

Abstract
Background and Purpose
We have shown that cholesterol is synthesized in the principal cells of renal cortical collecting ducts (CCD) and stimulates the epithelial sodium channels (ENaC). Here we have determined whether lovastatin, a cholesterol synthesis inhibitor, can antagonize the hypertension induced by activated ENaC, following deletion of the cholesterol transporter (ATP‐binding cassette transporter A1; ABCA1).
Experimental Approach
We selectively deleted ABCA1 in the principal cells of mouse CCD and used the cell‐attached patch‐clamp technique to record ENaC activity. Western blot and immunofluorescence staining were used to evaluate protein expression levels. Systolic BP was measured with the tail‐cuff method.
Key Results
Specific deletion of ABCA1 elevated BP and ENaC single‐channel activity in the principal cells of CCD in mice. These effects were antagonized by lovastatin. ABCA1 deletion elevated intracellular cholesterol levels, which was accompanied by elevated ROS, increased expression of serum/glucocorticoid regulated kinase 1 (Sgk1), phosphorylated neural precursor cell‐expressed developmentally down‐regulated protein 4‐2 (Nedd4‐2) and furin, along with shorten the primary cilium, and reduced ATP levels in urine.
Conclusions and Implications
These data suggest that specific deletion of ABCA1 in principal cells increases BP by stimulating ENaC channels via a cholesterol‐dependent pathway which induces several secondary responses associated with oxidative stress, activated Sgk1/Nedd4‐2, increased furin expression, and reduced cilium‐mediated release of ATP. As ABCA1 can be blocked by cyclosporine A, these results suggest further investigation of the possible use of statins to treat CsA‐induced hypertension.
Abbreviations
- ABCA1
ATP‐binding cassette transporter A1
- AQP‐2
aquaporin 2
- CCD
cortical collecting ducts
- Cho
cholesterol
- CsA
cyclosporine A
- DIDS
4,4′‐diisothiocyanostilbene‐2,2′‐disulfonic acid
- ENaC
epithelial sodium channel
- Nedd4‐2
neural precursor cell‐expressed developmentally down‐regulated protein 4‐2
- PO
open probability
- SBP
systolic BP
- Sgk1
serum/glucocorticoid regulated kinase 1.
What is already known
Elevated epithelial sodium channel activity contributes to increased blood pressure.
What this study adds
Selectively deletion of ABCA1 in CCD principal cells elevates intracellular cholesterol and causes hypertension by stimulating ENaC.
What is the clinical significance
Since ABCA1 can be blocked by CsA, this study sets a platform for further investigating the possible use of statins to treat CsA‐induced hypertension.
1. INTRODUCTION
The epithelial sodium channels (ENaC) play important parts in the regulation of total body Na+ homeostasis and salt‐sensitive hypertension (Peng et al., 2017; Wang et al., 2018). It is assembled from homologous gene products, mainly encoding α, β, and γ subunits (Canessa et al., 1994). Gain‐of‐function mutations of β and γ subunits cause hypertension, as seen in Liddle's syndrome (Schild et al., 1995; Shimkets et al., 1994). Previously, we have shown that cholesterol, which comprises more than 30% of the cell membrane components, stimulates ENaC in cultured distal nephron cells (Wei et al., 2007). However, cholesterol is tightly packed with other membrane lipids in the apical membrane of epithelial cells. Our previous studies have shown that the endogenous cholesterol is difficult to extract from the apical membrane of distal nephron cells and that the exogenous cholesterol is also difficult to incorporate into the apical membrane (Wei et al., 2007). Due to this, manipulations of the cholesterol in the outer leaflet of the cell membrane did not, or only slightly, affected ENaC activity (Balut et al., 2006; West & Blazer‐Yost, 2005). For a long time, it remained controversial whether cholesterol can regulate ENaC under physiological conditions. Cholesterol is not only located in the outer leaflet which is resistant to experimental manipulations but is also found in the inner leaflet. It is not known if the cholesterol in the inner leaflet is more important for ENaC activity than that in the outer leaflet.
The level of cholesterol in the inner leaflet is precisely controlled by the cholesterol transporter, ATP‐binding cassette transporter A1 (ABCA1; Wang, Silver, Thiele, & Tall, 2001). Therefore, loss of ABCA1 function should elevate cholesterol in the inner leaflet. If the cholesterol in the inner leaflet regulates ENaC, alterations of the ABCA1 function should affect ENaC activity. As ABCA1 transporters are blocked by cyclosporine A (CsA) and 4,4′‐diisothiocyanostilbene‐2,2′‐disulfonic acid (DIDS; Becq et al., 1997; Wang et al., 2009), blockade of ABCA1 with CsA or DIDS elevated intracellular cholesterol and stimulated ENaC in cultured distal nephron cells (Wang et al., 2009). These in vitro studies suggest that loss of the ABCA1 cholesterol transporting function may cause hypertension by stimulating ENaC. However, CsA is better known as a calcineurin inhibitor (Naesens, Kuypers, & Sarwal, 2009), and DIDS is most often used as an inhibitor of anion transporters (Ye et al., 2015). Therefore, molecular knockdown of ABCA1 expression is required to confirm the role of ABCA1 in the regulation of ENaC activity. The related studies are very significant because recent clinical studies suggest that loss of ABCA1 function due to reduced expression levels or polymorphisms parallels with hypertension (Xu, Zhou, Gu, & Li, 2009; Yamada et al., 2008). However, there is no direct evidence to show whether and how loss of ABCA1 function causes hypertension.
Statins are commonly used for reducing cholesterol levels in the blood, because these drugs can inhibit cholesterol synthesis in the liver. Our studies have shown that statins also inhibit cholesterol synthesis in the cortical collecting ducts (CCD) principal cells (Liu, Song, et al., 2013). We have also shown that reducing cholesterol levels by lovastatin decreases ENaC activity whereas exogenous cholesterol increases ENaC activity in excised inside‐out patches (Zhai et al., 2018, 10.1016/j.bbadis.2018.08.027), indicating that intracellular cholesterol is important for ENaC activity. In this study, we show that selective deletion of ABCA1 in CCD principal cells elevates intracellular cholesterol and causes hypertension by stimulating ENaC. Interestingly, lovastatin can reverse the hypertension caused by the deletion of ABCA1. There are two parallel pathways involved in the activation of ENaC by deletion of ABCA1. One is associated with activated serum/glucocorticoid regulated kinase 1 (Sgk1)/neural precursor cell‐expressed developmentally down‐regulated protein 4‐2 (Nedd4‐2) to elevate ENaC apical density, while the other is related to both increased furin expression and reduced ATP release, probably due to shortened primary cilia to increase ENaC open probability (P O).
2. METHODS
2.1. Animal experiments
All animal care and experimental procedures were approved by the Animal Care and Use Committee of Emory University and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology (McGrath & Lilley, 2015). Both aquaporin 2 (AQP2)‐Cre(B6.Cg‐Tg(Aqp2‐cre)1Dek/J; RRID:IMSR_JAX:006881) and ABCA1‐flox (B6.129S6‐Abca1 tm1Jp/J; RRID:IMSR_JAX:028266) mouse strains were purchased from Jackson Laboratory (Maine, USA), and a colony was established at Emory University. Conditional targeting of the mouse ABCA1 gene was achieved by flanking exons 45–46, which encode the second nucleotide‐binding fold, with loxP sites as previously described (Timmins et al., 2005). The mice with a floxed ABCA1 gene were crossed with mice expressing Cre downstream of the AQP2 promoter. The AQP2‐Cre gene is only expressed in the principal cells of the collecting duct, testis, and vas deferens (Nelson et al., 1998). To avoid possible complications resulting from the deletion of ABCA1 in the vas deferens and testis, only female AQP2‐Cre mice were used for breeding. Female AQP2‐Cre mice were mated with male homozygous floxed ABCA1 mice; female offspring heterozygous for both AQP2‐Cre and floxed ABCA1 were bred with males homozygous for floxed ABCA1. Littermates with Cre and homozygous for floxed ABCA1 were used as principal cell‐selective ABCA1 KO mice. Littermates without Cre but homozygous for floxed ABCA1 served as controls. Mice were genotyped using the following primers: ABCA1 13F (5′‐GGA GGT GAC TGA AAG GCA TCC ATC‐3′) and ABCA1 15R (5′‐CCT GTC TCA GCC CTG CAT GC‐3′), which amplify the region in the targeted allele spanning the loxP sites. The PCR product of the non‐recombined allele is 1,600 bp, whereas the recombined allele yields a 500‐bp product. PCR amplified for the AQP2‐Cre transgene using the primers mAQP2 F (5′‐CCT CTG CAG GAA CTG GTG CTG G‐3′) and CreTag R (5′‐GCG AAC ATC TTC AGG TTC TGC GG‐3′), which amplify the 671‐bp junction between the AQP2 promoter and the Cre gene (Strait, Stricklett, & Kohan, 2007).
The experiments were performed using 120 male mice aged 12 weeks (24–28 g) and housed in a temperature‐controlled environment (23 ± 2°C) with a 12‐hr light/dark cycle. To evaluate the effect of lovastatin on ENaC activity and systolic BP (SBP) in ABCA1 KO mice, animals were divided into control mice and ABCA1 KO mice groups. After basal SBP measurement, mice received lovastatin (20 mg·kg−1·day−1) by intraperitoneal injection for 2 weeks.
2.2. SBP measurement
SBP was measured by tail cuff as previously described (Bao et al., 2014). Blood pressures were measured on a warmed platform (BP‐2000, Visitech Systems), and the SBP of the mice was measured before the start of the lovastatin treatment (Week 0). The mice were placed in the holder for 15 min at 37°C. With this method, the reappearance of pulsation on a digital display of the BP cuff was detected by a pressure transducer and amplified and recorded as the SBP. During the measurement, 10 individual readings were obtained in rapid sequence. The highest and lowest readings were discarded, and the average of the remaining eight readings was used.
2.3. Single‐channel recordings of patch‐clamp technique
Cell‐attached patch clamp was used to assess ENaC activity in isolated, split‐open mice CCD as previously described (Bao et al., 2014). Principal cells were identified by their characteristic morphology in the split‐open tubule. Specifically, principal cells appear in the Hoffman modulation image to be large, polygonal, or round cells with concave surfaces; intercalated cells have asymmetric shapes with convex but convoluted surfaces. The CCD adhered to a cover glass coated with Cell‐Tak (Cat. No., 354240, Corning, New York, NY), and the cover glass was placed on a chamber mounted on an inverted Nikon Eclipse TE2000 microscope. The tubule was perfused with a bath solution containing (in mM) 140 NaCl, 5 KCl, 1 CaCl2, and 10 HEPES adjusted to pH 7.4 with NaOH. Patch pipettes were pulled from borosilicate glass with a Sutter P‐97 horizontal puller (Sutter, Novato, CA, USA), and the resistance of the pipettes ranged from 6 to 8 MΩ when filled with the pipette solution (in mM) 140 LiCl, 5 KCl, 1 CaCl2, and 10 HEPES adjusted to pH 7.4 with LiOH. To assess the ENaC activity, cell‐attached patches were formed under voltage‐clamp conditions (Vpipette = +40 mV) on the apical plasma membranes of principal cells. ENaC activity was determined during an at least 15‐min recording period. Only the patches with a seal resistance >2 GΩ were used. Single‐channel ENaC currents were recorded in a cell‐attached configuration with an Axon Multiclamp 200B amplifier (Axon Instruments, Foster City, CA, USA) interfaced via a Digidata 1420 (Axon Instruments) at room temperature (22–25°C). Data were sampling at 5 kHz with a low‐pass filter at 1 kHz using Clampex 10.2 software (Molecular Devices, Sunnyvale, CA, USA). Before analysis, the single‐channel traces were further filtered at 50 Hz. The single‐channel amplitude was constructed by all‐point amplitude histogram, and the histograms were fit using multiple Gaussians and optimized using a simplex algorithm. NP O, the product of the number of channels and the Po, was used to measure the channel activity within a patch. The total number of active channels (N) in the patch was determined by multiple channel events observed in a patch.
2.4. Immunohistochemistry and confocal microscopy
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Kidneys were fixed with 4% paraformaldehyde. For paraffin embedding, tissues were dehydrated in a series of graded ethanol followed by xylene and embedded in paraffin and then cut into 3‐μm sections. Sections were deparaffinized and rehydrated. To reveal antigens, sections were incubated in 1‐mM Tris solution (pH 9.0) supplemented with 0.5‐mM EGTA and heated in a microwave oven for 5 min. Non‐specific binding of IgG was prevented by incubating the sections with 1% BSA for 30 min. Sections were incubated overnight at 4°C with primary antibodies diluted in PBS supplemented with 1% BSA. The sections were washed in PBS‐T and incubated with Alexa Fluor 488 conjugated donkey anti‐rabbit IgG (A21206, RRID:AB_141708; 1:1,000; Invitrogen) or Alexa Fluor 568 conjugated donkey anti‐goat (A11057, RRID:AB_142581; 1:1,000; Invitrogen) for 1 hr. For filipin staining, frozen kidney sections were fixed in 4% paraformaldehyde and then incubated with 1.5 mg·ml−1 glycine. The kidney sections were incubated overnight at 4°C with AQP‐2 antibody and then with Alexa Fluor 568 conjugated donkey anti‐goat (A11057, RRID:AB_142581; 1:1,000; Invitrogen) for 1 hr at room temperature. After washing with PBS, filipin was incubated for 1 hr at room temperature. Kidney sections were mounted with vectashield antifade mounting medium (Cat. No., H‐1000; Vectashield; Vector Laboratories). Filipin staining was viewed by confocal microscope using DAPI filter. All slides were imaged using a confocal microscope (Olympus, Fluoview 1000, Japan). Identical acquisition settings were used for all images. Pixel intensity was quantified across a line drawn from the tubule lumen through the centre of individual cells without a visible nucleus and adjacent to the nucleus in cells with a visible nucleus using National Institutes of Health ImageJ software. The control fluorescent intensity is used as a calibrator, and relative fluorescent intensity is calculated against this calibrator. Each value reported was corrected by background signal and reflects the mean of measurements made in at least 20 tubules that were selected at random. To identify primary cilia, kidney sections cells were stained using an anti‐acetylated‐α‐tubulin antibody. The principal cells of CCD were identified using AQP‐2 antibody. Ciliary length was analysed using Olympus Fluoview FV1000 version 3.1 software.
2.5. Western blotting
Freshly isolated kidney cortex were minced and washed once with PBS and then homogenized using an Omni TH homogenizer (Warrenton, VA) with HEENG buffer (Grimm, Coleman, Delpire, & Welling, 2017) containing protease and phosphatase inhibitors (Cat. No., 1861280; Thermo Scientific). Tissue lysates were centrifuged at 10,000× g at 4°C for 10 min to remove debris. Protein concentration was determined using the BCA protein assay (Cat. No., 23223, Cat. No., 23224; Thermo Scientific). Samples (40 μg) of total protein were separated on 10% SDS‐polyacrylamide gels and transferred onto PVDF membranes, blocked by 5% nonfat dry milk or 3% fat free BSA for 1 hr, followed by incubating with primary antibody for overnight. After washing with TBS‐T, the membranes were incubated with HRP‐conjugated goat anti‐rabbit IgG secondary antibody (Bio‐Rad, Cat. No., 170‐6515; RRID:AB_11125142; 1:3,000 dilution, Hercules, CA, USA) for 1 hr; labelled proteins were visualized with enhanced chemiluminescence (Bio‐Rad, Cat. No., 170‐5061) and quantified by scanning densitometry (Bio‐Rad), as described (Liu, Han, et al., 2013; Yu et al., 2013). Raw densitometric data in different blots were transformed as fold change of the control mean, expressed in arbitrary units of OD. The housekeeping GAPDH or β‐actin proteins were used as loading controls. Quantification of each band was performed via densitometry using the ImageJ software (NIH ImageJ software, RRID:SCR_003070; National Biosciences, Lincoln, NE, USA).
2.6. Antibodies
The antibodies used in this study were as follows: rabbit anti‐ABCA1 (IF, 1:100; WB, 1:1,000, SAB4300712; RRID:AB_2801584; Sigma Aldrich), goat anti‐AQP2 (IF, 1:100, sc‐9882; RRID:AB_2289903 Santa Cruz Biotechnology), rabbit anti‐α‐ENaC (IF, 1:100; WB, 1:1,000, SPC‐403; RRID:AB_10640131; Stressmarq Biosciences), rabbit anti‐β‐ENaC (IF, 1:100;WB, 1:1,000, SPC‐404; RRID:AB_10644173; Stressmarq Biosciences), rabbit anti‐γ‐ENaC (IF, 1:100; WB, 1:1,000, SPC‐405; RRID:AB_10640369; Stressmarq Biosciences), rabbit anti‐4HNE (IF, 1:100; WB, 1:1,000, HNE11‐S; RRID:AB_2629282; Alpha Diagnostic), rabbit anti‐total Sgk1 (WB, 1:1,000, PA5‐21147; RRID:AB_11151949; Thermo), rabbit anti‐Phospho‐Sgk1 (WB, 1:1,000, 44‐1260G; RRID:AB_2533592; Thermo), rabbit anti‐total Nedd4‐2 (WB, 1:1,000; ab46521; RRID:AB_2149325; Abcam), rabbit anti‐Phospho‐Nedd4‐2 (WB, 1:1,000; ab168349; RRID:AB_2801582; Abcam), rabbit anti‐furin (WB, 1:1,000; ab183495; RRID:AB_2801581; Abcam), anti‐prostasin (WB, 1:1,000; 15527‐1‐AP; RRID:AB_2172168; Proteintech), mouse anti‐acetylated‐α‐tubulin antibody (IF, 1:1,000; T7451; RRID:AB_609894; Sigma), rabbit anti‐GAPDH (WB, 1:1,000, sc‐25778; RRID:AB_10167668; Santa Cruz Biotechnology), mouse anti‐β‐actin (WB, 1:1,000, sc‐8432; RRID:AB_626630 Santa Cruz Biotechnology), and rabbit anti‐β tubulin (WB, 1:2,000, bs‐4511R; RRID:AB_11114300; Beijing Bioss).
2.7. Urinary ATP measurement
Urine was collected in metabolic cages under mineral oil over 24 hr from ABCA1 KO mice and control mice, fed with normal chow. Urinary levels of ATP were quantified using a luciferase bioluminescence assay (ATP Determination Kit, A22066; Molecular Probes, Eugene, OR) following the manufacturer's instructions. The relative levels of ATP in each experiment were calibrated with standard curves.
2.8. Randomization and blinding
Animals were randomized for treatment. Tissue collection and preparation, and quantification of blots were carried out in a blind manner. Data collection and evaluation of all experiments were performed without knowledge of the group identity.
2.9. Data and statistical analyses
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). We designed the experiments using equal group sized; however, there were some experimental losses. The data were analysed by an investigator blinded to the experimental conditions. All data are shown as means ± SEM. According to our previous studies and/or preliminary experiments on protein expression for intra‐group variation and differences between group means, we calculated the group size and found that n = 5 was sufficient to detect a difference with 95% confidence and 80% power. All data were subjected to the Kolmogorov–Smirnov normality test and then parametric or non‐parametric analyses were chosen accordingly. If the data were not normally distributed, a non‐parametric test (Mann–Whitney test to compare two groups or Kruskal–Wallis test with Dunn's post hoc test to compare three groups) was performed. If the data were found to follow a Gaussian distribution, parametric tests were done (Student's two‐tailed t‐test for comparisons between two groups or one/two‐way ANOVA). Data subjected to ANOVA were followed by Bonferroni's post hoc tests only when the F value attained P < .05 and there was no significant inhomogeneity of variances. Differences were considered statistically significant at P < .05. GraphPad Prism 5 software (GraphPad, RRID:SCR_002798; La Jolla, CA) was used for all statistical calculations.
2.10. Materials
All compounds used in these experiments were purchased from Sigma‐Aldrich (St Louis, MO) except where specified.
2.11. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017; Alexander, Kelly et al., 2017a, b; Alexander, Peters et al., 2017)
3. RESULTS
3.1. Lovastatin corrects hypertension induced by selective deletion of ABCA1 in CCD principal cells
We have shown that pharmacological blockade of ABCA1 with CsA elevates intracellular cholesterol, increases ENaC activity, and induces hypertension (Wang et al., 2009; Zhai et al., 2018, 10.1016/j.bbadis.2018.08.027). To further determine the role of ABCA1, ABCA1 flox mice were crossed with AQP‐2 Cre mice to selectively delete ABCA1 in CCD principal cells. Confocal microscopy demonstrated that ABCA1, shown in green, was mainly expressed at the basolateral membrane of the kidney tubules, including CCD principal cells in control mice; AQP‐2 was used, after immunostaining (shown in red), as a biomarker for CCD principal cells (Figure 1a, upper panel). In contrast, in the principal cell‐selective ABCA1 KO mice, ABCA1 was only detected in the epithelial cells of other tubules, but not in the AQP‐2 positive cells, suggesting that ABCA1 was successfully knocked out in the principal cells (Figure 1a, lower panel). Genotypes were further confirmed by PCR analyses (Figure 1b). Furthermore, the total ABCA1 protein in the kidney cortex was also significantly reduced in the principal cell‐selective ABCA1 KO mice (Figure 1c,d). ABCA1 KO mice at 12‐week‐old and the age‐matched control mice were treated with lovastatin after the baseline SBP had been measured. The data show that the average SBP in ABCA1 KO mice was significantly higher than that of control mice. Interestingly, ABCA1 KO‐induced elevation of SBP was decreased gradually by application of lovastatin, while lovastatin had no effect on SBP of control mice (Figure 1e).
Figure 1.
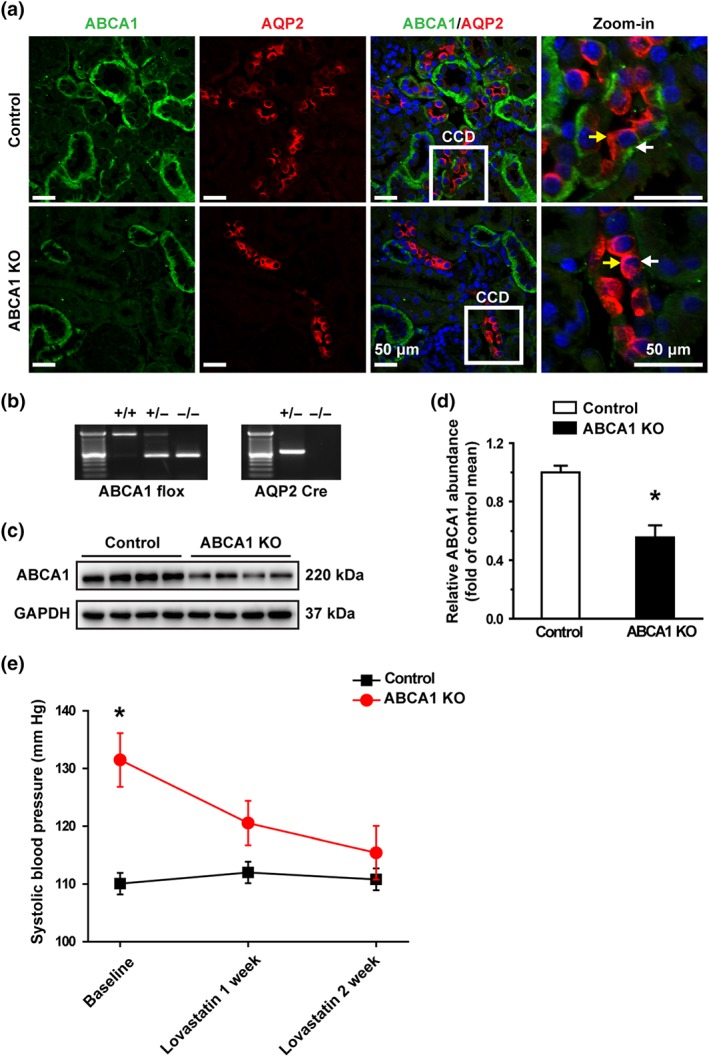
Lovastatin corrects hypertension caused by loss of ABCA1 function. (a) Confocal microscopy images of ABCA1 (green) in control and ABCA1 KO mice. The principal cells in CCD were labelled with an anti‐AQP2 antibody shown in red (also applied to other figures). ABCA1 was expressed in the basolateral membrane of the kidney cortex epithelial cells; AQP‐2 was expressed in the apical membrane of the CCD principal cells. Each experiment was repeated three times in each mouse. The images represent data from six mice, showing consistent results. Yellow arrowheads indicate apical membrane of the cell; white arrowheads indicate the basolateral membrane of the cell. Scale bars: 50 μm. (b) Genotyping analysis of 4‐week‐old mice. (c) Western blot of kidney cortex lysates from control and ABCA1 KO mice using antibodies against either ABCA1 or GAPDH as a loading control. (d) Summary data of western blots showing ABCA1 expression in kidney cortex from control and ABCA1 KO mice; n = 8 in each group. *P < .05, significantly different from control; Student's two‐tailed t‐test. (e) SBP from ABCA1 KO mice (n = 9 ) was significantly increased, compared with control mice (n = 7), while lovastatin attenuated the elevated SBP. *P < .05, significantly different from control; two‐way ANOVA followed by Bonferroni's post hoc test
Table 1.
ENaC functional studies in split‐open CCD in WT and ABCA1 KO mice
| Group | ENaC activity, NPo | ENaC open probability, Po | Number of active ENaC, N |
|---|---|---|---|
| Control mice | 0.56 ± 0.12 | 0.19 ± 0.03 | 2.80 ± 0.44 |
| ABCA1 KO mice | 2.33 ± 0.56* | 0.44 ± 0.06* | 5.00 ± 0.79* |
| Control mice + Lovastatin | 0.24 ± 0.08& | 0.12 ± 0.04 | 2.00 ± 0.24 |
| ABCA1 KO mice + Lovastatin | 0.38 ± 0.09 # | 0.16 ± 0.02 # | 2.40 ± 0.42 # |
Data presented are means ± SEM. Summary of total ENaC activity (NP O), the number of active ENaC, (N), and ENaC open probability (P O) in control mice and ABCA1 KO mice after 2 weeks of infusion with lovastatin.
P < .05, significantly different from control mice.
P < .05, significantly different from ABCA1 KO mice.
P < .05, significantly different from control mice.
3.2. Lovastatin reverses the increased ENaC activity in the ABCA1 KO mice
To test whether deletion of ABCA1 in CCD principal cells affects ENaC activity, we performed cell‐attached patch‐clamp experiments to examine ENaC activity in principal cells of the acutely split‐opened CCD isolated from 12‐week‐old mice (Figure 2a). Compared to control, deletion of ABCA1 significantly increased ENaC activity in the principal cells of isolated CCD, as reflected by greatly elevating P O, the number of active ENaC (N), and the total ENaC activity (NP O; Figure 2a–d). Importantly, the effects were abolished by chronic infusion of lovastatin, albeit lovastatin had a modest effect on ENaC activity in control mice (Figure 2a–d). These results strongly suggest that selective deletion of ABCA1 in principal cells increases SBP by stimulating ENaC via a cholesterol‐dependent mechanism.
Figure 2.
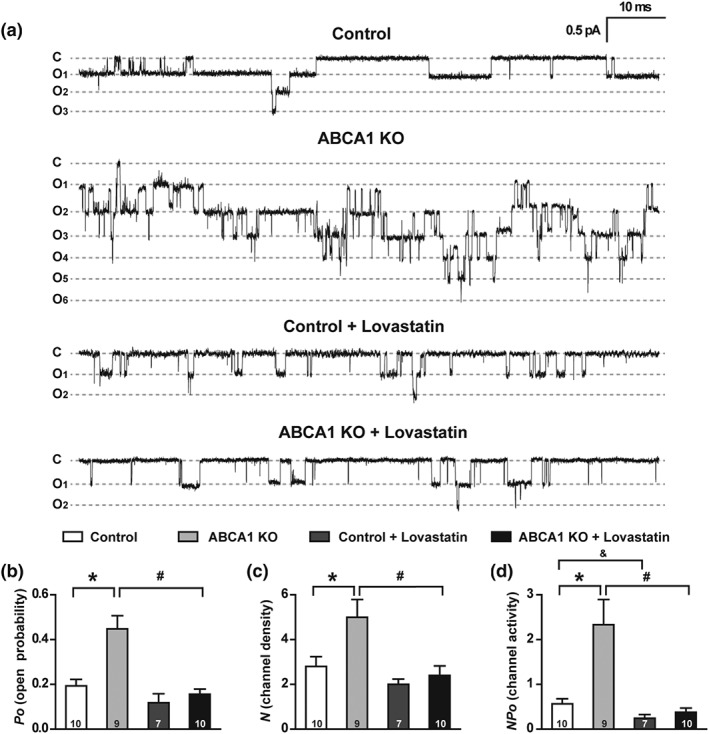
ABCA1 KO elevates both ENaC P O and the number of active ENaC (N) in the patch. (a) Representative single‐channel recordings of ENaC activity from cell‐attached patches in control and ABCA1 KO mouse with or without lovastatin treatment. Downward events show ENaC openings. Vpipette = +40 mV. (b–d) Summary data of ENaC P O (b), channel density (c), and NP O, which is N times P O (d); n is shown at the base of each bar. *P < .05, # P < .05, & P < .05, significantly different as indicated; one‐way ANOVA followed by Bonferroni post hoc test. Summary data of total ENaC activity, number of active ENaC, and ENaC open probability are shown in Table 1
3.3. Deletion of ABCA1 increases the expression levels of γ‐ENaC in CCD principal cells
To determine whether deletion of ABCA1 alters ENaC expression, both confocal microscopy and western blot experiments were performed. The data show that fluorescent intensities of α‐ENaC (Figure 3a,b) and β‐ENaC (Figure 3e,f) in the CCD principal cells and the protein levels of α‐ENaC (Figure 3c,d) and β‐ENaC (Figure 3g,h) in the kidney cortex were not altered in ABCA1 KO mice, suggesting that ABCA1 deletion did not affect the expression of α‐ENaC and β‐ENaC. In contrast, both fluorescent intensity of γ‐ENaC (Figure 4a,b) and the protein levels of full length γ‐ENaC (uncleaved) and cleaved γ‐ENaC (Figure 4c,d) in the kidney cortex were significantly increased in ABCA1 KO mice. These data suggest that deletion of ABCA1 stimulates ENaC by increasing the expression levels of both the full‐length γ‐ENaC and the cleaved γ‐ENaC.
Figure 3.
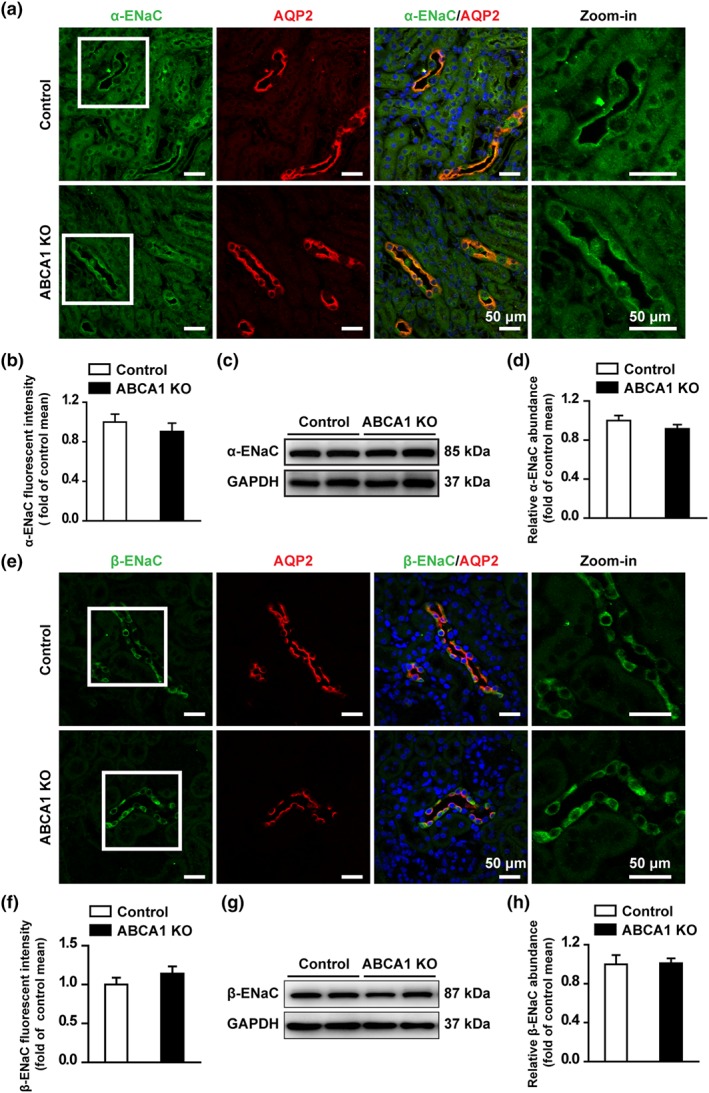
ABCA1 KO does not change the expression of α‐ENaC and β‐ENaC in the CCD of mouse kidney. (a) Confocal microscopy images of α‐ENaC (green) in kidney cortex from control and ABCA1 KO mice. Scale bars: 50 μm. (b) Summary data of α‐ENaC fluorescence intensity in kidney slices from control and ABCA1 KO mice. Each experiment was repeated three times in six mice; 20 images were used for the analysis. (Mann–Whitney test). (c) Western blots of kidney cortex lysates from control and ABCA1 KO mice using antibodies against either α‐ENaC or GAPDH as a loading control. (d) Summary data of Western blots, showing α‐ENaC expression in kidney cortex from control and ABCA1 KO mice (n = 8 in each group). Student's two‐tailed t‐test. (e) Confocal microscopy images of β‐ENaC (green) in kidney cortex from control and ABCA1 KO mice. Scale bars: 50 μm. (f) Summary data of β‐ENaC fluorescence intensity in kidney slices from control and ABCA1 KO mice. Each experiment was repeated three times in six mice; 20 images were used for analysis (Mann–Whitney test). (g) Western blot of kidney cortex lysates from control and ABCA1 KO mice, using antibodies against either β‐ENaC or GAPDH as a loading control. (h) Summary data of Western blots, showing β‐ENaC expression in kidney cortex from control and ABCA1 KO mice (n = 8 in each group). Student's two‐tailed t‐test
Figure 4.
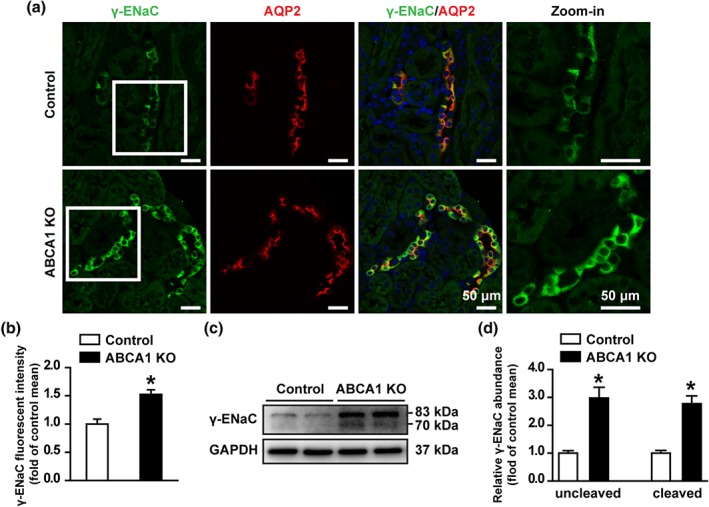
ABCA1 KO increases both full length and cleaved γ‐ENaC expression in mouse kidney CCD. (a) Confocal microscopy images of γ‐ENaC (green) in the kidney slices from control and ABCA1 KO mice. Scale bars: 50 μm. (b) Summary data of γ‐ENaC fluorescence intensity in kidney sections from control and ABCA1 KO mice. Each experiment was repeated three times in six mice; 20 images were used for analysis. *P < .05, significantly different from control; Student's two‐tailed t‐test. (c) Western blot of kidney cortex lysates from control and ABCA1 KO mice, using antibodies against either γ‐ENaC or GAPDH as a loading control. (d) Summary data of Western blots, showing γ‐ENaC expression in kidney cortex from control and ABCA1 KO mice; n = 6 in each group. *P < .05, significantly different from control; Student's two‐tailed t‐test
3.4. Deletion of ABCA1 elevates intracellular cholesterol and induces oxidative stress in CCD principal cells
To determine whether deletion of ABCA1 alters the levels of intracellular cholesterol in CCD principal cells, confocal microscopy experiments were performed using kidney slices, stained either with a fluorescent cholesterol‐binding compound, filipin, to evaluate the levels of intracellular cholesterol or with an antibody against AQP‐2 to label CCD principal cells. Compared with that of control mice, the fluorescent intensity of filipin in CCD principal cells of ABCA1 KO mice was significantly increased. In contrast, the fluorescent intensity of the cells in other nephron segments remained unchanged (Figure 5a,c). As we have shown that cholesterol elevates intracellular ROS in lymphoma cells and CCD principal cells (Liu, Song, et al., 2013; Song et al., 2014), we also tested if the elevated intracellular cholesterol due to deletion of ABCA1 can induce oxidative stress. 4‐hydroxynonenal (4‐HNE) was used as a marker of oxidative stress as previously reported (Luo et al., 2018). Confocal microscopy showed that the levels of 4‐HNE was significantly increased in CCD principal cells of ABCA1 KO mice (Figure 5b,d), where intracellular cholesterol was elevated. Furthermore, Western blots demonstrated that the expression levels of 4‐HNE were significantly increased in the kidney cortex of ABCA1 KO mice (Figure 5e,f). These data suggest that deletion of ABCA1 causes oxidative stress in CCD principal cells probably via elevation of intracellular cholesterol.
Figure 5.
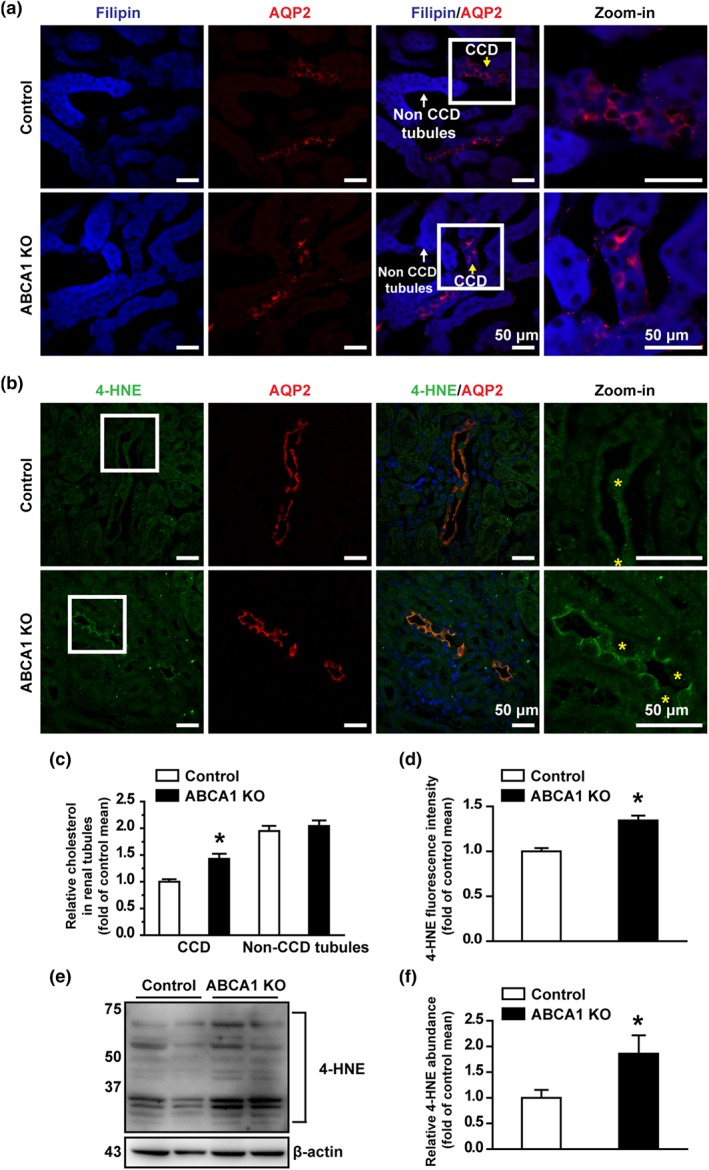
ABCA1 KO increases intracellular cholesterol in the mouse CCD. (a) Confocal microscopy images of cholesterol levels (blue, labelled with filipin) either in CCD or in other tubules from control and ABCA1 KO mice. Scale bars: 50 μm. (b) Confocal microscopy images of 4‐HNE levels (green) from control and ABCA1 KO mice. Yellow stars indicate the intercalated cells cell. Scale bars: 50 μm. (c) Summary data of fluorescence intensity either in CCD or in other tubules from control and ABCA1 KO mice. Each experiment was repeated three times in six mice; 20 images were used for analysis. *P < .05, significantly different from control; Student's two‐tailed t‐test. (d) Summary data of fluorescence intensity from control and ABCA1 KO mice. Each experiment was repeated three times in six mice; 20 images were used for analysis. *P < .05, significantly different from control; Student's two‐tailed t‐test. (e) Western blot of kidney cortex lysates from control and ABCA1 KO mice, using antibodies against 4‐HNE and β‐actin as a loading control. (f) Summary data of Western blots, showing 4‐HNE expression in kidney cortex from control and ABCA1 KO mice; n = 6 in each group. *P < .05, significantly different from control; Student's two‐tailed t‐test)
3.5. Deletion of ABCA1 activates Sgk1/Nedd4‐2 and increases furin expression
Previous studies have shown that the expression of Sgk1 is significantly increased in kidneys from obese mice (Huang et al., 2006) and that aldosterone increases the expression of Sgk1 by elevating ROS in cultured rat peritoneal fibroblasts (Yamahara et al., 2009). Therefore, we performed western blot experiments to determine whether the increased ROS due to deletion of ABCA1 can also stimulate Sgk1 expression. Indeed, both total Sgk1 and phosphorylated Sgk1 were significantly increased in kidney cortex of ABCA1 KO mice (Figure 6a,b). As a downstream protein of Sgk1 signalling, levels of Nedd4‐2 were also assessed. Although ABCA1 KO did not alter the expression of total Nedd4‐2, an E3 ubiquitin‐protein which controls ENaC surface expression by catalysing its ubiquitination and degradation (Figure 6c). However, the levels of phosphorylated Nedd4‐2, which reduces the interaction between Nedd4‐2 and ENaC and leads to elevated cell surface expression of ENaC, were significantly increased in ABCA1 KO mice (Figure 6d). As ABCA1 KO increased cleaved γ‐ENaC (as shown in Figure 4), the levels of two proteases, furin and prostasin which can cleave ENaC, were also examined. The data show that furin, but not prostasin, was significantly increased (Figure 6e,f). These data suggest that deletion of ABCA1 increases full length and cleaved γ‐ENaC by both initiating Sgk1/Nedd4‐2 signalling and increasing furin expression.
Figure 6.
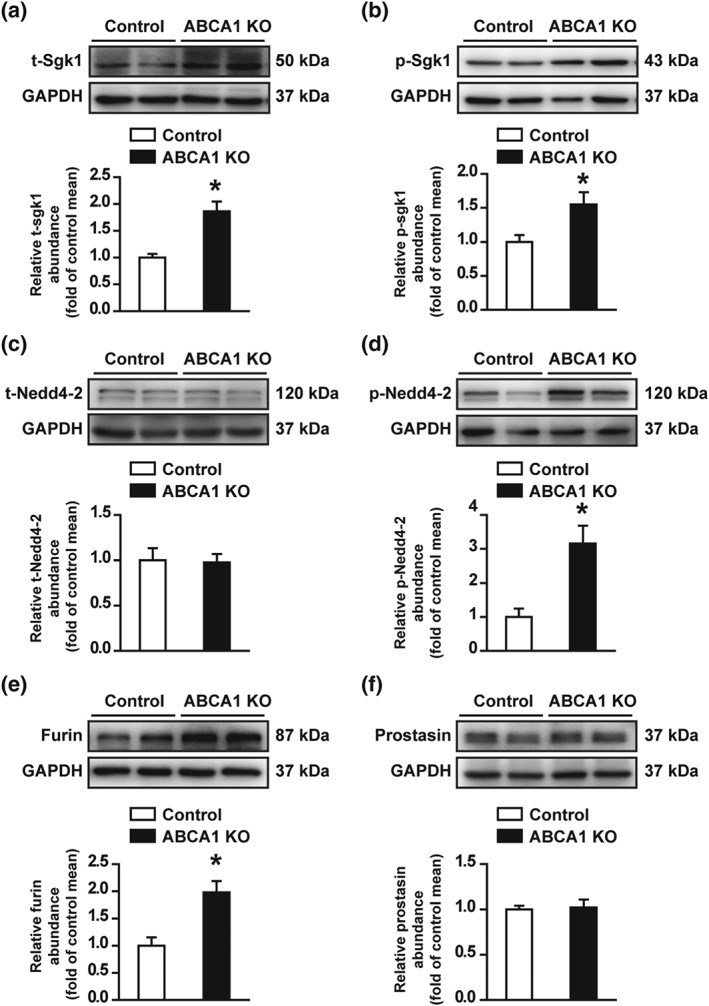
Deletion of ABCA1 increases both Sgk1 and furin expression in mouse kidney. (a and b) Western blots of total Sgk1 (t‐Sgk1) and phosphorylated Sgk1 (p‐Sgk1) in kidney cortex from control and ABCA1 KO mice. Summary data of Western blots, showing t‐Sgk1 and p‐Sgk1 in kidney cortex from control and ABCA1 KO mice; n = 8 in each group for t‐Sgk1; n = 6 in each group for p‐Sgk1. *P < .05, significantly different from control; Student's two‐tailed t‐test. (c and d) Western blot of total Nedd4‐2 (t‐Nedd4‐2) and phosphorylated Nedd4‐2 (p‐Nedd4‐2) in kidney cortex from control and ABCA1 KO mice. Summary data of Western blots, showing t‐Nedd4‐2 and p‐Nedd4‐2 from control and ABCA1 KO mice; n = 8 in each group; *P < .05, significantly different from control; Student's two‐tailed t‐test. (e and f) Western blot of furin and prostasin in kidney cortex from control and ABCA1 KO mice. Summary data of Western blots, showing the levels of furin and prostasin in kidney cortex from control and ABCA1 KO mice; n = 8 in each group. *P < .05, significantly different from control; Student's two‐tailed t‐test
3.6. Deletion of ABCA1 shortens ciliary length and inhibits ATP release
Recent studies have shown that cholesterol has an essential role in the formation and maintenance of primary cilia (Canterini et al., 2017; Formichi et al., 2018). As deletion of ABCA1 can elevate intracellular cholesterol, loss of ABCA1 function may alter the morphology of cilia in principal cells. Therefore, the cilia of CCD principal cells were immnofluorescently labelled with acetylated α‐tubulin. The data show that deletion of ABCA1 significantly reduced the length of cilia and that the effect was reversed by lovastatin (Figure 7a,b). Consistent with these data, application of exogenous cholesterol reduced the cilia length of mpkCCDc14 cells, which was restored by lovastatin (Figure S1). As previous studies have shown that cilia are required for flow‐induced ATP release (Hovater et al., 2008) and that ATP reduces ENaC P O through hydrolysis of phosphatidylinositol‐4,5‐bisphosphate (PIP2; Pochynyuk, Bugaj, Rieg, et al., 2008; Stockand et al., 2010), we also examined the ATP levels in the urine from control and ABCA1 KO mice. The data show that deletion of ABCA1 significantly reduced urinary ATP and that this effect was also reversed by lovastatin (Figure 7c). These results suggest that shortened cilia and subsequently reduced ATP release from CCD principal cells also contribute to increased ENaC P O found in ABCA1 KO mice.
Figure 7.
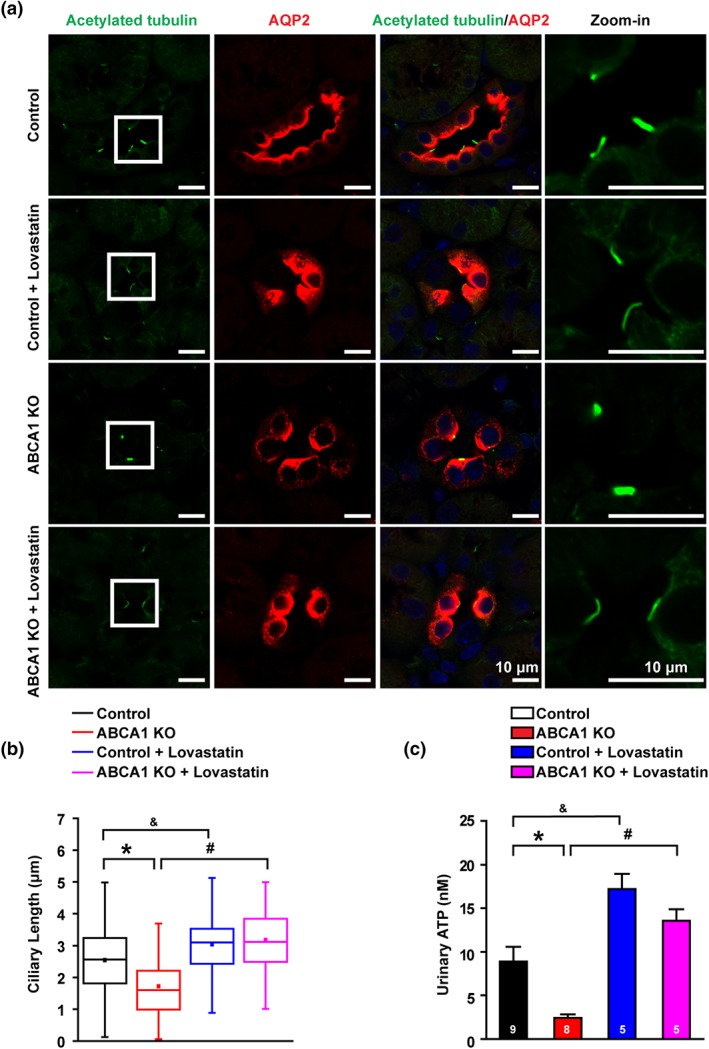
ABCA1 KO reduces ciliary length and reduces luminal ATP release. (A) Confocal images of kidney slices of either control or ABCA1 KO mice. Cilia were labelled with an anti‐acetylated α‐tubulin antibody shown in green. Scale bars: 10 μm. (b) Summary data of cilia length (the numbers of cilia from six separate experiments in each group were used for analysis: control mice group: 290 cilia from nine mice; control mice treated with lovastatin group: n = 82 cilia from five mice; ABCA1 KO mice group: n = 264 cilia from eight mice; and ABCA1 KO mice treated with lovastatin group: n = 83 cilia from five mice. Cilia in ABCA1 KO mice were stunted. *P < .05, # P < .05, & P < .05; significantly different as indicated; one‐way ANOVA followed by Bonferroni post hoc test. (c) Summary data of urinary ATP from control and ABCA1 KO mice with or without lovastatin treatment. *P < .05, # P < .05, & P < .05; significantly different as indicated; one‐way ANOVA followed by Bonferroni post hoc test
3.7. Lovastatin attenuates CsA‐induced increases in ENaC activity by inhibiting furin expression and Sgk1/Nedd4‐2 signalling
To determine whether lovastatin attenuated CsA‐induced increases in ENaC current, the cell‐attached patch‐clamp recording was performed on the apical membrane of A6 distal nephron cells. The data show that lovastatin attenuated CsA‐induced increases in both ENaC activity and percentage of patches containing active ENaC channels in A6 cells (Figure S2 A–C). Importantly, CsA significantly stimulates Sgk1/Nedd4‐2 signalling and furin expression, whereas the effects were abolished by lovastatin (Figure S3 A–E).
Our results demonstrate that deletion of ABCA1 in CCD principal cells increases intracellular cholesterol to enhance ENaC activity either directly or indirectly by shortening the primary cilium to reduce the release of ATP, a well‐known ENaC inhibitor which reduces ENaC activity by decreasing PIP2 via the P2Y2 receptor. It also activates Sgk1/Nedd4‐2 signalling, a well‐known ENaC regulator which increases ENaC density by reducing ENaC degradation from the apical membrane, and increases the expression of furin, an enzyme which increases ENaC activity by proteolytically removing the inhibitory domains from ENaC. Finally, the enhanced ENaC activity and expression due to loss of ABCA1 function result in hypertension, which was corrected by lovastatin (Figure 8).
Figure 8.
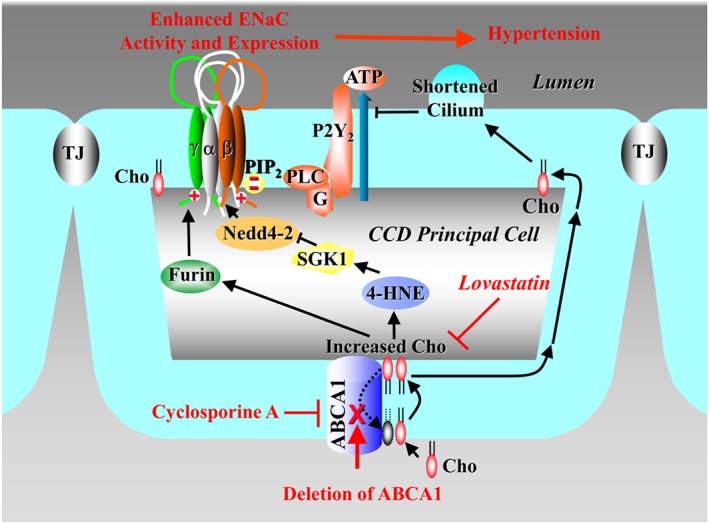
Mechanism by which statins antagonize hypertension induced by activated ENaC due to loss of ABCA1 function, either from pharmacological blockade with CsA or from genetic deletion. The signal transduction pathway is associated with oxidative stress, activated Sgk1/Nedd4‐2, increased furin expression, and reduced cilium‐mediated release of ATP. Cho, cholesterol
4. DISCUSSION
The major findings of the present study are as follows: (a) deletion of ABCA1 in principal cells increases SBP in mice which can be corrected by inhibition of cholesterol synthesis with lovastatin; (b) deletion of ABCA1 increases ENaC single‐channel activity in principal cells of split‐opened CCD; (c) deletion of ABCA1 increases ENaC apical density by activating Sgk1/Nedd4‐2 signalling; and (d) deletion of ABCA1 increases ENaC P O by both increasing furin expression and reducing urinary ATP, probably due to reduced primary cilia length.
This study is the first to show that selectively deleting ABCA1 in CCD principal cells can initiate several pathways which increase ENaC P O, apical density, and proteolytic cleavage. The observation that deletion of ABCA1 elevates cholesterol in the principal cells is not surprising, because ABCA1 functions as a cholesterol transporter to eliminate cholesterol by facilitating cholesterol movement from the inner leaflet to the outer leaflet membrane (Timmins et al., 2005; Vaughan & Oram, 2003). Our previous studies have shown that cholesterol elevates intracellular ROS in both lymphocytes and CCD principal cells (Liu, Song, et al., 2013; Song et al., 2014). We have also shown that hydrogen peroxide stimulates ENaC in cultured distal nephron cells (Ma, 2011) and that oxidized LDL significantly increased ENaC activity by elevating intracellular ROS (Liang et al., 2018). In this study, we show that increased intracellular cholesterol causes ROS probably by increasing 4‐HNE in ABCA1 KO mice. As previous studies have shown that elevated ROS increase Sgk1 expression (Yamahara et al., 2009), while reduced ROS decrease Sgk1 expression (Shibata, Nagase, Yoshida, Kawachi, & Fujita, 2007), we argue that the increased Sgk1 expression may be due to the oxidative stress induced by loss of ABCA1 function. Sgk1 is a protein kinase which stimulates ENaC by phosphorylating Nedd4‐2 and subsequently inhibiting ENaC degradation from the apical membrane (Debonneville et al., 2001). Our data show that deletion of ABCA1 only increases the levels of both full length and cleaved γ‐ENaC, but not α‐ and β‐ENaC. This is not surprising because previous studies also indicate that γ‐ENaC is the only subunit responding to the challenge with low sodium diet (Frindt & Palmer, 2009). However, our in vitro experiments show that cholesterol also alters α‐ENaC expression. Currently, we do not have a good explanation for the different results. It has been argued that the amount of the cleaved γ‐ENaC can accurately reflect the modulation of ENaC activity (Bruns et al., 2007; Rossier & Stutts, 2009). The increased γ‐ENaC appears to be associated with activating Sgk1/Nedd4‐2, whereas the increased ENaC activity is due to elevated furin expression, as furin is a protease which elevates ENaC P O by excising its inhibitory domains (Sheng, Carattino, Bruns, Hughey, & Kleyman, 2006; Snyder, 2005). Furin is up‐regulated in adipose tissue of obese mice (Enomoto et al., 2012), indicating that cholesterol may play a role in promoting furin expression.
The single‐channel data from split‐open CCD show that deletion of ABCA1 not only increases the active ENaC density in the apical membrane of CCD principal cells but also increases ENaC P O. The increased ENaC P O should be due to the reduced ATP release, because we and others have shown that ATP reduces ENaC P O by reducing the levels of PIP2 through the purinergic P2Y2 receptor (Ma, Chou, Wei, & Eaton, 2007; Ma, Saxena, & Warnock, 2002; Pochynyuk, Bugaj, Vandewalle, & Stockand, 2008; Pochynyuk, Tong, Staruschenko, Ma, & Stockand, 2006). In parallel with the increased intracellular cholesterol, we found that deletion of ABCA1 can shorten the primary cilia of principal cells. This is consistent with previous studies showing that cholesterol modulates the length of cilia (Canterini et al., 2017; Formichi et al., 2018). The cilia function as a sensor of distal nephron flow (Praetorius & Spring, 2003) to control ATP release from the principal cells (Hovater et al., 2008). Therefore, loss of the cilium‐controlled ATP release should account for the increased ENaC P O in ABCA1 KO mice. Using a cell culture model, our recent studies have already shown that depletion of cholesterol by lovastatin reduces ENaC activity by decreasing PIP2 in microvilli in cultured distal nephron cells (Zhai et al., 2018). These studies suggest that principal cells have a mechanism by which they control sodium reabsorption that depends upon distal nephron flow, the primary cilia, and ATP release from the cells. In other words, high flow‐induced deformation of cilia causes the release of ATP that strongly inhibits ENaC. This is a normal physiological mechanism that links hypovolemic states with reduced sodium reabsorption and increased urinary sodium (Pochynyuk et al., 2010). In addition to the physiological role, we show here that impaired sensing of the distal nephron flow by the shortened primary cilia due to loss of ABCA1 function also contiributes to the pathological elevation of ENaC P O. Furthermore, elevated intracellular ROS should also contribute to the increased ENaC P O in ABCA1 KO mice, because our previous studies have already shown that ROS stimulate ENaC in cultured distal nephron cells (Ma, 2011; Zhang et al., 2013). Because our earlier studies have shown that cholesterol elevates intracellular ROS both in lymphoma cells and in CCD principal cells (Liu, Song, et al., 2013; Song et al., 2014), we argue that cholesterol stimulates ENaC also by elevating intracellular ROS.
In parallel to the elevated ENaC activity, we show that deletion of ABCA1 in principal cells causes hypertension. It has long been noticed that the immunosuppressive drug CsA causes hypertension. The underlying mechanism remains unknown, even though it is commonly thought that the effects should be due to the inhibition of calcineurin (W. Zhang & Victor, 2000). However, CsA is also a potent ABCA1 blocker (Le Goff et al., 2004). Our data, for the first time, show that the hypertension induced by deletion of ABCA1 can be corrected by inhibiting cholesterol synthesis with lovastatin, indicating that CsA may cause hypertension by blocking ABCA1 function which leads to the accumulation of cholesterol in principal cells. Therefore, the present study provides a platform for further investigation of the possible use of statins to treat CsA‐induced hypertension
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Z.R.Z. and H.P.M. designed the study. M.M.W., B.L.S., C.L., Q.Y., Y.J.Z., V.L., N.N., X.Y., X.D.Y., B.L.Z., Q.S.W., L.Z., J.M., S.Z., and Y.X.C. carried out experiments. T.T. generated principal cell‐selective ABCA1 KO mice. M.M.W., Q.Y., R.S., and N.N. analysed the data. M.M.W., Z.R.Z., and H.P.M. made the figures. M.M.W., Z.R.Z., and H.P.M. drafted and revised the paper. All authors approved the final version of the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1.
Lovastatin increased, but cholesterol decreased, the cilia length of mpkCCDc14 cells.
Figure S2. Lovastatin attenuates CsA‐induced increases in ENaC activity in A6 cells.
Figure S3. Lovastatin attenuates CsA‐induced increases in ENaC activity by inhibiting furin expression and Sgk1/Nedd4‐2 signaling.
ACKNOWLEDGEMENTS
This work was supported by Key Project of Chinese National Program for Fundamental Research and Development (Grant 973 Program 2014CB542401 to Z.Z.), National Natural Science Foundation of China (Grants 81320108002 and 91639202 to Z.Z.; 81600224 to M.W.), National Institutes of Health (NIH) (Grants R01 DK 100582 to H.M. and R01 HL 102167 to R.L.S.), and the Natural Science Foundation of Heilongjiang Province (Grant H2016046 to M.W.). The study was also supported by Nn10 program of Harbin Medical University Cancer Hospital.
Wu M‐M, Liang C, Yu X‐D, et al. Lovastatin attenuates hypertension induced by renal tubule‐specific knockout of ATP‐binding cassette transporter A1, by inhibiting epithelial sodium channels. Br J Pharmacol. 2019;176:3695–3711. 10.1111/bph.14775
Contributor Information
Zhi‐Ren Zhang, Email: zhirenz@yahoo.com.
He‐Ping Ma, Email: heping.ma@emory.edu.
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Other ion channels. British Journal of Pharmacology, 174, S195–S207. 10.1111/bph.13881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. British Journal of Pharmacology, 174, S360–S446. 10.1111/bph.13883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balut, C. , Steels, P. , Radu, M. , Ameloot, M. , Driessche, W. V. , & Jans, D. (2006). Membrane cholesterol extraction decreases Na+ transport in A6 renal epithelia. American Journal of Physiology. Cell Physiology, 290(1), C87–C94. 10.1152/ajpcell.00184.2005 [DOI] [PubMed] [Google Scholar]
- Bao, H. F. , Thai, T. L. , Yue, Q. , Ma, H. P. , Eaton, A. F. , Cai, H. , … Eaton, D. C. (2014). ENaC activity is increased in isolated, split‐open cortical collecting ducts from protein kinase Cα knockout mice. American Journal of Physiology. Renal Physiology, 306(3), F309–F320. 10.1152/ajprenal.00519.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becq, F. , Hamon, Y. , Bajetto, A. , Gola, M. , Verrier, B. , & Chimini, G. (1997). ABC1, an ATP binding cassette transporter required for phagocytosis of apoptotic cells, generates a regulated anion flux after expression in Xenopus laevis oocytes. The Journal of Biological Chemistry, 272(5), 2695–2699. 10.1074/jbc.272.5.2695 [DOI] [PubMed] [Google Scholar]
- Bruns, J. B. , Carattino, M. D. , Sheng, S. , Maarouf, A. B. , Weisz, O. A. , Pilewski, J. M. , … Kleyman, T. R. (2007). Epithelial Na+ channels are fully activated by furin‐ and prostasin‐dependent release of an inhibitory peptide from the γ‐subunit. The Journal of Biological Chemistry, 282(9), 6153–6160. 10.1074/jbc.M610636200 [DOI] [PubMed] [Google Scholar]
- Canessa, C. M. , Schild, L. , Buell, G. , Thorens, B. , Gautschi, I. , Horisberger, J. D. , & Rossier, B. C. (1994). Amiloride‐sensitive epithelial Na+ channel is made of three homologous subunits. Nature, 367(6462), 463–467. 10.1038/367463a0 [DOI] [PubMed] [Google Scholar]
- Canterini, S. , Dragotto, J. , Dardis, A. , Zampieri, S. , De Stefano, M. E. , Mangia, F. , … Fiorenza, M. T. (2017). Shortened primary cilium length and dysregulated Sonic hedgehog signaling in Niemann‐Pick C1 disease. Human Molecular Genetics, 26(12), 2277–2289. 10.1093/hmg/ddx118 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debonneville, C. , Flores, S. Y. , Kamynina, E. , Plant, P. J. , Tauxe, C. , Thomas, M. A. , … Staub, O. (2001). Phosphorylation of Nedd4‐2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. The EMBO Journal, 20(24), 7052–7059. 10.1093/emboj/20.24.7052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto, T. , Shibata, R. , Ohashi, K. , Kambara, T. , Kataoka, Y. , Uemura, Y. , … Ouchi, N. (2012). Regulation of adipolin/CTRP12 cleavage by obesity. Biochemical and Biophysical Research Communications, 428(1), 155–159. 10.1016/j.bbrc.2012.10.031 [DOI] [PubMed] [Google Scholar]
- Formichi, P. , Battisti, C. , De Santi, M. M. , Guazzo, R. , Tripodi, S. A. , Radi, E. , … Federico, A. (2018). Primary cilium alterations and expression changes of Patched1 proteins in niemann‐pick type C disease. Journal of Cellular Physiology, 233(1), 663–672. 10.1002/jcp.25926 [DOI] [PubMed] [Google Scholar]
- Frindt, G. , & Palmer, L. G. (2009). Surface expression of sodium channels and transporters in rat kidney: Effects of dietary sodium. American Journal of Physiology. Renal Physiology, 297(5), F1249–F1255. 10.1152/ajprenal.00401.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm, P. R. , Coleman, R. , Delpire, E. , & Welling, P. A. (2017). Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol, 28(9), 2597–2606. 10.1681/ASN.2016090948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovater, M. B. , Olteanu, D. , Hanson, E. L. , Cheng, N. L. , Siroky, B. , Fintha, A. , … Schwiebert, E. M. (2008). Loss of apical monocilia on collecting duct principal cells impairs ATP secretion across the apical cell surface and ATP‐dependent and flow‐induced calcium signals. Purinergic Signal, 4(2), 155–170. 10.1007/s11302-007-9072-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, D. Y. , Boini, K. M. , Osswald, H. , Friedrich, B. , Artunc, F. , Ullrich, S. , … Lang, F. (2006). Resistance of mice lacking the serum‐ and glucocorticoid‐inducible kinase SGK1 against salt‐sensitive hypertension induced by a high‐fat diet. American Journal of Physiology. Renal Physiology, 291(6), F1264–F1273. 10.1152/ajprenal.00299.2005 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff, W. , Peng, D. Q. , Settle, M. , Brubaker, G. , Morton, R. E. , & Smith, J. D. (2004). Cyclosporin A traps ABCA1 at the plasma membrane and inhibits ABCA1‐mediated lipid efflux to apolipoprotein A‐I. Arteriosclerosis, Thrombosis, and Vascular Biology, 24(11), 2155–2161. 10.1161/01.ATV.0000144811.94581.52 [DOI] [PubMed] [Google Scholar]
- Liang, C. , Wang, Q. S. , Yang, X. , Niu, N. , Hu, Q. Q. , Zhang, B. L. , … Ma, H. P. (2018). Oxidized low‐density lipoprotein stimulates epithelial sodium channels in endothelial cells of mouse thoracic aorta. British Journal of Pharmacology, 175(8), 1318–1328. 10.1111/bph.13853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B. C. , Song, X. , Lu, X. Y. , Fang, C. Z. , Wei, S. P. , Alli, A. A. , … Ma, H. P. (2013). Lovastatin attenuates effects of cyclosporine A on tight junctions and apoptosis in cultured cortical collecting duct principal cells. American Journal of Physiology. Renal Physiology, 305(3), F304–F313. 10.1152/ajprenal.00074.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Han, Y. , Li, L. , Lu, H. , Meng, G. , Li, X. , … Ji, Y. (2013). The hydrogen sulfide donor, GYY4137, exhibits anti‐atherosclerotic activity in high fat fed apolipoprotein E(−/−) mice. British Journal of Pharmacology, 169(8), 1795–1809. 10.1111/bph.12246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, J. , Chen, G. , Liang, M. , Xie, A. , Li, Q. , Guo, Q. , … Cheng, J. (2018). Reduced expression of glutathione S‐transferase α 4 promotes vascular neointimal hyperplasia in CKD. Journal of the American Society of Nephrology, 29(2), 505–517. 10.1681/ASN.2017030290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H. P. (2011). Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3‐kinase‐dependent pathway. The Journal of Biological Chemistry, 286(37), 32444–32453. 10.1074/jbc.M111.254102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H. P. , Chou, C.‐F. , Wei, S.‐P. , & Eaton, D. C. (2007). Regulation of the epithelial sodium channel by phosphatidylinositides: Experiments, implications, and speculations. Pflügers Archiv‐European Journal of Physiology, 455(1), 169–180. 10.1007/s00424-007-0294-3 [DOI] [PubMed] [Google Scholar]
- Ma, H. P. , Saxena, S. , & Warnock, D. G. (2002). Anionic phospholipids regulate native and expressed epithelial sodium channel (ENaC). The Journal of Biological Chemistry, 277(10), 7641–7644. 10.1074/jbc.C100737200 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naesens, M. , Kuypers, D. R. , & Sarwal, M. (2009). Calcineurin inhibitor nephrotoxicity. Clinical Journal of the American Society of Nephrology, 4(2), 481–508. 10.2215/CJN.04800908 [DOI] [PubMed] [Google Scholar]
- Nelson, R. D. , Stricklett, P. , Gustafson, C. , Stevens, A. , Ausiello, D. , Brown, D. , & Kohan, D. E. (1998). Expression of an AQP2 Cre recombinase transgene in kidney and male reproductive system of transgenic mice. The American Journal of Physiology, 275(1 Pt 1), C216–C226. 10.1152/ajpcell.1998.275.1.C216 [DOI] [PubMed] [Google Scholar]
- Peng, K. , Lu, X. , Wang, F. , Nau, A. , Chen, R. , Zhou, S. F. , & Yang, T. (2017). Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II‐induced hypertension. American Journal of Physiology. Renal Physiology, 312(2), F245–F253. 10.1152/ajprenal.00178.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochynyuk, O. , Bugaj, V. , Rieg, T. , Insel, P. A. , Mironova, E. , Vallon, V. , & Stockand, J. D. (2008). Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. The Journal of Biological Chemistry, 283(52), 36599–36607. 10.1074/jbc.M807129200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochynyuk, O. , Bugaj, V. , Vandewalle, A. , & Stockand, J. D. (2008). Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. American Journal of Physiology. Renal Physiology, 294(1), F38–F46. 10.1152/ajprenal.00403.2007 [DOI] [PubMed] [Google Scholar]
- Pochynyuk, O. , Rieg, T. , Bugaj, V. , Schroth, J. , Fridman, A. , Boss, G. R. , … Vallon, V. (2010). Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2‐receptor tone. The FASEB Journal, 24(6), 2056–2065. 10.1096/fj.09-151506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochynyuk, O. , Tong, Q. , Staruschenko, A. , Ma, H.‐P. , & Stockand, J. D. (2006). Regulation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. American Journal of Physiology‐Renal Physiology, 290(5), F949–F957. 10.1152/ajprenal.00386.2005 [DOI] [PubMed] [Google Scholar]
- Praetorius, H. A. , & Spring, K. R. (2003). The renal cell primary cilium functions as a flow sensor. Current Opinion in Nephrology and Hypertension, 12(5), 517–520. 10.1097/00041552-200309000-00006 [DOI] [PubMed] [Google Scholar]
- Rossier, B. C. , & Stutts, M. J. (2009). Activation of the epithelial sodium channel (ENaC) by serine proteases. Annual Review of Physiology, 71, 361–379. 10.1146/annurev.physiol.010908.163108 [DOI] [PubMed] [Google Scholar]
- Schild, L. , Canessa, C. M. , Shimkets, R. A. , Gautschi, I. , Lifton, R. P. , & Rossier, B. C. (1995). A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proceedings of the National Academy of Sciences of the United States of America, 92(12), 5699–5703. 10.1073/pnas.92.12.5699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, S. , Carattino, M. D. , Bruns, J. B. , Hughey, R. P. , & Kleyman, T. R. (2006). Furin cleavage activates the epithelial Na+ channel by relieving Na+ self‐inhibition. American Journal of Physiology. Renal Physiology, 290(6), F1488–F1496. 10.1152/ajprenal.00439.2005 [DOI] [PubMed] [Google Scholar]
- Shibata, S. , Nagase, M. , Yoshida, S. , Kawachi, H. , & Fujita, T. (2007). Podocyte as the target for aldosterone: Roles of oxidative stress and Sgk1. Hypertension, 49(2), 355–364. 10.1161/01.HYP.0000255636.11931.a2 [DOI] [PubMed] [Google Scholar]
- Shimkets, R. A. , Warnock, D. G. , Bositis, C. M. , Nelson‐Williams, C. , Hansson, J. H. , Schambelan, M. , … Lifton, R. P. (1994). Liddle's syndrome: Heritable human hypertension caused by mutations in the β subunit of the epithelial sodium channel. Cell, 79(3), 407–414. 10.1016/0092-8674(94)90250-X [DOI] [PubMed] [Google Scholar]
- Snyder, P. M. (2005). Minireview: Regulation of epithelial Na+ channel trafficking. Endocrinology, 146(12), 5079–5085. 10.1210/en.2005-0894 [DOI] [PubMed] [Google Scholar]
- Song, X. , Liu, B. C. , Lu, X. Y. , Yang, L. L. , Zhai, Y. J. , Eaton, A. F. , … Shen, B. Z. (2014). Lovastatin inhibits human B lymphoma cell proliferation by reducing intracellular ROS and TRPC6 expression. Biochimica et Biophysica Acta, 1843(5), 894–901. 10.1016/j.bbamcr.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockand, J. D. , Mironova, E. , Bugaj, V. , Rieg, T. , Insel, P. A. , Vallon, V. , … Pochynyuk, O. (2010). Purinergic inhibition of ENaC produces aldosterone escape. J Am Soc Nephrol, 21(11), 1903–1911. 10.1681/ASN.2010040377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strait, K. A. , Stricklett, P. K. , & Kohan, D. E. (2007). Altered collecting duct adenylyl cyclase content in collecting duct endothelin‐1 knockout mice. BMC Nephrology, 8, 8 10.1186/1471-2369-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins, J. M. , Lee, J. Y. , Boudyguina, E. , Kluckman, K. D. , Brunham, L. R. , Mulya, A. , … Parks, J. S. (2005). Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA‐I. The Journal of Clinical Investigation, 115(5), 1333–1342. 10.1172/JCI200523915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, A. M. , & Oram, J. F. (2003). ABCA1 redistributes membrane cholesterol independent of apolipoprotein interactions. Journal of Lipid Research, 44(7), 1373–1380. 10.1194/jlr.M300078-JLR200 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhang, Z. R. , Chou, C. F. , Liang, Y. Y. , Gu, Y. , & Ma, H. P. (2009). Cyclosporine stimulates the renal epithelial sodium channel by elevating cholesterol. American Journal of Physiology. Renal Physiology, 296(2), F284–F290. 10.1152/ajprenal.90647.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N. , Silver, D. L. , Thiele, C. , & Tall, A. R. (2001). ATP‐binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. The Journal of Biological Chemistry, 276(26), 23742–23747. 10.1074/jbc.M102348200 [DOI] [PubMed] [Google Scholar]
- Wang, Z. R. , Liu, H. B. , Sun, Y. Y. , Hu, Q. Q. , Li, Y. X. , Zheng, W. W. , … Ma, H. P. (2018). Dietary salt blunts vasodilation by stimulating epithelial sodium channels in endothelial cells from salt‐sensitive Dahl rats. British Journal of Pharmacology, 175(8), 1305–1317. 10.1111/bph.13817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, S. P. , Li, X. Q. , Chou, C. F. , Liang, Y. Y. , Peng, J. B. , Warnock, D. G. , & Ma, H. P. (2007). Membrane tension modulates the effects of apical cholesterol on the renal epithelial sodium channel. The Journal of Membrane Biology, 220(1–3), 21–31. 10.1007/s00232-007-9071-7 [DOI] [PubMed] [Google Scholar]
- West, A. , & Blazer‐Yost, B. (2005). Modulation of basal and peptide hormone‐stimulated Na transport by membrane cholesterol content in the A6 epithelial cell line. Cellular Physiology and Biochemistry, 16(4–6), 263–270. 10.1159/000089852 [DOI] [PubMed] [Google Scholar]
- Xu, M. , Zhou, H. , Gu, Q. , & Li, C. (2009). The expression of ATP‐binding cassette transporters in hypertensive patients. Hypertension Research, 32(6), 455–461. 10.1038/hr.2009.46 [DOI] [PubMed] [Google Scholar]
- Yamada, Y. , Kato, K. , Yoshida, T. , Yokoi, K. , Matsuo, H. , Watanabe, S. , … Nozawa, Y. (2008). Association of polymorphisms of ABCA1 and ROS1 with hypertension in Japanese individuals. International Journal of Molecular Medicine, 21(1), 83–89. [PubMed] [Google Scholar]
- Yamahara, H. , Kishimoto, N. , Nakata, M. , Okazaki, A. , Kimura, T. , Sonomura, K. , … Mori, Y. (2009). Direct aldosterone action as a profibrotic factor via ROS‐mediated SGK1 in peritoneal fibroblasts. Kidney & Blood Pressure Research, 32(3), 185–193. 10.1159/000225379 [DOI] [PubMed] [Google Scholar]
- Ye, Z. , Wu, M. M. , Wang, C. Y. , Li, Y. C. , Yu, C. J. , Gong, Y. F. , … Zhang, Z. R. (2015). Characterization of cardiac anoctamin1 Ca(2)(+)‐activated chloride channels and functional role in ischemia‐induced arrhythmias. Journal of Cellular Physiology, 230(2), 337–346. 10.1002/jcp.24709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, S. S. , Cai, Y. , Ye, J. T. , Pi, R. B. , Chen, S. R. , Liu, P. Q. , … Ji, Y. (2013). Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF‐κB‐dependent transcriptional activity. British Journal of Pharmacology, 168(1), 117–128. 10.1111/j.1476-5381.2012.01903.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai, Y. J. , Liu, B. C. , Wei, S. P. , Chou, C. F. , Wu, M. M. , Song, B. L. , … Ma, H. P. (2018). Depletion of cholesterol reduces ENaC activity by decreasing phosphatidylinositol‐4,5‐bisphosphate in microvilli. Cellular Physiology and Biochemistry, 47(3), 1051–1059. 10.1159/000490170 [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Chen, S. , Liu, H. , Zhang, B. , Zhao, Y. , Ma, K. , … Zhang, Z. (2013). Hydrogen sulfide prevents hydrogen peroxide‐induced activation of epithelial sodium channel through a PTEN/PI(3,4,5)P3 dependent pathway. PLoS ONE, 8(5), e64304 10.1371/journal.pone.0064304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , & Victor, R. G. (2000). Calcineurin inhibitors cause renal afferent activation in rats: A novel mechanism of cyclosporine‐induced hypertension. American Journal of Hypertension, 13(9), 999–1004. 10.1016/S0895-7061(00)00288-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Lovastatin increased, but cholesterol decreased, the cilia length of mpkCCDc14 cells.
Figure S2. Lovastatin attenuates CsA‐induced increases in ENaC activity in A6 cells.
Figure S3. Lovastatin attenuates CsA‐induced increases in ENaC activity by inhibiting furin expression and Sgk1/Nedd4‐2 signaling.