formation (Figures 3J–3L) in the treated tumors. Furthermore, a comprehensive analysis of the tumor immune infiltrate upon CXCR2 blockade revealed that the frequency of CD45+CD11b+LY6G−F4/80+macrophages was not affected by the treatment (Figure S5A). Importantly, we observed an increased infiltration of CD4+ and CD8+ T cells in the treated tumors (Figures 3M and 3N; Figures S5A and S5B for the gating strategy), associated with activation of the CD8+ subset, as indicated by granzyme B staining (Figures S5C and S5D). Further analysis of the CD3+ infiltrate showed that the frequency of CD4+ regulatory cells was not significantly affected by the CXCR2 blockade (Figures S5E and S5F).
As an additional investigation of TAMs reprogramming upon CXCR2 blockade, we performed in vitro assays with tumor-conditioned macrophages exposed to αCXCR2. Our assays showed that the phagocytic activity of macrophages was increased upon CXCR2 blockade, thus confirming the ex vivo gene expression data (Figures S5G and S5H). Additionally, tumor-conditioned macrophages exposed to αCXCR2 were able to induce a senescence response in target tumor cells (Figures S5I and S5J). We next investigated whether the infiltration of CD11b+F480−Ly6G+myeloid cells was affected by αCXCR2 treatment. We found that CXCR2 blockade only slightly affected the recruitment of CD11b+F480neg Ly6G+ cells in Ptenpc−/−; Trp53pc−/− tumors (Figures S6A and S6B), probably because of the presence of additional cytokines capable of recruiting these immune cells as previously reported by others (Patnaik et al., 2017). To further explore the role of LY6G+ cells in our settings, we performed a pre-clinical trial combining aCXCR2 with a neutralizing antibody for LY6G. Our results indicated that CXCR2 blockade resulted in a strong tumor inhibition and macrophage reprogramming also in absence of Ly6G+ cells, as shown by flow cytometry and immunohistochemical analyses. Moreover, combination of aCXCR2 and anti-LY6G increased tumor growth inhibition compared with the single treatments (Figures 3O and 3P; Figures S6C and S6D). Altogether, these findings demonstrate that inhibition of the CXCR2 receptor in Ptenpc−/−; Trp53pc−/− tumors in vivo results in the re-education of tumor-infiltrating macrophages toward a pro-inflammatory and anti-tumorigenic functional state and in a strong activation of a p53-independent cellular senescence response.
Infusion of CXCR2-KO Monocytes Drives Tumor Inhibition and Senescence Induction in Ptenpc−/−; p53pc−/− Tumor-Bearing Mice
Autologous infusion of human IFNγ-activated monocytes was attempted in the past to treat cancer-bearing patients. However, such infusions resulted in poor clinical response, because monocytes reaching the tumors were polarized mainly toward alternatively activated TAMs. Indeed, high TNFα levels were never detected in the serum of treated patients even after several infusions (Lopez et al., 1992, Lopez et al., 1994, Stevenson et al., 1988). We therefore hypothesized that infusion of CXCR2-KO monocytes in tumor-bearing mice could overcome this problem. To test our hypothesis, we infused CXCR2 wild-type (WT) and CXCR2-KO bone marrow-derived monocytes in Ptenpc−/−; Trp53pc−/−mice (Figure 4A; see Figure S7A for the FACS characterization of infused cells). One million monocytes were injected intravenously every 3 days for a total of six injections, and tumors were collected the day after the last infusion. Infusion of mCherry-labeled monocytes, and the consequent FACS analysis of the tumor infiltrate, allowed the detection of these cells in the tumor and their differentiation from macrophages (see Figure S7B for the gating strategy). Strikingly, FACS analysis showed that CXCR2-KO monocytes infiltrating the tumor polarized mainly toward pro-inflammatory TAMs and that the frequency of TAMs was similar in both mice infused with CXCR2 WT and CXCR2-KO monocytes (Figures 4B and 4C; Figure S7C). Immunofluorescence staining on tumor sections further showed a significant increase of pro-inflammatory Inos-expressing macrophages upon infusion of CXCR2-KO monocytes (Figures 4D and 4E). Furthermore, RT-qPCR analysis performed on TAMs FACS-sorted from the tumors confirmed the re-education of TAMs toward pro-inflammatory functional state in mice infused with the CXCR2-KO monocytes, as shown by the decreased expression of both CD206 and arginase and increased TNFα expression (Figure 4F). Importantly, infusion of CXCR2-KO but not CXCR2 WT monocytes in Ptenpc−/−; Trp53pc−/− mice and the resulting enrichment in pro-inflammatory macrophages was associated with a strong tumor inhibition, induction of DNA damage, and senescence response, as shown by immunohistochemistry staining for Ki-67, pH2AX, p16, pHp1γ, and SA-β-galactosidase (Figures 4G–4L; Figures S7D and S7E). Western blot analysis for GATA4 levels further confirmed the induction of a senescence response in these tumors (Figure S7F). Of note, infusion of WT monocytes did not result in tumor growth advantage compared with age-matched tumor-bearing Ptenpc−/−; Trp53pc−/− untreated mice (Figure 4I). Altogether, these results indicate that inhibition of the CXCR2 receptor in TAMs promotes a re-education of these cells toward a pro-inflammatory state that induces a strong senescence response, even in Ptenpc−/−; Trp53pc−/−-null prostate tumors.
Figure 4.

CXCR2-Depleted Monocytes Infusion Drives TAM Re-education and Senescence Induction in Ptenpc−/−; p53pc−/− Tumors
(A) Schedule of treatment used in the pre-clinical trial with CXCR2 WT or CXCR2-KO infused monocytes (n = 6 mice per group).
(B and C) Representative FACS plots of immunophenotyping (B) and quantification (C) showing macrophages ratio of CD11c+CD206− pro-inflammatory and CD11c+/− CD206+ anti-inflammatory macrophages infiltrating the tumor upon infusion of CXCR2 WT or CXCR2-KO monocytes. Events are gated on CD45+CD11b+F4/80+ cells (n = 4 mice per group).
(D and E) Representative confocal immunofluorescence image (D) and quantification (E) of F4/80-positive (red) and Inos-positive (green) tumor-infiltrating macrophages. Cells were counterstained with the nuclear marker DAPI (blue). Scale bar, 15 μM.
(F) RT-qPCR gene expression analysis on CD45+F4/80+ macrophages sorted from tumors.
(G–J) H&E (G), Ki-67 IHC staining (original magnification, 20×) (H), relative anterior prostate lobe volume compared with untreated Ptenpc−/−; p53pc−/− mice (I), and Ki-67 quantification in Ptenpc−/−; p53pc−/− prostatic tumors upon infusion of CXCR2 WT or CXCR2-KO monocytes (J).
(K) Immunofluorescence staining of e-Cadherin-positive (red) and pHP1γ-positive (green) epithelial cells in Ptenpc−/−; p53pc−/− prostatic tumors upon infusion of CXCR2 WT or CXCR2-KO monocytes. Cells were counterstained with the nuclear marker DAPI (blue). Scale bar, 20 μm.
(L) Representative images of SA-βgalactosidase IHC staining on Ptenpc−/−; p53pc−/− prostatic tumors. Original magnification, 40×. Scale bar, 100 μm.
Error bars are mean ± SEM. ∗∗p < 0.01 and ∗∗∗p < 0.001.
Pro-inflammatory Macrophages Drive Senescence Induction in PTEN-Null Cells through TNFα
To broaden the translational relevance of these findings, we next investigated the mechanism by which αCXCR2-treated pro-inflammatory macrophages promote senescence inhuman PTEN-deficient prostate cancer cells. We therefore exposed human epithelial cancer cell lines to the c.m. of TAMs treated in presence or absence of the CXCR2 antagonist (tumor-conditioned macrophages). C.m. from pro-inflammatory (IFNγ/LPS) and anti-inflammatory (IL-4/IL-13) macrophages was used as control. Treatment with c.m. from αCXCR2-treated tumor-conditioned macrophages promoted a strong growth arrest and senescence response specifically in PTEN−/−; TRP53+/+ LnCaP and PTEN−/−; TRP53−/− PC3 cells, while PTEN+/+; TRP53+/+ 22RV1 control cells were not affected. In line with these findings, c.m. from IFNγ/LPS-treated pro-inflammatory macrophages also induced senescence and cell growth inhibition in LnCaP and PC3 cells, whereas c.m. from IL-4/IL-13-polarized macrophages slightly promoted proliferation in these cells (Figures 5A and 5B). Of note, deletion of Trp53 did not result in decreased sensitivity of PTEN-null cells to the administration of the c.m. from aCXCR2-TAMs. To then identify factors secreted by pro-inflammatory macrophages affecting the proliferation and senescence of PTEN-deficient cells, we performed an in vitro screening in MEFs with cytokines released by in vitro derived pro-inflammatory macrophages (Figure S8A). Among all the tested factors, TNFα was the only molecule capable of inducing growth inhibition and senescence in PTEN-null human cancer cells (Figures 5C and 5D) and decreasing tumor growth of prostate tumor organoids (Figure S8B). These findings were also validated in an isogenic TRAMP-C1 mouse epithelial prostate cancer cell line in which Pten was deleted by using a Crispr-Cas9 system (Figures S8C and S8D).
Figure 5.

Pro-inflammatory Macrophages Induce Growth Arrest and Senescence Enhancement in Pten-Null Tumors
(A and B) Bar graph showing fold change in cell proliferation (A) and percentage (B) of SA-b-galactosidase positive human cancer cells exposed to conditioned media from stimulated macrophages. Macrophages were either untreated or stimulated with the following: IFNγ/LPS, IL-4/IL-13, and conditioned media from PC3 cancer cells ± aCXCR2.
(C and D) Bar graph showing fold change in cell proliferation (C) and percentage (D) of SA-b-galactosidase positive cells from 22Rv1, LnCaP, and PC3 treated with recombinant TNFα.
(E) RT-qPCR analysis of TNFR1 expression in the three human cell lines.
(F) Bright-field images from three-dimensional (3D) culture of CRPC patient-derived organoids and cell line-derived organoids (hanging drop) acquired with Zeiss LSM700 confocal laser scanning microscope.
(G) RT-qPCR analysis showing the anti-correlation of PTEN and TNFR1 relative expression in human mCRPC patient-derived organoids. Bar chart showing fold change comparison between each gene in reference to a PTEN +/+ control (22Rv1) and Lamin A/C used as a housekeeping gene.
Error bars are mean ± SEM (n = 2). ∗p < 0.05, ∗∗p = 0.0071 (two-way ANOVA), and ∗∗∗p < 0.001.
To further dissect the specificity of TNFα for the PTEN−/−background, we analyzed the mRNA expression levels of TNFR1 in human prostate cancer cell lines and human tumor organoids. RT-qPCR analysis for TNFR1showed the existence of an inverse correlation between the expression of PTEN and TNFR1 in these settings (Figures 5E–5G), thereby potentially explaining the higher sensitivity of PTEN−/− cells to TNFα exposure. This was also confirmed in TRAMP-C1-Pten−/−prostate tumor cells (Figure S8E).
CXCR2 Blockade Reverts the Effect of Anti-CXCR2 Treatment in PTEN-Null Prostate Tumors In Vivo
To further investigate the impact of TNFα in tumor treated with anti-CXCR2 antagonist, we treated mice bearing Pten−/−-TRAMP-C1 allograft tumors with a combination of AZD5069 and a neutralizing antibody for TNFα. Strikingly, the anti-tumorigenic and pro-senescent effect of AZD5069 treatment was reverted upon blockade of TNFα. Indeed, tumor volume was significantly decreased upon administration of the CXCR2 antagonist over time, and its efficacy was dramatically reverted upon TNFα neutralization in vivo. These results were further validated in the Pten-null-TRAMP-C1 cells in which TNFR1 was deleted by using a Crispr-Cas9 system (Pten-null; TNFR1-null-TRAMP-C1). Of note, tumor size of Pten-null-TRAMP-C1 cells injected in hosting mice was not affected by AZD5069 treatment in cells depleted from the TNFR1, thus confirming the role played by TNFα in mediating the efficacy of the CXCR2 antagonist (Figure 6A). Furthermore, in accordance with the pro-senescent effect exerted by TNFα-releasing macrophages in Pten−/− cancers, TNFα neutralization and TNFR1 inactivation in Pten-null-TRAMP-C1 tumors resulted in the inhibition of senescence, as shown by a decreased expression of p16 levels in these tumors (Figures 6B and 6C).
Figure 6.

TNFα Neutralization Disrupts the Efficacy of CXCR2 Blockade in Syngeneic Prostate Tumors In Vivo
For the allograft experiments, 2.5 × 106 TRAMP-C1 cells were injected subcutaneously into the flank of male C57BL/6 mice. When tumors were approximately 200 mm3, mice were randomized to the treatment groups. Tumor growth was monitored daily by measuring the tumor size with caliper. Mice were treated with αCXCR2 (AZD5069 100 mg/kg) daily for 3 weeks.
(A) Bar graph showing the relative tumor volume (mm3) at days 10, 20, and 30 after injection in mice bearing TRAMPTRAMP-C1C1 Pten−/−; Trp53−/− allografts treated with αCXCR2 with or without combination with a monoclonal antibody for TNFα. The graph is also showing the tumor size of Pten−/−; Trp53−/− TNFR1−/− TRAMP-C1 cells in the presence or absence of αCXCR2 treatment, at the same time points. CRISPR/Cas9 was performed on TRAMP-C1 cells to delete TNFR1.
(B and C) Representative images of IHC staining (B) and quantification (C) of p16 in all the conditions.
(D) Schematic model showing the mechanism by which CXCR2 blockade reprograms TAMs in prostate tumors.
Error bars are mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Discussion
Altogether our findings disclose the mechanism by which CXCR2 antagonists exert an antitumor response. Here we show that the majority of TAMs infiltrating Pten-null prostate cancer expressed the C-X-C chemokine receptor type 2 (CXCR2). Pharmacological blockade of the CXCR2 receptor in tumor models in vivo, by a selective antagonist, promoted the re-education of TAMs toward a TNFα-releasing pro-inflammatory phenotype, which resulted in induction of senescence and tumor inhibition. Dissection of the mechanisms by which TAMs affect tumor progression has paved the way for the development of macrophage-targeting therapies. Indeed, multiple therapeutic strategies aimed at TAMs depletion have been developed. Among those, α-CSF-1R monoclonal antibodies have been shown to alter macrophage frequency in models of colorectal adenocarcinoma and fibrosarcoma (Ries et al., 2014). In addition, depletion of TAMs has been demonstrated to be a key mechanism mediating the anti-tumor activity of trabectedin in human liposarcoma (Germano et al., 2013). Importantly, recent findings also support the possibility to activate the anti-tumor activity of TAMs in cancer, rather than blocking their recruitment or localization. Administration of an agonistic α-CD40 antibody in a model of pancreatic cancer resulted in the re-education of TAMs toward a pro-inflammatory phenotype, leading to a reduction in tumor volume (Beatty et al., 2013). Furthermore, reprogramming of tumor-infiltrating macrophages toward an anti-cancer phenotype has also been achieved in murine tumor models through class IIa HDAC inhibition and via the expression of antiangiogenic and immunomodulatory protein histidine-rich glycoprotein (HRG) (Guerriero et al., 2017, Rolny et al., 2011). In this regard, in the present study, we took advantage of the AZD5069, a CXCR2 antagonist that, in contrast to therapies that deplete TAMs in tumors, blocks tumorigenesis by re-educating the TAMs toward an anti-tumorigenic and pro-senescent functional state without affecting the total number of TAMs in the tumor microenvironment. Given that the macrophage represents the most prominent population infiltrating the tumor in most cancers, our strategy may permit exploitation of the localization of macrophages at the tumor bed by triggering their anti-tumorigenic power. Interestingly, here we show that members of the CXCL family, known to bind the CXCR2 receptor, were increased in prostate cancer and correlate with cancer progression, thus supporting previous evidence demonstrating that the CXCL-CXCR pathway plays a substantial role in tumor development and further indicating that developing effective therapies aimed at the disruption of such pathway may be critical for tumor treatment in many cancer types. In addition, our findings provide evidence that support the use of autologous infusion of monocytes as a therapy for prostate cancer. Indeed, our findings show that infusion of CXCR2-KO monocytes in tumor-bearing animals resulted in tumor inhibition and senescence induction, accompanied by the re-education of the infiltrating macrophages toward a pro-inflammatory state. Of note, autologous infusion of human activated monocytes was attempted in the past but resulted in poor clinical response in cancer-bearing patients. Importantly, significant levels of pro-inflammatory cytokines were never detected in the serum of treated patients after monocytes infusions (Lopez et al., 1992, Lopez et al., 1994, Stevenson et al., 1988), suggesting that activated monocytes were reprogrammed toward an anti-inflammatory and therefore pro-tumorigenic phenotype once infused. Our results show that a more efficient strategy could be achieved in the clinic by combining infusion of monocytes with the αCXCR2 treatment, as this combination will skew the monocytes and derived macrophages toward an anti-tumorigenic functional state. Finally, given that CXCR2 antagonists are currently being evaluated in the clinic to treat different types of tumor, including prostate cancer, it will be imperative to investigate the clinical settings in which these compounds may have the largest impact. In this regard, our results indicate that tumors harboring Pten deletion are sensitive to αCXCR2 treatment and to the consequent TNFα-induced senescence, as these tumor cells upregulate TNFR1 (Figure 6D). Previous evidence supported the role of TNFα in triggering apoptosis of tumor cells and senescence induction (Beyne-Rauzy et al., 2004, Dumont et al., 2000, Guerriero et al., 2017). However, whether TNFα is capable to induce an antitumor response may depend on the genetic background of the tumors, as in certain tumor types, TNFα does not affect tumor growth and even increases tumorigenesis (Braumüller et al., 2013, Waters et al., 2013). Thus, clinical trials evaluating the efficacy of CXCR2 antagonists should take into account this information, and treated patients should be stratified for the level of PTEN in the tumors.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3ε (Clone 145-2C11) | Biolegend | Cat# 100434; Lot. B241616; RRID:AB_893324 |
| CD4 (Clone GK1.5) | Biolegend | Cat# 100434; Lot. B248433; RRID:AB_893324 |
| CD8a (Clone 53-6.7) | Biolegend | Cat# 100741; Lot. B272200; RRID:AB_11124344 |
| CD11b (Clone M1/70) | Biolegend | Cat# 101235; Lot. B251703; RRID:AB_10897942 |
| CD45 (Clone 30-F11) | Biolegend | Cat# 103155; Lot. B253523; RRID:AB_2650656 |
| Ly6C (Clone HK1.4) | Invitrogen | Ref. 25-5932-82; Lot. 1990189; RRID: AB_1724153 |
| Ly6G (Clone 1A8) | BD | Cat# 551459; Lot. 21064; RRID:AB_394206 |
| NK 1.1 (Clone PK136) | Invitrogen | Ref. 17-5941-81; Lot. 2003461; RRID: AB_469479 |
| CD206 (Clone C068C2) | Biolegend | Cat# 141707; Lot. B230155; RRID:AB_10896057 |
| F4/80 (Clone BM8) | Invitrogen | Ref. 47-4801-80; Lot. 4338512; RRID: AB_1548745 |
| Gr1 (Clone RB6-8C5) | Invitrogen | Ref. 12-5931-81; Lot. 4335548; RRID: AB_466045 |
| B220 (Clone RA3-6B2) | Biolegend | Cat# 103222; Lot. B24773; RRID:AB_313005 |
| CXCR2 (Clone SA045E1) | Biolegend | Cat# 149609; Lot. B258930; RRID:AB_2565689 |
| TNF-a (Clone MP6-XT22) | Biolegend | Cat# 506307; Lot. B230018; RRID:AB_315428 |
| Granzyme B (Clone GB11) | Biolegend | Cat# 515407; Lot. B260874; RRID:AB_2562195 |
| Purified anti-mouse CD16/32 (Clone 93) | Biolegend | Cat# 101301; Lot. B264872; RRID:AB_312800 |
| Dynabeads mouse T activator CD3/CD28 | GIBCO By ThermoFisher | Ref. 11452D; Lot. 00758725 |
| Cell Trace Violet Cell Proliferation Kit | Invitrogen | Cat. C34557; Lot. 1851453 |
| GATA-4 (D3A3M) | Cell Signaling | 36966; RRID: AB_2799108 |
| HSP-90 (C45G5) | Cell Signaling | 4877S; RRID: AB_2233307 |
| PTEN | Cell Signaling | 552S; RRID: AB_390810 |
| a-TUBULIN (9F3) | Cell Signaling | 2128; RRID: AB_1950376 |
| p-STAT6 (Tyr641) (D8S9Y) | Cell Signaling | 56554; RRID: AB_2799514 |
| STAT-6 | Cell Signaling | 9362s; RRID: AB_2271211 |
| TNFR1 (D3I7K) | Cell Signaling | 13377S; RRID: AB_2798194 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human Recombinant IL-13 | Peprotech | Cat. AF-200-13; Lot. 0512AFC23 H1913 |
| Human Recombinant IL-4 | Peprotech | Cat. AF-200-04; Lot. 0712AFC14 G1218 |
| Human Recombinant IFN-g | Peprotech | Cat. AF-300-02; Lot. 0412 AFC27 |
| Mouse Recombinant IFN-g | Peprotech | Cat. 315-05; Lot. 061398 |
| Mouse Recombinant CXCR2 | Peprotech | Cat. 250-15; Lot. 098152 E0913 |
| Mouse Recombinant IL-4 | Peprotech | Cat. 214-14; Lot. 081449 K1915 |
| Mouse Recombinant IL-13 | Peprotech | Cat. 210-13; Lot. 1111207 |
| BD Golgi Plug | BD | Cat. 51-230 1KZ; Lot. 4309737-1 |
| PMA (Phorbol 12-myristate 13-acetate) | Sigma-Aldrich | Cat. P8139-1MG |
| Ionomycin | Sigma-Aldrich | Cat. I0634 |
| Dnase I | Sigma-Aldrich | Cat. 4716728001 |
| Anti human TNF-a (recombinant) | Peprotech | Cat. AF-300-01A; Lot. 0609AFC25 |
| Anti mouse-TNF-a (Recombinant) | Peprotech | Cat. 500-P64; Lot. 1111207 |
| Critical Commercial Assays | ||
| Senescence β-Galactosidase Staining Kit | Cell Signaling Technology | Cat. #9860S |
| Proteome Profiler Mouse Cytokine Array Kit, Panel A | R&D Systems | Cat. ARY006 |
| EasySep PE Positive Selection Kit II | Stem Cell Technology | Catalog # 17684 |
| Deposited Data | ||
| Raw RNA-Seq Data | This Paper | GEO accession number: GSE125273https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125273 |
| Experimental Models: Cell Lines | ||
| LNCap/C4 | ATCC | ATCC® CRL-3313 |
| PC3 | ATCC | ATCC® CRL-1435 |
| 22RV1 | ATCC | ATCC® CRL-2505 |
| TRAMP-C1 | ATCC | ATCC® CRL-2730 |
| THP-1 | ATCC | ATCC® TIB-202 |
| Experimental Models: Organisms/Strains | ||
| Mouse; C57BL/6NCrl | Charles River | Strain Code: 027 |
| Mouse; Ptenpc−/− (C57BL/6J background) | Donation from the laboratory of Prof. Pier Paolo Pandolfi (Harvard) | N/A |
| Mouse; Ptenpc−/−; Trp53pc−/− (C57BL/6J background) | Donation from the laboratory of Prof. Pier Paolo Pandolfi (Harvard) | N/A |
| Oligonucleotides | ||
| PTEN CRISPR/Cas9 KO Plasmid (m) | Santa Cruz Biotechnology | sc-422475 |
| TNFR1 CRISPR/Cas9 KO Plasmid (m) | Santa Cruz Biotechnology | sc-423442-KO-2 |
| RT-qPCR primers, see Table S1 | “this paper” | Sigma Aldrich |
| Software and Algorithms | ||
| Flow Jo_v10 | Tree Star | https://flowjo.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Cytoscape | Cytoscape | https://cytoscape.org/ |
| Graph Pad Prism | GraphPad Software | https://www.graphpad.com |
Lead Contact and Materials Availability
There are restrictions to the availability of the following reagent: goat polyclonal anti-pSTAT6 (sc-11762, Santa Cruz Biotechnology). The reagent has been discontinued. Further information and requests for resources and reagents may be directed to and will be fulfilled by Lead Contact, Prof. Andrea Alimonti (andrea.alimonti@ior.usi.ch).
Experimental Model and Subject Details
Mice
All mice were maintained under specific pathogen-free conditions of the IRB institute and experiments were performed according to state guidelines and approved by the local ethical committee (“Dipartimento della Sanità e Socialità, Esperimenti su animali,” Canton Ticino). Ptenpc+/+, Ptenpc−/− and Ptenpc−/−; Trp53pc−/− mice (C57BL/6J background) were generated and genotyped as previously described12. Female PtenloxP/loxP and female PtenloxP/loxP; Trp53loxP/loxP mice were crossed with male PB-Cre4 transgenic mice and genotyped for Cre using following primers, primer1 (5′ TGATGGACATGTTCAGGGATC 3′) and primer2 (5′ CAGCCACCAGCTTGCATGA 3′). B6.129S2(C)-Cxcr2tm1Mwm/J (CXCR2 KO) mice were purchased from Jackson laboratory (Stock No: 006848). For the allograft experiments, 2.5x106 TRAMP-C1 cells were injected subcutaneously into the flank of male C57BL/6N mice. When tumors were approximately 200 mm3, mice were randomized to the treatment groups. Tumor growth was monitored daily by measuring the tumor size with caliper. In some experiments mice were treated with intraperitoneal injections of 100 μL of the vehicle or 100 μL αCXCR2 (AZD5069) from Astrazeneca, at a final concentration of 100mg/kg daily for a period of 3 weeks. For the anti-TNFα treatment, mice were injected intraperitoneally with the InVivoMAb anti-mouse TNFα from BioXCell (XT3.11) for a period of 3 weeks, 3 times a week. In some experiments mice were infused with bone marrow derived macrophages (BMDMs), differentiated in vitro as previously described (Murray et al., 2014). BMDMs were obtained from male donor CXCR2 WT mice or CXCR2 KO mice. Mice received 1x106 BMDMs for intravenous injection. Injections were administered every 3 days for a period of 2 weeks. Recipient mice were sacrificed 1 day post the last injection.
Cells
Primary MEFs (Mouse Embrionic Fibroblasts) were derived from littermate embryos and obtained by crossing Ptenlx/lx and Ptenlx/lx; Trp53lx/lx animals as previously described. Embryos were harvested at 13.5 days post coitum and individual MEFs were produced and cultured as previously described (Chen et al., 2005). Both female and male derived MEFs were utilized. At passage 2 cells were harvested for protein blot analysis. The LnCap (C4), 22Rv1 and PC3 cell lines were purchased from the American Type Culture Collection (ATCC). Cells were maintained at 5% CO2 at 37°C and cultured in RPMI with 10% heat inactivated FBS. All the human cell lines were used for in vitro experiments. The TRAMP-C1 cell line was purchased from American Type Culture Collection (ATCC). Cells were maintained at 5% CO2 at 37°C and cultured in DMEM with 10% heat inactivated FBS. For the allograft experiments, 2.5x106 TRAMP-C1 cells were injected subcutaneously into the flank of male C57BL/6N mice. The human monocytic cell line THP-1 was used to obtain human macrophages upon differentiation. Cells were maintained at 5% CO2 at 37°C and cultured in RPMI with 10% heat inactivated FBS. THP-1 cells were treated for 24 h with 100 ng/ml PMA to obtain differentiated macrophages(Starr et al., 2018). On the next day cells were washed and media was replaced.
CRISPR-Cas9 transfection
TRAMP-C1 cells were maintained in 75 cm2 flask to a 50%–60% confluency in DMEM media supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin and 2 mM L-glutamine. The transfection of the PTEN CRISPR/Cas9 KO plasmid (Santa Cruz Biotechnology) was performed using jetPRIME® transfection reagent according the manufactory protocol at the 1:2 DNA / jetPRIME® ratio. 24h after transfection, the GFP transduced cells were sorted to purity 99% and plated as single cell on 96-well plates. At day 7 after cell sorting the grown cell colonies were moved into 24-well plates for further expansion. The knock-down of Pten gene in each cell colony was confirmed by RT-qPCR and western blot.
Human organoids
Organoids were grown in 3D Matrigel® (cat.356231, Corning) under prostate epithelial conditions and lysed directly onto wells and RNA was purified using the Zymo-Spin Columns. RNA was extracted from 3D cultured organoids using Quick-RNA MicroPrep (cat.R1050, Zymo Research). RT-qPCR was carried out using RevertAid First Strand cDNA synthesis kit (cat.K1622, Thermo Scientific) following manufacturer’s protocol. PCR amplification was carried out using Brilliant III Ultra-Fast SYBR® Green qPCR mastermix (cat.600883, Agilent Technologies) and Agilent Mx3000P system. cDNA from PTEN−/− (PC3) and PTEN +/+ (22Rv1) was used as fold-change control and Lamin A/C as housekeeping gene and internal control. Fold change (relative expression) was calculated using ddCT method. Primer pairs were designed to span exon-exon junctions, useful for limiting the amplification only to mRNA. The following sequences were utilized: PTEN: fwd 5′-ACCCACCACAGCTAGAACTT-3′, rev 5′-GGGAATAGTTACTCCCTTTTTGTC-3′; TNFRI: fwd 5′-CTGGAGCTGTTGGTGGGAAT-3′, rev 5′-TGACCCATTTCCTTTCGGCA-3′; Lamin A/C: fwd 5′-TGCAGGAGCTCAATGATCGC-3′, rev 5′-CATTGTCAATCTCCACCAGTC-3′. In some experiments cell viability was measured using 3D CellTiter-Glo® 3D reagent (cat.G9681, Promega) by quantifying metabolically active cells releasing ATP. Cell line-derived organoids were plated at a density of 2000 cells per well in 96-well optical plates (cat.3610, Corning) embedded in Matrigel® as hanging drops (5 μL per well). Cells were treated with recombinant TNFα (cat 300-01A, PeproTech) at 10ng/ml and luminescence measurement was performed after 5 days in culture.
Method Details
Necropsy and histopathology
Animals were necropsied and all tissues were examined regardless of their pathological status. Tumor size was measured by a tumor caliper and then applying the following formula: Size = (Width2 × Length)/2. For the prostatic tumors, the size of two anterior lobes was considered. Normal and tumor tissue samples were fixed in 10% neutral-buffered formalin (Sigma) overnight. Tissues were processed by ethanol dehydration and embedded in paraffin according to standard protocols. Sections (5 μm) were prepared for antibody detection and hematoxylin and eosin (H&E) staining (Diapath, C0303) and (Diapath, C0363) respectively. To evaluate evidence of invasion, sections were cut at 20 μm intervals and H&E stained. Slides were prepared containing 3 to 5 of these interval sections. In all experiments the histology was evaluated blindly.
Immunohistochemistry and immunofluorescence
For immunohistochemistry (IHC), tissues were fixed in 10% formalin (Thermo Scientific, 5701) and embedded in paraffin in accordance with standard procedures. Preceding immunohistochemical staining, tumor sections were exposed to two washes with OTTIX plus solution (Diapath, X0076) and subsequent hydration with OTTIX shaper solution (Diapath, X0096) followed by deionized water. Antigen unmasking was performed by heating sections in the respective pH solutions based on the antibodies used at 98°C for 20 mins. Subsequently the sections were blocked for peroxidases and non-specific binding of antibodies using 3% H2O2 (VWR chemicals, 23615.248) and Protein-Block solution (DAKO Agilent technologies, X0909) respectively for 10 min each split by 0.5% PBST washing. Sections were stained for anti-p16 (M156; Santa Cruz), anti-Ki67 (Clone SP6; Lab Vision Corporation), anti-pH2AX and anti-CD31. Images were obtained using objectives of 5x and 20x magnification and Pixel image of 1.12 μm and 0.28 μm respectively. For the immunofluorescence (IF) staining, tissue paraffin embedded sections were stained for anti-E-Cadherin (BD Biosciences, 610181), anti-Vimentin (Dako), anti-F4/80 (Serotec), anti-pHp1gamma (Cell signaling). Confocal images were obtained with the Leica TCS SP5 confocal microscope or Leica TCS SP8II. In all experiments the histology was evaluated blindly.
Prostatic epithelial cell purification and cytokine array
Ptenpc+/+, Ptenpc−/− and Ptenpc−/−; Trp53pc−/− 12 weeks old mice were sacrificed and whole prostates were isolated and processed to single cell suspension for magnetic activated cells sorting (MACS). Single cells were stained with purified-anti-CD45 (leukocytes), and incubated 20 min on ice. Cells were then loaded into MS column (Miltenyi biotech) for MACS separation and unstained epithelial cells were collected in the negative fraction. Purified prostatic epithelial cell were processed as indicated in cytokine array kit (Proteome Profiler Mouse XL Cytokine Array, R&D Systems). Developed films were scanned and obtained images were analyzed using ImageJ 1.43u and background signals were subtracted from the experimental values. Experiments were performed in technical duplicate.
In vitro differentiation of Bone Marrow derived macrophages and Human THP-1-derived macrophages
Bone marrow derived macrophages (BMDMs) were differentiated in vitro as previously described (Fleetwood et al., 2009, Murray et al., 2014). Briefly, bone marrow precursors were flushed from long bones of C57BL/6N mice or CXCR2 KO mice and cultured in DMEM supplemented with 10% heat-inactivated-FCS media, in the presence of 10ng/ml of M-CSF or GM-CSF (Peprotech). At day 4 non-adherent cells were collected and cultured for a further 3 days in the presence of fresh media. On day 7, the media was replaced with complete media containing specific cytokines for macrophages polarization (pro-inflammatory phenotype: 10 ng/ml LPS and 10 ng/ml IFNγ; anti-inflammatory phenotype: 30 ng/ml IL-4, 30 ng/ml IL-13 and 100 ng/ml CXCL2). At day 10 cells were harvested and analyzed by flow cytometry and qRT-PCR. The human monocytic cell line THP-1 was treated for one day with PMA 100 ng/ml to obtain differentiated macrophages (Starr et al., 2018, Plos One). On the next day, cells were washed and media was replaced. For the production of the conditioned media, c.m. from human tumor cell line was added in presence or absence of αCXCR2 (1uM). After 48h cells were washed and media was replaced. After 48h c.m. from macrophages were collected and used to condition human tumor cell lines for 48h. Conditioned media from IFNγ/LPS and IL-4/IL-13 stimulated macrophages was used as control.
Western blot
Tissue and cell lysates were prepared with RIPA buffer (1x PBS, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Roche). Total protein concentration was measured using BCA Protein Assay Kit (Cat: 23225; Pierce, Rockford). Equal amounts of proteins were separated by SDS-PAGE and western blotted onto a 0.45 μm- nitrocellulose membrane. Membranes were blocked in 5% defatted milk in Tris-buffered saline containing 0.1% Tween-20 (TBST), probed with diluted antibodies and incubated at 4°C overnight. The following primary antibodies were utilized: rabbit polyclonal anti-Pten (1:1000 dilution, Cell Signaling), rabbit polyclonal anti-p16 (1:1000 dilution, clone M156; Santa Cruz Biotechnology), rabbit monoclonal anti-TNFR1 (1:1000 dilution, Cell Signaling), rabbit polyclonal anti-HSP90 (1:1000 dilution, Cell Signaling), mouse monoclonal anti-α-Tubulin (1:1000 dilution, Cell Signaling), rabbit monoclonal anti-STAT6 (1:1000 dilution, Cell Signaling), goat polyclonal anti-pSTAT6 (1:500 dilution, Santa Cruz Biotechnology). After washing in TBST, the membrane was incubated with secondary antibody conjugated with horseradish peroxidase (HRP) (dilution 1:5000, Cell Signaling). The protein bands were visualized using the ECL Western Blotting Substrate (Pierce).
Flow cytometry analysis
For phenotype analysis, isolated cells were re-suspended in PBS containing 1% FCS (Sigma-Aldrich) and were stained for 15 min at room temperature with anti–mouse monoclonal antibodies. Samples were acquired on a BD LSR-Fortessa flow cytometer (BD Biosciences) and a BD FACSymphony flow cytometer. When needed, cells were sorted from the prostate single-cell suspension using a FACSAria III cell sorter (BD Biosciences), after staining with specific antibodies for 30 min at 4°C in PBS containing 1% FCS. Data were analyzed using FlowJo software (TreeStar, Ashland, OR).
In vitro suppression assay
In vitro suppression assays were carried out in RPMI/10% FCS in 96-well U-bottom plates (Corning, NY). Either naive splenocytes, CD3+ cells isolated from murine lymph nodes or leucocytes isolated from murine peripheral blood were utilized to perform three different sets of suppression assays. In all sets of experiments isolated cells were labeled with 5 μM CFSE (Molecular Probes), and activated in vitro with anti-CD3 and anti-CD28 beads (Invitrogen) according to the manufacturer’s instructions. Condition media of polarized macrophages was added to the culture. After 3-5 days, cells were acquired by BD LSR Fortessa or BD FACS Symphony and the proliferation of CFSE-labeled CD8+ T cells was assessed upon staining with the following anti-mouse monoclonal antibodies: CD3 APC-Cy7 (clone B241616); CD4 PerCP-Cy5.5 (clone B240053); CD8 APC (clone 53-6.7). Analysis of the data was performed by FlowJo software.
Gene expression
Total RNA was quantified by NanoDrop ND-1000 Spectro-photometer (NanoDrop Technologies, Wilming-ton, DE) and RNA quality was assessed using Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA). Gene expression profiling was carried out using the one-color labeling method: labeling, hybridization, washing, and slide scanning were performed following the manufacturers protocols (Agilent Technologies). Briefly, 100 ng of total RNA were amplified, labeled with Cy3-CTP and purified with columns. Six-hundred ng of labeled specimens were hybridized on Agilent SurePrint G3 Mouse 8x60K Gene Expression arrays. After 17 h the slide was washed and scanned using the Agilent Scanner version C (G2505C, Agilent Technologies). Raw data were processed using the Bioconductor package Limma (Linear models for microarray analysis). Background correction was performed with the normexp method with an offset of 50, and quantile was used for the between-array normalization. The empirical Bayes method was used to compute a moderated t-statistics in order to identify differentially expressed genes in treated versus not treated mice. Data analysis and data visualization for the underlying GS-GS network and the superimposed GS-cluster network were carried out following the methodologies previously described(Delaleu et al., 2013). Parameters deviating from the original description are: i) GS collection updated to May 1st 2017, ii) gene ranking for GSEA based on fold change (genes 44064; n = 3 per group, Agilent SurePrint G3 Mouse GE 8x60K microarray), iii) Significance threshold for GSs being mapped: FDRq < 0.001 and TAGS ≥ 50, iv) layout algorithm: Cytoscape 3.5.0, yFiles organic layout 2 with edge connectivity threshold 0.065, v) inflation parameter for MCL clustering: 2.0 and vi). Cluster label annotation was supported by Auto Annotate(Su et al., 2014).
Real-time PCR
RNA was isolated by TRIzol reagent (QIAGEN) and retro-transcribed using SuperScriptIII (Invitrogen, 11752-250) according to the manufacturer’s instructions. Quantitative PCR (RT-qPCR) reactions (Bio-Rad) for each sample were done in triplicate using KAPA SYBR FAST qPCR green (KK4605) (Applied Biosystems). The primer sequences were obtained from PrimerBank (http://pga.mgh.harvard.edu/primerbank/index.html). Each value was adjusted by GADPH level as reference.
Screening of macrophage-derived factors
Pten+/+; Trp53+/+ and Pten−/−; Trp53−/− MEFs were plated at a density of 5 × 103 cells /ml. Both female and male derived MEFs were utilized. 2h after the cell plating, the following recombinant proteins were added to the cells as a single agent or in combination with another cytokine/chemokine: 100 ng/ml IL6, 100 ng/ml CXCL5, 100 ng/ml IL-1α, 10 ng/ml TNFα, 10 ng/ml GM-CSF, 100 ng/ml IL-12, 200 ng/ml CCL5, 250 ng/ml adiponectin. Recombinant cytokines were purchased from Peprotech. At day 3 the cell viability was tested by crystal violet staining proliferation assay. Briefly, conditional media was aspirated and the cells were gently washed twice with PBS. The cell fixation was performed by adding 10% Formalin for 10 min. The cells were gently washed with PBS and stained with 0.05% Crystal Violet for 20 min. After the staining, crystal violet was discarded; the cells were washed in water and dried at room temperature overnight. Absorbance was read using the Microplate Reader at 590 nm.
Co-culture of MEFs with murine macrophages conditioned media
In the co-culture experiments, conditioned media from either IFNγ/LPS or IL-4/IL-13 polarized macrophages was added to the MEFs 1 day after seeding. Co-cultures were stopped 72hrs later and cells harvested for protein extraction or stained for analysis. Senescence was assessed by mean of a Senescence β-Galactosidase Staining Kit (Cell Signaling, 9860).
Phagocytosis assay
Briefly, murine bone marrow derived macrophages were exposed to the conditioned media collected from TRAMP-C1 prostate cancer cells, to recapitulate the effect of cancer cells on infiltrating macrophages. Tumor-conditioned macrophages were co-coltured with RFP-labeled TRAMP-C1 cancer cells overnight, in presence or absence of αCXCR2 to assess the phagocytic activity versus tumor cells. Cells were then collected and analyzed by flow cytometry following staining for macrophage markers with the following anti-mouse monoclonal antibodies: CD11b APC (clone M1/70); F4/80 eFluor780 (clone BM8). The frequency of RFP+ F480+ cells was quantified to assess the phagocytic activity of macrophages.
Quantification and Statistical Analysis
Statistical analyses were performed using a two-tailed unpaired Student’s t test. Values are presented as mean ± SEM (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). For studies comparing more than two groups, 2-way ANOVA multiple comparisons by Prism6 was also utilized. Differences were considered significant when ∗p < 0.05 or are indicated as not significant (ns).
Data and Code Availability
The accession number for the RNA-sequencing dataset reported in this paper is GEO: GSE125273.
Acknowledgments
We thank all members of the IRB animal core facility for technical assistance and animal work. We thank Dr. D. Jarrossay for helping with the cell sorting experiments. We thank all members of the Dr. A. Alimonti laboratory for scientific discussions. This work was supported by a European Research Council (ERC) starting grant (261342) and an ERC consolidator (683136); grants from the Josef Steiner Foundation, the Helmut Horten Foundation, and Krebsliga (KFS 3505-08-2014); SNF (Ambizione) (PZ00P3_136612); and an AIRC Start-Up grant 2016 (19141).
Author Contributions
A.A. and D.D. developed the concept and designed the experiments. D.D., M. Mirenda, J.V., A.C., V.G., G.B., E.P., and A. Rinaldi performed experiments. D.D., N.D., G.C., P.O., and A. Rinaldi performed the gene expression-related experiments and analyzed the data. D.D., J.V., R.D., and M. Masetti established and carried out fluorescence microscopy. M.L. and S.M. performed immunohistochemical experiments and analysis. M.G. provided the EC cell line. L.G., D.W., and R.P.M. provided and analyzed the TMA and samples from prostate cancer patients. S.B. provided the AZD5069 compound. J.D.B. supervised the organoid experiments and interpreted the data. D.D., M. Mirenda, and A.A. interpreted the data. D.D. and A.A. wrote the paper.
Declaration of Interests
S.B. is employed in the Oncology Department of AstraZeneca, Li KaShing Centre, Cambridge, UK. A.A. and J.D.B. have received a research grant from AstraZeneca for the clinical development of AZD5069. J.D.B. has served on advisory boards for many companies, including AstraZeneca, Astellas, Bayer, Boehringer Ingelheim, Genentech/Roche, Genmab, GlaxoSmithKline, Janssen, Merck Serono, Merck Sharp & Dohme, Menarini/Silicon Biosystems, Orion, Pfizer, Sanofi-Aventis, and Taiho. The ICR has a commercial interest in abiraterone, PARP inhibition in DNA repair-defective cancers, and PI3K/AKT pathway inhibitors (no personal income). The ICR has received funding or other support for my research work from AstraZeneca, Astellas, Bayer, Genentech, GlaxoSmithKline, Janssen, Merck Serono, Merck Sharp & Dohme, Menarini/Silicon Biosystems, Orion, Sanofi-Aventis, and Taiho. J.D.B. has been the chief investigator (CI) or principal investigator (PI) of many industry sponsored clinical trials. All the other authors declare no competing interests.
Published: August 20, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.068.
Supplemental Information
References
- Alimonti A., Carracedo A., Clohessy J.G., Trotman L.C., Nardella C., Egia A., Salmena L., Sampieri K., Haveman W.J., Brogi E. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010;42:454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer C., Squadrito M.L., Laoui D., Thompson D., Hansen S.K., Kiialainen A., Hoves S., Ries C.H., Ooi C.H., De Palma M. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 2016;18:790–802. doi: 10.1038/ncb3371. [DOI] [PubMed] [Google Scholar]
- Beatty G.L., Torigian D.A., Chiorean E.G., Saboury B., Brothers A., Alavi A., Troxel A.B., Sun W., Teitelbaum U.R., Vonderheide R.H. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013;19:6286–6295. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyne-Rauzy O., Recher C., Dastugue N., Demur C., Pottier G., Laurent G., Sabatier L., Mansat-De Mas V. Tumor necrosis factor alpha induces senescence and chromosomal instability in human leukemic cells. Oncogene. 2004;23:7507–7516. doi: 10.1038/sj.onc.1208024. [DOI] [PubMed] [Google Scholar]
- Bingle L., Brown N.J., Lewis C.E. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J. Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- Braumüller H., Wieder T., Brenner E., Aßmann S., Hahn M., Alkhaled M., Schilbach K., Essmann F., Kneilling M., Griessinger C. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–365. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- Calcinotto A., Spataro C., Zagato E., Di Mitri D., Gil V., Crespo M., De Bernardis G., Losa M., Mirenda M., Pasquini E. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature. 2018;559:363–369. doi: 10.1038/s41586-018-0266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L., Fragkogianni S., Sims A.H., Swierczak A., Forrester L.M., Zhang H., Soong D.Y.H., Cotechini T., Anur P., Lin E.Y. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35:588–602. doi: 10.1016/j.ccell.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Trotman L.C., Shaffer D., Lin H.K., Dotan Z.A., Niki M., Koutcher J.A., Scher H.I., Ludwig T., Gerald W. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaleu N., Nguyen C.Q., Tekle K.M., Jonsson R., Peck A.B. Transcriptional landscapes of emerging autoimmunity: transient aberrations in the targeted tissue’s extracellular milieu precede immune responses in Sjögren’s syndrome. Arthritis Res. Ther. 2013;15:R174. doi: 10.1186/ar4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont P., Balbeur L., Remacle J., Toussaint O. Appearance of biomarkers of in vitro ageing after successive stimulation of WI-38 fibroblasts with IL-1alpha and TNF-alpha: senescence associated beta-galactosidase activity and morphotype transition. J. Anat. 2000;197:529–537. doi: 10.1046/j.1469-7580.2000.19740529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escamilla J., Schokrpur S., Liu C., Priceman S.J., Moughon D., Jiang Z., Pouliot F., Magyar C., Sung J.L., Xu J. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015;75:950–962. doi: 10.1158/0008-5472.CAN-14-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleetwood A.J., Dinh H., Cook A.D., Hertzog P.J., Hamilton J.A. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J. Leukoc. Biol. 2009;86:411–421. doi: 10.1189/jlb.1108702. [DOI] [PubMed] [Google Scholar]
- Germano G., Frapolli R., Belgiovine C., Anselmo A., Pesce S., Liguori M., Erba E., Uboldi S., Zucchetti M., Pasqualini F. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Guerriero J.L., Sotayo A., Ponichtera H.E., Castrillon J.A., Pourzia A.L., Schad S., Johnson S.F., Carrasco R.D., Lazo S., Bronson R.T. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543:428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann T., Biswas S.K., Lawrence T., Sica A., Lewis C.E. Regulation of macrophage function in tumors: the multifaceted role of NF-kappaB. Blood. 2009;113:3139–3146. doi: 10.1182/blood-2008-12-172825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S., Woo G., Nguyen A.V., Figueiredo C.C., Foubert P. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C., Xu Q., Martin T.D., Li M.Z., Demaria M., Aron L., Lu T., Yankner B.A., Campisi J., Elledge S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612. doi: 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti M., Masieri L., Raspollini M.R., Minervini A., Mari A., Comito G., Giannoni E., Carini M., Chiarugi P., Serni S. The role of M1 and M2 macrophages in prostate cancer in relation to extracapsular tumor extension and biochemical recurrence after radical prostatectomy. BioMed Res. Int. 2014;2014:486798. doi: 10.1155/2014/486798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M., Fechtenbaum J., David B., Martinache C., Chokri M., Canepa S., De Gramont A., Louvet C., Gorin I., Mortel O. Adoptive immunotherapy with activated macrophages grown in vitro from blood monocytes in cancer patients: a pilot study. J. Immunother. 1992;11:209–217. doi: 10.1097/00002371-199204000-00008. [DOI] [PubMed] [Google Scholar]
- Lopez M., Louvet C., Martinache C., Bony V., Scotto F., Barelaud S., Jiang R., Drapier J.C., Smadja V., De Gramont A. Infusion of large quantities of autologous blood monocyte-derived macrophages in two cancer patients did not induce increased concentration of IL-6, TNF-alpha, soluble CD14 and nitrate in blood plasma. Eur. Cytokine Netw. 1994;5:411–414. [PubMed] [Google Scholar]
- Mantovani A., Schioppa T., Porta C., Allavena P., Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–322. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez F.O., Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzieri R., Pucci F., Moi D., Zonari E., Ranghetti A., Berti A., Politi L.S., Gentner B., Brown J.L., Naldini L., De Palma M. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19:512–526. doi: 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J., Allen J.E., Biswas S.K., Fisher E.A., Gilroy D.W., Goerdt S., Gordon S., Hamilton J.A., Ivashkiv L.B., Lawrence T. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava Rodrigues D., Rescigno P., Liu D., Yuan W., Carreira S., Lambros M.B., Seed G., Mateo J., Riisnaes R., Mullane S. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Invest. 2018;128:4441–4453. doi: 10.1172/JCI121924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonomura N., Takayama H., Kawashima A., Mukai M., Nagahara A., Nakai Y., Nakayama M., Tsujimura A., Nishimura K., Aozasa K., Okuyama A. Decreased infiltration of macrophage scavenger receptor-positive cells in initial negative biopsy specimens is correlated with positive repeat biopsies of the prostate. Cancer Sci. 2010;101:1570–1573. doi: 10.1111/j.1349-7006.2010.01563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik A., Swanson K.D., Csizmadia E., Solanki A., Landon-Brace N., Gehring M.P., Helenius K., Olson B.M., Pyzer A.R., Wang L.C. Cabozantinib eradicates advanced murine prostate cancer by activating antitumor innate immunity. Cancer Discov. 2017;7:750–765. doi: 10.1158/2159-8290.CD-16-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pienta K.J., Machiels J.P., Schrijvers D., Alekseev B., Shkolnik M., Crabb S.J., Li S., Seetharam S., Puchalski T.A., Takimoto C. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest. New Drugs. 2013;31:760–768. doi: 10.1007/s10637-012-9869-8. [DOI] [PubMed] [Google Scholar]
- Pyonteck S.M., Akkari L., Schuhmacher A.J., Bowman R.L., Sevenich L., Quail D.F., Olson O.C., Quick M.L., Huse J.T., Teijeiro V. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B.Z., Pollard J.W. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath M., Müller I., Kropf P., Closs E.I., Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front. Immunol. 2014;5:532. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries C.H., Cannarile M.A., Hoves S., Benz J., Wartha K., Runza V., Rey-Giraud F., Pradel L.P., Feuerhake F., Klaman I. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Rolny C., Mazzone M., Tugues S., Laoui D., Johansson I., Coulon C., Squadrito M.L., Segura I., Li X., Knevels E. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19:31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Salvagno C., Ciampricotti M., Tuit S., Hau C.S., van Weverwijk A., Coffelt S.B., Kersten K., Vrijland K., Kos K., Ulas T. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat. Cell Biol. 2019;21:511–521. doi: 10.1038/s41556-019-0298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T., Bauler T.J., Malik-Kale P., Steele-Mortimer O. The phorbol 12-myristate-13-acetate differentiation protocol is critical to the interaction of THP-1 macrophages with Salmonella Typhimurium. PLoS ONE. 2018;13:e0193601. doi: 10.1371/journal.pone.0193601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele C.W., Karim S.A., Leach J.D.G., Bailey P., Upstill-Goddard R., Rishi L., Foth M., Bryson S., McDaid K., Wilson Z. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29:832–845. doi: 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson H.C., Lacerna L.V., Jr., Sugarbaker P.H. Ex vivo activation of killer monocytes (AKM) and their application to the treatment of human cancer. J. Clin. Apher. 1988;4:118–121. doi: 10.1002/jca.2920040216. [DOI] [PubMed] [Google Scholar]
- Su G., Morris J.H., Demchak B., Bader G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinformatics. 2014;47:8.13.1–8.13.24. doi: 10.1002/0471250953.bi0813s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Tanaka T., Shi W., Matsumoto M., Minami M., Kashiwamura S., Nakanishi K., Yoshida N., Kishimoto T., Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- Waters J.P., Pober J.S., Bradley J.R. Tumour necrosis factor and cancer. J. Pathol. 2013;230:241–248. doi: 10.1002/path.4188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-sequencing dataset reported in this paper is GEO: GSE125273.