

Abstract
Cerebrospinal fluid (CSF) is produced in the cerebral ventricles and circulates within the subarachnoid space (SAS) of the brain and spinal cord, where it exchanges with interstitial fluid of the parenchyma. The access of CSF to the entire central nervous system (CNS) makes it an attractive medium for drug delivery. However, few intrathecal (IT) therapies have reached the clinic due, in part, to limited distribution and rapid clearance. Given the success of nanoparticle (NP) carriers in prolonging circulation and improving delivery of systemically administered agents, we sought to evaluate the distribution of IT injected NPs within the CNS. We administered fluorescent, 100 nm PEGylated-NPs into the cisterna magna of healthy mice and studied their distribution along the brain and spinal cord. Our data demonstrate that NPs are capable of distributing rapidly through the SAS along the entire neuraxis with reproducible, anatomically defined patterns of delivery. NPs were well retained within the leptomeninges for over 3 weeks, showing preference for ventral surfaces and minimal penetration into the CNS parenchyma. Clearance of NPs occurred across the cribriform plate into the nasal mucosa, with a small fraction of NPs localizing with nerve roots exiting the spinal column. Larger 10 µm particles were also capable of moving through the SAS but did not achieve as widespread distribution. These studies demonstrate the ability of NPs to achieve widespread delivery along the neuraxis and highlight IT administration as a potentially significant route of administration for delivery of nanomedicine to the subarachnoid space.
Subject terms: Permeation and transport, Nanoparticles
Introduction
Intrathecal drug administration, i.e. the delivery of active agents directly into the CSF, has gained renewed attention as an approach that could circumvent systemic barriers to CNS drug delivery. IT administration achieves high concentrations of drug in the CSF that bathes the brain and spinal cord with minimal systemic exposure1–6. However, poor drug solubility, inadequate pharmacokinetics, limited tissue distribution, and neurotoxicity remain significant obstacles for most IT administered agents7–12. Ultimately, few drugs possess biophysical properties that are compatible with delivery via this route. Hydrophilic compounds typically distribute rapidly through the SAS but are cleared quickly with CSF turnover, requiring frequent dosing to maintain drug levels13–17. Hydrophobic/lipophilic compounds are often limited by poor solubility. Additionally, partitioning of small molecules into lipophilic cell membranes and binding to extracellular matrix proteins severely restricts their distribution away from the site of injection, making the approach unsuitable for treating diseases that affect non-focal regions of the CNS15,18–20.
Encapsulation of therapeutic molecules within polymeric or liposomal NPs is a strategy that has been utilized extensively to overcome biophysical and pharmacokinetic limitations of systemically administered agents21–26. The versatility of NP systems allows for a wide range of compounds to be effectively encapsulated and released over prolonged periods of time23. Importantly, intravenously administered NPs have been engineered to traverse a variety of biological barriers through optimization of properties such as size and surface features.
We hypothesized that IT administered NPs would be effectively distributed through the SAS by the convective forces of the CSF, enabling them to reach CSF-exposed surfaces of the brain and spinal cord. To test this hypothesis, we administered fluorescently labeled, non-degradable polystyrene NPs to healthy mice via direct injection into the cisterna magna. This model NP platform was selected for several reasons, including monodisperse size, stable internal fluorescent labeling, and surface chemistry that is amenable to covalent modification. NP delivery was evaluated by a number of approaches to characterize distribution both along the neuraxis and within the SAS. These studies show the potential of NPs to achieve widespread delivery along the neuraxis after IT administration and serve as a mechanistic foundation to guide the future design of NPs for IT delivery of therapeutics to treat diseases of the CNS.
Results
FNP PEGylation
Successful PEGylation of fluorescent, polystyrene nanoparticles (FNPs) via EDC chemistry was confirmed by the appearance of a peak at 3.6 in the pegylated FNPs when analyzed by 1H NMR (SI Fig. 1). Additionally, dynamic light scattering showed the expected increase in both average diameter and zeta potential. After PEGylation, the 100 nm FNP diameter increased to 122 ± 8.17 nm and the zeta potential increased from −45 ± 5.4 to −14 ± 2.2 mV. FNPs were confirmed to be stable in protein rich environments, since DLS measurements of FNPs incubated in 10% FBS at 37 °C showed no significant changes in size, PDI or zeta potential over one week (SI Fig. 2).
In Vivo FNP Tracking
To evaluate broad patterns of NP distribute following intracisternal injection, FNP distribution was monitored semi-quantitatively in vivo in healthy mice over 2 days using an IVIS imaging system. Due to the limitations of fluorescence penetration, IVIS imaging in live mice could only reliably detect the FNP signal along a short segment of the spinal column in the middle of the back, where the signal was not obstructed by the musculature of the shoulders and hindlimbs. At 5 min post-injection, the FNP signal was detected at the injection site only (Fig. 1). By 2 hours post-injection, the FNP signal was detected over the entire brain region and along the spinal cord, although the strongest signal remained focused at the injection site. From 2 to 48 hours post-injection, a small degree of redistribution of FNP signal from the brain to the spinal cord was observed. The brain AUC significantly decreased from 6.6 at 2 hrs to 2.7 AU*hrs by 48 hrs (p = 0.0098, Fig. 1C), with the greatest clearance observed from 2 to 6 hrs (6.6 to 4.8 AU*hrs). Although there was a modest decrease in FNP AUC along the spinal cord from 2 to 6 hrs (1.9 to 0.86 AU*hrs), the spinal signal AUC increased to 1.2 AU*hrs by 48 hours.
Figure 1.

Semi-quantitative IVIS tracking of FNP distribution after IT administration in intact healthy mice. FNPs can be observed with semi-quantitative measurement over the brain and spinal cord. (A) Representative images show the spread of FNPs away from the cisterna magna over time. FNPs were detectable along the spinal cord by 2 hours but only along the mid of the back unobstructed by the extra muscle of the forelimbs and hindlimbs. (B) A graph of FNP intensity along a line profile from nose to tail demonstrates a relative decrease in total signal along the dorsal brain with a slight increase in signal along the dorsal spine over time. (C) AUCs calculated from the brain signal (−20 to 10 mm) and spinal signal (10 to 40 mm) showed significant clearance of FNPs from the brain region from 2 to 48 hours (p < 0.05, 1-way ANOVA with Tukey’s multiple comparison test), while the FNP intensity along the spinal cord did not significantly change over the 48 hours. Mean + SD shown in B to help keep lines visible. AUC points show mean ± SD.
Ex Vivo Analysis of FNP Spinal Column Distribution
Transverse tissue slices, taken from known distances along the spinal column, were imaged by confocal microscopy to visualize FNP distribution within the SAS around and along the spinal cord in decalcified samples. IVIS imaging of a complete neuraxis before and after decalcification was examined to confirm the decalcification process did not produce a loss or redistribution of FNP signal (SI Fig. 3). Although the fluorescence signal was observed to increase after decalcification, presumably because the reduction of signal attenuation by bone, the spatial distribution of fluorescent signal along the neuraxis remained consistent before and after decalcification. In a subset of animals, we removed the bone and meninges prior to imaging. Removal of the meninges resulted in a loss of nearly all detectable FNP signal (data not shown), providing evidence that the FNPs are retained within the SAS. Although it is difficult to visualize the boundary between the pia matter and the parenchyma, we did not observe evidence for significant FNP penetration into the spinal cord parenchyma from the CSF. FNPs were observed to traverse the length of the spinal cord within 2 hours of injection, including sacral regions. FNP signal encompassed the entire circumference of the spinal cord (Figs 2, 3 and SI Fig. 4). FNPs were reliably localized with exiting nerve roots (Figs 2 and 3C). We consistently observed a small population of FNPs within the central canal at all time points except at 2 hours, especially in cervical and thoracic spinal sections. Three weeks after injection, the FNPs were still readily detected along the entire length of the spinal column, although they appeared more punctate with slightly larger foci compared to earlier time points (Fig. 2). To test if the rapid distribution of the FNPs to the sacral spinal cord was size dependent, we examined the distribution of 10 µm non-PEGylated FluoSphere microparticles (MPs) within the spinal cord SAS. The MPs could be found in the sacral region within 2 hours, albeit very infrequently (3 total in 54 slices observed, SI Fig. 5).
Figure 2.

Representative confocal images of FNP distribution along the spinal cord at 2 hours and 3 weeks. FNPs (red) distributed to the sacral regions of the spinal column within 2 hours and remained for over 3 weeks. FNPs could be observed following the meninges around spinal nerve roots (2 hr arrow), which may represent a clearance route for FNPs from the CNS. Even at later time points, the FNPs were mainly confined to meninges with minimal evidence for parenchymal penetration. FNPs appeared more punctate at later time points and could frequently be found within the central canal (3 wk arrows). The location of the slice along the spinal column is shown for each slice in the lower right. Zoomed in image of only FNP signal within the dashed box shown to the right of each tissue slice. Equivalent linear adjustments were made to the FNP signal in each image to enable better visualization. Cell nuclei (DAPI) are shown in blue. Scale bar = 200 µm.
Figure 3.

FNPs achieve widespread delivery through the SAS within 2 hours of IT administration. (A) Sagittal view of the entire neuraxis showing distribution of FNPs (red) from the olfactory bulb to the sacral spinal cord. (B) FNPs were able to access nearly all CSF exposed tissue, including into the sulci of the cerebellum. Along the spinal column, FNPs distributed around the entire circumference of the spinal cord all the way from the cervical (C) to the sacral (D) regions. Scale bar in B = 500 µm and in C&D = 200 µm.
Quantification of FNP intensity with ImageJ revealed a gradual exponential decay in fluorescent signal moving caudal from the injection site towards the sacral spinal section (Fig. 4 and SI Fig. 6). The general trends observed from quantification of tissue slices were in agreement with the semi-quantitative measurements generated by IVIS imaging. For the first 48 hours and even at 1 week, the total FNP delivery (AUC) to the spinal column did not significantly change (Fig. 3B). We did observe a similar trend as IVIS with a decrease in AUC from 2 to 6 hours (3.4E9 to 2.4E9 AU), followed by a slight increase in delivery from 24 to 48 hours (2.7E9 to 3.0E9 AU). FNP concentration along the spinal cord continued to trend upwards from 48 hours to 1 week (3.0E9 to 3.8E9 AU) and was significantly higher at 3 weeks post-injection (5.1E9 AU) compared to 6, 24 and 48 hours post-injection (p < 0.05).
Figure 4.

Quantification of FNP intensity along the spinal column following IT injection. (A) Exponential curve fits of FNP intensity along the spinal column showed relatively constant decay in FNP signal moving caudally down the spinal cord at all time points. (B) AUC of the curve fits for individual mice at each time point (n = 3/time point) shows FNP delivery to the spinal cord trended upwards over time and was significantly higher at 3 weeks compared to 6, 24 and 48 hours (p < 0.05, 1-way ANOVA with Bonferroni’s Multiple Comparison Test). Mean ± SD shown.
Ex Vivo Brain Distribution
To visualize the anterior/posterior distribution of FNPs around the complex geometries of the brain, the brain was sliced in sagittal sections (Fig. 5 and SI Fig. 7). Consistent with our observations along the spinal cord, we did not find evidence of FNP penetration across the pia mater into the brain parenchyma. In general, FNPs distributed throughout the entire SAS to reach all CSF exposed surfaces of the brain. FNPs were observed to follow the meninges into the sulci of the cerebellum (Fig. 3B) and were also observed along the trigeminal nerve (Fig. 5). In all mice, we observed the greatest concentrations of FNPs within concave structures around the brain, particularly within the supracerebellar cistern between the cortex and cerebellum on the dorsal side of the brain and the pituitary recess anterior of the pons on the ventral side of the brain. FNP delivery was consistently more concentrated along ventral surfaces of the brain compared to dorsal surfaces, and FNP signal was especially sparse along the dorsal prefrontal cortex region at all time points examined. To test whether the ventral preference was an artifact of the mouse lying in the prone position after injection, we evaluated FNP distribution in 3 mice that were maintained in a supine position for 2 hours post-injection and sacrificed immediately thereafter. Recovery position did not produce any identifiable changes to the FNP distribution, and delivery to the prefrontal cortex region remained sparse (SI Fig. 8). The distribution of 10 µm MPs within the SAS of the brain was more limited than FNPs. MPs did not distribute into the sulci of the cerebellum and their delivery to ventral surfaces of the brain was even more limited than FNPs (Fig. 6).
Figure 5.

Representative confocal images of FNP distribution around the brain. FNPs (red) were distributed around the entire brain and could be seen following the meninges into the sulci of the cerebellum. There was a strong preference for ventral distribution and consistently high delivery to the supracerebellar cistern (C) and pituitary recess (P). Clearance of FNPs across the cribriform plate into the nasal mucosa (N) was seen at all time points. Qualitatively, a general decrease in total FNP intensity was observed over time, consistent with IVIS data. Linear corrections were applied equally across images to better show FNP distribution. Cell nuclei (DAPI) are in blue to show the gross anatomy of the brain. Images without DAPI, to better see FNP signal, can be found in the Supplementary data. Scale bar = 500 µm.
Figure 6.
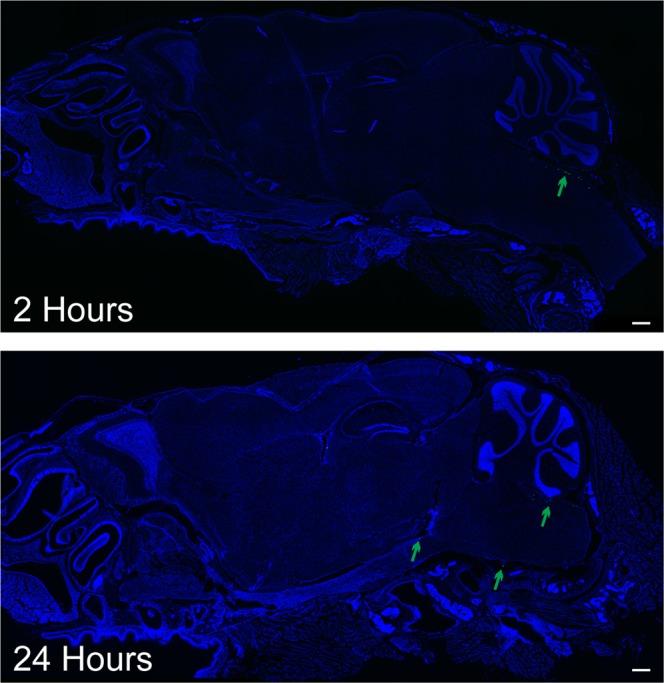
Representative confocal images of MP distribution around the brain. MPs (green) exhibited significantly less mobility around the brain compared to FNPs. Most MPs remained near the injection site, with a few moving along the ventral side of the brain. No MPs were found along the dorsal brain. Cell nuclei (DAPI) are in blue to show the gross anatomy of the brain. Scale bar = 500 µm.
CNS Clearance
We observed evidence for FNP transport across the cribriform plate into the nasal sinuses at all time points (Figs 5 and SI Fig. 6). To test whether IT administered FNPs were cleared into peripheral compartments, we examined the liver and spleen with confocal microscopy. There was no observable FNP signal above background in the liver at any of the time points (data not shown). FNPs were detectable within the spleen at all time points, with the greatest concentration of FNPs seen at 2 hours after injection (Fig. 7). Anatomically, FNPs appeared to be consolidated around the white pulps of the spleen.
Figure 7.
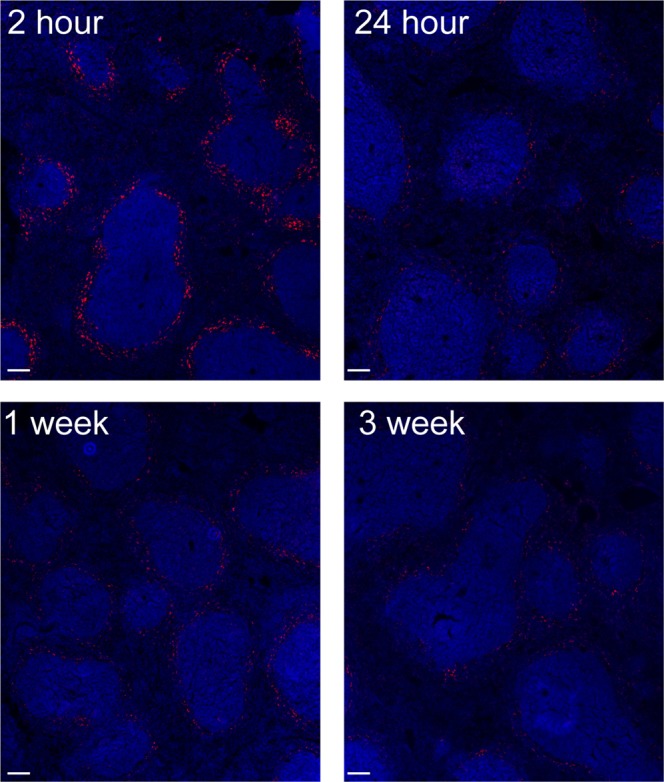
Representative confocal image of FNP distribution to the spleen over time. FNPs (red) appeared to be localized around the white pulp, with the greatest signal observed at 2 hours. Cell nuclei (DAPI) shown in blue. Scale bar = 100 µm.
Discussion
CSF is produced by the choroid plexus, after which it moves through the ventricles, where it distributes down the spinal cord and around the brain through the SAS27,28. The SAS exists between the arachnoid mater and pia mater around the brain and spinal cord. Thin, collagen trabeculae extend between the arachnoid mater and pia mater, generating a mesh-like network within the SAS through which the CSF moves. Although the overall magnitude of a bulk and net directional flow of CSF are debated, it is generally accepted that CSF moves in a oscillatory fashion due to the rhythmic forces generated by the cardiac and respiratory cycles29–32. Most therapies of interest for neurological disease achieve poor CSF distribution, and the use of IT administration in the clinic has been mainly limited to analgesics and a few water-soluble molecules. Hydrophilic molecules are known to distribute and clear rapidly as CSF turns over. SPECT studies by Wolf et al. suggest that the limited tissue distribution of hydrophobic molecules is due to binding of molecules to cellular and extracellular components of the leptomeninges and SAS, thus highlighting biophysical considerations that could limit the movement of IT administered molecules15. Limited distribution is advantageous for the translation of analgesics, where local delivery avoids possible detrimental respiratory side effects. However, limited distribution is not an advantage for lipophilic drugs that need to reach larger tissue volumes, such as chemotherapies.
Nanoparticles have been administered IT previously20,33–35. Dengler et al. found 230 nm, non-degradable, silica-lipid NPs resided within the meninges for at least 8 weeks after lumbar injection. Multiple reports have shown effective local nano/micro-particle mediated sustained delivery for the treatment of spinal cord injury36–38. Shyam et al. and Hagihara et al. both demonstrated the potential of NPs to transfect cells across multiple CNS regions after IT administration, and Kitamura et al. demonstrated that chemotherapy-loaded liposomes could prolong survival in a rat model of meningeal gliomatosis. While these studies establish the potential significance of the IT delivery route, little is known about the spatiotemporal distribution of NPs within the SAS. Thus, the objective of this study was to thoroughly characterize the distribution of solid NPs within the CNS following IT administration.
Injection of both 100 nm FNPs and 10 µm MPs into the cisterna magna was well tolerated in all mice. FNPs were observed to distribute through the meninges along the entire neuraxis, including into the sulci of the brain and along nerve bundles (trigeminal, optic and exiting spinal nerve roots). Distribution of FNPs to the sacral portion of the spinal column within 2 hours post administration is too rapid for Fickian diffusion alone, highlighting a key role for convective forces. Movement of the FNPs in all directions from the cisterna magna supports a model of CSF mixing as opposed to strictly directional laminar CSF flow29–31,39.
Recent studies highlight the significance of CSF mixing within the SAS of the spinal cord, with an expectation of decreased oscillation moving caudally along the spinal column32,40,41. In the current study, we observed that, while FNPs accessed distal regions of the spinal cord, a gradient of FNP concentration existed in the rostral-caudal direction. This gradient could represent an equilibrium established by decreasing oscillatory forces. The FNP signal became more punctate at 3 weeks, which could be indicative of FNP entrapment in microanatomy or aggregation over time. Based on prior works by Wolf et al. and Papisov et al., a moderate, exponential FNP gradient along the spinal column is consistent with the distribution of hydrophilic, low-binding small molecules injected intracisternally or intraventricularly14,15.
FNPs are clearly capable of redistributing in all directions away from the cisterna magna. The distribution of FNPs was symmetrical between the hemispheres, with both sagittal and coronal brain sections demonstrating that FNPs concentrate along the ventral surfaces of the brain. Lesser delivery of FNPs to the dorsal surface of the brain was observed here. This phenomenon does not appear to be dependent on the size of the infused agent, as it was similarly observed for both a small molecule (99mTc-DMSA, ~500 Da) and a protein (idursulfase, 76 kDa) following lumbar injection in healthy rats14,15, as well as for 10 µm MPs in this work. A relatively high FNP concentration was observed in the supracerebellar cistern at all time points, although a relatively small population of FNPs moved past the cistern towards the olfactory bulb. We saw a similarly pronounced concentration of FNPs within the pituitary recess on the ventral side of the brain, although significantly more FNPs moved rostrally beyond the recess. These data suggest there must be inherent differences in net movement/clearance of CSF or features of the trabecular mesh along the dorsal versus ventral surfaces and microanatomy of the brain. To our knowledge, this has not been directly observed in prior publications.
Although FNPs were capable of accessing all leptomeningeal surfaces, the concentration of FNPs was not uniform with respect to brain and spinal cord anatomy. This study does not assess whether the distinct patterns of delivery observed for FNPs could be generalized to other substances injected into the CSF, such as small molecules, proteins, or gene therapies. However, the relative scale of differences observed for FNP delivery were of a magnitude that would be expected to influence the activity of therapeutic molecules. These results highlight the need for additional work to further elucidate how microanatomy of the leptomeninges and regional CSF dynamics influence the distribution and activity of molecules delivered via the IT route.
CSF is continually produced by the choroid plexus and turns over multiple times a day (4×/day in human, 12×/day in mice)42,43. The half-life of IT administered molecules is typically on the order of a few hours17, and clearance remains a significant obstacle to effective drug delivery. CSF clearance is proposed to occur through multiple routes, including direct transfer to blood through arachnoid villi, movement across the cribriform plate into the nasal mucosa, and glymphatic clearance from perineural transport along both cranial and spinal nerve roots. The relative significance of each of these routes is often debated and varies by species31,42,44–47. Here, we found that nondegradable, FNPs remained distributed throughout the murine neuraxis at a relatively high concentration for over 3 weeks. Both IVIS and confocal data suggest the majority of FNP clearance occurs from the brain compartment, with minimal clearance along the spinal cord. In agreement with prior works in rodents, we saw FNP movement across the cribriform plate into the nasal mucosa at all time points. One possible explanation for the lack of FNPs along the dorsal brain could be clearance of FNP into venous circulation through arachnoid villi along the Superior Sagittal Sinus (SSS). However, recent works suggest direct CSF to blood clearance is minimal in mice45,46, and we failed to observe direct interactions of the FNPs within the SSS in coronal sections. Additionally, the lack of FNP signal within the liver suggests minimal to no clearance of FNP to the blood. Future studies using whole body imaging would be necessary to fully understand the contribution of each of these clearance routes in the context of NP distribution.
NP size is one of the most important biophysical parameters governing fate of NPs following their administration by various routes, including intravenous48,49, oral50,51, intranasal52,53, or convection enhanced delivery54,55. DepoCyt is a 10 µm multilamellar liposome that has been shown to reach lumbar spinal cord within a day following intraventricular administration in humans56. These data suggest that large particles are capable of navigating the SAS to reach distal regions of the CNS. However, because trabeculae form a porous mesh between the arachnoid mater and pia mater, we expected that the distribution of larger sized MPs would be restricted relative to smaller sized NPs, particularly within the smaller anatomy of the mouse57,58. In contrast to this expectation, we found MPs were capable of navigating the SAS to the sacral spinal cord within 2 hours of IT administration in mice, although their distribution was not as complete as smaller sized FNPs. Most distal slices contained no MPs and those that did never contained more than 1 MP/slice. Thus, while microparticle systems can help overcome the issue of rapid drug clearance through prolonged drug release, the minimal number of MPs in distal regions suggests that these systems must inevitably rely on movement of free drug from the particle through CSF to reach its target. Smaller NP systems, such as the FNPs used here, could overcome this delivery barrier to deposit drug more uniformly within the SAS. In addition, their small size means they can more easily interact with or be more easily taken up into cells, allowing for opportunities to increase delivery to specific cells or tissues through active targeting. The minimal penetration of 100 nm NPs across the pia mater or diffusion along perivascular routes in the parenchyma likely limits their use for applications requiring delivery to deep CNS regions. Although NPs as large as 100 nm can maneuver through brain perivascular spaces under convection enhanced delivery54, sub-50 nm and ideally >20 nm NPs are likely necessary to achieve significant penetration from the SAS into the brain59,60.
Ultimately, our data provide the first direct evidence that 100 nm NPs are capable of rapidly distributing through the SAS of the brain and spinal column following IT administration in healthy mice. The retention of 100 nm FNPs within the leptomeningeal space supports future work examining their potential for overcoming the pharmacokinetic limitations of therapies and imaging agents for treating and tracking diseases of the meninges, including meningitis, meningiomas and leptomeningeal metastasis from solid tumors61,62. Controlled release properties of nanocarriers could be utilized to provide prolonged drug exposure while reducing healthy tissue exposure to high peak drug concentrations. Although the non-biodegradable NPs used here were retained for over 3 weeks, many NPs used for drug delivery are degradable; thus, overall retention and efficacy of a nanomedicine could be dictated by both carrier degradation and drug release rates. IT administration has the further advantage of exposing nanoparticles more directly to target tissues and cells within the SAS, which could open new possibilities for targeted drug delivery and imaging. Taken in sum, these studies provide foundational characterization of the fate of nanoscale colloids following IT injection in healthy mice. This work opens new opportunities and potential challenges to consider for the delivery of drugs from NPs administered directly to the CSF to treat disease.
Materials and Methods
Materials
100 nm carboxylate-modified FluoSpheresTM (red fluorescent, Ex/Em 580/605 nm), 10 µm FluoSpheres (green fluorescent, Ex/Em 468/508 nm) and 20x Borate Buffer were purchased from ThermoFisher Scientific (Waltham, MA USA). Endotoxin-free water was obtained from G-Biosciences (St. Louis, MO, USA). Poly(ethylene glycol)-amine (mPEG-Amine, 2 kDa MW) was purchased from Creative PEGWorks (Chapel Hill, NC USA). Other chemicals or reagents were purchased from Sigma-Aldrich (St. Louis, MO USA) unless specified.
FluoSphere PEGylation and characterization
Carboxylate-modified FluoSphereTM NPs (FNP, 100 nm) were covalently modified with mPEG-Amine (2 kDa MW) by carbodiimide chemistry as previously described by Nance et al.54. FNPs were washed to remove sodium azide using 0.5 ml Amicon-Ultra centrifugation filters (10k MWCO). mPEG-amine (5x molar excess) was added and allowed to stir for 15 min. Next, 6.5 mg N-Hydroxysuccinimide (NHS) dissolved in 6 ml borate buffer (200 mM, pH8) was added, followed by 15.4 mg 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC). After 3 hours, the reaction was quenched with excess glycine (100 mM) for 30 min. Unreacted PEG, EDC, NHS and glycine were removed by dialysis (100k MWCO) for 24 hours. FNPs were washed and collected by centrifugation using Amicon-Ultra centrifugation filters (100k MWCO) and resuspended to 20 mg/ml in 1x PBS for storage at 4 °C.
FNP size and zeta potential was measured in 1 mM KCl before and after PEGylation using a NanoBrook 90Plus Zeta (Brookhaven Instruments, Holtsville, NY USA). Pegylated FS stability in 10% FBS at 37 °C was measure at 1, 2, 4, 12, 24 hours and 1 week. 1 mg of lyophilized sample was dissolved in 0.6 mL of chloroform-d (CDCl3). H-NMR spectra of the samples were obtained using a 300 MHz Bruker Avance NMR spectrometer at standard room temperature.
In Vivo studies
All experiments, procedures and animal care practices were approved by the Barrow Neurological Institute’s Institutional Animal Care and Use Committee and were performed in accordance with all relevant guidelines. Healthy, female C57 albino mice (Charles River) were used for in vivo experiments.
FNP Administration
FNPs were administered directly into the CSF of the cisterna magna through the atlanto-occipital membrane between the skull and C1 vertebrae. Mice were anesthetized with ketamine/xylazine (100/10 mg/kg) and mounted in a stereotaxic frame (Kopf Instruments, Tujunga, CA, USA). A Hamilton syringe (29 gauge needle, 30° beveled tip) was used to inject 2 µl FNPs over 1 min at a depth of 1 mm, and the needle was left in place for an additional 2 min before removing. All animals received a subcutaneous (SQ) injection of Buprenorphine SR (1 mg/kg) prior to surgery, and ibuprofen was provided in their water for 1 week to control pain.
In Vivo FNP Tracking
Macroscopic images of FNP distribution and semi-quantitative measurements of clearance kinetics (5 min, 2, 6, 24 and 48 hours after injection) were obtained using a Xenogen IVIS Spectrum In Vivo Imaging System (Ex 570/Em 620). Fur was removed from the neck and back prior to imaging. A line profile was drawn along the length of the neuraxis to quantify intensity. Background signal measured from a pre-injection image of each subject was subtracted from each time point. FNP intensity at each time point was normalized within each subject to the maximum measured intensity at 5 min.
Ex Vivo neuraxis collection
Mice were perfused transcardially 2, 6, 24 and 48 hours, or 1 and 3 weeks post FNP administration with PBS followed by 4% paraformaldehyde (PFA). The entire neuraxis from nasal sinuses to sacral spinal region was isolated intact, leaving the bone in place, and post-fixed in 4% PFA for an additional 48 hrs. The bone was decalcified over 5 days in a 4% HCl and 4% Formic Acid (v/v) solution. After cryoprotection in a 30% (w/v) sucrose solution for 48 hrs, the brain and spinal cord were cut at the C1 vertebrae to enable separate processing.
Brain and spinal column processing
OCT embedded spinal cord (coronal) and brain (sagittal) were sliced to a thickness of 20 µm and counterstained with Vectashield plus DAPI. Slides were imaged (FNPs: 560/605 nm, MPs: 468/508 nm) on an inverted confocal microscope. Laser intensities and gain were maintained across samples and time points. For quantification of the spinal cord distribution, an average background pixel intensity (non-injected controls) was subtracted from each image and the FNP intensity was measured by ImageJ (v1.47, NIH).
Statistical testing
All statistical tests were performed using GraphPad Prism 5. Comparisons of IVIS brain and spinal cord AUC over time were made with a 1-way ANOVA with Tukey’s Multiple comparison post hoc test. A 1-way ANOVA with Bonferroni multiple comparisons test was used to compare area under the curve (AUC) of spinal cord curve fits, expressed as arbitrary units (AU) * time (hrs). Statistical significance is reported for p < 0.05.
Supplementary information
Acknowledgements
We gratefully acknowledge funding from the Barrow Neurological Foundation, the University of Texas Houston Health Science Center, the National Institutes of Health (R01NS111292), the Ian’s Friends Foundation, and the Achievement Reward for College Scientists (ARCS) Foundation.
Author Contributions
K.T.H. wrote the main manuscript text. All authors reviewed the manuscript. K.T.H., D.I.S., R.J.W. and R.W.S. were all involved in the conception, design and interpretation of the experiments. K.T.H. and S.D. performed the experiments and analyzed results.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-49028-w.
References
- 1.Berg SL, Poplack DG. Treatment of Meningeal Malignancy. The Oncologist. 1996;1:56–61. [PubMed] [Google Scholar]
- 2.Bottros MM, Christo PJ. Current perspectives on intrathecal drug delivery. J. Pain Res. 2014;7:615–626. doi: 10.2147/JPR.S37591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandberg DI, et al. Methotrexate administration directly into the fourth ventricle in children with malignant fourth ventricular brain tumors: a pilot clinical trial. J. Neurooncol. 2015;125:133–141. doi: 10.1007/s11060-015-1878-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandberg DI, et al. Chemotherapy administration directly into the fourth ventricle in a nonhuman primate model. J. Neurosurg. Pediatr. 2012;9:530–541. doi: 10.3171/2012.1.PEDS11410. [DOI] [PubMed] [Google Scholar]
- 5.Sandberg DI, et al. Pharmacokinetic analysis of etoposide distribution after administration directly into the fourth ventricle in a piglet model. J. Neurooncol. 2010;97:25–32. doi: 10.1007/s11060-009-9998-x. [DOI] [PubMed] [Google Scholar]
- 6.Sandberg DI, et al. Safety and pharmacokinetic analysis of methotrexate administered directly into the fourth ventricle in a piglet model. J. Neurooncol. 2010;100:397–406. doi: 10.1007/s11060-010-0210-0. [DOI] [PubMed] [Google Scholar]
- 7.Beck M, et al. Autonomic dysfunction in ALS: A preliminary study on the effects of intrathecal BDNF. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2005;6:100–103. doi: 10.1080/14660820510028412. [DOI] [PubMed] [Google Scholar]
- 8.Ghersi-Egea JF, et al. Fate of cerebrospinal fluid-borne amyloid beta-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J. Neurochem. 1996;67:880–883. doi: 10.1046/j.1471-4159.1996.67020880.x. [DOI] [PubMed] [Google Scholar]
- 9.Nutt JG, et al. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60:69–73. doi: 10.1212/WNL.60.1.69. [DOI] [PubMed] [Google Scholar]
- 10.Kalra S, Genge A, Arnold DL. A prospective, randomized, placebo-controlled evaluation of corticoneuronal response to intrathecal BDNF therapy in ALS using magnetic resonance spectroscopy: feasibility and results. Amyotroph. Lateral Scler. Mot. Neuron Disord. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2003;4:22–26. doi: 10.1080/14660820310006689. [DOI] [PubMed] [Google Scholar]
- 11.Pardridge WM. Drug transport in brain via the cerebrospinal fluid. Fluids Barriers CNS. 2011;8:7. doi: 10.1186/2045-8118-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro WR, Schmid M, Glantz M, Miller JJ. A randomized phase III/IV study to determine benefit and safety of cytarabine liposome injection for treatment of neoplastic meningitis. J. Clin. Oncol. 2006;24:1528–1528. doi: 10.1200/JCO.2005.03.7689. [DOI] [Google Scholar]
- 13.Glantz MJ, et al. Randomized Trial of a Slow-Release Versus a Standard Formulation of Cytarabine for the Intrathecal Treatment of Lymphomatous Meningitis. J. Clin. Oncol. 1999;17:3110–3116. doi: 10.1200/JCO.1999.17.10.3110. [DOI] [PubMed] [Google Scholar]
- 14.Papisov MI, et al. Delivery of proteins to CNS as seen and measured by positron emission tomography. Drug Deliv. Transl. Res. 2012;2:201–209. doi: 10.1007/s13346-012-0073-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolf, D. A. et al. Dynamic dual-isotope molecular imaging elucidates principles for optimizing intrathecal drug delivery. JCI Insight1 (2016). [DOI] [PMC free article] [PubMed]
- 16.Kwong Y-L, Yeung DYM, Chan JCW. Intrathecal chemotherapy for hematologic malignancies: drugs and toxicities. Ann. Hematol. 2009;88:193–201. doi: 10.1007/s00277-008-0645-y. [DOI] [PubMed] [Google Scholar]
- 17.Zimm S, Collins JM, Miser J, Chatterji D, Poplack DG. Cytosine arabinoside cerebrospinal fluid kinetics. Clin. Pharmacol. Ther. 1984;35:826–830. doi: 10.1038/clpt.1984.120. [DOI] [PubMed] [Google Scholar]
- 18.Flack SH, Bernards CM. Cerebrospinal Fluid and Spinal Cord Distribution of Hyperbaric Bupivacaine and Baclofen during Slow Intrathecal Infusion in Pigs. J. Am. Soc. Anesthesiol. 2010;112:165–173. doi: 10.1097/ALN.0b013e3181c38da5. [DOI] [PubMed] [Google Scholar]
- 19.Ummenhofer WC, Arends RH, Shen DD, Bernards CM. Comparative Spinal Distribution and Clearance Kinetics of Intrathecally Administered Morphine, Fentanyl, Alfentanil, and Sufentanil. J. Am. Soc. Anesthesiol. 2000;92:739–753. doi: 10.1097/00000542-200003000-00018. [DOI] [PubMed] [Google Scholar]
- 20.Kitamura I, et al. Intrathecal chemotherapy with 1,3-bis(2-chloroethyl)-1-nitrosourea encapsulated into hybrid liposomes for meningeal gliomatosis: an experimental study. Cancer Res. 1996;56:3986–3992. [PubMed] [Google Scholar]
- 21.Bawa, R., Audette, G. F. & Rubinstein, I. Handbook of Clinical Nanomedicine: Nanoparticles, Imaging, Therapy, and Clinical Applications. (CRC Press, 2016).
- 22.Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016;1:10–29. doi: 10.1002/btm2.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swami, A. et al. Nanoparticles for Targeted and Temporally Controlled Drug Delivery. in Multifunctional Nanoparticles for Drug Delivery Applications (eds Svenson, S. & Prud’homme, R. K.) 9–29 (Springer US, 2012).
- 24.Tosi G, et al. Potential use of polymeric nanoparticles for drug delivery across the blood-brain barrier. Curr. Med. Chem. 2013;20:2212–2225. doi: 10.2174/0929867311320170006. [DOI] [PubMed] [Google Scholar]
- 25.Householder KT, et al. Intravenous delivery of camptothecin-loaded PLGA nanoparticles for the treatment of intracranial glioma. Int. J. Pharm. 2015;479:374–380. doi: 10.1016/j.ijpharm.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Householder KT, et al. pH driven precipitation of quisinostat onto PLA-PEG nanoparticles enables treatment of intracranial glioblastoma. Colloids Surf. B Biointerfaces. 2018;166:37–44. doi: 10.1016/j.colsurfb.2018.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakka L, Coll G, Chazal J. Anatomy and physiology of cerebrospinal fluid. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2011;128:309–316. doi: 10.1016/j.anorl.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Kelly EJ, Yamada S. Cerebrospinal Fluid Flow Studies and Recent Advancements. Semin. Ultrasound CT MRI. 2016;37:92–99. doi: 10.1053/j.sult.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Tangen KM, Hsu Y, Zhu DC, Linninger AA. CNS wide simulation of flow resistance and drug transport due to spinal microanatomy. J. Biomech. 2015;48:2144–2154. doi: 10.1016/j.jbiomech.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 30.Ridgway JP, Turnbull LW, Smith MA. Demonstration of pulsatile cerebrospinal-fluid flow using magnetic resonance phase imaging. Br. J. Radiol. 1987;60:423–427. doi: 10.1259/0007-1285-60-713-423. [DOI] [PubMed] [Google Scholar]
- 31.Brinker T, Stopa E, Morrison J, Klinge P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS. 2014;11:10. doi: 10.1186/2045-8118-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada S, et al. Visualization of Cerebrospinal Fluid Movement with Spin Labeling at MR Imaging: Preliminary Results in Normal and Pathophysiologic Conditions. Radiology. 2008;249:644–652. doi: 10.1148/radiol.2492071985. [DOI] [PubMed] [Google Scholar]
- 33.Dengler EC, et al. Mesoporous silica-supported lipid bilayers (protocells) for DNA cargo delivery to the spinal cord. J. Control. Release Off. J. Control. Release Soc. 2013;168:209–224. doi: 10.1016/j.jconrel.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagihara Y, Saitoh Y, Kaneda Y, Kohmura E, Yoshimine T. Widespread gene transfection into the central nervous system of primates. Gene Ther. 2000;7:759–763. doi: 10.1038/sj.gt.3301169. [DOI] [PubMed] [Google Scholar]
- 35.Shyam R, et al. Intraventricular Delivery of siRNA Nanoparticles to the Central Nervous System. Mol. Ther. — Nucleic Acids. 2015;4:e242. doi: 10.1038/mtna.2015.15. [DOI] [PubMed] [Google Scholar]
- 36.Cerqueira Susana R, et al. Microglia Response and In Vivo Therapeutic Potential of Methylprednisolone‐Loaded Dendrimer Nanoparticles in Spinal Cord Injury. Small. 2013;9:738–749. doi: 10.1002/smll.201201888. [DOI] [PubMed] [Google Scholar]
- 37.Donaghue IEH, Tator C, S. Shoichet M. Sustained delivery of bioactive neurotrophin-3 to the injured spinal cord. Biomater. Sci. 2015;3:65–72. doi: 10.1039/C4BM00311J. [DOI] [PubMed] [Google Scholar]
- 38.Tan J, et al. Changes in compressed neurons from dogs with acute and severe cauda equina constrictions following intrathecal injection of brain-derived neurotrophic factor-conjugated polymer nanoparticles. Neural Regen. Res. 2013;8:233–243. doi: 10.3969/j.issn.1673-5374.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papisov MI, Belov VV, Gannon KS. Physiology of the intrathecal bolus: the leptomeningeal route for macromolecule and particle delivery to CNS. Mol. Pharm. 2013;10:1522–1532. doi: 10.1021/mp300474m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta S, Soellinger M, Boesiger P, Poulikakos D, Kurtcuoglu V. Three-Dimensional Computational Modeling of Subject-Specific Cerebrospinal Fluid Flow in the Subarachnoid Space. J. Biomech. Eng. 2008;131:021010–021010. doi: 10.1115/1.3005171. [DOI] [PubMed] [Google Scholar]
- 41.Loth F, Yardimci MA, Alperin N. Hydrodynamic Modeling of Cerebrospinal Fluid Motion Within the Spinal Cavity. J. Biomech. Eng. 2000;123:71–79. doi: 10.1115/1.1336144. [DOI] [PubMed] [Google Scholar]
- 42.Simon MJ, Iliff JJ. Regulation of cerebrospinal fluid (CSF) flow in neurodegenerative, neurovascular and neuroinflammatory disease. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 2016;1862:442–451. doi: 10.1016/j.bbadis.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rudick RA, Zirretta DK, Herndon RM. Clearance of albumin from mouse subarachnoid space: a measure of CSF bulk flow. J. Neurosci. Methods. 1982;6:253–259. doi: 10.1016/0165-0270(82)90088-7. [DOI] [PubMed] [Google Scholar]
- 44.Spector R, Robert Snodgrass S, Johanson CE. A balanced view of the cerebrospinal fluid composition and functions: Focus on adult humans. Exp. Neurol. 2015;273:57–68. doi: 10.1016/j.expneurol.2015.07.027. [DOI] [PubMed] [Google Scholar]
- 45.Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat. Commun. 2017;8:1434. doi: 10.1038/s41467-017-01484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aspelund A, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015;212:991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. 2014;11:26. doi: 10.1186/2045-8118-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulkarni SA, Feng S-S. Effects of Particle Size and Surface Modification on Cellular Uptake and Biodistribution of Polymeric Nanoparticles for Drug Delivery. Pharm. Res. 2013;30:2512–2522. doi: 10.1007/s11095-012-0958-3. [DOI] [PubMed] [Google Scholar]
- 49.Yadav KS, Chuttani K, Mishra AK, Sawant KK. Effect of Size on the Biodistribution and Blood Clearance of Etoposide-Loaded PLGA Nanoparticles. PDA J. Pharm. Sci. Technol. 2011;65:131–139. [PubMed] [Google Scholar]
- 50.Date AA, Hanes J, Ensign LM. Nanoparticles for oral delivery: design, evaluation and state-of-the-art. J. Control. Release Off. J. Control. Release Soc. 2016;240:504–526. doi: 10.1016/j.jconrel.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ensign LM, Cone R, Hanes J. Oral Drug Delivery with Polymeric Nanoparticles: The Gastrointestinal Mucus Barriers. Adv. Drug Deliv. Rev. 2012;64:557–570. doi: 10.1016/j.addr.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mistry A, Stolnik S, Illum L. Nanoparticles for direct nose-to-brain delivery of drugs. Int. J. Pharm. 2009;379:146–157. doi: 10.1016/j.ijpharm.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 53.Mistry A, Stolnik S, Illum L. Nose-to-Brain Delivery: Investigation of the Transport of Nanoparticles with Different Surface Characteristics and Sizes in Excised Porcine Olfactory Epithelium. Mol. Pharm. 2015;12:2755–2766. doi: 10.1021/acs.molpharmaceut.5b00088. [DOI] [PubMed] [Google Scholar]
- 54.Nance EA, et al. A Dense Poly(ethylene glycol) Coating Improves Penetration of Large Polymeric Nanoparticles within Brain Tissue. Sci. Transl. Med. 2012;4:149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacKay JA, Deen DF, Szoka FC. Distribution in brain of liposomes after convection enhanced delivery; modulation by particle charge, particle diameter, and presence of steric coating. Brain Res. 2005;1035:139–153. doi: 10.1016/j.brainres.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 56.Kim S, et al. Extended CSF cytarabine exposure following intrathecal administration of DTC 101. J. Clin. Oncol. 1993;11:2186–2193. doi: 10.1200/JCO.1993.11.11.2186. [DOI] [PubMed] [Google Scholar]
- 57.Saboori P, Sadegh A. Histology and Morphology of the Brain Subarachnoid Trabeculae. Anat. Res. Int. 2015;2015:e279814. doi: 10.1155/2015/279814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lü J, Zhu XL. Characteristics of distribution and configuration of intracranial arachnoid membranes. Surg. Radiol. Anat. 2005;27:472. doi: 10.1007/s00276-005-0025-4. [DOI] [PubMed] [Google Scholar]
- 59.Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc. Natl. Acad. Sci. 2006;103:5567–5572. doi: 10.1073/pnas.0509425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lam MA, et al. The ultrastructure of spinal cord perivascular spaces: Implications for the circulation of cerebrospinal fluid. Sci. Rep. 2017;7:12924. doi: 10.1038/s41598-017-13455-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gwak H-S, et al. Recent Advancements of Treatment for Leptomeningeal Carcinomatosis. J. Korean Neurosurg. Soc. 2015;58:1–8. doi: 10.3340/jkns.2015.58.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bokstein F, Lossos A, Siegal T. Leptomeningeal metastases from solid tumors: a comparison of two prospective series treated with and without intra-cerebrospinal fluid chemotherapy. Cancer. 1998;82:1756–1763. doi: 10.1002/(SICI)1097-0142(19980501)82:9<1764::AID-CNCR24>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.