

Abstract
Introduction
Hereditary diffuse gastric cancer (HDGC) is a cancer syndrome associated with variants in E-cadherin (CDH1), diffuse gastric cancer and lobular breast cancer. There is considerable heterogeneity in its clinical manifestations. This study aimed to determine associations between CDH1 germline variant status and clinical phenotypes of HDGC.
Methods
One hundred and fifty-two HDGC families, including six previously unreported families, were identified. CDH1 gene-specific guidelines released by the Clinical Genome Resource (ClinGen) CDH1 Variant Curation Expert Panel were applied for pathogenicity classification of truncating, missense and splice site CDH1 germline variants. We evaluated ORs between location of truncating variants of CDH1 and incidence of colorectal cancer, breast cancer and cancer at young age (gastric cancer at <40 or breast cancer <50 years of age).
Results
Frequency of truncating germline CDH1 variants varied across functional domains of the E-cadherin receptor gene and was highest in linker (0.05785 counts/base pair; p=0.0111) and PRE regions (0.10000; p=0.0059). Families with truncating CDH1 germline variants located in the PRE-PRO region were six times more likely to have family members affected by colorectal cancer (OR 6.20, 95% CI 1.79 to 21.48; p=0.004) compared with germline variants in other regions. Variants in the intracellular E-cadherin region were protective for cancer at young age (OR 0.2, 95% CI 0.06 to 0.64; p=0.0071) and in the linker regions for breast cancer (OR 0.35, 95% CI 0.12 to 0.99; p=0.0493). Different CDH1 genotypes were associated with different intracellular signalling activation levels including different p-ERK, p-mTOR and β-catenin levels in early submucosal T1a lesions of HDGC families with different CDH1 variants.
Conclusion
Type and location of CDH1 germline variants may help to identify families at increased risk for concomitant cancers that might benefit from individualised surveillance and intervention strategies.
INTRODUCTION
Genotype-guided screening and surveillance have been proposed for improved individualised management of patients affected by cancer susceptibility syndromes.1 These include cancer risk assessment and prevention strategies in women who have inherited variants in BRCA1/BRCA2 genes and risk stratification in familial adenomatosis polyposis (FAP) families affected by APC gene variants.2–5 Hereditary diffuse gastric cancer (HDGC) has been associated with germline variants in CDH1.6,7 However, it is unclear how type and location of CDH1 germline variants are associated with cancer risk and neoplastic phenotypes observed in these families.
HDGC is a clinically defined cancer syndrome characterised by the early onset of diffuse gastric cancer (DGC) and lobular breast cancer (LBC).7–9 The clinical phenotype of HDGC shows considerable heterogeneity with type of cancer and age of onset.8–10 For example, gastric cancers in some families have been described in patients as young as 16 years old.7 In other families, the main cancer phenotype is LBC and either no family members or only older family members are affected by DGC.8,11–13 Some reports also suggest an association with colorectal cancer.8,14,15
Between 20% and 25% of families with HDGC who meet current International Gastric Cancer Linkage Consortium (IGCLC) clinical testing criteria of early-onset, multi-generational DGC and LBC (two gastric cancer cases regardless of age, one confirmed DGC, either in 1st or 2nd degree relative; one case of DGC <40 years; personal or family history of DGC and LBC, one diagnosed <50 years) harbour germline variants in the E-cadherin (CDH1; NM_004360) locus.9,16 The underlying mechanism for the variable clinical phenotype is poorly understood but might include the individual CDH1 germline variant,17,18 the type of second hit leading to biallelic CDH1 loss (ie, epigenetic changes vs loss of heterozygosity),19 variants that are able to activate cryptic or alternative splice sites20,21 or variable abilities to subject CDH1 mRNA transcripts to nonsense-mediated mRNA decay (NMD).22,23 To date, no association has been described between the underlying genetic alteration in the CDH1 gene and the clinical presentation within this cancer syndrome.24,25
Here, we examine 152 families with known CDH1 variants, classify pathogenicity of their CDH1 gene product and characterise associations between the location of CDH1 variant and clinical manifestation of the syndrome.
MATERIALS AND METHODS
Identification of HDGC families
We performed a systematic review in accordance with the Preferred Reporting Items for Systematic reviews and Meta-Analyses26 and conducted a database search by reviewing references published in the PubMed database between January 1998 and June 2018 using the following keywords: CDH1, E-cadherin, HDGC, germline variant and gastric cancer. References describing CDH1 variants collected in the sentinel reports of Corso et al,25 Benusiglio et al11 and Hansford et al9 (including supplemental materials) on HDGC were also included. References were individually screened and evaluated. Families were excluded if the germline CDH1 variant was not reported, if an accurate prediction of the resulting CDH1 protein product was not possible, if the reported substitution did not match the wild type sequence of the CDH1 gene (UniProt: P12830–1; Ensembl: ENST00000261769), if a non-coding germline variant in CDH1 with an unknown protein product was originally reported, if no family history on DGC and/or LBC was available or if family history did not align with the 2015 International Gastric Cancer Linkage Consortium (IGCLC) criteria for HDGC (at least one family member affected by DGC or LBC)7 (online supplementary figure 1). In case of duplicate reporting, the most up-to-date reference was selected. To classify pathogenicity of variants, the recently released specifications of the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) variant guidelines for the analysis of CDH1 germline variants by the Clinical Genome Resource CDH1 Variant Curation Expert Panel (ClinGen CDH1 VCEP) implemented by the ClinGen Hereditary Cancer Domain were applied.27,28 The updated ACMG/AMP codes included (A) PVS1 null variant (nonsense, frameshift, canonical ±1 or two splice sites, initiation codon, single or multiexon deletion) in a gene where loss of function is a known mechanism of disease applying CDH1 rule specifications/caveats (exception for canonical splice sites and truncations in NMD-resistant zone, strong; G to non-G variant disrupting the last nucleotide of an exon, moderate), (B) PM2 absent in population database (<one out of 100 000 alleles in gnomAD cohort, moderate) and (C) PM4 protein length changes as a result of in-frame deletions/insertions in a nonrepeat region or stop-loss variants (per original ACMG/AMP guidelines, moderate) (for comprehensive summary of all CDH1 rule specifications, please see Lee et al)28 PP5 ‘Reputable source recently reports variant as pathogenic’ was captured but not applied. Based on ClinGen CDH1 VCEP guidelines computational prediction supporting deleterious effect (PP3) was not applied to missense variants or variants affecting splice sites. In vitro or in vivo functional evidence supporting damaging effect on CDH1 gene function (PS3) was only applied for the classification of splice site variants. Final pathogenicity allocation and variant curation was made via the ACMG/AMP scoring combination criteria as described in the sentinel report of Richards and colleagues.27 We also included six previously unreported families from the National Cancer Institute’s prospective Hereditary Gastric Cancer Registry (IRB-approved protocol NCI-09-C-0079) (online supplementary figure 2).
CDH1 germline variant predictions and classification
We constructed a hand-curated database of all CDH1 variants as reported in the identified HDGC families identified in the literature search or our HDGC registry. All reported variants were mapped to the human CDH1 wild-type sequence (UniProt: P12830–1; Ensembl: ENST00000261769) to confirm concordance between reported substitutions and entries in the EMBL database. On collection, the variants were mapped to the high quality merged Ensembl/Havana transcript (Ensembl: CDH1–201) including their genomic position, amino acid (aa) position(s) and splice sites. Prediction software tools included VariantTaster MutationTaster (online version: http://www.mutationtaster.org/) and SIFT (classify coding insertion/deletions, or indels, online version: http://sift.bii.a-star.edu.sg). Variants were classified as missense when a single nucleotide variant (SNV) introduced a non-synonymous aa change, splice site variants when a SNV involved any exon-intron boundary within ±2 nucleotides or the donor or acceptor site of known splice sites within 50 nucleotides of termini of introns, and as truncating variants when SNVs including deletion/insertions introduced a premature stop codon. In line with CDH1 gene-specific variant curation by ClinGen CDH1 VCEP, the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/) was used as reference population database to determine allele frequency. The Human Genome Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php) for presence of variants in reputable database (PP5) was also screened. Classification tools such as LoFtool and PolyPhen-2 predictions (as implemented in the Ensembl VEP Human Slice Site Finder version 3 (HSF3); online: www.umd.be/HSF3), HGMD (ver. Professional 2015.3) and SPANR (Splicing-based Analysis of Variants; http://tools.genes.toronto.edu/)) were used to evaluate splice site variants as a truncating or missense variant. SIFT was used to predict if the concerned exon was affected and/or deleted. Both HSF3, MutationTaster and SPANR were also used to predict if truncating variants are subject to NMD or if premature stop codon variants are likely to evade this cellular surveillance mechanism. Truncating variants inducing a premature stop codon more than ~50 bps upstream of any exon-exon junction generally trigger NMD and were classified as NMD competent, variants inducing a termination codon within about ~50 to 55 nucleotides of the final exon-exon junction complex as NMD-deficient (online supplementary figure 3).23,29 For genotype-clinical phenotype associations in HDGC families, truncating variants were classified by location of the variant on the CDH1 gene locus and its relation to functionally known domains of the E-cadherin receptor from NCBI’s conserved domain database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi?seqinput=NP_004351.1) and UniProt database (http://www.uniprot.org/uniprot/P12830#family_and_domains). Online supplementary table 1 lists identified CDH1 variants and associated clinicopathological information of 152 HDGC families after exclusion of: (1) synonymous variants, (2) variants not aligned with CDH1 wild-type sequence or (3) variants whose protein products could not be predicted. Figure 1A shows a summary of location of truncating, missense and splice site variants in relation to E-cadherin domains.
Figure 1.
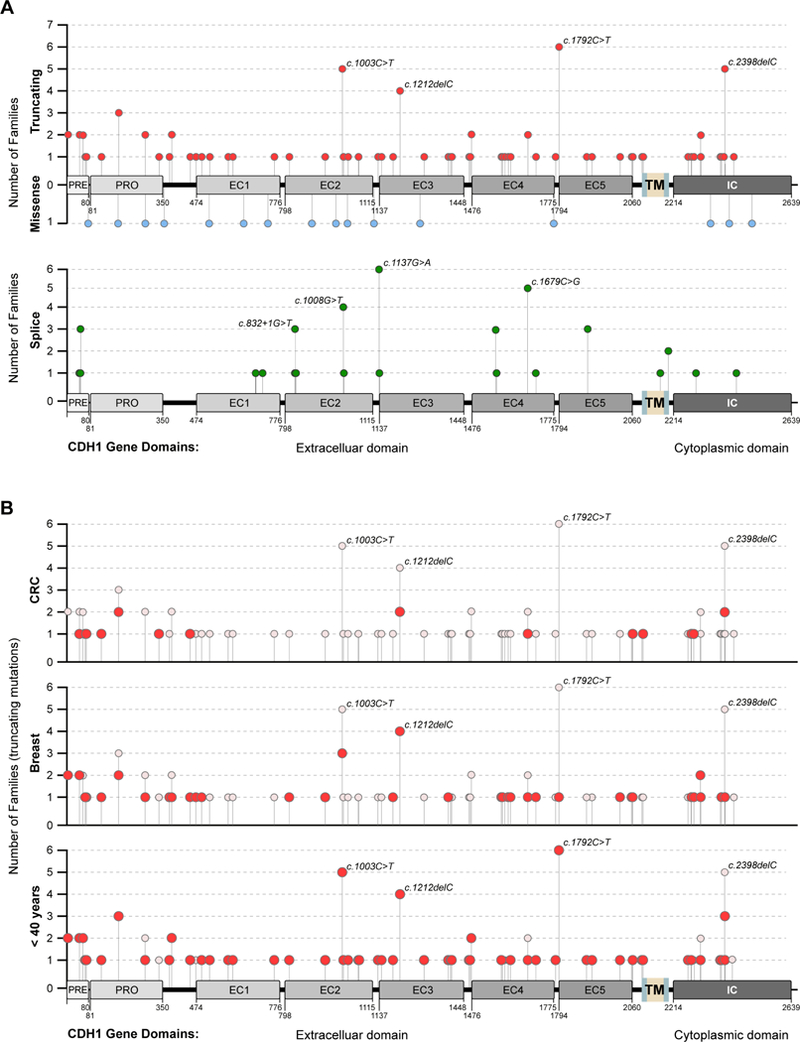
(A) Gene map depicting location of truncating (red), missense (blue) and splice site (green) cDH1 germline variants in relation to functional E-cadherin domains. Number of recurrent variants in independent families indicated on y-axis. (B) gene maps of truncating cDH1 variants of HDGC families with family members affected by colorectal (top), breast (mid) and early cancer (gastric cancer <40 years or lobular breast cancer <50 years) (bottom). Total number of families with individual cDH1 genotype shown on y-axis, and full circles indicate number of families with respective phenotype.
Gastric cancer, non-gastric cancer manifestations and clinical phenotypes of HDGC
The following data were extracted from individual reports: histopathological diagnosis of DGC or LBC in at least one affected family member, total number of family members affected by gastric cancer (including histologically confirmed diagnosis of ≥T1 a gastric cancers in risk-reducing gastrectomy specimens), number of family members diagnosed with breast cancer, age of diagnosis and gender. HDGC-associated cancer occurring at young age was defined as gastric cancer diagnosed at less than 40 years of age or LBC at less than 50 years.7 Family members affected by colon cancer were also captured, though not required to harbour histopathological criteria of signet ring cell morphology or mucinous differentiation. Online supplementary table 1 summarises clinicopathological and genetic information of the study population.
Immunohistochemistry
Immunohistochemical stains for β-Catenin (clone 17C2, Leica Biosystems Newcastle, Newcastle Upon Tyne, UK, #B-CAT-L-U, 1:100), E-cadherin (clone 36B5, Leica Biosystems Newcastle, #E-CAD-L-CE, 1:100) and Ki-67 (clone KI-67, #M7240, 1:300) were carried out after antigen retrieval. Antigen retrieval was carried out for 25 min. BondMax (#DS9800) was used with diaminobenzidine as chromogen. Immunohistochemical stains for phospho-mTOR Ser2448 (Cell Signal Technology, Danvers, Massachusetts, USA, Cat. #2971, 1:100), phospho-Akt S473 (Cell Signal Technology, Cat. #9721, 1:50), phospho-Akt T308 (Cell Signal Technology, Cat.# 9275, 1:100) and phospho-ERK (Cell Signal Technology, Clone 20G11, Cat.#4376, 1:500) were carried out after heat induced antigen retrieval using citrate buffer (10 mM citric acid, 0.05% Tween-20, pH10). The primary antibodies were incubated overnight at 4°C, detected using Envision + Dual labelled polymer (DAKO, K4061) and visualised with 3,3-diaminobenzadine (DAKO). Slides were lightly counterstained with haematoxylin, dehydrated in a series of graded ethanol, cleared in xylene and coverslipped. Histopathology and immunohistochemistry was reviewed by the three study pathologists (AP SH and MM).
statistical methods
Fisher’s exact test was used to compare the following phenotypes: family members affected by colorectal cancer and breast cancer or family member affected by cancer at a young age (gastric cancer at <40 years of age or LBC at <50 years). Mehta’s modification to Fisher’s exact test was used to test for an association between unordered categorical parameters and a dichotomous parameter.30 Associations between the phenotypes and the domain of variants were calculated using Fisher’s exact test.
Logistic regression analysis was conducted to examine if truncating variants in a given domain of CDH1 might be significantly associated with clinical phenotypes. ORs were calculated to quantify associations of truncating variants affecting prespecified domains of CDH1 and phenotypes compared with other domains of CDH1 (OR >1 indicates an increased risk of a clinical phenotype for a given domain compared with the risk for other domains).
Molecular modelling
Human CDH1 (UniProt ID: P12830–1) is 882 aa long and includes five cadherin domains (cadherin-1 to cadherin-5; EC1-EC5). Full-length experimental three-dimensional (3D) information is not available for CDH1, but crystal structures are available for cadherin domains 1 and 2 (155–367 aa) and the C-terminal end region, aa 756–773. To assess variantal impact on CDH1 function, we created full-length CDH1 protein models using I-TASSER online server, a bioinformatics tool for predicting 3D structure model of protein molecules from aa sequences (http://zhanglab.ccmb.med.umich.edu/I-TASSER). Homodimer CDH1 was created using two first-ranked (based on confidence score, C-Score) I-TASSER monomer models using the mouse X-ray crystal structure, PDB ID 3 3Q2V as the template. The structure overlay and the figures were created using the BIOVIA Discovery Studio Visualizer (versions 4.1 and 4.5; Accelrys/Biovia Software).
RESULTS
Phenotypes of HDGC families with pathogenic truncating and missense variants
Initially, a total of 195 unique CDH1 variants were identified throughout all 16 exons of the CDH1 gene from the selected 45 sources as the result of the systematic review (online supplementary figure 1). The four most common causes for exclusion after the initial screen (full text review) were: (1) lack of CDH1 variant testing in clinical reports of HDGC, (2) unavailability of family history information with reported CDH1 germline variant, (3) inability to accurately predict protein alteration and (4) the reported variant substitution did not match to the human CDH1 wild type sequence. After removal of large genomic deletion variants (six families) where in silico tools were unable to accurately classify the protein product and duplicate reports, a total of 92 families with truncating, 16 families with missense and 44 families with splice site germline CDH1 variants with complete information were identified (online supplementary table 1). Applying the criteria PVS1 (null variant), PM2 (absent in population databases), PM4 (protein length change) and for splice site variants only PS3 (in vitro or in vivo functional studies supportive of damaging effect on gene product) according to CDH1 variant curation specifications outlined by ClinGen CDH1 VCEP, all truncating variants were classified as pathogenic (P) or likely pathogenic (LP) compared with 23 out of 26 individual splice site and none of the missense variants (online supplementary table 1). With the applied criteria, the odds for a truncating CDH1 variant to be classified as pathogenic compared with splice site variants were 75.9 (Fisher’s exact test; p<0.001) and 7.67 (p = 0.0161) for either pathogenic or likely pathogenic, respectively.
There were no discernable differences in the clinical phenotypes between the three groups (table 1). About half of families with truncating variants also had at least one family member affected by breast cancer, whereas 19% of families with missense variants and 34% with splice site variants had one family member or more affected by breast cancer (p=0.06). Twenty per cent of HDGC families with truncating CDH1 variants also had one or more family members affected by CRC, compared with 33% of HDGC families with missense and 13% with splice site variants. (table 1).
Table 1.
Demographic characteristics of study subjects with truncating, missense and splice site CDH1 germline variants
| Truncating | Missense | splice | P value* | |
|---|---|---|---|---|
| Number of families | 92 | 16 | 44 | - |
| Mean age, youngest affected family member (years) | 31.3 | 38.3 | 31.5 | - |
| Families with ≥1 family member affected by cancer at young age (gastric cancer <40 or lobular breast cancer <50 years of age) | 82% (75/92) | 67% (10/15) | 81% (34/42) | 0.37 |
| Average number of affected family members by GC† | 4.05 | 3.75 | 5.55 | - |
| Families with ≥1 family member affected by BC | 48% (43/90) | 19% (3/16) | 34% (15/44) | 0.06 |
| Average number of family members affected by BC† | 1.98 | 1 | 1.67 | - |
| Families with ≥1 family member affected by CRC | 20% (17/86) | 33% (4/12) | 13% (5/38) | 0.3 |
| Average number of family members affected by CRC† | 1.41 | 1.75 | 1.6 | - |
p values were given by Fisher’s exact test.
mean number of affected family members of families with ≥1 affected family member of respective cancer.
GC, gastric cancer; BC, breast cancer; CRC, colorectal cancer.
Non-random distribution of CDH1 germline variants in HDGC families
Distribution of CDH1 germline variants across all CDH1 coding regions showed different regions of the CDH1 gene more frequently affected than others (figure 1A). The CDH1 coding regions are comprised of cadherin-pre and cadherin-pro signal peptides, five calcium-binding extracellular cadherin (EC) repeat domains (domains 1–5) and the intracellular cytoplasmatic region of CDH1 (including docking regions for p120 and β-catenin) (figure sourced from http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi?seqinput=NP_001304113.1). Variant rates of truncating variants per base pair (bp) varied from 0.01500 for the EC5 domain as the least affected to 0.10000 (p = 0.0059) for the cadherin-pre domain (PRE signal peptide; nucleotides 1–80) as the most frequently affected. Another site frequently affected by truncating variants were the linker regions, which do not involve known functional CDH1 domains (truncating variant rate per bp 0.05785, p = 0.0111) (online supplementary table 2). In contrast, splice site variants were enriched in the PRE (0.06250; p = 0.0097) and EC domain 2 (EC2) (0.03140; p = 0.0325) and EC domain 4 (EC4) (0.03330; p = 0.0226), the less frequent missense mutations did not show any enrichment.
While the majority of variants were found in single families, there were recurrent variants (‘hotspots’) reported in several locations involving at least four families. These included the truncating variants c.1003C>T, c.1212delC, c.1792C>T and c.2398delC and the splice site variants c.1008G>T, c.1137G>A and c.1679C>G (in ≥4 families) (figure 1A).
CDH1 genotype-cancer phenotype correlations in HDGC families afflicted by truncating CDH1 germline variants
We first examined potential associations between the three clinical phenotypes (breast cancer, colorectal cancer and diagnosis of cancer at young age (gastric cancer <40 years of age or LBC <50 years) in HDGC families with truncating CDH1 variants. There were no discernible associations between the three cancer phenotypes (online supplementary table 3). Next, we examined the location of truncating variants for any associations with the three clinical phenotypes shown in figure 1B. EC1 and EC2 were combined and analysed jointly, as well as EC domains 3–5 due to similar functions. EC1 and EC2 are involved in regulation of the cadherin dimerisation state by modulation of E-cadherin molecules homodimerisation. EC1 and EC2 are involved in regulation of the cadherin dimerisation state by modulation of E-cadherin molecules homodimerisation.31 C-terminal cadherin domains EC3, EC4 and EC5 are involved in heterodimerisation involving other adhesion molecules or receptor tyrosine kinases.32–34 PRE and PRO domains functioning as signal peptides and regulating intracellular trafficking were combined as well.
Family members with variants in the PRE-PRO region were more likely to be diagnosed with colorectal cancer, when compared with variants in other regions (p = 0.01) (online supplementary table 4). Families not affected by early cancer (gastric cancer <40 or LBC <50 years of age) more frequently had truncating CDH1 variants located in the intracellular (IC) domain or adjacent EC3/4/5 domains (p = 0.022). There was not a strong association between the location of truncating variants in a particular region and the frequency of breast cancer; breast cancer was more often reported in HDGC families with truncating CDH1 variants affecting the EC domains EC¾/5 and PRE-PRO regions compared with linker regions not involving functional domains (p = 0.125), which seemed to be protective (online supplementary table 4). Logistic regression analysis revealed an increased probability of colorectal cancer in families with truncating variants affecting the PRE-PRO region (OR 6.20, 95% CI 1.79 to 21.48; p = 0.004), a protective effect of variants occurring in the IC domain of the E-cadherin receptor for early cancer (gastric cancer diagnosed at <40 years of age or LBC at <50 years) (OR 0.2, 95% CI 0.06 to 0.64; p = 0.0071) and a decreased frequency of breast cancer when variants affecting linker regions between functional E-cadherin domains (OR 0.35, 95% CI 0.12 to 0.99; p = 0.0493) (table 2; figure 2).
Table 2.
OR of a given domain versus other domains including 95% CIs and p values by logistic regression of domain of truncating CDH1 variant and clinical phenotype
| Colorectal cancer | ||||
|---|---|---|---|---|
| Domain | Estimate | 2.5% | 97.5% | P value |
| PRE-PRO | 6.20 | 1.79 | 21.477 | 0.004 |
| EC 1 and 2 | NA | NA | NA | NA |
| EC 3, 4 and 5 | 0.872 | 0.252 | 3.021 | 0.8287 |
| IC | 1.622 | 0.445 | 5.91 | 0.4632 |
| Linker | 0.351 | 0.073 | 1.681 | 0.1902 |
| Breast cancer | ||||
| PRE-PRO | 2.545 | 0.793 | 8.171 | 0.1164 |
| EC 1 and 2 | 0.685 | 0.222 | 2.115 | 0.5104 |
| EC 3,4 and 5 | 2.038 | 0.775 | 5.36 | 0.1488 |
| IC | 0.821 | 0.277 | 2.437 | 0.7223 |
| Linker | 0.346 | 0.12 | 0.993 | 0.0493 |
| Affected family members at young age* | ||||
| PRE-PRO | 1.444 | 0.292 | 7.136 | 0.6518 |
| EC 1 and 2 | NA | NA | NA | NA |
| EC 3, 4 and 5 | 0.683 | 0.209 | 2.226 | 0.5269 |
| IC | 0.195 | 0.059 | 0.641 | 0.0071 |
| Linker | 2.333 | 0.486 | 11.213 | 0.2901 |
Gastric cancer at <40 years or lobular breast cancer <50 years.
EC, extracellular; IC, intracellular; NA, too few counts to calculate OR.
Figure 2.
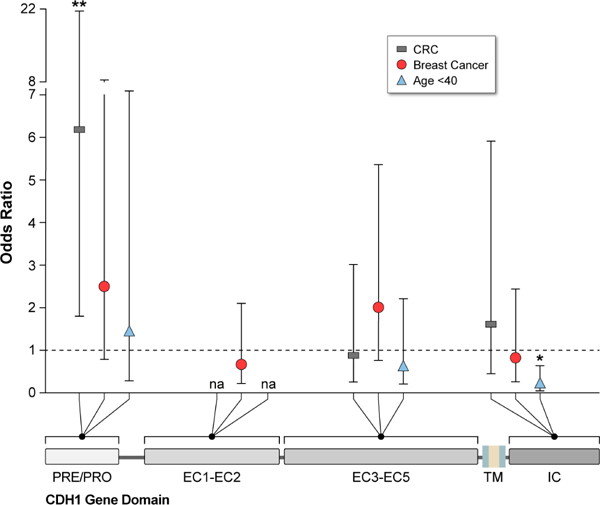
ORs of phenotypes colorectal cancer, breast cancer and cancer at young age (gastric cancer <40 years or lobular breast cancer <50 years) by location of truncating CDH1 germline variant (by CDH1 nucleotide position of functional domain) (*p<0.05; **p<0.01). EC, extracellular; IC, intracellular.
Association of predicted NMD of truncating CDH1 variants and clinical phenotypes
To assess if NMD was associated with clinical phenotype in HDGC families, truncating variants with premature termination codons (PTCs) located within 50 bps upstream of the last exon-exon junction were classified as NMD deficient and truncating variants with PTCs more than 50 bps upstream of the last exon-intron junction as NMDcompetent (online supplementary table 1; online supplementary figure 3). HDGC families with CDH1 germline variants predicted to undergo NMD more frequently had family members affected by early cancer (gastric cancer diagnosed at <40 years of age or LBC at <50 years) compared with families with CDH1 variants evading NMD (p = 0.029) (online supplementary table 5).
Impact of location of truncating CDH1 variant on signal transduction in early T1a lesions
To generate a molecular rationale for the observed genotype-phenotype associations of the enrichment of truncating CDH1 variants in known CDH1 functional domains and different clinical phenotypes, we first built three-dimensional comparative models using a novel hierarchical threading and assembly refinement approach adopted by software protein structure prediction tool I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). The first-rank confidence score model is shown in online supplementary figure 4A and B and was used as a representative model for CDH1. Within the predicted wild-type model, the cytoplasmatic p120 catenin and β-catenin binding domains were marked and the expanded views display the available cocrystallised experimental complexes of the human β-catenin-CDH1 and the homologous mouse p120 β-catenin-CDH1. The CDH1 model can be used to visualise how a c.187C>T substitution leads to a truncated protein (p.Ar-g63Term; CM980317) with the loss of all the key domains (EC1 until the C-terminal end) beyond position 63, whereas a variant within the IC domain like c.2310_2310delC will lead to a CDH1 mutant that might be able to be integrated into the cell membrane and bind to p120 but is devoid of β-catenin retention. Thus, different CDH1 variants impacting different CDH1 functions might translate into different clinical phenotypes.
To associate the different clinical manifestations with different signal transduction states measured by histopathology, we studied activation states of MAPK, PI3K-AKT and β-catenin signalling in early T1a lesions in prophylactic gastrectomy specimens of four HDGC families with CDH1 variants affecting different regions of CDH1 (table 3). T1a lesion from families with del124_126C-CCinsT (PRO region), c.521dupA (EC1), c.1565 + 1G>A (EC4) and 2295+2T>G (intracellular region) were examined for expression levels of phosphorylated AKT, mTOR and ERK, as well as Ki67, E-cadherin and cellular localisation of β-catenin. All T1a lesions were small, submucosal foci of signet ring cell clusters that showed no signs of proliferation as measured by Ki67 levels (figure 3). There were differences in phospho-mTOR expression levels and β-catenin activation states depending on CDH1 genotype. T1a lesions with del124_126CCCinsT (PRO region) variants showed activation of P-catenin and mTOR, whereas T1a lesions with the 2295+2T>G CDH1 variant (IC region) had no activation. Some activation of P-catenin (as measured by nuclear translocation) was observed in all families, with the exception of c.2295+2T>G mutant T1a gastric cancer.
Table 3.
Summary of immunolabeling of early HDGCs from families with germline c.del124_126CCCinsT, c.521dupA, c.1565+1G>A and 2295+2T>G CDH1 variants
| del124_126CCCinsT | c.521dupA* | c.521dupA* | c.1565+1G>A | 2295+2T>G | 2295+2T>G† | |
|---|---|---|---|---|---|---|
| ECAD | +/– | – | – | +/– | – | – |
| Ki-67 | – | – | – | – | – | – |
| β-catenin | ++ | + | + | ++ | – | + |
| p-AKT | – | – | – | – | – | +/– |
| p-mTOR | + | +/– | – | + | – | ++ |
| p-ERK | – | +/– | – | +/– | – | ++ |
Two different 521dupA CDH1 variant carriers from same HDGC family.
2295+2* 2295+2T>G CDH1 variant carrier from c.2295+2T>G CDH1 HDGC family with stage IV disease (peritoneal implant of right colon).
HDGC, hereditary diffuse gastric cancer.
Figure 3.
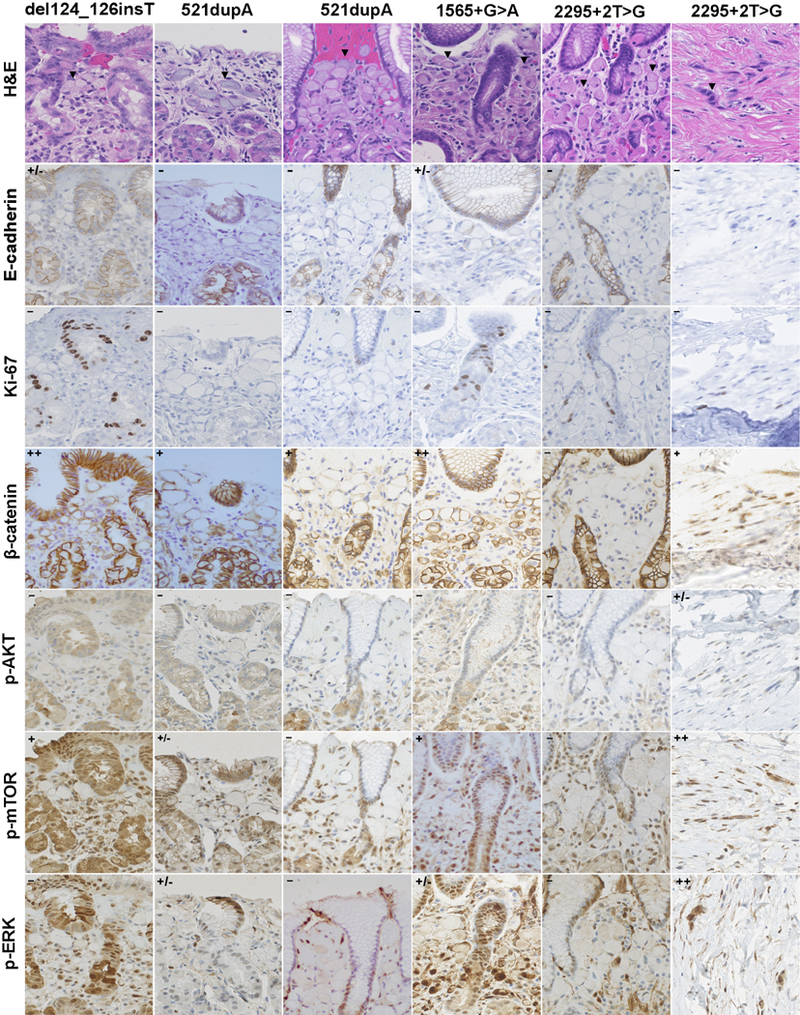
Signal transduction perturbations in early intramucosal T1a HDGC lesions from CDH1 carriers with truncating del124_126CCCinsT (PRO region), c.521dupA (EC1), c.1565+1G>A (EC4) and 2295+2T>G (1C) variants. c.521 dupA variant T1a lesions from two variant carriers are shown. For HDGC families with c.2295+2T>G variant, H&E and immunostains are shown for both early HDGC (T1a intramucosal lesion) and stage IV (serosal implant of peritoneal carcinomatosis; last column). c.2295+2T>G CDH1 germline variants were confirmed in both individuals. H&E shows dispersed intramural signet ring cell clusters (<20 cells) within mucosal glands, and arrow indicates signet ring cells with typical peripheral nucleus. p-catenin activation was scored on nuclear staining levels of early HDGC lesions, p-mTOR immunolabeling was found observed both in cytoplasmatic and nuclear cellular locations and p-ERK labelling (c.1 565+1G>A and 2295+2T>G CDH1 variant in stage IV lesion) was more pronounced in the nucleus (20x). EC, extracellular; IC, intracellular.
DISCUSSION
Several studies evaluating cancer predisposition syndromes have identified shared genotype-phenotype associations across families and affected family members; these associations can be used to derive and calculate risk estimates and recommend individualised surveillance and risk-reducing interventions based on identified selective genotypes. In BRCA2, multiple breast cancer and three ovarian cancer cluster regions are associated with relative hazard rations ranging from 0.57 to 2.3.2,3 In patients with FAP, distinct genotype-phenotype correlations have been described between APC gene variants and various extracolonic manifestations, severity of polyposis, risk of early development of colorectal cancer and death.4,5,35 Similarly, the 2015 clinical guidelines for management of HDGC note a persistent need for developing similar correlations in familial gastric cancer with germline CDH1 variants.7 IGCLC has developed screening guidelines for families at risk recommending germline CDH1 variant testing by the patient’s mid-20s; if tested positive, patients are recommended to consider breast and gastric surveillance with consideration of risk-reducing total gastrectomy before the age of 30 years.7,16,24 These recommendations are based on the observations that the cumulative life-time risk of gastric cancer before age 16 years is less than 1% but increased to more than 4% by age 30 years or an increase in breast cancer risk >50 years of age.8,9 These recommendations are currently applied indiscriminately to all CDH1 germline variant carriers irrespective of individual CDH1 variant genotype and/or family history.
There is increasing recognition that CDH1 germline variants are associated with multiple disorders of a wider disease spectrum beyond elevated susceptibility for diffuse gastric and LBC rather than one disease state.24,36,37 It however currently remains undetermined whether colorectal cancer is also a manifestation of HDGC: while several reports suggest an association with colorectal cancer, no increased overall incidence of CRC has been reported in patients with HDGC so far.7,10,14,15,38 The updated 2015 IGCLC guidelines recommend in HDGC families with CDH1 germline variants in which colon cancer is reported in variant carriers to collect age at diagnosis, variant status and whether the histopathology showed a mucinous component and/or signet ring cells.7 For such families, enhanced colonoscopy screening should be considered at age 40 years or 10 years younger than the youngest diagnosis of colon cancer, whichever is younger, and repeated at intervals of 3–5 years, whereas for families not affected by colorectal cancer national guidelines for colorectal cancer screening should be followed.7 Here, we noted significant enrichment of early truncating variants in the PRE-PRO region of CDH1 in HDGC families affected by CRC (0R=6.20, 95% CI 1.79 to 21.48; p=0.004), suggesting an association with increased risk of colorectal cancer in this patient subset. Together with the finding that 20% of HDGC families with truncating CDH1 variants, and between 13% and 33% of families with splice site and missense variants, had at least one family member affected by CRC, which is higher than population-based risks of colorectal cancer, appears to be consistent with prior reports that E-cadherin loss can accelerate colon cancer progression.39,40 In addition, this analysis also identified variants associated with breast and gastric cancer frequencies: truncating CDH1 variants in the linker region were associated with a reduced frequency of breast cancer and variants in the IC domain with a reduced frequency of early cancer (gastric cancer occurring at <40 or LBC occurring at <50 years of age).
There are a number of preclinical and clinical observations that support the finding of genotype-phenotype associations in HDGC due to CDH1 germline variants. Preclinically, variants disrupting different functional domains of E-cadherin have been associated with different tumour suppressor functions. Sasaki and colleagues were the first to show that the loss of EC domains abrogates cell-cell contact but suppresses tumour cell growth via redistribution of β-catenin to the cell membrane due to intact interactions with the catenin domain.41 Deletion variants in exons 8 and 9 affect the EC-binding domains of E-cadherin, subsequently increasing EGFR activation, and carry another distinct tumour suppressor function.33,42 Cancer cell lines derived from patients with missense variants in the extracellular domains of E-cadherin were correspondingly less able to suppress EGFR signalling compared with cell lines with wild type E-cadherin thus possibly driving a unique phenotype.33,34,41–43 It also can be speculated that the loss of signal peptide function of E-cadherin due to loss of the PRE-PRO region may be linked to a more virulent phenotype. The lack of targeting the nascent translated protein chain to the endoplasmatic reticulum might lead to aberrant intracellular trafficking with possible deleterious interference in protein homeostasis of the cell and the complete absence of any E-cadherin fragments reaching the cell surface may both aid malignant transformation. Thus, variants affecting different functional domains may generate loss of different tumour suppressor functions and possibly explain the different cancer manifestations within the syndrome.
To provide some molecular correlation for the observed genotype-phenotype associations and the heterogeneity in the clinical phenotype, we screened T1a lesions of four different HDGC families with different CDH1 germline variants for activation of β-catenin, PI3K-AKT or ERK signalling and identified different activation states in T1a lesions of gastrectomy specimens from different CDH1 germline variant carriers. Our group has previously shown that c.1380delA CDH1 (EC3) mutant gastric cancer cells derived from a HDGC patient with a germline c.1380delA variant showed increased sensitivity to anti-Src, FAK, mTOR and MEK class small molecule inhibitors which, in part, appears to align with the findings of elevated mTOR and p-ERK levels measured in above T1 lesions.44 Another possible explanation for the different intracellular transduction perturbations and variable cancer phenotypes might be differences in truncating CDH1 variants’ ability to be subjected to NMD.22 Our findings of HDGC families with truncating CDH1 germline variants subject to NMD being more frequently affected by cancer at a young age (gastric cancer <40 years or LBC <50 years of age) is in line with an earlier study that investigated an association of NMD and age of onset of gastric cancer and reported that carriers with truncating CDH1 variants escaping NMD are less affected by early onset of gastric cancer.23 Other observations that support that different CDH1 genotypes are associated with different biology and clinical manifestations in this cancer predisposition syndrome include the observation that somatic structural CDH1 alterations conferred a poorer outcome when compared with epigenetic silencing or differences in the second hit of the CDH1 allele in primary versus metastatic lesions.19,45
While important, the presented findings must be interpreted within the limitations of our study. Most critically, while we included six previously unreported HDGC families, the majority of our analysis is based on previously reported HDGC families and incurs the risk of observational and reporting bias. Under-reporting of HDGC families with germline CDH1 variants from developing countries, of families with recurrent CDH1 variants and reliance on recent reports could all have introduced bias and skewed results. Adoption of screening and prophylactic interventions are known to vary geographically, thus the variable age of youngest affected family by cancer has to be interpreted with caution.25 The lack of longitudinal information and control for ascertainment bias is yet another limitation. Some of the reports predated the inclusion of LBC as the second cancer phenotype of HDGC into IGCLC guidelines. Thus, information on the subtype of breast cancer was not always available. Equally, no histopathological information on the colorectal cancer cancer with regard to signet ring cell or mucinous differentiation was available. As comparisons between family history-based and population-based cancer risks are subject to many variations, all ORs are relative ORs to other functional regions of the CDH1 gene and not risk estimates compared with general population-based risks. When scoring variants for pathogenicity, we followed recently released guidelines of the ClinGen CDH1 VCEP implemented by the ClinGen Hereditary Cancer Domain, which recommended to use specifications of the ACMG and AMP for the classification of pathogenicity of CDH1 germline variants.27,28 Since only three of the listed criteria (PVS1, PM2 and PM4; and PS3 for splice site only) were consistently available in the identified reports, we might have under-reported pathogenic or likely pathogenic variants. In light of the universal scoring of truncating variants with the available criteria as pathogenic or likely pathogenic, and only a minority of splice site variants (3 out of total of 26 individual splice site variants) classified as of unknown significance, any impact onto the analysis is likely to be very small. As emphasised by ClinGen CDH1 VCEP, missense CDH1 variants to date have not been confidently proven to be disease causing, and these variants were not included into the analysis.28 Our exclusion criteria where no protein product could be predicted, where the index variants were incorrect or where large portions of the gene were deleted might have skewed the dataset, however, was in our opinion necessary in order to only include high confidence, accurately curated variants. Of note, two of the four described HDGC families with large genomic deletions involving coding sequences of the PRE-PRO region including the start codon (chrl 6: deletion 67 193 822–67 387 415 (del exon 1–2 [193 593 bp]) were reported to have family members affected by colorectal cancer, a finding that would have strengthened the association of loss-of-function variants affecting the PRE-PRO region and colorectal cancer risk further.9
In conclusion, we identified associations between locations of CDH1 truncating germline variants and cancer frequencies across CDH1 domains that may aid to individualise screening, surveillance and risk-reducing intervention strategies or design chemopreventive programmes aimed at high-risk subgroups. The association of colorectal cancer and variants in the PRE-PRO-region suggests a potential role for enhanced colonoscopic surveillance in certain families with HDGC. The inherent heterogeneity of the disease will require more collection of clinical and genetic information of HDGC families to further refine genotype-phenotype associations and to create risk prediction models.
Supplementary Material
Acknowledgements
We would like to thank Alan Hoofring from Medical illustration, NIH Medical Arts Branch, for his significant help with the figure development for this manuscript.
Funding This work was supported by the intramural research program from the National Cancer institute, National institutes of Health, under contract HHSN261200800001E.
Footnotes
Disclaimer The content of this publication does not reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available upon reasonable request to the corresponding author.
Additional material is published online only. To view please visit the journal online (http://dx.doi.org/10.1136/jmedgenet-2018-105361).
REFERENCES
- 1.Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol 2014;32:2001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gayther SA, Warren W, Mazoyer S, Russell PA, Harrington PA, Chiano M, Seal S, Hamoudi R, van Rensburg EJ, Dunning AM, Love R, Evans G, Easton D, Clayton D, Stratton MR, Ponder BA. Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nat Genet 1995;11:428–33. [DOI] [PubMed] [Google Scholar]
- 3.Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, Mazoyer S, Chenevix-Trench G, Easton DF, Antoniou AC, Nathanson KL, Laitman Y, Kushnir A, Paluch-Shimon S, Berger R, Zidan J, Friedman E, Ehrencrona H, Stenmark-Askmalm M, Einbeigi Z, Loman N, Harbst K, Rantala J, Melin B, Huo D, Olopade Oi, Seldon J, Ganz PA, Nussbaum RL, Chan SB, Odunsi K, Gayther SA, Domchek SM, Arun BK, Lu KH, Mitchell G, Karlan BY, Walsh C, Lester J, Godwin AK, Pathak H, Ross E, Daly MB, Whittemore AS, John EM, Miron A, Terry MB, Chung WK, Goldgar DE, Buys SS, Janavicius R, Tihomirova L, Tung N, Dorfling CM, van Rensburg EJ, Steele L, Neuhausen SL, Ding YC, Ejlertsen B, Gerdes AM, Hansen T, Ramón y Cajal T, Osorio A, Benitez J, Godino J, Tejada Mi, Duran M, Weitzel JN, Bobolis KA, Sand SR Fontaine A, Savarese A, Pasini B, Peissel B, Bonanni B, Zaffaroni D, Vignolo-Lutati F, Scuvera G, Giannini G, Bernard L, Genuardi M, Radice P, Dolcetti R, Manoukian S, Pensotti V, Gismondi V, Yannoukakos D, Fostira F, Garber J, Torres D, Rashid MU, Hamann U, Peock S, Frost D, Platte R, Evans DG, Eeles R, Davidson R, Eccles D, Cole T, Cook J, Brewer C, Hodgson S, Morrison PJ, Walker L, Porteous ME, Kennedy MJ, izatt L, Adlard J, Donaldson A, Ellis S, Sharma P, Schmutzler RK, Wappenschmidt B, Becker A, Rhiem K, Hahnen E, Engel C, Meindl A, Engert S, Ditsch N, Arnold N, Plendl HJ, Mundhenke C, Niederacher D, Fleisch M, Sutter C, Bartram CR, Dikow N, Wang-Gohrke S, Gadzicki D, Steinemann D, Kast K, Beer M, Varon-Mateeva R, Gehrig A, Weber BH, Stoppa-Lyonnet D, Sinilnikova OM, Mazoyer S, Houdayer C, Belotti M, Gauthier-Villars M, Damiola F, Boutry-Kryza N, Lasset C, Sobol H, Peyrat JP, Muller D, Fricker JP, Collonge-Rame MA, Mortemousque i, Nogues C, Rouleau E, isaacs C, De Paepe A, Poppe B, Claes K, De Leeneer K, Piedmonte M, Rodriguez G, Wakely K, Boggess J, Blank SV, Basil J, Azodi M, Phillips KA, Caldes T, de la Hoya M, Romero A, Nevanlinna H, Aittomäki K, van der Hout AH, Hogeivorst FB, Verhoef S, Collée JM, Seynaeve C, Oosterwijk JC, Gille JJ, Wijnen JT, Gómez Garcia EB, Kets CM, Ausems MG, Aalfs CM, Devilee P, Mensenkamp AR, Kwong A, Olah E, Papp J, Diez O, Lazaro C, Darder E, Blanco i, Salinas M, Jakubowska A, Lubinski J, Gronwald J, Jaworska-Bieniek K, Durda K, Sukiennicki G, Huzarski T, Byrski T, Cybulski C, Toloczko-Grabarek A, Zfowocka-Perfowska E, Menkiszak J, Arason A, Barkardottir RB, Simard J, Laframboise R, Montagna M, Agata S, Alducci E, Peixoto A, Teixeira MR, Spurdle AB, Lee MH, Park SK, Kim SW, Friebel TM, Couch FJ, Lindor NM, Pankratz VS, Guidugli L, Wang X, Tischkowitz M, Foretova L, Vijai J, Offit K, Robson M, Rau-Murthy R, Kauff N, Fink-Retter A, Singer CF, Rappaport C, Gschwantler-Kaulich D, Pfeiler G, Tea MK, Berger A, Greene MH, Mai PL, imyanitov EN, Toland AE, Senter L, Bojesen A, Pedersen iS, Skytte AB, Sunde L, Thomassen M, Moeller ST, Kruse TA, Jensen UB, Caligo MA, Aretini P, Teo SH, Selkirk CG, Hulick PJ, Andrulis i. CiMBA Consortium. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015;313:1347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soravia C, Berk T, Madlensky L, Mitri A, Cheng H, Gallinger S, Cohen Z, Bapat B. Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet 1998;62:1290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertario L, Russo A, Sala P, Varesco L, Giarola M, Mondini P, Pierotti M, Spinelli P, Radice P Hereditary Colorectal Tumor Registry. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol 2003;21:1698–707. [DOI] [PubMed] [Google Scholar]
- 6.Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature 1998;392:402–5. [DOI] [PubMed] [Google Scholar]
- 7.van der Post RS, Vogelaar iP, Carneiro F, Guilford P, Huntsman D, Hoogerbrugge N, Caldas C, Schreiber KE, Hardwick RH, Ausems MG, Bardram L, Benusiglio PR, Bisseling TM, Blair V, Bleiker E, Boussioutas A, Cats A, Coit D, DeGregorio L, Figueiredo J, Ford JM, Heijkoop E, Hermens R, Humar B, Kaurah P, Keller G, Lai J, Ligtenberg MJ, O’Donovan M, Oliveira C, Pinheiro H, Ragunath K, Rasenberg E, Richardson S, Roviello F, Schackert H, Seruca R, Taylor A, Ter Huurne A, Tischkowitz M, Joe ST, van Dijck B, van Grieken NC, van Hillegersberg R, van Sandick JW, Vehof R, van Krieken JH, Fitzgerald RC. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pharoah PD, Guilford P, Caldas C. international Gastric Cancer Linkage Consortium. incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001;121:1348–53. [DOI] [PubMed] [Google Scholar]
- 9.Hansford S, Kaurah P, Li-Chang H, Woo M, Senz J, Pinheiro H, Schrader KA, Schaeffer DF, Shumansky K, Zogopoulos G, Santos TA, Claro i, Carvalho J, Nielsen C, Padilla S, Lum A, Talhouk A, Baker-Lange K, Richardson S, Lewis i, Lindor NM, Pennell E, MacMillan A, Fernandez B, Keller G, Lynch H, Shah SP, Guilford P, Gallinger S, Corso G, Roviello F, Caldas C, Oliveira C, Pharoah PD, Huntsman DG. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol 2015;1:23–32. [DOI] [PubMed] [Google Scholar]
- 10.Kaurah P, MacMillan A, Boyd N, Senz J, De Luca A, Chun N, Suriano G, Zaor S, Van Manen L, Gilpin C, Nikkel S, Connolly-Wilson M, Weissman S, Rubinstein WS, Sebold C, Greenstein R, Stroop J, Yim D, Panzini B, McKinnon W, Greenblatt M, Wirtzfeld D, Fontaine D, Coit D, Yoon S, Chung D, Lauwers G, Pizzuti A, Vaccaro C, Redal MA, Oliveira C, Tischkowitz M, Olschwang S, Gallinger S, Lynch H, Green J, Ford J, Pharoah P, Fernandez B, Huntsman D. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007;297:2360–72. [DOI] [PubMed] [Google Scholar]
- 11.Benusiglio PR, Malka D, Rouleau E, De Pauw A, Buecher B, Noguès C, Fourme E, Colas C, Coulet F, Warcoin M, Grandjouan S, Sezeur A, Laurent-Puig P, Molière D, Tlemsani C, Di Maria M, Byrde V, Delaloge S, Blayau M, Caron O. CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: a multicentre study. J Med Genet 2013;50:486–9. [DOI] [PubMed] [Google Scholar]
- 12.Petridis C, Shinomiya i, Kohut K, Gorman P, Caneppele M, Shah V, Troy M, Pinder SE, Hanby A, Tomlinson i, Trembath RC, Roylance R, Simpson MA, Sawyer EJ. Germline CDH1 mutations in bilateral lobular carcinoma in situ. Br J Cancer 2014;110:1053–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corso G, Figueiredo J, La Vecchia C, Veronesi P, Pravettoni G, Macis D, Karam R, Lo Gullo R, Provenzano E, Toesca A, Mazzocco K, Carneiro F, Seruca R, Melo S, Schmitt F, Roviello F, De Scalzi AM, intra M, Feroce i, De Camilli E, Villardita MG, Trentin C, De Lorenzi F, Bonanni B, Galimberti V. Hereditary lobular breast cancer with an emphasis on E-cadherin genetic defect. J Med Genet 2018;55:431–41. [DOI] [PubMed] [Google Scholar]
- 14.Richards FM, McKee SA, Rajpar MH, Cole TR, Evans DG, Jankowski JA, McKeown C, Sanders DS, Maher ER. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet 1999;8:607–10. [DOI] [PubMed] [Google Scholar]
- 15.Brooks-Wilson AR, Kaurah P Suriano G, Leach S, Senz J, Grehan N, Butterfield YS, Jeyes J, Schinas J, Bacani J, Kelsey M, Ferreira P, MacGillivray B, MacLeod P Micek M, Ford J, Foulkes W, Australie K, Greenberg C, LaPointe M, Gilpin C, Nikkel S, Gilchrist D, Hughes R, Jackson CE, Monaghan KG, Oliveira MJ, Seruca R, Gallinger S, Caldas C, Huntsman D. Germline E-cadherin mutations in hereditary diffuse gastric cancer: assessment of 42 new families and review of genetic screening criteria. J Med Genet 2004;41:508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Post RS, Vogelaar iP, Manders P, van der Kolk LE, Cats A, van Hest LP, Sijmons R, Aalfs CM, Ausems MG, Gömez García EB, Wagner A, Hes FJ, Arts N, Mensenkamp AR, van Krieken JH, Hoogerbrugge N, Ligtenberg MJ. Accuracy of hereditary diffuse gastric cancer testing criteria and outcomes in patients with a germline mutation in CDH1. Gastroenterology 2015. 149:e19–. [DOI] [PubMed] [Google Scholar]
- 17.Pereira PS, Teixeira A, Pinho S, Ferreira P, Fernandes J, Oliveira C, Seruca R, Suriano G, Casares F. E-cadherin missense mutations, associated with hereditary diffuse gastric cancer (HDGC) syndrome, display distinct invasive behaviors and genetic interactions with the Wnt and notch pathways in drosophila epithelia. Hum Mol Genet 2006;15:1704–12. [DOI] [PubMed] [Google Scholar]
- 18.Figueiredo J, Söderberg O, Simoes-Correia J, Grannas K, Suriano G, Seruca R. The importance of E-cadherin binding partners to evaluate the pathogenicity of E-cadherin missense mutations associated to HDGC. Eur J Hum Genet 2013;21:301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oliveira C, Sousa S, Pinheiro H, Karam R, Bordeira-Carriço R, Senz J, Kaurah P Carvalho J, Pereira R, Gusmao L, Wen X, Cipriano MA, Yokota J, Carneiro F, Huntsman D, Seruca R. Quantification of epigenetic and genetic 2nd hits in CDH1 during hereditary diffuse gastric cancer syndrome progression. Gastroenterology 2009;136:2137–48. [DOI] [PubMed] [Google Scholar]
- 20.Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet 2016;17:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Yuan Y. Alternative RNA splicing and gastric cancer. Mutat Res 2017;773:263–73. [DOI] [PubMed] [Google Scholar]
- 22.Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 2015;16:665–77. [DOI] [PubMed] [Google Scholar]
- 23.Karam R, Carvalho J, Bruno i, Graziadio C, Senz J, Huntsman D, Carneiro F, Seruca R, Wilkinson MF, Oliveira C. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene 2008;27:4255–60. [DOI] [PubMed] [Google Scholar]
- 24.Kluijt i, Siemerink EJ, Ausems MG, van Os TA, de Jong D, Simoes-Correia J, van Krieken JH, Ligtenberg MJ, Figueiredo J, van Riel E, Sijmons RH, Plukker JT, van Hillegersberg R, Dekker E, Oliveira C, Cats A, Hoogerbrugge N. Dutch Working Group on Hereditary Gastric Cancer. CDH1-related hereditary diffuse gastric cancer syndrome: clinical variations and implications for counseling. Int J Cancer 2012;131:367–76. [DOI] [PubMed] [Google Scholar]
- 25.Corso G, Marrelli D, Pascale V, Vindigni C, Roviello F. Frequency of CDH1 germline mutations in gastric carcinoma coming from high-and low-risk areas: metanalysis and systematic review of the literature. BMC Cancer 2012;12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moher D, Liberati A, Tetzlaff J, Altman DG. PRiSMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRiSMA statement. PLoS Med 2009;6:e1000097. [PMC free article] [PubMed] [Google Scholar]
- 27.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee K, Krempely K, Roberts ME, Anderson MJ, Carneiro F, Chao E, Dixon K, Figueiredo J, Ghosh R, Huntsman D, Kaurah P Kesserwan C, Landrith T, Li S, Mensenkamp AR, Oliveira C, Pardo C, Pesaran T, Richardson M, Slavin TP, Spurdle AB, Trapp M, Witkowski L, Yi CS, Zhang L, Plon SE, Schrader KA, Karam R. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum Mutat 2018;39:1553–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivas MA, Pirinen M, Conrad DF, Lek M, Tsang EK, Karczewski KJ, Maller JB, Kukurba KR, DeLuca DS, Fromer M, Ferreira PG, Smith KS, Zhang R, Zhao F, Banks E, Poplin R, Ruderfer DM, Purcell SM, Tukiainen T, Minikel EV, Stenson PD, Cooper DN, Huang KH, Sullivan TJ, Nedzel J, Bustamante CD, Li JB, Daly MJ, Guigo R, Donnelly P, Ardlie K, Sammeth M, Dermitzakis ET, McCarthy Mi, Montgomery SB, Lappalainen T, MacArthur DG. GTEx Consortium Geuvadis Consortium. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 2015;348:666–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta CR, Patel NR. A network algorithm for performing fisher’s exact test in r x c contingency tables. Journal of the American Statistical Association 1983;78:427–34. [Google Scholar]
- 31.Sivasankar S, Zhang Y, Nelson WJ, Chu S. Characterizing the initial encounter complex in cadherin adhesion. Structure 2009;17:1075–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasmeen A, Bismar TA, Al Moustafa AE. ErbB receptors and epithelial-cadherin-catenin complex in human carcinomas. Future Oncol 2006;2:765–81. [DOI] [PubMed] [Google Scholar]
- 33.Bremm A, Walch A, Fuchs M, Mages J, Duyster J, Keller G, Hermannstädter C, Becker KF, Rauser S, Langer R, von Weyhern CH, Höfler H, Luber B. Enhanced activation of epidermal growth factor receptor caused by tumor-derived E-cadherin mutations. Cancer Res 2008;68:707–14. [DOI] [PubMed] [Google Scholar]
- 34.Li D, Lo W, Rudloff U. Merging perspectives: genotype-directed molecular therapy for hereditary diffuse gastric cancer (HDGC) and E-cadherin-EGFR crosstalk. Clin Transl Med 2018;7:7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newton KF, Mallinson EK, Bowen J, Lalloo F, Clancy T, Hill J, Evans DG. Genotype-phenotype correlation in colorectal polyposis. Clin Genet 2012;81:521–31. [DOI] [PubMed] [Google Scholar]
- 36.Frebourg T, Oliveira C, Hochain P, Karam R, Manouvrier S, Graziadio C, Vekemans M, Hartmann A, Baert-Desurmont S, Alexandre C, Lejeune Dumoulin S, Marroni C, Martin C, Castedo S, Lovett M, Winston J, Machado JC, Attie T, Jabs EW, Cai J, Pellerin P, Triboulet JP, Scotte M, Le Pessot F, Hedouin A, Carneiro F, Blayau M, Seruca R. Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J Med Genet 2006;43:138–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. E-cadherin alterations in hereditary disorders with emphasis on hereditary diffuse gastric cancer. Prog Mol Biol Transl Sci 2013;116:337–59. [DOI] [PubMed] [Google Scholar]
- 38.Salahshor S, Hou H, Diep CB, Loukola A, Zhang H, Liu T, Chen J, iselius L, Rubio C, Lothe RA, Aaltonen L, Sun XF, Lindmark G, Lindblom A. A germline E-cadherin mutation in a family with gastric and colon cancer. Int J Mol Med 2001;8:439–43. [DOI] [PubMed] [Google Scholar]
- 39.Kim SA, inamura K, Yamauchi M, Nishihara R, Mima K, Sukawa Y, Li T, Yasunari M, Morikawa T, Fitzgerald KC, Fuchs CS, Wu K, Chan AT, Zhang X, Ogino S, Qian ZR. Loss of CDH1 (E-cadherin) expression is associated with infiltrative tumour growth and lymph node metastasis. Br J Cancer 2016;114:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raskin L, Guo Y, Du L, Clendenning M, Rosty C, Lindor NM, Gruber SB, Buchanan DD. Colon Cancer Family Registry (CCFR). Targeted sequencing of established and candidate colorectal cancer genes in the colon cancer family registry cohort. Oncotarget 2017;8:93450–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sasaki CY, Lin H, Morin PJ, Longo DL. Truncation of the extracellular region abrogrates cell contact but retains the growth-suppressive activity of E-cadherin. Cancer Res 2000;60:7057–65. [PubMed] [Google Scholar]
- 42.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. Embo J 2004;23:1739–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression?. Oncogene 2008;27:6920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen i, Mathews-Greiner L, Li D, Abisoye-Ogunniyan A, Ray S, Bian Y, Shukla V, Zhang X, Guha R, Thomas C, Gryder B, Zacharia A, Beane JD, Ravichandran S, Ferrer M, Rudloff U. Transcriptomic profiling and quantitative high-throughput (qHTS) drug screening of CDH1 deficient hereditary diffuse gastric cancer (HDGC) cells identify treatment leads for familial gastric cancer. J Transl Med 2017;15:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corso G, Carvalho J, Marrelli D, Vindigni C, Carvalho B, Seruca R, Roviello F, Oliveira C. Somatic mutations and deletions of the E-cadherin gene predict poor survival of patients with gastric cancer. J Clin Oncol 2013;31:868–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.