

P. gingivalis is implicated in the onset and progression of periodontal disease and associated with some systemic diseases. Probiotic bacteria represent an attractive preventative therapy for periodontal disease. However, the efficacy of probiotic bacteria can be variable between studies. Our data support the known importance of selecting particular strains of bacteria for probiotic use, not simply a single species. Specifically, in the context of probiotic intervention of periodontitis, our data suggest that high-level expression of pyruvate oxidase with hydrogen peroxide production in L. delbrueckii could be an important characteristic for the design of a probiotic supplement or a microbial therapeutic.
KEYWORDS: Lactobacillus, Porphyromonas gingivalis
ABSTRACT
Despite a growing interest in using probiotic microorganisms to prevent disease, the mechanisms by which probiotics exert their action require further investigation. Porphyromonas gingivalis is an important pathogen implicated in the development of periodontitis. We isolated several strains of Lactobacillus delbrueckii from dairy products and examined their ability to inhibit P. gingivalis growth in vitro. We observed strain-specific inhibition of P. gingivalis growth in vitro. Whole-genome sequencing of inhibitory and noninhibitory strains of L. delbrueckii revealed significant genetic differences supporting the strain specificity of the interaction. Extracts of the L. delbrueckii STYM1 inhibitory strain contain inhibitory activity that is abolished by treatment with heat, proteinase K, catalase, and sodium sulfite. We purified the inhibitory protein(s) from L. delbrueckii STYM1 extracts using ammonium sulfate precipitation, anion-exchange chromatography, and gel filtration chromatography. Pyruvate oxidase was highly enriched in the purified samples. Lastly, we showed that purified, catalytically active, recombinant pyruvate oxidase is sufficient to inhibit P. gingivalis growth in vitro without the addition of cofactors. Further, using a saturated transposon library, we isolated transposon mutants of P. gingivalis in the feoB2 (PG_1294) gene that are resistant to killing by inhibitory L. delbrueckii, consistent with a mechanism of hydrogen peroxide production by pyruvate oxidase. Our results support the current understanding of the importance of strain selection, not simply species selection, in microbial interactions. Specific L. delbrueckii strains or their products may be effective in the treatment and prevention of P. gingivalis-associated periodontal disease.
IMPORTANCE P. gingivalis is implicated in the onset and progression of periodontal disease and associated with some systemic diseases. Probiotic bacteria represent an attractive preventative therapy for periodontal disease. However, the efficacy of probiotic bacteria can be variable between studies. Our data support the known importance of selecting particular strains of bacteria for probiotic use, not simply a single species. Specifically, in the context of probiotic intervention of periodontitis, our data suggest that high-level expression of pyruvate oxidase with hydrogen peroxide production in L. delbrueckii could be an important characteristic for the design of a probiotic supplement or a microbial therapeutic.
INTRODUCTION
Periodontitis, an inflammatory disease of the tissue that supports the teeth, is caused by a dysbiosis or imbalance between beneficial and pathogenic oral bacteria (1). This contributes to inflammation and, if left untreated, eventually results in alveolar bone and tooth loss. One of the major bacteria associated with periodontitis is Porphyromonas gingivalis, which is considered to be a keystone pathogen in periodontal disease (1). Current treatment for periodontitis involves intensive and expensive therapies such as scaling and root planing, as well as antibiotic treatment which are not preventative treatments. Currently available options for prevention are focused primarily on standard dental hygiene.
The FAO/WHO define probiotics as “live microorganisms which when administered in adequate amounts confer a health benefit on the host” (2). Some of the most well-studied probiotics include Lactobacillus, Bifidobacterium, and Lactococcus species that colonize the gut and have beneficial effects on celiac disease, obesity, irritable bowel syndrome, Campylobacter jejuni infection, and infant sepsis (3–10). Commonly found in yogurts and other fermented foods, many Lactobacillus strains are Generally Recognized As Safe (GRAS) by the U.S. Food and Drug Administration (FDA) for the use in specific food productions. Use of probiotic Lactobacillus and Lactococcus species is just beginning to be explored in the oral cavity (11–14). Several recent studies have examined the efficacy of probiotics in the treatment of dental caries. Many of these studies employed administration of different Lactobacillus species by different vehicles (i.e., milk, cheese, yogurt, tablets, etc.) for various time spans. These studies demonstrated a significant decrease in carriage of Streptococcus mutans (the predominant bacterium involved in dental caries), reduced caries risk, and reduced periodontal disease symptoms following treatment with the probiotic, thereby underscoring the feasibility of probiotic use in the oral cavity (12, 15).
Lactobacillus species have also been used in the treatment of periodontal disease. The effectiveness of probiotic Lactobacillus treatment has varied in clinical trials, which may be due to strain variability within the genera and specific species. Nonetheless, there are several studies demonstrating that Lactobacillus reuteri and Lactobacillus brevis decrease gingival bleeding, an important marker of disease (14, 16). In another study, Lactobacillus salivarius supplementation decreased the relative abundance of periodontal pathogens (17). Lactobacillus gasseri and Lactobacillus rhamnosus GG treatment reduce alveolar bone loss in mouse models of periodontitis (18, 19). These results are perhaps unsurprising given several studies demonstrating that individuals who consume more fermented dairy products like yogurt and kefir (which contain Lactobacillus species) have a lowered incidence of periodontal disease and/or decreased severity of disease (20, 21). One of the main Lactobacillus species found in fermented dairy products is Lactobacillus delbrueckii. Very few studies examine the role of this particular Lactobacillus species in periodontal disease.
One potential issue with the use of Lactobacillus species in the oral cavity is that their effect and colonization may be transient. However, L. salivarius, L. fermentum, and L. gasseri are found consistently in the oral cavity (22, 23). Furthermore, one recent study showed that L. delbrueckii from yogurt was detectable in the salivary microbiota up to 24 h after consumption of the yogurt (24).
In this study, we isolated strains of L. delbrueckii, a common probiotic found in yogurts, and identified strains that inhibited and did not inhibit the growth of P. gingivalis. We subsequently determined that the mechanism by which the inhibiting strain of L. delbrueckii exerted its effect was through the enzymatic activity of pyruvate oxidase. Understanding the mechanisms by which probiotic bacteria can inhibit growth of pathogenic strains may be important in the development of preventative strategies for periodontitis.
RESULTS
Inhibition of P. gingivalis by L. delbrueckii is strain specific.
We isolated two strains of L. delbrueckii from commercial yogurt products and two strains from raw cow milk. The commercial isolates were STYM1 and GVKM1, while the raw milk isolates were named SYB7 and SYB13. Their identity was confirmed by 16S rRNA sequencing (data not shown). We examined each of these strains, as well as the ATCC 11842 type strain, for its ability to inhibit P. gingivalis growth in an agar overlay assay and a spot assay. Both STYM1 and GVKM1 inhibit P. gingivalis growth in both assays, while SYB7 and SYB13 isolates and the 11842 strain have little impact on P. gingivalis growth (Fig. 1).
FIG 1.
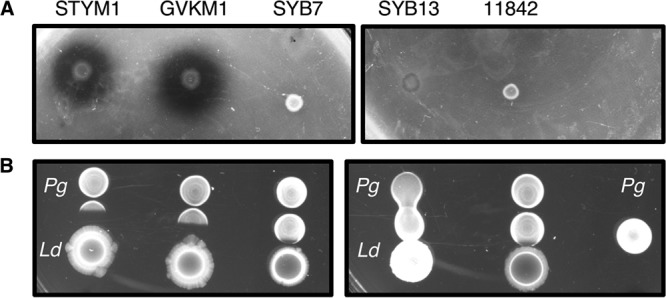
L. delbrueckii inhibition of P. gingivalis is strain specific. (A) Agar overlay assay with different strains of L. delbrueckii. L. delbrueckii strains were spotted and allowed to grow for 2 days anaerobically. The plates were moved to an aerobic environment, where the strains were killed by exposure to chloroform vapors and then overlaid with a soft agar inoculated with P. gingivalis, incubated, and imaged. (B) Spot assay with different strains of L. delbrueckii. L. delbrueckii strains were spotted and allowed to grow for 2 days anaerobically. The plates were moved to an aerobic environment, where P. gingivalis was spotted adjacent to the L. delbrueckii spots, allowed to dry, and incubated for 2 days anaerobically. Plates were imaged after the final 2 days of growth.
We focused on the characterization of the STYM1 strain since it showed the most inhibition of P. gingivalis. Since lactobacilli are known to secrete antimicrobial molecules, we tested whether the supernatant from STYM1 grown in brain heart infusion (BHI) broth inhibited P. gingivalis. Cell-free supernatants were harvested from either overnight or 48 h STYM1 cultures and tested for activity in a broth-based assay monitoring endpoint P. gingivalis growth. Only the 48-h supernatant inhibited growth of P. gingivalis (see Fig. S1A in the supplemental material). Since Lactobacillus species have a fermentative metabolism, we wanted to ensure that the inhibitory activity in 48-h supernatant was not due to lowered pH from increased acid end products. The pH was stable between 24 and 48 h of incubation in all strains, suggesting that pH was not involved in the inhibition (see Fig. S1B in the supplemental material). Further, treatment of the 48-h STYM1 supernatant with heat, proteinase K, or passage through a 10-kDa-MWCO (molecular weight cutoff) filter significantly reduced the inhibitory effect of the supernatant on P. gingivalis growth, suggesting that an inhibitory protein was in the STYM1 supernatant (Fig. S1C).
When examining the growth dynamics of STYM1 in BHI broth culture, we observed significant autolysis of the STYM1 culture during the period of 24 to 48 h of incubation indicated by a drop in the optical density at 600 nm (OD600) and an ∼10-fold reduction in CFU/ml (Fig. S2A and B). Previous research has demonstrated that Lactobacillus species can undergo autolysis in the late stationary phase, especially in response to carbon starvation (25, 26). BHI medium only contains 0.2% (wt/vol) glucose, so we tested whether glucose limitation induced autolysis under these conditions. Indeed, we found that the autolysis of STYM1 in BHI was due to carbon limitation (Fig. S2A). Considering that only the 48-h culture supernatant had inhibitory activity, we reasoned that the inhibitory molecule could be an intracellular component released into the supernatant via autolysis.
Cellular extracts of STYM1 contain an inhibitory protein.
To confirm that the inhibitory molecule is located intracellularly, we tested soluble cellular extract for inhibitory activity in an agar overlay assay. Only the STYM1 cellular extract had inhibitory activity, while the extracts from the noninhibitory strains had no effect on P. gingivalis growth (Fig. 2A). To determine the nature of the inhibitory activity in the STYM1 extract, we treated the extract with either heat, proteinase K, or passage through a 10-kDa-MWCO filter. Treatment with heat and proteinase K completely abolished inhibitory activity in STYM1 extract, and the 10-kDa-MWCO filter retained the majority of the inhibitory activity, suggesting that one or several proteins are responsible for the inhibition of P. gingivalis (Fig. 2B, C, and D).
FIG 2.
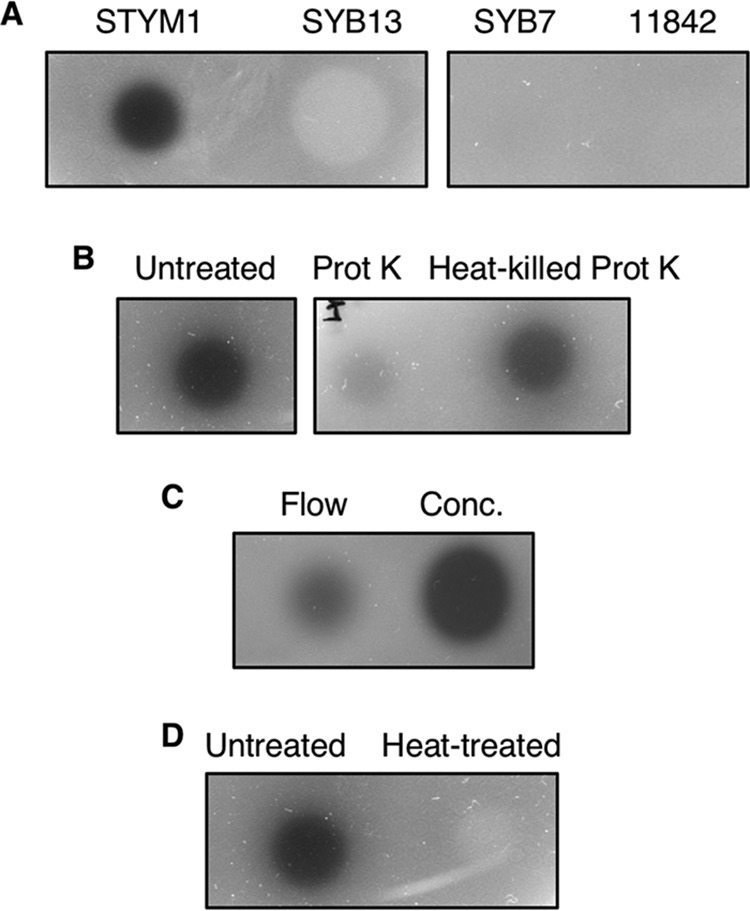
STYM1 cellular extracts have inhibitory protein. Extract was spotted onto plates, allowed to dry under aerobic conditions, and overlaid with a soft top agar inoculated with P. gingivalis. Plates were imaged 2 days after anaerobic growth. (A) Agar overlay assay with equal protein amounts of soluble cell extracts from STYM1, SYB13, SYB7, and 11842. (B to D) Agar overlay assays with STYM1 cellular extract either treated with proteinase K (Prot K) or heat-killed proteinase K (Heat-killed Prot K) (B), passage through a 10-kDa-MWCO filter (Flow and Conc.) (C), or heat treatment (95°C for 20 min) (D).
Fractionation of STYM1 extract.
To identify the inhibitory protein in the STYM1 extracts, we fractionated the extracts using ammonium sulfate precipitation, anion-exchange chromatography, and gel filtration. Fractions with inhibitory activity were pooled and used as input for the next fractionation step. After gel filtration, inhibitory activity peaked in fraction 9 (Fig. 3A and B). Proteins in this fraction were visualized by Coomassie staining after SDS-PAGE, which revealed only four bands, with one dominant band at ∼70 kDa (Fig. 3C). The dominant band at 70 kDa in fraction 9 was excised from the gel and analyzed by mass spectrometry (see Table S1 in the supplemental material). The top five most abundant proteins in the sample were glutamine-fructose-6-phosphate aminotransferase, pyruvate oxidase, pyruvate kinase, phosphoenolpyruvate-protein phosphotransferase, and molecular chaperone DnaK.
FIG 3.
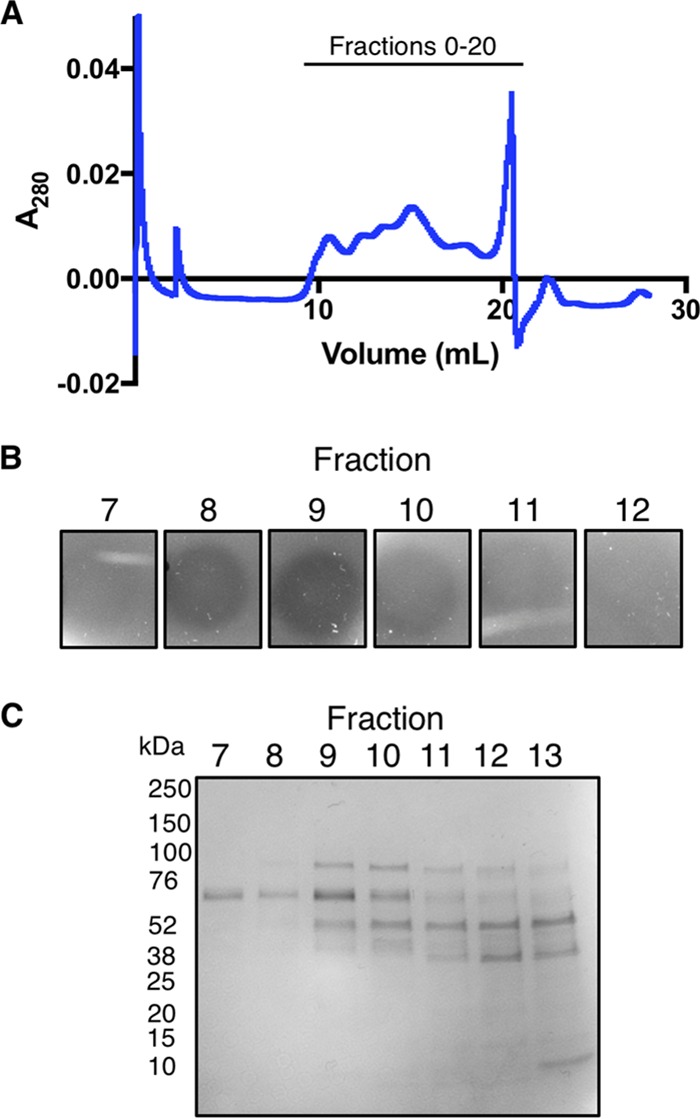
Fractionation of STYM1 cellular extracts. (A) Trace of the absorbance at 280 nm of the eluate of gel filtration of pooled inhibitory fractions from anion-exchange chromatography. Fractions were collected following the void starting at 10 ml in 500-μl volumes. (B) Agar overlay of gel filtration fractions 7 through 12 from panel A. Fractions were spotted onto plates, allowed to dry under aerobic conditions, and overlaid with a soft top agar inoculated with P. gingivalis. Plates were imaged 2 days after anaerobic growth. (C) Coomassie stain of gel filtration fractions 7 through 13 after separation by SDS-PAGE. The 70-kDa band from fraction 9 was excised for mass spectrometry analysis.
Genomic analysis of inhibitor versus noninhibitor strains.
We took a genomics approach to determine the differences between inhibitor versus noninhibitor strains of L. delbrueckii and to assess whether any proteins identified by mass spectrometry were unique to inhibitor strains. Whole-genome sequencing of STYM1 and two noninhibitor strains (SYB7 and ATCC 11842) revealed many differences and genomic rearrangements. The STYM1 genome is ∼400 kb larger than the 11842 genome and SYB7 genome and not surprisingly has 412 and 405 additional genes, respectively, that are unique; many of these encode transposases of metabolic function or hypothetical. The STYM1 genome is almost identical to the published L. delbrueckii strain ND02, having only 62 single nucleotide polymorphisms or small indels, so we use the ND02 gene locus nomenclature of LDBND (27).
We used our genome analysis to determine whether any of the proteins identified by mass spectrometry were unique to the inhibitor strain relative to noninhibitor strains. This analysis revealed that three oxidase genes identified by mass spectrometry—pyruvate oxidase (Pox), lactate oxidase (Lox), and multicopper oxidase (Mco)—are truncated in noninhibitory strains and likely produce nonfunctional peptides (Fig. 4). At the pox locus, the genomic arrangement is similar in inhibitor and noninhibitor strains, but there is a 283-bp deletion within the pox gene in the noninhibitor strains that results in the introduction of a premature stop codon (Fig. 4A). In the lox region, the 5′ region of lox, as well as the entire upstream lactate permease gene, is deleted in the noninhibitor strains (Fig. 4B). In the multicopper oxidase region, the STYM1 and SYB7 strains have an intact mco gene, but the 11842 strain has a 7-bp insertion that results in the introduction of a premature stop codon (Fig. 4C).
FIG 4.

L. delbrueckii genome heterogeneity of pox, lox, and mco regions. (A) Gene maps of the pox regions in STYM1, SYB7, and 11842. Indicated are the hypothetical membrane protein (orange), pyruvate oxidase (LDBND_1487) (pox, blue), and d-Ala-d-Ala carboxypeptidase (dacA, yellow). (B) Gene maps of the lox regions in STYM1, SYB7, and 11842. Indicated are the oligopeptidase F (pepF, red), lactate oxidase (lox, blue), lactate permease (lctP, green), zinc ribbon protein (dark purple), and an Mn/Zn permease (light purple). (C) Gene maps of the mco regions in STYM1, SYB7, and 11842. Indicated are the peptide release factor C (prfC, aqua), hypothetical proteins (orange), multicopper oxidase (mco, green), and ClpE protease (clpE, red). (D) Agar overlay of STYM1 extract either untreated or treated with 10 μg of catalase. Extract was spotted onto plates, allowed to dry under aerobic conditions, and overlaid with a soft top agar inoculated with P. gingivalis. Plates were imaged 2 days after anaerobic growth.
STYM1 extract produces hydrogen peroxide.
Since Pox, Lox, and some multicopper oxidases generate hydrogen peroxide, we tested whether addition of catalase to STYM1 cellular extracts could eliminate inhibitory activity. Indeed, catalase treatment of STYM1 extracts completely abolished inhibitory activity, demonstrating that hydrogen peroxide production, likely by one or more of the oxidases identified by mass spectrometry, is the mechanism of action (Fig. 4D).
We wanted to determine whether the inhibitory strains produce more hydrogen peroxide than noninhibitory strains due to the genomic differences. In addition, since it is common for oral streptococci to produce hydrogen peroxide, we tested the hydrogen peroxide production of S. sanguinis SK36 in our assay (28, 29). STYM1 and GVKM1 produced more hydrogen peroxide more quickly upon exposure to oxygen than the noninhibitory strains and S. sanguinis, as indicated by the greater diameter and intensity of the Prussian blue dye formation at both 2 and 20 h after exposure to oxygen (Fig. 5).
FIG 5.

Inhibitory strains produce more hydrogen peroxide more rapidly upon exposure to oxygen. Shown are images of each L. delbrueckii strain and S. sanguinis SK36 on Prussian blue agar plates after exposure to oxygen at 2 and 20 h after exposure. Dilutions of hydrogen peroxide are also shown. Shown are the mean measurements of the Prussian blue diameter from three independent experiments. Error bars represent standard errors of the mean. Statistical significance was determined by a one-way ANOVA corrected for multiple comparisons (*, P < 0.001). The asterisk indicates comparison and statistical significance between the asterisked bar and each of the SYB7, SYB13, 11842, and SK36 bars at the same time point.
Screening of P. gingivalis transposon library by exposure to STYM1 extracts.
Next, we determined whether hydrogen peroxide was the major component of STYM1 inhibitory activity and whether we could isolate P. gingivalis-resistant mutants. To do this, we took advantage of a transposon library constructed in the W83 strain of P. gingivalis (30). We exposed the P. gingivalis transposon library to STYM1 extract and then plated for survivors. We selected 36 surviving colonies from two separate experiments and sequenced their transposon-genome junctions to determine the location of transposon insertion. Remarkably, 34 of 36 clones had transposon insertions in the feoB2 gene (PG_1294) of W83, while the other two clones had transposon insertions in the predicted feoC homologue, FeoB-associated cysteine-rich membrane protein (Fig. 6A). FeoB2 (PG_1294) has been previously characterized as an iron transporter in P. gingivalis (31, 32). Deletion mutants of feoB2 (PG_1294) have lowered intracellular iron levels, are unable to grow in vivo in a mouse abscess model, and have increased resistance to hydrogen peroxide and atmospheric oxygen (31, 33). FeoB1 (PG_1043) has been established as a manganese transporter that is important for oxidative stress tolerance (31). Previous reports refer to PG_1043 as feoB2 and PG_1294 as feoB1, whereas National Center for Biotechnology Information (NCBI) annotation refers to PG_1043 as feoB1 and PG_1294 as feoB2. We chose to refer to PG_1294 as feoB2 and PG_1043 as feoB1, consistent with the NCBI annotation. When tested in isolation, the feoB2 transposon mutant displays significantly greater resistance to killing by STYM1 extract and complementation with the native feoB2 gene restores sensitivity (Fig. 6B). These data confirm that resistance is due to disruption of the feoB2 gene and not the presence of a secondary site mutation elsewhere in the genome or downstream polar effects.
FIG 6.

Transposon insertions in P. gingivalis feoB2 operon confer resistance to killing by STYM1 extracts. (A) Unique transposon insertion locations for transposon mutants isolated after exposure to STYM1 cellular extracts are indicated by blue arrows. Only unique insertion sites are marked since some of the isolated mutants had the same insertion sites. (B) Confirmation of feoB2 transposon mutant resistance phenotype. The number of CFU after 3 h of exposure to STYM1 extract is shown for the wild-type W83 parental strain, the transposon mutant in feoB2 (Tn-FeoB2), the Tn-FeoB2 strain complemented with the wild-type feoB2 gene (Tn-FeoB2 Comp), and the Tn-FeoB2 strain complemented with the empty pT-Cow vector (Tn-FeoB2 Empty). Data represent the averages of three independent experiments, and error bars represent the standard errors. Statistical significance was determined by one-way ANOVA (***, P < 0.001).
One of the main mechanisms by which hydrogen peroxide exerts antimicrobial effects is through the formation of reactive oxygen species via the Fenton reaction with intracellular iron (34). Because we only recovered transposon mutants in feoB2 that were resistant to killing by STYM1 extracts, we reasoned that hydrogen peroxide production must be the dominant function of the inhibitory protein in the STYM1 extracts.
Supplementation of STYM1 extracts with Pox substrates and cofactors enhances H2O2 production.
In Lactobacillus species, Pox catalyzes the conversion of pyruvate, phosphate, and oxygen to acetyl phosphate, carbon dioxide, and hydrogen peroxide. Pox employs thiamine pyrophosphate (TPP), flavin adenine dinucleotide (FAD), and Mn+2 to carry out this reaction (35, 36). Since Pox was one of the most enriched proteins in the inhibitory fractions after gel filtration, we determined whether we could increase hydrogen peroxide production in the STYM1 extracts by simply adding substrates for Pox. Prussian blue agar has been developed to detect oxidase activity in bacterial extracts (37). We adapted this system to be used with tryptic soy agar (TSA) plates so that we could detect hydrogen peroxide production in STYM1 extracts. Halos of Prussian blue form around wells in the agar plate filled with hydrogen peroxide. The reaction is specific, and the diameter of the halo is dependent on the concentration of hydrogen peroxide (Fig. 7A).
FIG 7.

Supplementation of STYM1 extract with pyruvate oxidase substrates and cofactors enhances H2O2 production. (A) Prussian blue agar plate with 50 μl of known concentrations of H2O2 in 6-mm-diameter wells after incubation at room temperature. (B) Prussian blue agar plate with STYM1 cellular extract alone or supplemented with Pox substrate, Pox substrate plus cofactors, or Lox substrate plus cofactors. Also included as negative controls are the 11842 strain cellular extract and no-extract controls with the indicated substrates or cofactors. Images are representative of three independent experiments. (C) Measurements of the Prussian blue diameter minus the inner well (6-mm diameter) for the STYM1 extract samples in panel B from three independent experiments. Error bars represent standard errors of the mean. Statistical significance determined by one-way ANOVA corrected for multiple comparisons (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
The STYM1 extract alone produces a moderate amount of hydrogen peroxide on Prussian blue plates in the range of 500 μM to 1 mM, while the 11842 strain extract produces no detectable hydrogen peroxide (Fig. 7B). However, upon supplementation with 50 mM pyruvate and phosphate, the hydrogen peroxide production significantly increased, as measured by the Prussian blue halo diameter. Hydrogen peroxide production was even further enhanced by the addition of the cofactors of the Pox enzyme: 10 mM Mn+2, 15 μM FAD, and 300 μM TPP. Supplementation of the STYM1 lysate with Lox substrate and cofactors 150 mM dl-lactate, 300 μM TPP, and 15 μM FAD resulted in a significant increase in hydrogen peroxide production relative to the extract alone. However, that increase was significantly less than the level observed with supplementation of Pox substrates and cofactors (Fig. 7C). These data suggest that Pox is likely the main producer of hydrogen peroxide in STYM1 extracts.
Multicopper oxidases are a large family of proteins consisting of tyrosinases, monooxygenases, dioxygenases, and laccases, among others (38). Most of these enzymes do not produce hydrogen peroxide. However, laccases have been characterized in some fungi, and several have been identified in bacterial species; bacterial laccases are able to produce hydrogen peroxide in some cases (39–41). Generally, the substrates of laccases are polyphenols and aromatic amines. To test whether laccase activity exists in STYM1 extracts, we examined the extract’s ability to oxidize the common laccase substrate syringalazine (42). STYM1 extract was unable to oxidize the syringalazine substrate, suggesting that the multicopper oxidase is not a laccase or at least cannot use syringalazine as a substrate.
Treatment of STYM1 extracts with oxidase inhibitors.
To further confirm the involvement of Pox in hydrogen peroxide production in STYM1 extracts, we took advantage of several inhibitors being described for Pox, Lox, and multicopper oxidases. Sodium sulfite is an inhibitor of flavin-dependent oxidases since the sulfite deactivates the flavin moiety by forming a covalent adduct to the molecule (43, 44). Both Pox and Lox are flavin-dependent oxidases, so both should be inhibited by sodium sulfite. Because Pox coordinates a metal ion, it can be inhibited by treatment with EDTA. Oxalate acts as a specific inhibitor of Lox, presumably by blocking the active site (45). Lastly, several multicopper oxidases are known to be inhibited by treatment with sodium azide, which disrupts the copper coordination in the protein (46).
We tested each of these inhibitors for its ability to abolish the inhibitory activity seen in STYM1 extracts. Sodium azide treatment had no effect on the inhibitory activity even when tested over a wide range of concentrations, suggesting that multicopper oxidase is not the inhibitory protein (Fig. 8A). Sodium oxalate treatment also had no effect on the STYM1 extract, indicating that lactate oxidase may not be the primary hydrogen peroxide-producing protein (Fig. 8B). However, treatment with sodium sulfite completely abrogated the inhibitory activity of STYM1 extract, suggesting that Pox and/or Lox are responsible for hydrogen peroxide production (Fig. 8C). Furthermore, EDTA treatment fully blocked the ability of STYM1 extract to inhibit P. gingivalis and supplementation of Mn+2 in excess of the EDTA concentration restored inhibitory activity in the extract (Fig. 8D). Taken together, these data suggest Pox is the primary hydrogen peroxide-producing enzyme in STYM1 extracts.
FIG 8.
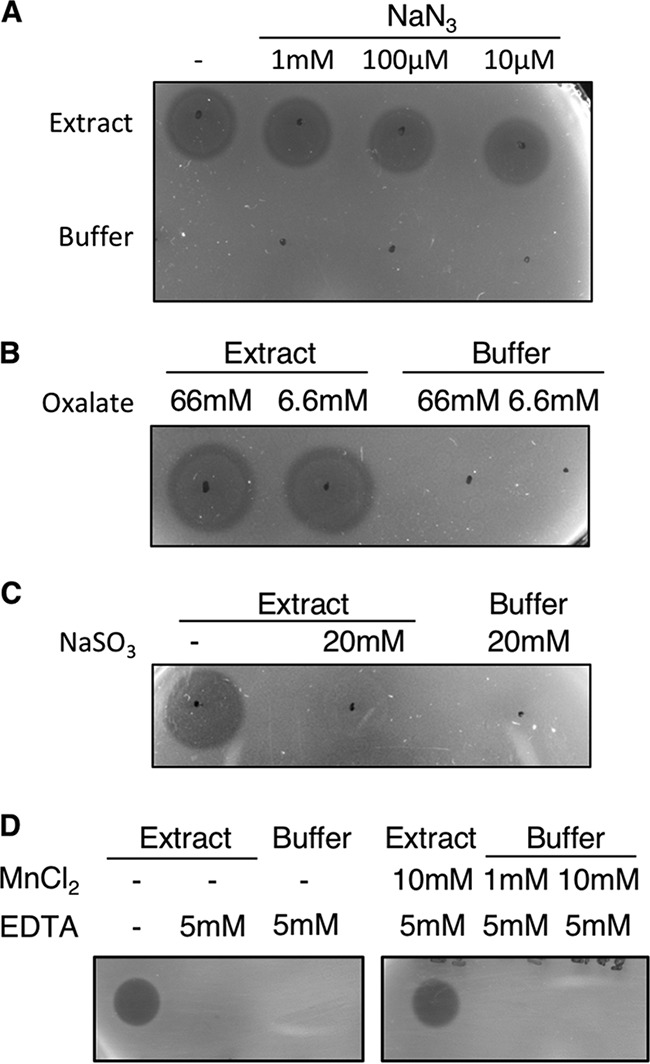
Treatment of STYM1 extracts with oxidase inhibitors. Extract was spotted onto plates, allowed to dry under aerobic conditions, and overlaid with a soft top agar inoculated with P. gingivalis. Plates were imaged 2 days after anaerobic growth. STYM1 extract was treated with the indicated concentration of either sodium azide (A), sodium oxalate (B), sodium sulfite (C), or EDTA (D) and tested in an agar overlay assay. In panel D, supplementation with MnCl2 was also included. Black dots indicate where the extract was added to the surface of the plate.
LDBND_1487 Pox is more highly expressed than LDBND_2051 Pox.
STYM1 encodes two predicted pox genes (LDBND_1487 and LDBND_2051), but only one (LDBND_1487) is unique to the STYM1 strain but not noninhibiting strains. Amino acid sequence alignment revealed that the protein sequences between the two genes are highly similar, with only four amino acid substitutions and one insertion of six amino acids (Fig. 9A). Promoter analysis with PePPER revealed the presence of the canonical −35 and −10 sequence upstream of LDBND_1487 but identified several mismatches with the canonical sequence upstream of LDBND_2051 (47) (Fig. 9B). Therefore, we decided to determine whether the two genes differ in their expression. Indeed, we found that LDBND_1487 was expressed at a significantly higher level than that of LDBND_2051 under the conditions tested (Fig. 9C).
FIG 9.

LDBND_1487 pox is more highly expressed than LDBND_2051 pox. (A) Alignment of the C-terminal region of LDBND_1487 Pox and LDBND_2051 Pox. The preceding N-terminal region is identical. (B) Alignment of the promoter regions of LDBND_1487 and LDBND_2051 with the PePPER-predicted −35 and −10 sites, the ribosome binding site, and the start codon indicated. (C) The expression of levels of the two pyruvate oxidase genes in STYM1 at the time of cellular extract harvest were measured by qRT-PCR. The graphs represent the mean expression level of LDBND_2051 relative to LDBND_1487 within each sample. The expression of LDBND_1487 was set to one in each sample. The data represent three independent experiments, and error bars represent standard errors of the mean. Statistical significance was determined by a two-tailed t test (**, P < 0.01).
Purified Pox is catalytically active and is sufficient to inhibit P. gingivalis growth in vitro.
Taken together, these data strongly suggest that LDBND_1487 Pox is the major, if not the sole, producer of hydrogen peroxide in STYM1 extracts. We attempted to generate a LDBND_1487 deletion strain multiple times with multiple different systems, but we were unable to isolate such a mutant. Although this may be a result of LDBND_1487 being an essential gene, we were unable to generate deletions in other genes or introduce plasmids into STYM1, suggesting that genetic manipulation of this strain failed due to technical reasons. Large variability in the genetic tractability of Lactobacillus species has been well documented (48). To circumvent this, we generated a His-tagged version of 1487-Pox and 2051-Pox and purified the enzymes on a Ni-NTA column (Fig. 10A). The eluted 1487-Pox and 2051-Pox fractions had a yellowish hue, which is characteristic of flavin-containing enzymes. The enzymes formed a population of dimers indicated by the band at ∼140 kDa, which is consistent with the oligomerization of other Pox enzymes (36, 49). We used an oxidative coupling reaction to assay for pyruvate oxidase activity of 1487-Pox and 2051-Pox, which results in the formation of a quinoneimine dye upon hydrogen peroxide production via Pox that is measurable by an increase in the absorbance at 550 nm. One unit of Pox activity is defined by the production of 1 μmol of H2O2 per min. Both 1487-Pox and 2051-Pox were catalytically active and did not have significantly different activity levels (Fig. 10B).
FIG 10.

Purified LDBND_1487 Pox is catalytically active and is sufficient to inhibit P. gingivalis in vitro. (A) Coomassie stain of purified 1487-Pox and 2051-Pox after SDS-PAGE. (B) Pyruvate oxidase activity was measured by the oxidative coupling reaction, which utilizes the horseradish peroxidase-catalyzed formation of a quinoneimine dye with an absorbance at 550 nm. The assay measures H2O2 production by the Pox enzyme. Pyruvate oxidase activity was determined by measuring the change in A550 per min and calculating the activity according to the equation in Materials and Methods. One unit of Pox activity is defined as the production of 1 μmol of H2O2 per min. The mean specific activity (U/mg) of 1487-Pox and 2051-Pox in at least three different purifications is shown. Error bars represent the standard errors of the mean, and significance was determined by a two-tailed t test (ns, not significant). (C) Agar overlay assay of purified 1487-Pox or 2051-Pox alone or purified 1487-Pox or 2051-Pox supplemented with substrate and/or substrate and cofactors. Buffer controls are also included. Enzyme mixtures were spotted onto plates, allowed to dry under aerobic conditions, and overlaid with a soft top agar inoculated with P. gingivalis. Plates were imaged 2 days after anaerobic growth.
We then tested the ability of purified recombinant 1487-Pox to inhibit P. gingivalis growth in an agar overlay assay. Aliquots of the Pox enzymes alone or supplemented with either substrate or substrate and FAD and TPP were spotted onto an agar plate and then overlaid with a soft agar inoculated with P. gingivalis. Purified 1487-Pox alone was unable to inhibit P. gingivalis growth in the overlay. However, 1487-Pox supplemented with only pyruvate and phosphate produced a clear zone of inhibition in the agar overlay, indicating that 1487-Pox was purified with native FAD and TPP tightly bound. Furthermore, 1487-Pox supplemented with substrate and FAD and TPP produced an even larger zone of inhibition. 2051-Pox inhibited P. gingivalis growth as well, but only with full supplementation (Fig. 10C). The substrates and cofactors alone had no effect on P. gingivalis growth, indicating that the inhibition is specific to the catalytic action of 1487-Pox (Fig. 10C). Together, these data demonstrate that Pox is sufficient to inhibit P. gingivalis growth in vitro.
DISCUSSION
In this report, we identify and describe the mechanism of antagonism between strains of L. delbrueckii and the oral pathogen P. gingivalis. The inhibitory capacity of L. delbrueckii is strain specific, underscoring the importance of strain selection and not simply species selection in developing microbial therapeutics. We demonstrate that the STYM1 strain of L. delbrueckii releases intracellular proteins via autolysis that can produce inhibitory amounts of hydrogen peroxide. We present strong evidence through studies using oxidase inhibitors, supplementation with enzyme substrates/cofactors, and purification of recombinant protein that the main, if not the sole, producer of hydrogen peroxide is the pyruvate oxidase (Pox, LDBND_1487).
Lactobacilli use multiple strategies, including the secretion of antimicrobial proteins called bacteriocins and the production of toxic metabolites such as hydrogen peroxide, to inhibit the growth of other bacteria (50). Hydrogen peroxide-mediated killing occurs through the production of superoxide and hydroxyl radicals via Fenton chemistry with intracellular iron, which ultimately damages the DNA, leading to cell death. Consistent with this, we recovered transposon mutants in the iron transporter feoB2 of P. gingivalis that are more resistant to killing by STYM1 extract, which is consistent with previous reports of hydrogen peroxide resistance feoB2 mutants (33). We did not recover transposon mutants in any other iron or heme uptake genes. This may have occurred for several reasons. The FeoB2 transporter is the only known ferrous iron transporter in P. gingivalis, and since the Fenton reaction requires ferrous iron, deletion of feoB2 would result in less toxicity than mutations in heme uptake genes. P. gingivalis likely uses iron from acquired heme. There are many heme acquisition mechanisms that have been detailed for P. gingivalis. One possibility is that iron may be disassociated from heme in the periplasm, perhaps by IhtA, and then transported into the cytoplasm via FeoB2 (51). In this case, deletion of feoB2 would impair iron transport much more than a single deletion in one of the many heme uptake systems. Lastly, it is possible that mutants in heme uptake have a more intermediate resistance phenotype than the feoB2 mutant. Since the selection for resistant mutants was strong (an ∼105-fold reduction in cell viability), the recovery of mutants with intermediate phenotypes would be much rarer.
P. gingivalis does not encode a catalase but does encode an alkyl hydroperoxidase for detoxifying hydrogen peroxide and also has a hydrogen peroxide-sensitive OxyR regulator (52, 53). It is possible that P. gingivalis can withstand low levels of hydrogen peroxide, but alkyl hydroperoxidases are generally only effective at low concentrations due to their rate limitation by NADH consumption (34). In fact, S. sanguinis inhibits P. gingivalis survival in a dual-species biofilm via hydrogen peroxide production from pyruvate oxidase, and this can only be mitigated by the presence of other bacterial species capable of detoxifying hydrogen peroxide (54). In our system, STYM1 clearly produces inhibitory levels of hydrogen peroxide, even more than S. sanguinis SK36, such that P. gingivalis’s defenses are inadequate.
Hydrogen peroxide can be generated in Lactobacillus species by several different enzymes including pyruvate oxidase, lactate oxidase, NADH oxidase, and NADH flavin-dependent reductases (35, 55, 56). Several Lactobacillus strains produce enough hydrogen peroxide to inhibit the growth of pathogens such as Listeria monocytogenes, Staphylococcus aureus, and Pseudomonas species (57–59).
The STYM1 strain encodes two pyruvate oxidase genes: LDBND_1487 and LDBND_2051. Genome analysis of STYM1 relative to noninhibitory strains revealed that LDBND_1487 is truncated in noninhibitory strains, but LDBND_2051 is intact. It is not uncommon for Lactobacillus species to carry multiple pyruvate oxidase genes in their genomes. For example, Lactobacillus plantarum encodes five pyruvate oxidase genes, only two of which are responsible for hydrogen peroxide production and Pox activity despite the other pox genes retaining amino acid residues important for catalysis and substrate and cofactor binding (60). This observation could be due to the expression levels of the different pox genes. In the STYM1 strain, LDBND_2051 Pox is catalytically active at a similar level to LDBND_1487 Pox. Purified LDBND_2051 Pox only inhibited P. gingivalis growth with full supplementation, suggesting that FAD and TPP may be bound less tightly than in LDBND_1487, although it is unclear whether this would be the case in vivo. Furthermore, LDBND_1487 pox is expressed more highly than LDBND_2051 pox, likely explaining how LDBND_1487 pox could be the main component of Pox activity in our samples. The expression data for LDBND_1487 and LDBND_2051 in the STYM1 strain are also confirmed by promoter analysis of the two genes. LDBND_1487 has −35 and −10 sites that are identical to the consensus sequence in E. coli and in Lactobacillus species (61–63). Alternatively, the LDBND_2051 pox promoter has several mismatches in the −35 and −10 sites in bases that have been shown to be critical for promoter binding by sigma factors (61, 64).
Interestingly, the regulation of LDBND_1487 pox in the STYM1 strain appears to be different than in several other organisms. In other organisms, pox is highly expressed in the stationary phase and under glucose limitation but is repressed under anaerobic conditions (35). This regulatory pattern of pox is consistent with our results in that we detected Pox by mass spectrometry in extracts from stationary-phase STYM1 L. delbrueckii grown in BHI, which is considered to be a low-glucose condition (0.2%) for Lactobacillus species. However, we did grow these cells under anaerobic conditions, which is a condition where pox is expected to be repressed. It is possible that the positive regulation of pox by growth phase and glucose limitation overrides the repression under anaerobic conditions. In fact, it is known that L. plantarum Pox can utilize alternative electron acceptors besides oxygen, which would possibly make Pox useful during anaerobic growth to increase energy production (36). This would make repression under anaerobic conditions dispensable.
It has also been shown that of two pox genes that are highly expressed in L. plantarum, one is strongly repressed in stationary phase, while the other’s expression is maintained (60). In addition, the repression of Pox under anaerobic conditions results in an approximately 50% decrease in expression levels in E. coli, but at a level that is still detectable, suggesting that while there is repression of Pox under anaerobic conditions, there is likely still some level of Pox produced (64). We have confirmed that LDBND_1487 is expressed and translated under the conditions of our assay based on our quantitative reverse transcription-PCR (qRT-PCR) and mass spectrometry data, indicating that this may be a novel regulatory pattern of pox under anaerobic conditions. Another explanation for the presence of two pox genes while only one constitutes the majority of total activity is that the two Pox enzymes may have different substrate specificities. It has been demonstrated that L. plantarum Pox can use the alternative substrates methylglyoxal and acetaldehyde, supporting this possibility (36).
The Pox enzyme has been characterized in other organisms. In L. plantarum, Pox is involved in acetate production during the stationary phase, where it converts pyruvate to acetylphosphate, which is converted to acetate by acetate kinase with the production of ATP (35, 60). This production of ATP is considered to be a major reason for the increased biomass during aerobic growth of L. plantarum (35). In Streptococcus pneumoniae, Pox accounts for the majority of hydrogen peroxide production and, similarly to L. plantarum, is involved in acetate and ATP production in concert with lactate oxidase, lactate dehydrogenase, and acetate kinase (65). In some L. delbrueckii strains, it is known that hydrogen peroxide can be produced by NADH oxidase, but STYM1 does not encode a predicted NADH oxidase (55). In Lactobacillus johnsonii, an NADH flavin reductase was found to be the major producer of hydrogen peroxide (56). Interestingly, STYM1 does encode homologues of the NADH flavin reductase (LDBND_1905 and 1906), but these genes are also present in the noninhibitory strains. Together, these observations suggest that different species can have distinct enzymes that are the main producers of hydrogen peroxide.
The role of hydrogen peroxide-producing enzymes in lactic acid bacteria remains an interesting question since these bacteria do not encode catalases, and many do not encode NADH peroxidases or alkyl hydroperoxide reductases, including the STYM1 strain. L. plantarum produces self-inhibitory levels of hydrogen peroxide upon aeration, which presumably would be a fitness disadvantage (60). However, hydrogen peroxide-producing lactobacilli have been associated with the maintenance of healthy gut and vaginal microbial communities and immune function. There is strong evidence that women with higher levels of hydrogen peroxide-producing lactobacilli have a decreased risk for developing bacterial vaginosis (66–70). In the oral cavity or in the vaginal tract, competition for glucose is likely high, so L. delbrueckii would experience carbon source limitation. We demonstrated that during carbon starvation L. delbrueckii undergoes autolysis, releasing pyruvate oxidase and other intracellular components. This may be a strategy for increasing the relative fitness of nonlysed L. delbrueckii in a form of microbial altruism. Therefore, retaining hydrogen peroxide-producing enzymes may help these bacteria maintain their specific niche and outcompete other organisms or invading pathogens in the community. Moreover, one study found that PPAR-γ, which is involved in anti-inflammatory responses and immune homeostasis, is activated by hydrogen peroxide produced by Lactobacillus crispatus (71). This aspect of hydrogen peroxide-producing bacteria is particularly attractive in the context of periodontitis since much of the pathophysiology is driven by an overactive immune response.
There is increasing interest in inhibiting P. gingivalis growth and colonization of the human oral cavity since the recent observation that P. gingivalis and its gingipains may be associated with Alzheimer’s disease and cardiovascular disease (72, 73). The characteristics of the STYM1 strain that we described make it a strong candidate for use as a probiotic strain in the treatment and/or prevention of periodontitis and P. gingivalis colonization. L. delbrueckii inhabits anaerobic or microaerobic environments since it is a facultative anaerobe. In a microaerobic environment such as at the interface between the gingival crevice and the tooth, L. delbrueckii produces hydrogen peroxide and would be in close enough proximity to P. gingivalis where hydrogen peroxide could freely diffuse to impact P. gingivalis growth. Previous studies have demonstrated that Lactobacillus species are stable colonizers of the oral cavity and that L. delbrueckii specifically can persist in the oral cavity well after the consumption of L. delbrueckii-containing food (22–24).
We also show that P. gingivalis-resistant mutants to STYM1 extracts are deficient in the feoB2 iron transporter, which has been shown to be an essential gene for in vivo survival in P. gingivalis strain W50 (31). This makes the development of resistance to STYM1-mediated or hydrogen peroxide-mediated killing in vivo very unlikely, furthering the attractiveness of STYM1 for probiotic use.
Many of the mechanisms of probiotic function remain poorly understood, hindering the rational design of probiotic strains of bacteria. In this report, we present the molecular mechanism by which a potential probiotic strain of L. delbrueckii, STYM1, inhibits the periodontal pathogen P. gingivalis. We purified the major hydrogen peroxide-producing enzyme pyruvate oxidase and showed that it inhibits P. gingivalis growth. The STYM1 strain could be developed into a useful probiotic strain for the treatment and/or prevention of periodontal disease and inform the design of other probiotic strains of bacteria.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
Bacterial strains, plasmids, and primers used in the study are listed in Tables 1 and 2. Bacterial stocks were stored in 20% glycerol at –80°C, and working cultures were inoculated from isolated colonies from agar plates. Purity was determined by Gram staining and microscopy. P. gingivalis strain W83 was grown on blood agar plates containing tryptic soy agar supplemented with 5% (vol/vol) defibrinated sheep’s blood (Hemostat, Dixon, CA), yeast extract (2 mg/ml; BD Biosciences, San Jose, CA), hemin (5 μg/ml; Sigma, St. Louis, MO), and menadione (0.5 μg/ml; Sigma, St. Louis, MO). Broth cultures of P. gingivalis were grown in BHI broth (BD Biosciences) supplemented with yeast extract (1 mg/ml), hemin (5 μg/ml), menadione (0.5 μg/ml), sodium bicarbonate (1 μg/ml; Fisher, Pittsburgh, PA), sodium thioglycolate (0.25 μg/ml; Fisher), and cysteine (0.5 μg/ml; Sigma). For agar overlay assays, tryptic soy agar plates supplemented with yeast extract (2 mg/ml), hemin (5 μg/ml), menadione (0.5 μg/ml), sodium bicarbonate (1 μg/ml), sodium thioglycolate (0.25 μg/ml), and cysteine (0.5 μg/ml) were used. Gentamicin (25 μg/ml; Fisher), erythromycin (5 μg/ml; Sigma), and tetracycline (1 μg/ml; Sigma) were used when appropriate for P. gingivalis growth and mutant selections. All P. gingivalis strains were grown at 37°C in GasPak EZ anaerobe pouch systems (BD Biosciences) for 48 h for broth cultures and 4 to 6 days for plate-based assays. L. delbrueckii strains were grown in MRS (BD Biosciences) broth or agar or in BHI broth described above. All L. delbrueckii were grown at 37°C GasPak EZ anaerobe pouch systems. Escherichia coli DH5α, S17-1λpir, and LOBSTR (Kerafast) strains were used for cloning, conjugation, and protein purification, respectively. Ampicillin (100 μg/ml; Fisher) was used when appropriate in LB agar or broth (Fisher).
TABLE 1.
Bacterial strains and plasmids used in the study
| Bacterial strain or plasmid | Description or purposea |
|---|---|
| Strains | |
| P. gingivalis | |
| W83 | Clinical isolate, also known as ATCC BAA-308 |
| Tn-FeoB2 | W83 background feoB2(PG1294) transposon mutant; Ermr |
| Tn-FeoB2 Comp | Complemented Tn-FeoB2 with pT-COW_FeoB2; Ermr Tetr |
| Tn-FeoB2 Empty | Complemented Tn-FeoB2 with pT-COW; Ermr Tetr |
| L. delbrueckii | |
| STYM1 | Wild-type inhibitor strain isolated from Stonyfield Greek yogurt |
| GVKM1 | Wild-type inhibitor strain isolated from Green Valley Farm kefir |
| SYB7 | Wild-type noninhibitor strain isolated from Smith Family Farm, ME raw milk |
| SYB13 | Wild-type noninhibitor strain isolated from Smith Family Farm, ME raw milk |
| ATCC 11842 | ATCC type strain |
| S. sanguinis | |
| SK36 | Wild type |
| E. coli | |
| Top10 | Cloning and plasmid maintenance |
| S17-1λpir | Conjugation into P. gingivalis |
| DH5α | Cloning and plasmid maintenance |
| LOBSTR | Low background strain for protein expression/purification |
| Plasmids | |
| pT-COW | Complementation vector without insert; Tetr Ampr |
| pT-COW_FeoB2 | pT-COW with wild-type W83 feoB2 and promoter |
| pFLAG-CTC_1487-Pox | E. coli expression plasmid with His6-tagged pyruvate oxidase (LDBND_1487); Ampr |
| pFLAG-CTC_2051-Pox | E. coli expression plasmid with His6-tagged pyruvate oxidase (LDBND_2051); Ampr |
Ermr, erythromycin resistance; Tetr, tetracycline resistance; Ampr, ampicillin resistance.
TABLE 2.
Primers used in this study
| Primer | Description or purpose | Sequence (5′–3′) |
|---|---|---|
| pSAM_WH2_seq1 | Semirandom PCR first round | CCCATTGGGAATAATAACCTTTATACCTG |
| Arb1 | Semirandom PCR first round | GGCCACGCGTGCACTAGTACN10TACNG |
| pSAM_WH2_seq2 | Semirandom PCR second round | GGTCTCTGCAATTGCTCGAG |
| Arb2 | Semirandom PCR second round | GGCCACGCGTGCACTAGTAC |
| pSAM_WH2_seq3 | Sequencing of semi-random PCR products | CAAGCAGAAGACGGCATACG |
| FeoB2_F_NheI | Amplification of W83 feoB2 and promoter region | GACCATGCTAGCTCTTTTGCCGCAGAGCTGATTC |
| FeoB2_R_SphI | Amplification of W83 feoB2 and promoter region | GACCATGCATGCTCAGAAGAAAAGTATTCCTATCCGGTAG |
| LDBND_1487_F_RT | qRT-PCR of LDBND_1487 pox | CTGGATGACCCAGAATTCGTGAAG |
| LDBND_1487_R_RT | qRT-PCR of LDBND_1487 pox | GAAGTTGCTTGAACTTGCAATGCTTC |
| LDBND_2051_F_RT | qRT-PCR of LDBND_2051 pox | GTTTCCCAGGTGGTTCTTTTGAC |
| LDBND_2051_R_RT | qRT-PCR of LDBND_2051 pox | CCATGCAGACACCAAGCTTG |
| 1487_pox_F_NdeI | Cloning of 1487-pox into pFLAG-CTC | AGATATCATATGGCAAAAATTAAGGGCGCAAAC |
| 1487_pox_R_XhoI | Cloning of 1487-pox into pFLAG-CTC, includes His6 tag and stop codon | AATTCCCTCGAGTTAGTGATGGTGATGGTGATGACTTCCGTGAGAAGCACCTGAAGTAGTGTC |
| 2051_pox_F_NdeI | Cloning of 2051-pox into pFLAG-CTC | AGATATCATATGGCAAAAATTAAGGGCGCAAACGCT |
| 2051_pox_R_XhoI | Cloning of 2051-pox into pFLAG-CTC, included His6 tag and stop codon | AATTCCCTCGAGTTAGTGATGGTGATGGTGATGACTTCCGTGAGAAGCACCTGAAC |
L. delbrueckii extracts and supernatants.
L. delbrueckii colonies were inoculated into BHI (10 ml) and grown anaerobically overnight at 37°C. For supernatants, the overnight culture was harvested by centrifugation for 10 min at 3,200 × g at room temperature. The supernatant was aspirated and passed through a 0.22-μm-pore size polyvinylidene difluoride filter to obtain cell-free supernatant. The pH was measured using a Beckman ϕ350 pH meter. For inhibitory activity testing, the supernatant was then combined 1:1 with fresh BHI medium and inoculated with P. gingivalis, and the final OD600 was recorded after 48 h of anaerobic growth. For cell extracts, four 10-ml cultures were combined and harvested by centrifugation for 10 min at 3,200 × g at room temperature. The cell pellet was washed once with 1 ml of 20 mM Bis-Tris (pH 7.0) and then centrifuged at 9,300 × g for 5 min. The cell pellet was resuspended in 1 ml of 20 mM Bis-Tris (pH 7.0; Sigma) with 1× cOmplete EDTA-free protease inhibitor cocktail mix (Roche, Sigma, St. Louis, MO). The cell suspension was sonicated on ice with three rounds of 15 × 1 s bursts. To remove cell debris, the suspension was centrifuged at 16,000 × g at 4°C for 15 min. The supernatant was aspirated and filtered through a 0.22-μm-pore size polyvinylidene difluoride filter, yielding the soluble cell extract, which was used in subsequent agar overlay assays and Prussian blue assays. The total protein concentration was determined by a BCA assay (Thermo Fisher, Waltham, MA).
For large-scale extraction for use in fractionation experiments, an overnight culture of L. delbrueckii grown in BHI was diluted 1:50 into 600 ml of BHI, followed by incubation overnight at 37°C. The same steps were performed as described above, except the buffer volumes were 40 ml.
Agar overlay assay and spot assay.
For agar overlays with live L. delbrueckii cultures, 2 to 5 μl of culture was spotted onto a BHI agar glass plate and allowed to dry under aerobic conditions. The plate was incubated anaerobically for 48 h, and then the L. delbrueckii cells were returned to an aerobic environment and killed by exposure to chloroform vapor for 3 h, allowed to degas for 30 min, and overlaid with 4 ml of soft BHI agar (0.7%) inoculated with the W83 strain of P. gingivalis to an OD600 of ∼0.1. The plates were incubated anaerobically for 2 to 3 days at 37°C, the zones of inhibition were analyzed, and the plates were imaged. For agar overlays with L. delbrueckii extracts, 10 to 30 μl of extract was spotted onto TSA (BD Biosciences) or BHI plates and allowed to dry in an aerobic environment. These plates were overlaid, incubated, and imaged as described above.
In the spot assays, 5-μl portions of L. delbrueckii culture were spotted onto BHI plates, allowed to dry under aerobic conditions, and incubated for 48 h anaerobically at 37°C. Then, 5 μl of P. gingivalis culture was spotted directly adjacent to the spot of L. delbrueckii growth, allowed to dry in an aerobic environment, and incubated anaerobically for 48 h at 37°C anaerobically. The plates were analyzed for zones of inhibition and imaged.
Growth curves.
L. delbrueckii was inoculated into 1 ml of BHI (which contains 0.2% glucose) or BHI supplemented with 2% glucose (Fisher) at an OD600 of 0.02, followed by incubation anaerobically at 37°C. Growth was monitored using a BioTek HT spectrophotometer. The OD600 was measured hourly over a 48-h period.
Biochemical characterization of STYM1 extract.
Prior to testing in an agar overlay assay, STYM1 extracts were treated with either heat, proteinase K, or passage through a 10-kDa-MWCO filter (Millipore, Burlington, MA). The STYM1 extract was heat inactivated at 95°C for 20 min. For proteinase K treatment, 2 μl of proteinase K (20 mg/ml; Qiagen, Germantown, MD) was added to 18 μl of extract, followed by incubation for 1 h at 37°C. Buffer was also added to a separate sample of extract and incubated for 1 h at 37°C. For heat-inactivated proteinase K, proteinase K was heated at 95°C for 20 min prior to the addition to STYM1 extract. For the 10-kDa-MWCO treatment, the STYM1 extract was flowed through a 10-kDa-MWCO filter according to the manufacturer’s instructions. The concentrate and the flowthrough were tested in an agar overlay assay.
STYM1 extracts were also treated with catalase and known oxidase inhibitors prior to testing in agar overlay assays. For catalase treatment, 10 μg of bovine liver catalase (Sigma) was added to STYM1 extracts before the agar overlay assay. To separate aliquots of STYM1 extract, sodium oxalate (Sigma) was added to final concentrations of 6.6 and 66 mM, sodium sulfite (Sigma) was added to a final concentration of 20 mM, and sodium azide (Fisher) was added to final concentrations of 1 mM, 100 μM, and 10 μM. For EDTA treatment, EDTA (Sigma) was added to a final concentration of 5 mM, followed by incubation for 1 h at room temperature prior to agar overlay or manganese supplementation. In some EDTA-treated samples, MnCl2 (Sigma) was added to final concentrations of 10 mM after the 1-h incubation with EDTA, and samples were incubated for an additional hour at room temperature before plating for the agar overlay assay.
Fractionation of STYM1 extracts.
Proteins from 40-ml cell extracts of STYM1 cultures were precipitated by stepwise ammonium sulfate precipitation. Briefly, ammonium sulfate (Sigma) was added to give 30, 40, 50, and 60% saturation during each step. After the addition of each step, the extract was rotated at 4°C for 1 h and then centrifuged at 10,000 × g for 20 min at 4°C. The supernatant was used for the next step-up of saturation, while the protein pellet was washed once in 40 ml of the same saturation of ammonium sulfate buffer and then solubilized in 1 to 2 ml of 20 mM Bis-Tris (pH 7.0) with cOmplete EDTA-free protease inhibitor cocktail. The solubilized pellet from each step was then tested in an agar overlay assay for inhibitory activity toward P. gingivalis.
Ammonium sulfate precipitation fractions with inhibitory activity were dialyzed overnight against 500 volumes of 20 mM Bis-Tris (pH 7.0) in 6- to 8-kDa-MWCO dialysis tubing. Anion exchange chromatography of these samples was carried out on Duo-Flow system (Bio-Rad, Hercules, CA) with a Hi-Trap Capto Q 1-ml column (GE Healthcare, Chicago, IL). The column was equilibrated with 5 ml of 20 mM Bis-Tris (pH 7.0); the sample was then applied to the column, and bound protein was eluted with a linear gradient of 0 to 1 M NaCl in 20 mM Bis-Tris (pH 7.0) over 25 ml at 1 ml/min flow rate. Each 1-ml fraction was tested for inhibitory activity in an agar overlay assay. Fractions with inhibitory activity were pooled and concentrated to 500 μl on 10-kDa-MWCO spin column (Millipore, Burlington, MA) prior to fractionation by size exclusion chromatography using a Superdex 200 column in 20 mM Bis-Tris (pH 7.0). Each 1-ml fraction was tested for inhibitory activity in an agar overlay assay. The protein content of each fraction was assessed by SDS-PAGE and Coomassie staining. The protein bands of interest were excised from the gel and analyzed by tandem liquid chromatography-mass spectrometry (LC-MS/MS). LC-MS/MS analysis was performed by the Taplin Biological Mass Spectrometry Facility at Harvard Medical School according to their standard protocol.
Transposon library screen and STYM1 extract exposure assays.
An aliquot of the P. gingivalis W83 transposon library was adjusted to an OD600 of 1.4 in 20 mM Bis-Tris (pH 7.0). For exposure experiments with W83 wild-type strain or individual transposon mutants, the strains were grown for 48 h in BHI, harvested by centrifugation at 9,300 × g for 5 min, and then resuspended in 20 mM Bis-Tris (pH 7.0) at an OD600 of 1.4. To 500-μl portions of these suspensions, STYM1 crude extract was added to a final concentration of 1 mg/ml of total protein. The suspension was incubated for 3 h at 37°C. Serial dilutions were plated on blood agar plates and incubated at 37°C for 6 days to enumerate the CFU. Surviving colonies of transposon mutants were reisolated on blood agar plates, and their genomic DNA was prepared with a DNeasy blood and tissue kit (Qiagen).
Nested semirandom PCR.
The transposon-genome junction of isolated transposon mutants was determined by nested semirandom PCR, which consists of two rounds of PCR. The first round used the primers pWH2_seq1 and arb1 and consisted of initial denaturation at 96°C for 3 min, followed by 5 cycles of 96°C for 1 min, annealing at 30°C for 1 min, and extension at 72°C for 1 min, followed by an additional 35 cycles but with the annealing step at 55°C and a 5-min extension at 72°C. The second round used 2 μl of the first reaction as the template and the primers pWH2_seq2 and arb2. The reaction consisted of an initial denaturation at 96°C for 3 min, followed by 35 cycles of 96°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 1 min, followed by a final extension at 72°C for 5 min. Samples were purified using a Promega Wizard kit (Promega, Madison, WI) according to the manufacturer’s instructions. Samples were sequenced with the pWH2_seq3 primer, and the sequence was aligned to the W83 genome to determine the location of transposon insertion.
Construction of feoB2 transposon mutant complement.
The wild-type feoB2 gene (PG_1294) and its native promoter (200 bp upstream) were PCR amplified, ligated into pT-COW (74), and transformed into E. coli Top10 (Thermo Fisher). Clones were confirmed by restriction digestion, PCR, and sequencing. The new plasmid, pT-COW_FeoB2, was transformed into E. coli S17-1λpir (75). The E. coli S17-1λpir harboring the pT-COW_FeoB2 plasmid was conjugated with the Tn-feoB2 strain by combining 750 μl of mid-logarithmic-phase cultures (OD600 0.4 to 0.8) of each strain and centrifugation at 9,600 × g for 2 min. The cell pellet was resuspended in 375 μl of sterile phosphate-buffered saline (PBS), transferred to a blood agar plate with no antibiotics, and incubated aerobically at 37°C for 5 h. The growth on the plate was scraped into 300 μl of PBS and then plated on blood agar plates containing gentamicin and tetracycline and incubated anaerobically at 37°C for 7 days. Complementation was confirmed by PCR and sequencing.
Prussian blue hydrogen peroxide detection.
Prussian Blue TSA plates were prepared as described previously (37). To assay for hydrogen peroxide production from L. delbrueckii strains and Streptococcus sanguinis SK36, 5-μl portions of broth cultures of each strain were spotted onto Prussian blue plates, allowed to dry, and incubated anaerobically at 37°C for 2 days. The plates were then exposed to atmospheric oxygen for 20 h, and images and Prussian blue diameters were analyzed at 2 and 20 h postexposure. To assay for hydrogen peroxide production, 6-mm wells were cut into the agar, and 50-μl portions of either different concentrations of hydrogen peroxide or test samples were added, followed by incubation for 30 min to 2 h at room temperature prior to imaging and measuring the diameter of the Prussian Blue halo. STYM1 extracts were supplemented with oxidase substrates and cofactors as follows. For pyruvate oxidase, the substrates sodium pyruvate (Sigma) and sodium phosphate (Fisher) were added to a final concentration of 50 mM each, and the cofactors thiamine pyrophosphate (TPP; Sigma), flavin adenine dinucleotide (FAD; Sigma), and MnCl2 were added to final concentrations of 300 μM, 15 μM, and 10 mM, respectively. For lactate oxidase, dl-sodium lactate (Fisher) was added to a final concentration of 150 mM to ensure there was 50 mM l-lactate available, and the cofactors TPP and FAD were added to final concentrations of 300 and 15 μM, respectively.
RNA isolation and qRT-PCR.
Total RNA from L. delbrueckii STYM1 was isolated after overnight growth in BHI medium. Then, 1 ml of the culture was centrifuged at 9,300 × g for 10 min at room temperature and resuspended in TRIzol reagent (Invitrogen, Carlsbad, CA). RNA was isolated according to the manufacturer’s instructions. Total RNA was examined for integrity by gel electrophoresis. The RNA was treated with DNase using a Turbo DNA-free kit (Invitrogen) according to the manufacturer’s instructions. cDNA was generated using the ImProm-II reverse transcription system (Promega) according to the manufacturer’s instructions.
qRT-PCR was performed with the iTaq Universal SYBR green supermix (Bio-Rad, Hercules, CA) on the CFX Connect real-time PCR detection system (Bio-Rad). Primer pairs amplifying a 150-bp product from the cDNA of LDBND_1487 pox and LDBND_2051 pox were added to a final concentration of 400 nM. The primer pairs were designed at the regions of the two genes that displayed substantial sequence variation between each other where one primer had at least six mismatches relative to the same location in the other pox gene. Thermal cycling consisted of initial denaturation of 95°C for 3 min, followed by 39 cycles of 95°C for 10 s and 60°C for 30 s. Melting curves for the products were examined to ensure that a single amplicon was produced. Samples were run in triplicate, and reverse transcriptase controls were included to confirm the absence of genomic DNA. A standard curve for each primer pair was generated using STYM1 genomic DNA. Expression was normalized to the 16S rRNA gene.
Purification of pyruvate oxidases.
LDBND_1487 and LDBND_2051 pyruvate oxidases were PCR amplified from STYM1 genomic DNA using the primers listed in Table 2. Primers for the amplification of LDBND_2051 were designed so that, at a minimum, the 3′ end had a mismatch with the sequence of LDBND_1487 to promote annealing to the LDBND_2051 pox gene. The C-terminal primer encoded a glycine-serine linker, followed by a His6 tag and a stop codon. The PCR product was ligated into the pFLAG-CTC vector (Sigma) using the NdeI and XhoI restriction sites and transformed into DH5α E. coli, resulting in the plasmid pFLAG-CTC-1487-Pox or pFLAG-CTC-2051-Pox. The constructs were confirmed by PCR and sequencing. pFLAG-CTC-1487-Pox or pFLAG-CTC-2051-Pox were transformed into LOBSTR E. coli, in which Pox was expressed as a C-terminal His6-Pox. A 6-ml starter culture was incubated at 37°C overnight in LB supplemented with 100 μg/ml of ampicillin. The starter culture was diluted 1:100 into 40 ml of LB supplemented with 100 μg/ml of ampicillin and grown at 37°C until an OD600 of 0.4 to 0.8 was reached. The temperature was then shifted to 25°C, and the cells were incubated with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma) for 20 h. The cells were harvested by centrifugation at 3,200 × g for 20 min, resuspended in 1 ml of PBS or 20 mM Bis-Tris/150 mM NaCl (pH 7) with EDTA-free cOmplete protease inhibitor cocktail, and lysed by sonication on ice, as described above. The soluble cell lysate was harvested by centrifugation at 16,000 × g for 15 min at 4°C and filtered through a 0.22-μm-pore size polyvinylidene difluoride filter to obtain cell-free soluble cell lysate. The soluble cell lysate was applied to a 1-ml bed volume of HisPure Ni-NTA resin (Thermo Fisher), and the eluate was applied a second time to the column to maximize protein binding. The column was washed twice with buffer containing 25 mM imidazole (Fisher), and then bound protein was eluted from the column in buffer containing 250 mM imidazole. Protein purity was assessed by SDS-PAGE and Coomassie staining (76).
Pyruvate oxidase activity assay.
Pyruvate oxidase activity of recombinant Pox was assessed by an oxidative coupling reaction. The reaction consisted of 50 mM sodium pyruvate (Sigma), 50 mM sodium phosphate (Fisher), 15 μM FAD, 300 μM TPP, 0.03% N-ethyl-N-(2-hydroxy-3-sulfopropyl)-m-toluidine (EHSTP; Sigma), 0.015% 4-aminoantipyrine (Sigma), and horseradish peroxidase (33 μg/ml; Sigma). Typically, 100 μl of a dilution of purified Pox enzyme was then added in a total reaction volume of 1 ml. Immediately upon addition of purified Pox, the formation of a quinoneimine dye was measured by an increase in the absorbance at 550 nm, which was recorded every 30 s at room temperature. Pox activity generates hydrogen peroxide, which is then used by peroxidase to oxidize EHSTP and 4-aminoantipyrine, forming the quinoneimine dye. Pox activity is assessed by determining the change in absorbance at 550 nm per min. One unit of Pox activity is defined as the production of 1 μmol of hydrogen peroxide per min. Activity was determined according to the following equation: specific activity = [(ΔA550/min)(dilution factor)(total reaction volume)]/[(36.88)(0.5)(path length)(sample volume)(C)], where the total reaction volume is 1 ml, the millimolar extinction coefficient of quinoneimine dye is 36.88, the factor of 0.5 was determined based on the fact that 1 mol of hydrogen peroxide produces 0.5 mol of quinoneimine dye, the light path length is 1 cm, the sample volume is 0.1 ml, and C is the concentration of undiluted enzyme in mg/ml.
Statistical analysis.
Data were analyzed using either a one-way analysis of variance (ANOVA) corrected for multiple comparisons or a two-tailed t test where appropriate. The data were drawn from three independent experiments, and error bars represent the standard error. P values of <0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Hu lab and Pooja Balani of the Duncan lab for informative discussions during these studies. We greatly thank Janna Bigalke, Andrea Koenigsberg, Claire Metrick, and Ellen White of the Heldwein lab for assisting in the biochemical fractionation experiments and protein purification.
This study was funded by R01 DE024308 (to L.T.H. and M.J.D.), F31 DE025523 (to L.P.C.), and F31 DE022491 (to B.A.K.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01271-19.
REFERENCES
- 1.Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 27:409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, Morelli L, Canani RB, Flint HJ, Salminen S, Calder PC, Sanders ME. 2014. Expert consensus document: the international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol 11:506–514. doi: 10.1038/nrgastro.2014.66. [DOI] [PubMed] [Google Scholar]
- 3.Moayyedi P, Ford AC, Talley NJ, Cremonini F, Foxx-Orenstein AE, Brandt LJ, Quigley E. 2010. The efficacy of probiotics in the treatment of irritable bowel syndrome: a systematic review. Gut 59:325–332. doi: 10.1136/gut.2008.167270. [DOI] [PubMed] [Google Scholar]
- 4.Delzenne NM, Neyrinck AM, Bäckhed F, Cani PD. 2011. Targeting gut microbiota in obesity: effects of prebiotics and probiotics. Nat Rev Endocrinol 7:639–646. doi: 10.1038/nrendo.2011.126. [DOI] [PubMed] [Google Scholar]
- 5.Yoo SR, Kim YJ, Park DY, Jung UJ, Jeon SM, Ahn YT, Huh CS, McGregor R, Choi MS. 2013. Probiotics L. plantarum and L. curvatus in combination alter hepatic lipid metabolism and suppress diet-induced obesity. Obesity 21:2571–2578. doi: 10.1002/oby.20428. [DOI] [PubMed] [Google Scholar]
- 6.de Sousa Moraes LF, Grzeskowiak LM, de Sales Teixeira TF, Gouveia Peluzio MDC. 2014. Intestinal microbiota and probiotics in celiac disease. Clin Microbiol Rev 27:482–489. doi: 10.1128/CMR.00106-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bermúdez-Humarán LG, Aubry C, Motta J-P, Deraison C, Steidler L, Vergnolle N, Chatel J-M, Langella P. 2013. Engineering lactococci and lactobacilli for human health. Curr Opin Microbiol 16:278–283. doi: 10.1016/j.mib.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Nishiyama K, Seto Y, Yoshioka K, Kakuda T, Takai S, Yamamoto Y, Mukai T. 2014. Lactobacillus gasseri SBT2055 reduces infection by and colonization of Campylobacter jejuni. PLoS One 9:e108827. doi: 10.1371/journal.pone.0108827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carreras NL, Martorell P, Chenoll E, Genovés S, Ramón D, Aleixandre A. 2018. Anti-obesity properties of the strain Bifidobacterium animalis subsp. lactis CECT 8145 in Zücker fatty rats. Benef Microbes 9:629–641. doi: 10.3920/BM2017.0141. [DOI] [PubMed] [Google Scholar]
- 10.Panigrahi P, Parida S, Nanda NC, Satpathy R, Pradhan L, Chandel DS, Baccaglini L, Mohapatra A, Mohapatra SS, Misra PR, Chaudhry R, Chen HH, Johnson JA, Morris JG, Paneth N, Gewolb IH. 2017. A randomized symbiotic trial to prevent sepsis among infants in rural India. Nature 548:407–412. doi: 10.1038/nature23480. [DOI] [PubMed] [Google Scholar]
- 11.Lin T-H, Lin C-H, Pan T-M. 2018. The implication of probiotics in the prevention of dental caries. Appl Microbiol Biotechnol 102:577–586. doi: 10.1007/s00253-017-8664-z. [DOI] [PubMed] [Google Scholar]
- 12.Gruner D, Paris S, Schwendicke F. 2016. Probiotics for managing caries and periodontitis: systematic review and meta-analysis. J Dent 48:16–25. doi: 10.1016/j.jdent.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Ahola AJ, Yli-Knuuttila H, Suomalainen T, Poussa T, Ahlström A, Meurman JH, Korpela R. 2002. Short-term consumption of probiotic-containing cheese and its effect on dental caries risk factors. Arch Oral Biol 47:799–804. doi: 10.1016/S0003-9969(02)00112-7. [DOI] [PubMed] [Google Scholar]
- 14.Krasse P, Carlsson B, Dahl C, Paulsson A, Nilsson A, Sinkiewicz G. 2006. Decreased gum bleeding and reduced gingivitis by the probiotic Lactobacillus reuteri. Swed Dent J 30:55–60. [PubMed] [Google Scholar]
- 15.Twetman S, Keller MK. 2012. Probiotics for caries prevention and control. Adv Dent Res 24:98–102. doi: 10.1177/0022034512449465. [DOI] [PubMed] [Google Scholar]
- 16.Riccia DD, Bizzini F, Perilli MG, Polimeni A, Trinchieri V, Amicosante G, Cifone MG. 2007. Anti-inflammatory effects of Lactobacillus brevis (CD2) on periodontal disease. Oral Dis 13:376–385. doi: 10.1111/j.1601-0825.2006.01291.x. [DOI] [PubMed] [Google Scholar]
- 17.Sajedinejad N, Paknejad M, Houshmand B, Sharafi H, Jelodar R, Shahbani Zahiri H, Noghabi KA. 2018. Lactobacillus salivarius NK02: a potent probiotic for clinical application in mouthwash. Probiotics Antimicrob Proteins 10:485–495. doi: 10.1007/s12602-017-9296-4. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi R, Kobayashi T, Sakai F, Hosoya T, Yamamoto M, Kurita-Ochiai T. 2017. Oral administration of Lactobacillus gasseri SBT2055 is effective in preventing Porphyromonas gingivalis-accelerated periodontal disease. Sci Rep 7:545. doi: 10.1038/s41598-017-00623-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gatej SM, Marino V, Bright R, Fitzsimmons TR, Gully N, Zilm P, Gibson RJ, Edwards S, Bartold PM. 2018. Probiotic Lactobacillus rhamnosus GG prevents alveolar bone loss in a mouse model of experimental periodontitis. J Clin Periodontol 45:204–212. doi: 10.1111/jcpe.12838. [DOI] [PubMed] [Google Scholar]
- 20.Kim HS, Kim YY, Oh JK, Bae KH. 2017. Is yogurt intake associated with periodontitis due to calcium? PLoS One 12:e0187258. doi: 10.1371/journal.pone.0187258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimazaki Y, Shirota T, Uchida K, Yonemoto K, Kiyohara Y, Iida M, Saito T, Yamashita Y. 2008. Intake of dairy products and periodontal disease: the Hisayama study. J Periodontol 79:131–137. doi: 10.1902/jop.2008.070202. [DOI] [PubMed] [Google Scholar]
- 22.Dal Bello F, Hertel C. 2006. Oral cavity as natural reservoir for intestinal Lactobacilli. Syst Appl Microbiol 29:69–76. doi: 10.1016/j.syapm.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Badet C, Thebaud NB. 2008. Ecology of Lactobacilli in the oral cavity: a review of literature. Open Microbiol J 2:38–48. doi: 10.2174/1874285800802010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dassi E, Ferretti P, Covello G, Bertorelli R, Denti MA, Sanctis VD, Tett A, Segata N. 2018. The short-term impact of probiotic consumption on the oral cavity microbiome. Sci Rep 8:10476. doi: 10.1038/s41598-018-28491-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pang XY, Cui WM, Liu L, Zhang SW, Lv J-P. 2014. Gene knockout and overexpression analysis revealed the role of N-acetylmuramidase in autolysis of Lactobacillus delbrueckii subsp. bulgaricus Ijj-6. PLoS One 9:e104829. doi: 10.1371/journal.pone.0104829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lortal S, Chapot-Chartier MP. 2005. Role, mechanisms and control of lactic acid bacteria lysis in cheese. Int Dairy J 15:857–871. doi: 10.1016/j.idairyj.2004.08.024. [DOI] [Google Scholar]
- 27.Sun Z, Chen X, Wang J, Zhao W, Shao Y, Guo Z, Zhang X, Zhou Z, Sun T, Wang L, Meng H, Zhang H, Chen W. 2011. Complete genome sequence of Lactobacillus delbrueckii subsp. bulgaricus strain ND02. J Bacteriol 193:3426–3427. doi: 10.1128/JB.05004-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L, Ge X, Dou Y, Wang X, Patel JR, Xu P. 2011. Identification of hydrogen peroxide production-related genes in Streptococcus sanguinis and their functional relationship with pyruvate oxidase. Microbiology 157:13–20. doi: 10.1099/mic.0.039669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng L, Itzek A, Chen Z, Kreth J. 2011. Oxygen dependent pyruvate oxidase expression and production in Streptococcus sanguinis. Int J Oral Sci 3:82–89. doi: 10.4248/IJOS11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein BA, Cornacchione LP, Collins M, Malamy MH, Duncan MJ, Hu LT. 2017. Using Tn-Seq to identify pigmentation-related genes of Porphyromonas gingivalis: characterization of the role of a putative glycosyltransferase. J Bacteriol 199:e00832-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dashper SG, Butler CA, Lissel JP, Paolini RA, Hoffmann B, Veith PD, O’Brien-Simpson NM, Snelgrove SL, Tsiros JT, Reynolds EC. 2005. A novel Porphyromonas gingivalis FeoB plays a role in manganese accumulation. J Biol Chem 280:28095–28102. doi: 10.1074/jbc.M503896200. [DOI] [PubMed] [Google Scholar]
- 32.He J, Miyazaki H, Anaya C, Yu F, Yeudall WA, Lewis JP. 2006. Role of Porphyromonas gingivalis FeoB2 in metal uptake and oxidative stress protection. Infect Immun 74:4214–4223. doi: 10.1128/IAI.00014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anaya-Bergman C, He J, Jones K, Miyazaki H, Yeudall A, Lewis JP. 2010. Porphyromonas gingivalis ferrous iron transporter FeoB1 influences sensitivity to oxidative stress. Infect Immun 78:688–696. doi: 10.1128/IAI.00108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorquet F, Goffin P, Muscariello L, Baudry J-B, Ladero V, Sacco M, Kleerebezem M, Hols P. 2004. Characterization and functional analysis of the poxB gene, which encodes pyruvate oxidase in Lactobacillus plantarum. J Bacteriol 186:3749–3759. doi: 10.1128/JB.186.12.3749-3759.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sedewitz B, Schleifer KH, Götz F. 1984. Purification and biochemical characterization of pyruvate oxidase from Lactobacillus plantarum. J Bacteriol 160:273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu Z, Zhou N, Zhao C, Qiu J. 2013. In-gel determination of l-amino acid oxidase activity based on the visualization of Prussian blue-forming reaction. PLoS One 8:e55548. doi: 10.1371/journal.pone.0055548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solomon EI, Sundaram UM, Machonkin TE. 1996. Multicopper oxidases and oxygenases. Chem Rev 96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 39.Claus H, Filip Z. 1997. The evidence of a laccase-like enzyme activity in a Bacillus sphaericus strain. Microbiol Res 152:209–216. doi: 10.1016/S0944-5013(97)80014-6. [DOI] [Google Scholar]
- 40.Hullo M-F, Moszer I, Danchin A, Martin-Verstraete I. 2001. CotA of Bacillus subtilis is a copper-dependent laccase. J Bacteriol 183:5426–5430. doi: 10.1128/JB.183.18.5426-5430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlosser D, Höfer C. 2002. Laccase-catalyzed oxidation of Mn2+ in the presence of natural Mn3+ chelators as a novel source of extracellular H2O2 production and its impact on manganese peroxidase. Appl Environ Microbiol 68:3514–3521. doi: 10.1128/aem.68.7.3514-3521.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harkin JM, Obst JR. 1973. Syringaldazine, an effective reagent for detecting laccase and peroxidase in fungi. Experientia 29:381–387. doi: 10.1007/BF01926734. [DOI] [Google Scholar]
- 43.Massey V, Müller F, Feldberg R, Schuman M, Sullivan PA, Howell LG, Mayhew SG, Matthews RG, Foust GP. 1969. The reactivity of flavoproteins with sulfite possible relevance to the problem of oxygen reactivity. J Biol Chem 244:3999–4006. [PubMed] [Google Scholar]
- 44.Müller F, Massey V. 1969. Flavin-sulfite complexes and their structures. J Biol Chem 244:4007–4016. [PubMed] [Google Scholar]
- 45.Ghisla S, Massey V. 1975. Mechanism of inactivation of the flavoenzyme lactate oxidase by oxalate. J Biol Chem 250:577–584. [PubMed] [Google Scholar]
- 46.Slomczynski D, Nakas JP, Tanenbaum SW. 1995. Production and characterization of laccase from Botrytis cinerea 61-34. Appl Environ Microbiol 61:907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Jong A, Pietersma H, Cordes M, Kuipers OP, Kok J. 2012. PePPER: a webserver for prediction of prokaryote promoter elements and regulons. BMC Genomics 13:299. doi: 10.1186/1471-2164-13-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aukrust TW, Brurberg MB, Nes IF. 1995. Transformation of Lactobacillus by electroporation, p 201–208. In Nickoloff JA. (ed), Electroporation protocols for microorganisms. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 49.Wang AY, Chang YY, Cronan JE. 1991. Role of the tetrameric structure of Escherichia coli pyruvate oxidase in enzyme activation and lipid binding. J Biol Chem 266:10959–10966. [PubMed] [Google Scholar]
- 50.Riley MA, Chavan MA. 2007. Bacteriocins: ecology and evolution. Springer, New York, NY. [Google Scholar]
- 51.Olczak T, Simpson W, Liu X, Genco CA. 2005. Iron and heme utilization in Porphyromonas gingivalis. FEMS Microbiol Rev 29:119–144. doi: 10.1016/j.femsre.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Johnson NA, Liu Y, Fletcher HM. 2004. Alkyl hydroperoxide peroxidase subunit C (AhpC) protects against organic peroxides but does not affect the virulence of Porphyromonas gingivalis W83. Oral Microbiol Immunol 19:233–239. doi: 10.1111/j.1399-302X.2004.00145.x. [DOI] [PubMed] [Google Scholar]
- 53.Diaz PI, Slakeski N, Reynolds EC, Morona R, Rogers AH, Kolenbrander PE. 2006. Role of OxyR in the oral anaerobe Porphyromonas gingivalis. J Bacteriol 188:2454–2462. doi: 10.1128/JB.188.7.2454-2462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu B, Macleod LC, Newsome E, Liu J, Xu P. 2019. Aggregatibacter actinomycetemcomitans mediates protection of Porphyromonas gingivalis from Streptococcus sanguinis hydrogen peroxide production in multi-species biofilms. Sci Rep 9:4944. doi: 10.1038/s41598-019-41467-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marty-Teysset C, de la Torre F, Garel J-R. 2000. Increased production of hydrogen peroxide by Lactobacillus delbrueckii subsp. bulgaricus upon aeration: involvement of an NADH oxidase in oxidative stress. Appl Environ Microbiol 66:262–267. doi: 10.1128/AEM.66.1.262-267.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hertzberger R, Arents J, Dekker HL, Pridmore RD, Gysler C, Kleerebezem M, Teixeira de Mattos MJ. 2014. H2O2 production in species of the Lactobacillus acidophilus group: a central role for a novel NADH-dependent flavin reductase. Appl Environ Microbiol 80:2229–2239. doi: 10.1128/AEM.04272-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tharrington G, Sorrells KM. 1992. Inhibition of Listeria monocytogenes by milk culture filtrates from Lactobacillus delbrueckii subsp. lactis. J Food Prot 55:542–544. doi: 10.4315/0362-028X-55.7.542. [DOI] [PubMed] [Google Scholar]
- 58.Dahiya RS, Speck ML. 1968. Hydrogen peroxide formation by Lactobacilli and its effect on Staphylococcus aureus. J Dairy Sci 51:1568–1572. doi: 10.3168/jds.S0022-0302(68)87232-7. [DOI] [PubMed] [Google Scholar]
- 59.Price RJ, Lee JS. 1970. Inhibition of Pseudomonas species by hydrogen peroxide producing Lactobacilli. J Milk Food Technol 33:13–18. doi: 10.4315/0022-2747-33.1.13. [DOI] [Google Scholar]
- 60.Goffin P, Muscariello L, Lorquet F, Stukkens A, Prozzi D, Sacco M, Kleerebezem M, Hols P. 2006. Involvement of pyruvate oxidase activity and acetate production in the survival of Lactobacillus plantarum during the stationary phase of aerobic growth. Appl Environ Microbiol 72:7933–7940. doi: 10.1128/AEM.00659-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee SJ, Gralla JD. 2001. Sigma38 (rpoS) RNA polymerase promoter engagement via −10 region nucleotides. J Biol Chem 276:30064–30071. doi: 10.1074/jbc.M102886200. [DOI] [PubMed] [Google Scholar]
- 62.Pouwels PH, Leer RJ. 1993. Genetics of Lactobacilli: plasmids and gene expression. Antonie Van Leeuwenhoek 64:85–107. [DOI] [PubMed] [Google Scholar]
- 63.Hawley DK, McClure WR. 1983. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res 11:2237–2255. doi: 10.1093/nar/11.8.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang Y-Y, Wang A-Y, Cronan JE. 1994. Expression of Escherichia coli pyruvate oxidase (PoxB) depends on the sigma factor encoded by the rpoS(katF) gene. Mol Microbiol 11:1019–1028. doi: 10.1111/j.1365-2958.1994.tb00380.x. [DOI] [PubMed] [Google Scholar]
- 65.Taniai H, Iida K, Seki M, Saito M, Shiota S, Nakayama H, Yoshida S. 2008. Concerted action of lactate oxidase and pyruvate oxidase in aerobic growth of Streptococcus pneumoniae: role of lactate as an energy source. J Bacteriol 190:3572–3579. doi: 10.1128/JB.01882-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abad CL, Safdar N. 2009. The role of Lactobacillus probiotics in the treatment or prevention of urogenital infections: a systematic review. J Chemother 21:243–252. doi: 10.1179/joc.2009.21.3.243. [DOI] [PubMed] [Google Scholar]
- 67.Cianci A, Cicinelli E, Leo VD, Fruzzetti F, Massaro MG, Bulfoni A, Parazzini F, Perino A. 2018. Observational prospective study on Lactobacillus plantarum P 17630 in the prevention of vaginal infections, during and after systemic antibiotic therapy or in women with recurrent vaginal or genitourinary infections. J Obstet Gynaecol 38:693–696. doi: 10.1080/01443615.2017.1399992. [DOI] [PubMed] [Google Scholar]
- 68.Kovachev S. 2018. Defence factors of vaginal Lactobacilli. Crit Rev Microbiol 44:31–39. doi: 10.1080/1040841X.2017.1306688. [DOI] [PubMed] [Google Scholar]
- 69.Eschenbach DA, Davick PR, Williams BL, Klebanoff SJ, Young-Smith K, Critchlow CM, Holmes KK. 1989. Prevalence of hydrogen peroxide-producing Lactobacillus species in normal women and women with bacterial vaginosis. J Clin Microbiol 27:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hawes SE, Hillier SL, Benedetti J, Stevens CE, Koutsky LA, Wølner-Hanssen P, Holmes KK. 1996. Hydrogen peroxide-producing lactobacilli and acquisition of vaginal infections. J Infect Dis 174:1058–1063. doi: 10.1093/infdis/174.5.1058. [DOI] [PubMed] [Google Scholar]
- 71.Voltan S, Martines D, Elli M, Brun P, Longo S, Porzionato A, Macchi V, D’Incà R, Scarpa M, Palù G, Sturniolo GC, Morelli L, Castagliuolo I. 2008. Lactobacillus crispatus M247-derived H2O2 acts as a signal transducing molecule activating peroxisome proliferator activated receptor-γ in the intestinal mucosa. Gastroenterology 135:1216–1227. doi: 10.1053/j.gastro.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 72.Kholy KE, Genco RJ, Van Dyke TE. 2015. Oral infections and cardiovascular disease. Trends Endocrinol Metab 26:315–321. doi: 10.1016/j.tem.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 73.Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RLM, Curtis MA, Dragunow M, Potempa J. 2019. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5:eaau3333. doi: 10.1126/sciadv.aau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gardner RG, Russell JB, Wilson DB, Wang GR, Shoemaker NB. 1996. Use of a modified Bacteroides-Prevotella shuttle vector to transfer a reconstructed β-1,4-d-endoglucanase gene into Bacteroides uniformis and Prevotella ruminicola B14. Appl Environ Microbiol 62:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 76.Brunelle JL, Green R. 2014. Coomassie blue staining Methods Enzymol 541:161–167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.