

Summary
It is well known that nuclear factor-kappaB (NF-κB) regulates neuronal structures and functions by nuclear transcription. Here, we showed that phospho-p65 (p-p65), an active form of NF-κB subunit, reversibly interacted with Nav1.7 channels in the membrane of dorsal root ganglion (DRG) neurons of rats. The interaction increased Nav1.7 currents by slowing inactivation of Nav1.7 channels and facilitating their recovery from inactivation, which may increase the resting state of the channels ready for activation. In cultured DRG neurons TNF-α upregulated the membrane p-p65 and enhanced Nav1.7 currents within 5 min but did not affect nuclear NF-κB within 40 min. This non-transcriptional effect on Nav1.7 may underlie a rapid regulation of the sensibility of the somatosensory system. Both NF-κB and Nav1.7 channels are critically implicated in many physiological functions and diseases. Our finding may shed new light on the investigation into the underlying mechanisms.
Subject Areas: Biological Sciences, Neuroscience, Molecular Neuroscience, Cellular Neuroscience
Graphical Abstract
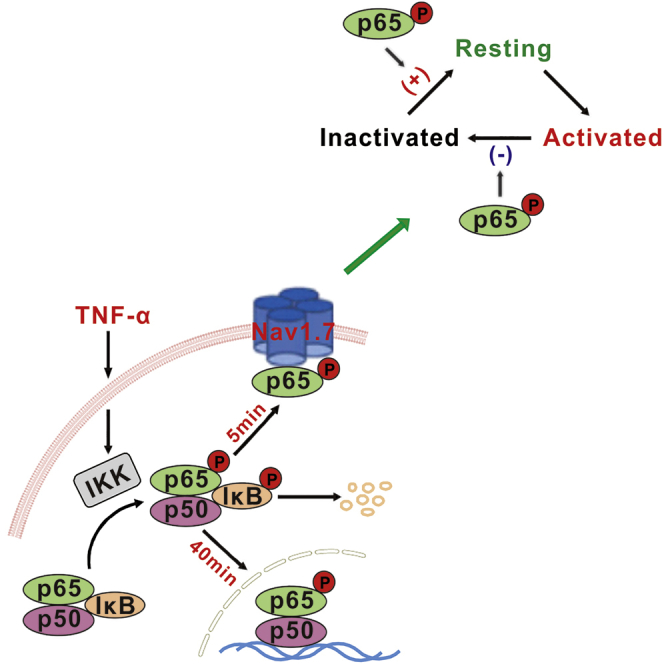
Highlights
-
•
NF-κB p-p65 interacts with Nav1.7 in the membrane of DRG neurons
-
•
The interaction is reversible, depending on the cytoplasmic p-p65 content
-
•
Reducing cytoplasmic p-p65 rapidly attenuates the interaction and Nav1.7 currents
-
•
The rapid effect on Nav1.7 channels is independent of p-p65 nuclear translocation
Biological Sciences; Neuroscience; Molecular Neuroscience; Cellular Neuroscience
Introduction
Nuclear factor-kappaB (NF-κB), a potent transcription factor, is highly conserved from insect to human and plays critical roles in a wide variety of physiological and pathological processes, such as memory storage (Meffert and Baltimore, 2005), immunity and cancer (Taniguchi and Karin, 2018), neurodegenerative diseases (Srinivasan and Lahiri, 2015), and chronic pain (Niederberger and Geisslinger, 2008). NF-κB p50/p65/inhibitor of NF-κB (IκB-α) complex is located in the cytoplasm. On activation, both p65 and IκB-α are phosphorylated, and then p-IκB-α is degenerated after ubiquitination, whereas phospho-p65 (p-p65) is translocated into the nucleus, where it regulates gene transcription (Niederberger and Geisslinger, 2008). At present, all the functions of NF-κB are attributed to the transcriptional effect in vertebrate (Salles et al., 2014). There are two NF-κB activation pathways, classical and alternative; tumor necrosis factor alpha (TNF-α) is critical in both of them (Wajant and Scheurich, 2011).
Dorsal root ganglion (DRG) neurons play an essential role in detecting the changes in the external environment. As a pseudounipolar neuron, its peripheral axon branches transfer different forms of sensory stimuli into action potentials (APs) and its central axon branches conduct the APs to spinal dorsal horn (Chahine and O'Leary, 2014). Activation of voltage-gated sodium (Nav) channels is indispensable for the initiation and conduction of APs. Among the nine subunits of Nav channels (Catterall et al., 2005), tetrodotoxin-sensitive (TTX-S) channels Nav1.3, Nav1.6, and Nav1.7 and TTX-resistant (TTX-R) channels Nav1.8 and Nav1.9 are proved important for the excitability of DRG neurons (Dib-Hajj et al., 2010). Previous works show that activation of TNF-α/NF-κB signaling leads to chronic hyperexcitability of DRG neurons (He et al., 2010, Huang et al., 2014, Tamura et al., 2014, Zang et al., 2010) and of cortical neurons (Chen et al., 2015) by transcriptional upregulation of sodium channels. In the present work, we provided evidence that p-p65 also enhances Na+ currents in a transcription-independent way. The data uncover a novel mechanism by which TNF-α/NF-κB signaling rapidly regulates cell excitability.
Results
p-p65 (s311) Is Located in the Membrane of DRG Neurons, and Reducing Cytoplasmic p-p65 Inhibits Na+ Currents within Minutes
Immunofluorescent staining showed that p-p65 (s311) was located not only in the nucleus but also in the membrane of DRG neurons in both naive and neuropathic rats, namely, vincristine (VCR, a chemotherapeutic agent)-induced peripheral neuropathy or lumbar 5 spinal nerve ligation (L5-SNL) (Figures 1A–1C). Western blots with membrane protein extract of DRGs revealed that the membrane p-p65 level was significantly higher in either VCR-treated or L5-SNL rats compared with vehicle-treated or sham-operated rats (Figures 1D and 1E). Double staining showed that p-p65 (s311) was colocalized with the markers of large neurons (NF-200) and of small neurons (CGRP and IB4) but not with the marker of satellite glial cells (GFAP) (Figure 1F). We quantified the levels of p-p65 in large (NF-200 positive) and small (NF-200 negative) DRG neurons. As shown in Figures 1G and 1H, the total gray value of p-p65 signal in large neurons was higher than that in small neurons, but the average gray value of p-p65 signal (total gray value/cell area) in large neurons was lower than that in small neurons. The data indicate that p-p65 is expressed in all types of DRG neurons, although expression is more intensive in small ones. Therefore, in the following experiments all sizes of DRG neurons were used.
Figure 1.

Phospho-p65 (s311) Is Located in the Membrane of Dorsal Root Ganglion Neurons and Is Increased in Neuropathic Conditions
(A–C) The representative confocal images show that p-p65 is located not only in the nucleus but also in the membrane of DRG neurons in naive (A), vincristine (VCR)-treated (B) and L5-spinal nerve ligation (L5-SNL) (C) rats. Scale bars: left 100 μm, right 20 μm.
(D and E) The western blots show that p-p65 in membrane is increased in VCR-treated (D) and L5-SNL (E) rats, compared with vehicle and sham rats. Samples were harvested after the last VCR injection or 10 days after L5-SNL. n = 6 in each group. ∗∗p < 0.01, ∗∗∗p < 0.001 compared with vehicle or sham group.
(F) The cell types that express p-p65 (s311) in DRG neurons. Scale bars: 50 μm.
(G and H) The total (G) and the average (H) gray value of p-p65 signal in NF-200 positive (larger-diameter) and NF-200-negative (small-diameter) neurons. n = 40 in NF200-negative group, n = 62 in NF200-positive group. ∗∗∗p < 0.001 compared with the corresponding NF200-negative group. Two-tailed t test. The specificity of the antibody for p-p65 (s311) was identified in Figure S2A. Data expressed as mean ± SD. The control of membrane fractionation process was shown in Figure S2C.
The primary function of DRG neurons is production of APs in response to various sensory stimuli, and opening of Nav channels is critical in this process. We, therefore, tested if membrane p-p65 might regulate the Nav channels. To do this, total Na+, TTX-S, or TTX-R currents were recorded in the DRG neurons of VCR-treated rats with microelectrodes containing p-p65 antibody (10 μg/mL) or IgG. TTX-S and TTX-R currents were isolated by blocking TTX-R channels with A-803467 (1 μM) (Jarvis et al., 2007) (Figure S1) and by blocking TTX-S channels with TTX (300 nM), respectively. As shown in Figures 2A–2C, compared with IgG control group, a significant reduction of total Na+, TTX-S, or TTX-R currents was evident at 6, 3, and 18 min after the onset of recordings. The results indicate that p-p65 facilitates Nav currents. To confirm this, pyrrolidinedithiocarbamate (PDTC, 10 nM), an NF-κB inhibitor that reduces intracellular p-p65 by inhibition of IκB-ubiquitin ligase activity (Hayakawa et al., 2003), was applied extracellularly 1 min after recordings. We found that PDTC inhibited total Na+ currents, TTX-S currents, or TTX-R currents within 6–9 min, compared with the vehicle group (Figures 2D–2F). We found that both intracellular anti-p-p65 and extracellular PDTC preferably inhibited TTX-S currents over TTX-R ones (Figures 2G and 2H).
Figure 2.

Intracellular Application of Anti-p-p65 (s311) or Extracellular Application of PDTC Preferably Reduces TTX-S Na+ Currents over TTX-R Ones within Minutes
(A–C) The effects of intracellular anti-p-p65 (10 μg/mL) on total (A), TTX-S (B) and TTX-R (C) Na+ currents. The raw traces were recorded immediately on onset (black) and 18 min (red) after recordings. n = 6 in each group. ∗p < 0.05, ∗∗∗p < 0.001 compared with the corresponding IgG group. (D–F) The effects of extracellular NF-κB inhibitor PDTC (10 nM) on total (D), TTX-S (E) and TTX-R (F) Na+ currents. The raw traces were recorded immediately on onset (black) and 18 min (red) after recordings. n = 6 in each group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the corresponding vehicle group.
(G and H) The histograms show the inhibitory rates of anti-p-p65 (G) and PDTC (H) on total, TTX-S, and TTX-R currents at 18 min after recordings. ∗p < 0.05, ∗∗p < 0.01 compared with total currents; ##p < 0.01, ###p < 0.001 compared with TTX-S currents. A-F, two-way repeated measures ANOVA followed by Bonferroni's multiple comparisons test; G and H, one-way ANOVA followed by Holm-Sidak's multiple comparisons test. Data expressed as mean ± SD.
Membrane p-p65 Gates Nav1.7 Channels by Protein-Protein Interaction
Our data that p-p65 was located in the membrane and that PDTC inhibited Nav currents within minutes suggested that p-p65 might regulate Nav channels, non-transcriptionally. To investigate the mechanisms underlying the rapid effect, we tested the possibility that p-p65 may gate sodium channels by protein-protein interaction. The co-immunoprecipitation (Co-IP) experiments with the protein extract of DRGs from VCR-treated rats showed that p-p65 interacted potently with Nav1.7, weakly with Nav1.6, and barely with Nav1.3, Nav1.8, or Nav1.9 (Figures 3A–3F). To confirm the p-p65-Nav1.7 interaction, we performed high-resolution images of structured illumination microscopy and found that 69.4 ± 11.1% of Nav1.7 was colocalized with p-p65, whereas only 30.3 ± 4.3% of p-p65 was colocalized with Nav1.7 in membrane of DRG neurons from VCR-treated rats (Figures 3G and 3H).
Figure 3.

p-p65 (s311) Interacts with Nav1.7 Channels in the Membrane of DRG Neurons
(A–F) Co-IP with DRG protein extract of VCR-treated rats shows that p-p65 strongly interacted with Nav1.7 (C and F), weakly with Nav1.6 (B), but not with Nav1.3 (A), Nav1.8 (D), or Nav1.9 (E).
(G) The structured illumination microscopies show that p-p65 is colocalized with Nav1.7 in DRG neuron membrane of VCR rat. Scale bars: left 10 μm, right 5 μm.
(H) Quantification data show colocalization rates of Nav1.7 with p-p65 (colocalized yellow spots/total Nav1.7 positive spots) and those of p-p65 with Nav1.7 (colocalized yellow spots/total p-p65 positive spots) in membrane. n = 4 in each group. ∗∗∗p < 0.001 compared with Nav1.7 with p-p65 group. The specificity of the antibody for Nav1.7 was identified (Figure S2B). Two-tailed t test. Data expressed as mean ± SD.
We then investigated the effect of the interaction on TTX-S Nav1.7 channels. If p-p65 was required for activation of Nav1.7 channels, blockade of Nav1.7 would occlude the inhibitory effect of PDTC on TTX-S currents as shown in Figures 2D–2F. Indeed, we found that PDTC (10 nM) failed to affect TTX-S currents when applied 15 min after extracellular application of ProTxII (5 nM), a selective Nav1.7 blocker (Schmalhofer et al., 2008), in DRG neurons of VCR-treated rats (Figures 4A and 4B). Conversely, ProTxII also failed to affect TTX-S Na+ currents when applied 15 min after PDTC (Figures 4C and 4D). To further study the effect of p-p65 on Nav1.7 channels, we repeated the experiments with ICA-121431 (5 μM), a potent rat Nav1.7 channel blocker (IC50: 37 nM) (McCormack et al., 2013), and found that the effects of ICA-121431 and PDTC on TTX-S currents were also occluded by each other (Figures 4E–4H). In addition, we found that intracellular application of anti-p-p65 also blocked the inhibitory effect of ProTxII on TTX-S currents (Figures 4I and 4J). The mutual inhibition between PDTC and Nav1.7 blockers on TTX-S currents was also observed in the DRG neurons from naive rats (Figures 5A–5H).
Figure 4.

NF-κB Inhibitor PDTC Inhibits Nav1.7 Channels by Reducing Nav1.7- p-p65 Interaction in Membrane of DRG Neurons
(A) The traces show TTX-S currents recorded in indicated conditions.
(B) ProTxII (5 nM) occludes the effect of PDTC (10 nM) on TTX-S currents of VCR rats. n = 7. ∗∗∗p < 0.001 compared with predrug control.
(C) The traces show TTX-S currents recorded in indicated conditions.
(D) PDTC (10 nM) occludes the effect of ProTxII (5 nM) on TTX-S currents of VCR rats. n = 6. ∗∗∗p < 0.001 compared with predrug control.
(E) The traces show TTX-S currents recorded in indicated conditions.
(F) ICA121431 (5 μM) occludes the effect of PDTC on TTX-S currents of VCR rats. n = 6. ∗∗∗p < 0.001 compared with predrug control.
(G) The traces show TTX-S currents recorded in indicated conditions.
(H) PDTC occludes the effect of ICA121431 on TTX-S currents of VCR rats. n = 6. ∗p < 0.05, ∗∗∗p < 0.001 compared with predrug control.
(I) The traces show TTX-S currents recorded in indicated conditions.
(J) Intracellular application of anti-p-p65 blocks the effect of ProTxII on TTX-S currents of VCR rats. n = 6. ∗∗∗p < 0.001 compared with the first recording.
(K and L) Thirty minutes after intrathecal injection of PDTC (15 μg/10 μL), both p-p65 (K) and Nav1.7 (L) in the membrane were tested. n = 6 in each group. ∗p < 0.05, ∗∗∗p < 0.001 compared with vehicle group; ##p < 0.01 compared with VCR group.
(M) The membrane Nav1.7 in DRGs of naive rats was measured 30 min after intrathecal injection of PDTC (15 μg/10 μL). n = 6 in each group.
(N) The interaction between p-p65 and Nav1.7 in DRGs of VCR-treated rats was reduced 30 min after intrathecal injection of PDTC (15 μg/10 μL). Nav1.7 was immunoprecipitated by p-p65 antibody. n = 3 in each group. ∗∗∗p < 0.001 compared with VCR + vehicle group.
(O and P) Intrathecal injection of PDTC (10 μL) dose-dependently reduced p-p65 levels in both membrane (P) and cytoplasm (O). The DRG tissues were harvested 30 min after injection. n = 4 in each group. ∗p < 0.05, ∗∗∗p < 0.001 compared with VCR + vehicle group. B, D, F, H, J, K, L, O, P, one-way ANOVA followed by Tukey's multiple comparisons test. M, N, Two-tailed t test. Data expressed as mean ± SD. N.S. mean not significant.
Figure 5.

The Effects of PDTC and Nav1.7 Blockers on TTX-S Channels Occlude Each Other in DRG Neurons of Naive Rats
(A) The traces show TTX-S currents recorded in indicated conditions.
(B) ProTxII blocks the effect of PDTC on TTX-S currents. n = 6. ∗∗p < 0.01, ∗∗∗p < 0.001 compared with predrug control.
(C) The traces show TTX-S currents recorded in indicated conditions.
(D) PDTC blocks the effect of ProTxII on TTX-S currents. n = 6. ∗∗∗p < 0.001 compared with predrug control.
(E) The traces show TTX-S currents recorded in indicated conditions.
(F) ICA-121431 occludes the effect of PDTC on TTX-S currents. n = 6. ∗∗∗p < 0.001 compared with predrug control.
(G) The traces show TTX-S currents recorded in indicated conditions.
(H) PDTC blocks the effect of ICA-121431 on TTX-S currents. n = 6. ∗p < 0.05, ∗∗∗p < 0.001 compared with predrug control. B, D, F, H, one-way ANOVA followed by Tukey's multiple comparisons test. Data expressed as mean ± SD. N.S. mean not significant.
To investigate how reducing intracellular p-p65 decreases Nav1.7 currents, we measured p-p65 and Nav1.7 in DRG membrane from VCR- and vehicle-treated rats. The western blots with membrane protein extract of DRGs revealed that both p-p65 and Nav1.7 were significantly increased in the VCR-treated group compared with the vehicle-treated group. Importantly, intrathecal injection of PDTC reduced p-p65 but did not affect Nav1.7 in cell membrane within 30 min (Figures 4K and 4L). The results indicate that the inhibitory effect of extracellular PDTC or intracellular anti-p-p65 on Na+ currents is due to the reduction of p-p65 but not of Nav1.7 in cell membrane. We found that PDTC also did not affect membrane Nav1.7 in DRGs of naive rats (Figure 4M). Our data that PDTC rapidly reduces TTX-S Na+ currents and membrane p-p65 suggested that the interaction between p-p65 and Nav1.7 in membrane might be reversible, depending on its intracellular content. To test this, we performed Co-IP with DRGs from VCR-treated rats and found that the p-p65-Nav1.7 interaction was substantially reduced 30 min after intrathecal injection of PDTC (Figure 4N). Consistently, intrathecal injection of PDTC reduced p-p65 in both cytoplasm and membrane dose-dependently (Figures 4O and 4P). The data indicate that p-p65 may regulate Nav1.7 channels by reversible interaction with the channel subtype.
TNF-α Enhances Membrane p-p65 (s311) and Nav1.7 Currents within Minutes in Cultured DRG Neurons
To test if nuclear transcription may also contribute to the rapid effect of p-p65 on Nav1.7 channels, we performed the experiments with cultured DRG neurons. As TNF-α plays a key role in both classical and alternative NF-κB activation pathways (Wajant and Scheurich, 2011), we incubated DRG neurons from naive rats with rat recombinant TNF-α (rrTNF-α, 100 ng/mL) and measured p-p65 levels in membrane and in nuclei at different time points afterward. A significant increase of p-p65 was detected in membrane within 5 min (Figure 6A), whereas in nuclei at 120 min but not within 40 min (Figures 6B and 6C). That is, in response to TNF-α stimulation, p-p65 membrane translocation is at least 40 min earlier than its nuclear translocation. Furthermore, we found that rrTNF-α did not affect membrane Nav1.7 level within 20 min (Figure 6D) but enhanced Nav1.7 currents, isolated by subtraction of the ProTxII-resistant Na+ currents from total Na+ currents (Li et al., 2018), within 5 min. The I-V curves (Figure 6E) showed that the peak Nav1.7 currents in the DRG neurons treated with rrTNF-α (100 ng/mL for 5 min) were significantly higher, compared with the neurons treated with the vehicle. To test if the effect of rrTNF-α on Nav1.7 currents is mediated by membrane p-p65, DRG neurons were incubated with rrTNF-α for 5 min and then with PDTC (10 nM) for 15 min. The peak currents in the rrTNF-α + PDTC group were significantly lower compared with the rrTNF-α alone group and were not different from the vehicle group (Figure 6E). Therefore, the rapid effect of TNF-α on Nav1.7 channels results from the p-p65-Nav1.7 interaction and is independent of p-p65 nuclear transcription and the membrane Nav1.7 level. To investigate the potential mechanism by which p-p65 regulates Nav1.7 currents, we performed experiments with HEK293 cells that express Nav1.7. The results showed that PDTC accelerated inactivation and delayed recovery but did not affect activation of Nav1.7 channels (Figures 6F–6H and Table 1). In consistence with the electrophysiological data, we found that intrathecal injection of PDTC alleviated the decrease in mechanical pain thresholds induced by VCR within 30 min (Figure 6I).
Figure 6.

TNF-α Enhances Membrane p-p65 (s311) and Nav1.7 Currents in Cultured DRG Neurons without Affecting Nuclear p-p65 and Membrane Nav1.7 within Minutes
(A–D) The western blots show the levels of p-p65 and Nav1.7 in membrane (A and D) or nucleus (B and C) of DRG neurons at indicated time points after application of rrTNF-α (100 ng/mL). n = 6 in each group. ∗∗∗p < 0.001 compared with vehicle group.
(E) The I-V curves show the peak Nav1.7 currents recorded under different potentials in indicated groups. n = 9, 8, and 9 in control, TNF-α, and TNF-α + PDTC group.
(F–H) The effects of PDTC (10 nM) on activation (F), inactivation (G), and recovery (H) of Nav1.7 channels in HEK293 cells. n = 6–7 in each group.
(I) Intrathecal injection of NF-κB inhibitor PDTC (10 μL) reduces mechanical allodynia in vincristine (VCR)-treated rats within 30 min. n = 5 in each group. ∗∗∗p < 0.001 compared with vehicle + VCR group. The data in A, B, C, D, and E were analyzed with one-way ANOVA followed by Tukey's multiple comparisons test, and in F, G, and H with two-tailed t test. I, two-way repeated measures ANOVA followed by Bonferroni's multiple comparisons test. Data expressed as mean ± SD. The control of nuclear fractionation process was shown in Figure S2C.
Table 1.
The Effects of PDTC on the Parameters of Activation, Inactivation, and Recovery of Nav1.7 Channels in HEK293 Cells
| Activation |
Inactivation |
Recovery |
|||
|---|---|---|---|---|---|
| V0.5 | k | V0.5 | k | τ | |
| Control | −33.6 ± 3.4 | 5.7 ± 1.7 | −77.6 ± 2.5 | 5.4 ± 0.2 | 4.5 ± 0.8 |
| PDTC | −34.8 ± 3.6 | 6.6 ± 1.0 | −84.0 ± 2.3a | 5.1 ± 0.4 | 9.2 ± 3.3b |
Mean values were derived from Boltzmann equation fits of individual data sets as described in the Methods.
p < 0.001 vs. corresponding control. Two-tailed paired student t test. Data expressed as mean ± SD.
p < 0.01
Discussion
As mentioned in the Introduction section, NF-κB plays important roles in many physiological and pathological processes. Up to date, all the functions of NF-κB are explained by its transcriptional effect. In this present work, we showed for the first time that p-p65 was also located in the membrane of DRG neurons and was increased in neuropathic conditions in vivo or in response to TNF-α stimulation in cultured DRG neurons. The membrane p-p65 regulated Nav1.7 channels by protein-protein interaction. In membrane of DRG neurons, ∼70% of Nav1.7 was colocalized with p-p65, whereas only ∼30% of p-p65 is colocalized with Nav1.7 (Figure 3H). The data suggested that p-p65 might also regulate cell function by interacting with other proteins in cell membranes. Further studies are needed to elucidate this issue. We also found that reduction of cytoplasmic p-p65 by intracellular anti-p-p65 or extracellular PDTC also inhibited TTX-R currents. As no interaction between p-p65 and TTX-R channels (Nav1.8 and Nav1.9) in DRG neurons was detected, the mechanisms underlying the effect remains elusive.
On activation, Nav channels go through rapid transitions from the resting to opening, inactivated state and eventually recover to the resting state (Aldrich et al., 1983). Nav1.7 is distinguished from other TTX-S channels by slow closed-state inactivation, which is suggested to determine action potential threshold by permitting to pass a current in response to small slow depolarization (see Dib-Hajj et al., [2007] for a review). Our data showed that PDTC reduced Nav1.7-p-p65 interaction (Figure 4N) and accelerated inactivation and delayed recovery but did not affect activation of Nav1.7 channels (Figures 6F–6H and Table 1). That is, p-p65-Nav1.7 interaction may increase Nav1.7 currents by slowing inactivation and facilitating recovery from inactivation, leading to an increase of the resting state of Nav1.7 that is ready for opening.
Previous works show that NF-κB is expressed at the synapses and neuromuscular junction and in neuronal fibers, and the local NF-κB is also believed to regulate gene expression (Dresselhaus et al., 2018, Meffert and Baltimore, 2005, Salles et al., 2014). The synaptic NF-κB is also speculated to function locally. Up to date, however, no direct evidence is available in vertebrate (Salles et al., 2014). In drosophila, it has been shown that Dorsal (homolog of NF-κB) in neuromuscular junction regulates glutamate receptor density in a transcription-independent way (Heckscher et al., 2007).
Nav1.7 has been intensively studied in the sensory system. In human, loss of function of Nav1.7 leads to complete inability to sense pain (Cox et al., 2006) and odors (Weiss et al., 2011), whereas gain of its function results in paroxysmal extreme pain disorder (Fertleman et al., 2006). In rodents, deletion of Nav1.7 in mouse DRG neurons attenuates acute nociception and nerve injury-, inflammation- and burn injury-induced pain hypersensitivity (Minett et al., 2012, Minett et al., 2014, Nassar et al., 2004, Shields et al., 2012). Blockage of Nav1.7 significantly alleviates neuropathic pain induced by the chemotherapeutic drug paclitaxel (Li et al., 2018). A recent study shows that mutation of Nav1.7 in human patients leads to functional absence of nociceptors (McDermott et al., 2019). Accordingly, Nav1.7 is a prominent target for treating chronic pain. Our finding that p-p65 gates Nav1.7 channels in naive and neuropathic rats raises a possibility that selective blockage of the interaction between p-p65 and Nav1.7 channels may treat the chronic pain resulting from gain of Nav1.7 function. The new strategy can avoid the side effects of NF-κB inhibitor owing to its transcriptional inhibition.
In addition, global deletion of Nav1.7 in mice leads to death shortly after birth (Nassar et al., 2004), indicating that the channel subtype should play important roles in other vital systems. Consistently, Nav1.7 is found in the brain regions that regulate autonomic and endocrine systems of rats (Morinville et al., 2007) and in airway parasympathetic ganglia of mice, guinea pig, and human (Kocmalova et al., 2017). Furthermore, Nav1.7 is also expressed in human immature dendritic cells (Zsiros et al., 2009). Nav1.7 expressed in gastric cancer cells and human non-small cell lung cancer cells promotes cancer progression or invasion (Campbell et al., 2013, Xia et al., 2016). It is interesting to know if NF-κB also non-transcriptionally regulates the Nav1.7 in these cells.
It has been well established that TNF-α produces a persistent hyperexcitability through gene transcription in DRG (He et al., 2010, Huang et al., 2014, Tamura et al., 2014, Zang et al., 2010) and in cortical neurons (Chen et al., 2015). TNF-α has also been repeatedly demonstrated to induce an acute excitation of DRG neurons (Liu et al., 2002, Zhang et al., 2002) and of subfornical organ neurons (Simpson and Ferguson, 2017). TNF-α enhances sodium currents in DRG neurons within minutes (Jin and Gereau, 2006). The mechanism underlying the rapid effect is unknown. Our result that p-p65 regulates Nav1.7 channels by protein-protein interaction may explain the TNF-α-induced acute excitation. A previous work shows that NF-κB in DRG neurons is activated by noxious electrical, chemical, and thermal stimulation of peripheral tissues within minutes, and the physiological significance of this rapid NF-κB activation is not clarified (Fujikawa et al., 2011). We propose that NF-κB may rapidly regulate somatosensory function via gating of ion channels in DRG neurons.
Together, activation of TNF-α/NF-κB signaling induces not only the chronic hypersensitivity of DRG neurons by nuclear transcription but also an acute excitation of the neurons by protein-protein interaction. In this process, NF-κB functions not only as a transcription factor but also as an ion channel modulator. Our finding may open up a new arena for investigating mechanisms by which NF-κB regulates cell functions.
Limitations of the Study
We explored that p-p65 rapidly regulates Nav1.7 by protein-protein interaction in DRG neurons, whereas the mechanism underlying the interaction was not investigated. NF-κB is also highly expressed in the neurons of the central nervous system and is activated by basal synaptic transmission, glutamate, and depolarization (Lilienbaum and Israel, 2003, Meffert and Baltimore, 2005). Whether it also non-transcriptionally regulates the functions of the central neurons remains elusive.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The study was supported by National Natural Science Foundation of China (31771166 to X.-G.L; 81801112 to M.-X.X).
Author Contributions
M.-X.X. performed the electrophysiology experiments, analyzed the data, and assisted in drafting the manuscript. X.-L.Z. performed western blot, co-immunoprecipitation, and microscopy and analyzed the data. J.X. performed western blot and behavioral tests and analyzed the data. D.L. performed microscopy. T.X. performed microscopy. W.-A.Z. and R.-P.P. analyzed the data. K.M. designed the experiments and helped to write the manuscript. X.-G.L. conceived the project, designed the experiments, and drafted the manuscript.
Declaration of Interests
The authors declare that they have no conflict of interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.08.017.
Contributor Information
Ke Ma, Email: marke72@163.com.
Xian-Guo Liu, Email: liuxg@mail.sysu.edu.cn.
Supplemental Information
References
- Aldrich R.W., Corey D.P., Stevens C.F. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Campbell T.M., Main M.J., Fitzgerald E.M. Functional expression of the voltage-gated Na(+)-channel Nav1.7 is necessary for EGF-mediated invasion in human non-small cell lung cancer cells. J. Cell Sci. 2013;126:4939–4949. doi: 10.1242/jcs.130013. [DOI] [PubMed] [Google Scholar]
- Catterall W.A., Goldin A.L., Waxman S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Chahine M., O'Leary M.E. Regulation/modulation of sensory neuron sodium channels. Handb. Exp. Pharmacol. 2014;221:111–135. doi: 10.1007/978-3-642-41588-3_6. [DOI] [PubMed] [Google Scholar]
- Chen W., Sheng J., Guo J., Gao F., Zhao X., Dai J., Wang G., Li K. Tumor necrosis factor-alpha enhances voltage-gated Na(+) currents in primary culture of mouse cortical neurons. J. Neuroinflammation. 2015;12:126. doi: 10.1186/s12974-015-0349-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.J., Reimann F., Nicholas A.K., Thornton G., Roberts E., Springell K., Karbani G., Jafri H., Mannan J., Raashid Y. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj S.D., Cummins T.R., Black J.A., Waxman S.G. From genes to pain: Nav1.7 and human pain disorders. Trends Neurosci. 2007;30:555–563. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj S.D., Cummins T.R., Black J.A., Waxman S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- Dresselhaus E.C., Boersma M.C.H., Meffert M.K. Targeting of NF-kappaB to dendritic spines is required for synaptic signaling and spine development. J. Neurosci. 2018;38:4093–4103. doi: 10.1523/JNEUROSCI.2663-16.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertleman C.R., Baker M.D., Parker K.A., Moffatt S., Elmslie F.V., Abrahamsen B., Ostman J., Klugbauer N., Wood J.N., Gardiner R.M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Fujikawa M., Nishitani N., Ibuki T., Kobayashi S., Matsumura K. Sensory stimuli induce nuclear translocation and phosphorylation of nuclear factor kappa B in primary sensory neurons of mice. Neurosci. Res. 2011;71:178–182. doi: 10.1016/j.neures.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Hayakawa M., Miyashita H., Sakamoto I., Kitagawa M., Tanaka H., Yasuda H., Karin M., Kikugawa K. Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J. 2003;22:3356–3366. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.H., Zang Y., Chen X., Pang R.P., Xu J.T., Zhou X., Wei X.H., Li Y.Y., Xin W.J., Qin Z.H. TNF-alpha contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain. 2010;151:266–279. doi: 10.1016/j.pain.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Heckscher E.S., Fetter R.D., Marek K.W., Albin S.D., Davis G.W. NF-kappaB, IkappaB, and IRAK control glutamate receptor density at the Drosophila NMJ. Neuron. 2007;55:859–873. doi: 10.1016/j.neuron.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Zang Y., Zhou L., Gui W., Liu X., Zhong Y. The role of TNF-alpha/NF-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem. Int. 2014;75:112–119. doi: 10.1016/j.neuint.2014.05.012. [DOI] [PubMed] [Google Scholar]
- Jarvis M.F., Honore P., Shieh C.C., Chapman M., Joshi S., Zhang X.F., Kort M., Carroll W., Marron B., Atkinson R. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. U S A. 2007;104:8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X., Gereau R.W., 4th Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J. Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocmalova M., Kollarik M., Canning B.J., Ru F., Adam Herbstsomer R., Meeker S., Fonquerna S., Aparici M., Miralpeix M., Chi X.X. Control of neurotransmission by NaV1.7 in human, Guinea pig, and mouse airway parasympathetic nerves. J. Pharmacol. Exp. Ther. 2017;361:172–180. doi: 10.1124/jpet.116.238469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., North R.Y., Rhines L.D., Tatsui C.E., Rao G., Edwards D.D., Cassidy R.M., Harrison D.S., Johansson C.A., Zhang H. DRG voltage-gated sodium channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J. Neurosci. 2018;38:1124–1136. doi: 10.1523/JNEUROSCI.0899-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilienbaum A., Israel A. From calcium to NF-kappa B signaling pathways in neurons. Mol. Cell. Biol. 2003;23:2680–2698. doi: 10.1128/MCB.23.8.2680-2698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Li H., Brull S.J., Zhang J.M. Increased sensitivity of sensory neurons to tumor necrosis factor alpha in rats with chronic compression of the lumbar ganglia. J. Neurophysiol. 2002;88:1393–1399. doi: 10.1152/jn.2002.88.3.1393. [DOI] [PubMed] [Google Scholar]
- McCormack K., Santos S., Chapman M.L., Krafte D.S., Marron B.E., West C.W., Krambis M.J., Antonio B.M., Zellmer S.G., Printzenhoff D. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl. Acad. Sci. U S A. 2013;110:E2724–E2732. doi: 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott L.A., Weir G.A., Themistocleous A.C., Segerdahl A.R., Blesneac I., Baskozos G., Clark A.J., Millar V., Peck L.J., Ebner D. Defining the functional role of NaV1.7 in human nociception. Neuron. 2019;101:905–919.e8. doi: 10.1016/j.neuron.2019.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert M.K., Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Minett M.S., Falk S., Santana-Varela S., Bogdanov Y.D., Nassar M.A., Heegaard A.M., Wood J.N. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 2014;6:301–312. doi: 10.1016/j.celrep.2013.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett M.S., Nassar M.A., Clark A.K., Passmore G., Dickenson A.H., Wang F., Malcangio M., Wood J.N. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 2012;3:791. doi: 10.1038/ncomms1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinville A., Fundin B., Meury L., Jureus A., Sandberg K., Krupp J., Ahmad S., O'Donnell D. Distribution of the voltage-gated sodium channel Na(v)1.7 in the rat: expression in the autonomic and endocrine systems. J. Comp. Neurol. 2007;504:680–689. doi: 10.1002/cne.21484. [DOI] [PubMed] [Google Scholar]
- Nassar M.A., Stirling L.C., Forlani G., Baker M.D., Matthews E.A., Dickenson A.H., Wood J.N. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. U S A. 2004;101:12706–12711. doi: 10.1073/pnas.0404915101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederberger E., Geisslinger G. The IKK-NF-kappaB pathway: a source for novel molecular drug targets in pain therapy? FASEB J. 2008;22:3432–3442. doi: 10.1096/fj.08-109355. [DOI] [PubMed] [Google Scholar]
- Salles A., Romano A., Freudenthal R. Synaptic NF-kappa B pathway in neuronal plasticity and memory. J. Physiol. 2014;108:256–262. doi: 10.1016/j.jphysparis.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Schmalhofer W.A., Calhoun J., Burrows R., Bailey T., Kohler M.G., Weinglass A.B., Kaczorowski G.J., Garcia M.L., Koltzenburg M., Priest B.T. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 2008;74:1476–1484. doi: 10.1124/mol.108.047670. [DOI] [PubMed] [Google Scholar]
- Shields S.D., Cheng X., Uceyler N., Sommer C., Dib-Hajj S.D., Waxman S.G. Sodium channel Na(v)1.7 is essential for lowering heat pain threshold after burn injury. J. Neurosci. 2012;32:10819–10832. doi: 10.1523/JNEUROSCI.0304-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson N.J., Ferguson A.V. The proinflammatory cytokine tumor necrosis factor-alpha excites subfornical organ neurons. J. Neurophysiol. 2017;118:1532–1541. doi: 10.1152/jn.00238.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M., Lahiri D.K. Significance of NF-kappaB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer's disease and multiple sclerosis. Expert Opin. Ther. Targets. 2015;19:471–487. doi: 10.1517/14728222.2014.989834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura R., Nemoto T., Maruta T., Onizuka S., Yanagita T., Wada A., Murakami M., Tsuneyoshi I. Up-regulation of NaV1.7 sodium channels expression by tumor necrosis factor-alpha in cultured bovine adrenal chromaffin cells and rat dorsal root ganglion neurons. Anesth. Analg. 2014;118:318–324. doi: 10.1213/ANE.0000000000000085. [DOI] [PubMed] [Google Scholar]
- Taniguchi K., Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol. 2018;18:309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- Wajant H., Scheurich P. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J. 2011;278:862–876. doi: 10.1111/j.1742-4658.2011.08015.x. [DOI] [PubMed] [Google Scholar]
- Weiss J., Pyrski M., Jacobi E., Bufe B., Willnecker V., Schick B., Zizzari P., Gossage S.J., Greer C.A., Leinders-Zufall T. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature. 2011;472:186–190. doi: 10.1038/nature09975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J., Huang N., Huang H., Sun L., Dong S., Su J., Zhang J., Wang L., Lin L., Shi M. Voltage-gated sodium channel Nav 1.7 promotes gastric cancer progression through MACC1-mediated upregulation of NHE1. Int. J. Cancer. 2016;139:2553–2569. doi: 10.1002/ijc.30381. [DOI] [PubMed] [Google Scholar]
- Zang Y., He X.H., Xin W.J., Pang R.P., Wei X.H., Zhou L.J., Li Y.Y., Liu X.G. Inhibition of NF-kappaB prevents mechanical allodynia induced by spinal ventral root transection and suppresses the re-expression of Nav1.3 in DRG neurons in vivo and in vitro. Brain Res. 2010;1363:151–158. doi: 10.1016/j.brainres.2010.09.048. [DOI] [PubMed] [Google Scholar]
- Zhang J.M., Li H., Liu B., Brull S.J. Acute topical application of tumor necrosis factor alpha evokes protein kinase A-dependent responses in rat sensory neurons. J. Neurophysiol. 2002;88:1387–1392. doi: 10.1152/jn.2002.88.3.1387. [DOI] [PubMed] [Google Scholar]
- Zsiros E., Kis-Toth K., Hajdu P., Gaspar R., Bielanska J., Felipe A., Rajnavolgyi E., Panyi G. Developmental switch of the expression of ion channels in human dendritic cells. J. Immunol. 2009;183:4483–4492. doi: 10.4049/jimmunol.0803003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.