

Abstract
Mild traumatic brain injury (mTBI) is a risk factor for neurodegenerative disorders, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). TBI-derived neuropathologies are promoted by inflammatory processes: chronic microgliosis and release of pro-inflammatory cytokines that further promote neuronal dysfunction and loss. Herein, we evaluated the effect on pre-programmed cell death/neuroinflammation/synaptic integrity and function of (−)-Phenserine tartrate (Phen), an agent originally developed for AD. This was studied at two clinically translatable doses (2.5 and 5.0 mg/kg, BID), in a weight drop (concussive) mTBI model in wild type (WT) and AD APP/PSEN1 transgenic mice. Phen mitigated mTBI-induced cognitive impairment, assessed by Novel Object Recognition and Y-maze behavioral paradigms, in WT mice. Phen fully abated mTBI-induced neurodegeneration, evaluated by counting Fluoro-Jade C-positive (FJC+) cells, in hippocampus and cortex of WT mice. In APP/PSEN1 mice, degenerating cell counts were consistently greater across all experimental groups vs. WT mice. mTBI elevated FJC+ cell counts vs. the APP/PSEN1 control (sham) group, and Phen similarly mitigated this. Anti-inflammatory effects on microglial activation (IBA1-immunoreactivity (IR)) and the pro-inflammatory cytokine TNF-α were evaluated. mTBI increased IBA1-IR and TNF-α/IBA1 colocalization vs. sham, both in WT and APP/PSEN1 mice. Phen decreased IBA1-IR throughout hippocampi and cortices of WT mice, and in cortices of AD mice. Phen, likewise, reduced levels of IBA1/TNF-α-IR colocalization volume across all areas in WT animals, with a similar trend in APP/PSEN1 mice. Actions on astrocyte activation by mTBI were followed by evaluating GFAP, and were similarly mitigated by Phen. Synaptic density was evaluated by quantifying PSD-95+ dendritic spines and Synaptophysin (Syn)-IR. Both were significantly reduced in mTBI vs. sham in both WT and APP/PSEN1 mice. Phen fully reversed the PSD-95+ spine loss in WT and Syn-IR decrease in both WT and APP/PSEN1 mice. To associate immunohistochemical changes in synaptic markers with function, hippocampal long term potentiation (LTP) was induced in WT mice. LTP was impaired by mTBI, and this impairment was mitigated by Phen. In synopsis, clinically translatable doses of Phen ameliorated mTBI-mediated pre-programmed cell death/neuroinflammation/synaptic dysfunction in WT mice, consistent with fully mitigating mTBI-induced cognitive impairments. Phen additionally demonstrated positive actions in the more pathologic brain microenvironment of AD mice, further supporting consideration of its repurposing as a treatment for mTBI.
Keywords: (−)-Phenserine, Traumatic brain injury, Alzheimer’s disease, Neurodegeneration, Neuroinflammation, TNF-α, Synaptic proteins, Long term potentiation, Cognitive impairment, Therapeutic intervention
1. Introduction
The occurrence of single or repeated episodes of traumatic brain injury (TBI) is associated with disability and death, and impacts 10 million individuals worldwide annually (Hyder et al., 2007; Ruff et al., 2012). TBI is estimated to constitute the third largest portion of global disease by the year 2020 (WHO, 2006). Within the US, some 1.7 million people sustain a TBI annually, and approximately 5.3 million people live with a TBI-induced disability (Langlois et al., 2006; Prins and Giza, 2012). Generally assessed using the Glasgow Coma Scale (GCS), the majority of TBIs (80–95%) are considered mild (GCS 13–15) to moderate (GCS 9–12) in nature, with severe TBI (GCS ≤8) comprising the remainder (Tagliaferri et al., 2006; Teasdale and Jennett, 1974).
Mild TBI (mTBI) commonly causes a broad spectrum of neurological symptoms including headache, sleep and vision disturbances, depression, anxiety, changes in mood and impaired cognitive function (Dixon, 2017; Tweedie et al., 2016a). Despite improvements in survival rate following initial injury, TBI can give rise to substantial and lifelong cognitive, physical, and behavioral impairments that necessitate long-term access to health care and disability services (Tagliaferri et al., 2006; Diaz-Arrastia et al., 2014). The elderly are especially susceptible to TBI, as the same insult generally gives rise to greater disability and dramatically increases the risk of developing neurodegenerative (Barnes et al., 2014; Gardner et al., 2014) and neuropsychiatric disorders (Chen et al., 2011). Although TBI symptoms may sometimes resolve within the first year after injury, a substantial number of patients exhibit protracted and sometimes permanent dysfunction, particularly emotional and cognitive problems (Pagulayan et al., 2006). TBI is hence considered to represent a ‘progression of events’, and emerging evidence indicates that this process can lead to an early onset of dementia (Barnes et al., 2014; Gardner et al., 2014) and neurodegenerative pathologies such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Chiu et al., 2016; Greig et al., 2014; Daneshvar et al., 2011). A recent study from Fann and collaborators, in a population-based cohort of nearly 2.8 million adults, showed that even a single event of mild TBI increases the risk to develop dementia and to acquire it earlier in life (Fann et al., 2018). Recent gene expression studies have demonstrated that mTBI can induce an upregulation of pathways that lead to AD and PD (Tweedie et al., 2013a, 2013b; Goldstein et al., 2012; Greig et al., 2014). In light of the lack of available therapeutic options (Moppett, 2007), it is important to understand the mechanisms that underlie the neuronal dysfunction and loss following head injury to aid the development of new therapeutic strategies, and to facilitate the evaluation of existing compounds.
TBI neuropathology develops in association with two major biochemical phases. The first phase includes the acute damage that occurs at the moment of the trauma. It is characterized by mechanical damage of the tissue, involving shearing, contusion and laceration, often causing vascular and axonal damage and blood-brain barrier (BBB) breakdown, which result in diffuse and immediate necrotic cell death (LaPlaca et al., 2007; Greig et al., 2014). This acute phase is followed by a longer-lasting secondary cascade of biological processes including neuroinflammatory and neurotoxic mechanisms, such as free radical generation, oxidative stress, excitotoxicity, microgliosis and release of pro-inflammatory cytokines (Maas et al., 2008; Bains and Hall, 2012; Das et al., 2012; Zhang et al., 2008; Chiu et al., 2016; Witcher et al., 2018), with consequent neuronal dysfunction and apoptosis (Greve and Zink, 2009; Barkhoudarian et al., 2011; Morganti-Kossmann et al., 2002; Schmidt et al., 2005). Our recent studies, together with those of others, suggest that such secondary processes may be halted and potentially reversed by mechanism-based treatments, if administered sufficiently early (Yang et al., 2016; Nudo, 2013; Moppett, 2007; Greig et al., 2014).
Extensive studies support the hypothesis that the unregulated release of inflammatory cytokines and chemokines are critical to the pathophysiology of TBI (Acosta et al., 2013; Lozano et al., 2015). Whereas the inflammatory response is fundamental to the initiation of neuro-reparative mechanisms that follow TBI (McCoy and Tansey, 2008; Schmidt et al., 2005), if excessive or unregulated, these same processes can drive neuronal dysfunction and degeneration by provoking a self-propagating pathological inflammatory cascade (McCoy and Tansey, 2008; Frankola et al., 2011). Following a TBI, there is a marked elevation in the generation and release of proinflammatory cytokines from activated microglia, particularly tumor necrosis factor-α (TNFα). In line with this, brain levels of TNFα mRNA and protein are raised in humans and rodents following TBI, and have been noted to precede the appearance of multiple other inflammatory cytokines (Feuerstein et al., 1994; Frugier et al., 2010; Woodcock and Morganti-Kossmann, 2013). Depending on the signaling pathway initiated, TNF-α can exacerbate oxidative stress, contribute to glutamate release, induce synaptic loss and exacerbate BBB dysfunction, each of which can lead to neuronal dysfunction and initiation of preprogrammed cell death (Frankola et al., 2011; McCoy and Tansey, 2008). Relatively few drugs have the potential to positively impact these multiple pathways, each leading to degenerative processes; this likely has, in large part, underpinned the failure of many drugs that have a single mechanism of action in clinical TBI studies, since no two TBI events are entirely alike in humans and all involve the instigation of multiple neuropathological cascades.
Our recent studies have highlighted the ability of the experimental drug (−)-phenserine tartrate (Phen) to provide neuroprotection and neurotrophic actions via multiple mechanisms (Lilja et al., 2013; Chang et al., 2017; Hoffer et al., 2017) following a brain insult. The focus of the present study was to ascertain whether such mechanisms are pertinent to mTBI in both normal and pathological brain environments. To determine this, we evaluated clinically translatable doses of Phen in both wild type (WT) and transgenic AD mice subjected to mild concussive head injury, with the latter providing a more complex brain milieu that may better parallel the complexity of the aged human brain (Becker and Greig, 2019).
2. Materials & methods
2.1. mTBI induction and drug treatment
For induction of mTBI, 6 to 8 weeks-old WT CD1 and 52 to 69 weeks-old APP/PSEN1 AD transgenic (Tg) male mice were anesthetized with isoflurane and placed under a metal tube device where the opening was positioned directly over the animal’s head, just anterior to the right ear. mTBI was induced by dropping a blunted cylindrical metal weight (30 g) down the metal tube (inner diameter 13 mm) from a height of 80 cm. Phen was administered I.P. twice a day, starting 30 min after the injury (0.1 ml/10 g body weight in double distilled H2O). Using behavioral and immunohistochemical techniques, we evaluated Phen at 2 clinically translatable doses in separate cohorts of WT mice (2.5 and 5.0 mg/kg BID post mTBI) and in APP/PSEN1 mice at 5 mg/kg BID post mTBI. For behavioral studies, Phen was administered for 5 consecutive days following TBI, allowing a 2-day washout period prior to initiation of cognitive evaluation to ensure that possible cholinergic systemic effects were not a confound in the behavioral paradigms. Immunohistochemical and LTP studies were performed at 72 h following mTBI, with an 18 h washout period following the final Phen or vehicle dose (our prior studies have demonstrated that cholinesterase inhibition and cholinergic effects are negligible after these washout periods (Greig et al., 2000; Greig et al., 2005). Prior to mTBI induction, mice were randomly selected into mTBI and sham groups, and within these groups mice were then randomly selected to receive either Phen or vehicle treatment. Sham animals underwent the exact same procedure as mTBI challenged ones, except without the weight drop. Vehicle-treated mice received 0.1 ml/10 g body weight double distilled H2O on the same schedule as Phen-treated mice.
All studies were implemented under experimental protocols that were fully approved by either the Animal Care and Use Committee of the Intramural Research Program, National Institute on Aging (Baltimore, MD, USA) (Protocol No. 438-TGB-2019) or by the Sackler Faculty of Medicine Ethics Committee (Tel Aviv, Israel) (Protocol No. M-11–086). All animal study methods were performed in accord with the National Institutes of Health (DHEW publication 85–23, revised, 1995). The animal numbers evaluated in each assessment group (Table 1), and the selected experimental measures and times at which they were performed were based on our prior studies. An analysis of the variance of data from these prior studies was evaluated in order to minimize the numbers of animals used based on power calculations (α = 0.05 and 1-β = 0.80).
Table 1.
Animal numbers associated with experimental procedures and groups. Notably, there was no mortality or morbidity of mice across groups.
| Wild type mice | AD Tg mice | ||||
|---|---|---|---|---|---|
| NOR | Y-maze | IMHC | EPhysiol | IMHC | |
| Total No. | 50 | 56 | 20 | 25 | 15 |
| Control | 12 | 17 | 5 | 5 | 5 |
| mTBI | 16 | 18 | 5 | 7 | 5 |
| mTBI+ Phen 2.5 | 11 | 10 | 5 | - | - |
| mTBI+ Phen 5 | 11 | 11 | 5 | 8 | 5 |
| Phen alone | - | - | - | 5 | - |
NOR: Novel object recognition paradigm; IMHC: Immunohistochemistry; EPhysiol: Electrophysiology studies.
2.2. Behavioral tests of cognitive function
As previously reported, mice subjected to mTBI were indistinguishable from those subjected to the sham procedure when assessed at 1 or 24 h later in relation to a series of evaluations focused on health and well-being; subjective measures that included grooming and appearance, righting skills, ambulation, and blinking reflex were combined with objective ones comprising weight and body temperature (Baratz et al., 2015). An evaluation of body temperature was undertaken in pilot studies across animal groups prior to and following mTBI, and demonstrated no statistically significant change. The cognitive abilities of mice were then quantitatively assessed by the novel object recognition (NOR) and the Y-maze paradigms to evaluate visual and spatial memory, respectively, 7 days following mTBI challenge (for animal numbers see Table 1). All equipment used for behavioral testing was cleansed with 70% ethanol between testing sessions to eliminate any potential olfactory-dependent cognitive influences on mouse behavior.
2.2.1. NOR paradigm
The NOR paradigm evaluates recognition memory, as previously described (Messier, 1997; Baratz et al., 2011; Edut et al., 2011). This task is based on the tendency of rodents to explore unfamiliar objects within their environment. The use of this natural propensity allows quantitative assessment of whether a mouse is able to discriminate between a familiar and a novel object. Mice were individually habituated to an open field black Plexiglass box (59 × 59 × 20 cm size) for 5 min, 48 h prior to the test. 24 h later, animals were allowed to explore a set of two identical objects for a 5 min acquisition phase. On the day of the test, animals were presented with a set of objects in the same environment, where one object was novel to them. Each mouse was allowed to freely explore the objects again for a 5 min period. The times spent exploring the familiar and the novel objects were recorded.
2.2.2. Y maze paradigm
The Y maze paradigm evaluates spontaneous exploration, responsiveness to novel environments and spatial memory function, as previously described (Dellu et al., 1992; Baratz et al., 2010). The apparatus consisted of a three-armed black Plexiglass maze with each arm (8 × 30 × 15 cm size) separated by 120° and distinctly marked with a different spatial cue (a triangle, a square, or a circle). A start arm for each experiment was chosen randomly. Each mouse was introduced into the Y maze on two occasions separated by a 2 min interval, during which the mouse was returned to its home cage. During the first 5 min trial, one of the two arms was randomly blocked. During the second 2 min trial, all arms were open for exploration and the total amount of time the mouse explored within each arm was measured.
2.3. Immunohistochemical studies
At 72 h following mTBI or the sham procedure, animals were anesthetized with a combination of ketamine (100 mg/kg) and xylazine (10 mg/kg), and perfused transcardially with 10 ml phosphate buffered saline (PBS) followed by 20 ml of 4% paraformaldehyde (PFA) Brains were removed from the skull and post-fixed with 4% PFA overnight. Coronal sections from the dorsal hippocampus and the posterior parietal cortex were cut at 40-μm thickness and stored for immunohistochemical procedures. N = 5 per group (Table 1). This 72 h time was selected based on our prior studies demonstrating substantial degeneration and neuroinflammation at this interval (Tashlykov et al., 2007; Tashlykov et al., 2009; Deselms et al., 2016).
Across our studies in both WT and APP/PSEN1 mouse models, immunohistochemical evaluation of mTBI damage was undertaken in hippocampus and cortex ipsilateral to injury. The brain regions evaluated; specifically, the CA1, CA3 and dentate gyrus areas of the hippocampus and the posterior parietal cortex (cortex) are shown in Supplemental Fig. 1 in relation to a Figure plate schematic obtained from the Paxinos and Franklin’s mouse brain atlas (Paxinos and Franklin, 2012). To evaluate the existence of contrecoup damage, immunohistochemical evaluations also were undertaken in contralateral brain regions to allow direct ipsi- and contralateral comparisons (Supplemental Fig. 2).
2.4. Fluoro-Jade C (FJC) staining
The number of degenerating cells was assessed by using Fluoro-Jade C (FJC; Millipore, AG325- 30MG). Specifically, brain slices were air-dried on a slide warmer at 50 °C for at least 1 h, and then were immersed into a 1% sodium hydroxide and 80% ethanol solution for 5 min and into 70% alcohol for 2 min followed by 2 min in distilled water + PBS (pH 7.4, 50:50). Next, the slices were incubated with a solution of 0.06% potassium permanganate for 10 min, then washed and immersed into a 0.1% acetic acid and 0.01% FJC solution for 30 min. The slides were then rinsed, dried for 5–10 min, and incubated in xylene for 1 min. DPX non-fluorescent mounting medium was used for mounting slides. Finally, images of FJC-positive cells within the hippocampus and cortex were captured using a FITC filter on a confocal laser-scanning microscope (FV 1000, Olympus, Japan). The number of FJC-positive cells was analyzed and counted using NIH software Image J 1.43 m.
2.5. Immunofluorescence
For Iba1 and GFAP single staining and for Iba1/TNFα, MAP2/PSD-95 and MAP2/Synaptophysin (Syn) double staining, brain samples were immunoreacted for 48 h with the following primary antibodies: anti-Iba1 1:200 (goat polyclonal anti-Iba1, Abcam, USA, #ab5076), anti-GFAP 1:2500 (chicken polyclonal ant-GFAP, Abcam, USA, #ab4674), Iba1 plus anti- TNFα antibody 1:200 (rabbit polyclonal anti-TNFα, Abbiotec, USA, #251900); anti-MAP2 1:500 (mouse monoclonal anti-MAP2, Abcam, USA, #ab11267) plus anti-PSD-95 antibody (rabbit polyclonal anti-PSD-95, Thermo Fisher, USA, #51–6900); anti-MAP2 plus anti-Synaptophysin antibody (rabbit polyclonal anti-SYN, Thermo Fisher, USA, #MA5–14532). After PBS washing, sections were incubated with a donkey anti-goat secondary antibody AlexaFluor® 555-conjugated IgG (H + L) cross-adsorbed 1:400 (ThermoFisher, USA, #A-21432) for detection of Iba1, with a goat anti-chicken secondary antibody AlexaFluor® 488-conjugated IgY (H + L) 1:500 (Thermofisher, USA, #A-11039) for detection of GFAP, and with a goat anti-mouse secondary antibody AlexaFluor® 594-conjugated IgG (H + L) cross-adsorbed 1:400 (ThermoFisher, USA, #A-11005) for detection of MAP2. A three-step detection was used to increase the signal of TNFα, PSD-95 and Synaptophysin by use of biotinylated anti-rabbit IgG (H + L) 1:200 (Vector Labs., USA, #BA-1000) and streptavidin–-fluorescein 1:400 (Vector Labs., USA, #SA-5001). Controls consisted of omitting the primary antibody.
2.6. Laser scanning confocal microscopy
Imaging of immunofluorescence staining was performed using an Olympus FV1000MPE confocal laser scanning microscope. Confocal images were acquired by a PlanApoN, ×60, 1.42 oil objective, a × 40 or a × 20 objective.
2.7. Immunofluorescence analysis and quantification
Iba1 analysis: Iba1-positive microglia were identified at ×60, 1.42 oil magnification. For each animal, three fields of hippocampus (CA1, CA3 and DG) and one of cortex were captured from both the left and right hemispheres and analyzed. For each microglial cell, the body and primary processes were outlined. The mean area/cell was then measured using NIH software ImageJ 1.43 m.
GFAP quantification: For each animal, one field of hippocampus (CA1) and one of cortex were captured from both the left and right hemispheres using a × 63 oil objective and analyzed. The total area occupied by GFAP IR was quantified using NIH software ImageJ 1.43 m. The result is expressed as percentage of change vs. control group.
Iba1/TNF-α colocalization analysis: For each animal, three fields of hippocampus (CA1, CA3 and DG) and one of cortex were captured from both the left and right hemispheres and analyzed. The volume of colocalized elements was determined as follows: for each dataset (20–30 images, step size: 1 μm), a colocalization channel between the red channel for Iba1 IR and the green channel for TNF-α IR was automatically composed by Imaris 7.3. In the resulting stacks, the volume of the elements of interest was calculated, summed and expressed as volume/μm3.
PSD-95 quantification: Changes in the expression of the post-synaptic marker PSD-95 were evaluated by PSD-95/MAP2 double immunostaining. Confocal images were taken using a × 60, 1.42 oil objective (x1.8 zoom). For each animal, n = 20 MAP2 positive dendritic segments (about 10 um of length) from each field of hippocampus and cortex were randomly chosen and the PSD-95 positive (+) spines along the dendrite were manually (Imaris 7.3) counted. The result is expressed as spine density/μm.
Synaptophysin quantification: For the pre-synaptic marker Synaptophysin, three fields of hippocampus and one of cortex were captured from both the left and right hemisphere, and then analyzed. The total volume occupied by Syn immunoreactivity was automatically quantified using Imaris 7.3 software. Results are expressed as IR Volume (μm3).
2.8. Long Term Potentiation (LTP) evaluation in hippocampal slices
For slice preparation, mice were anesthetized with isoflurane and decapitated at 72 h post TBI or sham procedure. The brain was rapidly removed and submerged in a modified artificial cerebrospinal fluid (m-aCSF) containing, in millimolar, NaCl 92; KCl, 2.5; NaH2PO4, 1.2; NaH2PO4 1.2; NaHCO3, 30; HEPES, 20; glucose, 25; sodium ascorbate, 5; sodium pyruvate, 3; MgCl2, 10; CaCl2, 0.5. Transverse hippocampal slices (280 μm) were prepared using a vibrating tissue slicer (Leica VT1200S, Leica Biosystems, Nussloch, Germany), and then stored in a holding chamber containing standard aCSF (containing, in millimolar, NaCl, 126; KCl, 3; CaCl2, 2.4; MgCl2, 1.5; NaH2PO4, 1.2; NaHCO3, 26; and glucose, 11). Caffeine (50 μM) was added in order to limit the influence of endogenous adenosine on synaptic transmission. After a 20 min incubation at 35 °C, slices were allowed to gradually equilibrate to room temperature for at least 30min prior to initiating recording. All solutions were constantly oxygenated with 95% O2/5% CO2. On average, n = 4 slices were used from a single animal per day with N = 13 slices total per group on average. Table 1 provides a breakdown of animal No. evaluated across groups.
Hippocampal brain slices were submerged in a chamber (RC-26, Warner Instruments, Hamden, CT, USA), and continuously perfused with aCSF (2ml/min) using a peristaltic pump (Cole-Parmer Instruments, Vernon Hills, IL, USA). Bath temperature was maintained at 30–32 °C by passing the aCSF through an in-line heater (TC324-C and SH27-B, Warner Instruments). Borosilicate glass electrodes (1.5mm o.d. × 0.86 mm i.d., Sutter Instruments, Novato, CA, USA) were fabricated using a horizontal puller (P-97, Sutter Instruments) and filled with aCSF. Electrodes were connected to the headstage of an AC amplifier (Model 1800, A-M Systems, Sequim, WA, USA). The tip of a bipolar-stimulating electrode consisting of twisted formvar-insulated nichrome wire (50 μm diameter; A-M Systems), connected to a constant current stimulus isolation unit (DS3, Digitimer LLC, Ft. Lauderdale, FL), was positioned in hippocampal area CA3 to activate Schaffer collateral axons forming synapses on CA1 pyramidal neuron dendrites. The recording electrode was positioned in CA1 stratum radiatum using a manual micromanipulator, and gradually lowered while monitoring field excitatory postsynaptic potential (fEPSP) responses, so that the largest fEPSP for a given stimulus intensity was obtained. Recording electrodes were fixed at this depth within the slice and input–output (I/O) curves relating fEPSP amplitude to stimulus intensity (20–200 μA, 0.1 ms) were generated. Baseline fEPSP responses of 30–50% of maximum were then obtained using a stimulus intensity determined from the I/O relationship (which did not indicate any statistically significant differences among groups, data not shown). Field EPSPs were then obtained every 30 s for at least 10 min during the baseline period. After recording a stable baseline, high frequency stimulation (HFS; comprised of three consecutive 100-Hz, 1-s trains, delivered 10s apart) was delivered, and then fEPSP responses were continuously monitored at 30 s intervals. The stimulation, data acquisition and signal analyses were performed online using an A/D board (PCIe-6321, National Instruments, Austin, TX, USA) and WinLTP software (https://www.winltp.com/; Anderson and Collingridge, 2007).
2.9. Analysis and statistics
All behavioral results are given as mean ± S.E.M values and were analyzed by SPSS V24. A paired t-test was used to determine significance between the familiar and novel object/arm within each group. Statistical significance was set at p < .05.
All immunohistochemistry/immunofluorescence results are shown as mean ± S.E.M values. The data from the analysis of FJC+ cells, IBA1-IR, GFAP-IR and IBA1/TNFα colocalization, PSD-95 and Synaptophysin in WT mice were statistically analyzed with a one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. Due to the greater variance across the broader age range among APP/PSEN1 mice, a Mann-Whitney test was used to analyze data from these markers in AD Tg mice. The level of significance was set at p < .05.
For LTP measurement, data are likewise presented as mean ± S.E.M values. Comparisons among experimental groups were made using ANOVA tests, with a critical value for statistical significance set at p < .05. A Dunnett’s multiple comparisons test was used to measure the mean level of LTP between 50 and 60 min (i.e., last 10 min of recording), following HFS.
3. Results
3.1. mTBI challenge and (−)-Phenserine were well tolerated across groups of mice
Mice exposed to mTBI and/or Phen were indistinguishable from sham animals when appraised at 1 and 24 h for evaluations of well-being; these consisted of subjective measures such as grooming and appearance, righting skills, ambulation, and blinking reflex combined with body weight measurements (Baratz et al., 2015). Additionally, no adverse or visually evident pharmacological actions were noted for either dose of Phen (2.5 and 5.0 mg/kg BID) throughout the study. Finally, there was no mortality or evident morbidity across all groups of sham and mTBI challenged mice.
3.2. (−)-Phenserine treatment attenuated mTBI-induced cognitive deficits
The NOR paradigm was performed to probe for impairments in visual memory in WT mice 7 days following mTBI challenge and 5 day Phen treatment. As illustrated in Fig. 1A, sham-mice spent a significantly longer time exploring the novel compared to the familiar object (p < .001), whereas mTBI-challenged mice failed to differentiate between the two items. mTBI-mice subsequently treated with Phen performed in a manner similar to the sham group and spent significantly more time exploring the novel object (2.5 mg/kg: p < .05; 5 mg/kg: p < .001), showing mitigation of the mTBI-induced visual memory impairment.
Fig. 1.
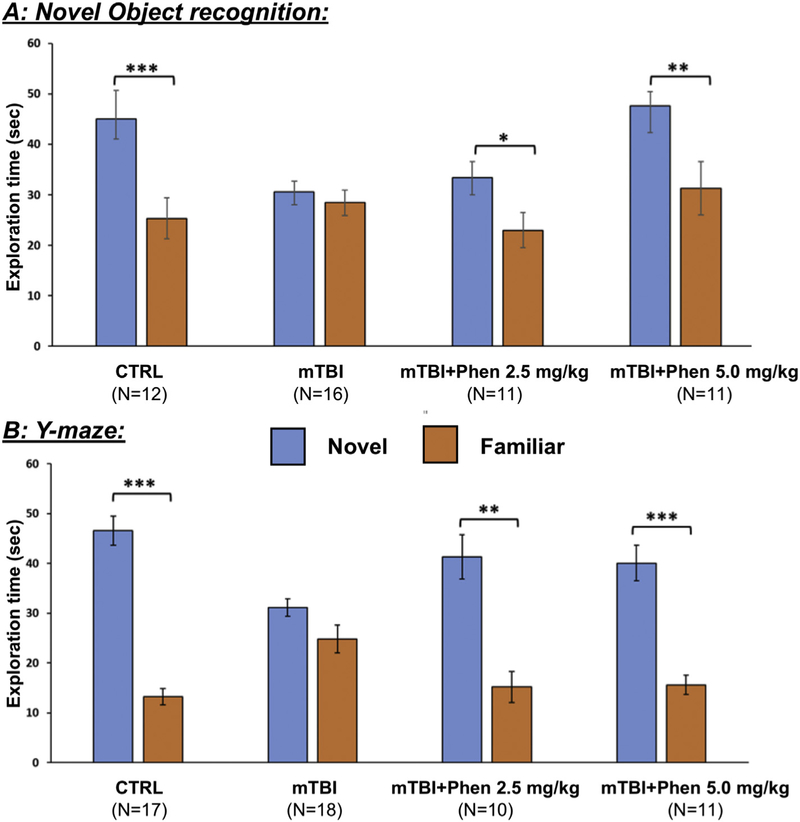
Post-injury treatment with Phen mitigates mTBI-induced cognitive impairments, as evaluated by NOR and Y-maze paradigms 7 days following injury. mTBI challenge induced significant behavioral deficits in NOR (A) and Y-maze (B) with mTBI-vehicle mice failing to differentiate between the novel and familiar objects or arms, respectively. In contrast, animals treated with Phen (2.5 and 5 mg/kg, BID x 5 days) explored the novel object/arm for significantly longer than the familiar one, similar to sham animals. Data are presented as mean ± S.E.M. of N observations, *p < .05, **p < .01, ***p < .001 comparing the novel to familiar object/arm.
The Y-maze paradigm was implemented to evaluate the spatial memory of the same mice following mTBI vehicle or Phen treatments. Sham-mice demonstrated a significant preference for the novel over the familiar arm of the maze (p < .001), whereas mice subjected to mTBI and treated with vehicle (mTBI-Veh) explored the novel and familiar arms with equal preference. In contrast, mTBI mice receiving Phen treatment showed no impairment in spatial memory, performing similar to sham-mice (2.5 mg/kg: p < .01; 5 mg/kg: p < .001) (Fig. 1B).
3.3. (−)-Phenserine prevented mTBI-induced neurodegeneration in the hippocampus and lateral cortex across groups of mice
To evaluate mechanisms underpinning mTBI-induced cognitive impairments and its remediation, cellular loss was quantified in ipsilateral brain 72 h following mTBI injury. Evaluation of FJC+ cells in WT mice demonstrated a significant increase in the number of degenerating neurons following mTBI/vehicle treatment across all brain areas studied, as compared to control (CTRL) sham-treated mice (p < .001 in CA1, CA3 and DG; p < .0001 in CTX). Treatment with Phen (5 mg/kg, BID) fully mitigated mTBI-induced neurodegeneration across all brain areas (Fig. 2A; p < .01 CA3 and DG; p < .001 CA1 and CTX). In APP/PSEN1 mice, the number of degenerating cells was consistently greater across all the experimental groups, as compared to WT mice (data not shown). After mTBI, a further elevation of FJC+ cells vs. the control (sham) group was evident, reaching statistical significance in area CA1 of the hippocampus (p < .05). Similar to WT mice, treatment of mTBI APP/PSEN1 mice with Phen (5 mg/kg, BID) resulted in a degeneration cell count in both the hippocampus and cortex that was not significantly different from control (sham) mice (Fig. 2B).
Fig. 2.

Phen reverses mTBI-induced neuronal loss in WT and APP/PSEN1 mice. Degenerating neuronal cells were quantified using the marker Fluorojade C (FJC: green). (A) An increased number of FJC+ cells were observed in vehicle administered mTBI (mTBI-VEH) vs. sham control (CTRL) WT mice across the hippocampus (CA1, CA3 and DG) and cerebral cortex (***p < .001, ****p < .0001 vs. CTRL by Tukey’s post hoc test). (B) A similar elevation was induced by mTBI in APP/PSEN1 vehicle administered mice that reached significance in the hippocampus (#p < .05 vs CTRL by Mann-Whitney rank test in CA1). Post treatment with Phen abated the neuronal loss, in which FJC+ cell counts were no different from values of sham control (CTRL) mice. Importantly in WT animals, values in the Phen 5 mg/kg group were significantly lower than the mTBI vehicle group (^^p < .01, ^^^p < .001 vs. mTBI by Tukey’s post hoc test). Representative images and graphs from hippocampus and cortex. Data shown as mean ± S.E.M. Scale bar = 30 μm.
3.4. (−)-Phenserine treatment of mTBI mice inhibited microglial and astroglial activation in the hippocampus and cortex of WT and AD mice
To evaluate the role of neuroinflammation on mTBI-mediated cognitive impairment and cellular loss, microglial activation state was appraised by quantifying IBA1 immunoreactivity (IR; a microglial cell marker) and TNF-α (Fig. 3). Following mTBI injury, IBA1-IR was increased in mTBI vehicle vs. control (sham) mice, in all analyzed brain regions of WT animals (p < .05 in CA1, p < .0001 in DG, CA3 and CTX) and in the CA1 region of the hippocampus of APP/PSEN1 mice (p < .05). In this regard, IBA1-IR was measured as the specific area occupied by IBA1+ cells in the analyzed brain regions. In the control group, microglial cells displayed a resting morphology with a small soma, and long and thin processes. In contrast, microglia showed an activated morphology in response to mTBI, characterized by a larger body with shorter and thicker processes. In WT mice, Phen (2.5 and 5 mg/kg BID) inhibited microglial activation throughout the hippocampus and cortex. In AD transgenic mice, a similar trend was evident, reaching statistical significance in the cortex (p < .05).
Fig. 3.

Phen ameliorates mTBI-induced neuroinflammation in WT and APP/PSEN1 mice. (A) Following mTBI injury, ionized calcium binding adaptor molecule 1 (IBA1) immunoreactivity (IR) (a microglia/macrophage-specific marker) was increased in mTBI vehicle vs. sham control (CTRL) mice, in all analyzed brain regions of WT animals and in the hippocampus of APP/PSEN1 mice. In contrast, IBA1 IR values throughout hippocampal and cortical areas for mTBI challenged mice administered Phen were not statistically different from their respective sham (CTRL) groups across WT and APP/PSEN1 mice. In mTBI-challenged WT mice, Phen (2.5 and 5 mg/kg BID) significantly lowered microglial activation throughout hippocampus and lateral cortex compared to the vehicle group. In APP/PSEN1 mTBI mice, this was achieved by Phen in cortex. IBA1 IR was measured as the area occupied by IBA1+ cells in the analyzed brain regions. Graphs from hippocampus (CA1 for APP/PSEN1 mice) and cortex.
(B) Glial fibrillary acidic protein (GFAP) IR (an astrocyte marker) was significantly increased in hippocampus and cortex of mTBI-challenged WT mice, and a similar trend, although not statistically significant, was evident in APP/PSEN1. Treatment with Phen fully reversed this GFAP IR increase in hippocampus and cortex of WT mice. A similar trend that did not reach statistical significance was evident in APP/PSEN1 transgenic animals. GFAP IR was quantified as total area occupied by GFAP + fluorescent signal in hippocampus and cortex. Representative confocal images from hippocampus - CA1 of WT mice. Graphs from hippocampus - CA1 and cortex. Scale bar = 100 μm.
*p < .05, **p < .01, ****p < .0001 vs. CTRL by Tukey’s post hoc test; ^p < .05, ^^p < .01, ^^^p < .001, ^^^^p < .0001 vs. mTBI by Tukey’s post hoc test.
#p < .05 vs. CTRL by Mann-Whitney rank test; @p < .05 vs. mTBI by Mann-Whitney rank test. Data shown as mean ± S.E.M.
Astroglial activation was evaluated by measuring the total area occupied by GFAP-IR in the hippocampus (CA1) and cortex (Fig. 3B)). Within the mTBI group, GFAP-IR was increased as compared to control (sham) mice in the hippocampus (CA1) and cortex of WT mice (p < .01 in CA1, p < .05 in cortex); a similar trend was evident in APP/PSEN1 mice, although it did not reach statistical significance. In WT mice, treatment with Phen (2.5 and 5 mg/kg BID) fully mitigated the astroglial activation induced by mTBI throughout the hippocampus and cortex. In APP/PSEN1 mice, an analogous trend for Phen was observable in both the CA1 and cortex (p = .0519 in CA1).
3.5. (−)-Phenserine inhibited the mTBI-induced production of TNF-α in microglial cells in WT and AD mice
The immunoreactivity (IR) for the pro-inflammatory cytokine TNF-α was increased within IBA1+ cells in the mTBI-vehicle group, as compared to the control (sham) group, in the hippocampus (p < .01 in CA1 and DG, p < .001 in CA3) and cortex (p < .0001) of WT, as well as in the cortex of APP/PSEN1 (p < .01; p = .057 in hippocampus) (Fig. 4). Notably, administration of Phen (2.5 and 5 mg/kg, BID) reduced the levels of IBA1/TNF-α IR co-localization volume across hippocampal (DG and CA3) and cortical areas in WT mice; Phen-treated AD mTBI animals, likewise, were not significantly different from the AD sham control group. Results are expressed as percentage of co-localization of TNF-α IR in IBA1+ cells.
Fig. 4.

Phen inhibits mTBI-induced TNF-α generation within activated microglial in WT and APP/PSEN1 mice. mTBI induced an elevation in immunoreactivity for the pro-inflammatory cytokine TNF-α within IBA1+ cells across all analyzed brain areas in WT animals (Fig. 4A) and in the cortex of APP/PSEN1 mice (Fig. 4B), as evaluated by TNF-α/Iba1 co-localization. By contrast, mTBI Phen-treated mTBI mice had values no different from the sham (CTRL) group. Administration of Phen (2.5 and 5 mg/kg, BID) reduced the levels of IBA1/TNF-α IR co-localization volume across all hippocampal and cortical areas in WT mTBI-challenged mice, as compared to the mTBI vehicle group. Representative confocal images showing co-localized elements (yellow) in IBA1 (red) positive cells in cortex. Percentage of co-localization of TNF-α IR in IBA1 positive cells. **p < .01, ***p < .001, ****p < .0001 vs. CTRL by Tukey’s post hoc test; ^^p < .01, ^^^^p < .0001 vs. mTBI by Tukey’s post hoc test. ##p < .01 vs. CTRL by Mann-Whitney rank test. Data shown as mean ± S.E.M. Scale bar = 30 μm.
3.6. (−)-Phenserine mitigated the mTBI-induced loss of pre- and post-synaptic proteins in WT and AD mice
To define changes in pre- and post-synaptic elements potentially underpinning mTBI-induced cognitive impairment, levels of PDS-95 and synaptophysin were evaluated in the hippocampus and cerebral cortex ipsilateral to injury. The post-synaptic marker PSD-95 was measured by counting PSD-95+ dendritic spines in MAP2+ dendrites (expressing data as the number of PSD-95+ dendritic spines/μm). The mTBI vehicle group showed a significant loss in the number of PSD-95+ dendritic spines in WT mice (p < .0001 in hippocampus-CA1 and cortex) as well as in APP/PSEN1 animals (p < .01 in hippocampus-CA1). Treatment with Phen (2.5 and 5 mg/kg) reversed spine loss in WT mice in a dose-dependent manner across both regions; the same trend was seen in mTBI-challenged AD mice (Fig. 5A).
Fig. 5.

Phen mitigates mTBI-induced loss of pre- and post synaptic elements in WT and APP/PSEN1 mice.
(A) Postsynaptic density protein 95 (PSD-95): mTBI induced a loss of PSD-95+ dendritic spines across all analyzed areas in WT mice, and in hippocampus of AD mice, as compared to the sham (CTRL) group. By contrast, Phen treated mTBI mice were not statistically different from the sham (CTRL) group. WT mTBI mice treated with Phen (2.5 and 5 mg/kg) possessed a greater number of PSD-95+ dendritic spines across both hippocampus and cortex, as compared to the mTBI vehicle group. Representative images showing PSD-95+ spines (green) in MAP2+ dendrites. Data are expressed as number of PSD-95+ dendritic spines/μm.
(B) Synaptophysin: the total volume occupied by the presynaptic marker synaptophysin IR was evaluated across WT and APP/PSEN1 mice and found to be significantly reduced in the mTBI vehicle group, as compared to their respective sham (CTRL) group. In contrast, mTBI Phen treated mice had synaptophysin IR levels no different from sham (CTRL) mice. Importantly, Phen treatment of mTBI-challenged mice resulted in significantly higher amounts of synaptophysin IR, compared to the mTBI vehicle group, across all analyzed brain areas in both WT and AD mice. *p < .05, **p < .01, ****p < .0001 vs. CTRL by Tukey’s post hoc test; ^p < .05, ^^p < .01, ^^^^p < .0001 vs. mTBI by Tukey’s post hoc test. ##p < .01 vs. CTRL by Mann-Whitney rank test. Data shown as mean ± S.E.M. @@p < .01 vs. mTBI by Mann-Whitney rank test. Data shown as mean ± S.E.M. Scale bar = 20 μm.
Changes in the expression of the pre-synaptic protein, synaptophysin, were evaluated in terms of total volume (μm3) occupied by synaptophysin-IR, whose hippocampal and cortical expression was significantly reduced in WT and AD animals challenged with mTBI (p < .01 in CA1 and p < .05 in cortex of WT; p < .01 in CA1 and cortex of APP/PSEN1). Administration of Phen at the dose of 5 mg/kg fully mitigated mTBI-induced synaptophysin IR reductions in both regions in WT and APP/PSEN1 mice (p < .05 and p < .01, respectively).
3.7. Contrecoup injury and mitigation by (−)-Phenserine
To evaluate potential contrecoup injury, FJC+ cells and markers of neuroinflammation (IBA1, IBA1/TNF-α co-localization volume) and synaptic integrity (PDS-95, synaptophysin) were quantified in the hemisphere contralateral to mTBI injury. Notably, a similar elevation across all markers was evident within each of the brain areas evaluated on ipsilateral and contralateral sides; specifically, there was no statistical difference between the mTBI-induced damage between the ipsilateral and contralateral brain areas. Furthermore, as reported above for brain areas ipsilateral to mTBI, Phen provided similar amelioration on the contralateral side (Supplemental Fig. 2).
3.8. (−)-Phenserine mitigated mTBI-induced LTP impairment in mice
To define possible physiological consequences of mTBI-mediated reductions in pre- and post-synaptic markers, we evaluated LTP in hippocampal brain slices obtained from sham and mTBI mice treated with either vehicle or Phen (5 mg/kg, BID). To minimize cholinergic effects of Phen on LTP, all slices were prepared 18 h after the final Phen treatment (in line with cholinesterase inhibition and cholinergic actions being negligable (Greig et al., 2000; Greig et al., 2005)). In sham (CTRL) brain slices, HFS caused a stable increase in fEPSP slope over a 60 min time course. In contrast, LTP was significantly smaller in slices obtained from mTBI-Veh mice (Fig. 6A–B), relative to the sham (CTRL) group (p < .001, Fig. 6B). Importantly, treatment with Phen mitigated this LTP deficit, with the mTBI+Phen animals demonstrating post-HFS fEPSP slopes that were not statistically different from the sham (CTRL) group (p = .1503). The action of Phen in sham mice was similarly evaluated and found to have no impact on LTP compared to sham vehicle (CTRL) animals (p = .6579, Fig. 6B). Additionally, the I/O (input-output) curves were not different across treatments groups.
Fig. 6.

Phen mitigates mTBI-induced deficits in LTP in WT mice.
(A) Time course of the mTBI-induced impairment in LTP (initiated by HFS at time = 0 on the x-axis) across the 60 min time course. The fEPSP slope is expressed as a % of the control baseline, collected prior to HFS. (B) LTP magnitude, evaluated over the last 10 min of recording in each of the groups shown in A. (C) Representative averaged traces of a control slice, a slice from a mTBI mouse, a slice from a mTBI+Phen 5 mg/kg mouse, and a slice from control mouse treated with 5 mg/kg Phen showing the mitigating effect of Phen treatment on the mTBI-induced LTP impairment. Traces are averaged from the period immediately prior to (pre) and 60min following (post) high-frequency stimulation. LTP in mTBI-Veh mice was significantly decreased as compared with the sham (CTRL) group. Note that treatment of mTBI-challenged mice with Phen (5 mg/kg, BID) mitigated the LTP impairment, and that the mTBI Phen group did not statistically differ from sham (CTRL) animals. Data shown as mean ± S.E.M. ***p < .001, one-way ANOVA (F (3,49) = 6.053), and Tukey’s post-hoc test.
4. Discussion
TBI is extremely common, with a lifetime prevalence of up to 40% among adults (Whiteneck et al., 2016), appears to be increasing in occurrence and is a leading cause of disability and death worldwide. Some 2.8 million Americans seek medical attention for TBI yearly (Taylor et al., 2017) at an annual cost in excess of $76 billion (CDC Grand Rounds: Reducing Severe Traumatic MBrain Injury in the United States [WWW Document], 2013). Unfortunately, current TBI treatment options are limited, and the development of efficacious drugs to mitigate the broad range of mTBI-mediated impairments continues to be a critical and unmet current medical need. In the present study, we evaluated the AD drug candidate Phen, since our prior studies demonstrated its ability to mitigate many of the cascades associated with TBI. This included glutamate excitotoxicity and oxidative stress, as well as the down regulation of TBI-induced gene pathways that lead to neurodegeneration (Hoffer et al., 2017; Tweedie et al., 2016b). Here we demonstrate that Phen ameliorates key aspects of neuroinflammation and synaptic and neuronal loss following mTBI in both WT and aged AD transgenic mice. Moreover, the beneficial effects of Phen on these neuronal markers extended to an improvement in synaptic function and plasticity, as LTP and cognitive impairments were fully mitigated by Phen treatment after mTBI. Our studies reinforce the significant role of neuroinflammation and both synaptic changes and neuronal loss in the degenerative processes, and support the possibility of reversing such neuropathology with appropriate intervention in humans.
Despite substantial progress in elucidated the pathophysiology of TBI and dissecting the molecular cascades underpining the neuronal dysfunction and death evident in the secondary phase of TBI-induced damage (e.g., glutamate excitotoxicity, ischemia, intracellular calcium dysregulation, oxidative stress, neuroinflammation, and other factors), identifying and pharmacologically targeting such mechanisms has not yet translated into clinical improvement for humans challenged with TBI (Janowitz and Menon, 2010; Loane and Faden, 2010; Bolouri and Zetterberg, 2015; Hawryluk and Bullock, 2016). Despite the investment of billions of dollars into dozens of Phase III clinical trials for TBI, not a single neuroprotective agent has entered into clinical practice (Stein and Sayeed, 2018), representing a 100% clinical drug development failure rate. Among many responsible factors that likely account for this failure is that the molecular pathology mechanisms that have directed ‘target-based’ TBI drug screening likely do not appear singularly, and their temporal profiles and outcomes almost certainly differ between preclinical models and human TBI. The emerging consensus is that multiple TBI-associated cascades are triggered in parallel and, if true, the multifactorial nature of TBI would make the discovery of a single effective mechanism-targeted drug improbable. To counter this, phenotypic screens can be used in drug development to identify compounds to mitigate a critical step associated with TBI, such as neuronal cell death, which is agnostic to any specific mechanism(s) underpinning this event. Thus, appropriate phenotypic-screening can identify drugs effective against a range of mechanisms, rather than against a single molecular target as occurs in target-based drug discovery screening (Swinney, 2013). This advantage of phenotypic drug screening may account for it being associated with the highest number of first-in-class drug approvals by the FDA between 1999 and 2008 (Swinney and Anthony, 2011).
Phen was originally developed through target-based screening as an anticholinesterase with additional non-cholinergic mediated amyloid precursor protein/amyloid-β peptide (APP/Aβ) lowering actions as a treatment for AD (Greig et al., 2005). The agent proved well-tolerated, was safely administered to 645 human subjects for up to and beyond one year with no drug associated severe adverse events and, when administered at therapeutic doses, demonstrated efficacy in AD (Winblad et al., 2010; Kadir et al., 2008; Nordberg et al., 2015). Notably, more recent preclinical studies support Phen as a compound of interest in phenotypic-based screens to mitigate neuronal cell dysfunction/death instigated by glutamate and oxidative stress in primary and human immortal neuronal cultures (Lilja et al., 2013; Hoffer et al., 2017). Cellular and animal studies demonstrated Phen’s direct activity against mechanisms associated with programmed (apoptotic) cell death. Specifically, Phen mitigated neuronal cell death, lowered proapoptotic proteins (Bax and activated-caspase 3) as well as APP, and elevated levels of pro-survival proteins (Bcl2 and BDNF) in cellular and animal models of ischemia/anoxia; thereby, mitigating behavioral impairments in the animals (Chang et al., 2017). Phen, likewise, mitigated programmed neuronal cell death in rodents challenged with soman toxicity, and increased survival by upregulating a core set of neuroprotective genes termed Activity-regulated Inhibitor of Death (AID) genes (Chen et al., 2014). In line with this, in the present study Phen (5 mg/kg) substantially mitigated mTBI-induced cellular degeneration, as evaluated by FJC. Although not exclusive, FJC predominantly labels degenerating neuronal cells (Schmued et al., 2005). Notable in Fig. 2, the mTBI-induced cellular degeneration was diffuse and abated by Phen in not only young WT but also aged APP/PSEN1 mice. Cellular loss was evident across the ipsilateral and, contrecoup, contralateral brain, and both were equally mitigated by Phen.
Synaptic proteins perform a critical role in optimizing neuronal cell signaling function by maintaining normal dendritic morphology and numbers of spines. Alterations in synaptic markers in hippocampus and cortex have been demonstrated in several animal models of TBI (Rachmany et al., 2017; Merlo et al., 2014; Ansari et al., 2008; Przekwas et al., 2016; Joo et al., 2018) as well as in AD, in both preclinical and clinical studies (Cambon et al., 2000; Heinonen et al., 1995; Selkoe, 2002; Berezcki et al., 2016). TBI, in particular, has been reported to acutely induce extensive dendritic de-arborization and a reduction in synapse number (Winston et al., 2013; Wen et al., 2017). Such a decline in synaptic strength has been proposed to underpin, at least in part, TBI mediated impairments in LTP and cognition (Liu et al., 2017). Indeed, synaptic loss, rather than either neurodegeneration or the formation of amyloid pathology, has been demonstrated to best associate with cognitive impairment (Selkoe, 2002; D’Amelio et al., 2011; Marchetti and Marie, 2011). As markers of neuronal connectivity, levels of the pre- and post-synaptic elements, synaptophysin and PSD-95, were quantified in our mouse study and were consistently reduced within hippocampus and cerebral cortex following mTBI. Synaptophysin, a calcium-binding protein localized to the membrane of presynaptic vesicles and implicated in vesicular trafficking, docking, synaptic reorganization and synaptogenesis (Südhof, 1995; Valtorta et al., 2004; Kwon and Chapman, 2011), has been widely employed as a marker to quantify synapse number following injury (Feng et al., 2016). In contrast, PSD-95 is richly expressed on post-synaptic synapses and has a regulatory role in ion-channel function, synaptic maturation, dendritic spine formation, and its levels, like synaptophysin, impact learning and memory deficits following TBI (Feng et al., 2016), in large part, by affecting the stabilization of synaptic alterations in LTP (Ansari et al., 2008; Ehrlich et al., 2007; Meyer et al., 2014). Notably, in both WT and APP/PSEN1 AD mice the post-mTBI administration of Phen consistently mitigated injury-induced losses of both these pre- and post-synaptic markers.
LTP entails the long-lasting enhancement of synaptic strength after repetitive activation of central glutamatergic synapses (reviewed by Nicoll, 2017), and is important for hippocampal processing and the encoding of memory (Lynch, 2004). Moreover, LTP impairment correlates with memory deficits in rodents (Morris, 2003). To examine the association between changes in synaptic protein expression and cellular mechanisms of learning and memory related to mTBI-induced cognitive impairment observed in our behavioral experiments, we evaluated hippocampal LTP in these animals. In accord with the literature, mice treated with vehicle demonstrated an impairment in the expression of LTP, 72 h after mTBI, coinciding with mTBI-induced declines in synaptic proteins (Ben Shimon et al., 2017; Chen et al., 2018). Moreover, in line with the changes in synaptic proteins, Phen mitigated the mTBI-induced LTP deficit, suggesting a direct functional consequence of these molecular alterations. Also, as Phen was without effect on LTP in sham controls, it appears that the rescue of LTP is related to the effects of Phen on the TBI-induced damage, and not to a non-specific enhancement of synaptic function.
The presence of activated microglial and astrocytic cells, a hallmark of neuroinflammation, has been reported across animal models of TBI (Acosta et al., 2013; Perez-Polo et al., 2013; Chiu et al., 2016; Bader et al., 2019; Glushakova et al., 2018). Consistent with this, mTBI induced an elevation in IBA1-IR and GFAP, markers of microglia and astrocytes, respectively, throughout the hippocampus and adjoining cerebral cortex. In contrast to the typical resting morphology of microglia with small somata, and long, thin processes in control brain, mTBI-related microglia manifested larger cell bodies and shorter, thicker processes characteristic of an activated state (Kreutzberg, 1996). Levels of the co-localized pro-inflammatory cytokine TNF-α, a chief mediator of acute neuroinflammation, likewise were elevated by mTBI (Fig. 4). TNF-α plays a critical role in triggering the activation of surrounding glial cells (Shohami et al., 1996; Kirkley et al., 2017; Liddelow et al., 2017). These can then generate and release additional TNF-α, nitric oxide, IL-1β as well as trigger the complement cascade to further exacerbate neuronal cell dysfunction, tissue injury and ultimately programmed neuronal cell death (Lynch et al., 2004; Craft et al., 2005; Chiu et al., 2016; Kirkley et al., 2017; Liddelow et al., 2017; Rizzo et al., 2018). Heightened expression of TNF-α mRNA and protein has been reported within 30 min of injury and to last for up to 24 h in TBI rodent models (Vitarbo et al., 2004). These changes often resolve in that time (Baratz et al., 2015; Woodcock and Morganti-Kossmann, 2013), but in some cases can last longer still up to 1–7 days post-TBI (Su et al., 2014; Tsai et al., 2015). Likewise, in humans, elevations in TNF-α appear to precede and trigger the appearance of other inflammatory cytokines, and have been reported as early as 17 min after injury in patients who die shortly after TBI (Frugier et al., 2010). Sustained elevation of TNF-α levels have also been reported in human CSF and serum at 24 h and later, for up to 3 weeks, following TBI (Woodcock and Morganti-Kossmann, 2013).
Several lines of evidence support elevated TNF-α acting as the primary cytokine aberrantly impacting both LTP and synaptic scaling during injury (Rizzo et al., 2018), and providing the molecular underpinning to cognitive impairment. TNF-α links the neuroinflammatory and excitotoxic processes across neurodegenerative disorders, and has been reported to enhance glutamate-mediated cytotoxicity by two parallel routes effected initially, by directly inducing the surface expression of Ca+2 permeable-AMPA receptors and NMDA receptors, and concomitantly by reducing inhibitory GABAA receptors on neurons. Indirectly, TNF-α inhibits glutamate transport in astrocytes (Fine et al., 1996). Through these mechanisms, heightened TNF-α changes the balance between synaptic excitation and inhibition (Stellwagen et al., 2005; Ogoshi et al., 2005) and, consequent to excessive Ca input into neurons, can induce neuronal cell dysfunction and death. This can create disproportionately elevated reactive oxygen species that disrupts glutamate transport in surrounding astrocytes, and thereby augments further neuronal cell dysfunction and microglial activation. Phen can likely mitigate this process at multiple levels; it lowers mTBI-induced microglial activation and TNF-α generation, as in the current study, and reduces both glutamate- and oxidative stress-mediated neuronal cell death (Lilja et al., 2013; Tweedie et al., 2016a).
A caveat in our study is that we evaluated only male mice. Albeit that the human incidence of TBI is two-fold higher in males vs. females and the former are more likely to under report an injury event, there is a growing literature indicating gender differences in post-injury outcomes and prognosis (Mollayeva et al., 2018). Whereas the female sex hormones oestrogen and progesterone are considered neuroprotective and both gender- and age-related differences in the expression of other neuroprotective and neurotrophic factors have been described, there are also mixed reports of differences in immune and neuroinflammatory responses as well as behavioral outcomes between males and females in preclinical and clinical studies (Caplan et al., 2017; Mollayeva et al., 2018). This warrants further analyses of treatments, such a Phen, in female preclinical models. Also, to be appreciated is that most animal models are limited, in that they do not mirror the mainstream pattern and duration of TBI challenge in humans. Nevertheless, the signal of efficacy of Phen in the current mTBI study provides impetus for further investigation.
In the light of the many drugs that demonstrate activity in a rodent model of TBI but then fail to show efficacy in humans, we additionally evaluated the ability of Phen to mitigate mTBI-induced changes in aged AD Tg mice. We hypothesized that these mice possess a particularly adverse brain microenvironment that may have more parallels to elderly humans (Bickford et al., 2017; von Bernhardi et al., 2015; d’Avila et al., 2018), and that TBI-induced changes would prove more of a challenge to mitigate (Becker and Greig, 2019). Although not a focus of the present study and not directly compared, aged AD Tg sham mice appeared to have a heightened level of neuroinflammation in contrast to that evident in younger WT mice in relation to IBA1-IR and colocalization with TNF-α. Similar to WT animals, Phen mitigated mTBI-induced elevations in neuroinflammatory markers in aged AD Tg mice.
In addition, Phen (2.5 mg/kg) was recently reported to mitigate neuronal loss, neuroinflammation and behavioral impairments in mice following controlled cortical impact-induced moderate TBI (Hsueh et al., 2019); thereby demonstrating activity across TBI preclinical models. Phen 2.5 and 5 mg/kg BID doses, evaluated in the present study, are translatable to a clinical dose following appropriate body surface area normalization across species based on FDA guidelines (U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER), 2005).
In summary, with a brain/plasma concentration ratio of 8:1, and primary metabolites possessing a ratio greater than unity (Greig et al., 2005), Phen can readily enter the brain irrespective of whether or not mTBI-induced changes in blood-brain barrier permeability occur. Following recent TBI clinical drug development failures, it has been proposed that either relatively “dirty” drugs possessing several mechanisms of action or combinations of multiple drugs with discrete but complementary mechanisms could provide a more favorable treatment approach (Janowitz and Menon, 2010). Phen appears to fit this guideline and, at clinically translatable doses, mitigates mTBI-induced neuronal cell death, losses in synaptic connectivity and neuroinflammation across brain regions in not only young WT mice but also aged AD Tg mice. Such actions translate into improved outcome measures in functional assays using LTP as well as visual and spatial memory. Many of these findings have been confirmed in separate studies of Phen across cellular and animal models of neurodegeneration (Chen et al., 2014; Chang et al., 2017; Hsueh et al., 2019). In the light of its capability to downregulate mTBI-induced gene pathways potentially leading to AD in prior preclinical studies (Tweedie et al., 2016) and its favorable tolerability in human clinical trials involving dosing for up to a year, there is increasing interest for the evaluation of Phen as a clinical candidate for mTBI.
Similarly, IL and CL levels were alike within the CA3 and DG brain regions across groups (not shown), and across all brain regions evaluated in sham control mice without head injury (not shown). Together, these results suggest that our mTBI models results in contrecoup damage, and that this – similar to ipsilateral damage – is mitigated by Phen.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This research was supported in part by (i) the Intramural Research Program of the National Institute on Aging, National Institutes of Health, USA, (ii) NIH grant AG057028 to BJH, (iii) MOST 106–2917-1–038-001, and (iv) the Ari and Regine Aprijaskis Fund, at Tel-Aviv University, and the Dr. Miriam and Sheldon G. Adelson Chair for the Biology of Addictive Diseases, Tel-Aviv University, Tel-Aviv, Israel to CGP.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2019.104528.
Declaration of competing interest
Becker RE has an issued patent on the use of (−)-Phenserine in neurodegenerative disorders. All other authors report no conflicts of interest.
References
- Acosta SA, Tajiri N, Shinozuka K, Ishikawa H, Grimmig B, Diamond DM, Sanberg PR, Bickford PC, Kaneko Y, Borlongan CV, 2013. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS One 8, e53376 10.1371/journal.pone.0053376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WW, Collingridge GL, 2007. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J. Neurosci. Methods 162, 346–356. 10.1016/J.JNEUMETH.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW, 2008. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J. Neurotrauma 25, 513–526. 10.1089/neu.2007.0451. [DOI] [PubMed] [Google Scholar]
- Bader M, Li Y, Lecca D, Rubovitch V, Tweedie D, Glotfelty E, Rachmany L, Kim HK, Choi H-I, Hoffer BJ, Pick CG, Greig NH, Kim DS, 2019. Pharmacokinetics and efficacy of PT302, a sustained-release Exenatide formulation, in a murine model of mild traumatic brain injury. Neurobiol. Dis 124, 439–453. 10.1016/J.NBD.2018.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains M, Hall ED, 2012. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 1822, 675–684. 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratz R, Rubovitch V, Frenk H, Pick CG, 2010. The influence of alcohol on behavioral recovery after mTBI in mice. J. Neurotrauma 27, 555–563. 10.1089/neu.2009.0891. [DOI] [PubMed] [Google Scholar]
- Baratz R, Tweedie D, Rubovitch V, Luo W, Yoon JS, Hoffer BJ, Greig NH, Pick CG, 2011. Tumor necrosis factor-α synthesis inhibitor, 3,6′-dithiothalidomide, reverses behavioral impairments induced by minimal traumatic brain injury in mice. J. Neurochem 118, 1032–1042. 10.1111/j.1471-4159.2011.07377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratz R, Tweedie D, Wang J-Y, Rubovitch V, Luo W, Hoffer BJ, Greig NH, Pick CG, 2015. Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J. Neuroinflammation 12, 45 10.1186/s12974-015-0237-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkhoudarian G, Hovda DA, Giza CC, 2011. The molecular pathophysiology of concussive brain injury. Clin. Sports Med. 30 (33–48), vii–viii. 10.1016/j.csm.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K, 2014. Traumatic brain injury and risk of dementia in older veterans. Neurology 83, 312–319. 10.1212/WNL.0000000000000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RE, Greig NH, 2019. Can we prevent dementia and not prevent neurons from dying? J. Alzheimer’s Dis. Preprint 1–4. 10.3233/JAD-181300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Shimon M, Zeimer T, Shavit Stein E, Artan-Furman A, Harnof S, Chapman J, Eisenkraft A, Pick CG, Maggio N, 2017. Recovery from trauma induced amnesia correlates with normalization of thrombin activity in the mouse hippocampus. PLoS One 12, e0188524 10.1371/journal.pone.0188524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereczki E, Francis PT, Howlett D, Pereira JB, Höglund K, Bogstedt A, Cedazo-Minguez A, Baek JH, Hortobágyi T, Attems J, Ballard C, Aarsland D, 2016. Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimers Dement. 12 (11), 1149–1158. 10.1016/j.jalz.2016.04.005.27224930. [DOI] [PubMed] [Google Scholar]
- Bickford PC, Flowers A, Grimmig B, 2017. Aging leads to altered microglial function that reduces brain resiliency increasing vulnerability to neurodegenerative diseases. Exp. Gerontol 94, 4–8. 10.1016/j.exger.2017.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolouri H, Zetterberg H, 2015. Animal models for concussion: molecular and cognitive assessments—relevance to sport and military concussions. In: Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. [PubMed] [Google Scholar]
- Cambon K, Davies H, Stewart M, 2000. Synaptic loss is accompanied by an increase in synaptic area in the dentate gyrus of aged human Apolipoprotein E4 transgenic mice. Neuroscience 97, 685–692. 10.1016/S0306-4522(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Caplan HW, Cox CS, Bedi SS, 2017. Do microglia play a role in sex differences in TBI? J. Neurosci. Res 95, 509–517. 10.1002/jnr.23854. [DOI] [PubMed] [Google Scholar]
- CDC Grand Rounds: Reducing Severe Traumatic MBrain Injury in the United States [WWW Document]. URL. https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6227a2.htm (accessed 2.22.19). [PMC free article] [PubMed]
- Chang C-F, Lai J-H, Wu JC-C, Greig NH, Becker RE, Luo Y, Chen Y-H, Kang S-J, Chiang Y-H, Chen K-Y, 2017. (−)-Phenserine inhibits neuronal apoptosis following ischemia/reperfusion injury. Brain Res. 1677, 118–128. 10.1016/J.BRAINRES.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Kang J-H, Lin H-C, 2011. Patients with traumatic brain injury. Stroke 42, 2733–2739. 10.1161/STROKEAHA.111.620112. [DOI] [PubMed] [Google Scholar]
- Chen J, Pan H, Chen C, Wu W, Iskandar K, He J, Piermartiri T, Jacobowitz DM, Yu Q-S, McDonough JH, Greig NH, Marini AM, 2014. (−)-Phenserine attenuates soman-induced neuropathology. PLoS One 9, e99818 10.1371/journal.pone.0099818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Kuo T-T, Yi-Kung Huang E, Hoffer BJ, Chou Y-C, Chiang Y-H, Ma H-I, Miller JP, 2018. Profound deficits in hippocampal synaptic plasticity after traumatic brain injury and seizure is ameliorated by prophylactic levetiracetam. Oncotarget 9, 11515–11527. 10.18632/oncotarget.23923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C, Liao Y, Yang L, Wang J, Tweedie D, Hanuma K, Greig NH, Wang J, Mei C, Section D, Branch TG, 2016. Neuroinflammation in an animal model of TBI. J. Neurosci. Methods 272, 38–49. 10.1016/j.jneumeth.2016.06.018.Neuroinflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Watterson M, Van Eldik LJ, 2005. Expert Opinion on Therapeutic Targets Neuroinflammation: A Potential Therapeutic Target Neuroinflammation: A Potential Therapeutic Target. 10.1517/14728222.9.5.887. [DOI] [PubMed]
- D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F, 2011. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci 14, 69–76. 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- Daneshvar DH, Riley DO, Nowinski CJ, McKee AC, Stern RA, Cantu RC, 2011. Long-term consequences: effects on normal development profile after concussion. Phys. Med. Rehabil. Clin. N. Am 22 (683–700), ix 10.1016/j.pmr.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M, Mohapatra S, Mohapatra SS, 2012. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J. Neuroinflammation 9, 236 10.1186/1742-2094-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Avila JC, Siqueira LD, Mazeraud A, Azevedo EP, Foguel D, Castro-Faria-Neto HC, Sharshar T, Chrétien F, Bozza FA, 2018. Age-related cognitive impairment is associated with long-term neuroinflammation and oxidative stress in a mouse model of episodic systemic inflammation. J. Neuroinflammation 15, 28 10.1186/s12974-018-1059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellu F, Mayo W, Cherkaoui J, Le Moal M, Simon H, 1992. A two-trial memory task with automated recording: study in young and aged rats. Brain Res. 588, 132–139. 10.1016/0006-8993(92)91352-F. [DOI] [PubMed] [Google Scholar]
- Deselms H, Maggio N, Rubovitch V, Chapman J, Schreiber S, Tweedie D, Kim DS, Greig NH, Pick CG, 2016. Novel pharmaceutical treatments for minimal traumatic brain injury and evaluation of animal models and methodologies supporting their development. J. Neurosci. Methods 272, 69–76. 10.1016/j.jneumeth.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx CE, Grimes CJB, Loh LTCY, Adam LTCGE, Oskvig D, Curley KC, Salzer W, 2014. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J. Neurotrauma 31, 135–158. 10.1089/neu.2013.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon KJ, 2017. Pathophysiology of traumatic brain injury. Phys. Med. Rehabil. Clin. N. Am 28, 215–225. 10.1016/j.pmr.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Edut S, Rubovitch V, Schreiber S, Pick CG, 2011. The intriguing effects of ecstasy (MDMA) on cognitive function in mice subjected to a minimal traumatic brain injury (mTBI). Psychopharmacology 214, 877–889. 10.1007/s00213-010-2098-y. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R, 2007. PSD-95 is required for activity-driven synapse stabilization. Proc. Natl. Acad. Sci. U. S. A 104, 4176–4181. 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann JR, Ribe AR, Pedersen HS, Fenger-Grøn M, Christensen J, Benros ME, Vestergaard M, 2018. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry 5, 424–431. 10.1016/S2215-0366(18)30065-8. [DOI] [PubMed] [Google Scholar]
- Feng Y, Cui Y, Gao J-L, Li R, Jiang X-H, Tian Y-X, Wang K-J, Li M-H, Zhang H-A, Cui J-Z, 2016. Neuroprotective effects of resveratrol against traumatic brain injury in rats: involvement of synaptic proteins and neuronal autophagy. Mol. Med. Rep 13, 5248–5254. 10.3892/mmr.2016.5201. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ, Liu T, Barone FC, 1994. Cytokines, inflammation, and brain injury: role of tumor necrosis factor-alpha. Cerebrovasc. Brain Metab. Rev 6, 341–360. [PubMed] [Google Scholar]
- Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA, 1996. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J. Biol. Chem 271, 15303–15306. 10.1074/JBC.271.26.15303. [DOI] [PubMed] [Google Scholar]
- Frankola KA, Greig NH, Luo W, Tweedie D, 2011. Targeting TNF-α to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 10, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier T, Morganti-Kossmann MC, O’Reilly D, McLean CA, 2010. In Situ detection of inflammatory mediators in Post Mortem human brain tissue after traumatic injury. J. Neurotrauma 27, 497–507. 10.1089/neu.2009.1120. [DOI] [PubMed] [Google Scholar]
- Gardner AJ, Iverson GL, Williams WH, Baker S, Stanwell P, 2014. A systematic review and meta-analysis of concussion in Rugby union. Sports Med. 44, 1717–1731. 10.1007/s40279-014-0233-3. [DOI] [PubMed] [Google Scholar]
- Glushakova OY, Glushakov AO, Borlongan CV, Valadka AB, Hayes RL, Glushakov AV, 2018. Role of caspase-3-mediated apoptosis in chronic caspase-3-cleaved tau accumulation and blood-brain barrier damage in the corpus callosum after traumatic brain injury in rats. J. Neurotrauma 35, 157–173. 10.1089/neu.2017.4999. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang X-L, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC, 2012. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med 4, 134ra60 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig NH, De Micheli E, Holloway HW, Yu QS, Utsuki T, Perry TA, Brossi A, Ingram DK, Deutsch J, Lahiri DK, Soncrant TT, 2000. The experimental Alzheimer drug phenserine: preclinical pharmacokinetics and pharmacodynamics. Acta Neurol. Scand. Suppl 2000 (176), 74–84. [DOI] [PubMed] [Google Scholar]
- Greig NH, Ruckle J, Comer P, Brownell L, Holloway H, Flanagan D Jr., Canfield C, Burford R, 2005. Anticholinesterase and pharmacokinetic profile of phenserine in healthy elderly human subjects. Curr. Alzheimer Res 2, 483–492. 10.2174/156720505774330564. [DOI] [PubMed] [Google Scholar]
- Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, Chiang Y-H, Hoffer BJ, Miller J, Lahiri DK, Sambamurti K, Becker RE, Pick CG, 2014. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers Dement. 10, S62–S75. 10.1016/j.jalz.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greve MW, Zink BJ, 2009. Pathophysiology of traumatic brain injury. Mt. Sinai J. Med. A J. Transl. Pers. Med 76, 97–104. 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- Hawryluk GWJ, Bullock MR, 2016. Past, present, and future of traumatic brain injury research. Neurosurg. Clin. N. Am 27, 375–396. 10.1016/J.NEC.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Heinonen O, Soininen H, Sorvari H, Kosunen O, Paljarvi L, Koivisto E, Riekkinen PJ, 1995. Loss of synaptophysin-like immunoreactivity in the hippocampal formation is an early phenomenon in Alzheimer’s disease. Neuroscience 64, 375–384. 10.1016/0306-4522(94)00422-2. [DOI] [PubMed] [Google Scholar]
- Hoffer BJ, Pick CG, Hoffer ME, Becker RE, Chiang Y-H, Greig NH, 2017. Repositioning drugs for traumatic brain injury - N-acetyl cysteine and Phenserine. J. Biomed. Sci 24, 71 10.1186/s12929-017-0377-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh S-C, Lecca D, Greig NH, Wang J-Y, Hoffer BJ, Miller JP, Chiang Y-H, 2019. June 10 (−)-Phenserine ameliorates contusion volume, neuroinflammation, and behavioral impairments induced by traumatic brain injury in mice. Cell Transplant. 10.1177/0963689719854693. ([Epub ahead of print] PMID: 31177840). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC, 2007. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation 22, 341–353. [PubMed] [Google Scholar]
- Janowitz T, Menon DK, 2010. Exploring new routes for neuroprotective drug development in traumatic brain injury. Sci. Transl. Med 2, 27rv1 10.1126/scitranslmed.3000330. [DOI] [PubMed] [Google Scholar]
- Joo H, Bae J, Lee J-S, Bang Y, Lee B-J, Park J-W, Lee K, Cho J-H, Bu Y, 2018. Icariin improves functional behavior in a mouse model of traumatic brain injury and promotes synaptic plasticity markers. Planta Med. 10.1055/a-0753-0400. [DOI] [PubMed] [Google Scholar]
- Kadir A, Andreasen N, Almkvist O, Wall A, Forsberg A, Engler H, Hagman G, Lärksäter M, Winblad B, Zetterberg H, Blennow K, Långström B, Nordberg A, 2008. Effect of phenserine treatment on brain functional activity and amyloid in Alzheimer’s disease. Ann. Neurol 63, 621–631. 10.1002/ana.21345. [DOI] [PubMed] [Google Scholar]
- Kirkley KS, Popichak KA, Afzali MF, Legare ME, Tjalkens RB, 2017. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflammation 14, 99 10.1186/s12974-017-0871-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW, 1996. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318. 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kwon SE, Chapman ER, 2011. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70, 847–854. 10.1016/j.neuron.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Wald MM, 2006. The epidemiology and impact of traumatic brain injury: a brief overview. J. Head Trauma Rehabil. 21, 375–378. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Simon CM, Prado GR, Cullen DK, 2007. CNS injury biomechanics and experimental models. Prog. Brain Res. 161, 13–26. 10.1016/S0079-6123(06)61002-9. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541 (7638), 481–487. 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilja AM, Luo Y, Yu Q, Röjdner J, Li Y, Marini AM, Marutle A, Nordberg A, Greig NH, 2013. Neurotrophic and neuroprotective actions of (−)- and (+)-phenserine, candidate drugs for Alzheimer’s disease. PLoS One 8, e54887 10.1371/journal.pone.0054887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Wang L, Cao Y, Xu W, Shi F, Tian Q, Zhou X, 2017. Hypothermia pre- treatment improves cognitive impairment via enhancing synaptic plasticity in a traumatic brain injury model. Brain Res. 1672, 18–28. 10.1016/J.BRAINRES.2017.07.008. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Faden AI, 2010. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci 31, 596–604. 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV, 2015. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat 11, 97–106. 10.2147/NDT.S65815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA, 2004. Long-term potentiation and memory. Physiol. Rev 84, 87–136. 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Lynch NJ, Willis CL, Nolan CC, Roscher S, Fowler MJ, Weihe E, Ray DE, Schwaeble WJ, 2004. Microglial activation and increased synthesis of complement component C1q precedes blood–brain barrier dysfunction in rats. Mol. Immunol 40, 709–716. 10.1016/J.MOLIMM.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Maas AIR, Stocchetti N, Bullock R, 2008. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 7, 728–741. 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- Marchetti M, Marie H, 2011. Hippocampal synaptic plasticity in Alzheimer’s disease: what have we learned so far from transgenic models? Rev. Neurosci 22, 373–402. 10.1515/RNS.2011.035. [DOI] [PubMed] [Google Scholar]
- McCoy MK, Tansey MG, 2008. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J. Neuroinflammation 5, 45 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo L, Cimino F, Angileri FF, La Torre D, Conti A, Cardali SM, Saija A, Germanò A, 2014. Alteration in synaptic junction proteins following traumatic brain injury. J. Neurotrauma 31, 1375–1385. 10.1089/neu.2014.3385. [DOI] [PubMed] [Google Scholar]
- Messier C, 1997. Object recognition in mice: improvement of memory by glucose. Neurobiol. Learn. Mem 67, 172–175. 10.1006/NLME.1996.3755. [DOI] [PubMed] [Google Scholar]
- Meyer D, Bonhoeffer T, Scheuss V, 2014. Balance and stability of synaptic structures during synaptic plasticity. Neuron 82, 430–443. 10.1016/j.neuron.2014.02.031. [DOI] [PubMed] [Google Scholar]
- Mollayeva T, Mollayeva S, Colantonio A, 2018. Traumatic brain injury: sex, gender and intersecting vulnerabilities. Nat. Rev. Neurol 4 (12), 711–722. 10.1038/s41582-018-0091-y. [DOI] [PubMed] [Google Scholar]
- Moppett IK, 2007. Traumatic brain injury: assessment, resuscitation and early management. Br. J. Anaesth 99, 18–31. 10.1093/bja/aem128. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel P, Kossmann T, 2002. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr. Opin. Crit. Care 8, 101–105. [DOI] [PubMed] [Google Scholar]
- Morris RGM, 2003. Long-term potentiation and memory. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci 358, 643–647. 10.1098/rstb.2002.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, 2017. A brief history of long-term potentiation. Neuron 93, 281–290. 10.1016/J.NEURON.2016.12.015. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Kadir A, Andreasen N, Almkvist O, Wall A, Blennow K, Långström B, Zetterberg H, 2015. Correlations between Alzheimer’s disease cerebrospinal fluid biomarkers and cerebral glucose metabolism after 12 months of Phenserine treatment. J. Alzheimers Dis 47, 691–704. 10.3233/JAD-132474. [DOI] [PubMed] [Google Scholar]
- Nudo RJ, 2013. Recovery after brain injury: mechanisms and principles. Front. Hum. Neurosci 7, 887 10.3389/fnhum.2013.00887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoshi F, Yin HZ, Kuppumbatti Y, Song B, Amindari S, Weiss JH, 2005. Tumor necrosis-factor-alpha (TNF-α) induces rapid insertion of Ca2+ −permeable α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp. Neurol 193, 384–393. 10.1016/J.EXPNEUROL.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Pagulayan KF, Temkin NR, Machamer J, Dikmen SS, 2006. A longitudinal study of health-related quality of life after traumatic brain injury. Arch. Phys. Med. Rehabil 87, 611–618. 10.1016/j.apmr.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ, 2012. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, 4th edition Academic Press, NY. [Google Scholar]
- Perez-Polo JR, Rea HC, Johnson KM, Parsley MA, Unabia GC, Xu G, Infante SK, DeWitt DS, Hulsebosch CE, 2013. Inflammatory consequences in a rodent model of mild traumatic brain injury. J. Neurotrauma 30, 727–740. 10.1089/neu.2012.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Giza CC, 2012. Repeat traumatic brain injury in the developing brain. Int. J. Dev. Neurosci 30, 185–190. 10.1016/j.ijdevneu.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Przekwas A, Somayaji MR, Gupta RK, 2016. Synaptic mechanisms of blast-induced brain injury. Front. Neurol 7, 2 10.3389/fneur.2016.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachmany L, Tweedie D, Rubovitch V, Li Y, Holloway HW, Kim DS, Ratliff WA, Saykally JN, Citron BA, Hoffer BJ, Greig NH, Pick CG, 2017. Exendin-4 attenuates blast traumatic brain injury induced cognitive impairments, losses of synaptophysin and in vitro TBI-induced hippocampal cellular degeneration. Sci. Rep 7, 3735 10.1038/s41598-017-03792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo FR, Musella A, De Vito F, Fresegna D, Bullitta S, Vanni V, Guadalupi L, Stampanoni Bassi M, Buttari F, Mandolesi G, Centonze D, Gentile A, 2018. Tumor necrosis factor and interleukin-1β modulate synaptic plasticity during neuroinflammation. Neural Plast. 2018, 8430123 10.1155/2018/8430123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff RL, Riechers RGII, Wang X-F, Piero T, Ruff SS, 2012. A case-control study examining whether neurological deficits and PTSD in combat veterans are related to episodes of mild TBI. Neurol. Res 10.1136/bmjopen-2011-000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt OI, Heyde CE, Ertel W, Stahel PF, 2005. Closed head injury—an inflammatory disease? Brain Res. Rev 48, 388–399. 10.1016/J.BRAINRESREV.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L, 2005. Fluoro-jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 1035, 24–31. 10.1016/J.BRAINRES.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 2002. Alzheimer’s disease is a synaptic failure. Science (80-. ) 298, 789–791. 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shohami E, Bass R, Wallach D, Yamin A, Gallily R, 1996. Inhibition of tumor necrosis factor alpha (TNFα) activity in rat brain is associated with cerebroprotection after closed head injury. J. Cereb. Blood Flow Metab. 16, 378–384. 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- Stein DG, Sayeed I, 2018. Repurposing and repositioning neurosteroids in the treatment of traumatic brain injury: a report from the trenches. Neuropharmacology. 10.1016/J.NEUROPHARM.2018.04.006. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC, 2005. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor. J. Neurosci 25, 3219–3228. 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Fan W, Ma Z, Wen X, Wang W, Wu Q, Huang H, 2014. Taurine improves functional and histological outcomes and reduces inflammation in traumatic brain injury. Neuroscience 266, 56–65. 10.1016/J.NEUROSCIENCE.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Südhof TC, 1995. The synaptic vesicle cycle: a cascade of protein–protein interactions. Nature 375, 645–653. 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Swinney DC, 2013. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther 93, 299–301. 10.1038/clpt.2012.236. [DOI] [PubMed] [Google Scholar]
- Swinney DC, Anthony J, 2011. How were new medicines discovered? Nat. Rev. Drug Discov. 10, 507–519. 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J, 2006. A systematic review of brain injury epidemiology in Europe. Acta Neurochir. 148, 255–268. 10.1007/s00701-005-0651-y. [DOI] [PubMed] [Google Scholar]
- Tashlykov V, Katz Y, Gazit V, Zohar O, Schreiber S, Pick CG, 2007. Apoptotic changes in the cortex and hippocampus following minimal brain trauma in mice. Brain Res. 1130, 197–205. 10.1016/J.BRAINRES.2006.10.032. [DOI] [PubMed] [Google Scholar]
- Tashlykov V, Katz Y, Volkov A, Gazit V, Schreiber S, Zohar O, Pick CG, 2009. Minimal traumatic brain injury induce apoptotic cell death in mice. J. Mol. Neurosci 37, 16–24. 10.1007/s12031-008-9094-2. [DOI] [PubMed] [Google Scholar]
- Taylor CA, Bell JM, Breiding MJ, Xu L, 2017. Traumatic brain injury–related emergency department visits, hospitalizations, and deaths — United States, 2007 and 2013. MMWR Surveill. Summ 66, 1–16. Doi: 10.15585/mmwr.ss6609a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teasdale G, Jennett B, 1974. Assessment of coma and impaired consciousness. Lancet 304, 81–84. 10.1016/S0140-6736(74)91639-0. [DOI] [PubMed] [Google Scholar]
- Tsai M-C, Chang C-P, Peng S-W, Jhuang K-S, Fang Y-H, Lin M-T, Tsao TC-Y, 2015. Therapeutic efficacy of neuro AiD™ (MLC 601), a traditional Chinese medicine, in experimental traumatic brain injury. J. NeuroImmune Pharmacol. 10, 45–54. 10.1007/s11481-014-9570-0. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Rachmany L, Rubovitch V, Lehrmann E, Zhang Y, Becker KG, Perez E, Miller J, Hoffer BJ, Greig NH, Pick CG, 2013a. Exendin-4, a glucagon-like peptide-1 receptor agonist prevents mTBI-induced changes in hippocampus gene expression and memory deficits in mice. Exp. Neurol 239, 170–182. 10.1016/j.expneurol.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Rachmany L, Rubovitch V, Zhang Y, Becker KG, Perez E, Hoffer BJ, Pick CG, Greig NH, 2013b. Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol. Dis 54, 1–11. 10.1016/j.nbd.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Fukui K, Li Y, Yu Q, Barak S, Tamargo IA, Rubovitch V, Holloway HW, Lehrmann E, Wood WH, Zhang Y, Becker KG, Perez E, Van Praag H, Luo Y, Hoffer BJ, Becker RE, Pick CG, Greig NH, 2016a. Cognitive impairments induced by concussive mild traumatic brain injury in mouse are ameliorated by treatment with phenserine via multiple non-cholinergic and cholinergic mechanisms. PLoS One 11, e0156493 10.1371/journal.pone.0156493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Rachmany L, Kim DS, Rubovitch V, Lehrmann E, Zhang Y, Becker KG, Perez E, Pick CG, Greig NH, 2016b. Mild traumatic brain injury-induced hippocampal gene expressions: the identification of target cellular processes for drug development. J. Neurosci. Methods 272, 4–18. 10.1016/j.jneumeth.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER), 2005. Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. https://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf%23search=%27guidekines+for+industry+sfe+starting%27.
- Valtorta F, Pennuto M, Bonanomi D, Benfenati F, 2004. Synaptophysin: leading actor or walk-on role in synaptic vesicle exocytosis? BioEssays 26, 445–453. 10.1002/bies.20012. [DOI] [PubMed] [Google Scholar]
- Vitarbo EA, Chatzipanteli K, Kinoshita K, Truettner JS, Alonso OF, Dietrich WD, 2004. Tumor necrosis factor alpha expression and protein levels after fluid percussion injury in rats: the effect of injury severity and brain temperature. Neurosurgery 55, 416–424 (discussion 424–5). [DOI] [PubMed] [Google Scholar]
- Wen Z, Li D, Shen M, Chen G, 2017. Therapeutic potentials of synapses after traumatic brain injury: a comprehensive review. Neural Plast. 2017, 4296075 10.1155/2017/4296075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteneck GG, Cuthbert JP, Corrigan JD, Bogner JA, 2016. Prevalence of self-reported lifetime history of traumatic brain injury and associated disability: a statewide population-based survey. J. Head Trauma Rehabil. 31, E55–E62. 10.1097/HTR.0000000000000140. [DOI] [PubMed] [Google Scholar]
- Winblad B, Giacobini E, Frölich L, Friedhoff LT, Bruinsma G, Becker RE, Greig NH, 2010. Phenserine efficacy in Alzheimer’s disease. J. Alzheimers Dis. 22, 1201–1208. 10.3233/JAD-2010-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston CN, Chellappa D, Wilkins T, Barton DJ, Washington PM, Loane DJ, Zapple DN, Burns MP, 2013. Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. J. Neurotrauma 30, 1966–1972. 10.1089/neu.2013.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher KG, Bray CE, Dziabis JE, McKim DB, Benner BN, Rowe RK, Kokiko-Cochran ON, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP, 2018. Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia 66, 2719–2736. 10.1002/glia.23523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock T, Morganti-Kossmann MC, 2013. The role of markers of inflammation in traumatic brain injury. Front. Neurol 4, 18 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, 2006. Neurological disorders public health challenges. In: WHO Library Cataloguing-in-Publication Data, Retrieved from. http://www.who.int/mental_health/neurology/neurological_disorders_report_web.pdf.
- Yang L-Y, Greig NH, Huang Y-N, Hsieh T-H, Tweedie D, Yu Q-S, Hoffer BJ, Luo Y, Kao Y-C, Wang J-Y, 2016. Post-traumatic administration of the p53 inactivator pifithrin-α oxygen analogue reduces hippocampal neuronal loss and improves cognitive deficits after experimental traumatic brain injury. Neurobiol. Dis 96, 216–226. 10.1016/j.nbd.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, West EJ, Van KC, Gurkoff GG, Zhou J, Zhang X-M, Kozikowski AP, Lyeth BG, 2008. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 1226, 181–191. 10.1016/j.brainres.2008.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.