

ABSTRACT
Osteogenesis imperfecta (OI) is a monogenic bone fragility disorder that usually is caused by mutations in one of the two genes coding for collagen type I alpha chains, COL1A1 or COL1A2. Mutations in at least 18 other genes can also lead to an OI phenotype. As genetic testing is more widely used, mutations in these genes are also more frequently discovered in individuals who have a propensity for fractures, but who do not have other typical clinical characteristics of OI. Intravenous bisphosphonate therapy is still the most widely used drug treatment approach. Preclinical studies in OI mouse models have shown encouraging effects when the antiresorptive effect of a bisphosphonate was combined with bone anabolic therapy using a sclerostin antibody. Other novel experimental treatment approaches include inhibition of transforming growth factor beta signaling with a neutralizing antibody and the inhibition of myostatin and activin A by a soluble activin receptor 2B. © 2019 The Authors. JBMR Plus published by Wiley Periodicals, Inc. on behalf of American Society for Bone and Mineral Research
Keywords: COLLAGEN TYPE I, FRACTURES, MUSCLE, OSTEOBLAST, OSTEOGENESIS IMPERFECTA, SEQUENCING
Introduction
Osteogenesis imperfecta (OI) is a heritable skeletal disorder that, as the name implies, is caused by defective bone formation.1, 2 This defect is caused by dominant or recessive mutations that lead to bone fragility and other skeletal manifestations, such as short stature and bone deformities. Extraskeletal tissues and organs can also be involved.3 Apart from bone fragility, the classical description of the OI phenotype includes blue or grey discoloration of the sclera and abnormalities of tooth structure called dentinogenesis imperfecta. The prevalence of OI has been estimated at 1 in 13,500 and 1 in 9,700 in two recent population‐based studies from Scandinavia.4, 5
OI is a rare, but frequently reviewed condition. Several recent general introductions to OI and excellent overviews on the genetic causes and pathomechanisms of OI are available.1, 2, 3, 6, 7 The present review focuses on recent clinical and translational studies on OI that are of relevance for metabolic bone specialists.
Clinical Types of Osteogenesis Imperfecta
As the phenotypic severity of OI varies widely, it can be useful to categorize individuals with similar clinical characteristics into more narrowly defined OI types. The 2015 Nosology and Classification of Genetic Skeletal Disorders distinguishes five clinically recognizable OI types (Fig. 1).8 OI type I is the mildest phenotype that is usually associated with straight limbs and a body height within or slightly below the reference range; median adult height Z‐scores range between −1.1 and −1.5.9, 10 OI type II represents the most severe end of the phenotypic spectrum and usually leads to death from respiratory failure shortly after birth. OI type III is the most severe form of OI in individuals surviving the neonatal period. Individuals with OI type III almost always have restricted mobility, develop scoliosis, and are very short, with a final height Z‐score typically between −8 and −9.9, 10, 11, 12, 13 The disease severity of OI type IV is intermediate between OI types I and III. With adequate care, most individuals with OI type IV are ambulatory, but more than half develop scoliosis.12, 14 Short stature is very common in OI type IV, with mean adult height Z‐scores between −3.6 and −4.6.9, 10 The skeletal features of the much rarer OI type V often resemble those of OI type IV, but OI type V is associated with additional distinctive characteristics, such as hyperplastic callus formation (observed in about two‐thirds of patients) and ossification of the interosseous membrane of the forearms (which eventually develops in almost all individuals with OI type V).15 Among the various OI types, OI type I is by far the most prevalent, comprising 70% of the entire cohort in a population‐based study.4
Figure 1.
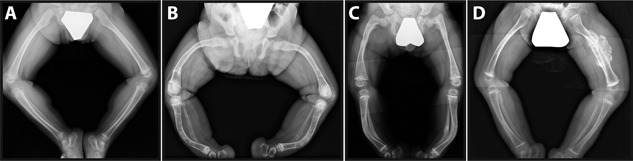
Lower extremity radiographs. (A) A 21‐month‐old boy with osteogenesis imperfecta (OI) type I caused by a frameshift mutation in COL1A1. (B) A 3‐year‐old girl with OI type III caused by a glycine substitution in COL1A2. Note the severe bowing of both femurs and tibias, and healing fracture of the distal right femur. (C) A 3‐year‐old boy with OI type IV caused by a valine deletion in COL1A2. Note the deformities of both femurs and tibias, and healing fracture of the left fibula. (D) A 19‐month‐old boy with OI type V caused by a IFITM5 mutation. Note the hyperplastic callus formation of the left femoral shaft and radiodense metaphyseal bands adjacent to the growth plate.
In addition to OI types I to V, many higher‐number OI types have been proposed based on genetic test results rather than phenotypic features. For example, the Online Mendelian Inheritance of Man database lists a new OI type for each newly discovered gene that is linked to an OI phenotype (http://www.ncbi.nlm.nih.gov/omim/). The drawback of this approach is that recoding the involved gene as an OI type with an arbitrary number adds a layer of complexity to the classification without providing additional information; it would be simpler to just state the name of the gene involved. Describing OI by a combination of the clinical OI phenotype (I to V) and the affected gene as proposed by the 2015 Nosology and Classification of Genetic Skeletal Disorders,(8) provides more useful information to clinicians.
Genetic Causes of Osteogenesis Imperfecta
The majority of individuals with OI have a disease‐causing mutation in one of the two genes that code for collagen type I alpha chains, COL1A1 and COL1A2. Collagen type I is the main component of the organic bone matrix and therefore plays a key role in the integrity of bone tissue.1, 2, 6 Mutations in 18 genes other than COL1A1 and COL1A2 have been associated with OI phenotypes and are listed in the OI mutation database (https://oi.gene.le.ac.uk). These genes are all expressed in osteoblasts, and most of them are directly involved in collagen type I metabolism, even though some of these genes seem to play a role in other aspects of osteoblast function such as Wnt signaling.1 Defects in the newer OI‐related genes usually lead to recessive forms of OI, but two genes (IFITM5, P4HB) are associated with dominant OI, and two genes (PLS3, MBTPS2) lead to X‐linked bone fragility.
Almost all individuals with a typical clinical presentation of OI have a detectable mutation in one of the currently known OI‐associated genes. A recent sequencing study in close to 600 individuals with a clinical diagnosis of OI found disease‐causing mutation in 97% of patients with OI type I (all of whom had COL1A1 or COL1A2 mutations) and in 99% of individuals with the more severe OI types (77% had COL1A1 or COL1A2 mutations).16 Thus, even though some OI genes remain to be discovered, they can be expected to affect only a small number of individuals with a typical OI phenotype.
Regarding genotype–phenotype correlations, COL1A1 mutations leading to haploinsufficiency of the collagen type I alpha 1 chain consistently give rise to OI type I, with mild bone fragility, blue/grey sclera, and normal‐looking teeth. Haploinsufficiency can result not only from COL1A1 stop or frameshift mutations,17 but also from some splice site mutations and deletions of the entire COL1A1 gene.18, 19 These mutations lead to decreased collagen type I production by osteoblasts and other cells and therefore have also been called quantitative collagen mutations.17 Mutations that change the amino acid sequence of the collagen type I alpha chains can be called qualitative mutations. They are frequently caused by glycine substitutions in the triple helical domain of the alpha 1 or alpha 2 chains. Such glycine substitutions can cause the entire range of phenotypic severity of OI, from mild to lethal.20
Mutations in genes other than COL1A1 and COL1A2 are usually associated with a moderate to very severe phenotype (OI type II, III, IV, or V). However, there are some exceptions. Some recessive BMP1 mutations are associated with a mild disease course that is similar to OI type I.21 PLS3 mutations also lead to a clinical picture that can resemble OI type I.22
Genetic Testing for Osteogenesis Imperfecta
Elucidating the disease‐causing mutation is useful in patients who have a clinical diagnosis of OI, as it provides information about the risk of recurrence in a family and allows for the identification of affected family members. Genetic testing can also have implications for clinical management. For example, finding the OI type V specific IFITM5 mutation indicates that the patient has a high risk of developing hyperplastic callus,23 radial head dislocation,24 and abnormalities in the cranio–cervical junction.25 Mutations affecting the C‐propeptide of the collagen type I alpha 1 chain are frequently associated with hip dysplasia,26 and glycine substitutions caused by mutations in exon 49 of COL1A2 may predispose to intracranial hemorrhage.27
Genetic testing can also be useful when the diagnosis is not obvious from the clinical picture. For example, it can sometimes be difficult to distinguish OI type I from other causes of recurrent fractures in children and adolescents.28 This situation was investigated in a study of 94 individuals less than 21 years of age who had a significant fracture history (one or more long‐bone fracture of the lower extremities, two or more long‐bone fractures of the upper extremities, one or more vertebral compression fracture: all in the absence of major trauma), but had white sclera and no signs of dentinogenesis imperfecta; therefore, they did not have unequivocal signs of OI.29 Sequence analysis of a panel of OI‐associated genes found disease‐causing mutations in 26 (28%) of these individuals. Hence, a proportion of children and adolescents with recurrent fractures have OI even if the family history is negative and the phenotypic appearance does not clearly suggest a diagnosis of OI.
As genetic testing is more widely used in research and clinical practice, it is becoming apparent that individuals with a typical OI phenotype only represent the severe end of the spectrum of bone disorders that are caused by mutations affecting collagen type I. As noted, “typical OI mutations” (glycine substitutions in the triple helical domain of the collagen type I alpha 1 and alpha 2 chains) are far more frequent in exome sequencing databases than is expected from the population prevalence of OI, suggesting that the majority of OI mutations do not lead to a readily recognizable OI phenotype.30 Examples for this are two COL1A2 mutations (p.Gly496Ala and p.Gly703Ser) that are found in the Icelandic population with relatively high frequency.31 These mutations are associated with low BMD, fractures, and a slightly below‐average height, but not with other clinical features of OI, such as dentinogenesis imperfecta or blue/grey sclera. Another study has shown that some glycine substitutions caused by COL1A2 mutations can have such mild effects that they do not cause a detectable phenotype in the heterozygous state, but lead to OI only when homozygous.32 It is likely that more examples of partial OI phenotypes will be found when sequencing studies are more frequently performed in adults with a diagnosis of osteoporosis.
Because OI usually is associated with low bone mass, one might choose to limit sequencing for OI mutations to individuals with low BMD. However, this strategy would fail to correctly identify some individuals with OI. Mutations that affect the C‐propeptide cleavage site are associated with frequent fractures in the presence of elevated BMD.33 In a series of 29 individuals with C‐propeptide cleavage site mutations, the mean lumbar spine BMD Z‐score was +2.9, whereas a control group with COL1A1 haploinsufficiency mutations had a mean lumbar spine BMD Z‐score of −2.2.34 Similar observations have been made in patients with mutations affecting BMP1, the enzyme that is responsible for C‐propeptide cleavage.21 These findings highlight the utility of sequence analysis of OI genes in young individuals with recurrent low‐trauma fractures, even if BMD is normal or elevated.
Fractures
The high fracture incidence in OI has been recognized since the first medical description of the disorder in 1690,35 but the fracture epidemiology across the lifespan has been described only recently. A population‐based study in Denmark found that individuals with a diagnosis of OI have eightfold higher fracture rates (all skeletal sites combined) than the general population.36 The relative fracture risk in OI as compared to the general population varied considerably with age (11‐fold higher in individuals with OI below 20 years of age, 6‐fold from 20 to 54 years, and 4‐fold in individuals aged 55 years and higher). As in the general population, women with OI had a higher fracture incidence during menopause than premenopausal women. OI was associated with a particularly high relative risk of femur and lower leg fractures, suggesting that fractures are not only more frequent in OI, but also more severe than in the general population. This study did not have information on OI types, but given that a population‐based cohort was investigated, the majority of study participants must have had OI type I. A limitation of the study is that spine radiographs were not obtained systematically; it is therefore likely that the incidence of vertebral fractures was underestimated.
Atypical femur fractures?
A study in children and adolescents with OI type I caused by COL1A1 haploinsufficiency mutations suggested that the rate of femur and tibia fractures was about 90 times higher than in their healthy peers.37 In adults with OI, it has been estimated that the incidence of femoral shaft fractures is about 35 times higher than in the general population.38
Diaphyseal femur fractures are thus common in OI. Such fractures often occur after minimal trauma, are often transverse, are almost always complete, and are non‐ or minimally comminuted, thereby fulfilling four major criteria for establishing a diagnosis of atypical femur fractures, as defined for fractures that occur in the context of postmenopausal osteoporosis.39 By extension, some case reports and small case series on OI have recently labeled transverse femur fractures as “atypical femur fractures,” especially when they occurred after bisphosphonate use.40
However, transverse diaphyseal femur fractures have been one of the most common types of fracture in OI even before the bisphosphonate era (Fig. 2).41 We found that about 25% of fractures occurring in the nondeformed femurs of individuals with OI fulfill the criteria of atypical femur fractures, regardless of whether they had been exposed to bisphosphonates.42 Overall, systematic studies on more than 300 femur fractures have failed to detect a relationship between bisphosphonate use and diaphyseal femur fractures in OI.42, 43, 44 These studies concurred that the main risk factor for such femur fractures in OI was the severity of the underlying disease rather than bisphosphonate treatment history. We submit that labeling diaphyseal femur fractures that are common in OI as “atypical” because they are rare in the context of postmenopausal osteoporosis is confusing and should be avoided.
Figure 2.
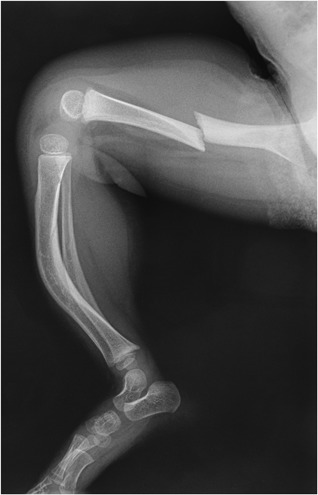
Lateral view of a diaphyseal femur fracture in a 21‐month‐old boy with osteogenesis imperfecta type IV, without previous deformities of the femur and in the absence of prior bisphosphonates treatment.
Other Disease Manifestations
Collagen type I is found in many tissues; therefore, collagen type I mutations might affect those tissues directly. In addition, abnormal bone shape and restricted mobility can lead to secondary problems in extraskeletal tissues. Thus, it is not surprising that many organ systems can be directly or indirectly affected in individuals with OI.3 Here we focus on a few topics that have been highlighted by recent studies.
A population‐based study in Denmark showed that individuals with OI have an increased risk of death at any age, leading to decreased life expectancy.45 The median age of death was 72.4 years for males and 77.4 years for females with OI, which was 10 and 7 years, respectively, earlier than death in the control population. In particular, the OI group had a higher risk of death from respiratory disease, gastrointestinal (GI) disease, and trauma. Although death after trauma can be assumed to be related to fractures, respiratory and GI disease may not be directly linked to bone fragility. The exact nature of these respiratory and GI disorders could not be determined in that study, but the authors speculated that increased use of nonsteroidal anti‐inflammatory drugs could play a role in GI disorders. Another common GI problem in OI is chronic constipation that can be severe in patients with pelvic deformity because of acetabular protrusion.46, 47, 48 Respiratory issues are a well‐known problem in the context of OI. Pulmonary hypoplasia and parenchymal abnormalities are thought to be the cause of death in OI type II.49, 50 Lung histology is also abnormal in several mouse models of OI.51, 52, 53 In addition, lung function can be affected by scoliosis.52, 54, 55
Sleep apnea, a disorder characterized by pauses in breathing, affects 1% to 6% of adults and 2% of children in the general population.56 Sleep apnea can lead to serious health issues, such as cardiovascular disease; through increased daytime sleepiness, it increases the risk of accidents. Sleep apnea appears to be more frequent in OI. A web‐based survey found that self‐reported sleep apnea was present in 32% of adults with severe OI, in 17% with moderate OI, and in 9% with mild OI.57 In a Finnish study, 15% of adults with OI (all types combined) had “diagnosed sleep apnea and used a positive airway pressure ventilator during sleep.”58 In a French cohort of 188 children with OI, 6.4% had sleep‐disordered breathing as documented by polysomnography.59 Thus, sleep apnea may be a potentially serious, but little‐studied aspect of OI.
Dental and craniofacial issues are also a source of major concern in OI. In a recent survey that largely included more severely affected individuals with OI, 75% of respondents noted that dental and craniofacial issues impacted their quality of life.57 A frequent dental abnormality in OI is dentinogenesis imperfecta, which is caused by dysplastic dentin and can lead to dental discoloration, tooth fracture, and attrition.60 Genotype–phenotype correlation studies show that the large majority of patients with OI types III and IV caused by qualitative mutations have dentinogenesis imperfecta, whereas only a small minority of individuals with COL1A1 haploinsufficiency mutations have dentinogenesis imperfecta that is visible on clinical inspection.37, 61, 62, 63 Beyond dentinogenesis imperfecta, tooth agenesis is a common finding in OI.64 These dental abnormalities may contribute to dysplasia of the mandible and maxilla, which frequently lead to malocclusion in individuals with OI types III or IV.65, 66
Cranial base abnormalities are an important complication of OI and can lead to compression of the structures of the posterior fossa, Chiari malformation, spinal cord syrinx formation, and hydrocephalus.67, 68 Such abnormalities are uncommon in mild OI, but develop in more than half of individuals with OI type III.25, 69 Bisphosphonate treatment may slow down the development of cranial base abnormalities, but it does not seem to prevent this complication.25, 70
Several studies have shown that the muscle system is involved in OI in humans and in mouse models.71 Muscle mass is decreased in children and adolescents with OI, even when their smaller body size is taken into account.72 Dynamic muscle tests in children with OI type I and IV have revealed functional deficits that cannot be explained by low muscle mass alone.73, 74 There are many potential reasons for muscle deficits in OI, including a direct effect of collagen type I mutations on muscle,75 joint hyperlaxity,76 and physical inactivity.
Treatment
The clinical management of OI depends on the severity of the phenotype. In uncomplicated OI type I, physical activity may be similar to the general population.77 Treatment needs may be limited to fracture management with the main purpose of medical follow‐up to screen for complications such as vertebral compression fractures, which, if present, may be an indication for i.v. bisphosphonate treatment.30, 78, 79 In more‐severe OI, where long‐bone deformities, scoliosis, and reduced mobility are major concerns, a multidisciplinary orthopedic and rehabilitation intervention program may be required.80
Ensuring adequate vitamin D intake is often recommended,81, 82 but how much vitamin D intake is needed in OI is not well‐established. A recent randomized controlled trial compared two doses of vitamin D supplementation, 400 IU and 2000 IU, in 60 children with OI, most of whom were vitamin D sufficient at baseline [mean serum 25(OH) vitamin D concentration: 67 nmol/L].83 No differences in lumbar spine areal BMD or any other outcome measures other than 25(OH) vitamin D serum levels were found between the two doses of vitamin D supplementation.
Bisphosphonates
Bisphosphonate therapy is the most widely used medical treatment in OI. The use of bisphosphonates in OI has been the focus of several recent reviews30, 84, 85; therefore, only a brief summary is given here. All studies agree that bisphosphonate treatment increases BMD in individuals with OI. However, the few randomized controlled trials that have been performed on bisphosphonate treatment in OI were not powered to assess outcomes other than BMD and had short treatment durations. Systematic reviews of randomized trials in OI consequently are unanimous in their verdict that there is not enough evidence to judge whether bisphosphonate therapy improves outcomes other than BMD.86, 87, 88 Nevertheless, long‐term follow‐up data from observational studies provide some information on clinically relevant treatment outcomes.
When i.v. bisphosphonate treatment is given to growing children, vertebra with deformities from compression fractures can gain a more normal shape through growth (Fig. 3).79, 89, 90, 91 The potential for vertebral reshaping depends on how much growth remains at the time when bisphosphonate treatment is started. In a study that assessed patients who had their first bisphosphonate infusion before 5 years of age, the majority of compressed vertebra had regained a normal shape by the time the children had reached final height.91 However, despite the positive effect on the shape of individual vertebra, bisphosphonates do not seem to have a major effect on the development of scoliosis. Two large studies found that i.v. bisphosphonate treatment slowed down the progression rate of scoliosis in the most severely affected patients, but the prevalence of scoliosis at maturity was not influenced by bisphosphonate treatment history.12, 14
Figure 3.
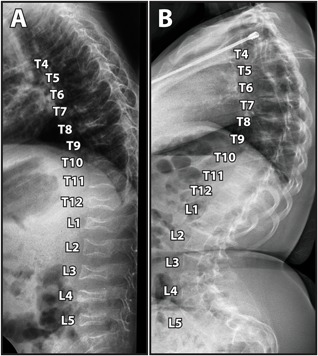
Lateral spine radiograph of a girl with osteogenesis imperfecta type IV showing reshaping of vertebral bodies during bisphosphonate treatment. (A) Age 2 years. (B) Age 15 years.
Bisphosphonate treatment seems to decrease long‐bone fracture rates by 30% to 60% in children with OI.78, 91, 92, 93 Given the high baseline fracture rates in OI, this means that many long‐bone fractures occur despite bisphosphonate treatment. Nevertheless, it has been observed that i.v. bisphosphonate treatment can improve mobility, especially when started early in life.79, 90 Long‐term follow‐up suggests that most children with OI type IV, but not those with OI type III, achieve the ability to walk independently.11
Regarding potential adverse events of bisphosphonates, delayed healing of osteotomy sites is frequently observed in children with OI who have intramedullary rodding procedures. Intravenous bisphosphonate therapy appears to increase the risk for this complication.94 Avoiding bisphosphonate treatment in the 4 months following surgery seems to decrease the percentage of osteotomy sites that heal with delay or not at all.95 Another potential adverse event linked to bisphosphonate therapy is osteonecrosis of the jaw. However, systematic reviews did not identify any confirmed occurrence of this problem in OI.96, 97
Drugs other than bisphosphonates
Many individuals with OI have fractures despite bisphosphonate treatment. Long‐bone deformities, short stature, and scoliosis continue to occur in children with severe OI even if treatment is started in infancy.12, 91 More effective treatment options are therefore needed.
Denosumab is an antiresorptive drug that uses an antibody against RANKL to inhibit osteoclast differentiation and activity.98 Denosumab is approved for the treatment of postmenopausal osteoporosis and other skeletal disorders in adults. Studies in children with OI are being conducted. A few published case series on denosumab treatment in children with OI found a decrease in bone metabolism markers and an increase in BMD.99, 100
Compared to bisphosphonates, denosumab has a much shorter duration of action, but it is not clear how long the antiresorptive effect of denosumab persists in children. The clearance of denosumab seems to depend on the amount of available RANKL.101 The amount of RANKL produced by children is not well‐established. Once the antiresorptive effect of a denosumab injection has run its course, hypercalcemia can develop very quickly in children, as evidenced in an 8‐year‐old girl who received denosumab for juvenile Paget's disease and who had a very high serum calcium concentration only days after a blood test had shown normocalcemia.102 Hypercalcemia and hypercalciuria have also been reported in children with OI receiving denosumab.103 It is possible that the long‐term antiresorptive action of a bisphosphonate is still needed in children receiving denosumab to prevent intermittent hypercalciuria and hypercalcemia and to prevent rapid bone loss once denosumab is discontinued.
Teriparatide, a bone anabolic agent, has been assessed in a randomized controlled trial in adults with OI.104 This showed increased BMD in mild OI, but was less effective in moderate‐to‐severe OI. In accordance with these findings, positive effects of teriparatide were observed in two studies that focused on adults with OI type I.105, 106 It therefore appears that teriparatide can be useful for the treatment of mild OI in adults. However, the use of teriparatide in children is contraindicated, as studies in growing rats have shown an increased risk of osteogenic sarcoma.107
Drugs on the horizon
A variety of novel pharmaceutical compounds have been assessed, mainly in preclinical studies. The OI mouse models that have been used for most of these studies are summarized in Table 1.
Table 1.
Mouse Models of Osteogenesis Imperfecta Used for Preclinical Studies of Novel Drug Treatments
| Mouse | Gene | Nucleotide | Protein | Bone phenotype |
|---|---|---|---|---|
| Dominant | ||||
| Brtl 127 | Col1a1 | c.1546G>T | Triple‐helical glycine substitution (p.Gly349Cys) | 30% perinatal lethality, small body size, rib fractures, long‐bone deformity, bone fragility, reduced BMD; increased bone turnover because of increased osteoclast precursors and reduced osteoblast activity |
| Jrt 128 | Col1a1 | Splice mutation | Exon 9 skipping (deletion of 18 triple‐helical amino acids) | Small body size, short long‐bones, low BV/TV and BMD, spontaneous fractures; reduced tensile properties in the skin, tail tendon tissue reduced |
| G610C 129 | Col1a2 | c.2098G>T | Triple‐helical glycine substitution (p.Gly610Cys) | Moderately severe phenotype, depending on genetic background. Reduced body size, BV/TV, BMD and bone strength |
| oim+/−130 | Col1a2 | c.3983delG | proα2(I) decreased by 50% | Intermediate between oim‐/‐ and wild‐type; normal body size, normal cortical thickness, lower mechanical strength |
| Recessive | ||||
| oim−/−131, 132 | Col1a2 | c.3983delG | proα2(I) absent | Small body size, fractures, limb deformities, cortical thinning, joint laxity, osteopenia, reduced BV/TV and BMD, kyphosis, increased osteoclast activity |
| Crtap−/− 133, 134 | Crtap | CRTAP absent | Moderate phenotype: growth delay, skeletal deformity, kyphosis, reduced BV/TV but high bone mineralization, cartilage dysplasia, decreased material properties of the skin |
+/− = heterozygous for the mutation; −/− = homozygous for the mutation; BMD = bone mineral density; BV/TV = bone volume per tissue volume.
Antibody‐mediated sclerostin inhibition is an approach to stimulate bone formation that has been used in clinical studies to treat osteoporosis in adults.98 Regarding OI, sclerostin antibody treatment increased bone mass and strength in several mouse models such as Brtl, G610C, and Crtap‐/‐,108, 109, 110 but was less beneficial in the Jrt mouse.111 Short‐term treatment with sclerostin antibody stimulated bone formation, suppressed bone resorption, and increased BMD in a small group of adults with OI.112
One issue with short‐acting treatment agents such as sclerostin antibody is that the bone will rapidly revert towards its baseline status once the treatment is discontinued, as has been noted in adults with osteoporosis.113 In the Brtl mouse, bone loss after the discontinuation of sclerostin antibody treatment could be prevented by subsequent administration of a bisphosphonate.114 It might also be advantageous to give bisphosphonates concomitant with sclerostin antibody, at least during growth. Studies in both G610C and Brtl mice have shown that the metaphyseal trabecula of growing mice were retained through the antiresorptive action of bisphosphonates and could serve as templates for the formation of new bone that was stimulated by sclerostin inhibition.115, 116
Transforming growth factor beta (TGFβ) plays an important role in determining bone mass and quality.117 Mice overexpressing TGFβ in bone have osteoporosis,118 whereas mice with genetic TGFβ inhibition have stronger bones.119 TGFβ signaling is increased in Crtap‐/‐ and G610C mice, and pharmacologic inhibition of TGFβ with a neutralizing antibody led to higher bone mass and stronger bones.120 TGFβ inhibition also improved the histological abnormalities of lung tissue in Crtap‐/‐ mice. Jrt mice also seem to have increased TGFβ signaling in bone tissue, but treatment with TGFβ neutralizing antibody did not have an obvious effect on bones or lungs.53, 121 It therefore appears possible that the effect of TGFβ inhibition in OI varies with the disease‐causing mutation and the related bone phenotype.
The muscle phenotype that is seen in human OI is replicated in some mouse models, such as oim and Jrt mice.71 Given the close correlation between muscle and bone,122 this led to the hypothesis that treatments targeting muscle could be beneficial for both muscle and bone in OI. Myostatin and activin A are two members of the TGFβ family that inhibit muscle growth by signaling through the activin receptor 2B.123 A soluble activin receptor 2B that inhibits both myostatin and activin A led to a marked increase in muscle and bone mass in oim mice.124 A slightly different soluble activin receptor 2B had similarly beneficial effects on muscles and bones in oim and G610C mice.125, 126 Inhibition of myostatin and activin A warrants further exploration as novel treatment approaches in OI.
Disclosures
The authors state that they have no conflicts of interest.
Acknowledgments
This study was supported by the Shriners of North America and the Fonds de recherche du Québec–Santé. Frank Rauch received a study grant from PreciThera Inc.
References
- 1. Kang H, Aryal ACS, Marini JC. Osteogenesis imperfecta: new genes reveal novel mechanisms in bone dysplasia. Transl Res. 2017;181:27–48. [DOI] [PubMed] [Google Scholar]
- 2. Lim J, Grafe I, Alexander S, Lee B. Genetic causes and mechanisms of osteogenesis imperfecta. Bone. 2017;102:40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marini JC, Forlino A, Bachinger HP, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. [DOI] [PubMed] [Google Scholar]
- 4. Lindahl K, Astrom E, Rubin CJ, et al. Genetic epidemiology, prevalence, and genotype‐phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet. 2015;23:1042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Folkestad L. Mortality and morbidity in patients with osteogenesis imperfecta in Denmark. Dan Med J. 2018;65. [PubMed] [Google Scholar]
- 6. Morello R. Osteogenesis imperfecta and therapeutics. Matrix Biol. 2018;71‐72:294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palomo T, Vilaca T, Lazaretti‐Castro M. Osteogenesis imperfecta: diagnosis and treatment. Curr Opin Endocrinol Diabetes Obes. 2017;24:381–8. [DOI] [PubMed] [Google Scholar]
- 8. Bonafe L, Cormier‐Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167A:2869–92. [DOI] [PubMed] [Google Scholar]
- 9. Germain‐Lee EL, Brennen FS, Stern D, et al. Cross‐sectional and longitudinal growth patterns in osteogenesis imperfecta: implications for clinical care. Pediatr Res. 2016;79:489–95. [DOI] [PubMed] [Google Scholar]
- 10. Jain M, Tam A, Shapiro JR, et al. Growth characteristics in individuals with osteogenesis imperfecta in North America: results from a multicenter study. Genet Med. 2018. Jul 4. doi: 10.1038/s41436-018-0045-1. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Montpetit K, Palomo T, Glorieux FH, Fassier F, Rauch F. Multidisciplinary treatment of severe osteogenesis imperfecta: functional outcomes at skeletal maturity. Arch Phys Med Rehabil. 2015;96:1834–9. [DOI] [PubMed] [Google Scholar]
- 12. Sato A, Ouellet J, Muneta T, Glorieux FH, Rauch F. Scoliosis in osteogenesis imperfecta caused by COL1A1/COL1A2 mutations—genotype‐phenotype correlations and effect of bisphosphonate treatment. Bone. 2016;86:53–7. [DOI] [PubMed] [Google Scholar]
- 13. Barber LA, Abbott C, Nakhate V, Do AND, Blissett AR, Marini JC. Longitudinal growth curves for children with classical osteogenesis imperfecta (types III and IV) caused by structural pathogenic variants in type I collagen. Genet Med. 2018. Oct 1. doi: 10.1038/s41436-018-0307-y. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anissipour AK, Hammerberg KW, Caudill A, et al. Behavior of scoliosis during growth in children with osteogenesis imperfecta. J Bone Joint Surg Am. 2014;96:237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rauch F, Moffatt P, Cheung M, et al. Osteogenesis imperfecta type V: marked phenotypic variability despite the presence of the IFITM5 c.‐14C>T mutation in all patients. J Med Genet. 2013;50:21–4. [DOI] [PubMed] [Google Scholar]
- 16. Bardai G, Moffatt P, Glorieux FH, Rauch F. DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int. 2016;27:3607–13. [DOI] [PubMed] [Google Scholar]
- 17. Willing MC, Deschenes SP, Scott DA, et al. Osteogenesis imperfecta type I: molecular heterogeneity for COL1A1 null alleles of type I collagen. Am J Hum Genet. 1994;55:638–47. [PMC free article] [PubMed] [Google Scholar]
- 18. Schleit J, Bailey SS, Tran T, et al. Molecular outcome, prediction, and clinical consequences of splice variants in COL1A1, which encodes the proalpha1(I) chains of type I procollagen. Hum Mutat. 2015;36:728–39. [DOI] [PubMed] [Google Scholar]
- 19. Bardai G, Lemyre E, Moffatt P, et al. Osteogenesis imperfecta type I caused by COL1A1 deletions. Calcif Tissue Int. 2016;98:76–84. [DOI] [PubMed] [Google Scholar]
- 20. Marini JC, Forlino A, Cabral WA, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fahiminiya S, Al‐Jallad H, Majewski J, et al. A polyadenylation site variant causes transcript‐specific BMP1 deficiency and frequent fractures in children. Hum Mol Genet. 2015;24:516–24. [DOI] [PubMed] [Google Scholar]
- 22. van Dijk FS, Zillikens MC, Micha D, et al. PLS3 mutations in X‐linked osteoporosis with fractures. N Engl J Med. 2013;369:1529–36. [DOI] [PubMed] [Google Scholar]
- 23. Cheung MS, Glorieux FH, Rauch F. Natural history of hyperplastic callus formation in osteogenesis imperfecta type V. J Bone Miner Res. 2007;22:1181–6. [DOI] [PubMed] [Google Scholar]
- 24. Fassier AM, Rauch F, Aarabi M, Janelle C, Fassier F. Radial head dislocation and subluxation in osteogenesis imperfecta. J Bone Joint Surg Am. 2007;89:2694–704. [DOI] [PubMed] [Google Scholar]
- 25. Cheung MS, Arponen H, Roughley P, et al. Cranial base abnormalities in osteogenesis imperfecta: Phenotypic and genotypic determinants. J Bone Miner Res. 2011;26:405–13. [DOI] [PubMed] [Google Scholar]
- 26. Kishta W, Abduljabbar FH, Gdalevitch M, Rauch F, Hamdy R, Fassier F. Hip dysplasia in children with osteogenesis imperfecta: association with collagen type I C‐propeptide mutations. J Pediatr Orthop. 2017;37:479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Faqeih E, Roughley P, Glorieux FH, Rauch F. Osteogenesis imperfecta type III with intracranial hemorrhage and brachydactyly associated with mutations in exon 49 of COL1A2. Am J Med Genet A. 2009;149A:461–5. [DOI] [PubMed] [Google Scholar]
- 28. Fiscaletti M, Coorey CP, Biggin A, et al. Diagnosis of recurrent fracture in a pediatric cohort. Calcif Tissue Int. 2018;103:529–39. [DOI] [PubMed] [Google Scholar]
- 29. Bardai G, Ward LM, Trejo P, Moffatt P, Glorieux FH, Rauch F. Molecular diagnosis in children with fractures but no extraskeletal signs of osteogenesis imperfecta. Osteoporos Int. 2017;28:2095–101. [DOI] [PubMed] [Google Scholar]
- 30. Trejo P, Rauch F. Osteogenesis imperfecta in children and adolescents‐new developments in diagnosis and treatment. Osteoporos Int. 2016;27:3427–37. [DOI] [PubMed] [Google Scholar]
- 31. Styrkarsdottir U, Thorleifsson G, Eiriksdottir B, et al. Two rare mutations in the COL1A2 gene associate with low bone mineral density and fractures in Iceland. J Bone Miner Res. 2016;31:173–9. [DOI] [PubMed] [Google Scholar]
- 32. Costantini A, Tournis S, Kampe A, et al. Autosomal recessive osteogenesis imperfecta caused by a novel homozygous COL1A2 mutation. Calcif Tissue Int. 2018;103:353–8. [DOI] [PubMed] [Google Scholar]
- 33. Lindahl K, Barnes AM, Fratzl‐Zelman N, et al. COL1 C‐propeptide cleavage site mutations cause high bone mass osteogenesis imperfecta. Hum Mutat. 2011;32:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cundy T, Dray M, Delahunt J, et al. Mutations that alter the carboxy‐terminal‐propeptide cleavage site of the chains of type I procollagen are associated with a unique osteogenesis imperfecta phenotype. J Bone Miner Res. 2018;33:1260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Charlier P, Perciaccante A, Bianucci R. Oldest medical description of osteogenesis imperfecta (17th Century, France). Clin Anat. 2017;30:128–9. [DOI] [PubMed] [Google Scholar]
- 36. Folkestad L, Hald JD, Ersboll AK, et al. Fracture rates and fracture sites in patients with osteogenesis imperfecta: a nationwide register‐based cohort study. J Bone Miner Res. 2017;32:125–34. [DOI] [PubMed] [Google Scholar]
- 37. Ben Amor IM, Roughley P, Glorieux FH, Rauch F. Skeletal clinical characteristics of osteogenesis imperfecta caused by haploinsufficiency mutations in COL1A1. J Bone Miner Res. 2013;28:2001–7. [DOI] [PubMed] [Google Scholar]
- 38. Goudriaan WA, Harsevoort GJ, van Leeuwen M, Franken AA, Janus GJM. Incidence and treatment of femur fractures in adults with osteogenesis imperfecta: an analysis of an expert clinic of 216 patients. Eur J Trauma Emerg Surg. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shane E, Burr D, Abrahamsen B, et al. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the american society for bone and mineral research. J Bone Miner Res. 2014;29:1–23. [DOI] [PubMed] [Google Scholar]
- 40. Nguyen HH, D MvdL, Verkerk AJ, Milat F, Zillikens MC, Ebeling PR. Genetic risk factors for atypical femoral fractures (AFFs): A systematic review. JBMR Plus. 2018;2:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dent JA, Paterson CR. Fractures in early childhood: osteogenesis imperfecta or child abuse? J Pediatr Orthop. 1991;11:184–6. [DOI] [PubMed] [Google Scholar]
- 42. Trejo P, Fassier F, Glorieux FH, Rauch F. Diaphyseal femur fractures in osteogenesis imperfecta: Characteristics and relationship with bisphosphonate treatment. J Bone Miner Res. 2017;32:1034–9. [DOI] [PubMed] [Google Scholar]
- 43. Vuorimies I, Arponen H, Valta H, et al. Timing of dental development in osteogenesis imperfecta patients with and without bisphosphonate treatment. Bone. 2017;94:29–33. [DOI] [PubMed] [Google Scholar]
- 44. Andersen JD, Bunger MH, Rahbek O, Hald JD, Harslof T, Langdahl BL. Do femoral fractures in adult patients with osteogenesis imperfecta imitate atypical femoral fractures? A case series. Osteoporos Int. 2018. [DOI] [PubMed] [Google Scholar]
- 45. Folkestad L, Hald JD, Canudas‐Romo V, et al. Mortality and causes of death in patients with osteogenesis imperfecta: A register‐based nationwide cohort study. J Bone Miner Res. 2016;31:2159–66. [DOI] [PubMed] [Google Scholar]
- 46. Lee JH, Gamble JG, Moore RE, Rinsky LA. Gastrointestinal problems in patients who have type‐III osteogenesis imperfecta. J Bone Joint Surg Am. 1995;77:1352–6. [DOI] [PubMed] [Google Scholar]
- 47. Violas P, Fassier F, Hamdy R, Duhaime M, Glorieux FH. Acetabular protrusion in osteogenesis imperfecta. J Pediatr Orthop. 2002;22:622–5. [PubMed] [Google Scholar]
- 48. Fiegel MJ. Cesarean delivery and colon resection in a patient with type III osteogenesis imperfecta. Semin Cardiothorac Vasc Anesth. 2011;15:98–101. [DOI] [PubMed] [Google Scholar]
- 49. Shapiro JR, Burn VE, Chipman SD, et al. Pulmonary hypoplasia and osteogenesis imperfecta type II with defective synthesis of alpha I(1) procollagen. Bone. 1989;10:165–71. [DOI] [PubMed] [Google Scholar]
- 50. Thibeault DW, Pettett G, Mabry SM, Rezaiekhaligh MM. Osteogenesis imperfecta type IIA and pulmonary hypoplasia with normal alveolar development. Pediatr Pulmonol. 1995;20:301–6. [DOI] [PubMed] [Google Scholar]
- 51. Baldridge D, Lennington J, Weis M, et al. Generalized connective tissue disease in Crtap‐/‐ mouse. PLoS ONE. 2010;5:e10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thiele F, Cohrs CM, Flor A, et al. Cardiopulmonary dysfunction in the Osteogenesis imperfecta mouse model Aga2 and human patients are caused by bone‐independent mechanisms. Hum Mol Genet. 2012;21:3535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baglole CJ, Liang F, Traboulsi H, et al. Pulmonary and diaphragmatic pathology in collagen type I alpha1 mutant mice with osteogenesis imperfecta. Pediatr Res. 2018;83:1165–71. [DOI] [PubMed] [Google Scholar]
- 54. Wekre LL, Kjensli A, Aasand K, Falch JA, Eriksen EF. Spinal deformities and lung function in adults with osteogenesis imperfecta. Clin Respir J. 2014;8:437–43. [DOI] [PubMed] [Google Scholar]
- 55. Tam A, Chen S, Schauer E, et al. A multicenter study to evaluate pulmonary function in osteogenesis imperfecta. Clin Genet. 2018;94:502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Levy P, Kohler M, McNicholas WT, et al. Obstructive sleep apnoea syndrome. Nat Rev Dis Primers. 2015;1:15015. [DOI] [PubMed] [Google Scholar]
- 57. Tosi LL, Oetgen ME, Floor MK, et al. Initial report of the osteogenesis imperfecta adult natural history initiative. Orphanet J Rare Dis. 2015;10:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arponen H, Waltimo‐Siren J, Valta H, Makitie O. Fatigue and disturbances of sleep in patients with osteogenesis imperfecta—a cross‐sectional questionnaire study. BMC Musculoskelet Disord. 2018;19:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leotard A, Taytard J, Aouate M, Boule M, Forin V, Lallemant‐Dudek P. Diagnosis, follow‐up and management of sleep‐disordered breathing in children with osteogenesis imperfecta. Ann Phys Rehabil Med. 2018;61:135–9. [DOI] [PubMed] [Google Scholar]
- 60. Malmgren B, Lindskog S. Assessment of dysplastic dentin in osteogenesis imperfecta and dentinogenesis imperfecta. Acta Odontol Scand. 2003;61:72–80. [DOI] [PubMed] [Google Scholar]
- 61. Rauch F, Lalic L, Roughley P, Glorieux FH. Genotype‐phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. Eur J Hum Genet. 2010;18:642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Andersson K, Dahllof G, Lindahl K, et al. Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta ‐ A retrospective cohort study. PLoS One. 2017;12:e0176466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hald JD, Folkestad L, Swan CZ, et al. Osteogenesis imperfecta and the teeth, eyes, and ears‐a study of non‐skeletal phenotypes in adults. Osteoporos Int. 2018;29:2781–9. [DOI] [PubMed] [Google Scholar]
- 64. Malmgren B, Andersson K, Lindahl K, et al. Tooth agenesis in osteogenesis imperfecta related to mutations in the collagen type I genes. Oral Dis. 2017;23:42–9. [DOI] [PubMed] [Google Scholar]
- 65. Rizkallah J, Schwartz S, Rauch F, et al. Evaluation of the severity of malocclusions in children affected by osteogenesis imperfecta with the peer assessment rating and discrepancy indexes. Am J Orthod Dentofacial Orthop. 2013;143:336–41. [DOI] [PubMed] [Google Scholar]
- 66. Bendixen KH, Gjorup H, Baad‐Hansen L, et al. Temporomandibular disorders and psychosocial status in osteogenesis imperfecta—a cross‐sectional study. BMC Oral Health. 2018;18:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ibrahim AG, Crockard HA. Basilar impression and osteogenesis imperfecta: a 21‐year retrospective review of outcomes in 20 patients. J Neurosurg Spine. 2007;7:594–600. [DOI] [PubMed] [Google Scholar]
- 68. Menezes AH. Specific entities affecting the craniocervical region: osteogenesis imperfecta and related osteochondrodysplasias: medical and surgical management of basilar impression. Childs Nerv Syst. 2008;24:1169–72. [DOI] [PubMed] [Google Scholar]
- 69. Arponen H, Makitie O, Haukka J, et al. Prevalence and natural course of craniocervical junction anomalies during growth in patients with osteogenesis imperfecta. J Bone Miner Res. 2012;27:1142–9. [DOI] [PubMed] [Google Scholar]
- 70. Arponen H, Vuorimies I, Haukka J, Valta H, Waltimo‐Siren J, Makitie O. Cranial base pathology in pediatric osteogenesis imperfecta patients treated with bisphosphonates. J Neurosurg Pediatr. 2015;15:313–20. [DOI] [PubMed] [Google Scholar]
- 71. Phillips CL, Jeong Y. Osteogenesis imperfecta: muscle‐bone interactions when bi‐directionally compromised. Curr Osteoporos Rep. 2018. [DOI] [PubMed] [Google Scholar]
- 72. Palomo T, Glorieux FH, Schoenau E, Rauch F. Body composition in children and adolescents with osteogenesis imperfecta. J Pediatr. 2016;169:232–7. [DOI] [PubMed] [Google Scholar]
- 73. Veilleux LN, Darsaklis VB, Montpetit K, Glorieux FH, Rauch F. Muscle function in osteogenesis imperfecta type IV. Calcif Tissue Int. 2017;101:362–70. [DOI] [PubMed] [Google Scholar]
- 74. Veilleux LN, Lemay M, Pouliot‐Laforte A, Cheung MS, Glorieux FH, Rauch F. Muscle anatomy and dynamic muscle function in osteogenesis imperfecta type I. J Clin Endocrinol Metab. 2014;99:E356–62. [DOI] [PubMed] [Google Scholar]
- 75. Gillies AR, Lieber RL. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve. 2011;44:318–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Brizola E, Staub AL, Felix TM. Muscle strength, joint range of motion, and gait in children and adolescents with osteogenesis imperfecta. Pediatr Phys Ther. 2014;26:245–52. [DOI] [PubMed] [Google Scholar]
- 77. Pouliot‐Laforte A, Veilleux LN, Rauch F, Lemay M. Physical activity in youth with osteogenesis imperfecta type I. J Musculoskelet Neuronal Interact. 2015;15:171–6. [PMC free article] [PubMed] [Google Scholar]
- 78. Lindahl K, Kindmark A, Rubin CJ, et al. Decreased fracture rate, pharmacogenetics and BMD response in 79 Swedish children with osteogenesis imperfecta types I, III and IV treated with Pamidronate. Bone. 2016;87:11–8. [DOI] [PubMed] [Google Scholar]
- 79. Alcausin MB, Briody J, Pacey V, et al. Intravenous pamidronate treatment in children with moderate‐to‐severe osteogenesis imperfecta started under three years of age. Horm Res Paediatr. 2013;79:333–40. [DOI] [PubMed] [Google Scholar]
- 80. Mueller B, Engelbert R, Baratta‐Ziska F, et al. Consensus statement on physical rehabilitation in children and adolescents with osteogenesis imperfecta. Orphanet J Rare Dis. 2018;13:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saraff V, Hogler W. Osteoporosis in children: diagnosis and management. Eur J Endocrinol. 2015;173:R185–97. [DOI] [PubMed] [Google Scholar]
- 82. Thomas IH, DiMeglio LA. Advances in the classification and treatment of osteogenesis imperfecta. Curr Osteoporos Rep. 2016;14:1–9. [DOI] [PubMed] [Google Scholar]
- 83. Plante L, Veilleux LN, Glorieux FH, Weiler H, Rauch F. Effect of high‐dose vitamin D supplementation on bone density in youth with osteogenesis imperfecta: A randomized controlled trial. Bone. 2016;86:36–42. [DOI] [PubMed] [Google Scholar]
- 84. Biggin A, Munns CF. Long‐term bisphosphonate therapy in osteogenesis imperfecta. Curr Osteoporos Rep. 2017;15:412–8. [DOI] [PubMed] [Google Scholar]
- 85. Simm PJ, Biggin A, Zacharin MR, et al. Consensus guidelines on the use of bisphosphonate therapy in children and adolescents. J Paediatr Child Health. 2018;54:223–33. [DOI] [PubMed] [Google Scholar]
- 86. Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2014;7:Cd005088. [DOI] [PubMed] [Google Scholar]
- 87. Hald JD, Evangelou E, Langdahl BL, Ralston SH. Bisphosphonates for the prevention of fractures in osteogenesis imperfecta: meta‐analysis of placebo‐controlled trials. J Bone Miner Res. 2015;30:929–33. [DOI] [PubMed] [Google Scholar]
- 88. Rijks EB, Bongers BC, Vlemmix MJ, et al. Efficacy and safety of bisphosphonate therapy in children with osteogenesis imperfecta: a systematic review. Horm Res Paediatr. 2015;84:26–42. [DOI] [PubMed] [Google Scholar]
- 89. Semler O, Beccard R, Palmisano D, et al. Reshaping of vertebrae during treatment with neridronate or pamidronate in children with osteogenesis imperfecta. Horm Res Paediatr. 2011;76:321–7. [DOI] [PubMed] [Google Scholar]
- 90. Astrom E, Jorulf H, Soderhall S. Intravenous pamidronate treatment of infants with severe osteogenesis imperfecta. Arch Dis Child. 2007;92:332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Palomo T, Fassier F, Ouellet J, Sato A, Montpetit K, Glorieux FH, Rauch F. Intravenous bisphosphonate therapy of young children with osteogenesis imperfecta: skeletal findings during follow up throughout the growing years. J Bone Miner Res. 2015;30:2150–7. [DOI] [PubMed] [Google Scholar]
- 92. Sakkers R, Kok D, Engelbert R, et al. Skeletal effects and functional outcome with olpadronate in children with osteogenesis imperfecta: a 2‐year randomised placebo‐controlled study. Lancet. 2004;363:1427–31. [DOI] [PubMed] [Google Scholar]
- 93. Bishop N, Adami S, Ahmed SF, et al. Risedronate in children with osteogenesis imperfecta: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2013;382:1424–32. [DOI] [PubMed] [Google Scholar]
- 94. Munns CF, Rauch F, Zeitlin L, Fassier F, Glorieux FH. Delayed osteotomy but not fracture healing in pediatric osteogenesis imperfecta patients receiving pamidronate. J Bone Miner Res. 2004;19:1779–86. [DOI] [PubMed] [Google Scholar]
- 95. Anam EA, Rauch F, Glorieux FH, Fassier F, Hamdy R. Osteotomy healing in children with osteogenesis imperfecta receiving bisphosphonate treatment. J Bone Miner Res. 2015;30:1362–8. [DOI] [PubMed] [Google Scholar]
- 96. Hennedige AA, Jayasinghe J, Khajeh J, Macfarlane TV. Systematic review on the incidence of bisphosphonate related osteonecrosis of the jaw in children diagnosed with osteogenesis imperfecta. J Oral Maxillofac Res. 2013;4:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hernandez M, Phulpin B, Mansuy L, Droz D. Use of new targeted cancer therapies in children: effects on dental development and risk of jaw osteonecrosis: a review. J Oral Pathol Med. 2017;46:321–6. [DOI] [PubMed] [Google Scholar]
- 98. Khosla S, Hofbauer LC. Osteoporosis treatment: recent developments and ongoing challenges. Lancet Diabetes Endocrinol. 2017;5:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hoyer‐Kuhn H, Netzer C, Koerber F, Schoenau E, Semler O. Two years' experience with denosumab for children with osteogenesis imperfecta type VI. Orphanet J Rare Dis. 2014;9:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hoyer‐Kuhn H, Franklin J, Allo G, et al. Safety and efficacy of denosumab in children with osteogenesis imperfecta—a first prospective trial. J Musculoskelet Neuronal Interact. 2016;16:24–32. [PMC free article] [PubMed] [Google Scholar]
- 101. Sutjandra L, Rodriguez RD, Doshi S, et al. Population pharmacokinetic meta‐analysis of denosumab in healthy subjects and postmenopausal women with osteopenia or osteoporosis. Clin Pharmacokinet. 2011;50:793–807. [DOI] [PubMed] [Google Scholar]
- 102. Grasemann C, Schundeln MM, Hovel M, et al. Effects of RANK‐ligand antibody (denosumab) treatment on bone turnover markers in a girl with juvenile Paget's disease. J Clin Endocrinol Metab. 2013;98:3121–6. [DOI] [PubMed] [Google Scholar]
- 103. Trejo P, Rauch F, Ward L. Hypercalcemia and hypercalciuria during denosumab treatment in children with osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact. 2018;18:76–80. [PMC free article] [PubMed] [Google Scholar]
- 104. Orwoll ES, Shapiro J, Veith S, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest. 2014;124:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Leali PT, Balsano M, Maestretti G, et al. Efficacy of teriparatide vs neridronate in adults with osteogenesis imperfecta type I: a prospective randomized international clinical study. Clin Cases Miner Bone Metab. 2017;14:153–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gatti D, Rossini M, Viapiana O, et al. Teriparatide treatment in adult patients with osteogenesis imperfecta type I. Calcif Tissue Int. 2013;93:448–52. [DOI] [PubMed] [Google Scholar]
- 107. Vahle JL, Sato M, Long GG, et al. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1‐34) for 2 years and relevance to human safety. Toxicol Pathol. 2002;30:312–21. [DOI] [PubMed] [Google Scholar]
- 108. Sinder BP, Eddy MM, Ominsky MS, Caird MS, Marini JC, Kozloff KM. Sclerostin antibody improves skeletal parameters in a Brtl/+ mouse model of osteogenesis imperfecta. J Bone Miner Res. 2013;28:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jacobsen CM, Barber LA, Ayturk UM, et al. Targeting the LRP5 pathway improves bone properties in a mouse model of osteogenesis imperfecta. J Bone Miner Res. 2014;29:2297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Grafe I, Alexander S, Yang T, et al. Sclerostin antibody treatment improves the bone phenotype of Crtap(‐/‐) mice, a model of recessive osteogenesis imperfecta. J Bone Miner Res. 2016;31:1030–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Roschger A, Roschger P, Keplingter P, et al. Effect of sclerostin antibody treatment in a mouse model of severe osteogenesis imperfecta. Bone. 2014;66:182–8. [DOI] [PubMed] [Google Scholar]
- 112. Glorieux FH, Devogelaer JP, Durigova M, et al. BPS804 anti‐sclerostin antibody in adults with moderate osteogenesis imperfecta: results of a randomized phase 2a trial. J Bone Miner Res. 2017;32:1496–504. [DOI] [PubMed] [Google Scholar]
- 113. Recknor CP, Recker RR, Benson CT, et al. The effect of discontinuing treatment with blosozumab: follow‐up results of a phase 2 randomized clinical trial in postmenopausal women with low bone mineral density. J Bone Miner Res. 2015;30:1717–25. [DOI] [PubMed] [Google Scholar]
- 114. Perosky JE, Khoury BM, Jenks TN, et al. Single dose of bisphosphonate preserves gains in bone mass following cessation of sclerostin antibody in Brtl/+ osteogenesis imperfecta model. Bone. 2016;93:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Little DG, Peacock L, Mikulec K, et al. Combination sclerostin antibody and zoledronic acid treatment outperforms either treatment alone in a mouse model of osteogenesis imperfecta. Bone. 2017;101:96–103. [DOI] [PubMed] [Google Scholar]
- 116. Olvera D, Stolzenfeld R, Marini JC, Caird MS, Kozloff KM. Low dose of bisphosphonate enhances sclerostin antibody‐induced trabecular bone mass gains in Brtl/+ osteogenesis imperfecta mouse model. J Bone Miner Res. 2018;33:1272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Grafe I, Alexander S, Peterson JR, Snider TN, Levi B, Lee B, Mishina Y. TGF‐beta family signaling in mesenchymal differentiation. Cold Spring Harb Perspect Biol. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Erlebacher A, Derynck R. Increased expression of TGF‐beta 2 in osteoblasts results in an osteoporosis‐like phenotype. J Cell Biol. 1996;132:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Filvaroff E, Erlebacher A, Ye J, et al. Inhibition of TGF‐beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development. 1999;126:4267–79. [DOI] [PubMed] [Google Scholar]
- 120. Grafe I, Yang T, Alexander S, et al. Excessive transforming growth factor‐beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20:670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Tauer JT, Abdullah S, Rauch F. Effect of anti‐TGFbeta treatment in a mouse model of severe osteogenesis imperfecta. J Bone Miner Res. Oct 24. doi: 10.1002/jbmr.3617. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 122. Veilleux LN, Pouliot‐Laforte A, Lemay M, Cheung MS, Glorieux FH, Rauch F. The functional muscle‐bone unit in patients with osteogenesis imperfecta type I. Bone. 2015;79:52–7. [DOI] [PubMed] [Google Scholar]
- 123. Latres E, Mastaitis J, Fury W, et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun. 2017;8:15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. DiGirolamo DJ, Singhal V, Chang X, Lee SJ, Germain‐Lee EL. Administration of soluble activin receptor 2B increases bone and muscle mass in a mouse model of osteogenesis imperfecta. Bone Res. 2015;3:14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Jeong Y, Daghlas SA, Kahveci AS, et al. Soluble activin receptor type IIB decoy receptor differentially impacts murine osteogenesis imperfecta muscle function. Muscle Nerve. 2018;57:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Jeong Y, Daghlas SA, Xie Y, H et al. Skeletal response to soluble activin receptor type IIB in mouse models of osteogenesis imperfecta. J Bone Miner Res. 2018;33:1760–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Forlino A, Porter FD, Lee EJ, Westphal H, Marini JC. Use of the Cre/lox recombination system to develop a non‐lethal knock‐in murine model for osteogenesis imperfecta with an alpha1(I) G349C substitution. Variability in phenotype in BrtlIV mice. J Biol Chem. 1999;274:37923–31. [DOI] [PubMed] [Google Scholar]
- 128. Chen F, Guo R, Itoh S, et al. First mouse model for combined osteogenesis imperfecta and Ehlers‐Danlos syndrome. J Bone Miner Res. 2014;29:1412–23. [DOI] [PubMed] [Google Scholar]
- 129. Daley E, Streeten EA, Sorkin JD, et al. Variable bone fragility associated with an Amish COL1A2 variant and a knock‐in mouse model. J Bone Miner Res. 2010;25:247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Saban J, Zussman MA, Havey R, Patwardhan AG, Schneider GB, King D. Heterozygous oim mice exhibit a mild form of osteogenesis imperfecta. Bone. 1996;19:575–9. [DOI] [PubMed] [Google Scholar]
- 131. Chipman SD, Sweet HO, McBride DJ, Jr. , et al. Defective pro alpha 2(I) collagen synthesis in a recessive mutation in mice: a model of human osteogenesis imperfecta. Proc Natl Acad Sci U S A. 1993;90:1701–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Phillips CL, Bradley DA, Schlotzhauer CL, et al. Oim mice exhibit altered femur and incisor mineral composition and decreased bone mineral density. Bone. 2000;27:219–26. [DOI] [PubMed] [Google Scholar]
- 133. Morello R, Bertin TK, Chen Y, et al. CRTAP is required for prolyl 3‐ hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. [DOI] [PubMed] [Google Scholar]
- 134. Fratzl‐Zelman N, Morello R, Lee B, et al. CRTAP deficiency leads to abnormally high bone matrix mineralization in a murine model and in children with osteogenesis imperfecta type VII. Bone. 2010;46:820–6. [DOI] [PMC free article] [PubMed] [Google Scholar]