

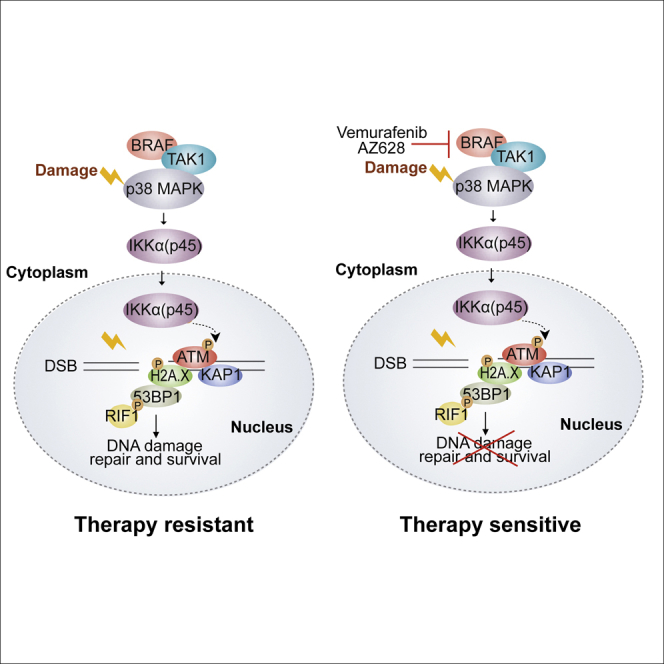
Summary
Phosphorylated IKKα(p45) is a nuclear active form of the IKKα kinase that is induced by the MAP kinases BRAF and TAK1 and promotes tumor growth independent of canonical NF-κB signaling. Insights into the sources of IKKα(p45) activation and its downstream substrates in the nucleus remain to be defined. Here, we discover that IKKα(p45) is rapidly activated by DNA damage independent of ATM-ATR, but dependent on BRAF-TAK1-p38-MAPK, and is required for robust ATM activation and efficient DNA repair. Abolishing BRAF or IKKα activity attenuates ATM, Chk1, MDC1, Kap1, and 53BP1 phosphorylation, compromises 53BP1 and RIF1 co-recruitment to sites of DNA lesions, and inhibits 53BP1-dependent fusion of dysfunctional telomeres. Furthermore, IKKα or BRAF inhibition synergistically enhances the therapeutic potential of 5-FU and irinotecan to eradicate chemotherapy-resistant metastatic human tumors in vivo. Our results implicate BRAF and IKKα kinases in the DDR and reveal a combination strategy for cancer treatment.
Keywords: IKK, BRAF, combination therapy, cancer treatment, ATM, phosphorylation, DNA-damage Repair, therapy resistance, patient-derived organoids, ortho-xenografts
Graphical Abstract
Highlights
-
•
IKKα kinase is activated by BRAF-TAK1-p38-MAPK in response to DNA damage
-
•
Loss of IKKα or BRAF attenuates ATM signaling and compromises DNA repair
-
•
Loss of IKKα or BRAF in combination with DNA damage potentiates tumor eradication
-
•
Combination treatment of patient-derived tumors prolongs survival in mice
Colomer et al. discover that IKKα kinase contributes to the chemo- and radio-resistance of cancer cells by facilitating ATM activation and DNA repair. BRAF inhibitors prevent damage-induced IKKα activation, leading to the attenuation of ATM signaling and DNA repair. IKKα depletion or BRAF inhibitors combined with 5-FU and irinotecan synergistically enhance the killing of patient-derived xenograft tumors.
Introduction
The DNA damage response (DDR) maintains genome stability by coordinating the cell cycle, DNA repair, and apoptosis in response to DNA lesions. Following DNA double-strand breaks (DSBs), the DDR is activated by ATM-dependent phosphorylation of numerous targets, including the effector kinase Chk2 and the histone H2A.X (called γH2A.X when phosphorylated). Dependent on cell-cycle stage, ATM coordinates the recruitment of 53BP1 or BRCA1 to damaged DNA, which determines DSB repair pathway choice and whether the lesion is repaired by non-homologous end joining (NHEJ) or homologous recombination (HR). In G1, 53BP1, RIF1, and the Sheldin co-factors oppose DSB resection, thereby favoring NHEJ and opposing HR (Panier and Boulton, 2014). Conversely, in S-G2, when an intact sister chromatid is available as a template following the completion of S phase, BRCA1 antagonizes 53BP1 and co-factors, thereby promoting a switch in DSB repair pathway to favor HR (Bothmer et al., 2010, Bothmer et al., 2011, Chapman et al., 2012a, Chapman et al., 2012b, Escribano-Díaz et al., 2013, Zimmermann et al., 2013). In non-proliferating cells (cells in G0-G1 phases), classical NHEJ is the preferred option for DSB repair since there is no homology donor for HR. From yeast to mammals, multiple genotoxic agents such as UV, ionizing radiation (IR), reactive oxygen species, and 5-fluorouracil (5-FU) also induce the protein kinase p38, which plays an essential role in the regulation of cellular checkpoints (Alao and Sunnerhagen, 2008, Bulavin et al., 2001, Preta et al., 2010, Rouse et al., 1994). Recently, it was demonstrated that p38α modulates the ATR pathway through direct phosphorylation of CtIP, which promotes therapy resistance in cancer cells (Canovas et al., 2018).
Contemporary treatments for most solid cancers involve surgery, radiotherapy, and combinations of chemotherapies, such as 5-FU, oxaliplatin, and irinotecan (Iri), which eradicate tumors by inducing DSBs in highly proliferative cells (reviewed in Brenner et al., 2014). However, it is well established that tumors also contain low proliferating and quiescent cells that are therapy resistant and contribute to tumor relapse and metastasis (Batlle and Clevers, 2017). Beyond non-specific DNA-damaging agents, antibodies targeting epidermal growth factor receptor (EGFR) constitute a second therapeutic option for cancer treatment, with mutations in KRAS and BRAF (two essential elements of the EGFR signaling pathway) that are predictive of treatment failure (Amado et al., 2008, Di Nicolantonio et al., 2008) and poor prognosis (Richman et al., 2009). Based on the structural characteristics of mutated RAS, the possibility of developing small molecules that revert its activation remains a significant challenge (Vetter and Wittinghofer, 2001). Thus, there is a growing interest in developing BRAF inhibitors and other inhibitors of the mitogen-activated protein kinase (MAPK) pathway that are being used for treating BRAF-mutated metastatic melanoma (Hu-Lieskovan et al., 2015, Ribas et al., 2014). However, recent BRAF inhibitor trials in cancer patients carrying BRAF-mutated tumors have produced largely negative results (Hong et al., 2016).
The nuclear factor κB (NF-κB) signaling pathway regulates innate and acquired immunity and is essential for most physiological processes but also for cancer progression (reviewed in Zhang et al., 2017). Multiple extracellular stimuli, including the inflammatory cytokines tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β), induce NF-κB through a series of signaling events that lead to the phosphorylation and activation of a kinase complex that consists of IKKα, IKKβ, and IKKγ/NEMO. The IKKα subunit is dispensable for NF-κB activation, but it has been found to exert multiple pro-tumorigenic functions. Several studies have demonstrated that IKKα enhances the metastatic activity of prostate tumors (Luo et al., 2007) and squamous cell carcinomas (Toll et al., 2015) by regulating the Maspin gene. Epithelial IKKα is required for the initiation and progression of intestinal adenomas (Colomer et al., 2018) and lung adenocarcinomas (Vreka et al., 2018) in mice. Recently, we identified a nuclear active form of the IKKα kinase, IKKα(p45), which is localized in the nuclear compartment of cancer cells. IKKα(p45) induces the phosphorylation of histone H3 and nuclear co-repressors, which is dependent on its interaction with non-activated full-length IKKα and promotes tumor growth independent of canonical NF-κB signaling (Margalef et al., 2012). The phosphorylation and activation of IKKα(p45) require the MAP kinases BRAF and TAK1 and take place in proximity to the endosomal compartment (Margalef et al., 2012, Margalef et al., 2015). The kinase p38α has also been linked to TAK1 (MAP3K7) and IKK activation in several situations, including the DDR (Chen et al., 2015, Hindi et al., 2018, Yang et al., 2011). In fact, exposure to chemotherapeutic agents leads to the activation of canonical NF-κB, downstream of ATM and NEMO (Wu et al., 2006).
Here, we make the unexpected discovery that the activity of BRAF and IKKα kinases is important for a proper DDR and for efficient DNA repair. In response to DNA damage, IKKα(p45) is rapidly activated by phosphorylation on Ser180, it translocates to the nucleus, and it co-localizes with 53BP1 at sites of damage. BRAF inhibition or loss of IKKα(p45) attenuates ATM activation and downstream checkpoint signaling, reduces the phosphorylation of key DDR factors, and compromises DNA repair, including 53BP1-dependent end joining. Finally, we show that BRAF inhibition or IKKα(p45) depletion synergizes with DNA-damaging chemotherapeutic agents to induce tumor eradication in mice, thus revealing a potential therapeutic strategy for cancer treatment.
Results
IKKα(p45) Is Rapidly Activated in Response to DNA Damage
IKKα(p45) is a largely uncharacterized kinase with no known substrates. In an attempt to identify processes that are regulated by IKKα, we sought to identify its kinase substrates through quantitative phospho-proteomic analysis of control and IKKα-knockdown HT29 cells. Unexpectedly, this approach identified several DDR pathway components, including 53BP1 and KAP1 (also known as TRIM28 or transcription intermediary factor 1β [TIF-1β]), as proteins whose phosphorylation depends on IKKα (see experimental strategy in Figure S1A and the results in Table S1). This observation raised the possibility that IKKα may function at some level in the DDR.
To examine this possibility, we exposed HT29 cells to UV light and performed western blot analysis at different time points. UV treatment rapidly induced the phosphorylation of nuclear IKKα(p45) on Ser180 (referred to as p-IKKα(p45)), which preceded the phosphorylation of canonical DDR proteins, including Chk1, KAP1, and H2A.X (Figure 1A). Different UV doses comparably increased p-IKKα(p45) levels in the nucleus (Figure S1B), and this occurred in a range of different cancer cells independent of the mutational status of KRAS or BRAF (Figures 1B and 1C). To determine whether the induction of nuclear p-IKKα(p45) extends beyond UV to other DNA-damaging agents, we also examined p-IKKα(p45) in response to IR and the topoisomerase II inhibitor, etoposide. p-IKKα(p45) was rapidly induced in response to all of the tested DNA-damaging agents, although the largest effect was produced by UV treatment (Figure 1D). Since p-IKKα(p45) is the nuclear form of activated IKKα, we asked whether it is also recruited to sites of DNA damage. Immunofluorescence analysis revealed that p-IKKα(p45) co-localized with 53BP1 in laser-induced stripes (Figure 1E). These results reveal that p-IKKα(p45) is rapidly induced by and accumulates at sites of DNA damage, which raised the possibility that p-IKK (p45) may contribute in some way to the regulation of the DDR pathway.
Figure 1.

Nuclear IKKα Is Activated by Different DNA-Damaging Agents
(A) Western blot (WB) analysis of HT29 exposed to UV light (130 mJ) and collected at the indicated time points.
(B) WB analysis of various CRC cells left untreated or collected 20 min after UV (130 mJ) exposure. K and B indicate the presence of mutations in KRAS (K) or BRAF (B) in the different cell lines.
(C) WB analysis of different colorectal (HT29), breast (MCF7), bladder (T24), melanoma (SKMEL 131), and pancreatic (RWP1) cancer cell lines collected 30 min after UV exposure (130 mJ).
(D) WB analysis of HT29 and WiDr cells treated with the indicated DNA-damaging agents and collected at different time points.
(E) Immunofluorescence analysis of HT29 cells to determine the co-localization of 53BP1 and p-IKK in laser-induced stripes.
p38-MAPK Activates IKKα in Response to DNA Damage
The activation of ATM or ATR is an early event in the cellular response to DNA damage, both kinases being essential for initiating the DDR signaling cascade in response to different DNA lesions. To determine whether ATM or ATR contributes to the induction of nuclear p-IKKα(p45) in response to DNA damage, we treated UV-irradiated cells with selective ATM or ATR inhibitors and blotted for IKKα(p45) Ser180 phosphorylation. While ATM and ATR inhibitors effectively abolished the phosphorylation of their downstream effector kinases Chk2 (pT68) and Chk1 (pS345; Figure 2A), this had no impact on the damage-induced Ser180 phosphorylation of IKKα(p45). We also considered the possibility that the DNA-dependent protein kinase, catalytic subunit (DNA-PKcs) may act redundantly to phosphorylate IKKα(p45) in response to damage. However, the combined inhibition of ATM and DNA-PKc also had no effect on the induction of p-IKKα(p45) following damage (Figure S2A). These data, coupled with the fact that p-IKKα(p45) induction precedes many other early damage markers, including γH2A.X and Chk1 (Figure 1A), suggested that IKKα(p45) phosphorylation either occurs upstream of or parallel to ATM-ATR activation.
Figure 2.

Activation of IKKα by DNA Damage Is Dependent on BRAF and p38α-MAPK Activity
(A) WB analysis of HT29 pretreated with ATM or ATR inhibitors and then exposed to UV (130 mJ).
(B) WB analysis of HT29 cells treated with the indicated inhibitors (p38i = SB203580, 10 μM; BRAFi = AZ628, 10 μM; TAKi = 5Z-7-oxozeaneol, 10 μM; MEKi = trametinib, 10 μM; bafilomycin, 10 nM) 16 h before UV exposure (130 mJ) and collected 30 min after treatment.
(C) WB analysis of p38α wild-type (WT) and knockout (KO) MEFs collected 30 min after UV exposure (130 mJ).
(D) WB analysis of WT MEFs treated with p38i for 16 h and then exposed to UV, as indicated.
(E) WB of different CRC cell lines treated with the BRAF inhibitor AZ628 (16 h, 10 μM) before UV light (130 mJ) exposure and collected 30 min after treatment.
(F) WB of cytoplasmic (C), nuclear (N), and chromatin (Chr) extracts from HT29 cells collected at different time points after UV treatment.
See also Figure S2.
Since IKKα is known to be activated by BRAF, TAK1, and p38-MAPK, we explored whether these kinases may also promote p-IKKα(p45) activation following UV treatment. Selective inhibitors of TAK1, BRAF, or p38-MAPK (but not MEKi) effectively abolished IKKα(p45) Ser180 phosphorylation in response to UV (Figure 2B). Of these kinases, only p38-MAPK and IKKα(p45) were observed to be significantly induced in response to UV treatment (Figure 2B). Nevertheless, TAK1 and BRAF inhibitors prevented the activation of p38-MAPK in response to UV, indicating that the basal activity of TAK1 and BRAF are required to prime p38-MAPK for subsequent damage-induced activation (Figure 2B).
To add further support to these findings and to exclude potential off-target effects of the p38-MAPK inhibitors, we examined IKKα(p45) phosphorylation in UV-treated p38α-MAPK knockout cells (Adams et al., 2000). In agreement with our inhibitor experiments, p38α-MAPK knockout cells failed to induce IKKα(p45) phosphorylation in response to UV (Figure 2C). p38α-MAPK inhibition also prevented IKKα(p45) Ser180 phosphorylation at multiple time points following UV treatment (Figure 2D). Moreover, BRAF inhibition was found to abrogate p-IKKα(p45) induction independent of BRAF mutational status (Figure 2E), irrespective of DNA-damaging stimulus (Figure S2B), and at a range of different time points (Figure S2C). Comparable effects on p-IKKα(p45) induction were also observed using two different BRAF inhibitors, vemurafenib and sorafenib (Figures S2D and S2E), which are used in clinical practice. These results establish that IKKα(p45) is rapidly activated in response to DNA-damaging agents downstream of TAK1, BRAF, and p38-MAPK. Notably, we did not detect evidence of nuclear TAK1, BRAF, or p38-MAPK following UV treatment (Figure 2F), suggesting that IKKα is principally activated in the cytoplasm, translocates to the nucleus, and accumulates on chromatin.
IKKα and BRAF Facilitate ATM Activation and Downstream DDR Signaling
In light of our findings that IKKα(p45) is rapidly induced by DNA damage, we consider the possibility that p-IKKα(p45) may contribute to the DDR. To investigate this possibility further, we conducted a phospho-proteomic analysis of control and IKKα-knockdown HT29 cells either untreated, as before, or subject to UV irradiation for 30 min. We observed that the UV-induced phosphorylation of several DDR components, including 53BP1, MDC1, and KAP1, were compromised in cells lacking IKKα (Figure 3A; Table S2). Western blot analysis of IKKα-knockdown cells exposed to UV at different time points further confirmed that Chk1 phosphorylation on Ser345 and γH2A.X induction is significantly attenuated in IKKα-depleted cells (Figure S3A). In contrast, knocking down IKKβ or NEMO had no measurable effect on Chk1, Ser345, and histone H2A.X phosphorylation upon UV treatment (Figure S3B).
Figure 3.

IKKα Downstream of BRAF Is Required for the Phosphorylation of Specific DDR Elements after DNA Damage
(A) Network of selected proteins related to DNA damage with phosphosites increasing upon UV irradiation in control (q < 0.15, positive log2 [fold-change]) but not in IKKα-knockdown cells (delta log2 [fold-change] > 0.5). Color fills represent the differential fold-change upon UV treatment between control and IKKα-knockdown cells, and the width of the phosphosite borders represents the significance of the change in control cells upon UV irradiation (bold: q < 0.05, medium: 0.05 < q < 0.1, light: 0.1 < q < 0.15).
(B) WB analysis of IKKα WT (+/+) and IKKα KO (−/−) cells treated with UV light (130 mJ).
(C) WB analysis of UV-treated IKKα KO cells (−) and the same cells transduced with Cherry-IKKα expression vector (+).
(D) WB analysis of HT29 pretreated with the BRAF inhibitor AZ638 (16 h, 10 μM) and then exposed to UV and collected at the indicated time points.
(E) Immunoprecipitation assay with anti-IKKα(p45) antibody from HT29 cells treated as indicated.
(F and G) In vitro kinase assay using glutathione S-transferase (GST) or GST-ATM (amino acids [aa] 1,854–2,063) as substrate and recombinant active IKKα (F) or lysates from IKKα WT (+/+) and IKKα KO (−/−) cells untreated or treated with UV for 15 min (G).
(H) Mass spectrometry analysis of the GST-ATM peptide phosphorylated in vitro with recombinant IKKα.
Since H2A.X, MDC1, 53BP1, and KAP1 are known substrates of ATM, we considered the possibility that IKKα(p54) may facilitate ATM activation after UV exposure. Auto-phosphorylation of ATM at Ser1981, which is an established marker of ATM activation, was significantly reduced in IKKα knockout cells exposed to UV (Figure 3B). IKKα knockout cells were also compromised for ATM activation following IR (Figure S3C) or doxorubicin treatment (Figure S3D) and at multiple time points tested. Impaired ATM activation in IKKα knockout cells was also associated with attenuated p-Chk2 (T68), p-KAP1 (S824), and γH2A.X levels. Impaired ATM activation following UV exposure was rescued by lentiviral transduction of mCherry-IKKα in the IKKα-deficient cells (Figure 3C).
Since BRAF is required for IKKα(p45) activation following DNA damage, we assessed the impact of BRAF inhibitors on ATM activation. In agreement with our previous data, inhibition of BRAF significantly reduced ATM activation, as measured by the induction of ATM Ser1981 auto-phosphorylation, and also compromised downstream phosphorylation of DDR markers in UV- (Figure 3D) and IR-treated (Figure S3E) HT29 cells. Similar results were also observed in breast, pancreatic, and melanoma cancer cell lines (Figure S3F).
Since the MRE11, RAD50, and NBS1 (MRN) complex is responsible for the recruitment of ATM to sites of DNA damage, we investigated whether IKKα deficiency or BRAF inhibition may affect MRN complex stability. Western blot analysis revealed that the protein levels of the MRN complex were unaffected both in IKKα-deficient cells (Figure S3G) and after BRAF inhibition (Figure S3H). We also investigated the impact of IKKα knockdown in Rad50s/s mutant cells, which exhibit a hypermorphic signaling phenotype that suppresses ATM deficiency by activating other phosphatidylinositol 3-kinase (PI3K)-like kinases (Morales et al., 2005). In contrast to wild-type cells, knocking down IKKα in Rad50s/s mutant cells did not prevent KAP1 and CHK1 phosphorylation after UV exposure, but instead resulted in the robust activation of these DDR markers (Figure S3I). These data suggest that similar to ATM deficiency, the Rad50s/s allele is capable of suppressing the signaling defects caused by IKKα knockdown, potentially through the activation of other PI3K-like kinases.
IKKα Interacts with and Directly Phosphorylates ATM in Response to DNA Damage
Next, we examined whether IKKα and ATM interact upon DNA damage. Immunoprecipitation experiments with an anti-IKKα(p45) antibody detected an interaction between endogenous ATM and both full-length and IKKα(p45), 15 min after UV treatment (Figure 3E). This observation raised the possibility that IKKα(p45) may directly phosphorylate ATM in response to DNA damage, potentially contributing to ATM activation. Consistent with this possibility, kinase assays revealed that recombinant IKKα is able to phosphorylate a fragment of ATM comprising amino acids 1,911–2,063 in vitro (Figure 3F). The same fragment was also phosphorylated in lysates from IKKα wild-type (WT) but not from IKKα-deficient cells (Figure 3G). Mass spectrometry analysis of the in vitro phosphorylated ATM fragment showed that S1974, S1987, S2058, and T2059 are direct substrates of the IKKα kinase (Figure 3H). It should be noted that once activated, ATM auto-phosphorylates itself on Ser367, Ser1893, and Ser1981, with the latter commonly used as a marker of ATM activation in vivo. However, these auto-phosphorylation sites are dispensable for ATM activation following DNA damage based on the fact that knockin mice mutated for these three sites exhibit normal downstream DDR signaling (Daniel et al., 2008). Since the loss of IKKα impairs robust ATM activation, our data raise the possibility that IKKα(p45) phosphorylation of these distinct sites in ATM may facilitate its activation.
IKKα and BRAF Contributes to DNA Damage Resolution
Since ATM activation is important for coordinating DNA repair, we next tested the possible impact of BRAF and IKKα inhibition in promoting lesion resolution. Inhibiting BRAF did not impair the formation of γH2A.X/53BP1 foci at sites of IR-induced DNA damage, but instead imposed a significant delay in lesion resolution (Figure 4A), consistent with defective or attenuated DNA repair. Phosphorylation of 53BP1 by ATM is required to recruit Rap1 interacting factor 1 (RIF1) to sites of DNA damage to counteract DSB end resection to promote NHEJ in G1 (Chapman et al., 2012a). In agreement with our previous observations, co-recruitment of 53BP1 and its cofactor RIF1 was attenuated following BRAF inhibition (Figure 4B) or in IKKα knockout mouse embryonic fibroblasts (MEFs) (Figure 4C). IKKα-deficient cells or cells subject to BRAF inhibition also exhibited a significant increase in the levels of DNA breaks in UV-treated MEFs as determined by comet assay (Figures 4D and S4A). These results suggest that the loss or inhibition of IKKα or BRAF compromises ATM activation and hinders downstream DNA repair.
Figure 4.

IKKα Downstream of BRAF Functionally Contributes to DNA Damage Resolution
(A) Immunofluorescence analysis of 53BP1 and γH2A.X in control or BRAF inhibitor-treated MEFs exposed to IR (2 Gy) and quantification from 3 independent experiments performed.
(B and C) Double immunofluorescence analysis of 53BP1 and RIF1 and quantification of the number of co-localizing foci per nucleus in BRAF-treated (2 h, 10 μM) (B) and IKKα wild-type (+/+) or KO (−/−) MEFs (C).
(D) Comet assay of IKKα wild-type (+/+) or KO (−/−) MEFs 8 and 24 h after DNA damage exposure (UV, 130 mJ).
(E and F) Representative images of FISH analysis from control and BRAF-inhibited Trf2-deficient MEFs using a telomeric probe (green) (E) or IKKα knockdown (small interfering IKKα [siIKKα]) cells (F). Quantification of the relative number of metaphases containing fused chromosomes from three independent experiments performed is shown at right.
See also Figure S4.
To further examine the contribution of IKKα and BRAF to 53BP1-dependent DNA repair, we used conditional Trf2FL/FL MEFs; inactivation of the shelterin subunit telomeric repeat-binding factor 2 (TRF2) leads to telomere deprotection and chromosome end-to-end fusions mediated by 53BP1-dependent NHEJ (Chapman et al., 2013, Celli and de Lange, 2005). Following TRF2 inactivation by adenoviral CRE transduction, MEFs were left untreated or treated with BRAF or MEK inhibitors (which do not prevent damage-induced IKKα(p45) phosphorylation; see Figure 2D) for 72 h and then the cells were processed for metaphase visualization. Using telomere-specific fluorescence in situ hybridization (FISH), cells were scored for the presence of chromosome end-to-end fusions. Consistent with our previous findings, IKKα knockdown (Figure 4E) or treatment with BRAF inhibitor (Figure 4F) significantly reduced chromosome fusions after Trf2 deletion. In contrast, MEK inhibition did not affect chromosome fusions in Trf2 null MEFs (Figure S4B), even though this had a comparable effect on cell-cycle position when compared to the BRAF inhibitor (Figure S4C) and IKKα knockdown (Figure S4D). Consistent with impaired ATM function, BRAF inhibition precluded RIF1 recruitment to the telomeres after Trf2 deletion (Figure S4E). These results further suggest that BRAF and IKKα affect 53BP1 and RIF1 function, thereby allowing the effective resolution of DNA damage by NHEJ in G1.
BRAF and IKKα Inhibition Synergize with DNA Damage-Based Therapy in Patient-Derived Tumoroids
Current protocols for cancer treatment rely to a large extent on DNA-damaging agents that selectively kill highly proliferative tumor cells but impose a lesser effect on low proliferating cancer cells. Furthermore, PI3K-like kinase inhibitors, including antagonists of ATM, ATR, and DNA-PKcs, are in clinical development to exploit their impact on the DDR. In light of our findings that BRAF and IKKα are required for efficient ATM signaling and DNA repair, we hypothesized that the inhibition of these kinases may synergize with DNA-damaging drugs to promote tumor cell killing. To test this possibility, we treated patient-derived colorectal cancer (CRC) tumoroids (Figure 5A), which have been recently validated as models for therapy prescription (Sato et al., 2011, Vlachogiannis et al., 2018), with suboptimal doses of 5-FU+irinotecan (Iri), alone or in combination with the BRAF inhibitors vemurafenib or AZ628. Consistent with our cellular studies, BRAF inhibition significantly reduced ATM activation and Chk1 phosphorylation in response to damage (Figure 5B), with the combination treatment imposing a striking synergistic effect on the eradication of tumoroids compared with single treatments alone (Figure 5C). Comparable synergistic effects with BRAF inhibitors were also observed when treating tumoroids with increasing doses of 5-FU+Iri (Figures 5D and 5E) or γ-irradiation (Figure S5A), or using the DNA-damaging agent doxorubicin (Figure S5B), which is commonly used in breast cancer treatment but is primarily ineffective against CRC tumors as a single agent.
Figure 5.

BRAF Inhibition Enhances the Effect of DNA Damage-Based Therapy in CRC Patient-Derived Tumoroids
(A) Experimental design used for the expansion of primary CRC tumors, generation of patient-derived tumoroids, and drug testing.
(B) Western blot analysis of control and BRAF-inhibited IMIM-TD#5, collected at the indicated time points after IR treatment.
(C) Quantification of tumoroid viability after treatment with the indicated compounds as a single treatment or in combination.
(D and E) Dose-response curves of two different tumoroids, IMIM-TD#6 (D) and IMIM-TD#5 (E), treated as indicated.
(F and G) Representative images of γH2A.X and cleaved caspase 3 staining in a representative tumoroid treated as indicated (F) and quantification of the percentage of positive cells from 20 tumoroids per condition counted (G).
(H) Comet assay of IMIM-TD#9, treated as indicated.
(I and J) Western blot analysis of control and IKKα-depleted IMIM-TD#9 using CRISPR-Cas9 technology (I) and quantification of cell viability after 72 h of culture with the indicated treatments (J).
The statistical analysis in (C) and (I) was performed by unpaired t test, comparing the combination treatments with single 5FU+irinotecan (red) or BRAF/IKK inhibitors (green); ∗∗∗∗p < 0.0001, n.s., non-significant. For the statistical analysis in (G), we used two-way ANOVA, and the p values are indicated as ∗∗p < 0.01 and ∗∗∗∗p < 0.0001. Vem, vemurafenib; 5FU, 5-fluorouracil; Dox, doxorubicin; and Iri, irinotecan.
See also Figure S5.
The loss of CRC tumoroid viability in the combination treatments was also associated with the massive accumulation of DNA damage and induction of apoptosis as determined by γH2A.X and cleaved caspase 3 levels, respectively (Figures 5F, 5G, S5C, and S5D). The enhanced accumulation of DNA breaks in tumoroids subject to the combination treatment was further confirmed by comet assays (Figures 5H and S5E). Most important, tumoroids subjected to the partial depletion of IKKα by CRISPR-Cas9 and treated with 5-FU+Iri displayed reduced ATM activation (Figure 5I) with no significant cell-cycle alterations (Figure S5F), and were not further sensitized by BRAF inhibitors (Figure 5J). IKKα-depleted tumoroids do not show any reduction in IKKβ levels or activity (Figures 5I and S5G). These data suggest that BRAF and IKKα inhibition could be exploited to improve tumor killing in combination with DNA-damaging chemotherapy or radiotherapy.
BRAF Inhibition Synergizes with DNA Damage-Based Therapy in a Patient-Derived Xenograft Model In Vivo
To further explore the therapeutic potential of drug combinations involving BRAF inhibitors and DNA-damaging agents, we used two independent patient-derived metastatic tumors (IMIM-X#1 and IMIM-X#3) that possess acquired resistance to regimes of DNA damage-based and EGFR antibody-based therapies (Montagut et al., 2012). Equivalent volumes of tumor cells were implanted in the cecum of nude mice, and tumor growth was monitored by palpation. At the time of tumor detection, animals were randomly ascribed to the treatment groups: control, 5-FU+Iri, vemurafenib, or 5-FU+Iri plus vemurafenib. The combination of 5-FU+Iri plus vemurafenib significantly reduced tumor growth in vivo compared with single-agent treatments (Figures 6A and 6B), and this was associated with the presence of extensive areas of necrosis and fibrosis (Figure 6C). Morphologically, residual neoplastic cells displayed a severe pleomorphism after combination treatment, but not with other conditions tested (Figure 6C), and was associated with the accumulation of DNA damage in the epithelial tumor component (Figures 6D and 6E). Notably, the combination treatment did not lead to measurable toxicity, as indicated by the overall aspect of the animals and the absence of anomalous cleaved caspase 3 staining in the colonic tissue adjacent to the implanted tumors (Figure S6A). The combination treatment may also affect the invasive nature of these tumors, as we detected only 1 animal with peritoneal implants from 5 animals in this group compared with the controls (5/5 animals with implants) and the single treated animals (5/6 and 3/6 for vemurafenib and 5-FU+Iri, respectively) (Figure S6B).
Figure 6.

BRAF Inhibition Enhances the Effect of DNA Damage-Based Therapy in Orthotopic Xenograft Model from Metastatic CRC
(A) Photograph of the tumors recovered at the end of the experiment with the indicated treatments.
(B) Quantification of the weight of the tumors in the different groups of treatment from two independent experiments performed using IMIM-X#1 and IMIM-X#3. Two-way ANOVA was used for statistical analysis, and the p values are indicated as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001.
(C) H&E staining of representative tumors treated as indicated. Note the various scales shown for the different treated tumors.
(D and E) Immunohistochemistry analysis of γH2A.X in IMIM-X#1 tumors obtained at the time of sacrifice (D) and quantification of the percentage of γH2A.X+ tumor cells (E) from a minimum of 10 fields (20×) counted per group of treatment. Two-way ANOVA was used for statistical analysis, and the p values are indicated as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001.
(F and G) Kaplan-Meier curves for mice bearing human tumor IMIM-X#1, treated as indicated. Overall (F) and cancer-related (G) survival analysis for the indicated treatments. Statistical significance was determined using the Mantel-Cox log-rank test. Vem, vemurafenib; 5FU, 5-fluorouracil; Dox, doxorubicin; and Iri, irinotecan.
See also Figure S6.
Finally, we examined the long-term therapeutic potential of the vemurafenib and 5-FU+Iri combination treatment of IMIM-X#1 tumors in vivo. After a 3-month follow-up, we observed a significant impact on the survival of animals treated with 5-FU+Iri in combination with vemurafenib when compared with vehicle-treated, vemurafenib-only, and 5-FU+Iri-treated animals. All of the mice treated with 5-FU+Iri or vemurafenib only died during the course of the experiment due to IMIM-X#1-derived tumor growth. In contrast, six of seven animals treated with 5-FU+Iri plus vemurafenib survived for the duration of the study and were tumor-free at the end of the experiment. There was no evidence of tumor growth in the only mouse that succumbed in this treatment group, which appeared to have died from other causes. The resulting Kaplan-Meier curves (Figures 6F and 6G) demonstrate the potent effect of combining BRAF inhibitors with DNA-damaging agents in promoting tumor eradication and long-term survival in vivo.
Discussion
IKKα was previously found to influence tumor initiation and cancer progression through NF-κB-dependent and-independent mechanisms. However, insights into the substrates of IKKα and the pathways that IKKα regulates were unknown. Using an unbiased proteomic analysis to identify IKKα substrates, we have uncovered an unappreciated role for IKKα and its activating kinase BRAF in the DDR pathway. Loss or inhibition of these kinases compromised ATM activation and downstream DDR signaling, impaired DNA repair by 53BBP1-dependent end joining, and synergized with commonly used chemotherapies to induce tumor eradication in vivo. Since these tumors are refractory to killing by single-agent treatments, our proposed combination strategy may have important implications for cancer treatment.
A role for IKKα in the DDR is supported by our findings that IKKα is rapidly activated by phosphorylation on Ser180 in response to a range of DNA-damaging agents and proceeds to accumulate on chromatin at sites of DNA damage. Damage-induced IKKα activation was found to occur independently of the canonical damage-responsive kinases ATM and ATR, but is instead dependent on BRAF, TAK1, and p38-MAPK kinases. We also showed that IKKα interacts with ATM shortly after the introduction of DNA damage, which raised the possibility that IKKα may directly regulate ATM function. Recombinant IKKα was found to directly phosphorylate ATM on four sites distinct from the known ATM auto-phosphorylation sites, including the commonly used activation maker pSer1981. IKKα-deficient cell extracts were also compromised for ATM phosphorylation in vitro, and the loss of IKKα or the inhibition of BRAF was found to attenuate ATM activation both in cells and in tumoroids. In agreement with the observed impairment in ATM activation, IKKα-deficient cells or inhibition of BRAF also resulted in the diminished phosphorylation of Chk1, Chk2, γH2A.X, MDC1, 53BP1, and Kap1 following the induction of DNA damage. We propose that IKKα affects the DDR by facilitating ATM activation. Since ATM auto-phosphorylation sites are dispensable for its activation (Daniel et al., 2008), future studies should assess the importance of the ATM residues phosphorylated by IKKα.
DDR signaling also plays an important role in coordinating the repair of DNA lesions. The loss or inhibition of IKKα or BRAF was found to delay the resolution of DNA damage, which manifests as the persistence of H2A.X and 53BP1 foci at late time points following the induction of damage. The inhibition of BRAF or loss of IKKα also resulted in persistent tail moments after damage in comet assays. Attenuated damage signaling in these cells is the likely cause of the DNA repair defects, as IKKα-deficient cells exhibit impaired recruitment of RIF1 to the sites of DNA damage, which is dependent on the phosphorylation of 53BP1 by ATM (Chapman et al., 2013). A role for BRAF and IKKα in promoting efficient ATM activation and 53BP1-dependent end joining was further supported by experiments in conditional Trf2 knockout MEFs. Conditional inactivation of the Shelterin subunit TRF2 results in telomere deprotection, which normally leads to “spaghetti chromosomes,” formed as a consequence of the fusion of dysfunctional telomeres by 53BP1-dependent end joining (Chapman et al., 2013). We found that treating Trf2-deficient MEFs with BRAF inhibitors (but not MEKi) or depletion of IKKα reduced telomere-telomere fusions. We attribute this effect to the reduction in the co-recruitment of 53BP1 and RIF1 at dysfunctional telomeres. These results highlight the importance of BRAF and IKKα signaling for efficient ATM signaling and subsequent repair following DNA damage.
Finally, we present evidence that clinically approved BRAF inhibitors could be repurposed for use in combination with DNA-damaging chemotherapies to target relevant cancers. This conclusion is based on our findings that the BRAF inhibitors vemurafenib or AZ628, when combined with 5-FU+Iri, effectively eradicate two different metastatic tumors that had acquired therapeutic resistance in human patients, although we cannot directly ascribe these results to the effect of BRAF inhibition on IKKα. Until now, the clinical use of BRAF inhibitors has focused on BRAF mutant (primarily BRAFV600E) tumors. Our findings demonstrating that BRAF inhibition synergizes with DNA-damaging drugs independent of BRAF mutation status therefore expands the potential target population that would benefit from this therapeutic approach. In addition, we have shown that compounds that are primarily ineffective in CRC but are used in other types of cancer (e.g., doxorubicin) should be reconsidered in combination treatments with BRAF inhibitors. Our results will prompt further preclinical and clinical studies to understand the broader utility of BRAF inhibitors beyond their current applications.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p-IKK s180 | Santa Cruz | sc-23470; RRID: AB_2122159 |
| IKKα | Merck Millipore | OP133; RRID: AB_10681621 |
| IKKα | Abcam | ab32041; RRID: AB_733070 |
| IKKα(p45) | Merck Millipore | MABF222; RRID: not available |
| p-Chk1 S345 | Cell Signaling | #2348; RRID: AB_331212 |
| Chk1 | Cell Signaling | #2360; RRID: AB_2080320 |
| p-KAP1 S824 | Bethyl Laboratories | A300-767A; RRID: AB_669740 |
| KAP1 | Cell Signaling | #4123; RRID: AB_2256670 |
| ɣH2A.X | Cell Signaling | #2577; RRID: AB_2118010 |
| ɣH2A.X | Merck Millipore | 05-636; RRID: AB_309864 |
| p-ERK 1/2 | Cell Signaling | #4370; RRID: AB_2315112 |
| ERK 1/2 | Cell Signaling | #4696; RRID: AB_390780 |
| Histone H3 | Abcam | ab1791; RRID: AB_302613 |
| Lamin B | Santa Cruz | sc-6216; RRID: AB_648156 |
| α-Tubulin | Sigma | T6074; RRID: AB_477582 |
| IKKβ | Abcam | ab32135; RRID: AB_733071 |
| NEMO | Santa Cruz | sc-8330; RRID: AB_2124846 |
| Cleaved caspase 3 | Cell Signaling | #9661; RRID: AB_2341188 |
| p-ATM S1981 | Merck Millipore | 05-740; RRID: AB_309954 |
| p-53BP1 S1618 | Cell Signaling | #6209; RRID: AB_11220229 |
| 53BP1 | Abcam | ab21083; RRID: AB_722496 |
| Anti-Rabbit-HRP (2ary) | DAKO | P0448; RRID: AB_2617138 |
| Anti-Mouse-HRP (2ary) | DAKO | P0260; RRID: AB_2636929 |
| Anti-Goat-HRP (2ary) | DAKO | P0449; RRID: AB_2617143 |
| 53BP1 | Bethyl Laboratories | A300-272A; RRID: AB_185520 |
| RIF1 | Santa Cruz | sc-65191; RRID: AB_2126820 |
| Alexa Fluor 488 donkey anti-rabbit (2ary) | Invitrogen | A21206; RRID: AB_141708 |
| Alexa Fluor 488 donkey anti-mouse (2ary) | Invitrogen | A21202; RRID: AB_141607 |
| Alexa Fluor 546 donkey anti-rabbit (2ary) | Invitrogen | A10040; RRID: AB_2534016 |
| Alexa Fluor 546 donkey anti-goat (2ary) | Invitrogen | A11056; RRID: AB_142628 |
| ATM | Santa Cruz | sc-135663; RRID: AB_2062962 |
| Biological Samples | ||
| PDXIMIM#1-6 | Hospital del Mar (Barcelona) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Sigma | N/A |
| Advanced DMEM/F12 | GIBCO | 12634028 |
| AZ628 (BRAFi) | Selleckchem | S2746 |
| Vemurafenib | Selleckchem | S1267 |
| Sorafenib | Selleckchem | S7397 |
| 5Z-7-oxozeaneol (TAKi) | Selleckchem | 499610 |
| Trametinib (MEKi) | Selleckchem | S2673 |
| 5-Fluorouracil (5-FU) | Accord | 606544.3 |
| Irinotecan | Frasenius Kabi | 687014.3 |
| Doxorubicin | Accord | 174247 |
| Matrigel® | Corning | 354234 |
| 1x B27 supplement | GIBCO | 17504044 |
| 1x N-2 supplement | GIBCO | 17502048 |
| Y-27632 (ROCK inhibitor) | Sigma | Y0503 |
| EGF | Sigma | E9644 |
| Human Noggin | PeproTech | 120-10C |
| Human R-spondin 1 | PeproTech | 120-38 |
| Nicotinamide | Sigma | N3376 |
| A8301 (ALK inhibitor) | Sigma | SML0788 |
| SB202190 | Sigma | S70677 |
| Prostaglandin E2 | Tocris | 2296 |
| Gastrin I human | Tocris | 3006 |
| Ad-GFP | Vector Biolabs | 1060 |
| Ad-GFP-Cre | Vector Biolabs | 1700 |
| PhosSTOP phosphatase inhibitor cocktail | Roche | PHOSS-RO |
| Complete Mini protease inhibitor cocktail | Roche | 11-836-170-001 |
| TAMRA-TelG 5′-(TTAGGG)3-3′ PNA probe | PNA Bio-synthesis | F1006 |
| Critical Commercial Assays | ||
| Envision+ System HRP Labeled Polymer anti-Rabbit | DAKO | K4003 |
| Envision+ System HRP Labeled Polymer anti-Mouse | DAKO | K4001 |
| 3,3′-diaminobenzidine (DAB) | DAKO | K3468 |
| ProLong. Diamond with DAPI | Thermo Scientific | P36971 |
| ECL solution | Biological Industries | 20-500-120 |
| ECL Prime Western Blotting Detection Reagent | GE Healthcare | RPN2232 |
| CometAssay Kit | Trevigen | 4250-050-K |
| DharmaFECT 1 transfection reagent | Dharmacon | T-2001-03 |
| Deposited Data | ||
| MS data from shC and shIKKα in UV/non-UV HT29 cells | PRIDE EBI-EMBL | PXD008932 |
| Raw data from our manuscript is available at http://dx.doi.org/10.17632/k6xkjc2bf7.2 | N/A | N/A |
| Experimental Models: Cell Lines | ||
| HT29 | ATCC | HTB-38D |
| WiDr | ATCC | CCL-218 |
| HCT116 | ATCC | CCL-247 |
| LIM1215 | ECACC | 10092301 |
| DLD1 | ATCC | CCL-221 |
| SW480 | ATCC | CCL-228 |
| MCF7 | ATCC | HTB-22 |
| T24 | ATCC | HTB-4 |
| SKMEL131 | ATCC | N/A |
| RWP1 | ATCC | N/A |
| Mouse Embryonic Fibroblasts p38α KO | Gift from A. Nebreda. | N/A |
| Mouse Embryonic Fibroblasts Trf2FL/FL | Gift from T. de Lange. | N/A |
| Experimental Models: Organisms/Strains | ||
| Athymic nude, nu/nu mice | Jackson Laboratories | 002019 |
| Oligonucleotides | ||
| ON-TARGETplus SMART pool siRNA Chuk | Dharmacon | SO-2621140G |
| ON-TARGETplus Non-targeting Control Pool | Dharmacon | D-001810-10 |
| MISSION shRNA, TRCN0000000508 (human) (shIKKα 1) | Sigma | N/A |
| MISSION shRNA, TRCN0000199496 (human) (shIKKα 4) | Sigma | N/A |
| TRC2 pLKO.5-puro Nonmammalian shRNA Control | Sigma | SHC202 |
| MISSION shRNA, TRCN00001897 (human) (shIKKβ) | Sigma | N/A |
| MISSION shRNA, TRCN000022146 (human) (shNEMO) | Sigma | N/A |
| Recombinant DNA | ||
| pMD2.G plasmid | Addgene | #12259 |
| pCMV-dR8.2 dvpr plasmid (human) | Addgene | #8455 |
| Software and Algorithms | ||
| GraphPad Prism 6 | Graphpad | https://www.graphpad.com/ |
| Volocity 6.3 | PerkinElmer | http://cellularimaging.perkinelmer.com/downloads/detail.php?id=14 |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Lluís Espinosa (lespinosa@imim.es) and Simon J. Boulton (simon.boulton@crick.ac.uk).
Experimental Model and Subject Details
Animal Studies
Fragments of human colorectal tumors obtained from MARbiobank with the informed consent of patients and following all recommendations of Hospital del Mar’ Ethics Committee, the Spanish regulations, and the Helsinki declaration’s Guide were transplanted and expanded in the cecum of nude mice as orthoxenografts. To perform in vivo drug testing, equivalent pieces of individual tumors were implanted orthotopically in the wall of the cecum of nude mice. When tumors were detectable by palpation (4-5 weeks), animals were randomly ascribed to the different groups of treatment. Vemurafenib (50mg/kg) was administered orally every day, and 5-FU and irinotecan (50mg/kg each) every 4 days intravenously. After 21 days of treatment, mice were euthanized and tumors collected, photographed, measured and processed for immunohistochemistry examination. In a parallel experiment, animals were equally treated and left for survival analysis. In all our procedures, animals were kept under pathogen-free conditions, and animal work was conducted according to the guidelines from the Animal Care Committee at the Generalitat de Catalunya. The Committee for Animal Experimentation at the Institute of Biomedical Research of Bellvitge (Barcelona) approved these studies.
Patient-derived tumoroids
For tumoroids generation, primary or xenografted tumors were disaggregated in 1mg/mL collagenase II (Sigma) and 20 μg/mL hyaluronidase (Sigma), filtered in 100 μm cell strainer, and seeded in Matrigel (BD Biosciences) as described(Sato et al., 2011). Tumoroids were expanded by serial passaging and kept frozen in liquid Nitrogen for being used in subsequent experiments. Mutations identified in the different tumors were: IMIM#1, EGFR(S464L) and TP53(I254T); IMIM#3, NRAS(Q61K), TP53(R175H), and EGFR(E928K); IMIM#5, KRAS(G12D); IMIM#6, KRAS; and IMIM#9, KRAS, NRAS and BRAF WT.
Cell lines
CRC cell lines LIM1215 (KRAS and BRAF wild-type), Caco2, DLD1, SW480 and HCT116 (KRAS mutated), WiDr and HT29 (BRAF mutated) were obtained from the ATCC. IKKα wild-type and knock out MEFs were kindly provided by Michael Karin (UCSD, La Jolla). SV40-LT-immortalized TRF2FL/FL were previously described (Celli and de Lange, 2005). All cells were grown in Dulbecco’s modified Eagle’s medium (Invitrogen) plus 10% fetal bovine serum (Biological Industries) and were maintained in a 5% CO2 incubator at 37°C.
Method Details
Tumoroid viability assays
600 single tumoroid cells were plated in 96-well plates in Matrigel. After 4 days in culture, we treated growing tumoroids with 5-FU, Irinotecan, doxorubicin, AZ628, vemurafenib or combinations for 72 hours at the indicated concentrations. Cell viability was determined using the CellTiter-Glo® 3D Cell Viability Assay (Promega) following manufacturer’s instructions in an Orion II multiplate luminometer (Berthold detection systems). Data were calculated as mean ± standard deviation from 3 independent experiments conducted in triplicates.
Cell lysis and Western Blot (WB)
Cells were lysed 20 min at 4°C in 300 μL of PBS plus 0.5% Triton X-100, 1 mM EDTA, 100 mM NA-orthovanadate, 0.25 mM phenylmethylsulfonyl fluoride, and complete protease inhibitor cocktail (Roche). Lysates were analyzed by western blotting using standard SDS–polyacrylamide gel electrophoresis (SDS-PAGE) techniques. In brief, protein samples were boiled in Laemmli buffer, run in polyacrylamide gels, and transferred onto polyvinylidene difluoride membranes. The membranes were incubated overnight at 4°C with the appropriate primary antibodies. After being washed, the membranes were incubated with specific secondary horseradish peroxidase–linked antibodies from Dako and visualized using the enhanced chemiluminescence reagent from Amersham.
Cell fractionation
For cytoplasm/nuclear/chromatin separations, cells were lysed in 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, and 0.05% NP-40 (pH 7.9) for 10 min on ice and centrifuged at 3,000 rpm. Supernatants were recovered as the cytoplasmic fraction, and the pellets were lysed in 5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, and 26% glycerol and sonicated for 5 min three times to recover the soluble nuclear fractions. The remaining pellet included the chromatin fraction. Lysates were run in SDS-PAGE and transferred onto Immobilon-P transfer membranes (Millipore) for western blot analysis.
immunoprecipitation Assay
For precipitation assay, control and UV-treated cells were crosslinked with dithiobis succinimidyl propionate (DSP, Pierce) 10 min at room temperature and then disrupted in RIPA buffer plus protease inhibitor cocktail (Roche). Cells were then centrifuged at 13,000 rpm for 15 min, and supernatants were incubated with 5 μg of anti-IKKα(p45) antibody. Precipitates were captured with 35 mL of protein A-Sepharose, extensively washed, and analyzed by WB. In most of the experiments, we used the Clean-Blot IP Detection Kit as secondary antibody.
Immunohistochemical Staining (IHC)
Tissues were fixed in 4% formaldehyde overnight at room temperature and embedded in paraffin. 4 μm paraffin embedded sections were first deparaffinized in xylene. IHC was performed following standard techniques with EDTA- or citrate-based antigen retrieval and developed with the Envision+ System HRP Labeled Polymer anti-Rabbit or anti-Mouse and 3,3′-diaminobenzidine (DAB) from DAKO. Images were obtained with an Olympus BX61 microscope.
Immunofluorescence (IF) analysis
For cell lines and tumoroid immune-fluorescence, cells were directly fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100 (Pierce), washed and incubated overnight with the corresponding primary antibodies. Secondary antibodies were the Alexa Fluor™ from Invitrogen. ProLong™ Diamond Antifade Mountant plus DAPI was used as mounting medium. Images were taken in an SP5 upright confocal microscope (Leica).
Peptide Nucleic Acid (PNA) Fluorescence In Situ Hybridization (FISH) and IF-FISH
Cells were treated with 0.2 μg/ml of colcemid for 90 minutes to arrest cells in metaphase. Trypsinized cells were incubated in 75 mM KCL, fixed with methanol:acetic acid (3:1), and spread on glass slides. To preserve chromosome architecture the slides were rehydrated in PBS for 5 minutes, fixed in 4% formaldehyde for 5 minutes, treated with 1 mg/ml of pepsin for 10 minutes at 37°C, and fixed in 4% formaldehyde for 5 minutes. Next, slides were dehydrated in 70%, 85%, and 100% (v/v) ethanol for 15 minutes each and then air-dried. Metaphase chromosome spreads were hybridized with telomeric TAMRA-TelG 5′-(TTAGGG)3-3′ PNA probe (Bio-synthesis) and slides were mounted using ProLong Gold antifade with DAPI (Life Technologies). Chromosome images and telomere signals were captured using Zeiss Axio Imager M1 microscope equipped with an ORCA-ER camera (Hamamatsu) controlled by Volocity 6.3 software (Improvision). For IF-FISH, cells grown on #1.5 glass coverslips were fixed for 5 minutes in cold methanol. Cells were washed twice for 5 min in PBS, incubated for 30 min in blocking solution (1 mg/ml BSA, 3% goat serum, 0.1% Triton X-100, 1 mM EDTA in PBS), and then incubated overnight with primary antibody against 53BP1 and secondary antibody anti-rabbit Alexa Fluor 488 secondary antibody for 1 hr and 30 min, in blocking solution with 5 min washes in PBS in-between. After dehydration of the cells, FISH experiments were performed as described above. Slides were mounted with ProLong Gold antifade containing DAPI and images were acquired with an Olympus FLV1000 inverted microscope equipped with a 63X oil objective.
Laser microirradiation induced DNA damage
HT29 cells were seeded on 35 mm glass bottom dish (Ibidi, 81158) and pre-sensitized for 48h with 10 μM BrdU. Cells were transferred to Olympus FV1000 confocal LSM with heated stage. Laser microirradition was performed in a stripe shape with a 405 nm laser focused through 40x objective (400mW at objective, 50 scans). 1 hour after DNA damage induction, cells were fixed and processed for IF.
Knock down and knock out assays
Lentiviral particles including shRNAs against IKK subunits were obtained from Sigma (table). The TRC2 pLKO.5-puro non-mammalian shRNA plasmid (SHC202) was used as negative control. siRNAs employed were ON-TARGET plus siRNA SMARTpool purchased from GE Dharmacon. RNA interference (RNAi) transfections were performed using Dharmafect Trasfection Reagent (Dharmacon) in a forward transfection mode using manufacturer’s guidelines.
Deletion of floxed alleles in Trf2FL/FL cells was carried out with either Ad-GFP or Ad-GFP-Cre adenovirus (Vector Biolabs, ref. 1060 and 1700) and checked by WB.
Cell cycle analysis
Cell cycle was determined by flow cytometry using the standard Propidium Iodide staining-based protocol in the LSR-Fortessa analyzer (BD Biosciences).
Comet Assay
Comet assays were performed using CometAssay® Trevigen Kit (4250-050-K) following manufacturer’s instructions. Pictures were taken using a Nikon Eclipse Ni-E epifluorescence microscope and tail moment was calculated using the OPENCOMET plugin for Fiji.
Kinase Assay
In vitro kinase assays were performed as previously described (Espinosa et al., 2003). In brief, 5 μg of GST or GST-ATM (Aa1911-2063) were incubated with 200 ng of recombinant human IKKα (ab102103) or 10 μg of cell lysates from WT or IKKα KO cells at 30°C for 30 minutes in the presence of ATPγP32. Reactions were stopped by adding loading buffer, run in a polyacrylamide gel and developed in autoradiograph film.
Mass Spectrometry Analysis
Cell lysates obtained in the different experimental conditions were processed and digested with trypsin and endoproteinase LysC with a ratio enzyme:sample of 1:10 for both enzymes (w:w). Samples were then subjected to phospho-peptide enrichment using titanium dioxide (TiO2) beads, and phospho-enriched samples were analyzed by LC-MS/MS. To identify IKKα-dependent phosphopeptides, samples were injected with a 120-minute chromatographic gradient in an Orbitrap Velos Pro with a data-dependent acquisition method using CID fragmentation for the top 20 most intense precursor ions and multistage activation. In the UV-activation experiment, samples were acquired with a 90-minute gradient in an Orbitrap Fusion Lumos with a data-dependent acquisition method using top speed, HCD fragmentation and ion-trap detection. In both cases, the resulting data were analyzed with the Proteome Discoverer software v1.4, using the search algorithm Mascot (v2.5) against a Human protein database (Uniprot, v2015) with oxidation (Met), and phosphorylation (Ser, Thr, Tyr) as variable modifications. Carbamidomethylation (Cys) was set as fixed modification and a mass tolerance of 7 ppm (MS1) and 0.5 Da (MS2) were used. Only peptides with a false discovery rate below 5% were considered for quantitative analysis. Peptides relative abundance was estimated with the area under the curve of extracted ion chromatograms. Protein network was generated using cytoscape software (https://cytoscape.org).
Quantification and Statistical Analysis
Statistical parameters, including number of events quantified, standard deviation, and statistical significance are reported in the figures and in the figure legends. Statistical analysis has been performed using GraphPad Prism6 software (GraphPad) and p < 0.05 is considered significant. Two-sided Student’s t test was used to compare differences between two groups and Two-Way ANOVA test was used to compare differences among multiple groups. Each experiment has been repeated at least twice.
Data and Code Availability
MS data are available at PRIDE EBI-EMBL database with identifier PRIDE: PXD008932.
Row data from our manuscript is available at https://doi.org/10.17632/k6xkjc2bf7.1
Acknowledgments
We want to thank the Bigas and Boulton lab members for constructive discussions and suggestions and Kitty van Zwieten, Grazzia Giuffrida, and Marta Garrido for technical support. This work was funded by grants from Instituto de Salud Carlos III FEDER (PIE15/00008 and PI16/00437), Generalitat de Catalunya 2017SGR135, and the “Xarxa de Bancs de Tumors” sponsored by Pla Director d’Oncologia de Catalunya (XBTC). C.C. is supported by FPI BES-2014-068451 and the EMBO Short-Term Fellowship (na7084). P.M. is supported by funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement #702430. The Boulton lab is supported by The Francis Crick Institute, which receives its core funding from Cancer Research UK (FC0010048), the UK Medical Research Council (FC0010048), and the Wellcome Trust (FC0010048). S.J.B. is funded by European Research Council (ERC) Advanced Investigator Grants (TelMetab) and a Wellcome Trust Senior Investigator Grant. The Centre de Regulació Genòmica/Universitat Pompeu Fabra Proteomics Unit is part of the “Plataforma de Recursos Biomoleculares y Bioinformáticos (ProteoRed)” supported by grant PT13/0001 of Instituto de Salud Carlos III from the Spanish government and “Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat de Catalunya” (2014SGR678). We acknowledge support from the Spanish Ministry of Economy and Competitiveness and “Centro de Excelencia Severo Ochoa 2013-2017” (SEV-2012-0208).
Author Contributions
A.B., S.J.B., and L.E. conceptualized the study, designed the experiments, and wrote the manuscript. E.B. and E.S. designed the proteomic analysis and evaluated the results. C.C., P.M., A. Villanueva, A. Vert, I.P., J.B., and M.M.-I. performed the biochemical assays and the in vitro and in vivo drug testing experiments. C.C. prepared the figures. M.G-.F. and M.I. performed the clinicopathological characterization of human tumors. C.M. provided clinical advice.
Declaration of Interests
The authors declare no competing interests.
Published: July 10, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.05.036.
Contributor Information
Anna Bigas, Email: abigas@imim.es.
Simon J. Boulton, Email: simon.boulton@crick.ac.uk.
Lluís Espinosa, Email: lespinosa@imim.es.
Supplemental Information
References
- Adams R.H., Porras A., Alonso G., Jones M., Vintersten K., Panelli S., Valladares A., Perez L., Klein R., Nebreda A.R. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell. 2000;6:109–116. [PubMed] [Google Scholar]
- Alao J.P., Sunnerhagen P. Rad3 and Sty1 function in Schizosaccharomyces pombe: an integrated response to DNA damage and environmental stress? Mol. Microbiol. 2008;68:246–254. doi: 10.1111/j.1365-2958.2008.06147.x. [DOI] [PubMed] [Google Scholar]
- Amado R.G., Wolf M., Peeters M., Van Cutsem E., Siena S., Freeman D.J., Juan T., Sikorski R., Suggs S., Radinsky R. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- Batlle E., Clevers H. Cancer stem cells revisited. Nat. Med. 2017;23:1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- Bothmer A., Robbiani D.F., Feldhahn N., Gazumyan A., Nussenzweig A., Nussenzweig M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A., Robbiani D.F., Di Virgilio M., Bunting S.F., Klein I.A., Feldhahn N., Barlow J., Chen H.T., Bosque D., Callen E. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner H., Kloor M., Pox C.P. Colorectal cancer. Lancet. 2014;383:1490–1502. doi: 10.1016/S0140-6736(13)61649-9. [DOI] [PubMed] [Google Scholar]
- Bulavin D.V., Higashimoto Y., Popoff I.J., Gaarde W.A., Basrur V., Potapova O., Appella E., Fornace A.J., Jr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001;411:102–107. doi: 10.1038/35075107. [DOI] [PubMed] [Google Scholar]
- Canovas B., Igea A., Sartori A.A., Gomis R.R., Paull T.T., Isoda M., Perez-Montoyo H., Serra V., Gonzalez-Suarez E., Stracker T.H., Nebreda A.R. Targeting p38alpha Increases DNA Damage, Chromosome Instability, and the Anti-tumoral Response to Taxanes in Breast Cancer Cells. Cancer Cell. 2018;33:1094–1110.e8. doi: 10.1016/j.ccell.2018.04.010. [DOI] [PubMed] [Google Scholar]
- Celli G.B., de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- Chapman J.R., Sossick A.J., Boulton S.J., Jackson S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012;125:3529–3534. doi: 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J.R., Taylor M.R., Boulton S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Chapman J.R., Barral P., Vannier J.B., Borel V., Steger M., Tomas-Loba A., Sartori A.A., Adams I.R., Batista F.D., Boulton S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I.T., Hsu P.H., Hsu W.C., Chen N.J., Tseng P.H. Polyubiquitination of Transforming Growth Factor β-activated Kinase 1 (TAK1) at Lysine 562 Residue Regulates TLR4-mediated JNK and p38 MAPK Activation. Sci. Rep. 2015;5:12300. doi: 10.1038/srep12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer C., Margalef P., Gonzalez J., Vert A., Bigas A., Espinosa L. IKKα is required in the intestinal epithelial cells for tumour stemness. Br. J. Cancer. 2018;118:839–846. doi: 10.1038/bjc.2017.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel J.A., Pellegrini M., Lee J.H., Paull T.T., Feigenbaum L., Nussenzweig A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J. Cell Biol. 2008;183:777–783. doi: 10.1083/jcb.200805154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nicolantonio F., Martini M., Molinari F., Sartore-Bianchi A., Arena S., Saletti P., De Dosso S., Mazzucchelli L., Frattini M., Siena S., Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- Escribano-Díaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J.T., Tkáč J., Cook M.A., Rosebrock A.P., Munro M., Canny M.D. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Espinosa L., Inglés-Esteve J., Aguilera C., Bigas A. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J. Biol. Chem. 2003;278:32227–32235. doi: 10.1074/jbc.M304001200. [DOI] [PubMed] [Google Scholar]
- Hindi S.M., Sato S., Xiong G., Bohnert K.R., Gibb A.A., Gallot Y.S., McMillan J.D., Hill B.G., Uchida S., Kumar A. TAK1 regulates skeletal muscle mass and mitochondrial function. JCI Insight. 2018;3:98441. doi: 10.1172/jci.insight.98441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D.S., Morris V.K., El Osta B., Sorokin A.V., Janku F., Fu S., Overman M.J., Piha-Paul S., Subbiah V., Kee B. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016;6:1352–1365. doi: 10.1158/2159-8290.CD-16-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lieskovan S., Mok S., Homet Moreno B., Tsoi J., Robert L., Goedert L., Pinheiro E.M., Koya R.C., Graeber T.G., Comin-Anduix B., Ribas A. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015;7:279ra41. doi: 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.L., Tan W., Ricono J.M., Korchynskyi O., Zhang M., Gonias S.L., Cheresh D.A., Karin M. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature. 2007;446:690–694. doi: 10.1038/nature05656. [DOI] [PubMed] [Google Scholar]
- Margalef P., Fernández-Majada V., Villanueva A., Garcia-Carbonell R., Iglesias M., López L., Martínez-Iniesta M., Villà-Freixa J., Mulero M.C., Andreu M. A truncated form of IKKα is responsible for specific nuclear IKK activity in colorectal cancer. Cell Rep. 2012;2:840–854. doi: 10.1016/j.celrep.2012.08.028. [DOI] [PubMed] [Google Scholar]
- Margalef P., Colomer C., Villanueva A., Montagut C., Iglesias M., Bellosillo B., Salazar R., Martínez-Iniesta M., Bigas A., Espinosa L. BRAF-induced tumorigenesis is IKKα-dependent but NF-κB-independent. Sci. Signal. 2015;8:ra38. doi: 10.1126/scisignal.2005886. [DOI] [PubMed] [Google Scholar]
- Montagut C., Dalmases A., Bellosillo B., Crespo M., Pairet S., Iglesias M., Salido M., Gallen M., Marsters S., Tsai S.P. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012;18:221–223. doi: 10.1038/nm.2609. [DOI] [PubMed] [Google Scholar]
- Morales M., Theunissen J.W., Kim C.F., Kitagawa R., Kastan M.B., Petrini J.H. The Rad50S allele promotes ATM-dependent DNA damage responses and suppresses ATM deficiency: implications for the Mre11 complex as a DNA damage sensor. Genes Dev. 2005;19:3043–3054. doi: 10.1101/gad.1373705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Preta G., de Klark R., Chakraborti S., Glas R. MAP kinase-signaling controls nuclear translocation of tripeptidyl-peptidase II in response to DNA damage and oxidative stress. Biochem. Biophys. Res. Commun. 2010;399:324–330. doi: 10.1016/j.bbrc.2010.06.133. [DOI] [PubMed] [Google Scholar]
- Ribas A., Gonzalez R., Pavlick A., Hamid O., Gajewski T.F., Daud A., Flaherty L., Logan T., Chmielowski B., Lewis K. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet Oncol. 2014;15:954–965. doi: 10.1016/S1470-2045(14)70301-8. [DOI] [PubMed] [Google Scholar]
- Richman S.D., Seymour M.T., Chambers P., Elliott F., Daly C.L., Meade A.M., Taylor G., Barrett J.H., Quirke P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J. Clin. Oncol. 2009;27:5931–5937. doi: 10.1200/JCO.2009.22.4295. [DOI] [PubMed] [Google Scholar]
- Rouse J., Cohen P., Trigon S., Morange M., Alonso-Llamazares A., Zamanillo D., Hunt T., Nebreda A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- Sato T., Stange D.E., Ferrante M., Vries R.G., Van Es J.H., Van den Brink S., Van Houdt W.J., Pronk A., Van Gorp J., Siersema P.D., Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- Toll A., Margalef P., Masferrer E., Ferrándiz-Pulido C., Gimeno J., Pujol R.M., Bigas A., Espinosa L. Active nuclear IKK correlates with metastatic risk in cutaneous squamous cell carcinoma. Arch. Dermatol. Res. 2015;307:721–729. doi: 10.1007/s00403-015-1579-6. [DOI] [PubMed] [Google Scholar]
- Vetter I.R., Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernández-Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920–926. doi: 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreka M., Lilis I., Papageorgopoulou M., Giotopoulou G.A., Lianou M., Giopanou I., Kanellakis N.I., Spella M., Agalioti T., Armenis V. Iκβ kinase αis required for development and progression of KRAS-mutant lung adenocarcinoma. Cancer Res. 2018;78:2939–2951. doi: 10.1158/0008-5472.CAN-17-1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.H., Shi Y., Tibbetts R.S., Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- Yang Y., Xia F., Hermance N., Mabb A., Simonson S., Morrissey S., Gandhi P., Munson M., Miyamoto S., Kelliher M.A. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol. Cell. Biol. 2011;31:2774–2786. doi: 10.1128/MCB.01139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Lenardo M.J., Baltimore D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell. 2017;168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M., Lottersberger F., Buonomo S.B., Sfeir A., de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MS data are available at PRIDE EBI-EMBL database with identifier PRIDE: PXD008932.
Row data from our manuscript is available at https://doi.org/10.17632/k6xkjc2bf7.1