

Abstract
Recent studies showed that the activation of prostaglandin (PG) receptor EP1 promotes cell migration and invasion in different cancers. The aim of this study was to investigate the role of EP1 in the proliferation of osteosarcoma (OS) cells in vitro and in vivo. EP1 mRNA and protein levels were analyzed by real-time RT-PCR and Western blot, respectively in human OS cell lines MG63, OS732, U-2OS, and 143B compared to human fetal osteoblastic hFOB 1.19 cells. MG63 cells were treated with PGE2, EP1 specific agonist 17-PT-PGE2, 17-PT-PGE2 + EP1 specific antagonist SC51089, or DMSO (control). EP1R-siRNA or a non-silencing irrelevant RNA duplex (negative control) were used for the transfection of MG63 cells, followed by PGE2 treatment. Nude mice carrying MG63 xenografts were treated with SC51089 (2 mg/kg/day). MG63 cells/xenografts were analyzed by MTT assay, TUNEL assay, PKC enzyme activity assay, and Western blot (EP1 and apoptotic proteins), and tumor growth/volume was evaluated in mice. EP1 levels were significantly higher in OS cells compared to osteoblasts. PGE2 or 17-PT-PGE2 treatment increased the proliferation and decreased the apoptosis of MG63 cells. Inhibition of EP1 by SC51089 or siRNA markedly decreased the viability of MG63 cells. Similarly, SC51089 treatment significantly inhibited MG63 cell proliferation and promoted apoptosis in vivo. The silencing of EP1 receptor by siRNA or blockade of EP1 signaling by SC51089 activated extrinsic and intrinsic apoptotic pathways both in vivo and in vitro, as evidenced by increased levels of Bax, cyt c, cleaved caspase-3, caspase-8 and caspase-9. EP1 appears to be involved in PGE2-induced proliferative activity of MG63 cells. Antagonizing EP1 may provide a novel therapeutic approach to the treatment of OS.
Keywords: Osteosarcoma, PGE2, EP1, SC51089, prostaglandin
INTRODUCTION
Osteosarcoma (OS) is the most frequent type of primary (non-hematopoietic) bone tumors occurring predominantly in young adults and adolescents. At present, available treatment strategies for OS include surgical operation, radiotherapy, and chemotherapy [1,2]. However, the prognosis of malignant OS is very poor and the 5-year overall survival (OS) is still low. The possible reasons include invasion into the surrounding tissue and/or distant organ metastasis early in tumor progression, which prevents surgical resection as the treatment option. Furthermore, malignant OS tumors are frequently resistant to all conventional chemotherapies and, in addition, chemotherapy can induce serious systemic toxicity and side effects in OS patients [3-5]. Thus, there is an urgent need to explore alternative therapeutic targets for effective OS management.
Prostaglandin E2 (PGE2), one of the metabolites of arachidonic acid, is a naturally occurring prostaglandin and has an important role in the regulation of physiological and pathological processes. PGE2 exerts its biological action through binding to specific G-protein-coupled receptors (GPCRs) known as prostaglandin receptors EP1, EP2, EP3, and EP4 [6]. It is established that the activation of these receptors by PGE2 induces different intracellular signaling pathways, which explains diverse and sometimes opposite effects of PGE2. The EP1 receptor is Gαq-coupled to mediate the mobilization of cytosolic Ca2+ and activation of protein kinase C (PKC) [6,7]. At present, the EP1 receptor has been found in various kinds of tumor cells, including breast cancer, hepatocellular carcinoma (HCC), colon cancer, and cholangiocarcinoma cells. Recent studies showed that the EP1 receptor contributes to carcinogenesis, aggressiveness, and tumor progression in a number of cancers. PGE2 could increase survivin expression, phosphorylation of focal adhesion kinase (FAK), and enhance cell adhesion and migration in HCC cells via the EP1 receptor [8,9]. Moreover, the upregulation of the EP1 receptor by PGE2 promotes skin tumor progression [10,11]. However, the pathological significance of the EP1 receptor in human OS has not been reported yet.
In the present study, we compared the expression of the EP1 receptor between several human OS cell lines and a human fetal osteoblastic cell line. Furthermore, we investigated the pathological role of EP1 in OS and the effect of EP1 on the survival of OS cells both in vivo and in vitro.
MATERIALS AND METHODS
Cell lines and culture
Human OS cell lines MG63, OS732, U-2OS, and143B and human fetal osteoblastic hFOB 1.19 cells were provided by American Type Culture Collection (ATCC, USA). The cells were cultured in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), 2.5 mmol/L L-glutamine (Invitrogen, Carlsbad, CA, USA), and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin) in a humidified incubator at 37°C with 5% CO2.
Real-time reverse transcription (RT)-PCR
The total RNA from human OS and hFOB 1.19 cells was isolated with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The RT reaction was performed using the PrimeScript™ RT Master Mix (Takara, Japan) in a final volume of 10 µl containing 0.5 µg total RNA, 2 µl 5X PrimeScript buffer, 0.5 µl Oligo (dT) primer, 0.5 µl PrimeScript RT enzyme, and RNase-free water. The primers used for the amplification of EP1 were as follows: forward 5′-GAGAAGGCTACTGCGGAGGA-3′; reverse 5′-CGGCCACA AAGTCGAAATC-3′. β-actin was used as the internal control for normalization of sample amounts, with the following primers: forward 5′-TCCTCCCTG GAGAAGAGCTA-3′; reverse 5′-CCAGACAGCACTGTGTTGGC-3′.
Small interfering RNA (siRNA) targeting EP1
We used siRNA targeting the EP1 receptor (EP1R-siRNA; ACUUCUAAGCACAACCAGAtt, Ambion, Austin, TX, USA) for the transfection of MG63 cells. MG63 cells were grown as a monolayer in 12-well plates (5×104 cells per well) for 24 hours, resulting in 30–50% confluency. The cells were then transfected with EP1R-siRNA or a non-silencing irrelevant RNA duplex (negative control) using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA). The cells were collected at 48 hours after transfection and subjected to Western blotting immediately.
Western blotting
Human OS and hFOB 1.19 cells were lysed with RIPA lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA) on ice. Membrane, cytoplasmic, and mitochondrial lysates were extracted by commercial extraction kits (Beyotime Institute of Biotechnology, Shanghai, China), and protein concentrations were determined by the bicinchoninic acid (BCA) assay using bovine serum albumin (BSA) as the standard. A total of 20 µg of protein samples were loaded on 12% sodium dodecyl sulfate (SDS) polyacrylamide gels and separated by electrophoresis. The proteins were transferred to polyvinylidene fluoride membranes and incubated with antibodies against EP1 (catalog number: sc-22646), Bcl2-associated X [Bax] (catalog number: sc-70407), B-cell lymphoma 2 [Bcl-2] (catalog number: sc-23960), cytochrome c [cyt c] (catalog number: sc-13560), cleaved caspase 3 (catalog number: sc-373730), cleaved caspase 8 (catalog number: sc-5263), and cleaved caspase 9 (catalog number: sc-7885) overnight at 4°C; 1:1000 dilution was used for all antibodies (Santa Cruz, CA, USA). The membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling Technology, MA, USA) at room temperature for 1 hour and visualized using enhanced chemiluminescence reagents (Beyotime, China). All bands were quantified by densitometry using ImageJ software.
Cell viability by MTT assay
The viability of MG63 cells was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. MG63 cells were cultured in 96-well plates (1×104 cells/well) for 24 hours and treated with PGE2 (5 µM, Cayman Chemical Co, Ann Arbor, MI, USA), EP1 specific agonist 17-PT-PGE2 (5 µM, Cayman Chemical), or 17-PT-PGE2 (5 µM) + EP1 specific antagonist SC51089 (2 µM, Sigma-Aldrich, St. Louis, MO, USA). The vehicle control cells were treated with 0.5% dimethyl sulfoxide (DMSO). Moreover, EP1-silenced MG63 cells and negative control MG63 cells were treated with PGE2 (5 µM) 48 hours after transfection. Forty-eight hours after treatments, 10 µl MTT was added to each well and samples were incubated at 37°C for 4 hours. Culture medium was then replaced with an equal volume of DMSO to dissolve formazan crystals. After shaking the mixture for 10 minutes at room temperature, the absorbance of each well was determined at 490 nm using a microplate reader (BioTek, Winooski, VT, USA).
Quantification of apoptosis
Quantitative assessment of apoptotic cells in vivo and in vitro was performed by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay, according to the manufacturer’s instructions (Promega, Madison, WI, USA). Briefly, MG63 cells were seeded in 24-well plates (2×104 cells/well) for 24 hours and then treated with PGE2 (5 µM), 17-PT-PGE2 (5 µM), or 17-PT-PGE2 (5 µM) + SC51089 (2 µM), while the vehicle control cells were treated with 0.5% DMSO. Moreover, EP1-silenced MG63 cells and negative control MG63 cells were treated with PGE2 (5 µM) 48 hours after transfection. Forty-eight hours after treatments, 4′,6-diamidino-2-phenylindole (DAPI) and TUNEL were used for nuclear DNA staining. The cells were analyzed using confocal fluorescence microscopy (Leica, Germany). The apoptotic index was calculated as the average percentage of positive nuclei, including apoptotic bodies, divided by the total number of nuclei from five random fields at 200× magnitude.
PKC measurement
MG63 cells were seeded in 24-well plates (2×104 cells/well) for 24 hours and then treated with PGE2 (5 µM), 17-PT-PGE2 (5 µM), or 17-PT-PGE2 (5 µM) + SC51089 (2 µM), while the vehicle control cells were treated with 0.5% DMSO. Forty-eight hours after treatment, PKC assay was carried out to evaluate the activation of EP1-PKC signaling pathway using a PKC activity assay kit (Millipore, Billerica, MA, USA), according to the manufacturer’s instructions. The counts per minute (cpm) of the samples containing PKC were compared to cpm of control samples containing no enzyme.
Mice MG63 xenograft
Animal experiments were approved by the Experimental Ethics Committee of Soochow University and performed according to the institutional Guidelines for Care and Use of Laboratory Animals. Human osteosarcoma xenografts were established in 4–6 week old male nude mice (Model Animal Research Center, NanJing University, China) by subcutaneous inoculation of 5×107 MG63 cells into the axilla of each mouse. After tumors reached 3–5 mm in diameter, the mice were randomly divided into control and SC51089 group (2 mg/kg/day) which received oral treatment for 21 days. Tumor growth was monitored every three days for 21 days. Measurements were made using calipers, and tumor volumes were calculated as follows: volume = length × width2 × 0.52.
Statistical analysis
The data are presented as mean ± SD. Statistical analysis was performed using SPSS for Windows, Version 15.0. (SPSS Inc., Chicago, USA). One-way analysis of variance (ANOVA) and Dunnett’s test were used for multiple comparisons. A difference was considered statistically significant when p values were less than 0.05.
RESULTS
Membrane expression of EP1 in OS cells
EP1 mRNA and protein levels were analyzed by real-time RT-PCR and Western blot, respectively in human OS cell lines MG63, OS732, U-2OS, and 143B compared to human fetal osteoblastic hFOB 1.19 cells. As seen in Figure 1A, the mRNA levels of EP1 were significantly higher in MG63, U-2OS, 143B, and OS732 cell lines compared to hFOB 1.19 cells. Similarly, the protein levels of EP1 were significantly higher in MG63, U-2OS, 143B, and OS732 cell lines compared to hFOB 1.19 [p < 0.01] (Figure 1B-C). Moreover, the expression of EP1 was the highest in MG63 cells. Therefore, MG63 cell line was used in the subsequent experiments to elucidate the pathological role of EP1 in the development of OS.
FIGURE 1.
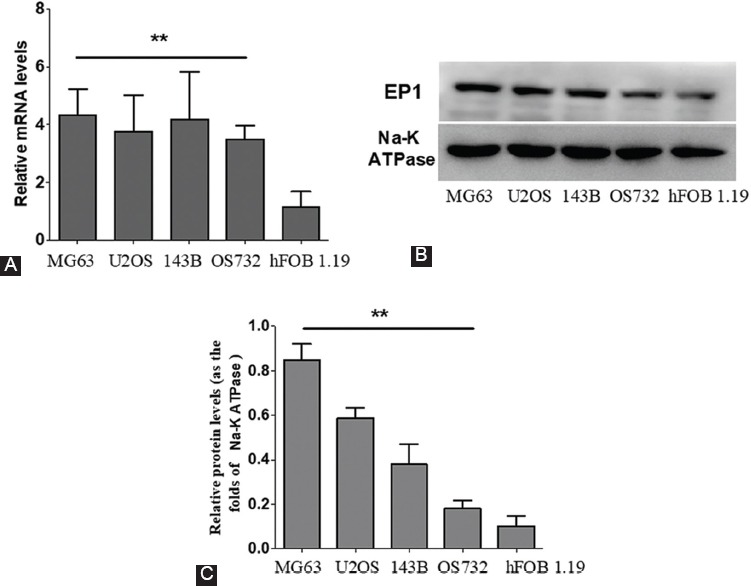
EP1 levels in human osteosarcoma (OS) cell lines vs. human fetal osteoblastic hFOB 1.19 cells. Each sample was prepared in triplicate and the experiment was repeated three times. All bar graphs represent the mean ± SD. **p < 0.01 vs. hFOB 1.19. A) The mRNA levels of EP1 were significantly higher in MG63, U-2OS, 143B, and OS732 compared to hFOB 1.19 cells. B-C) Similarly, the protein levels of EP1 were significantly higher in MG63, U-2OS, 143B, and OS732 cell lines compared to hFOB 1.19.
EP1 receptor is involved in PGE2-induced proliferation of OS cells
To investigate the role of EP1 in the proliferation of OS cells, we first treated MG63 cells with PGE2 or EP1 specific agonist (17-PT-PGE2). As seen in Figure 2A, both PGE2 and 17-PT-PGE2 increased the activity of PKC (a downstream molecule of EP1) compared to DMSO-treated control cells (p < 0.01), suggesting that the activation of EP1 by PGE2 and 17-PT-PGE2 occurred. Furthermore, the treatment with PGE2 or 17-PT-PGE2 increased the proliferative ability of MG63 cells compared to control cells [p < 0.01] (Figure 2B). By contrast, the treatment with PGE2 or 17-PT-PGE2 significantly decreased the apoptosis of MG63 cells (Figure 2C-D, p < 0.01).
FIGURE 2.
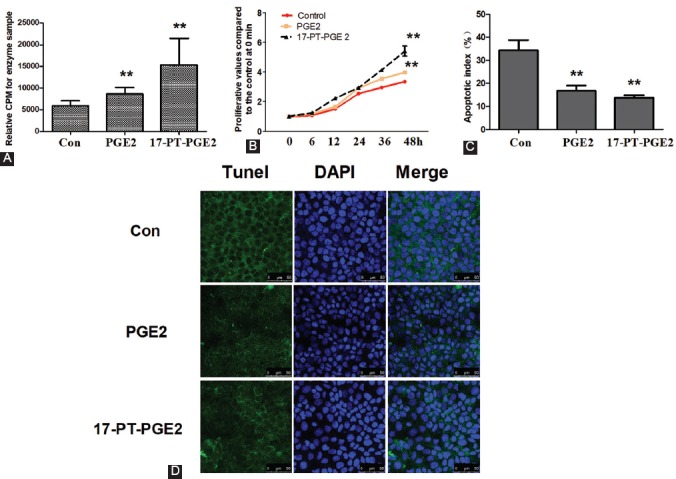
Activation of the EP1 receptor promoted proliferation in osteosarcoma (OS) cells. MG63 cells were cultured with PGE2 (5 µM) or 17-PT-PGE2 (5 µM) for 48 hours. A) PKC activity assay. MG63 cells were treated with 17-PT-PGE2 for 48 hours. Equal amounts of total proteins (30 mg) were added to microcentrifuge tubes and assayed for PKC levels using a PKC activity assay kit. B) Cell viability was tested by MTT assay. C-D) Following the treatment, MG63 cells were fixed and stained with DAPI and TUNEL. The apoptotic nuclei were visualized and photographed using a laser confocal microscope. Each sample was prepared in triplicate and all experiments were repeated three times. All bar/line graphs represent the mean ± SD. **p < 0.01 vs. DMSO-treated control (con). Both PGE2 and 17-PT-PGE2 increased the activity of PKC compared to control cells. Furthermore, the treatment with PGE2 and 17-PT-PGE2 increased the proliferation and decreased the apoptosis of MG63 cells compared to control cells. PGE2: Prostaglandin E2; PKC: Protein kinase C.
Blockade or silencing of the EP1 receptor inhibited 17-PT-PGE2-induced cell proliferation of MG63 cells
To further verify the role of EP1 in the proliferation of OS cells, we blocked and silenced EP1 receptor in MG63 cells. As seen in Figure 3A, the EP1 specific antagonist SC51089 significantly blocked 17-PT-PGE2-induced activation of the EP1/PKC pathway. In addition, MG63 cells exposed to 17-PT-PGE2 + SC51089 showed a decreased proliferation and increased apoptosis compared to MG63 cells exposed to 17-PT-PGE2 only (Figure 3B-C, p < 0.01). To further investigate the specific role of EP1 in MG63 cell proliferation and apoptosis, MG63 cells were transfected with EP1R-siRNA. As shown in Figure 3D-G, depletion of the EP1 receptor greatly reduced the viability and increased apoptosis of MG63 cells (p < 0.01).
FIGURE 3.
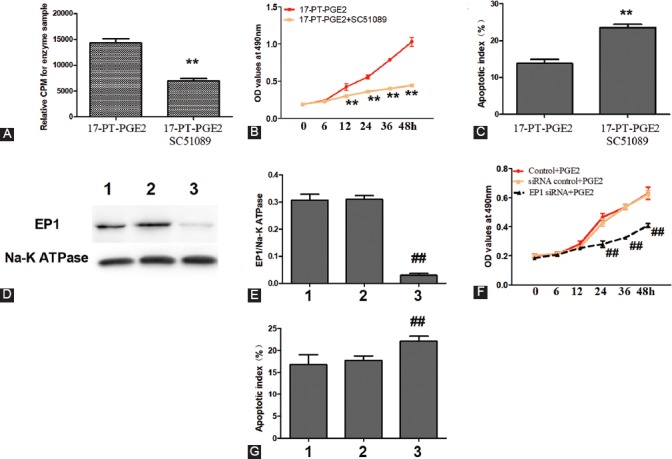
Blockade or silencing of the EP1 receptor decreased MG63 cell viability. MG63 cells were co-cultured with 17-PT-PGE2 (5 µM) and SC51089 (2 µM) for 48 hours. A) PKC activity assay. B) Cell viability was tested by MTT assay. C) MG63 cells were fixed and stained with DAPI and TUNEL. SC51089 significantly blocked 17-PT-PGE2-induced activation of the EP1/PKC pathway. In addition, MG63 cells exposed to 17-PT-PGE2 + SC51089 showed a decreased proliferation and increased apoptosis compared to MG63 cells exposed to 17-PT-PGE2 only. D-G) MG63 cells transfected with an EP1R-siRNA were exposed to PGE2 (5 µM) for 48 hours. The silencing of EP1 receptor suppressed PGE2-induced proliferation in MG63 cells. Knockout efficiency was measured by Western blotting; cell viability was tested by MTT assay; and apoptosis of MG63 cells was assessed by DAPI and TUNEL staining. 1: Control+PGE2; 2: siRNAControl+PGE2; 3: EP1R-siRNA+PGE2. Each sample was prepared in triplicate and all experiments were repeated three times. All bar/line graphs represent the mean ± SD. **p < 0.01 vs. 17-PT-PGE2; ##p < 0.01 vs. siRNA Control+PGE2. PGE2: Prostaglandin E2; PKC: Protein kinase C; EP1R-siRNA: siRNA targeting the EP1 receptor; siRNAControl: Non-silencing irrelevant RNA duplex.
Silencing of the EP1 receptor activated extrinsic and intrinsic apoptotic pathways
To determine whether the silencing of EP1 receptor by EP1R-siRNA was associated with the regulation of extrinsic and intrinsic apoptotic pathways, we investigated the effect of EP1 receptor depletion on the levels of apoptosis-related molecules. As shown in Figure 4, a significant increase in the level of Bax, cytoplasmic cyt c, and cleaved caspase-3, caspase-8 and caspase-9 was observed in EP1R-siRNA-transfected MG63 cells compared to the cells transfected with irrelevant RNA duplex (p < 0.01). By contrast, the protein expression of Bcl-2 and mitochondrial cyt c was downregulated by EP1 silencing (p < 0.01).
FIGURE 4.
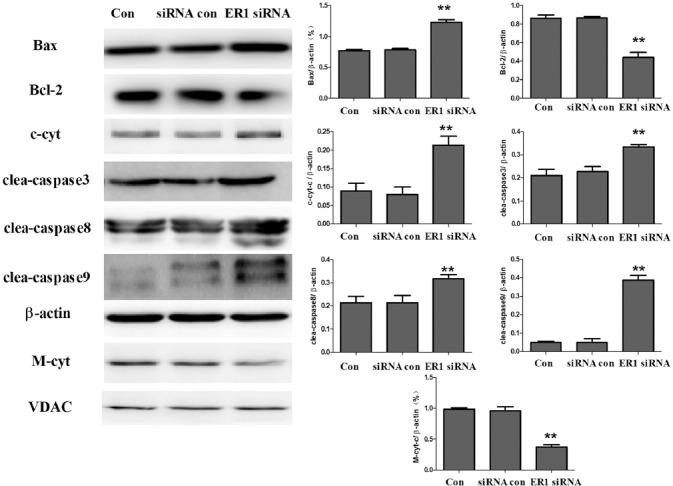
Silencing of the EP1 receptor in MG63 cells affected the levels of apoptotic molecules. MG63 cells were transfected with EP1R-siRNA. After transfection (48 hours), the cells were exposed to PGE2 (5 µM) for 48 hours. The proteins in cytoplasmic and mitochondrial lysates were extracted and assayed by Western blotting. Each sample was prepared in triplicate and the experiment was repeated three times. All bar graphs represent the mean ± SD. **p vs. siRNA control (Con). A significant increase in the level of Bax, cytoplasmic cyt c, and cleaved caspase-3, caspase-8 and caspase-9 was observed in EP1R-siRNA-transfected MG63 cells compared to the cells transfected with siRNA control. By contrast, the protein expression of Bcl-2 and mitochondrial cyt c was downregulated by EP1 silencing. c-cyt: Cytoplasmic cytochrome c; M-cyt: Mitochondrial cytochrome c; clea: cleaved; VDAC: Voltage-dependent anion channel; EP1R-siRNA: siRNA targeting the EP1 receptor; siRNA control: Non-silencing irrelevant RNA duplex.
EP1 antagonist SC51089 significantly inhibited MG63 proliferation in vivo
To further verify the pathological role of EP1 in OS, we treated nude mice carrying MG63 xenografts with the EP1 antagonist SC51089. As shown in Figure 5A-C, SC51089 treatment decreased PKC activity, reduced tumor weight, and inhibited the growth of MG63 cells in vivo. Moreover, TUNEL staining revealed that the positive area was lower in the tumor xenograft samples from SC51089 group compared to DMSO-treated control group (Figure 5D, p < 0.01).
FIGURE 5.
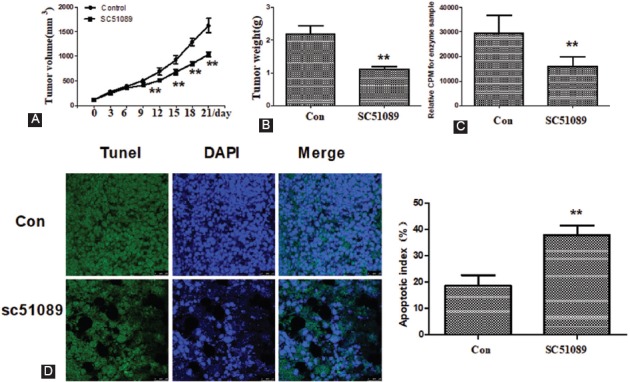
SC51089 significantly inhibited MG63 proliferation in vivo. A) Tumor growth curves in nude mice with MG63 xenograft tumors. The curves of tumor volume were generated every three days for 21 days. B) The weight of excised tumors was measured at the end of experiment. C) PKC activity. D) TUNEL and DAPI staining. All bar/line graphs represent the mean ± SD (n = 6). **p vs. DMSO-treated control (Con). SC51089 treatment decreased PKC activity, reduced tumor weight, and inhibited the growth of MG63 cells in vivo. TUNEL staining revealed that the positive area was lower in the tumor xenograft samples from SC51089 group compared to DMSO-treated control group. PKC: Protein kinase C.
EP1 antagonist SC51089 regulated apoptotic molecules in MG63 transplanted mice
To determine the effect of EP1 antagonist SC51089 on the extrinsic and intrinsic apoptotic pathways in mice transplanted with MG63 cells, we analyzed the levels of apoptosis-related molecules in the tumor tissue. As shown in Figure 6, SC51089 treatment caused a significant increase in the protein levels of Bax, cytoplasmic cyt c, and cleaved caspase 3, caspase 8 and caspase 9 in the tumor tissue from MG63 transplanted mice (p < 0.01). Moreover, SC51089 decreased the protein levels of Bcl-2 and mitochondrial cyt c in the tumor tissue (p < 0.01).
FIGURE 6.
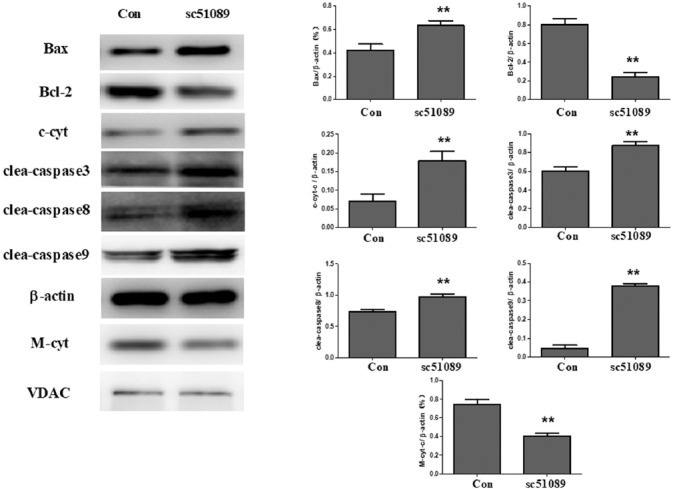
SC51089 affected the levels of apoptotic molecules in MG63 transplanted mice. The samples in cytoplasmic and mitochondrial lysates were extracted and assayed by Western blotting. SC51089 treatment caused a significant increase in the protein levels of Bax, cytoplasmic cyt c, and cleaved caspase 3, caspase 8 and caspase 9 in the tumor tissue from MG63 transplanted mice. Moreover, SC51089 decreased the protein levels of Bcl-2 and mitochondrial cyt c in the tumor tissue. All bar graphs represent the mean ± SD of the experiment (n = 6). **p vs. DMSO-treated control (Con). c-cyt: Cytoplasmic cytochrome c; M-cyt: Mitochondrial cytochrome c; clea: cleaved; VDAC: Voltage-dependent anion channel.
DISCUSSION
The pathway involving the EP1 receptor and its downstream molecules is a well-known signaling pathway considered to play an important role in PGE2-induced proliferation, growth, and migration of malignant cells. Previous studies showed that the activation of EP1 promotes migration and proliferation of HCC, skin cancer, colon cancer, and non-small cell lung cancer cells [8-13]. This renders the EP1-mediated signaling pathway a potential target for the development of novel cancer therapeutic approaches. However, the pathological significance of the EP1 receptor in human OS is still not clear. In the current study, we investigated the effects of EP1 on the proliferation and growth of OS cells both in vitro and in vivo. We found that the expression of EP1 was significantly higher in MG63, U2OS, 143B, and OS732 compared to hFOB 1.19 cells. Moreover, the level of EP1 was significantly higher in MG63 cells compared to the other OS cells. Therefore, in the subsequent experiments, we used MG63 cells to study the pathological role of EP1 in OS development. We showed that the activation of the EP1 receptor by PGE2 or EP1 specific agonist 17-PT-PGE2 effectively activated the EP1/PKC signaling pathway and promoted the proliferation of MG63 cells in vitro. On the other hand, the inactivation of the EP1 receptor by the specific antagonist SC51089 or silencing of EP1 by siRNA reduced PKC activity and promoted apoptosis in MG63 cells in vivo and in vitro. Although in the studies of Surh et al. [10,11] BK5.EP1 transgenic mice overexpressing EP1 had a reduced tumor multiplicity after 7,12-dimethyl-benz[a]anthracene (DMBA)/12-O-Tetradecanoylphorbol-13-acetate (TPA) treatment compared to wild type mice, which was possibly due to the downregulation of PKC, the BK5.EP1 transgenic mice showed higher papilloma to carcinoma conversion rate. Using HUH-7 and HepG2 cells, Bai et al. [9] demonstrated that PGE2 treatment increases survivin expression and that this effect is mimicked by EP1 receptor transfection or treatment with a selective EP1 agonist. Moreover, the PGE2-induced upregulation of survivin is blocked by a selective EP1 antagonist or siRNA against the EP1 receptor [9]. Consistent with these findings, our results showed that PGE2-induced activation of EP1 plays an important role in OS development.
The activation of EP1, which is coupled to Gαq, by PGE2 activates phosphatidylcholine-phospholipase C and ultimately leads to an increase in intracellular Ca2+ and activation of PKC [14]. Recent studies found that the activation of the EP1/PKC pathway induces gene transcription and protein synthesis of several key proteins that are involved in cell migration and invasion. Briefly, signaling through the EP1 receptor activated by PGE2 has been shown to enhance integrin expression, induce the phosphorylation and activation of FAK in cancer cells as well as induce c-Src activation, epidermal growth factor receptor (EGFR) transactivation, and cyclic adenosine monophosphate (cAMP) response element-binding (CREB) phosphorylation [7,8,15,16]. Interestingly, a previous study [17] found that PGE2 stimulates Fas ligand expression via the EP1 receptor in colon cancer cells, suggesting the effect of PGE2 on apoptosis pathways. Apoptosis is a programmed cell death mechanism initiated by the extrinsic or intrinsic pathways [18]. In the extrinsic apoptotic pathway, cell death signals are mediated by proapoptotic death receptors (DRs) such as those belonging to tumor necrosis factor (TNF) receptor superfamily, and this pathway is characterized by the activation of caspase-8 [19]. In the intrinsic pathway, apoptosis is controlled by Bcl-2 family proteins which have either antiapoptotic (such as Bcl-2) or proapoptotic functions (such as Bax). The oligomerization of proapoptotic Bax and Bak in the mitochondrial outer membrane leads to its permeabilization and subsequent release of cyt c, followed by the conversion of procaspase-9 to active, cleaved caspase-9. Either activated caspase-8 in the extrinsic pathway or cyt c-activated caspase-9 in the intrinsic pathway may activate executioner caspase-3, resulting in apoptosis [20]. In order to understand the relationship between the EP1 receptor and extrinsic and intrinsic apoptotic pathways, we investigated the effect of depletion of the EP1 receptor by siRNA or blockade of the EP1/PKC signaling by SC51089 on apoptosis-related molecules. As expected, we found that both EP1R-siRNA and SC51089 intervention activated the extrinsic and intrinsic apoptotic pathways, which was evidenced by increased levels of Bax, cytoplasmic cyt c, and cleaved caspase 3, caspase 8 and caspase 9 in MG63 cells and MG63 transplanted mice. Our results indicate that the EP1 receptor is involved in PGE2-induced extrinsic and intrinsic apoptotic pathways in MG63 cells.
CONCLUSION
In the present study, we demonstrated that PGE2 can promote proliferation and growth of MG63 cells via EP1 both in vivo and in vitro. The extrinsic and intrinsic apoptotic pathways are involved in EP1 receptor-mediated viability of MG63 cells. Our findings provide new information regarding the putative role of the EP1 receptor in the development of OS and suggest that blocking EP1 may represent a new therapeutic strategy for OS.
ACKNOWLEDGMENTS
This work was supported by the Science and Technology development plan of Suzhou (No. SZS201720) and Key disciplines of medicine of Suzhou (No. Szxk201802).
DECLARATION OF INTERESTS
The authors declare no conflict of interests.
REFERENCES
- 1.Moore DD, Luu HH. Osteosarcoma. Cancer Treat Res. 2014;162:65–92. doi: 10.1007/978-3-319-07323-1_4. https://doi.org/10.1007/978-3-319-07323-1_4. [DOI] [PubMed] [Google Scholar]
- 2.Ferrari S, Serra M. An update on chemotherapy for osteosarcoma. Expert Opin Pharmacother. 2015;16(18):2727–36. doi: 10.1517/14656566.2015.1102226. https://doi.org/10.1517/14656566.2015.1102226. [DOI] [PubMed] [Google Scholar]
- 3.Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma:current treatment and a collaborative pathway to success. J Clin Oncol. 2015;33(27):3029–35. doi: 10.1200/JCO.2014.59.4895. https://doi.org/10.1200/JCO.2014.59.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daqian W, Chuandong W, Xinhua Q, Songtao A, Kerong D. Chimaphilin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines through insulin-like growth factor-I receptor (IGF-IR) signaling. Chem Biol Interact. 2015;237:25–30. doi: 10.1016/j.cbi.2015.05.008. https://doi.org/10.1016/j.cbi.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Longhi A, Ferrari S, Tamburini A, Luksch R, Fagioli F, Bacci G, et al. Late effects of chemotherapy and radiotherapy in osteosarcoma and Ewing sarcoma patients:the Italian Sarcoma Group Experience (1983-2006) Cancer. 2012;118(20):5050–9. doi: 10.1002/cncr.27493. https://doi.org/10.1002/cncr.27493. [DOI] [PubMed] [Google Scholar]
- 6.Jiang J, Qiu J, Li Q, Shi Z. Prostaglandin E2 signaling:alternative target for glioblastoma? Trends Cancer. 2017;3(2):75–8. doi: 10.1016/j.trecan.2016.12.002. https://doi.org/10.1016/j.trecan.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai X, Wang J, Guo Y, Pan J, Yang Q, Zhang M, et al. Prostaglandin E2 stimulates β1-integrin expression in hepatocellular carcinoma through the EP1 receptor/PKC/NF-κB pathway. Sci Rep. 2014;4:6538. doi: 10.1038/srep06538. https://doi.org/10.1038/srep06538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bai X, Wang J, Zhang L, Ma J, Zhang H, Xia S, et al. Prostaglandin E₂receptor EP1-mediated phosphorylation of focal adhesion kinase enhances cell adhesion and migration in hepatocellular carcinoma cells. Int J Oncol. 2013;42(5):1833–41. doi: 10.3892/ijo.2013.1859. https://doi.org/10.3892/ijo.2013.1859. [DOI] [PubMed] [Google Scholar]
- 9.Bai XM, Jiang H, Ding JX, Peng T, Ma J, Wang YH, et al. Prostaglandin E2 upregulates survivin expression via the EP1 receptor in hepatocellular carcinoma cells. Life Sci. 2010;86(5-6):214–23. doi: 10.1016/j.lfs.2009.12.009. https://doi.org/10.1016/j.lfs.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 10.Surh I, Rundhaug J, Pavone A, Mikulec C, Abel E, Fischer SM. Upregulation of the EP1 receptor for prostaglandin E2 promotes skin tumor progression. Mol Carcinog. 2011;50(6):458–68. doi: 10.1002/mc.20730. https://doi.org/10.1002/mc.20730. [DOI] [PubMed] [Google Scholar]
- 11.Surh I, Rundhaug JE, Pavone A, Mikulec C, Abel E, Simper M, et al. The EP1 receptor for prostaglandin E2 promotes the development and progression of malignant murine skin tumors. Mol Carcinog. 2012;51(7):553–64. doi: 10.1002/mc.20820. https://doi.org/10.1002/mc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Callaghan G, Ryan A, Neary P, O'Mahony C, Shanahan F, Houston A. Targeting the EP1 receptor reduces Fas ligand expression and increases the antitumor immune response in an in vivo model of colon cancer. Int J Cancer. 2013;133(4):825–34. doi: 10.1002/ijc.28076. https://doi.org/10.1002/ijc.28076. [DOI] [PubMed] [Google Scholar]
- 13.Pan J, Yang Q, Shao J, Zhang L, Ma J, Wang Y, et al. Cyclooxygenase-2 induced β1-integrin expression in NSCLC and promoted cell invasion via the EP1/MAPK/E2F-1/FoxC2 signal pathway. Sci Rep. 2016;6:33823. doi: 10.1038/srep33823. https://doi.org/10.1038/srep33823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee IT, Lin CC, Lin WN, Wu WL, Hsiao LD, Yang CM. Lung inflammation caused by adenosine-5'-triphosphate is mediated via Ca2+/PKCs-dependent COX-2/PGE2 induction. Int J Biochem Cell Biol. 2013;45(8):1657–68. doi: 10.1016/j.biocel.2013.05.006. https://doi.org/10.1016/j.biocel.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Cheng S, Zhang M, Ma X, Zhang L, Wang Y, et al. Prostaglandin E2 promotes hepatocellular carcinoma cell invasion through upregulation of YB-1 protein expression. Int J Oncol. 2014;44(3):769–80. doi: 10.3892/ijo.2013.2234. https://doi.org/10.3892/ijo.2013.2234. [DOI] [PubMed] [Google Scholar]
- 16.Sun B, Rong R, Jiang H, Zhang H, Wang Y, Bai X, et al. Prostaglandin E2 receptor EP1 phosphorylate CREB and mediates MMP2 expression in human cholangiocarcinoma cells. Mol Cell Biochem. 2013;378(1-2):195–203. doi: 10.1007/s11010-013-1610-1. https://doi.org/10.1007/s11010-013-1610-1. [DOI] [PubMed] [Google Scholar]
- 17.O'Callaghan G, Kelly J, Shanahan F, Houston A. Prostaglandin E2 stimulates Fas ligand expression via the EP1 receptor in colon cancer cells. Br J Cancer. 2008;99(3):502–12. doi: 10.1038/sj.bjc.6604490. https://doi.org/10.1038/sj.bjc.6604490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaman S, Wang R, Gandhi V. Targeting the apoptosis pathway in hematologic malignancies. Leuk Lymphoma. 2014;55(9):1980–92. doi: 10.3109/10428194.2013.855307. https://doi.org/10.3109/10428194.2013.855307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sayers TJ. Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol Immunother. 2011;60(8):1173–80. doi: 10.1007/s00262-011-1008-4. https://doi.org/10.1007/s00262-011-1008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jendrossek V. The intrinsic apoptosis pathways as a target in anticancer therapy. Curr Pharm Biotechnol. 2012;13(8):1426–38. doi: 10.2174/138920112800784989. https://doi.org/10.2174/138920112800784989. [DOI] [PubMed] [Google Scholar]