

Abstract
Mouse models provide important resources for many areas of vision research, pertaining to retinal development, retinal function and retinal disease. The Translational Vision Research Models (TVRM) program uses chemical mutagenesis to generate new mouse models for vision research. In this chapter, we report the identification of mouse models for Grml, Grkl and Lrit3. Each of these is characterized by a primary defect in the electroretinogram. All are available without restriction to the research community.
Keywords: Mutagenesis, Electroretinogram, Photoreceptor, Mice, Retina
24.1. Introduction
Mouse models of retinal diseases are an important genetic resource for furthering our understanding of molecules necessary for vision. This reflects, in part, our ability to develop and discover mouse models that bear disruption in genes implicated in human conditions and that replicate key features of human disease. The Translational Models for Vision Research (TMVR) program, sited at The Jackson Laboratory (JAX), uses chemical mutagenesis followed by high throughput eye-specific screens to identify mouse models bearing mutations that lead to ocular phenotypes (Won et al. 2011, 2012). The purpose of this report is to describe three new models that extend allelic series for genes known to play important roles in the outer retina.
24.2. Materials and Methods
24.2.1. Mouse Mutagenesis, Husbandry, and Ocular Screening
As described in detail (Won et al. 2011), N-ethyl-N-nitrosourea (ENU) was administered to male C57BL/6J mice. G3 offspring generated using a three-generation backcross mating scheme (Won et al. 2011) were examined at 12 weeks of age. All mice underwent screening by indirect ophthalmoscopy (Hawes et al. 1999). A subset of mice were also screened by ERG, using a previously described system and protocol (Hawes et al. 2000). In brief, after a minimum of 2 h of dark adaptation, mice were anesthetized with ketamine (16 mg/kg) and xylazine (80 mg/kg) diluted in normal saline. Strobe stimuli were presented in darkness and again after 10 min of light adaption. In depth ERG studies were conducted at the Cleveland Clinic, using published protocols (Yu et al. 2012).
24.2.2. Genetic Mapping and Mutational Analysis
Tvrm207 and Tvrm257 mutants with abnormal ERGs were mated with DBA/2J mice. Resulting F1 offspring were intercrossed to generate a segregating F2 population. Each F2 progeny underwent ERG testing and genome wide scans using a DNA pooling strategy, and genotyping with simple sequence length polymorphism markers was carried out. Tvrm207 was found to map to Chr. 8 proximal to marker D8Mit124 while Tvrm257 mapped to Chr. 3 between markers D3Mit348 and D3Mit14. The mapping was based on 154 and 704 meioses, respectively.
Exome capture libraries prepared from Tvrm84 and Tvrm207 mutant DNAs were subject to high throughput sequencing. Mutations within candidate genes were identified by comparison of mutant and WT sequences and verified in 10 affected and 10 unaffected mice from the inbred Tvrm84 and Tvrm207 colonies. Without exception, affected mice were homozygous for the mutation while unaffected mice were either heterozygous or WT for the mutations.
In the case of Tvrm257, primers within introns flanking exons in candidate genes were generated to amplify each exon from genomic DNA. DNA of mice from the mapping population (10 affected and 10 unaffected) and inbred Tvrm257 (5 affected and 5 unaffected) colonies were amplified, sequenced and compared using published procedures (Won et al. 2011). Without exception, affected mice were homozygous for the mutation while unaffected mice were either heterozygous or WT for the mutation.
24.2.3. Histological Analysis
Mice were euthanized by CO2 inhalation and eyes were enucleated. Eyes were fixed overnight in cold methanol/acetic acid solution (3:1, v/v). Paraffin embedded eyes were cut into 6 μm sections, stained by hematoxylin and eosin (H&E), and examined by light microscopy.
24.3. Results
24.3.1. A New Allele of Grm1tvrm84
The glutamate receptor, metabotropic 1 (GRM1) is widely expressed in the central nervous system (CNS). GRM1 mutations or copy number variations may predispose to a variety of conditions including schizophrenia (Ayoub et al. 2012) or depression (Menke et al. 2012). Previously described Grml mouse mutants include the recoil wobbler (Grm1rcw) (Sachs et al. 2007), Grm1nmf373 (Sachs et al. 2007), Grm1crv4 (Conti et al. 2006) and Grm1−/− (Conquet et al. 1994) models. All have reduced body size, a neurological phenotype including ataxic gait, tremor, skeletal defects, and learning abnormalities in the absence of gross structural defects of the CNS.
Affected tvrm84 mice were identified based on their ataxic phenotype. Subsequent ERG testing showed the presence of a normal ERG waveform of reduced amplitude. Affected mice present with a normal fundus appearance and retinal histology. Comparison of high throughout sequencing data between mutant and WT mice indicated a c.1607T > A mutation in Grml. The Grm1tvrm84 mutation is predicted to lead to a point mutation: p.Iso536Lys.
Figure 24.1 summarizes the ERG analysis. The upper panels present representative ERGs obtained under dark-adapted (Fig. 24.1a) or light-adapted (Fig. 24.1b) conditions. The overall ERG waveform is maintained in Grm1tvrm84 mice, but is reduced under dark-adapted conditions. The lower panels present average (± sem) measures of the major ERG components. The reduction of the dark-adapted ERG is seen across the stimulus range used (Fig. 24.1c), while the light-adapted data superimpose (Fig. 24.1d). No gross morphological abnormalities were observed in homozygous Grm1tvrm84 mice (data not shown).
Fig. 24.1.
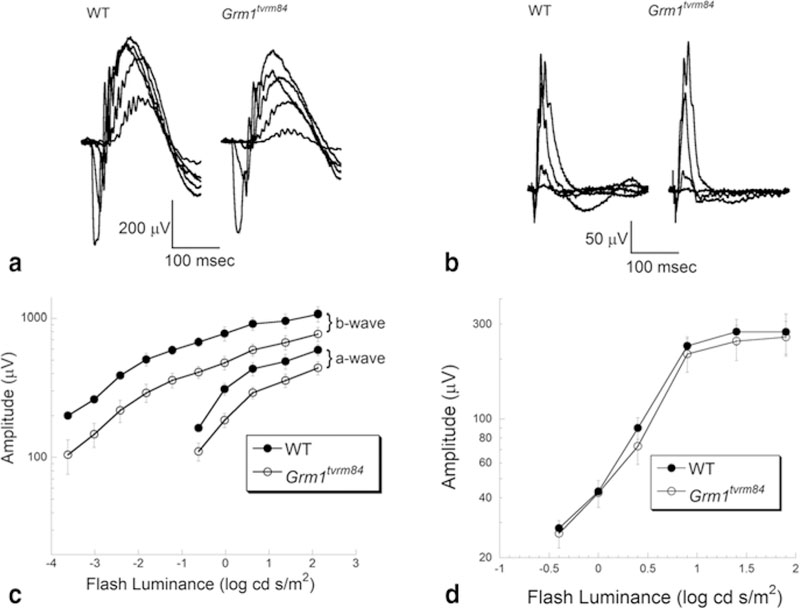
ERG characteristics of Grm1tvrm84 mutant. Representative ERGs obtained from 1 month old mice under dark-adapted (a) and light-adapted (b) stimulus conditions. Summary response functions for the major components of the dark-adapted (c) and light-adapted (d) ERG. Symbols indicate average ± s.e.m. of 8–9 mice
24.3.2. A New Allele for Grk1twm207
Rhodopsin kinase, encoded by GRK1/Grk1, is responsible for the initial steps by which light-activated rhodopsin is returned to an inactive state. Rhodopsin kinase accomplishes this through phosphorylation of a series of serine residues on the C-terminus of rhodopsin (Mendez et al. 2000). In humans, GRK1 mutations cause Oguchi’s Disease (Yamamoto et al. 1997; Cideciyan et al. 1998). In mice, single cell studies of Grk1−/− rods reveal abnormal phototransduction deactivation kinetics (Chen et al. 1999). Grk1−/− mice have a modest loss of cells in the outer nuclear layer, but a more rapid loss of outer segment length (Fan et al. 2010).
The tvrm207 line was identified by a reduced amplitude ERG and this feature was used to map tvrm207 to Chr. 8. Comparison of exome sequences of Grkl identified a nucleotide transition: c.1088T > C. The Grk1tvrm207 mutation is predicted to lead to an amino acid change: p.Leu363Pro. As shown in Fig. 24.2a, ERG amplitudes are significantly reduced in Grk1tvrm207 mice. The amplitude reduction is present as early as P19, and there is relatively little progression up to 4 months of age (Fig. 24.2b). Consistent with the ERG data, the outer nuclear layer of Grk1tvrm207 mice changes little in overall thickness over the first 3 months. Photoreceptors are absent, however, by 1 year of age (Fig. 24.2c). The loss of photoreceptors may be due to the extended exposure to vivarium lighting over the lifetime of the animals, as Grk1−/− mice are sensitive to light induced damage (Chen et al. 1999).
Fig. 24.2.
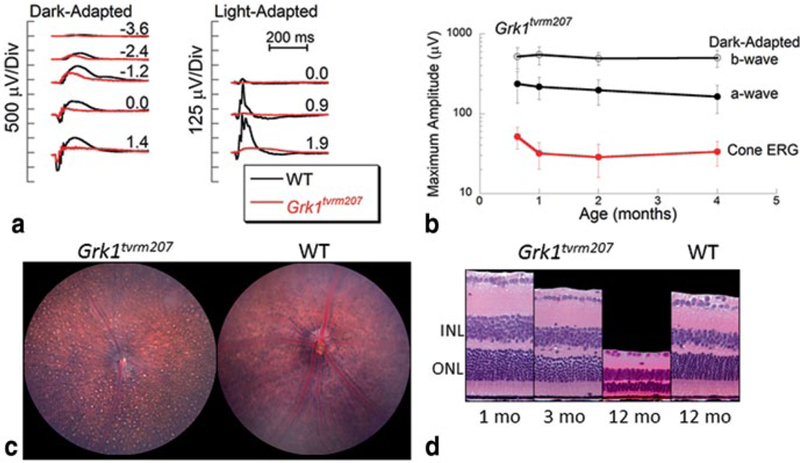
Characteristics of Grm1tvrm207 mutant. a ERGs obtained from 1-month-old mice under dark-adapted (left) and light-adapted (right) stimulus conditions. b The reduction in ERG amplitude is present at an early age and remains stable across the age-range examined. c Fundus photo of a 12-old-mutant indicate retinal spotting and granular appearance in comparison to a 3-month-old C57BL/6J mouse. d Representative retinal cross-sections obtained from 1-, 3-, and 12-month-old mutant and 12-month-old control mice
24.3.3. A New Allele for Lrit3tvrm257
Leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 (LR1T3) is the most recently identified gene in which mutations cause complete congenital stationary night blindness (cCSNB) (Zeitz et al. 2013). This discovery was followed by the description of a null mutant for Lrit3, which has a preserved ERG a- wave, an absent ERG b-wave, and normal retinal morphology (Neuille et al. 2014). The tvrm257 line was identified based on an absent ERG b-wave, and this feature was mapped to the Lrit3 locus. Direct sequencing of amplified exons of Lrit3 from tvrm257 mice identified a nucleotide transition: c.401T > C. The Lrit3tvrm257 mutation is predicted to lead to a point mutation: p.Leu134Pro.
ERG studies document the presence of a normal a-wave without a subsequent b-wave in Lrit3tvrm257 mice (Fig. 24.3). The cone ERG is also abnormal. Gross morphological abnormalities are not observed in Lrit3tvrm257 mutants up to 7 months of age, the oldest age examined (data not shown). Overall this phenotype matches that of the Lrit3−/− mouse (Neuillé et al. 2014) and other mouse models involving proteins expressed in depolarizing bipolar cells (Pardue and Peachey 2014).
Fig. 24.3.
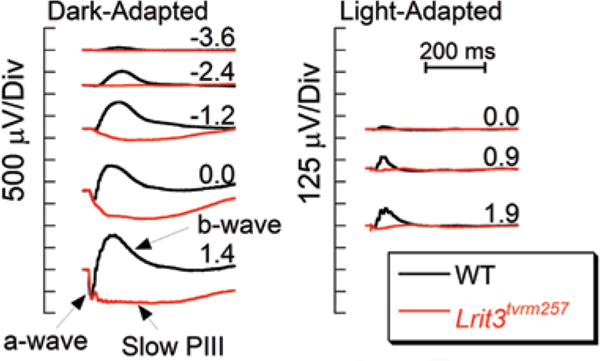
ERG Characteristics of Lrit3tvrm257 mutant. ERGs obtained from 1 month old mice under dark-adapted ( left) and light-adapted ( right) stimulus conditions
24.4. Discussion
We report the identification of three new mouse strains with disruption in Grml, Grkl or Lrit3. The main retinal abnormality is an abnormal ERG, although the nature of this abnormality differs across the three mouse models. Unlike the knockout models that are currently available, these mouse lines all involve missense mutations which are not expected to abrogate protein translation, and all mutations are coisogenic on the C57BL/6J background. Overall, such point mutants are rare, as knockout targeting vectors are designed to ensure total loss of expression and absence of the encoded protein. Point mutants can provide information about domain functions and may exhibit different phenotypes compared to their knock-out counterparts (e.g., Peachey et al. 2012). These mice, therefore, provide or expand the allelic series for the genes involved. As is the case for other TVRM models, these mice are available without restriction to the research community.
Acknowledgments
This work was supported by NIH grants (R01EY16501; P30CA34196), Foundation Fighting Blindness, VA Medical Research Service, and Research to Prevent Blindness.
Contributor Information
Jeremy R. Charette, The Jackson Laboratory, Bar Harbor, ME 04609, USA
Ivy S. Samuels, Louis Stokes Cleveland VA Medical Center, Cleveland, OH 44106, USA Cole Eye Institute, Cleveland Clinic, Cleveland, OH 44195, USA.
Minzhong Yu, Cole Eye Institute, Cleveland Clinic, Cleveland, OH 44195, USA.
Lisa Stone, The Jackson Laboratory, Bar Harbor, ME 04609, USA.
Wanda Hicks, The Jackson Laboratory, Bar Harbor, ME 04609, USA.
Lan Ying Shi, The Jackson Laboratory, Bar Harbor, ME 04609, USA.
Mark P. Krebs, The Jackson Laboratory, Bar Harbor, ME 04609, USA
Jürgen K Naggert, The Jackson Laboratory, Bar Harbor, ME 04609, USA.
Patsy M. Nishina, The Jackson Laboratory, Bar Harbor, ME 04609, USA Cole Eye Institute, Cleveland Clinic, Cleveland, OH 44195, USA.
Neal S. Peachey, Louis Stokes Cleveland VA Medical Center, Cleveland, OH 44106, USA Cole Eye Institute, Cleveland Clinic, Cleveland, OH 44195, USA; Department of Ophthalmology, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH, USA.
References
- Ayoub MA, Angelicheva D, Vile D et al. (2012) Deleterious GRM1 mutations in schizophrenia. PLoS ONE 7:e32849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Burns ME, Spencer M et al. (1999) Abnormal photoresponses and light-induced apopto- sis in rods lacking rhodopsin kinase. Proc Natl Acad Sci U S A 96:3718–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Zhao X, Nielsen L et al. (1998) Null mutation in the rhodopsin kinase gene slows recovery kinetics of rod and cone phototransduction in man. Proc Natl Acad Sci U S A 95:328–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH et al. (1994) Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature 372:237–243 [DOI] [PubMed] [Google Scholar]
- Conti V, Aghaie A, Cilli M et al. (2006) crv4, a mouse model for human ataxia associated with kyphoscoliosis caused by an mRNA splicing mutation of the metabotropic glutamate receptor 1 (Grm1). Int J Mol Med 18:593–600 [PubMed] [Google Scholar]
- Fan J, Sakurai K, Chen CK et al. (2010) Deletion of GRK1 causes retina degeneration through a transducin-independent mechanism. J Neurosci 30:2496–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes NL, Smith RW, Chang B et al. (1999) Mouse fundus photography and angiography: a catalogue of normal and mutant phenotypes. Mol Vis 5:22. [PubMed] [Google Scholar]
- Hawes NL, Chang B, Hageman GS et al. (2000) Retinal degeneration 6 (rd6): a new mouse model for human retinitis punctata albescens. Invest Ophthalmol Vis Sci 41:3149–3157 [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A et al. (2000) Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 28:153–164 [DOI] [PubMed] [Google Scholar]
- Menke A, Sämann P, Kloiber S et al. (2012) Polymorphisms within the metabotropic glutamate receptor 1 gene are associated with depression phenotypes. Psychoneuroendocrinol 37:565–575 [DOI] [PubMed] [Google Scholar]
- Neuillé M, El Shamieh S, Orhan E et al. (2014) Lrit3 deficient mouse (nob6): a novel model of complete congenital stationary night blindness (cCSNB). PLoS ONE 9:e90342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardue MT, Peachey NS (2014) Mouse b-wave mutants. Doc Ophthalmol 128:77–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peachey NS, Pearring JN, Bojang P Jr et al. (2012) Depolarizing bipolar cell dysfunction due to a Trpml point mutation. J Neurophysiol 108:2442–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AJ, Schwendinger JK, Yang AW et al. (2007) The mouse mutants recoil wobbler and nmf373 represent a series of Grm1 mutations. Mamm Genome 18:749–756 [DOI] [PubMed] [Google Scholar]
- Won J, Shi LY, Hicks W et al. (2011) Mouse models for vision research. J Ophthalmol 2011:391384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J, Shi LY, Hicks W et al. (2012) Translational vision research models program. Adv Exp Med Biol 723:391–397 [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Sippel KC, Berson EL, Dryja TP (1997) Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 15:175–178 [DOI] [PubMed] [Google Scholar]
- Yu M, Sturgill-Short G, Ganapathy P et al. (2012) Age-related changes in visual function in cystathionine-beta-synthase mutant mice, a model of hyperhomocysteinemia. Exp Eye Res 96:124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitz C, Jacobson SG, Hamel CP et al. (2013) Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 92:67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]