

Summary
Hyperekplexia disease is usually caused by naturally occurring point mutations in glycine receptors (GlyRs). However, the γ-aminobutyric acid type A receptor (GABAAR) seems to be also involved regarding the therapeutic basis for hyperekplexia using benzodiazepines, which target GABAARs but not GlyRs. Here, we show that the function of GABAARs was significantly impaired in the hypoglossal nucleus of hyperekplexic transgenic mice. Such impairment appeared to be mediated by interaction between GABAAR and mutant GlyR. The GABAAR dysfunction was caused only by mutant GlyR consisting of homomeric α1 subunits, which locate primarily at pre- and extra-synaptic sites. In addition, the rescue effects of diazepam were attenuated by Xli-093, which specifically blocked diazepam-induced potentiation on α5-containing GABAAR, a major form of pre- and extra-synaptic GABAAR in the brainstem. Thus, our results suggest that the pre- and extra-synaptic GABAARs could be a potential therapeutic target for hyperekplexia disease caused by GlyR mutations.
Subject Areas: Neurogenetics, Molecular Interaction, Molecular Neuroscience
Graphical Abstract
Highlights
-
•
Hyperekplexic mutant GlyRs interact with GABAARs and disrupt the GABAAR function
-
•
Pre- and extra-synaptic GABAARs are deficient in the hyperekplexia disease
-
•
α5-Containing GABAAR is a potential therapeutic target for the hyperekplexia disease
Neurogenetics; Molecular Interaction; Molecular Neuroscience
Introduction
γ-Aminobutyric acid (GABA) and glycine are the major inhibitory neurotransmitters in the brain (Nemecz et al., 2016). Glycine receptor (GlyR) and GABA type A receptor (GABAAR) are members of a large Cys-loop superfamily and are structurally similar ligand-gated ion channels (Langosch et al., 1990, Jacob et al., 2008). On activation, the GlyR and GABAAR selectively conduct Cl− through central pores, leading to neuron hyperpolarization and inhibitory neurotransmission in the central nervous system (Nemecz et al., 2016). These receptors are usually localized at the synapse postsynaptically (Essrich et al., 1998, Langosch et al., 1988). Emerging evidence has suggested that certain isoforms of GABAAR, including α5 subunit-containing receptors, can be found pre-synaptically and extra-synaptically (Brickley and Mody, 2012, Castro et al., 2011, Delgado-Lezama et al., 2013, Jia et al., 2005, Hauser et al., 2005). GlyR is widely distributed in the central nervous system, particularly in the brainstem and spinal cord (Hruskova et al., 2012, Xiong et al., 2014). To date, four α-subunits (α1-4) and one β-subunit of GlyR have been identified. All GlyR α subunits can form functional homomeric channels that are mainly located on the pre- and extra-synaptic membrane of a synapse (Hruskova et al., 2012, Xiong et al., 2014, McCracken et al., 2017, Turecek and Trussell, 2001). However, after co-assembling with the α subunits, the β subunit can form functional postsynaptic heteromeric αβ channels (Pribilla et al., 1992, Xiong et al., 2014).
Hyperekplexia is a human genetic neurological disorder usually caused by point mutations in α1 GlyRs (Shiang et al., 1993). Although rare, this disease can be life-threatening in children and is characterized by exaggerated startle response and muscle stiffness following an unexpected stimulus. Numerous point mutations in the GlyR α1 subunit have been identified and characterized as hyperekplexic mutations disrupting GlyR function (Bode and Lynch, 2014). Among them, the R271Q was the most common dominant GlyRα1 mutation identified in patients with hyperekplexia (Thomas et al., 2013). Despite strong evidence suggesting that the point mutations in the α1 GlyR are strongly associated with hyperekplexia, the primary therapeutic agent effectively used to treat hyperekplexia in humans is benzodiazepines (Dijk and Tijssen, 2010, Garg et al., 2008, Tijssen et al., 1997), which selectively enhances GABAAR functioning (Dray and Straughan, 1976, Macdonald and Barker, 1978). Thus, GABAARs seems to be the primary therapeutic target in hyperekplexia. Consistently, a previous investigation revealed a deficiency in both glycinergic and GABAergic transmission in the spinal cord of R271Q mutant mice (Becker et al., 2002, Von Wegerer et al., 2003). Unfortunately, the cellular and molecular mechanisms underlying the GABAAR deficiency in hyperekplexia remains unclear. Such deficiency is not caused by the posttranslational modification of either GlyR or GABAAR protein since radioligand binding to these receptors was unaffected (Becker et al., 2002). The speculation that GlyR can cross-talk or interact with GABAAR has been long-standing (Wojcik et al., 2006). These receptors are abundant in the spinal cord and brainstem where the neurotransmitters GABA and glycine are colocalized and co-released from the same vesicles at many motoneuron synapses (Jonas et al., 1998). Strong evidence suggests that a substantial proportion of spinal cord inhibitory synapses host both GlyRs and GABAARs. Nevertheless, direct evidence and the in vivo consequence of the potential GlyR-GABAAR interaction have not been reported. Considering these questions, we conducted experiments using various approaches to explore the nature of the interaction through which hyperekplexic mutations in the GlyR α1 subunits disrupt GABAAR functioning at synapses.
Results
GABAARs Deficiency in the Brainstem of Hyperekplexic Mutant Mice
To determine whether the hyperekplexic point mutations in the α1 GlyR could affect GABAergic transmission, we measured GABA release and GABAAR functioning using patch clamp recording in the hypoglossal nucleus slices from two transgenic mouse lines carrying GlyRα1 R271Q and S267Q hyperekplexic point mutations. Another mouse line carrying GlyRα1 M287L point mutation was set as a negative control since this mutation was not found in patients with hyperekplexia and has been previously shown to scarcely change the function of GlyR in mice (Bode and Lynch, 2014, Xiong et al., 2014). Both the frequency and amplitude of GABAergic spontaneous inhibitory postsynaptic currents (sIPSCs) were remarkably attenuated in the hypoglossal nucleus of GlyRα1R271Q and GlyRα1S267Q but not in GlyRα1M287L mutant mice (Figures 1A and S1). Consistently, the electrical stimulation-evoked GABAergic IPSCs (eIPSCs) and the puffing GABA-induced currents were both significantly reduced in the GlyRα1R271Q and GlyRα1S267Q mutant mice (Figures 1B and 1C).
Figure 1.

Dysfunction of GABAARs in the Hyperekplexic Mutant Mice
(A) Trace records, average frequency, and amplitude of GABAergic sIPSCs in brainstem hypoglossal nucleus slices from WT, GlyRα1R271Q, and GlyRα1S267Q mutant mice.
(B) Trace records and average amplitude of GABAergic eIPSCs in brainstem hypoglossal nucleus slices from WT and GlyRα1R271Q mutant mice.
(C) Trace records and average values of GABA maximal current induced by puffing 1 mM GABA in brainstem hypoglossal nucleus slices from WT and GlyRα1S267Q mutant mice.
(D) Trace records, average frequency, and amplitude of GABAergic mIPSCs in brainstem hypoglossal nucleus slices from WT and GlyR α1S267Q mutant mice.
(E) Trace records and average values of BSTC in brainstem hypoglossal nucleus slices from WT and GlyRα1S267Q mutant mice.
All digits within the columns represent numbers of cells measured from at least three mice. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 based on unpaired t tests; ns, not significant (p > 0.05).
Next, we separately examined the function of GABAARs at various synaptic locations including the pre-, post-, and extra-synapses. Here the GlyRα1S267Q mice were used as a representative. The frequency but not the amplitude of the GABAergic mIPSCs was significantly decreased in the brainstem hypoglossal nucleus of the GlyRα1S267Q mice compared with that in the wild-type (WT) littermates (Figure 1D). This suggests that a pre- but not post-synaptic impairment exists in GABAergic transmission. Then, we tested the bicuculline-sensitive tonic current (BSTC), which represents extra-synaptic GABAAR activity. The amplitude of the BSTC was also significantly reduced in the GlyRα1S267Q mice compared with that in the WT littermates (Figure 1E).
Hyperekplexic GlyRα1 Mutations Cause GABAAR Deficiency when Co-expressed in HEK-293 Cells
Mutant GlyRs may disrupt the function of GABAARs in the same neuron since the preponderance of evidence has indicated a wide colocalization of GlyRs and GABAARs in brainstem neurons (Muller et al., 2004, Muller et al., 2006, Lorenzo et al., 2006, Lorenzo et al., 2007, Waldvogel et al., 2019). Next, we investigated whether the GlyRα1 mutations could induce GABAAR deficiency if these receptors were co-expressed in HEK-293 cells. The GlyRα1R271Q and GlyRα1S267Q hyperekplexic point mutations significantly reduced the maximal amplitudes of the current (Imax) activated by puffing glycine (Figures 2A and 2B) and GABA (Figures 2A and 2C). GlyRα1R271Q and GlyRα1S267Q, but not GlyRα1M287L, mutations shifted the dose-response curve of the GABA current to the right (Figures 2D and 2E) and increased the half-maximal effective concentration (EC50) values of the GABAARs (Figure 2E). Interestingly, the other two GlyR α subunits, including α2 and α3, exhibited the same characteristics as α1 subunit in impairing GABAAR functions. For instance, the point mutations in the GlyRα2 (R305Q) and GlyRα3 (R320Q) subunits corresponding to R271Q of GlyRα1 not only reduced the glycine Imax (Figure 2F) when expressed alone but also inhibited GABA Imax when co-expressed with GABAARs (Figure 2G).
Figure 2.

Impairment in GABAARs by Hyperekplexic GlyR α1 Mutations in HEK-293 Cells
(A) Representative trace records of glycine Imax (up) and GABA Imax (down) separately induced by 1 mM glycine and GABA in HEK-293 cells co-expressing GABAARs (α1β2γ2) and various hyperekplexic mutant α1 GlyRs.
(B) The average values of glycine Imax induced by 1 mM glycine in HEK-293 cells co-expressing GABAARs (α1β2γ2) and various hyperekplexic mutant α1 GlyRs. The data were normalized to the Imax of the GlyRα1WT group.
(C) Average values of GABA Imax induced by 1 mM GABA in HEK-293 cells co-expressing GABAARs and various hyperekplexic mutant α1 GlyRs. The data were normalized to their respective controls (GlyRα1WT group).
(D and E) Dose-response curves of IGABA in HEK-293 cells co-expressing GABAARs (α1β2γ2) and either WT, α1R271Q, α1S267Q, or α1M287L GlyRs. The data were normalized to Imax of the GlyRα1WT group (D) or its own group (E).
(F) The average values of glycine Imax in HEK-293 cells expressing WT or α2R305Q or α3R320Q GlyRs.
(G) The average values of GABA Imax in HEK-293 cells co-expressing GABAARs (α1β2γ2) and WT or α2R305Q or α3R320Q GlyRs.
All digits within the columns represent numbers of cells measured. Data are represented as mean ± SEM. **p < 0.01, ***p < 0.001 based on unpaired t tests; ns, not significant (p > 0.05).
At high concentrations, GABA can also activate GlyRs (Figure S2) (Singer, 2008). Thus, we examined the efficacy of muscimol, which is a full agonist specific for GABAAR but not GlyR (Snodgrass, 1978) (Figure S3A), in activating GABAAR-GlyR complexes. Consistent with our observation in the above-mentioned experiments using GABA, the amplitude of muscimol (100 μM)-induced maximal current (Imax) was also significantly decreased in HEK-293 cells co-expressing GABAARs with GlyRα1R271Q or GlyRα1S267Q (Figure S3B).
The Protein-Protein Interactions between GABAAR and Hyperekplexic Mutant GlyR
First, we investigated whether the hyperekplexic mutations will affect the membrane trafficking of GlyR and GABAAR. The western-blotting results showed that both the S267Q and R271Q point mutations in the GlyRα1 subunit did not affect the protein expression level of GlyR or GABAAR in plasma membranes extracted from HEK-293 cells co-transfected with the cDNA of GlyRα1WT, GlyRα1R271Q, and GlyRα1S267Q with or without GABAARs (Figure S4).
A possible mechanism of the GABAAR deficiency in the presence of mutant GlyRs is that there may exist an interaction between GlyR and GABAAR proteins. To test this hypothesis, we conducted a co-immunoprecipitation (Co-IP) assay of mutant or WT GlyRα1 subunits and GABAARs (α1β2γ2) co-expressed in HEK-293 cells. The point mutations R271Q and S267Q, but not M287L, significantly increased the amount of GlyR protein co-immunoprecipitated with GABAAR proteins from whole cell lysates (Figures 3A, S5, and S6) and plasma membrane preparations (Figure S7). Similar results were observed in vivo in transgenic mice carrying GlyRα1 mutations. The association between the GlyRs and GABAARs was remarkably enhanced in the brainstem of the GlyRα1R271Q (Figures 3B and S8A–S8C) and GlyRα1S267Q (Figures 3C and S8D–S8F), but not GlyRα1M287L, mutant mice (Figures 3D and S8G–S8I).
Figure 3.

Identification of Interaction between GABAAR and Hyperekplexic Mutant GlyRs
(A) GlyRα1 was purified using GABAAR α1 antibodies in HEK-293 cells co-expressing GABAARs (α1β2γ2) and WT/mutant α1 GlyRs, and the co-precipitating proteins were detected by immunoblotting. Inputs are immunoblots of the same protein in cell lysates before Co-IP. Quantification of WT and mutant GlyR α1 binding to GABAAR α1 subunits (n = 3). The data were normalized to the WT group.
(B and C) Endogenous brainstem GlyRα1 of WT and GlyRα1 R271Q (B) or S267Q (C) KI mice was purified using GABAAR α1 antibodies, and the co-precipitating proteins were detected by immunoblotting. Inputs are immunoblots of the same protein in tissue lysates before Co-IP. Quantification of WT and R271Q (B) or S267Q (C) mutant GlyRα1 binding to GABAARα1 (n = 3 mice).
(D) Endogenous brainstem GlyRα1 of WT and GlyRα1 M287L KI mice was purified using GABAAR α1 antibodies, and the co-precipitating proteins were detected by immunoblotting. Inputs are immunoblots of the same protein in tissue lysates before Co-IP. Quantification of WT and M287L mutant GlyRα1 binding to GABAARα1 (n = 3 mice). The data were normalized to the WT group.
Data are represented as mean ± SEM. *p < 0.05, ***p < 0.001 based on unpaired t tests; ns, not significant (p > 0.05).
The Site R271 Is Critical for the Interaction between GABAAR and Hyperekplexic Mutant GlyR
Subsequently, we conducted a molecular dynamic simulation to evaluate the interaction between the subunits of GABAARs (Miller and Aricescu, 2014) and hyperekplexic mutant GlyR subunits (Huang et al., 2017) in different combinations of dimers. First, all simulations were reliable since no unexpected collapse of protein structures were observed (Figure S9). Then, the binding affinities between the subunits in the dimers, including GABAAR homer-dimers (GB/GB) and GABAAR bound with GlyR (GB/GR) or GlyR mutant (GB/GRM), were analyzed and compared. Among the WT receptor combinations, two subunits in the GB/GB complex showed the strongest binding affinity with a binding free energy (BFE) of −121.9 ± 4.3 kcal/mol. However, the binding affinities in the GB/GR complex were much weaker (−89.2 ± 8.9 kcal/mol). A significant decrease in BFE was observed between the subunits in GB/GRM (Figure 4B). Notably, the BFE value in GB/GRM was as low as −123.4 ± 14.3 kcal/mol, which is highly similar to the value observed in GB/GB. Further analysis indicates that the mutation of R271Q may lead to hydrogen bonding with TYR227 instead of GLN231 on GABAAR (Figure 4A). The R271Q mutation promotes a conformational change in the GB and GR subunits, leading to more intensive H-bond formation and a larger contact surface area between the subunits (Figures 4C and 4D).
Figure 4.

Molecular Dynamic Simulation, Mutagenesis, and Correlation Analysis
(A) Overview of residues forming H-bond between GB chain and GR chain in the GB/GR and GB/GRM complexes at the end of the simulation. GB chain and residue labels are colored in cyan. GR chain and residue labels are colored in pink. H-bonds are shown by the red dashed line.
(B) Binding energy (kcal/mol) between subunits in various composing form of dimers.
(C) Number of H-bonds formed between GB chain and GR chain in the GB/GR and GB/GRM complexes. The data are shown as averages of each 200 ps. Data are represented as mean ± SD.
(D) VDW contact surface between GB chain and GR chain in the GB/GR and GB/GRM complexes. Proteins are displayed in lines. Contact surfaces were mapped and colored according to the distances between two chains.
(E) Average values of GABA Imax induced by 1 mM GABA in HEK-293 cells co-expressing GABAARs and various R271 site mutant GlyR α1 subunits. All data were normalized to their respective controls (WT group).
(F) Correlation analysis of CoMSIA values of various amino acids at 271 and the percentage inhibition of GABA Imax.
(G) GlyRα1 was purified using GABAAR α1 antibodies in HEK-293 cells co-expressing GABAARs (α1β2γ2) and GlyRα1 carrying various R271 mutations, and the co-precipitating proteins were detected by immunoblotting. Inputs are immunoblots of the same protein in cell lysates before Co-IP. Quantification of WT and R271 mutant GlyRα1 binding to GABAAR α1 (n = 3). Data were normalized to the WT group.
(H) Correlation analysis of the percentage decrease in GABA Imax and amount of R271 mutant α1 GlyRs co-immunoprecipitated with GABAARs.
All digits within the columns represent numbers of cells measured. Data are represented as mean ± SEM. **p < 0.01, ***p < 0.001 based on unpaired t tests; ns, not significant (p > 0.05).
To obtain further molecular insight into the role of site R271 in the association between GABAARs and GlyRs, we used mutagenesis to analyze the interrelationship between the function of GABAARs and the biophysical properties of the amino acid residue at 271 of the GlyRα1. The mutation-induced decrease in glycine Imax (Figure S10A) and GABA Imax (Figure 4E) varied substantially. No correlation was observed between the percentage inhibition of glycine Imax and that of GABA Imax (Figure S10B), suggesting that the dysfunction in GABAAR does not depend on the efficacious levels of GlyRs. Then, to examine the biophysical properties of the amino acid residue at 271 of the GlyRα1, we performed a comparative molecular similarity index analysis (CoMSIA), which is a comprehensive method evaluating polarity, electrostatic potential, and steric property. A strong correlation was observed between the CoMSIA values of various amino acids at 271 and the function of GlyRs (Figure S10C) or GABAARs (Figure 4F). Combined with the results of the molecular dynamics simulation, the R271Q point mutation likely suppresses the function of GlyR by altering the protein conformational change required for channel gating. This point mutation also disrupted GABAAR functioning by enhancing the interaction between GlyR and GABAAR. To further test this hypothesis, we performed a Co-IP assay to examine the interaction between the mutant R271E/L/K/G α1 GlyRs and GABAARs co-expressed in HEK-293 cells. Both the GlyRα1 subunits and GABAARα1 subunits were identified in the co-immunoprecipitants pulled down by the GABAARα1 antibodies (Figures 4G and S11A–S11C). Among the four GlyRα1 R271 mutations, the R271L and R271E mutations appeared to enhance the binding of GlyR to GABAAR. The protein levels of GlyRα1R271X bound to GABAARα1 were significantly and positively correlated with the extent of the GABAAR deficiency, although their levels substantially varied (Figure 4H).
GlyR β Subunits Restore Dysfunction of GABAARs Caused by GlyR α1 Mutations
The above-mentioned results have suggested that the pre- and extra- but not post-synaptic GABAARs were impaired in hyperekplexia disease. It is worth mentioning that GlyR homomers (α/α) have been found to primarily reside at pre/extra-synaptic sites, whereas GlyR heteromers (α/β) are mostly post-synaptic (Betz et al., 1991, Hruskova et al., 2012, Xiong et al., 2014, McCracken et al., 2017, Turecek and Trussell, 2001). Thus, a possible scenario is that different combinations of GlyR subunits may have distinct abilities to interact with GABAARs. To test this hypothesis, we performed the electrophysiological experiments and Co-IP assay. Addition of the GlyR β subunit indeed prevents the hyperekplexic point mutations in the α1 subunit from hijacking the GABAARs because no functional disruption in the GABAAR was observed after co-expressing the GlyR β subunits with the GlyRα1R271Q/GABAAR complexes in HEK-293 cells (Figures 5A and 5B). Furthermore, the GlyR β subunits also significantly interrupted the association between the mutant α1 GlyRs and GABAARs in HEK-293 cells (Figures 5C and S12). These observations may hint at why only pre- and extra-synaptic GABAARs have been impaired in hyperekplexia.
Figure 5.

GlyR β Subunits Restore GABAARs Functioning by Interrupting the Interaction between GABAARα1 and GlyRα1 Subunits
(A) Average glycine Imax values in HEK-293 cells co-expressing GABAARs (α1β2γ2) and either homomeric or heteromeric hyperekplexic mutant α1/β GlyRs.
(B) Average GABA Imax values in HEK-293 cells co-expressing GABAARs (α1β2γ2) and either homomeric or heteromeric hyperekplexic mutant α1/β GlyRs.
(C) GlyRα1 was purified using GABAAR α1 antibodies in HEK-293 cells co-expressing GABAARs (α1β2γ2) and homomeric or heteromeric hyperekplexic mutant α1/β GlyRs, and the co-precipitating proteins were detected by immunoblotting. Inputs are immunoblots of the same protein in cell lysates before Co-IP. Quantification of WT and mutant GlyRα1 binding to GABAAR α1 (n = 3). Data were normalized to the WT group.
All digits within the columns represent numbers of cells measured. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 based on unpaired t tests; ns, not significant (p > 0.05).
Colocalization and Interaction of α5-Containing GABAARs and Hyperekplexic Mutant GlyRs
Emerging evidence suggests that α5 subunits-containing GABAAR (α5βxγx) is the primary form of pre- and extra-synaptic GABAARs in several brain regions, including the hippocampus, spinal cord, and brainstem (Brickley and Mody, 2012, Castro et al., 2011, Delgado-Lezama et al., 2013, Jia et al., 2005). Genetic deletion of α5-containing GABAA receptor could also cause severe convulsive seizure (Galanopoulou, 2008). Here, using RNAscope techniques, we conducted in situ hybridization and observed a high degree of colocalization of GlyRα1 and GABAARα5 subunit mRNAs in neurons in the hypoglossal nucleus of the brainstem in both the GlyRα1S267Q and WT mice (Figures 6A, 6B, and S13). Therefore, we next examined whether the hyperekplexic mutant GlyRs could also affect the α5-containing GABAARs. The GABA Imax was significantly decreased when the α1S267Q mutant GlyRs were co-expressed with α5β2γ2 GABAARs in HEK-293 cells (Figure 6C). Compared with the WT, the S267Q point mutation significantly increased the amount of GlyRs co-immunoprecipitated with α5β2γ2 GABAARs in both the HEK-293 cells (Figures 6D and S14A–S14C) and the brainstem of GlyRα1S267Q mutant mice (Figures 6E and S14D–S14F).
Figure 6.

Interaction between α5-Containing GABAAR and Hyperekplexic Mutant GlyR
(A) Representative confocal imaging showing the colocalization of GABAARα5 and GlyRα1 subunit mRNAs in the GlyRα1S267Q mouse brainstem using RNAscope technology (scale bar, 25 μm).
(B) Left, percentage of GlyRα1 mRNA–positive neurons that co-express GABAARα5 mRNA. Right, percentage of GABAARα5 mRNA–positive neurons that co-express GlyRα1 mRNA (n = 3 mice).
(C) Trace records and average values of GABA Imax activated by 1 mM GABA in HEK-293 cells co-expressing GABAARs (α5β2γ2) and WT or α1S267Q mutant GlyRs. The digits within the columns represent numbers of cells measured.
(D) GABAARα5 was purified using GlyRα1 antibodies in HEK-293 cells co-expressing GABAARs (α5β2γ2) and WT/S267Q mutant GlyRα1, and the co-precipitating proteins were detected by immunoblotting. Inputs are immunoblots of the same protein in cell lysates before Co-IP. Quantification of GABAAR α5 binding to WT and S267Q mutant GlyR α1 (n = 3 mice).
(E) Endogenous brainstem GABAAR α5 in WT and GlyRα1 S267Q KI mice were purified using GlyRα1 antibodies, and the co-precipitating proteins were detected by immunoblotting. Inputs are immunoblots of the same protein in tissue lysates before Co-IP. Quantification of mouse brainstem GABAAR α5 binding to WT and S267Q mutant GlyR α1 (n = 3 mice).
Data are represented as mean ± SEM. **p < 0.01, ***p < 0.001 based on unpaired t tests; ns, not significant (p > 0.05).
Pre- and Extra-synaptic α5-Containing GABAAR Is a Therapeutic Target of Diazepam for Hyperekplexia Disease
Benzodiazepines (BZDs) have always been used as the first-line medication to treat patients with hyperekplexia in the clinic (Dijk and Tijssen, 2010, Garg et al., 2008, Tijssen et al., 1997). Therefore, we next assessed whether diazepam (DIA), the most common BZD, could rescue the pre- and extra-synaptic GABAAR deficiency in the brainstem hypoglossal nucleus of GlyRα1S267Q KI mice. We conducted the following electrophysiological recordings, Co-IP experiments, and behavioral tests using homozygous and heterozygous GlyRα1S267Q KI mice because most GlyRα1R271Q KI mice died within 2–3 weeks (Figure S15). DIA significantly rescued the reduced frequency of GABA mIPSCs (Figure 7A) and the attenuated amplitude of the BSTC (Figure 7B) in the brainstem hypoglossal nucleus of the GlyRα1S267Q mutant mice. Consistently, DIA also significantly restored the attenuated GABA Imax in HEK-293 cells co-expressing α5-containing GABAARs and α1S267Q GlyRs (Figure S17A). These effects of DIA were remarkably diminished by Xli-093 (Figures 7A and 7B), which could specifically block DIA-induced potentiation on α5- (Figure S16) but not α1/α2-containing GABAARs (Clayton et al., 2015) (Figures S17B and S17C).
Figure 7.

DIA Rescues Dysfunction of Pre- and Extra-synaptic α5-Containing GABAARs and Exaggerated Startle Responses in Hyperekplexic Mutant Mice
(A) Trace records, average frequency, and amplitude of GABAergic mIPSCs in brainstem hypoglossal nucleus slices from WT and GlyR α1 S267Q mutant mice with or without diazepam (10 μM) and/or Xli-093 (1 μM) pre-incubation.
(B) Trace records and average values of bicuculline-sensitive tonic currents (BSTC) in brainstem hypoglossal nucleus slices from WT and GlyR α1 S267Q mutant mice with or without diazepam (10 μM) and/or Xli-093 (1 μM) pre-incubation.
(C) Hind feet clenching behavior in GlyRα1S267Q mutant mice and effect of DIA (i.p. 10 mg/kg) and Xli-093 (intra-brainstem hypoglossal nucleus injection, 5 μg) on this behavior.
(D) Average values of startle responses induced by white noise at 85, 90, and 95 dB in WT (n = 8) and GlyRα1S267Q (n = 8) mice.
(E) Average values of startle response activated by white noise at 85 dB in WT and GlyRα1S267Q mutant mice with or without diazepam (i.p. 10 mg/kg) and/or Xli-093 (intra-brainstem hypoglossal nucleus injection, 5 μg) treatments.
(F) Correlation analysis of fold increases in startle response, percentage decreases in mIPSC frequency, and amount of mutant α1 GlyRs co-immunoprecipitated with GABAARs in hyperekplexic mutant mice.
All digits within the columns represent numbers of cells or mice measured. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 based on unpaired t tests; ns, not significant (p > 0.05).
Next, we investigated whether the restoration of pre- and extra-synaptic GABAAR functioning by DIA in the brainstem hypoglossal nucleus was sufficient to treat hyperekplexia. An intraperitoneal (i.p.) injection of DIA markedly alleviated hind feet clenching behaviors and exaggerated tremors in the GlyRα1S267Q KI mice when the animals were picked up by their tails (Figure 7C). The therapeutic effect of DIA was completely abolished by an intra-brainstem hypoglossal nucleus microinjection of Xli-093 (Figure 7C). The GlyRα1S267Q mutant mice displayed exaggerated startle reflexes in response to various acoustic stimuli (Figure 7D). The systemic administration of DIA significantly inhibited the exaggerated startle responses of the GlyRα1S267Q KI mice. This DIA therapeutic effect was remarkably suppressed by an intra-brainstem hypoglossal nucleus injection of Xli-093 (Figure 7E). The startle reactions of the WT and various hyperekplexic mutant mice were significantly correlated with their brainstem hypoglossal nucleus presynaptic GABAARs deficiency (expressed as percentage decreases in mIPSC frequency) and the bonding strength between the GlyR and GABAAR (expressed as normalized GlyR-GABAAR CO-IP) (Figure 7F). Altogether, our results reveal that the pre- and extra-synaptic α5-containing GABAAR may be the major acting target of BDZ to treat hyperekplexia disease.
Discussion
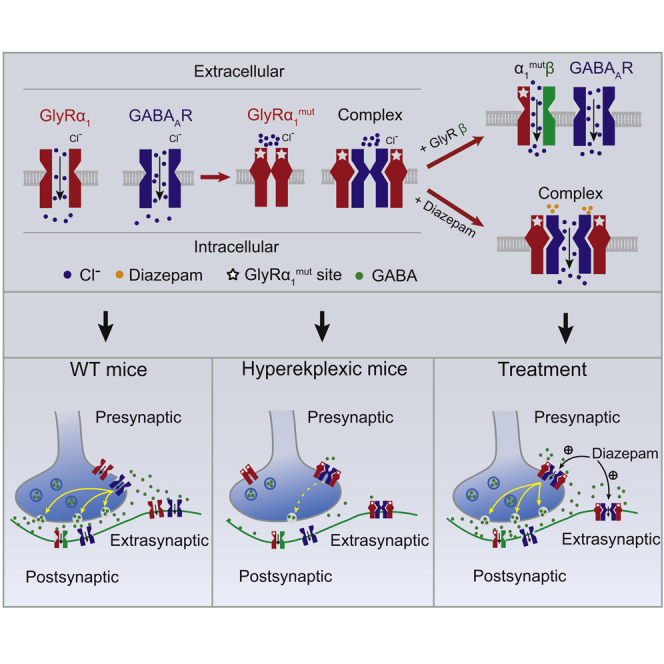
Both GABAAR and GlyR mediate rapid synaptic transmissions in the central nervous system (Jacob et al., 2008, Langosch et al., 1990). Despite the widespread speculation that cross talk exists between these two types of receptors (Schmieden et al., 1993, Shrivastava et al., 2011, Maric et al., 2011), knowledge regarding the nature of such an interaction is limited. The data presented in this study provided several lines of evidence that primary hyperekplexic point mutations in the GlyR α1 subunit can suppress GABAAR functioning by hijacking GABAARs via protein interaction both in vitro and in vivo. This interaction underlies the pathological mechanism of hyperekplexia (Figure 8). First, hyperekplexic mutations in GlyR α1 subunits impair the functioning of both GlyRs and GABAARs in HEK293 cells and the mouse brainstem hypoglossal nucleus. Second, the mutant GlyRs are highly capable of forming hetero-oligomers with certain types of GABAARs. The R271Q point mutation increased the binding free energy, contact surface area, and number of hydrogen bonds between GABAARα1 and GlyRα1 protein. Third, the signal intensity of such GlyR-GABAAR complexes is highly correlated with the severity of the GABAAR deficiency and exaggerated startle responses in hyperekplexic mice.
Figure 8.

Schematic of Mechanisms in which Hyperekplexic Mutant GlyRs Disrupt Inhibitory Neurotransmission by Interacting with Pre- and Extra-synaptic GABAARs
(A) Under normal conditions, presynaptic GABAARs facilitate GABA release from GABAergic neuron terminals, activating postsynaptic GABAARs to inhibit neurons in the brainstem hypoglossal nucleus. The extra-synaptic GABAARs mediate the chronic inhibition of postsynaptic neurons in the brainstem hypoglossal nucleus.
(B) In hyperekplexia disease, the mutant GlyRα1 binds to pre- and extra-synaptic GABAARs and, therefore, reduce GABA release and the chronic inhibition. The postsynaptic GlyRβ subunits prevent the mutant GlyRα1 from binding to the GABAARs.
(C) DIA exerts its therapeutic effect by allosterically potentiating pre- and extra-synaptic α5-containing GABAARs in the brainstem hypoglossal nucleus.
In this study, weak binding between the WT GlyRs and GABAARs was observed in both the HEK-293 cells and brainstem tissues. This weak bonding is unlikely to affect the functioning of both ion channels because the glycine and GABA-activated currents did not show differences when the WT GlyRs and GABAARs were either separately expressed or co-expressed in the HEK-293 cells. In contrast, this weak binding may provide a possible explanation for the synergistic effects of glycine and GABA that have been observed in several previous reports (Li and Yang, 1998, Rogers et al., 2016). For instance, a strong synergistic interaction has been observed between GABA and glycine in acutely isolated crucian carp retina neurons. The co-application of both agonists resulted in much larger responses (current >400 pA) than either GABA or glycine alone (current <20 pA) (Li and Yang, 1998). Another report also demonstrated that GABA and glycine can act synergistically at the spinal cord to generate a tonic inhibition of the micturition reflex pathway (Rogers et al., 2016). However, such bonding between GlyR and GABAAR does not appear to always be a good thing. In fact, the hyperekplexic mutations in GlyR caused stronger binding with GABAAR but remarkably impaired the functioning of both channels.
Site mutations generally attenuate the interaction between two associated proteins (Salpietro et al., 2019, Smets et al., 2017, Bizarro and Meier, 2017). However, our findings reveal an entirely opposite pattern in the modulation of protein-protein interactions, particularly under pathological conditions. For instance, several hyperekplexic site mutations in GlyR α1, such as R271Q and S267Q, enhance its bonding interaction with GABAAR and therefore induce dysfunction in GABAAR. This mechanism may be universal since a similar pattern has been observed in several previous studies investigating the molecular and cellular mechanisms of various diseases. For instance, the R882H mutation in DNA (cytosine-5-)-methyltransferase 3α (DNMT3A) enhances its binding to polycomb repressive complex 1 (PRC1) protein and causes transcriptional silencing, suggesting that PRC1 favors R882 mutants over WT as binding partners in DNMT3A-mutated leukemia disease (Koya et al., 2016). Furthermore, the H443P mutant NOD-like receptor (NLR) protein NLRC4 more strongly interacts with 19S proteasome ATPase Sug1 and ubiquitinated proteins in auto-inflammatory syndrome. This enhanced interaction triggers the constitutive caspase-8-mediated cell death (Raghawan et al., 2017).
The hijacking of GABAARs by mutant GlyRs also results in a deficiency in major inhibitory neurotransmission. This finding is consistent with a previous study showing that the R271Q point mutation causes the hyperekplexia phenotype and impairs glycine and GABA transmission in mice (Becker et al., 2002, Von Wegerer et al., 2003). The GlyR β subunit greatly reduces the formation of the GlyR-GABAAR complex, suggesting that the hijacking of the GABAAR by the mutant GlyRα1 subunits likely occurs in pre- or extra-synaptic sites where the GlyR β subunit is absent. Consistently, the low levels of the GlyR β subunit were associated with the hyperekplexic phenotype in mice (Becker et al., 2000). This hypothesis was tested and supported by the subsequent electrophysiological recordings, which indicated that only pre- and extra-synaptic GABAARs were impaired in the brainstem hypoglossal nucleus of hyperekplexic mice. Therefore, this study reveals that the pre- and extra-synaptic GABAARs, specifically the α5 subunit-containing GABAARs primarily located in brainstem hypoglossal nucleus, are novel primary targets in hyperekplexia. This hypothesis is supported by our finding that the GABAARα5 and GlyRα1 subunits are colocalized in the brainstem hypoglossal nucleus in GlyRα1 S267Q and WT mutant mice as revealed by RNAscope technology. DIA, which has been widely used to treat hyperekplexia in the clinic (Garg et al., 2008, Becker et al., 2000, Tijssen et al., 1997), indeed specifically rescued the deficiency of pre- and extra-synaptic α5-containing GABAARs in the HEK-293 cells and mouse brainstem hypoglossal nucleus and restored the exaggerated startle reflex behaviors in the hyperekplexic mutant mice. Thus, developing specific GABAARα5 agonists or modulators may be critical for the treatment of hyperekplexia without producing the major psychoactive or sedative side effects that are associated with benzodiazepines, such as DIA. Such dynamic changes in pre- and extra-synaptic GlyR-GABAAR complexes may also contribute to various physiological and pathological processes, such as pain, anxiety, and sleep disorders (Botta et al., 2015, Bravo-Hernandez et al., 2016, Crestani et al., 2002, Xiong et al., 2011, Xiong et al., 2012). Thus, this GlyR-GABAAR interaction not only leads to human hyperekplexia but also may contribute to various neurological disorders involving GlyR and GABAAR deficiency.
Limitations of the Study
Although we identified the interaction between GlyR and GABAAR in the brain of hyperekplexic transgenic mice, the detailed interaction pattern and interaction sites between both receptors remain unsolved in the present study. Future research should consider utilizing more advanced molecular biology approaches to clarify the detailed mechanisms involved.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Yuri Blednov and Adron Harris (University of Texas at Austin, Texas) for providing the α1 S267Q and α1 M287L mutant mice. We thank Hans Weiher (University of Applied Sciences Bonn-Rhein-Sieg, Germany) for providing the α1 R271Q mutant mice. We thank Pearce RA (University of Wisconsin Madison) for providing plasmids of GABAARs. We thank Dr. David Lovinger (National Institute on Alcohol Abused and Alcoholism, National Institutes of Health, USA) for helpful comments on the manuscript. We acknowledge support from National Key R&D Program of China (2016YFC1300500-2, 2014AA020535), National Natural Science Foundation of China (Grants 91849206, 91649121 & 81901157), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDPB1005), the Fundamental Research Funds for the Central Universities, the Major Program of Development Foundation of Hefei Center for Physical Science and Technology (2017FXZY006), CAS Interdisciplinary Innovation Team (JCTD-2018-20), and Users with Excellence Program/Project of Hefei Science Center CAS.
Author Contributions
W.X. initiated, designed, and supervised the project; G.Z., Q.C., and H.P. conducted electrophysiological recordings. G.Z. conducted western blot experiments; K.C., Y.G., and D.L. conducted molecular dynamic simulation; L.Z. conducted in situ hybridization using RNAscope; G.Z., X.Z., and Y.H. conducted animal behavioral tests; T.P. synthesized Xli-093; G.Z. and W.X. analyzed data; W.X. and G.Z. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.08.018.
Data and Code Availability
The raw data that support the findings of this study are available from the corresponding authors, upon request.
Supplemental Information
References
- Becker L., Hartenstein B., Schenke J., Kuhse J., Betz H., Weiher H. Transient neuromotor phenotype in transgenic spastic mice expressing low levels of glycine receptor β-subunit: an animal model of startle disease. Eur. J. Neurosci. 2000;12:27–32. doi: 10.1046/j.1460-9568.2000.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker L., Von Wegerer R., Schenkel J., Zeilhofer H.U., Swandulla D., Weiher H. Disease-specific human glycine receptor α1 subunit causes hyperekplexia phenotype and impaired glycine- and GABAA-receptor transmission in transgenic mice. J. Neurosci. 2002;22:2505–2512. doi: 10.1523/JNEUROSCI.22-07-02505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz H., Kuhse J., Schmieden V., Malosio M.L., Langosch D., Prior P., Schmitt B., Kirsch J. How to build a glycinergic postsynaptic membrane. J. Cell Sci. Suppl. 1991;15:23–25. doi: 10.1242/jcs.1991.supplement_15.4. [DOI] [PubMed] [Google Scholar]
- Bizarro J., Meier U.T. Inherited SHQ1 mutations impair interaction with NAP57/dyskerin, a major target in dyskeratosis congenita. Mol. Genet. Genomic Med. 2017;5:805–808. doi: 10.1002/mgg3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode A., Lynch J.W. The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol. Brain. 2014;7:2. doi: 10.1186/1756-6606-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botta P., Demmou L., Kasugai Y., Markovic M., Xu C., Fadok J.P., Lu T., Poe M.M., Xu L., Cook J.M. Regulating anxiety with extrasynaptic inhibition. Nat. Neurosci. 2015;18:1493. doi: 10.1038/nn.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Hernandez M., Corleto J.A., Barragan-Iglesias P., Gonzalez-Ramirez R., Pineda-Farias J.B., Felix R., Calcutt N.A., Delgado-Lezama R., Marsala M., Granados-Soto V. The α5 subunit containing GABAA receptors contribute to chronic pain. Pain. 2016;157:613–626. doi: 10.1097/j.pain.0000000000000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley S.G., Mody I. Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron. 2012;73:23–34. doi: 10.1016/j.neuron.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro A., Aguilar J., Gonzalez-Ramirez R., Loeza-Alcocer E., Canto-Bustos M., Felix R., Delgado-Lezama R. Tonic inhibition in spinal ventral horn interneurons mediated by α5 subunit-containing GABAA receptors. Biochem. Biophys. Res. Commun. 2011;412:26–31. doi: 10.1016/j.bbrc.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Clayton T., Poe M.M., Rallapalli S., Biawat P., Savic M.M., Rowlett J.K., Gallos G., Emala C.W., Kaczorowski C.C., Stafford D.C. A review of the updated pharmacophore for the α5 GABAA benzodiazepine receptor model. Int. J. Med. Chem. 2015;2015:430248. doi: 10.1155/2015/430248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F., Keist R., Fritschy J.M., Benke D., Vogt K., Prut L., Bluthmann H., Mohler H., Rudolph U. Trace fear conditioning involves hippocampal α5-GABAA receptors. Proc. Natl. Acad. Sci. U S A. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Lezama R., Loeza-Alcocer E., Andres C., Aguilar J., Guertin P., Felix R. Extrasynaptic GABAA receptors in the brainstem and spinal cord: structure and function. Curr. Pharm. Des. 2013;19:4485–4497. doi: 10.2174/1381612811319240013. [DOI] [PubMed] [Google Scholar]
- Dijk J.M., Tijssen M.A. Management of patients with myoclonus: available therapies and the need for an evidence-based approach. Lancet Neurol. 2010;9:1028–1036. doi: 10.1016/S1474-4422(10)70193-9. [DOI] [PubMed] [Google Scholar]
- Dray A., Straughan D.W. Benzodiazepines: GABA and glycine receptors on single neurons in the rat medulla. J. Pharm. Pharmacol. 1976;28:314–315. doi: 10.1111/j.2042-7158.1976.tb04163.x. [DOI] [PubMed] [Google Scholar]
- Essrich C., Lorez M., Benson J.A., Fritschy J.M., Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the γ 2 subunit and gephyrin. Nat. Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Galanopoulou A.S. GABA(A) receptors in normal development and seizures: friends or foes? Curr. Neuropharmacol. 2008;6:1–20. doi: 10.2174/157015908783769653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R., Ramachandran R., Sharma P. Anaesthetic implications of hyperekplexia–'startle disease'. Anaesth. Intensive Care. 2008;36:254–256. doi: 10.1177/0310057X0803600217. [DOI] [PubMed] [Google Scholar]
- Hauser J., Rudolph U., Keist R., Mohler H., Feldon J., Yee B.K. Hippocampal alpha5 subunit-containing GABAA receptors modulate the expression of prepulse inhibition. Mol. Psychiatry. 2005;10:201–207. doi: 10.1038/sj.mp.4001554. [DOI] [PubMed] [Google Scholar]
- Hruskova B., Trojanova J., Kulik A., Kralikova M., Pysanenko K., Bures Z., Syka J., Trussell L.O., Turecek R. Differential distribution of glycine receptor subtypes at the rat calyx of Held synapse. J. Neurosci. 2012;32:17012–17024. doi: 10.1523/JNEUROSCI.1547-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Chen H., Shaffer P.L. Crystal structures of human GlyRα3 bound to ivermectin. Structure. 2017;25:945–950. doi: 10.1016/j.str.2017.04.007. [DOI] [PubMed] [Google Scholar]
- Jacob T.C., Moss S.J., Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F., Pignataro L., Schofield C.M., Yue M., Harrison N.L., Goldstein P.A. An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J. Neurophysiol. 2005;94:4491–4501. doi: 10.1152/jn.00421.2005. [DOI] [PubMed] [Google Scholar]
- Jonas P., Bischofberger J., Sandkühler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- Koya J., Kataoka K., Sato T., Bando M., Kato Y., Tsuruta-Kishino T., Kobayashi H., Narukawa K., Miyoshi H., Shirahige K. DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat. Commun. 2016;7:10924. doi: 10.1038/ncomms10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langosch D., Becker C.M., Betz H. The inhibitory glycine receptor: a ligand-gated chloride channel of the central nervous system. Eur. J. Biochem. 1990;194:1–8. doi: 10.1111/j.1432-1033.1990.tb19419.x. [DOI] [PubMed] [Google Scholar]
- Langosch D., Thomas L., Betz H. Conserved quaternary structure of ligand-gated ion channels: the postsynaptic glycine receptor is a pentamer. Proc. Natl. Acad. Sci. U S A. 1988;85:7394–7398. doi: 10.1073/pnas.85.19.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Yang X.L. Strong synergism between GABAA and glycine receptors on isolated carp third-order neurons. Neuroreport. 1998;9:2875–2879. doi: 10.1097/00001756-199808240-00036. [DOI] [PubMed] [Google Scholar]
- Lorenzo L.E., Barbe A., Portalier P., Fritschy J.M., Bras H. Differential expression of GABAA and glycine receptors in ALS-resistant vs. ALS-vulnerable motoneurons: possible implications for selective vulnerability of motoneurons. Eur. J. Neurosci. 2006;24:1506. doi: 10.1111/j.1460-9568.2006.04863.x. [DOI] [PubMed] [Google Scholar]
- Lorenzo L.E., Russier M., Barbe A., Fritschy J.M., Bras H. Differential organization of gamma-aminobutyric acid type A and glycine receptors in the somatic and dendritic compartments of rat abducens motoneurons. J. Comp. Neurol. 2007;504:112–126. doi: 10.1002/cne.21442. [DOI] [PubMed] [Google Scholar]
- Macdonald R., Barker J.L. Benzodiazepines specifically modulate GABA-mediated postsynaptic inhibition in cultured mammalian neurones. Nature. 1978;271:563–564. doi: 10.1038/271563a0. [DOI] [PubMed] [Google Scholar]
- Maric H.M., Mukherjee J., Tretter V., Moss S.J., Schindelin H. Gephyrin-mediated gamma-aminobutyric acid type A and glycine receptor clustering relies on a common binding site. J. Biol. Chem. 2011;286:42105–42114. doi: 10.1074/jbc.M111.303412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken L.M., Lowes D.C., Salling M.C., Carreau-Vollmer C., Odean N.N., Blednov Y.A., Betz H., Harris R.A., Harrison N.L. Glycine receptor α 3 and α2 subunits mediate tonic and exogenous agonist-induced currents in forebrain. Proc. Natl. Acad. Sci. U S A. 2017;114:E7179–E7186. doi: 10.1073/pnas.1703839114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P.S., Aricescu A.R. Crystal structure of a human GABAA receptor. Nature. 2014;512:270–275. doi: 10.1038/nature13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller E., Triller A., Legendre P. Glycine receptors and GABA receptor alpha 1 and gamma 2 subunits during the development of mouse hypoglossal nucleus. Eur. J. Neurosci. 2004;20:3286–3300. doi: 10.1111/j.1460-9568.2004.03785.x. [DOI] [PubMed] [Google Scholar]
- Muller E., Corronc L., Triller A., Legendre P. Developmental dissociation of presynaptic inhibitory neurotransmitter and postsynaptic receptor clustering in the hypoglossal nucleus. Mol. Cell. Neurosci. 2006;32:254–273. doi: 10.1016/j.mcn.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Nemecz A., Prevost M.S., Menny A., Corringer P.J. Emerging molecular mechanisms of signal transduction in pentameric ligand-gated ion channels. Neuron. 2016;90:452–470. doi: 10.1016/j.neuron.2016.03.032. [DOI] [PubMed] [Google Scholar]
- Pribilla I., Takagi T., Langosch D., Bormann J., Betz H. The atypical M2 segment of the beta subunit confers picrotoxinin resistance to inhibitory glycine receptor channels. EMBO J. 1992;11:4305–4311. doi: 10.1002/j.1460-2075.1992.tb05529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghawan A.K., Sripada A., Gopinath G., Pushpanjali P., Kumar Y., Radha V., Swarup G. A disease-associated mutant of NLRC4 shows enhanced interaction with SUG1 leading to constitutive FADD-dependent caspase-8 activation and cell death. J. Biol. Chem. 2017;292:1218–1230. doi: 10.1074/jbc.M116.763979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M.J., Shen B., Reese J.N., Xiao Z.Y., Wang J.C., Lee A., Roppolo J.R., Groat W.C., Tai C.F. Role of glycine in nociceptive and non-nociceptive bladder reflexes and pudendal afferent inhibition of these reflexes in cats. Neurourol. Urodyn. 2016;35:798–804. doi: 10.1002/nau.22821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpietro V., Malintan N.T., Llano-Rivas I., Spaeth C.G., Efthymiou S., Striano P., Vandrovcova J., Cutrupi M.C., Chimenz R., David E. Mutations in the neuronal vesicular SNARE VAMP2 affect synaptic membrane fusion and impair human neurodevelopment. Am. J. Hum. Genet. 2019;104:721–730. doi: 10.1016/j.ajhg.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieden V., Kuhse J., Betz H. Mutation of glycine receptor subunit creates beta-alanine receptor responsive to GABA. Science. 1993;262:256–258. doi: 10.1126/science.8211147. [DOI] [PubMed] [Google Scholar]
- Shiang R., Ryan S.G., Zhu Y.Z., Hahn A.F., O'Connell P., Wasmuth J.J. Mutations in the α1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat. Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- Shrivastava A.N., Triller A., Sieghart W. GABAA receptors: post-synaptic co-localization and cross-talk with other receptors. Front. Cell. Neurosci. 2011;5:7. doi: 10.3389/fncel.2011.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer J.H. GABA is an endogenous ligand for synaptic glycine receptors. Neuron. 2008;57:475–477. doi: 10.1016/j.neuron.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Smets M., Link S., Wolf P., Schneider K., Solis V., Ryan J., Meilinger D., Qin W., Leonhardt H. DNMT1 mutations found in HSANIE patients affect interaction with UHRF1 and neuronal differentiation. Hum. Mol. Genet. 2017;26:1522–1534. doi: 10.1093/hmg/ddx057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snodgrass S.R. Use of 3H-muscimol for GABA receptor studies. Nature. 1978;273:392–394. doi: 10.1038/273392a0. [DOI] [PubMed] [Google Scholar]
- Thomas R.H., Chung S.K., Wood S.E., Cushion T.D., Drew C.J., Hammond C.L., Vanbellinghen J.F., Mullins J.G., Rees M.I. Genotype-phenotype correlations in hyperekplexia: apnoeas, learning difficulties and speech delay. Brain. 2013;136:3085–3095. doi: 10.1093/brain/awt207. [DOI] [PubMed] [Google Scholar]
- Tijssen M.A., Schoemaker H.C., Edelbroek P.J., Roos R.A., Cohen A.F., vanDijk J.G. The effects of clonazepam and vigabatrin in hyperekplexia. J. Neurol. Sci. 1997;149:63–67. doi: 10.1016/s0022-510x(97)05378-1. [DOI] [PubMed] [Google Scholar]
- Turecek R., Trussell L.O. Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- Von Wegerer J., Becker K., Glockenhammer D., Becker C.M., Zeilhofer H.U., Swandulla D. Spinal inhibitory synaptic transmission in the glycine receptor mouse mutant spastic. Neurosci. Lett. 2003;345:45–48. doi: 10.1016/s0304-3940(03)00499-3. [DOI] [PubMed] [Google Scholar]
- Waldvogel H.J., Biggins F.M., Singh A., Arasaratnam C.J., Faull R.L.M. Variable colocalisation of GABAA receptor subunits and glycine receptors on neurons in the human hypoglossal nucleus. J. Chem. Neuroanat. 2019;97:99–111. doi: 10.1016/j.jchemneu.2019.02.005. [DOI] [PubMed] [Google Scholar]
- Wojcik S.M., Katsurabayashi S., Guillemin I., Friauf E., Rosenmund C., Brose N., Rhee J.S. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Xiong W., Cheng K.J., Cui T.X., Godlewski G., Rice K.C., Xu Y., Zhang L. Cannabinoid potentiation of glycine receptors contributes to cannabis-induced analgesia. Nat. Chem. Biol. 2011;7:296–303. doi: 10.1038/nchembio.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W., Cui T.X., Cheng K.J., Yang F., Chen S.R., Willenbring D., Guan Y., Pan H.L., Ren K., Xu Y. Cannabinoids suppress inflammatory and neuropathic pain by targeting alpha 3 glycine receptors. J. Exp. Med. 2012;209:1121–1134. doi: 10.1084/jem.20120242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W., Chen S.R., He L.M., Cheng K.J., Zhao Y.L., Chen H., Li D.P., Homanics G.E., Peever J., Rice K.C. Presynaptic glycine receptors as a potential therapeutic target for hyperekplexia disease. Nat. Neurosci. 2014;17:232–239. doi: 10.1038/nn.3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding authors, upon request.