

Abstract
Background and Purpose
Parkinson's disease (PD) involves an initial loss of striatal dopamine terminals evolving into degeneration of dopamine neurons in substantia nigra (SN), which can be modelled by 6‐hydroxydopamine (6‐OHDA) administration. Adenosine A2A receptor blockade attenuates PD features in animal models, but the source of the adenosine causing A2A receptor over‐activation is unknown. As ATP is a stress signal, we have tested if extracellular catabolism of adenine nucleotides into adenosine (through ecto‐5′‐nucleotidase or CD73) leads to A2A receptor over‐activation in PD.
Experimental Approach
Effects of blocking CD73 with α,β‐methylene ADP (AOPCP) were assayed in 6‐OHDA‐treated rats and dopamine‐differentiated neuroblastoma SH‐SY5Y cells.
Key Results
6‐OHDA increased ATP release and extracellular conversion into adenosine through CD73 up‐regulation in SH‐SY5Y cells. Removing extracellular adenosine with adenosine deaminase, blocking CD73 with AOPCP, or blocking A2A receptors with SCH58261 were equi‐effective in preventing 6‐OHDA‐induced damage in SH‐SY5Y cells. In vivo striatal exposure to 6‐OHDA increased ATP release and extracellular formation of adenosine from adenosine nucleotides and up‐regulated CD73 and A2A receptors in striatal synaptosomes. Intracerebroventricular administration of AOPCP phenocopied effects of SCH58261, attenuating 6‐OHDA‐induced (a) increase of contralateral rotations after apomorphine, (b) reduction of dopamine content in striatum and SN, (c) loss of TH staining in striatum and SN, (d) motor dysfunction in the cylinder test, and (e) short‐term memory impairment in the object recognition test.
Conclusion and Implications
Our data indicate that increased ATP‐derived adenosine formation is responsible for A2A receptor over‐activation in PD, suggesting CD73 as a new target to manage PD.
Abbreviations
- 6‐OHDA
6‐hydroxydopamine
- ADA
adenosine deaminase
- AOPCP
α,β‐methylene ADP
- DOPAC
3,4‐dihydroxyphenylacetic acid
- MTT
3‐(4,5‐dimethylthiazole‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- PD
Parkinson's disease
- SN
substantia nigra
What is already known
Antagonists of adenosine A2A receptors alleviate motor symptoms of Parkinson's disease.
What this study adds
A2A receptor overfunction requires increased ATP release and CD73‐mediated catabolism into adenosine.
What is the clinical significance
CD73 is a key controller of A2A receptors and a novel target in Parkinson's disease.
1. INTRODUCTION
ATP is a well‐established stress signal in the brain (Rodrigues, Tomé, & Cunha, 2015). There is concurrent evidence that ATP can either signal directly through its P2 receptors or alternatively signal through adenosine receptors after the extracellular catabolism of ATP into adenosine by ecto‐nucleotidases (see Rodrigues et al., 2015). In animal models of Parkinson's disease (PD), ATP can signal directly, namely, through P2X7 receptors to control neurodegeneration and motor impairments (Carmo et al., 2014; Marcellino et al., 2010). However, the best documented purinergic receptors controlling PD are adenosine A2A receptors (see Morelli, Carta, & Jenner, 2009). In fact, not only does the consumption of caffeine (an adenosine receptor antagonist) correlate inversely with the incidence of PD, but there is also robust evidence obtained in different animal models that the excessive function of A2A receptors is deleterious, as demonstrated by the ability of pharmacological and genetic deletion of these receptors to attenuate the evolution of motor symptoms and of dopaminergic neurodegeneration (reviewed in Morelli et al., 2009). Although the source of the adenosine responsible for the over‐activation of A2A receptors has not been determined, the recently described tight physical and functional coupling between A2A receptors and CD73 or ecto‐5′‐nucleotidase (the enzyme forming adenosine from extracellular adenine nucleotides) in the striatum (Augusto et al., 2013; Ena, De Backer, Schiffmann, & de Kerchove d'Exaerde, 2013) prompts the hypothesis that the release of ATP as a stress signal might be the source of the adenosine responsible for the over‐activation of A2A receptors contributing to the development of PD. Furthermore, there is previous evidence to suggest that extracellular ATP increases neurodegeneration (see Rodrigues et al., 2015) and that the activity of CD73 can affect neurodegeneration (Boeck, Kroth, Bronzatto, & Vendite, 2007; Meng et al., 2019) both directly through the control of neuronal survival (Heilbronn, Maienschein, Carstensen, Gann, & Zimmermann, 1995) and indirectly through its ability to control inflammatory responses (see Antonioli, Pacher, Vizi, & Haskó, 2013).
Combining the use of simple cellular models with rodent models of PD, we now directly tested if the extracellular catabolism of adenine nucleotides into adenosine is the underlying mechanism responsible for the over‐activation of A2A receptors causing PD evolution. We found that CD73 is up‐regulated in cellular and animal models of PD and the inhibition of CD73 phenocopied the neuroprotective effects of A2A receptor antagonists, indicating that increased ATP‐derived adenosine formation is responsible for over‐activation of A2A receptors in PD.
2. METHODS
2.1. Animals and drug administration
We follow the Editorial guidelines on experimental design and analysis in pharmacology. All animal care and experimental procedures in this study were conducted in accordance with the principles and procedures outlined as “3Rs” in the guidelines of the European Union (2010/63/EU) and FELASA and were approved by the Portuguese Ethical Committee (DGAV) and by the Ethics Committee on Animal Experimentation of the Federal University of Ceará (CEPA). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the Editorial guidelines made by the British Journal of Pharmacology. Adult male Wistar rats (220–250 g; RGD_13508588) were obtained from either the animal house of the Federal University of Ceará or Charles River (Barcelona, Spain) and were maintained at 23–25°C, with 12‐hr light to 12‐hr dark cycle, and standard diet and tap water ad libitum. Rats were anaesthetized with a combination of ketamine (100 mg·kg−1, i.p.) and xylazine (20 mg·kg−1, i.p.), and unilateral intra‐striatal 6‐hydroxydopamine (6‐OHDA) injections (18 μg/3 μl in a 0.02% ascorbate‐saline solution) were performed through a 5‐μl Hamilton® syringe using a stereotaxic apparatus (Stoelting, USA) at the following coordinates (mm): Site 1: L: −2.5, AP: +0.5, V: +5.0; Site 2: L: −3, AP: −0.5, V: +6.0; and Site 3: L: −3.7, AP: −0.9, V: +6.5 from bregma. Sham‐operated rats received vehicle and were used as controls. Behavioural tests were carried out from 9 a.m. until 4 p.m., from the 15th until the 18th day after surgery, and animals were killed on the 19th day. All behavioural tests were carried out in a sound‐attenuated room with an 8‐lx illumination, to which the animals were previously habituated. All apparatus was cleaned with 20% ethyl alcohol to remove any odours after testing each animal.
When we tested the A2A receptor antagonist SCH58261, it was administered intraperitoneally daily at 7 p.m., starting in the day of 6‐OHDA administration until the animals were killed. The CD73 inhibitor α,β‐methylene ADP (AOPCP; 100 μM) was administered (0.25 μl·hr−1 for 14 days) directly into the right lateral ventricle through osmotic minipumps (Model 1002; Alzet Corporation), placed in a subcutaneous pocket in the dorsal region, and connected via polyethelene tubing to an intracranial cannulae (Alzet Brain Infusion Kit II) targeting the following coordinates relative to bregma: 1.5 mm posterior, 1.0 mm lateral, and 3.7 mm below the horizontal plane of bregma. Control animals received a similar intracerebroventricular administration of saline.
2.2. Open field test
The rats were tested for locomotor activity 15 days after surgery using an open field apparatus as previously described (e.g., Carmo et al., 2014), which consisted of a black acrylic chamber (50 × 50 cm), with 50‐cm‐high walls, and the floor was divided into four squares of equal size. Each rat was positioned in the centre of the arena and allowed to explore freely. The number of crossings and rearings was scored for 5 min.
2.3. Object recognition test
On the next morning, rats performed the object recognition test in the same arena to probe short‐term memory (Bevins & Besheer, 2006). Rats first underwent a training/acquisition session, in which they were exposed during 10 min to two identical objects in the arena (two 100‐ml blue bottles), placed 7 cm away from the walls of the open field. The test session was performed 60 min after training where one object was replaced (a familiar and a novel one, i.e., the 100‐ml blue bottle and a 50‐ml green tube). The time spent exploring the new (T novel) and familiar (T familiar) object was recorded during 5 min. Object exploration was defined as the orientation of the nose to the object at a distance ≤2 cm, touching with forepaws or nose and sniffing and biting the objects, but climbing on the objects was not considered. Recognition memory was expressed as a recognition index calculated as (Tnovel)/(Tnovel+Tfamiliar), as previously described (Canas et al., 2009).
2.4. Limb use asymmetry (cylinder test)
The test was carried out 17 days after surgery, as previously described (Carmo et al., 2014). Rats were placed in a transparent cylinder (30 cm high and 20 cm in diameter) to record the use of each forelimbs during rearing behavior, over 5 min. Behaviour was expressed in terms of (a) per cent use of the contralateral (impaired) forelimb, (b) per cent use of the ipsilateral (non‐impaired) forelimb, and (c) per cent simultaneous use of both limbs, relative to the total number of paws used during rearing activity.
2.5. Rotational behaviour
Eighteen days after surgery (after all other behavioural tests), animals were tested for rotational behaviour after receiving an intraperitoneal bolus injection of apomorphine hydrochloride (0.6 mg·kg−1; Sigma‐Aldrich). Rotational testing was conducted as previously described (Carmo et al., 2014), by placing rats inside a cylindrical container (33‐cm diameter and 35‐cm height) and counting the number of contralateral rotations (number of 360° contralateral turns) during 60 min.
2.6. Determination of monoamine levels
The measurement of dopamine and 3,4‐dihydroxyphenylacetic acid (DOPAC) in striatal and mesencephalic tissue (n = 6 per group) was carried out by HPLC, as previously described (Borycz et al., 2007). Tissue homogenates (10% w/v) were sonicated in 0.1‐M HClO4 for 30 s, centrifuged at 4°C for 15 min at 9,000× g, and the supernatant was filtered (0.2 μm, Millipore). A 20‐μl sample was then injected into the C‐18 HPLC column. The mobile phase was 0.163‐M citric acid (pH 3.0), containing 0.02‐mM NaCl with 0.69‐mM sodium octanesulfonic acid as the ion pairing reagent, 4% v/v acetonitrile and 1.7% v/v tetrahydrofuran. Dopamine and DOPAC were electrochemically detected, using an amperometric detector (Shimadzu). The amount of monoamines was determined by comparison with freshly prepared standards, and their concentrations were expressed as ng·mg−1 of tissue.
2.7. Immunohistochemical analysis
The antibody‐based procedures used in this study comply with the Editorial guidelines made by the British Journal of Pharmacology. Immunohistochemistry was essentially carried out as previously described (Carmo et al., 2014). Briefly, the animals were anaesthetized with sodium thiopental and transcardially perfused with ice‐cold PBS followed by 4% paraformaldehyde in PBS. The brains were removed, post‐fixed in 4% paraformaldehyde for 16–24 hr, and cryoprotected in 30% sucrose for 48 hr at 4°C. The brain was then frozen in dry ice, and 50‐μm coronal sections were prepared using a cryostat (Leica CM3050 S) at −21°C. TH immunodetection was performed in a one‐in‐six series of 50‐μm free‐floating sections. The sections were washed three times for 10 min with PBS and incubated with PBS supplemented with 10% methanol and 1.05% hydrogen peroxide for 40 min at room temperature, to block endogenous peroxidase‐like activities. After washing three times for 10 min with PBS and blocking endogenous proteins with 10% normal goat serum in PBS supplemented with Triton X‐100 (blocking solution) for 2 hr at room temperature, the sections were incubated with the primary antibody (rabbit anti‐TH, 1:1,000; ref. AB152 from Merck‐Millipore; RRID:AB_390204) diluted in blocking solution at 4°C for 48 hr. The sections were then washed with PBS before incubation for 2 hr at room temperature with a secondary goat anti‐rabbit biotinylated antibody (1:200, Vector Labs; RRID:AB_2313606) diluted in blocking solution and washed with PBS. The avidin‐biotin‐HRP conjugate (ABC Staining System, Santa Cruz Biotechnology) was used for 40 min at room temperature for amplification of the signal and revealed with DAB Peroxidase Substrate Kit (Vector labs). The reaction was stopped by washing with PBS before mounting on gelatin‐coated slides, dried, dehydrated by a gradient of ethanol, and cleared with xylene. Finally, the sections were coverslipped with Entellan (Merck). The stained brain sections were visualized using an Olympus BX41 epi‐fluorescent microscope equipped with an Olympus DP71 camera. Immunoreactivity was measured by semi‐quantitative densitometric analysis using an image‐analysis program (ImageJ software; RRID:SCR_003070). With freehand selection, we measured the OD of both striata (ipsilateral and contralateral) using the anterior commissure as a negative control. The values obtained for the sham‐operated group were averaged, and the values of other groups calculated as a percentage of that mean.
2.8. Preparation of striatal synaptosomes
Striatal synaptosomes (purified synapses) were prepared as previously described (Canas et al., 2009). After decapitation, the two striata were dissected and homogenized in sucrose (0.32 M) solution containing 1‐mM EDTA, 10‐mM HEPES, and 1 mg·ml−1 BSA (Sigma), pH 7.4 at 4°C, supplemented with a protease inhibitor, PMSF (0.1 mM), a cocktail of inhibitors of proteases (CLAP 1%, Sigma), and the antioxidant DTT (1 μM). The homogenate was centrifuged at 3,000× g for 10 min at 4°C, and the resulting supernatant was further centrifuged at 14,000× g for 12 min at 4°C. The resulting pellet (P2 fraction) was resuspended in 1 ml of a 45% (v/v) Percoll solution in HEPES buffer (140‐mM NaCl, 5‐mM KCl, 25‐mM HEPES, 1‐mM EDTA, and 10‐mM glucose, pH 7.4). After centrifugation at 14,000× g for 2 min at 4°C, the white top layer was collected (synaptosomal fraction), resuspended in 1‐ml HEPES buffer, and further centrifuged at 14,000× g for 2 min at 4°C. The pellet was then resuspended in appropriate solutions for binding assays or Western blot analysis. The purity of this synaptic fraction has been previously quantified as >95% (Canas et al., 2009).
2.9. Neurotoxicity in SH‐SY5Y cells
SH‐SY5Y cells (LGC Promochem; RRID:CVCL_0019) were cultured in a 1:1 mixture of Ham's F12 and DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) and a mix of antibiotic and antimycotic (Invitrogen), in a humidified atmosphere of 5% of CO2 in air at 37°C. Differentiation was induced by lowering the FBS in culture medium to 1% and adding 10‐μM retinoic acid during 7 days (see Lopes et al., 2010). After differentiation, cells were resuspended in Krebs‐HEPES (125‐mM NaCl, 3‐mM KCl, 10‐mM glucose, 10‐mM HEPES, 2.2‐mM KH2PO4, 1.2‐mM MgCl2, and 2‐mM CaCl2, pH 7.4) and treated for 30 min with 2 U·ml−1 adenosine deaminase (ADA), 100‐μM AOPCP, or 100 nM SCH58261 before exposure to 30‐μM 6‐OHDA during 24 hr in the continuous presence of the tested drugs. 6‐OHDA was freshly prepared in 0.1% ascorbic acid to avoid oxidation.
The activity of cellular dehydrogenases, taken as an index of cellular dysfunction (Mosmann, 1983), was estimated by the quantification of the ability of the cells to reduce 3‐(4,5‐dimethylthiazole‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) to form a blue formazan product. At the end of the exposure to drugs, cells were incubated with 1 mg of MTT (from a recently prepared stock of 5 mg·ml−1 in PBS), and the cultures were returned to the incubator for 3 hr to allow MTT reduction to proceed. After the incubation media were removed, the purple formazan crystals were dissolved in 1‐ml isopropanol during 30 min on a shaking table. The resulting purple solution was spectrophotometrically measured at 540 nm.
Cellular damage was additionally evaluated by measuring the amount of cytoplasmic LDH released into the medium, using an assay based on the reduction of NAD+ by the action of LDH. At the end of the treatments, the medium from each well was transferred to clean flat‐bottom plate to proceed with enzymic analysis, according to the in vitro toxicology assay kit (Hospitex Diagnostics). The effect of 6‐OHDA (30 μM) on SH‐SY5Y cells was further confirmed by quantification of the number of cells labelled with Hoescht 33258. Twenty‐four hours after beginning 6‐OHDA treatment, cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature, followed by a 10‐min incubation with Hoescht 33258 dye (1 mg·ml−1, Invitrogen) at room temperature. Cells were then washed and maintained in PBS and imaged on a Zeiss Axiovert 200 microscope (Zeiss) using the 20× objective to count the average in three different fields of the total number of stained cells as well as the number of apoptotic‐like cells, characterized by displaying brighter spots of Hoescht 33258 staining.
2.10. Extracellular ATP quantification
Samples (50 μl) were collected at different time points (2 min up to 6 hr) from the incubation medium of cultured cells and stored at −80°C. ATP quantification was carried out using a luciferin–luciferase bioluminescence assay, as previously described (Ferreira et al., 2015). Samples were mixed with 50 μl of the luciferin–luciferase bioluminescent ATP assay mix from Sigma (FLAAM), and the light emitted at 565 nm was quantified using a luminometer (either a Perkin Elmer Victor3 or a Wallac 1250). For the quantification of ATP outflow from synaptosomes (Ferreira et al., 2015), a suspension containing synaptosomes, an ATP assay mix (with luciferin and luciferase; from Sigma), and Krebs‐HEPES solution was equilibrated at 25°C up to 10 min to ensure the functional recovery of the nerve terminals. After 60 s to measure the basal outflow, the evoked release of ATP was triggered with 32 mM of KCl (isomolar substitution of NaCl in the Krebs‐HEPES solution). After this chemical stimulation, the light emitted was recorded for an additional 200 s. The evoked release of ATP was calculated by integration of the area of the peak upon subtraction of the estimated basal ATP outflow.
ATP levels were quantified using a calibration curve of ATP standards. The quantification in each condition and time point was performed in triplicates.
2.11. Extracellular catabolism of ATP and of AMP
The extracellular catabolism of ATP or of AMP was evaluated as previously described (Cunha, Sebastião, & Ribeiro, 1992). When using synaptosomes, they were maintained in an incubation vial with Krebs‐HEPES solution at 30.5°C. After 10 min of incubation, either ATP (10 μM) or AMP (10 μM) was added, without or with AOPCP (100 μM), and aliquots were collected from the reaction medium (60 μl) at 0, 2, 5, 10, and 15 min. When using differentiated SH‐SY5Y cells, these were incubated for 24 hr in Krebs‐HEPES solution without or with 6‐OHDA (30 μM), the medium was changed, and either ATP (10 μM) or AMP (10 μM) was added, without or with AOPCP (100 μM), and aliquots were collected from the reaction medium (60 μl) at 0, 2, 5, 10, 15, and/or 20 min. Each aliquot was centrifuged at 14,000× g (15 s at 4°C), and the supernatant was stored at −20°C for HPLC quantification of adenosine (Cunha et al., 1992). The remaining synaptosomes or cells in each assay were either pelleted or scraped and homogenized in 2% (v/v) Triton X‐100, to quantify protein with the BCA assay.
2.12. Western blot analysis
Membranes from either striatal synaptosomes or differentiated SH‐SY5Y cells were resuspended in RIPA (50‐mM Tris, 150‐mM NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 1‐mM EDTA, and 0.1% sodium dodecyl sulphate) plus cOmplete tablets EDTA‐FREE Easy pack (Roche), 0.1‐mM DTT, and 0.1‐mM PMSF, and the amount of protein was determined using the bicinchoninic acid method (Pierce) to carefully dilute all preparations to a final concentration of 2 μg protein·μl−1 in Laemmli buffer. The analysis of CD73 and A2A receptor density was carried out as previously described (Augusto et al., 2013) using mouse anti‐A2AR (1:1,000, Merck‐Millipore, clone 7F6‐G5‐A2), and goat anti‐murine CD73 (1:1,000, Santa Cruz Biotechnology, sc‐25603), followed by the appropriate secondary antibody conjugated with alkaline phosphatase (Amersham) before revealing the membranes with ECF (Amersham). Membranes were visualized with an imaging system (VersaDoc 3000, Bio‐Rad) and quantified with the Quantity One software (Bio‐Rad; RRID:SCR_005375). The membranes were re‐probed and tested for α‐tubulin immunoreactivity (1:1,000; Sigma‐Aldrich, ref. T6074) to confirm that similar amounts of protein were applied to the gels (Canas et al., 2009).
2.13. Receptor binding assay
The binding assays were performed as previously described (Cunha, Canas, Oliveira, & Cunha, 2006). Briefly, synaptosomes were vigorously resuspended in a pre‐incubation solution (containing 50‐mM Tris, 1‐mM EDTA, and 2‐mM EGTA, pH 7.4) at 4°C and centrifuged at 25,000× g for 20 min at 4°C; the supernatants were discarded, and the pellet, corresponding to the synaptosomal membranes, was resuspended in the pre‐incubation solution, and a sample was collected to determine the protein concentration using the bicinchoninic acid assay (Pierce). ADA (2 U·ml−1, Roche) was added, and the membranes were incubated for 30 min at 37°C to remove endogenous adenosine. The mixtures were centrifuged at 25,000× g for 20 min at 4°C, and the pelleted membranes were resuspended in Tris‐Mg solution (containing 50‐mM Tris and 10‐mM MgCl2, pH 7.4) with 4 U·ml−1 of ADA. Binding with 3 nM of the selective A2A receptor antagonist, 3H‐SCH58261 (specific activity of 77 Ci·mmol−1; prepared by GE Healthcare and offered by E. Ongini, Schering‐Plough, Italy) was performed for 1 hr at room temperature with 54–75 μg of protein, with constant swirling. The binding reactions were stopped by addition of 4 ml of ice‐cold Tris‐Mg solution and filtration through Whatman GF/C glass microfibre filters (GE Healthcare) in a filtration system (Millipore). The radioactivity was measured with 2 ml of scintillation liquid (AquaSafe 500Plus, Zinsser Analytic). The specific binding was expressed as fmol·mg−1 protein and was estimated by subtraction of the non‐specific binding, which was measured in the presence of 12 μM of xanthine amine congener (Sigma), an antagonist of adenosine receptors. All binding assays were performed in duplicate.
2.14. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. All data are presented as the mean ± SEM of n experiments. All animals were randomized before the first drug treatment, and the experimenters carrying out the behavioural, neurochemical, and biochemical studies were blind to the treatments of the animals, ex vivo preparations, or cells. We selected the number of animals per group based on our previous experience of the variability of the measurements without carrying out a formal power analysis. The time‐dependent effects of 6‐OHDA on ATP release was first assessed with a one‐way ANOVA followed by a Dunnett's post hoc test to compare different groups with the control group. In the in vitro studies, the effect of drugs was analysed using a two‐tailed unpaired t test. When testing the impact of a tested drug on the effects of 6‐OHDA in in vivo or ex vivo experiments, the data were first analysed with a two‐way ANOVA eventually followed by a Newman–Keuls post hoc test. All tests were performed using GraphPad Prism 6.0 software (RRID:SCR_002798) considering significance at a 95% confidence interval.
2.15. Materials
The following compounds were supplied as follows: SCH58261 (Tocris); AOPCP, apomorphine HCl, ATP. AMP, 6‐OHDA (Sigma).
2.16. Nomenclature of targets and ligands
Key protein and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/2018: (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017; Alexander, Peters et al., 2017).
3. RESULTS
3.1. 6‐OHDA bolsters ATP release and formation of extracellular adenosine in SH‐SY5Y cells
SH‐SY5Y cells differentiated with retinoic acid to acquire a dopaminergic phenotype become susceptible to 6‐OHDA‐induced damage (Carmo et al., 2014; Sun et al., 2016). We tested a concentration of 6‐OHDA of 30 μM as it caused a ~50% reduction of cellular functionality and viability within the tested concentration range of 3–300 μM (data not shown). In accordance with the concept that ATP is a stress signal associated with neuronal damage (Rodrigues et al., 2015), the exposure of differentiated SH‐SY5Y cells to 6‐OHDA (30 μM) triggered a rapid and sustained increase of the extracellular levels of ATP (Figure 1a). Compared to the levels of extracellular ATP at the time of addition of 6‐OHDA (Time 0; 1.98 ± 0.28 nM, n = 10), which were similar to the average levels of extracellular ATP in SH‐SY5Y cells incubated in the absence of 6‐OHDA (1.74 ± 0.19 nM, n = 10), the levels of extracellular ATP were significantly higher 5 min after the addition of 6‐OHDA and reached a maximal value after 90 min, remaining at this elevated levels up to 6 hr after the addition of 6‐OHDA (Figure 1a).
Figure 1.
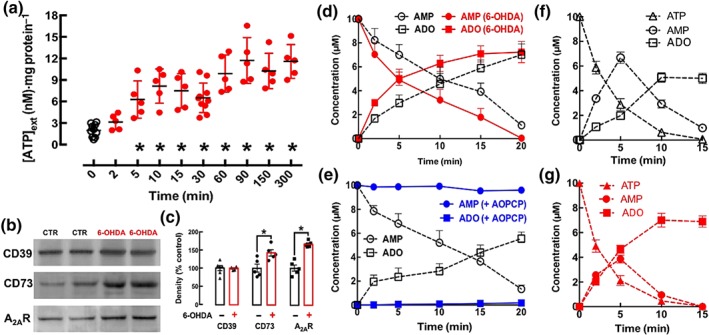
6‐Hydroxydopamine (6‐OHDA, 30 μM) leads to a rapid and sustained increase of extracellular ATP and an up‐regulation of both A2A receptors and of the ecto‐5′‐nucleotidase (CD73) responsible for an increased formation of extracellular adenosine (ADO) derived from adenine nucleotides in differentiated SY5Y neuroblastoma cells. (a) The extracellular levels of ATP were rapidly (within 5 min) increased in a sustained manner (up to 6 hr) after the exposure to 6‐OHDA. (b, c) Western blot analysis showed an increased density of immuno‐reactive A2A receptors (A2AR) and of CD73 but not of the ecto‐nucleoside triphosphate diphosphohydrolase‐1 (NTPDase1 / CD39) at 2 hr after adding 6‐OHDA. (d) The extracellular catabolism of AMP (10 μM, added at Time 0) and the formation of adenosine were greater in cells exposed for 2 hr to 6‐OHDA, compared those when cells were exposed to Krebs solution only. (e) α,β‐Methylene ADP (AOPCP, 100 μM) blocked the extracellular hydrolysis of AMP (10 μM, added at Time 0) to adenosine. (f, g) The extracellular catabolism of ATP (10 μM, added at Time 0) was similar in the control (f) and 6‐OHDA‐treated cells (g), but the formation of extracellular AMP was lower and the formation of ADO was higher in cells exposed for 2 hr to 6‐OHDA (g) compared with data from cells exposed to Krebs solution only (f). Except for the representative Western blots (b), the data shown are means ± SEM of n = 5–10 in (a), n = 5–6 in (c), n = 4 (d), n = 3 (e), and n = 4 (f, g). *P < .05, significantly different from control; Dunnett's test in (a) and a two‐tailed unpaired Student's t test in (c)
As the exposure of SH‐SY5Y cells to 6‐OHDA (3–300 μM) only triggers apoptosis and cellular death after 2–4 hr (Sun et al., 2016), we investigated adaptive changes of the purinergic system in SH‐SY5Y cells after 2 hr of exposure to 6‐OHDA (30 μM). As illustrated in Figure 1b, the exposure to 6‐OHDA did not change the density of CD39, the ecto‐enzyme hydrolyzing ATP to AMP, but caused an early up‐regulation of CD73, the ecto‐enzyme hydolysing AMP to adenosine formation (see Cunha, 2001), as well as an up‐regulation of A2A receptors. Thus, whereas there was no significant variation of CD39 immunoreactivity in 6‐OHDA‐treated SH‐SY5Y cells compared to control, there was an increase of the immunoreactivity of CD73 and of A2A receptors (Figure 1c).
This increased density of CD73 was paralleled by an increased extracellular conversion of AMP into adenosine typified by a faster catabolism of extracellularly added AMP and a larger formation of extracellular adenosine (Figure 1d). This extracellular catabolism of AMP and formation of adenosine in SH‐SY5Y cells was indeed mediated by CD73 as its selective inhibitor, AOPCP, used at a concentration (100 μM) previously validated to be supra‐maximal and selective in different preparations (Cunha et al., 1992; Cunha, Sebastião, & Ribeiro, 1998; George et al., 2015), virtually blocked the extracellular catabolism of AMP and formation of adenosine in the medium of SH‐SY5Y cells (Figure 1e). This shows AOPCP to be a useful tool to explore the role of ATP‐derived adenosine in SH‐SY5Y cells. The increased activity of CD73 without alteration of CD39 activity after exposure to 6‐OHDA allowed a more efficient ATP‐derived adenosine formation. As illustrated in Figure 1f,g, after exposure to 6‐OHDA, there was a similar extracellular catabolism of ATP accompanied by a lower accumulation of extracellular AMP at 5 min and a larger formation of extracellular adenosine, at 10 min.
3.2. Role of ATP‐derived adenosine and A2A receptors in 6‐OHDA‐induced dysfunction and damage of SH‐SY5Y cells
We next investigated the effects of A2A receptors and of ATP‐derived adenosine in the cellular dysfunction and damage caused by exposure of SH‐SY5Y cells to 6‐OHDA. As shown in Figure 2a, the exposure for 24 hr to 6‐OHDA (30 μM) decreased the reduction potential of SH‐SY5Y cells, as assessed by the decreased MTT reduction, a measure of cellular dysfunction (Mosmann, 1983). Blockade of A2A receptors with a supramaximal concentration of the selective A2A receptor antagonist SCH58261 (100 nM; Lopes et al., 2004; Rebola, Lujan, Cunha, & Mulle, 2008) attenuated 6‐OHDA‐induced cellular dysfunction and a similar attenuation was observed upon removal of endogenous extracellular adenosine with a maximally effective and selective concentration (2 U·ml−1; see Cunha et al., 1998) of ADA (Figure 2a). Notably, the elimination of ATP‐derived adenosine extracellular formation with 100‐μM AOPCP was equi‐effective to attenuate 6‐OHDA‐induced cellular dysfunction (Figure 2a), whereas none of these tested drugs affected MTT reduction by SH‐SY5Y cells in the absence of 6‐OHDA (Figure 2a). Moreover, there were no additive effects neither of ADA nor of AOPCP in the simultaneous presence of SCH58261 (n = 4; Figure 2a), suggesting that ATP‐derived adenosine is the main source of the extracellular adenosine responsible for the over‐activation of A2A receptors contributing for the 6‐OHDA‐induced cellular dysfunction in SH‐SY5Y cells.
Figure 2.
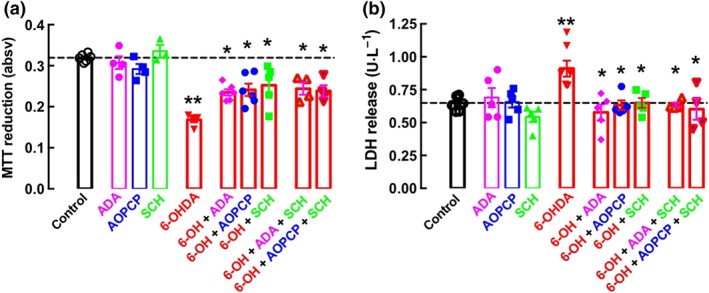
Removing extracellular adenosine, blocking ATP‐derived formation of extracellular adenosine, or antagonizing A2A receptors (A2AR) are equi‐effective in attenuating the dysfunction and damage in differentiated SY5Y neuroblastoma cells exposed to 6‐hydroxydopamine (6‐OHDA). Dopamine‐differentiated SY5Y cells were incubated for 24 hr in the absence or presence of 6‐OHDA (30 μM) without or with either adenosine deaminase (ADA, 2 U·ml−1, which converts adenosine into inosine) or α,β‐methylene ADP (AOPCP, 100 μM, which inhibits the extracellular formation of adenosine from adenine nucleotides) or SCH58261 (100 nM, a selective antagonist of A2A receptors) to measure either cellular dysfunction (assessed by a decrease of the reduction activity of cells using the colorimetric indicator MTT) in (a) or cellular damage (assessed by the release of the cytosolic enzyme, LDH) in (b). The data shown are means ± SEM of n = 3–6 in (a) and n = 4–8 in (b). **P < .05, significantly different from control; *P < .05, significantly different from 6‐OHDA; two‐tailed unpaired Student's t test
Cellular dysfunction caused by the exposure of SH‐SY5Y cells to 6‐OHDA was accompanied by cellular damage, as evaluated by the release to the extracellular medium of the cytosolic marker LDH. Thus, the exposure for 24 hr to 6‐OHDA (30 μM) damaged SH‐SY5Y cells, as shown by increased extracellular LDH activity (Figure 2b). Cellular damage was further confirmed by quantification of SH‐SY5Y cells stained with Hoescht 33258, exposure for 24 hr to 6‐OHDA (30 μM) decreasing the total number of cells per field and increasing the number of apoptotic‐like cells per field. Equally effective in attenuating 6‐OHDA‐induced cellular damage (Figure 2b) were SCH58261 (100 nM), ADA (2 U·ml−1), and AOPCP (100 μM), whereas none of these tested drugs altered extracellular LDH activity in the absence of 6‐OHDA (Figure 2b). Moreover, there were no additive effects, either of ADA or of AOPCP, in the simultaneous presence of SCH58261 (Figure 2b), further strengthening the conclusion that ATP‐derived adenosine is the main source of the extracellular adenosine responsible for the over‐activation of A2A receptors contributing to 6‐OHDA‐induced cellular damage in SH‐SY5Y cells.
3.3. 6‐OHDA‐induced motor dysfunction is accompanied by a striatal up‐regulation of A2A receptors, whose over‐activation is critical for the expression of damage and dysfunction
There is robust evidence in different animal models of PD that blockade of A2A receptors ameliorates neurodegeneration and dysfunction associated with PD (see Morelli et al., 2009). Indeed, 6‐OHDA‐lesioned rats display a significant increase of the number of apomorphine‐induced contralateral rotations (measured during 60 min) compared to sham‐operated rats (Figure 3a). This response was attenuated, by more than 50%, upon treatment with the selective A2A receptor antagonist SCH58261 (0.1 mg·kg−1·day−1), whereas SCH58261 was devoid of effects in sham‐operated rats (Figure 3a).
Figure 3.
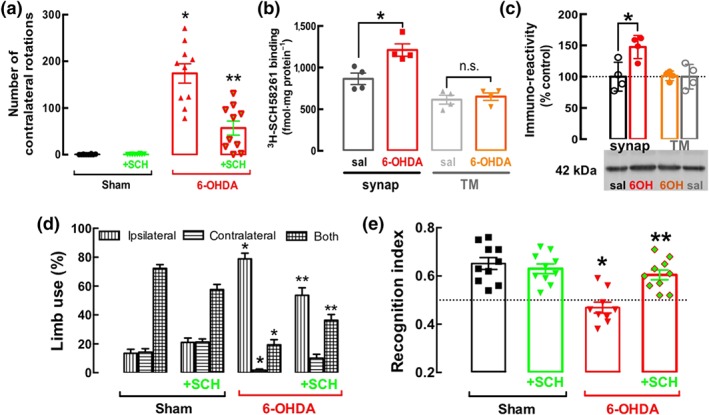
The unilateral administration of 6‐OHDA in the striatum of rats triggers a hemiparkinsonian state together coupled to an increased density of A2AR receptors selectively in synapses, and blockade of these receptors attenuates motor and memory dysfunction caused by 6‐OHDA. (a) The A2A receptor antagonist SCH58261 (0.1 mg·kg−1, i.p. daily) attenuated the apomorphine‐induced rotational behaviour in 6‐OHDA‐lesioned rats, whereas it was without effect in saline‐injected rats, as shown by the number of contralateral rotations counted for 60 min after the administration of apomorphine (0.6 mg·kg−1), evaluated 18 days after unilateral administration of 6‐OHDA in the striatum. There was an up‐regulation of A2A receptors in the striatum of rats 19 days after the administration of 6‐OHDA, selectively in synaptosomal membranes (synap) but not in total membranes (TMs), as evaluated by the specific binding of 3H‐SCH58261 (6 nM) (b) or by Western blot analysis (c). (d) The unilateral injection of 6‐OHDA decreased the use of the contralateral limb, especially together with the ipsilateral limb, and this was recovered upon treatment with SCH58261 (0.1 mg·kg−1, i.p. daily). (e) SCH58261 also prevented the decreased recognition memory of rats intra‐striatally injected with 6‐OHDA, as evaluated in the novel object recognition task: All groups of rats investigated equally both objects in the training phase (not shown), but only 6‐OHDA‐lesioned rats failed to discriminate the novel object during the test phase (e), without alteration of the total distance travelled during the test session between the four groups (not shown). The data shown are means ± SEM of n = 10 rats per group in all behavioural tests in (a, d, e), n = 4 in the binding assays in (b), and n = 5 in the Western blot analysis in (c). *P < .05, significantly different from control; two‐tailed unpaired Student's t test (b, c) or two‐way ANOVA with Newman–Keuls post hoc test (a, d, e). **P < .05, significantly different from 6‐OHDA; two‐way ANOVA with Newman–Keuls post hoc test.
This dopamine‐related maladaptive motor control was accompanied by an up‐regulation of A2A receptors mainly in synapses. The specific binding density of 3H‐SCH58261 to striatal synaptosomal membranes was increased, by about 50% of the control value, in 6‐OHDA‐treated rats, whereas there was no significant difference of 3H‐SCH58261 binding to striatal total membranes (which predominantly include non‐synaptic and non‐neuronal membranes) of control and 6‐OHDA‐treated rats (Figure 3b). Consistent with these data, there was a similar increase of A2A receptor immunoreactivity in striatal synaptosomal membranes of 6‐OHDA‐treated rats, whereas there was no significant modification of A2A receptor immunocontent in total striatal membranes ( Figure 3c).
This increased density of synaptic A2A receptors translated into an A2A receptor overfunction leading to abnormal behaviour, as demonstrated by the ability of SCH58261 treatment to normalize abnormal motor behaviour and deficient recognition memory in 6‐OHDA‐treated rats (Figure 3d,e). Compared with control rats, 6‐OHDA‐treated rats favoured the ipsilateral, rather than the contralateral, paw in their rearing movements in the cylinder test, and SCH58261 treatment attenuated this asymmetry in 6‐OHDA‐treated rats, without major effects in control rats (Figure 3d). 6‐OHDA‐treated rats also displayed a lower recognition index compared to controls and this effect was prevented by treatment with SCH58261 (Figure 3e).
3.4. 6‐OHDA‐induced dysfunction increases ATP release and CD73‐mediated formation of extracellular adenosine in the striatum
Striatal nerve terminals (synaptosomes) derived from rats treated with 6‐OHDA displayed a larger depolarization‐induced release of ATP than that from synaptosomes of control rats (Figure 4a). These 6‐OHDA‐treated rats also displayed an increased CD73 immunoreactivity in their striatal synapses (Figure 4b).
Figure 4.
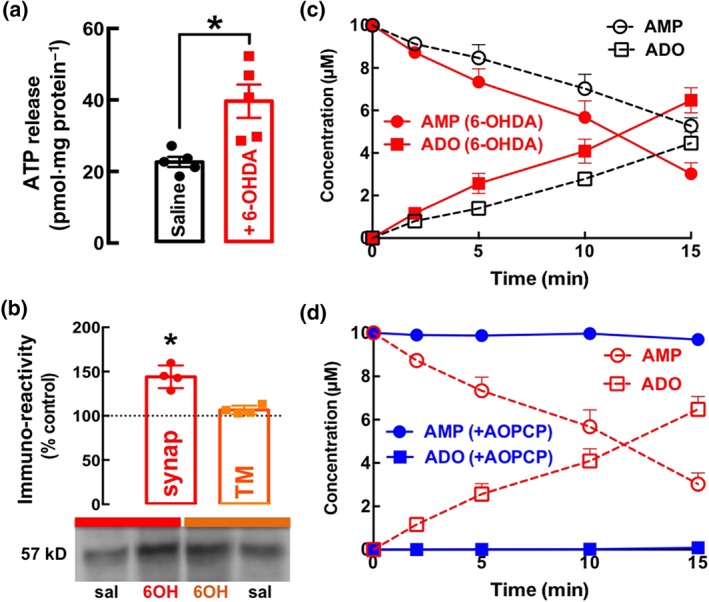
The unilateral administration of 6‐OHDA in the striatum of rats increases the evoked release of ATP from depolarized synapses, up‐regulates ecto‐5′‐nucleotidase (CD73) selectively in synapses, enhancing the formation of extracellular adenosine (ADO) from adenine nucleotides. (a) The high K+‐evoked release of ATP from striatal synaptosomes was greater in synaptosomes derived from mice killed 19 days after being challenged with 6‐OHDA intra‐striatally, compared with saline treated (control). (b) The density of immuno‐reactive CD73 was selectively enhanced in membranes from synaptosomes (synap), but not in total membranes (TMs), from 6‐OHDA (6OH)‐treated rats, compared with those from saline (sal)‐treated rats. (c) The extracellular catabolism of AMP (10 μM, added at Time 0) and the formation of ADO were greater in striatal synaptosomes from 6‐OHDA‐treated rats than in those from saline‐treated rats. (d) α,β‐Methylene ADP (AOPCP, 100 μM) blocked the extracellular catabolism of AMP (10 μM, added at Time 0) into adenosine in synaptosomes from 6‐OHDA‐treated rats. The data shown are means ± SEM of n = 4–5. *P < .05, significantly different from control; Dunnett's test in (a) and significantly different from saline; two‐tailed unpaired Student's t test
This increased density of CD73 was paralleled by an increased extracellular conversion of AMP into adenosine typified by a faster catabolism of extracellularly added AMP and a larger formation of extracellular adenosine in striatal synaptosomes from 6‐OHDA‐treated rats compared to control rats (Figure 4c). This increased extracellular catabolism of AMP and formation of adenosine was mediated by CD73 as its competitive inhibitor, AOPCP (100 μM), almost abolished the extracellular catabolism of AMP and the formation of adenosine in striatal synaptosomes from 6‐OHDA‐treated rats (Figure 4d). These findings confirm the value of AOPCP as a probe of the role of extracellular ATP‐derived adenosine in the abnormal over‐activation of A2A receptors leading to motor symptoms characteristic of PD.
3.5. Inhibition of CD73‐mediated formation of extracellular adenosine in the striatum prevents 6‐OHDA‐induced neurotoxicity and motor dysfunction
Administration of 6‐OHDA in the striatum is known to induce lesions in both the dopaminergic fibres in the striatum and the cell bodies in the substantia nigra (SN; Deumens, Blokland, & Prickaerts, 2002). Indeed, 19 days after the 6‐OHDA challenge, rats displayed a loss of TH immunoreactivity, as shown in Figure 5, in the ipsilateral striatum as well as in the SN. Upon infusion of AOPCP through the mini‐pumps in the striatum, the 6‐OHDA‐induced decrease of TH immunoreactivity in the striatum was no longer observed and there was a significant attenuation of the effect of 6‐OHDA on TH immunoreactivity in the SN (Figure 5). The contralateral striatum was not affected by 6‐OHDA (Figure 5), as noted by others (see Deumens et al., 2002).
Figure 5.
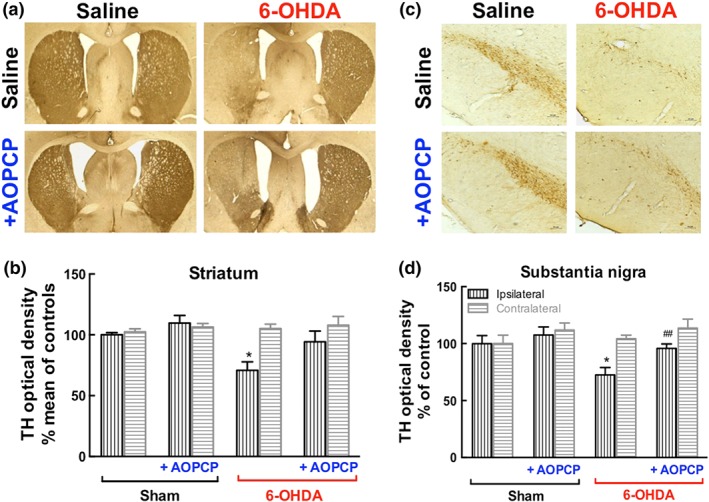
The elimination of ATP‐derived formation of extracellular adenosine abrogates 6‐OHDA‐induced dopaminergic neurodegeneration in the striatum and nigra. Representative coronal sections of the dorsolateral striatum (a) or substantia nigra (c) stained TH from saline‐treated rats and 6‐OHDA‐treated rats without or with the CD73 inhibitor α,β‐methylene ADP (AOPCP, 100 μM continuous intracerebroventricular infusion). Immunohistochemical quantification of TH density in the striatum (b) or substantia nigra (d). The data are means ± SEM (n = 6). *P < .05, significantly different from saline; # P < .05, significantly different from 6‐OHDA; one‐way ANOVA with Newman–Keuls post hoc test
This morphological evidence of the ability of AOPCP to prevent 6‐OHDA‐induced neurotoxicity was confirmed by the neurochemical quantification of the levels of dopamine and its metabolites (Table 1). Thus, 6‐OHDA administration decreased dopamine levels in the striatum and mesencephalon, compared with control rats as well as decreasing DOPAC levels in the striatum and mesencephalon, compared with controls. The treatment with AOPCP prevented the 6‐OHDA‐induced decrease in the content of dopamine and DOPAC in the striatum and mesencephalon and did not cause any significant changes in the sham group (Table 1).
Table 1.
Effects of the CD73 inhibitor, α,β‐methylene ADP (AOPCP, 100 μM administered intracerebroventricularly at 0.25 μl·hr−1 for 14 days) on the decreased levels of dopamine and its metabolite (ng·mg−1 tissue) in the rat striatum and mesencephalon after 6‐OHDA administration
| Striatum | Sham | Sham + AOPCP | 6‐OHDA | 6‐OHDA + AOPCP | |
| DA | 1,704 ± 197.0 | 1,202 ± 178.9 | 52.8 ± 28.7* | 111.5 ± 57.6 | |
| DOPAC | 513.6 ± 82.9 | 717.5 ± 64.0 | 85.0 ± 27.6* | 139.0 ± 69.5 | |
| Mesencephalon | Sham | Sham + AOPCP | 6‐OHDA | 6‐OHDA + AOPCP | |
| DA | 151.0 ± 81.0 | 115.4 ± 29.8 | 14.9 ± 4.6* | 35.2 ± 8.5 | |
| DOPAC | 49.2 ± 28.6 | 88.0 ± 27.2 | 10.1 ± 4.3* | 18.8 ± 8.0 |
Data shown are mean ± SEM of six rats per group.
P < .05, significantly different from sham; one‐way ANOVA followed by Tukey's post hoc test. Abbreviations: DA, dopamine; DOPAC, 3,4‐dihydroxyphenylacetic acid.
Administration of AOPCP directly into the right lateral ventricle through osmotic mini‐pumps also significantly attenuated apomorphine‐induced contralateral rotations in 6‐OHDA‐lesioned rats but was devoid of effects in sham‐operated rats (Figure 6a). In accordance with this modified dopaminergic function, the voluntary motor activity deteriorated in rats treated with 6‐OHDA, as concluded from the decreased use of the paw contralateral to 6‐OHDA administration either alone or together with the ipsilateral paw. Both deficits were attenuated by AOPCP treatment (Figure 6b). Notably, the exploratory behaviour (measured during 5 min) was not affected by either 6‐OHDA administration or AOPCP treatment, when evaluated 16 days after surgery, as the number of crossings and rearings in the open field test (Figure 6c).
Figure 6.
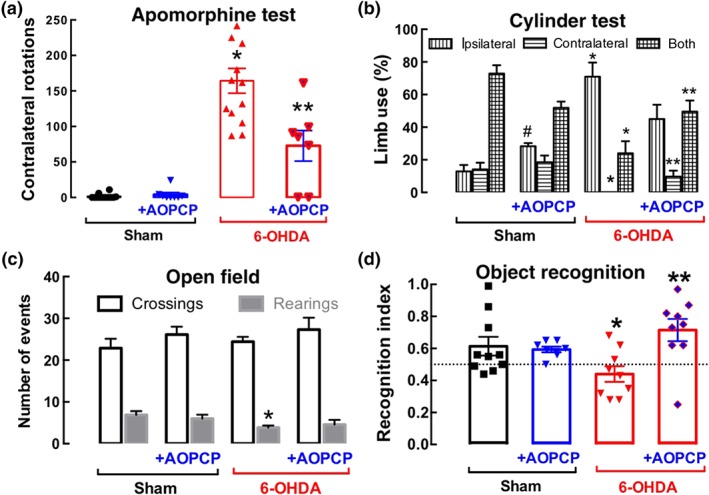
Blocking the formation of extracellular adenosine derived from ATP prevents 6‐OHDA‐induced motor and memory dysfunction. (a) The continuous intracerebroventricular infusion of the CD73 inhibitor α,β‐methylene ADP (AOPCP, 100 μM) attenuated the apomorphine‐induced contralateral rotation in rats unilaterally challenged with 6‐OHDA. (b) The unilateral injection of 6‐OHDA decreased the use of the contralateral limb, especially together with the ipsilateral limb, and this was restored after treatment with AOPCP (100 μM continuous intracerebroventricular infusion). (c) Neither the unilateral injection of 6‐OHDA nor the treatment with AOPCP altered the spontaneous locomotion in the open field. (d) AOPCP also prevented the decreased recognition memory of rats intra‐striatally injected with 6‐OHDA, as evaluated in the novel object recognition task: All groups of rats investigated equally both objects in the training phase (not shown), but only 6‐OHDA‐lesioned rats failed to discriminate the novel object during the test phase (e), without alteration of the total distance travelled during the test session between the four groups (not shown). Data shown are means ± SEM (n = 10 rats per group). *P < .05, significantly different from saline; **P < .05 significantly different from 6‐OHDA; # P < .05 significantly different from saline; two‐way ANOVA with Newman–Keuls post hoc test
Finally, recognition memory was significantly impaired in 6‐OHDA‐lesioned rats compared with the sham‐operated group and AOPCP treatment prevented this 6‐OHDA‐induced memory deficit without affecting the memory performance of the control group (Figure 6d).
4. DISCUSSION
The present study shows that the critical role of A2A receptors in the evolution of striatal dopamine deficits and motor and memory impairments in a model of PD is due to both (a) an up‐regulation of these receptors and (b) an increased ATP release and extracellular formation of ATP‐derived adenosine, selectively directed to sustain an over‐activation of A2A receptors. This conclusion extends to a pathophysiological context of PD the previously demonstrated association between the extracellular formation of ATP‐derived adenosine and the selective activation of A2A receptors in hippocampal synapses (Rebola et al., 2008), in cultured cerebellar neurons (Boeck et al., 2007), and in motor nerve endings (Magalhães‐Cardoso et al., 2003). This further confirms the previous proposals that different sources of extracellular adenosine are directed to the selective activation of different adenosine receptors (see Cunha, 2016).
This selective association of ATP‐derived formation of extracellular adenosine provides a functional rationale to the previously observed close physical association in the striatum of A2A receptors and CD73 (Augusto et al., 2013), the rate‐limiting enzyme in the extracellular formation of adenosine from adenine nucleotides (see Cunha, 2001). This close association seems to be even more marked in pathological conditions, as shown by the parallel up‐regulation of A2A receptors and of CD73 in the striatum of this animal model of PD. Previous studies had already reported that the treatment of rodents with 6‐OHDA leads to an increased expression of the mRNA for A2A receptors (Pinna et al., 2002) and to an increased density of A2A receptors (Bhattacharjee et al., 2011) in the affected striatum. Additionally, the present study is the first to report an up‐regulation and an increased activity of CD73 in an animal model of PD, which occurs in parallel to the changes in A2A receptors. A similar parallel up‐regulation of A2A receptors and CD73 was also observed in the limbic cortex of both animal models of epilepsy (see Bonan et al., 2000; Rebola et al., 2003, 2005), and in patients with temporal lobe epilepsy (Barros‐Barbosa et al., 2016). The same changes occur in the hippocampus upon brain ischaemia (Braun, Zhu, Krieglstein, Culmsee, & Zimmermann, 1998; Ye et al., 2018), repeated stress (Cunha et al., 2006; Fontella et al., 2004), or aging (Cunha, Almeida, & Ribeiro, 2001). Although it has been reported that A2A receptors and CD73 mRNA expression is regulated in parallel (Napieralski, Kempkes, & Gutensohn, 2003), common regulatory elements eventually present in the promoters of both genes have not yet been characterized, probably due to the complexity of the promoter of A2A receptors. leading to the expression of different transcripts in different cell types (Lee et al., 2003; Yu et al., 2004). It is worth noting that the use of a simple cellular model of toxicity (dopamine‐differentiated SH‐SY5Y cells) allowed us to show that the parallel up‐regulation of A2A receptors and CD73, as well as of the release of ATP, occurs before the onset of overt neurodegeneration. This strongly suggests that this gain of function of the purinergic system is not a late consequence of neurotoxicity but is more likely to represents an early adaptive event, probably contributing in a critical manner to the emergence of neurotoxicity.
Remarkably, the up‐regulation of A2A receptors and of CD73 in early PD was most evident in synaptic membranes rather than in whole membranes of the striatum, which suggests that the deleterious effects of over‐activation of the A2A receptors, due to increased ATP‐derived formation of extracellular adenosine has a primary effect on synapses. This lends additional support to the increasingly accepted idea that synaptotoxicity (or axonopathy) might be an initial process in the evolving neurodegeneration characteristic of PD, in a retrograde or dying‐back mechanism (reviewed in Calo, Wegrzynowicz, Santivañez‐Perez, & Grazia Spillantini, 2016; Picconi, Piccoli, & Calabresi, 2012). In fact, there is an axonopathy that predates overt neurotoxicity in different animal models of PD (see Chung, Koprich, Siddiqi, & Isacson, 2009; Decressac, Mattsson, Lundblad, Weikop, & Björklund, 2012; Kitada et al., 2007; Nordströma et al., 2015; Tagliaferro et al., 2015) as well as in PD patients (Chu et al., 2012; Kordower et al., 2013; Sossi et al., 2004). Notably, early synaptotoxicity predating overt neuronal damage seems to be a feature of other neurodegenerative disorders (see Lepeta et al., 2016). This involvement of synaptotoxicity in the early phases of PD is particularly relevant in view of the proposed association of increased levels and activity of CD73 with sites of altered synaptic plasticity (Lie et al., 1999). Likewise, it has also been proposed that A2A receptors are mainly up‐regulated in synapses in different pathological brain conditions such as Alzheimer's disease, convulsions, or upon aging, and blockade of these receptors prevented synaptotoxicity, thus alleviating phenotypic traits of these different conditions (see Cunha, 2016). Thus, it is also tempting to propose that the A2A receptors may critically unbalance synaptic function in the striatum (see Schiffmann, Fisone, Moresco, Cunha, & Ferré, 2007) to prevent PD‐associated neurodegeneration and motor dysfunction.
Finally, the present findings also reinforce the importance of ATP as a stress signal in brain dysfunction (see Rodrigues et al., 2015). Previous studies have already shown that noxious insults, such as hypoxia, trigger an increase of the extracellular levels of ATP in the striatum (Lutz & Kabler, 1997; Melani et al., 2012). The role of ATP as an effector of neuronal damage in the striatum (Ryu et al., 2002) seems to be exerted in different and parallel ways. In fact, we have previously shown that the blockade of P2X7 receptors was sufficient to prevent PD neurodegeneration and associated symptoms (Carmo et al., 2014). We now show that the extracellular catabolism of adenosine nucleotides, as well as the blockade of A2A receptors, is also sufficient to prevent PD neurodegeneration and associated symptoms. This shows that ATP has a dual, parallel, and equi‐effective role in the control of PD neurodegeneration and associated symptoms. It induces neurodegeneration directly through P2X7 receptors and, indirectly, through A2A receptors after its extracellular conversion into adenosine. Notably, there is a striking parallel between the control operated by P2X7 receptors and by A2A receptors of different processes contributing tor neurodegeneration: Thus, the antagonism of either P2X7 or A2A receptors directly prevents the damage of neurons (Ohishi et al., 2016; Silva, Porciúncula, Canas, Oliveira, & Cunha, 2007) as well as astrocytosis (Apolloni, Amadio, Montilli, Volonté, & D'Ambrosi, 2013; Matos et al., 2012) and microgliosis (Gomes et al., 2013; He, Taylor, Fourgeaud, & Bhattacharya, 2017). However, it is currently unknown if there is any interaction between A2A and P2X7 receptors, apart from their parallel functioning in the control of brain damage (see Rodrigues et al., 2015; see also Ye et al., 2018). Further work is certainly required to unravel the organization of these different layers of purinergic modulation in terms of their timing of action, their site of action, and their putative interaction.
In conclusion, the present study shows that there is a parallel up‐regulation of CD73 and A2A receptors in models of early PD and the deleterious role of the adenosine modulation system is due to the simultaneous up‐regulation of A2A receptors together with an increased CD73‐mediated formation of extracellular adenosine, selectively activating A2A receptors. Consistent with these findings, we showed that inhibiting ATP‐derived formation of extracellular adenosine phenocopies the neuroprotection and motor benefits afforded by A2A receptor antagonists in an animal model of PD. This observed critical dependence of A2A receptor overfunction on ATP‐derived adenosine in PD also prompts considering the interest in using CD73‐dependent pro‐drugs acting on A2A receptors (see Flögel et al., 2012) to manage this neurodegenerative disorder.
CONFLICT OF INTEREST
R.A.C. is a scientific advisor of the Institute for Scientific Information (ISIC). All other authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
M.C. and F.D.F. performed the behavioural analysis and the striatal morphological analysis. F.Q.G. and J.P.O. did the ATP release experiments. J.P.O., A.R.T., and R.A.C. did the kinetic experiments. F.V.D., C.M.P., and P.A. performed the studies with cultured cells. P.M.C. and M.C. did the fractionation analysis and Western blot analysis, and R.A.C. performed binding assays. R.A.C. and G.M.A. provided supervision and guided the project, and R.A.C. wrote the manuscript. All authors commented on the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This study adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
ACKNOWLEDGEMENTS
This study was supported by Santa Casa da Misericórdia, CAPES‐FCT, Centro 2020 (CENTRO‐01‐0145‐FEDER‐000008:BrainHealth 2020 and CENTRO‐01‐0246‐FEDER‐000010) and through Fundação para a Ciência e a Tecnologia (PTDC/NEU‐NMC/4154/2014 and POCI‐01‐0145‐FEDER‐03127).
Carmo M, Gonçalves FQ, Canas PM, et al. Enhanced ATP release and CD73‐mediated adenosine formation sustain adenosine A2A receptor over‐activation in a rat model of Parkinson's disease. Br J Pharmacol. 2019;176:3666–3680. 10.1111/bph.14771
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonioli, L. , Pacher, P. , Vizi, E. S. , & Haskó, G. (2013). CD39 and CD73 in immunity and inflammation. Trends in Molecular Medicine, 19, 355–367. 10.1016/j.molmed.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apolloni, S. , Amadio, S. , Montilli, C. , Volonté, C. , & D'Ambrosi, N. (2013). Ablation of P2X7 receptor exacerbates gliosis and motoneuron death in the SOD1‐G93A mouse model of amyotrophic lateral sclerosis. Human Molecular Genetics, 22, 4102–4116. 10.1093/hmg/ddt259 [DOI] [PubMed] [Google Scholar]
- Augusto, E. , Matos, M. , Sévigny, J. , El‐Tayeb, A. , Bynoe, M. S. , Müller, C. E. , … Chen, J. F. (2013). Ecto‐5′‐nucleotidase (CD73)‐mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. The Journal of Neuroscience, 33, 11390–11399. 10.1523/JNEUROSCI.5817-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros‐Barbosa, A. R. , Ferreirinha, F. , Oliveira, Â. , Mendes, M. , Lobo, M. G. , Santos, A. , … Correia‐de‐Sá, P. (2016). Adenosine A2A receptor and ecto‐5′‐nucleotidase/CD73 are upregulated in hippocampal astrocytes of human patients with mesial temporal lobe epilepsy (MTLE). Purinergic Signal, 12, 719–734. 10.1007/s11302-016-9535-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins, R. A. , & Besheer, J. (2006). Object recognition in rats and mice: a one‐trial non‐matching‐to‐sample learning task to study 'recognition memory'. Nature Protocols, 1, 1306–1311. 10.1038/nprot.2006.205 [DOI] [PubMed] [Google Scholar]
- Bhattacharjee, A. K. , Lang, L. , Jacobson, O. , Shinkre, B. , Ma, Y. , Niu, G. , … Kiesewetter, D. O. (2011). Striatal adenosine A2A receptor‐mediated positron emission tomographic imaging in 6‐hydroxydopamine‐lesioned rats using [18F]‐MRS5425. Nuclear Medicine and Biology, 38, 897–906. 10.1016/j.nucmedbio.2011.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeck, C. R. , Kroth, E. H. , Bronzatto, M. J. , & Vendite, D. (2007). Effect of the L‐ or D‐aspartate on ecto‐5′nucleotidase activity and on cellular viability in cultured neurons: Participation of the adenosine A2A receptors. Amino Acids, 33, 439–444. 10.1007/s00726-006-0455-2 [DOI] [PubMed] [Google Scholar]
- Bonan, C. D. , Walz, R. , Pereira, G. S. , Worm, P. V. , Battastini, A. M. , Cavalheiro, E. A. , … Sarkis, J. J. (2000). Changes in synaptosomal ectonucleotidase activities in two rat models of temporal lobe epilepsy. Epilepsy Research, 39, 229–238. 10.1016/S0920-1211(00)00095-4 [DOI] [PubMed] [Google Scholar]
- Borycz, J. , Pereira, M. F. , Melani, A. , Rodrigues, R. J. , Köfalvi, A. , Panlilio, L. , … Ferré, S. (2007). Differential glutamate‐dependent and glutamate‐independent adenosine A1 receptor‐mediated modulation of dopamine release in different striatal compartments. Journal of Neurochemistry, 101, 355–363. 10.1111/j.1471-4159.2006.04386.x [DOI] [PubMed] [Google Scholar]
- Braun, N. , Zhu, Y. , Krieglstein, J. , Culmsee, C. , & Zimmermann, H. (1998). Upregulation of the enzyme chain hydrolyzing extracellular ATP after transient forebrain ischemia in the rat. The Journal of Neuroscience, 18, 4891–4900. 10.1523/JNEUROSCI.18-13-04891.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo, L. , Wegrzynowicz, M. , Santivañez‐Perez, J. , & Grazia Spillantini, M. (2016). Synaptic failure and α‐synuclein. Movement Disorders, 31, 169–177. 10.1002/mds.26479 [DOI] [PubMed] [Google Scholar]
- Canas, P. M. , Porciúncula, L. O. , Cunha, G. M. , Silva, C. G. , Machado, N. J. , Oliveira, J. M. , … Cunha, R. A. (2009). Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by β‐amyloid peptides via p38 mitogen‐activated protein kinase pathway. The Journal of Neuroscience, 29, 14741–14751. 10.1523/JNEUROSCI.3728-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo, M. R. , Menezes, A. P. , Nunes, A. C. , Pliássova, A. , Rolo, A. P. , Palmeira, C. M. , … Andrade, G. M. (2014). The P2X7 receptor antagonist Brilliant Blue G attenuates contralateral rotations in a rat model of Parkinsonism through a combined control of synaptotoxicity, neurotoxicity and gliosis. Neuropharmacology, 81, 142–152. 10.1016/j.neuropharm.2014.01.045 [DOI] [PubMed] [Google Scholar]
- Chu, Y. , Morfini, G. A. , Langhamer, L. B. , He, Y. , Brady, S. T. , & Kordower, J. H. (2012). Alterations in axonal transport motor proteins in sporadic and experimental Parkinson's disease. Brain, 135, 2058–2073. 10.1093/brain/aws133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, C. Y. , Koprich, J. B. , Siddiqi, H. , & Isacson, O. (2009). Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α‐synucleinopathy. The Journal of Neuroscience, 29, 3365–3373. 10.1523/JNEUROSCI.5427-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha, G. M. , Canas, P. M. , Oliveira, C. R. , & Cunha, R. A. (2006). Increased density and synapto‐protective effect of adenosine A2A receptors upon sub‐chronic restraint stress. Neuroscience, 141, 1775–1781. 10.1016/j.neuroscience.2006.05.024 [DOI] [PubMed] [Google Scholar]
- Cunha, R. A. (2001). Regulation of the ecto‐nucleotidase pathway in rat hippocampal nerve terminals. Neurochemical Research, 26, 979–991. 10.1023/A:1012392719601 [DOI] [PubMed] [Google Scholar]
- Cunha, R. A. (2016). How does adenosine control neuronal dysfunction and neurodegeneration? Journal of Neurochemistry, 139, 1019–1055. 10.1111/jnc.13724 [DOI] [PubMed] [Google Scholar]
- Cunha, R. A. , Almeida, T. , & Ribeiro, J. A. (2001). Parallel modification of adenosine extracellular metabolism and modulatory action in the hippocampus of aged rats. Journal of Neurochemistry, 76, 372–382. 10.1046/j.1471-4159.2001.00095.x [DOI] [PubMed] [Google Scholar]
- Cunha, R. A. , Sebastião, A. M. , & Ribeiro, J. A. (1992). Ecto‐5′‐nucleotidase is associated with cholinergic nerve terminals in the hippocampus but not in the cerebral cortex of the rat. Journal of Neurochemistry, 59, 657–666. 10.1111/j.1471-4159.1992.tb09420.x [DOI] [PubMed] [Google Scholar]
- Cunha, R. A. , Sebastião, A. M. , & Ribeiro, J. A. (1998). Inhibition by ATP of hippocampal synaptic transmission requires localized extracellular catabolism by ecto‐nucleotidases into adenosine and channeling to adenosine A1 receptors. The Journal of Neuroscience, 18, 1987–1995. 10.1523/JNEUROSCI.18-06-01987.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac, M. , Mattsson, B. , Lundblad, M. , Weikop, P. , & Björklund, A. (2012). Progressive neurodegenerative and behavioural changes induced by AAV‐mediated overexpression of α‐synuclein in midbrain dopamine neurons. Neurobiology of Disease, 45, 939–953. 10.1016/j.nbd.2011.12.013 [DOI] [PubMed] [Google Scholar]
- Deumens, R. , Blokland, A. , & Prickaerts, J. (2002). Modeling Parkinson's disease in rats: An evaluation of 6‐OHDA lesions of the nigrostriatal pathway. Experimental Neurology, 175, 303–317. 10.1006/exnr.2002.7891 [DOI] [PubMed] [Google Scholar]
- Ena, S. L. , De Backer, J. F. , Schiffmann, S. N. , & de Kerchove d'Exaerde, A. (2013). FACS array profiling identifies ecto‐5′‐nucleotidase as a striatopallidal neuron‐specific gene involved in striatal‐dependent learning. The Journal of Neuroscience, 33, 8794–8809. 10.1523/JNEUROSCI.2989-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, S. G. , Gonçalves, F. Q. , Marques, J. M. , Tomé, Â. R. , Rodrigues, R. J. , Nunes‐Correia, I. , … Köfalvi, A. (2015). Presynaptic adenosine A2A receptors dampen cannabinoid CB1 receptor‐mediated inhibition of corticostriatal glutamatergic transmission. British Journal of Pharmacology, 172, 1074–1086. 10.1111/bph.12970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flögel, U. , Burghoff, S. , van Lent, P. L. , Temme, S. , Galbarz, L. , Ding, Z. , … Schrader, J. (2012). Selective activation of adenosine A2A receptors on immune cells by a CD73‐dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis. Science Translational Medicine, 4, 146ra108. [DOI] [PubMed] [Google Scholar]
- Fontella, F. U. , Bruno, A. N. , Crema, L. M. , Battastini, A. M. , Sarkis, J. J. , Netto, C. A. , & Dalmaz, C. (2004). Acute and chronic stress alter ecto‐nucleotidase activities in synaptosomes from the rat hippocampus. Pharmacology, Biochemistry, and Behavior, 78, 341–347. 10.1016/j.pbb.2004.04.005 [DOI] [PubMed] [Google Scholar]
- George, J. , Gonçalves, F. Q. , Cristóvão, G. , Rodrigues, L. , Meyer Fernandes, J. R. , Gonçalves, T. , … Gomes, C. A. (2015). Different danger signals differently impact on microglial proliferation through alterations of ATP release and extracellular metabolism. Glia, 63, 1636–1645. 10.1002/glia.22833 [DOI] [PubMed] [Google Scholar]
- Gomes, C. , Ferreira, R. , George, J. , Sanches, R. , Rodrigues, D. I. , Gonçalves, N. , & Cunha, R. A. (2013). Activation of microglial cells triggers a release of brain‐derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A2A receptor‐dependent manner: A2A receptor blockade prevents BDNF release and proliferation of microglia. Journal of Neuroinflammation, 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y. , Taylor, N. , Fourgeaud, L. , & Bhattacharya, A. (2017). The role of microglial P2X7: Modulation of cell death and cytokine release. Journal of Neuroinflammation, 14, 135 10.1186/s12974-017-0904-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilbronn, A. , Maienschein, V. , Carstensen, K. , Gann, W. , & Zimmermann, H. (1995). Crucial role of ecto‐5′‐nucleotidase in differentiation and survival of developing neural cells. Neuroreport, 7, 257–261. [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada, T. , Pisani, A. , Porter, D. R. , Yamaguchi, H. , Tscherter, A. , Martella, G. , … Shen, J. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1‐deficient mice. Proceedings of the National Academy of Sciences of the United States of America, 104, 11441–11446. 10.1073/pnas.0702717104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower, J. H. , Olanow, C. W. , Dodiya, H. B. , Chu, Y. , Beach, T. G. , Adler, C. H. , … Bartus, R. T. (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain, 136, 2419–2431. 10.1093/brain/awt192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. C. , Chien, C. L. , Sun, C. N. , Huang, C. L. , Huang, N. K. , Chiang, M. C. , … Chern, Y. (2003). Characterization of the rat A2A adenosine receptor gene: A 4.8‐kb promoter‐proximal DNA fragment confers selective expression in the central nervous system. The European Journal of Neuroscience, 18, 1786–1796. 10.1046/j.1460-9568.2003.02907.x [DOI] [PubMed] [Google Scholar]
- Lepeta, K. , Lourenco, M. V. , Schweitzer, B. C. , Martino Adami, P. V. , Banerjee, P. , Catuara‐Solarz, S. , … Seidenbecher, C. (2016). Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. Journal of Neurochemistry, 138, 785–805. 10.1111/jnc.13713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie, A. A. , Blümcke, I. , Beck, H. , Wiestler, O. D. , Elger, C. E. , & Schoen, S. W. (1999). 5′‐Nucleotidase activity indicates sites of synaptic plasticity and reactive synaptogenesis in the human brain. Journal of Neuropathology and Experimental Neurology, 58, 451–458. 10.1097/00005072-199905000-00004 [DOI] [PubMed] [Google Scholar]
- Lopes, F. M. , Schröder, R. , da Frota, M. L. Jr. , Zanotto‐Filho, A. , Müller, C. B. , Pires, A. S. , … Klamt, F. (2010). Comparison between proliferative and neuron‐like SH‐SY5Y cells as an in vitro model for Parkinson disease studies. Brain Research, 1337, 85–94. 10.1016/j.brainres.2010.03.102 [DOI] [PubMed] [Google Scholar]
- Lopes, L. V. , Halldner, L. , Rebola, N. , Johansson, B. , Ledent, C. , Chen, J. F. , … Cunha, R. A. (2004). Binding of the prototypical adenosine A2A receptor agonist CGS 21680 to the cerebral cortex of adenosine A1 and A2A receptor knockout mice. British Journal of Pharmacology, 141, 1006–1014. 10.1038/sj.bjp.0705692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz, P. L. , & Kabler, S. (1997). Release of adenosine and ATP in the brain of the freshwater turtle (Trachemys scripta) during long‐term anoxia. Brain Research, 769, 281–286. 10.1016/S0006-8993(97)00719-1 [DOI] [PubMed] [Google Scholar]
- Magalhães‐Cardoso, M. T. , Pereira, M. F. , Oliveira, L. , Ribeiro, J. A. , Cunha, R. A. , & Correia‐de‐Sá, P. (2003). Ecto‐AMP deaminase blunts the ATP‐derived adenosine A2A receptor facilitation of acetylcholine release at rat motor nerve endings. The Journal of Physiology, 549, 399–408. 10.1113/jphysiol.2003.040410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellino, D. , Suárez‐Boomgaard, D. , Sánchez‐Reina, M. D. , Aguirre, J. A. , Yoshitake, T. , Yoshitake, S. , … Rivera, A. (2010). On the role of P2X7 receptors in dopamine nerve cell degeneration in a rat model of Parkinson's disease: Studies with the P2X7 receptor antagonist A‐438079. Journal of Neural Transmission, 117, 681–687. 10.1007/s00702-010-0400-0 [DOI] [PubMed] [Google Scholar]
- Matos, M. , Augusto, E. , Machado, N. J. , dos Santos‐Rodrigues, A. , Cunha, R. A. , & Agostinho, P. (2012). Astrocytic adenosine A2A receptors control the amyloid‐β peptide‐induced decrease of glutamate uptake. Journal of Alzheimer's Disease, 31, 555–567. 10.3233/JAD-2012-120469 [DOI] [PubMed] [Google Scholar]
- Melani, A. , Corti, F. , Stephan, H. , Müller, C. E. , Donati, C. , Bruni, P. , … Pedata, F. (2012). Ecto‐ATPase inhibition: ATP and adenosine release under physiological and ischemic in vivo conditions in the rat striatum. Experimental Neurology, 233, 193–204. 10.1016/j.expneurol.2011.09.036 [DOI] [PubMed] [Google Scholar]
- Meng, F. , Guo, Z. , Hu, Y. , Mai, W. , Zhang, Z. , Zhang, B. , … Gao, Z. (2019). CD73‐derived adenosine controls inflammation and neurodegeneration by modulating dopamine signalling. Brain, 142, 700–718. 10.1093/brain/awy351 [DOI] [PubMed] [Google Scholar]
- Morelli, M. , Carta, A. R. , & Jenner, P. (2009). Adenosine A2A receptors and Parkinson's disease. Handbook of Experimental Pharmacology, 193, 589–615. 10.1007/978-3-540-89615-9_18 [DOI] [PubMed] [Google Scholar]
- Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. Journal of Immunological Methods, 65, 55–63. 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
- Napieralski, R. , Kempkes, B. , & Gutensohn, W. (2003). Evidence for coordinated induction and repression of ecto‐5′‐nucleotidase (CD73) and the A2A adenosine receptor in a human B cell line. Biological Chemistry, 384, 483–487. 10.1515/BC.2003.054 [DOI] [PubMed] [Google Scholar]
- Nordströma, U. , Beauvais, G. , Ghosh, A. , Pulikkaparambil Sasidharan, B. C. , Lundblad, M. , Fuchs, J. , … Brundin, P. (2015). Progressive nigrostriatal terminal dysfunction and degeneration in the engrailed1 heterozygous mouse model of Parkinson's disease. Neurobiology of Disease, 73, 70–82. 10.1016/j.nbd.2014.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohishi, A. , Keno, Y. , Marumiya, A. , Sudo, Y. , Uda, Y. , Matsuda, K. , … Nagasawa, K. (2016). Expression level of P2X7 receptor is a determinant of ATP‐induced death of mouse cultured neurons. Neuroscience, 319, 35–45. 10.1016/j.neuroscience.2016.01.048 [DOI] [PubMed] [Google Scholar]
- Picconi, B. , Piccoli, G. , & Calabresi, P. (2012). Synaptic dysfunction in Parkinson's disease. Advances in Experimental Medicine and Biology, 970, 553–572. 10.1007/978-3-7091-0932-8_24 [DOI] [PubMed] [Google Scholar]
- Pinna, A. , Corsi, C. , Carta, A. R. , Valentini, V. , Pedata, F. , & Morelli, M. (2002). Modification of adenosine extracellular levels and adenosine A2A receptor mRNA by dopamine denervation. European Journal of Pharmacology, 446, 75–82. 10.1016/S0014-2999(02)01818-6 [DOI] [PubMed] [Google Scholar]
- Rebola, N. , Coelho, J. E. , Costenla, A. R. , Lopes, L. V. , Parada, A. , Oliveira, C. R. , … Cunha, R. A. (2003). Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. The European Journal of Neuroscience, 18, 820–828. 10.1046/j.1460-9568.2003.02815.x [DOI] [PubMed] [Google Scholar]
- Rebola, N. , Lujan, R. , Cunha, R. A. , & Mulle, C. (2008). Adenosine A2A receptors are essential for long‐term potentiation of NMDA‐EPSCs at hippocampal mossy fiber synapses. Neuron, 57, 121–134. 10.1016/j.neuron.2007.11.023 [DOI] [PubMed] [Google Scholar]
- Rebola, N. , Porciúncula, L. O. , Lopes, L. V. , Oliveira, C. R. , Soares‐da‐Silva, P. , & Cunha, R. A. (2005). Long‐term effect of convulsive behavior on the density of adenosine A1 and A2A receptors in the rat cerebral cortex. Epilepsia, 46(Suppl.5), 159–165. 10.1111/j.1528-1167.2005.01026.x [DOI] [PubMed] [Google Scholar]
- Rodrigues, R. J. , Tomé, A. R. , & Cunha, R. A. (2015). ATP as a multi‐target danger signal in the brain. Frontiers in Neuroscience, 9, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, J. K. , Kim, J. , Choi, S. H. , Oh, Y. J. , Lee, Y. B. , Kim, S. U. , & Jin, B. K. (2002). ATP‐induced in vivo neurotoxicity in the rat striatum via P2 receptors. Neuroreport, 13, 1611–1615. 10.1097/00001756-200209160-00008 [DOI] [PubMed] [Google Scholar]
- Schiffmann, S. N. , Fisone, G. , Moresco, R. , Cunha, R. A. , & Ferré, S. (2007). Adenosine A2A receptors and basal ganglia physiology. Progress in Neurobiology, 83, 277–292. 10.1016/j.pneurobio.2007.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, C. G. , Porciúncula, L. O. , Canas, P. M. , Oliveira, C. R. , & Cunha, R. A. (2007). Blockade of adenosine A2A receptors prevents staurosporine‐induced apoptosis of rat hippocampal neurons. Neurobiology of Disease, 27, 182–189. 10.1016/j.nbd.2007.04.018 [DOI] [PubMed] [Google Scholar]
- Sossi, V. , de la Fuente‐Fernández, R. , Holden, J. E. , Schulzer, M. , Ruth, T. J. , & Stoessl, J. (2004). Changes of dopamine turnover in the progression of Parkinson's disease as measured by positron emission tomography: Their relation to disease‐compensatory mechanisms. Journal of Cerebral Blood Flow and Metabolism, 24, 869–876. 10.1097/01.WCB.0000126563.85360.75 [DOI] [PubMed] [Google Scholar]
- Sun, X. , Shi, X. , Lu, L. , Jiang, Y. , & Liu, B. (2016). Stimulus‐dependent neuronal cell responses in SH‐SY5Y neuroblastoma cells. Molecular Medicine Reports, 13, 2215–2220. https://doi: 10.3892/mmr.2016.4759 [DOI] [PubMed] [Google Scholar]
- Tagliaferro, P. , Kareva, T. , Oo, T. F. , Yarygina, O. , Kholodilov, N. , & Burke, R. E. (2015). An early axonopathy in a hLRRK2(R1441G) transgenic model of Parkinson disease. Neurobiology of Disease, 82, 359–371. 10.1016/j.nbd.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, X. C. , Hu, J. X. , Li, L. , Li, Q. , Tang, F. L. , Lin, S. , … Xiong, W. C. (2018). Astrocytic Lrp4 (low‐density lipoprotein receptor‐related protein 4) contributes to ischemia‐induced brain injury by regulating ATP release and adenosine‐A2AR (adenosine A2AR receptor) signaling. Stroke, 49, 165–174. 10.1161/STROKEAHA.117.018115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Frith, M. C. , Suzuki, Y. , Peterfreund, R. A. , Gearan, T. , Sugano, S. , … Chen, J. F. (2004). Characterization of genomic organization of the adenosine A2A receptor gene by molecular and bioinformatics analyses. Brain Research, 1000, 156–173. 10.1016/j.brainres.2003.11.072 [DOI] [PubMed] [Google Scholar]