

Abstract
A defining feature of HIV-associated neurocognitive disorder (HAND) is the loss of excitatory synaptic connections. Synaptic changes that occur during exposure to HIV appear to result, in part, from a homeostatic scaling response. Here we discuss the mechanisms of these changes from the perspective that they might be part of a coping mechanism that reduces synapses to prevent excitotoxicity. In transgenic animals expressing the HIV proteins Tat or gp120, the loss of synaptic markers precedes changes in neuronal number. In vitro studies have shown that HIV-induced synapse loss and cell death are mediated by distinct mechanisms. Both in vitro and animal studies suggest that HIV-induced synaptic scaling engages new mechanisms that suppress network connectivity and that these processes might be amenable to therapeutic intervention. Indeed, pharmacological reversal of synapse loss induced by HIV Tat restores cognitive function. In summary, studies indicate that there are temporal, mechanistic and pharmacological features of HIV-induced synapse loss that are consistent with homeostatic plasticity. The increasingly well delineated signaling mechanisms that regulate synaptic scaling may reveal pharmacological targets suitable for normalizing synaptic function in chronic neuroinflammatory states such as HAND.
Keywords: HIV-1, homeostatic plasticity, NMDA receptor, synaptic scaling, HIV-associated neurocognitive disorder
1. Introduction
HIV-associated neurocognitive disorder (HAND) afflicts almost half of HIV-infected individuals [1]. Cognitive deficits in HAND correlate with the loss of synaptic connections [2–4] and patients with mild neurocognitive impairment have an increased risk of further cognitive decline [5]. Because HIV does not infect neurons, HIV neurotoxicity is indirect and thought to be mediated by a neuroinflammatory response to viral proteins and inflammatory cytokines released by infected microglia and macrophages [1, 5, 6]. Here we discuss the effects of HIV neurotoxins on synaptic function and evidence that suggests early stage synapse loss might result from homeostatic plasticity.
2. Mechanism of Synapse Loss
Neurons scale their synaptic input in response to both low and high levels of activity [7]. Whether these same processes are responsible for the synapse loss observed during neurodegenerative conditions remains unclear. A dying cell will retract dendrites and lose network connections leading to overt synapse loss. In contrast, homeostatic scaling is a neurophysiological process that neurons use to stabilize their activity around a functional operating point. In the early stages of HAND, synapse loss precedes overt neuronal death by months to years [8], suggesting that synaptic changes may be a coping mechanism. The viral proteins and inflammatory cytokines elevated by HIV in the brain result in excessive activation of receptors for the neurotransmitter glutamate. Homeostatic scaling down of glutamatergic synapses under excitotoxic conditions may be a neuroprotective response, rather than a symptom of the overt synapse loss that accompanies a cell’s demise. Furthermore, this type of scaling may be reversible during the early stages of the disease. This homeostatic plasticity model posits that HIV tips the precarious balance neurons maintain between full integration into synaptic networks and the risk of excitotoxicity (Fig. 1).
Fig. 1:
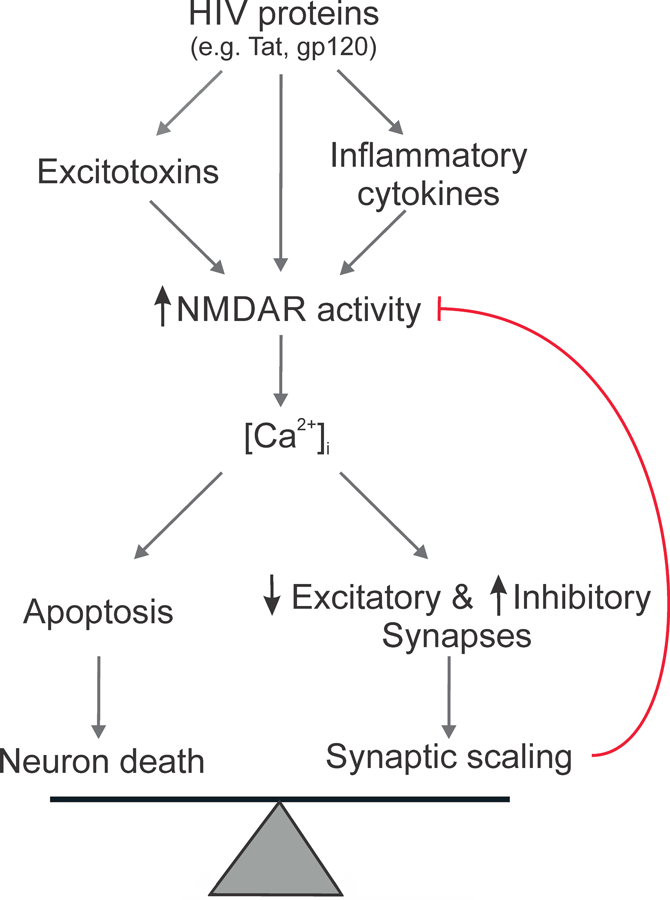
Balancing survival and function. Neurotoxic factors elevated by HIV act via various upstream mechanisms to converge on the potentiation of NMDARs. Neurons and networks then adapt to the presence of this stimulus by scaling synapses to attenuate Ca2+-mediated toxicity.
Neuroinflammation is a contributing factor to synapse loss in many neurodegenerative diseases [9–11]. In the context of the neuroinflammatory response in HAND, key factors include secretion of inflammatory cytokines, release of HIV proteins, activation of glia, and release of reactive oxygen species. Inflammatory cytokines are elevated in the CSF of HIV patients with cognitive impairment [12, 13]. Synapse loss induced by cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor α (TNFα) has been described in a number of models and has been the subject of thorough reviews [14,15]. Reactive oxygen species impair synaptic function [14] and are released following exposure to HIV proteins and antiretroviral drugs [15–18]. However, in the context of HIV, reactive oxygen species have been linked to cell death [19, 20] but their role in synapse loss has not been explored.
Exposure to the HIV envelope glycoprotein gp120 and the HIV protein Tat (transactivator of transcription) produces synapse loss both in vitro and in animal models [21–25]. Several observations are consistent with the idea that these synaptic changes might be part of a neuroprotective scaling response. The synapse loss induced by HIV proteins occurs prior to cell death in in vitro studies [21, 24, 26, 27] and in transgenic animals expressing Tat or gp120 the loss of synaptic markers precedes changes in neuronal number [23, 28–30]. Pharmacological reversal of synapse loss induced by the HIV protein Tat is consistent with the idea that synaptic changes may result from homeostatic plasticity [21, 31, 32]. Indeed, different mechanisms are responsible for HIV Tat-induced synapse loss and cell death; cell death can be prevented without affecting synapse loss and preventing synapse loss actually sensitizes neurons to cell death [21].
Both cell death and synaptic changes are initiated by over activation of NMDARs, but the downstream signaling pathways diverge to produce distinct outcomes [6, 33] (Fig. 2). HIV proteins and inflammatory cytokines produce a rapid initial potentiation of NMDAR activity [34–37]. High concentrations of HIV Tat (>1 µM) act directly on NMDARs to increase activity [38], lower concentrations more subtly potentiate NMDAR function through kinase signaling cascades including the Src [39] and protein kinase C [35] pathways. Exposure to gp120IIIB potentiates NMDARs via protein kinase A dependent phosphorylation [40] and Src activation [36, 41, 42]. It appears that phosphorylation-mediated potentiation of the NMDAR is a common step that initiates synaptic changes (Fig. 2), consistent with protection of synapses afforded by NMDAR antagonists [21, 24, 31]. This initial over activation of NMDARs is followed by down regulation [43–45] and loss of excitatory synapses [21–24].
Fig. 2:

HIV neurotoxins potentiate NMDARs to evoke distinct synaptic scaling and neuronal death pathways. GluN2A-containing NMDARs promote anti-apoptotic pathways and regulate changes to excitatory and inhibitory synapses. GluN2B-containing NMDARs promote pro-apoptotic pathways and prevent synapse recovery. Figure modified from [60].
NMDAR subtypes play specific roles in the Tat-induced loss of synapses connecting hippocampal neurons in culture. NMDARs are tetramers composed of two obligatory GluN1 subunits and two GluN2 (A-D) or GluN3 (A-B) subunits. The predominant GluN2 subtypes in the hippocampus are GluN2A and GluN2B [46]. Activation of GluN2A-containing NMDARs promotes neuronal survival, whereas activation of GluN2B-containing NMDARs can activate pro-apoptotic pathways [47, 48]. Inhibition of GluN2A, but not GluN2B-containing NMDARs prevents Tat-induced synapse loss which is consistent with the pro-survival role of GluN2A-containing NMDARs, because early synapse loss appears to be a homeostatic mechanism to prevent cell death. Furthermore, inhibition of GluN2B, but not GluN2A-containing NMDARs prevents Tat-induced cell death [31]. Application of an inhibitor selective for GluN2B-containing NMDARs after synapse loss has already occurred restores synaptic density. Thus, the pharmacology changes during exposure to the HIV neurotoxin such that the sustained activation of GluN2B-containing NMDARs prevents recovery of synapses. Several factors may contribute to the selective roles of NMDAR subtypes. Cellular location may account for some specificity; GluN2A-containing NMDARs are predominantly synaptic and their activation is pro-survival, whereas GluN2B-containing NMDARs are predominantly extrasynaptic and their activation is pro-apoptotic [47, 49]. Another distinction between the GluN2 subtypes is their intracellular C-terminal domains which couple to different downstream signaling pathways [50]. Thus, the processes activated by Ca2+ influx via NMDARs display a source specificity.
Pathological activation of NMDARs produces excessive Na+ and Ca2+ influx that elicits toxicity via mitochondrial generated reactive oxygen species [51], dendritic swelling [52], and activation of the pro-apoptotic caspase cascade [53]. In addition to NMDAR, HIV proteins potentiate other Ca2+ permeable channels including L type calcium channels, TRPC channels, and ryanodine- and IP3-receptor Ca2+ release channels [33]. These channels have not been linked directly to HIV induced synaptic changes, although their effects on Ca2+-mediated changes in metabolism, neurotransmitter release and network excitability could indirectly affect NMDAR signaling. HIV also affects gap junctions and hemi-channels [54]. Recent studies have shown metabotropic NMDAR activity is important for regulating pannexin 1 gap junctions [55], excitotoxicity [56] and synaptic plasticity [57]. Proteomic analysis of synaptosomes derived from HIV infected individuals found synapsin Ib levels were inversely related to HIV-1 loads [58]. Loss of this presynaptic marker could be a form of scaling, which in vitro studies indicate is dependent on NMDARs situated on the postsynaptic dendrite [59]. Experiments using chelators with different Ca2+ binding kinetics suggest that the Ca2+-sensitive target that drives loss of glutamatergic synapses following exposure to HIV Tat is close to the mouth of the NMDA-gated channel [60].
Ca2+ entry through the NMDAR can activate a number of signaling pathways that modulate synaptic function [7, 61]. Calmodulin-dependent kinase II (CamKII) is localized in close proximity to the mouth of the NMDAR [62, 63]. Ca2+ binding to calmodulin with subsequent activation of CamKII is a key pathway regulating synaptic plasticity, including induction of LTP through phosphorylation of AMPA receptors and changes in gene expression following activation of CREB [64–66]. Indeed, inhibition of CamKII prevents Tat-induced synapse loss [60]. Additionally, NMDAR activation and HIV gp120 upregulate matrix metalloprotease-2 (MMP-2), an enzyme that breaks down the extracellular matrix to facilitate spine remodeling [67, 68]. Down regulation of synaptic strength is linked to NMDA-dependent activation of calcineurin [69]. However, inhibition of this Ca2+-sensitive phosphatase with FK506 failed to prevent changes in synapse number evoked by HIV Tat [60], suggesting that the death-associated protein kinase pathway is not responsible for the initial loss of synapses. Ca2+ entry via NMDARs, particularly during synaptic activity, induces robust activation of CREB and its co-activator CREB-binding protein, resulting in the transcription of many pro-survival genes [70], including Brain-derived neurotrophic factor (BDNF).
BDNF is a growth factor secreted by excitatory neurons; depending the specific brain region it can modulate synaptic upscaling and downscaling [71, 72]. gp120 decreases furin levels reducing pro-BDNF processing, which is consistent with BDNF levels in tissue from postmortem HIV-positive subjects [73]. Both gp120 and TNF-α reduce anterograde transport and intracellular stores of BDNF [74]. Additionally, Tat induced microRNA-132 expression down-regulates a number of genes, including BDNF [75]. In patients with HAND, CSF levels of BDNF and fibroblast growth factor 2 (FGF2) are lower than control patients [76] and the severity of neurological disease correlates with BDNF and neurotrophin-3 in the CSF [77]. Whether reduced BDNF levels in HAND result from a decrease in the number of excitatory synapses in impaired patients or whether reduced BDNF is a result of HIV neurotoxins disrupting normal BDNF processing is unclear. Either scenario implies strategies that correct BDNF levels might restore synaptic function.
GluN2A-containing NMDARs can activate the kinase Akt to promote neuronal survival [47, 56]. Tat activates Akt [68] and Akt is necessary for Tat-induced downregulation of NMDAR activity [43]. Mass spectrometry quantification of synaptic proteins in brains from HIV gp120 transgenic mice and human HAND patients found lowered pAkt/Akt ratio consistent with dysregulation of this pathway [78]. Tat-activated Akt phosphorylates the E3 ligase MDM2 (murine double minute 2), increasing its activity [79]. Inhibition of MDM2 prevents both Tat and gp120-induced synapse loss [21, 24]. MDM2 plays a key role in activity dependent homeostatic plasticity [80], consistent with the idea that fundamental scaling mechanisms are recruited during HIV neurotoxicity. Changes in proteasome subunit composition correlate with HIV loads in brain tissue, further implicating changes in protein turnover as a contributing factor to HAND [81]. MDM2 ubiquitinates many target proteins including p53, MAP2 and PSD-95 [82–84]. PSD-95 is a scaffolding protein that tethers excitatory receptors such as NMDARs and AMPARs to the post-synaptic membrane. Mutation of the PEST sequence at the N terminus of PSD-95, a site that regulates its stability and ubiquitination [85], prevents Tat-induced synapse loss [21]. Because PSD-95 stabilizes spines, its ubiquitination by MDM2 may lead to retraction of the spine and synapse loss [82]. Indeed, knockdown of the MAGUK family of scaffolding proteins, which includes PSD-95, produces a synaptic consolidation similar to the homeostatic process evoked by HIV proteins [86]. Tat also causes a proteasome-mediated degradation of MAP2 and the collapse of cytoskeletal filaments [84]. Indeed, MAP2 levels are reduced in SIV models [87] and MAP2-positive neurons are selectively vulnerable in HIV patients [88]. p53, another target of MDM2, regulates transcription which may affect synapse density by changing the expression of synaptic proteins, such as ephrin B2 [89], a receptor involved in the formation of glutamatergic synapses that is reduced in postmortem brains of HIV-infected subjects [90]. However, p53 levels were elevated in an SIV model [91]. The lower pAkt/Akt ratio and elevated p53 levels observed in models with chronic exposure to virus or toxins may result from measurements taken at a later stage of the disease. In summary, the MDM2 pathway provides a clear example of a signaling cascade activated by the increase in NMDAR activity induced by HIV neurotoxins and leads to downscaling of glutamatergic synapses.
Changes in the release or reuptake of glutamate produced by HIV may also contribute to the over activation of NMDARs responsible for synapse loss. Indeed, CSF glutamate levels correlate with cognitive decline in HIV-infected patients [92]. During SIV infection excitatory amino acid transporters (EAATs) are disrupted [93]. The HIV proteins Tat and gp120 reduce expression of EAAT2 in astrocytes, reducing uptake of glutamate [94–97]. TNFα, a pro-inflammatory cytokine produced by HIV infected macrophages and microglia, inhibits glutamate uptake into primary human astrocytes [98]. Thus, multiple mechanisms activated by HIV in the brain lead to impaired glutamate uptake. HIV proteins also increase the release of glutamate. HIV proteins evoke the release of cytokines, such as IL-1β, which increase the production of prostaglandins that act presynaptically to increase glutamate release [99]. Similarly, platelet-activating factor, which is elevated in inflammatory conditions such as HIV-associated dementia (HAD), produces synaptic facilitation and sensitization to dendritic damage by increasing glutamate release [100–102]. Tat also acts directly on presynaptic terminals to increase glutamate release [103, 104]. The phosphorylation of tyrosines on the NMDAR induced by HIV proteins and inflammatory cytokines does not activate it directly, phosphorylation sensitizes NMDARs to glutamate [105]. Thus, elevated extracellular glutamate synergizes with the increased sensitivity of the receptor likely contributing to the altered network activity observed in the presence of HIV [106–109]. Networks adapt to altered activity levels through homeostatic scaling mechanisms. Thus, synaptic scaling may be an attempt to normalize activity levels in networks that have been excited by persistent elevation of extracellular glutamate due to the presence of HIV.
Many HAND patients exhibit movement disorders and sub-cortical pathology consistent with abnormal dopaminergic synaptic markers in striatal extracts from patients with HIV encephalitis (HIVE) [110]. Tat decreases dopamine (DA) transporter expression and function and also inhibits the vesicular monoamine transporter [111–114]. Impaired DA transport may elevate DA levels early in infection possibly leading to DA induced toxicity apparent at later stages of disease [115]. Changes in DA metabolism may account for some of the pronounced adverse effects produced by drugs of abuse in HAND patients [116]. While HAND neuropathology includes the loss of dopaminergic synapses this loss appears to result from a degenerative process, possibly resulting from oxidative damage produced by excess DA. Elevated DA levels may affect the plasticity of glutamatergic and GABAergic synapses, although the degree to which these neurotransmitter systems interact to scale synapses in HIV is unclear.
Activation of microglia is a key feature of HAND [117]. Contact with microglial filopodia is an early step in synapse elimination following injection of HIV Tat protein into the CNS [118, 119]. Recent studies have shown that microglia use complement-based tagging to prune synapses during development [120] and neurodegenerative disease [121]. Microglial mediated synaptic pruning occurs in animal models of HAND [122] and the level of complement factors (C1q/C3) detected in the CSF of HIV infected individuals correlates with a marker for neuronal damage [123]. However, the phagocytic ability of monocytes and macrophages [124, 125] is decreased following HIV infection or exposure to Tat [126]. Thus, the role of synaptic pruning by microglia in HIV-induced synapse loss is unclear, although its participation in refining neural circuits during development would seem to indicate that this process may be regulated by synaptic activity. The degree to which microglia contribute to the removal of synaptic structures will influence whether recovery of synaptic density involves the formation of new synapses.
In summary, while some studies indicate that synapse loss induced by HIV is part of the agonal event, other results, particularly from models in which discrete application of HIV neurotoxins mimic the early phase of infection, appear to support a mechanism more similar to homeostatic scaling. Scaling normalizes cellular excitability and balances network activity so that information encoded in synaptic connections is maintained without saturating the dynamic range of the network or inducing excitotoxicity. Thus, homeostatic scaling mechanisms may participate in adaptation to the presence of HIV neurotoxins. Mapping these neurophysiological mechanisms to the pathological neuroinflammatory state produced by HIV in the brain may inform the development of therapeutic approaches to prevent or correct this overcompensation.
3. Increased Inhibition to counter HIV-induced excitotoxicity
Network scaling in response to excess excitation increases GABAergic tone in addition to decreasing glutamatergic activity. Increased number of inhibitory synapses or levels of gephyrin, a scaffolding protein found at GABAergic synapses, have been observed in hippocampal cultures treated with Tat protein and in CA1 hippocampal neurons from mice expressing HIV Tat [30, 60]. The formation of new inhibitory synapses is more consistent with a coping mechanism than part of the death process. The increase in the number of inhibitory synapses during exposure to HIV neurotoxins follows a time course similar to the loss of excitatory synapses and the adaptation of NMDA receptors [21, 127]. Like the loss of excitatory synapses, the increase in inhibitory synapses is initiated by Ca2+ influx through GluN2A-containing NMDA receptors and subsequent activation of CaMKII [21, 24, 60] (Fig. 2). Increased excitatory neurotransmission can activate Npas4 which increases the expression of genes that control inhibitory synapse development [128], although whether this homeostatic process participates in network adaptation to HIV neurotoxins is not known. Thus, increased inhibitory synaptic transmission is part of a neuroprotective pathway distinct from that regulating glutamatergic synapses. However, in striatum and prefrontal cortex exposed to HIV proteins GABAergic neurotransmission is inhibited [129, 130]. The same Tat expressing transgenic mice that exhibited increased gephyrin immunostaining in the hippocampus, also exhibited decreased levels of synaptotagmin 2, a marker for GABAergic presynaptic terminals [30]. GABA-A receptors are down-regulated in frontal neocortex from patients with HIV encephalitis [131]. The reason for the increased GABAergic signaling in some HIV models and decreased signaling in others is unclear but, might result from synaptic adaptations that are unique to specific brain regions, the use of different methods for detecting inhibitory synapses, and the timing of the measurement with early detection favoring scaling mechanisms and later stages of disease showing loss of synapses and neurons. There is precedent in other neurodegenerative disorders that GABAergic over compensation leads to impaired function. Excessive inhibition creates an excitatory-inhibitory imbalance that impairs cognitive function in Down syndrome [132–134]. Increased tonic inhibition impairs memory in an Alzheimer’s disease model [135]. Inhibition of GABA transporters, a target of some antiepileptic drugs, produces cognitive deficits [136]. Whether excess inhibition contributes to cognitive impairment in HAND is not known.
4. Preventing HIV neurotoxicity
The recognition that loss of synaptic connections underlies neurologic disease in HIV infected patients brought the synapse to the forefront as a target for neuroprotective strategies in HAND [8]. Rather than review the many compounds tested for neuroprotective effects in models of HAND, we focus on several drugs that highlight the types of mechanisms that can be modulated to prevent or normalize synaptic scaling. These include NMDA receptors, microglia-mediated inflammatory processes, neurotrophic growth factors, and the neuroprotective endocannabinoid system.
NMDA receptors play a major role in pathological neuronal excitation, and based on the results that have been discussed, NMDAR antagonism may attenuate HIV neuropathological processes. Memantine is an uncompetitive blocker of NMDA-gated channels, which preferentially inhibits extrasynaptic NMDARs because of its fast off-rate [137]. In vitro and in vivo studies with memantine have demonstrated protective effects against neurotoxicity evoked by HIV gp120 and Tat [138–140]. The NMDAR antagonist dizocilpine protects against HIV protein-induced synapse loss and death in vitro [31]. While the use of broad spectrum glutamate receptor antagonists in patients is fraught with adverse effects [141, 142], drugs targeting NMDAR subtypes, particularly those that contain the GluN2B subunit, show promise in animal models of HAND [32].
Microglia involvement in HIV-related neurotoxicity includes synaptic pruning and release of cytokines, chemokines, prostaglandins, and nitric oxide [117]. Anti-inflammatory drugs are an increasingly attractive adjunctive therapy for the treatment of HAND. Minocycline is a broad-spectrum tetracycline antibiotic derivative that is neuroprotective in models of neurodegenerative disease [143–145]. Minocycline inhibits HIV production in infected microglial cultures without being toxic to microglia themselves [146] and minocycline-treated macaques showed less severe encephalitis, reduced CNS expression of neuroinflammatory markers, less axonal degeneration, and lower CNS virus replication [147]. These promising results did not translate in clinical trials of minocycline for the treatment of HAND [148, 149], despite being safe and well-tolerated among subjects. This discrepancy between the effects of minocycline in the SIV encephalitis model and HIV-positive individuals warrants further studies to define the precise underlying neuroprotective mechanism in the former in order to facilitate development of more efficacious therapeutics for HAND. Drugs that suppress the function of microglia act upstream of NMDAR potentiation to prevent the excitation that subsequently engages homeostatic processes.
Levels of neurotrophic factors are suppressed in HIV neurotoxicity models and HAND patients (section 2). Replacing this deficit with exogenous neurotrophins increases neuronal survival and normalizes synaptic connections [150]. FGF1 protected primary human cultures from HIV gp120 [151] and overexpression of FGF1 prevented neurodegeneration in gp120 expressing transgenic mice [152]. BDNF is neuroprotective against gp120 in vitro and in vivo [153]. Normalizing neurotrophin levels demonstrates the utility of modulating the mechanisms responsible for synaptic scaling.
The endocannabinoid (eCB) system provides on demand protection from excitotoxicity and neuroinflammation [154–157]. Cannabinoid type 1 receptors (CB1R) are expressed in neurons and modulate synaptic transmission. CB2R are primarily expressed in cells of the immune system and modulate inflammation. CB2R agonists are neuroprotective in several models of HIV neurotoxicity [158]. CB2R agonists protect synaptic networks from gp120 by inhibiting release of inflammatory cytokines from microglia [24] and they restore suppressed neurogenesis in gp120 transgenic mice [159]. In a mouse model of HIVE a CB2R agonist reduced the levels of immune activation, infiltration of human cells into the brain, and downregulated neuroinflammation [160]. Impaired function of the eCB system may disrupt its neuroprotective capability; thus, upregulating this system may attenuate HIV neurotoxicity. Inhibition of the 2-arachidonyl glycerol (2-AG) hydrolytic enzyme monoacylglycerol lipase (MGL) by the selective, irreversible inhibitor JZL184 prevents gp120-induced synapse loss [42]. Inhibition of MGL by JZL184 has also been demonstrated to suppress proinflammatory cascades that underlie neurodegenerative disorders, as demonstrated in an MPTP mouse model of Parkinson’s disease [161]. Because inhibition of MGL only potentiates endogenously produced 2-AG, receptor activation is dependent on a stimulus, enabling prolonged, low-dose MGL inhibition to elicit an anti-inflammatory effect without producing CB1R tolerance or cannabinoid dependence [162]. Furthermore, because brain arachidonic acid production is primarily dependent on MGL, in contrast to the gut, drugs that inhibit MGL can reduce brain prostaglandin levels without the gastrointestinal side effects produced by nonsteroidal anti-inflammatory drugs [163]. Thus, MGL inhibition increases 2-AG levels and reduces prostaglandin production to reduce HIV-induced excitotoxicity and neuroinflammation.
5. Reversing synapse loss and recovering function
The idea that synaptic changes early in the course of HAND might result from homeostatic scaling to adapt to the presence of HIV neurotoxins is supported by results demonstrating that these changes are reversible. Upon initiation of cART, HIV infected patients with neurocognitive impairment exhibit longitudinal improvement in neuropsychological function and regression of neuropathology [164, 165]. These studies suggest that HAND, at least in part, can be reversed. More recent work has focused on reversing the synaptic deficits in HAND patients in which viral titer is suppressed by cART.
In vitro synaptic networks exposed to HIV toxins initially display an excitotoxic state characterized by potentiated NMDAR activity, followed by compensatory decreases in the number of excitatory synapses and increases in the number of inhibitory synapses (see sections 2 and 3). Generally, manipulations that block the initial excitatory state (see section 4) are distinctly different from treatments that restore synaptic function. One treatment that both prevents and reverses synapse loss is receptor associated protein (RAP), a chaperone protein that binds to the lipoprotein related protein (LRP). Polymorphisms in the genes for LRP, and its ligands apolipoprotein E and α-macroglobulin are linked to Alzheimer’s disease [166] and HAD [167]. RAP prevents Tat internalization [168] preventing potentiation of the NMDAR and subsequent loss of synapses [21]. Intriguingly, RAP also rescues lost synapses following Tat exposure, suggesting an LRP-dependent mechanism may be involved in Tat-induced spine suppression [21]. Note that most treatments that attenuate synapse loss evoked by HIV neurotoxins are not able to induce recovery once loss has occurred, consistent with the idea that a new state has developed in which the synaptic network has adapted to the presence of the excitotoxin, changing the pharmacology. For example, excitatory signaling through GluN2A-containing NMDARs plays a critical role in spine loss as indicated by block by the GluN2A antagonists TCN201 and NVP-AAM077 [31, 43]. In contrast, ifenprodil and memantine, antagonists of GluN2B-containing NMDARs, do not block the initial effects of Tat. However, following synapse loss (16–24 h Tat exposure) these drugs rescue synapses [31]. This finding suggests that Tat-exposure may alter synapse regulation such that GluN2B-containing NMDARs are negatively coupled to spine regulation. Collectively these in vitro findings suggest that HIV toxins induce an excitotoxic state that drives synaptic scaling and that reversing these adaptations may be a viable approach to restore cognitive function in HAND.
Despite variability in animal models of HAND, in vivo work suggests that there are potential treatment targets for HIV neurotoxicity, such as K+ channels, estrogen receptors, and NMDARs. In a HIVE model where immunosuppressed SCID mice were injected with HIV-infected primary human macrophages, the K+ channel blocker 4-aminopyrdine (4-AP) ameliorated HIVE-associated deficits in long-term potentiation, changes in synaptic structure, and improved spatial learning and memory assessed by radial arm maze [169]. Another study targeted estrogen receptors with the legume isoflavone daidzein and the liquorice extract liquiritigenin, which both act as estrogen receptor agonists [27]. They found that these compounds reversed HIV-Tat induced loss of F-actin labeled puncta, a marker for synaptic spines, but did not assess effects on cognition. Nakanishi and colleagues (2015) examined the effects of blocking NMDARs on neuropathology in gp120-expressing mice and reported that the NMDA antagonist nitromemantine reversed both dendritic damage assessed with MAP2 immunoreactivity and loss of presynaptic terminals assessed by synaptophysin immunoreactivity [170]. In an SIV model, treatment with memantine upregulated BDNF and diminished dopamine deficits [171]. Further, our lab has recently used within-subject multiphoton imaging to demonstrate that HIV-1 Tat infusion decreases dendritic spine density, as well as cognitive function in mice, and that administration of ifenprodil, a selective antagonist for GluN2B-containing NMDARs, rescues both dendritic spine density and cognitive function (Fig. 3). These findings collectively suggest that small molecule pharmacotherapy can restore lost synapses and function in HAND, supporting the idea that the initial cognitive deficits resulting from HIV exposure may be due to aberrant homeostatic scaling mechanisms that are potentially controllable.
Fig. 3:

Synaptic loss and cognitive impairment induced by HIV proteins can be pharmacologically reversed. Infusion of HIV Tat into the lateral ventricle decreased spine density in the retrosplenial cortex and impaired cognitive function. A single infusion of Tat produced a prolonged decrease in spines (horizontal red arrow). Spines lost during Tat exposure are shown in red at baseline. Systemic administration of ifenprodil (blue horizontal bar) rescued spines (new spines are blue) and cognitive function. Note that after clearance of ifenprodil spine density and cognitive function returned to levels observed before drug administration. Figure modified from [32].
One of the first clinical trials for HAND treatment used the voltage-gated calcium channel blocker nimodipine [172]. No effects on cognition were reported although, this early trial was conducted against a very different treatment backdrop than that of the current cART era [173], and faced a clinical population with worsened neurocognitive outlook. Later trials have focused on the NMDA antagonist memantine, which has shown some promise in ameliorating Alzhiemer’s disease symptoms [174]. An initial study administered memantine to subjects with stage one AIDS dementia complex, within the Adult AIDS Clinical Trial Group (AACTG) [175]. Unfortunately they found no significant effect of memantine on the NPZ-8 neuropsychological index, but did report a promising trend in some clinical subgroups, as well as marked variability. The fact that memantine, an NMDA antagonist, is well tolerated and shows trends towards amelioration of HAND symptoms is promising in light of recent preclinical findings with selective NMDAR targeting drugs, such as those acting on GluN2b-containing NMDARs and negative allosteric modulators. Overall, these reports highlight the challenges facing clinical trials for treatment of progressive disease states; future efforts will need to pay particular attention to disease progression, intervention timing, and treatment duration to demonstrate effectiveness of HAND treatments.
6. Synthesis and Conclusions
Synaptic changes that occur during exposure to HIV have a component that appears to result from a homeostatic scaling response. HIV-induced synaptic changes share common mechanisms with this form of plasticity. Synaptic scaling is most readily apparent in in vitro studies where the toxicity is evoked by an acute challenge and a clear reversal of HIV induced synaptic damage can be demonstrated. Interestingly, the over activation of NMDARs drives both the early reversible synaptic changes and delayed cell death processes, though these signaling pathways diverge and may be triggered by different subtypes of NMDARs. More slowly developing toxicity accurately reflects the chronic nature of HAND, but in these models it is more difficult to differentiate an early plastic response from the agonal event. cART improves the neurocognitive function in patients beginning treatment, suggesting an initial synaptic resilience, but reversing cognitive impairment in clinical trials has not been encouraging. In vitro and animal studies suggest that following HIV-induced synaptic scaling new mechanisms suppress network connectivity that might be amenable to therapy.
7. Future Directions
There is currently no effective treatment for HAND. The idea that HIV-induced synapse loss is due, at least in part, to homeostatic scaling both raises concerns and is grounds for optimism. Two important issues warrant investigation. First, our understanding of synaptic adaptations that occur during exposure to HIV neurotoxins, their underlying mechanisms and the time course of their expression is incomplete. Knowing when specific neuronal changes occur during HIV infection is essential to designing effective pharmacotherapy for HAND. Second, if loss of glutamatergic synapses is a mechanism to cope with excess excitation brought on by HIV, it is important to determine whether lost connectivity is a necessary cost for neuronal survival or whether persistent synapse loss is a protective mechanism gone awry. We are optimistic that the increasingly well delineated signaling mechanisms that regulate homeostatic scaling will reveal pharmacological targets suitable for normalizing synaptic function in chronic neuroinflammatory states such as HAND.
Acknowledgments.
This work was supported by the National Institute on Drug Abuse - National Institutes of Health grant DA07304.
Footnotes
Conflict of Interest. The authors declare they have no competing interests.
References
- 1.Ellis R, Langford D, Masliah E (2007) HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci 8:33–44 [DOI] [PubMed] [Google Scholar]
- 2.Ellis RJ, Calero P, Stockin MD (2009) HIV infection and the central nervous system: a primer. Neuropsychol Rev 19:144–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E (1999) Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol 9:209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ru W, Tang SJ (2017) HIV-associated synaptic degeneration. Mol Brain 10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC (2016) HIV-associated neurocognitive disorder - pathogenesis and prospects for treatment. Nature reviews Neurology 12:234–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410:988–994 [DOI] [PubMed] [Google Scholar]
- 7.Turrigiano GG (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135:422–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellizzi MJ, Lu SM, Gelbard HA (2006) Protecting the synapse: evidence for a rational strategy to treat HIV-1 associated neurologic disease. J Neuroimmune Pharmacol 1:20–31 [DOI] [PubMed] [Google Scholar]
- 9.Mandolesi G, Gentile A, Musella A, Fresegna D, De Vito F, Bullitta S, Sepman H, Marfia GA, Centonze D (2015) Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nature reviews Neurology 11:711–724 [DOI] [PubMed] [Google Scholar]
- 10.Rao JS, Kellom M, Kim HW, Rapoport SI, Reese EA (2012) Neuroinflammation and synaptic loss. Neurochem Res 37:903–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bandaru VV, McArthur JC, Sacktor N, Cutler RG, Knapp EL, Mattson MP, Haughey NJ (2007) Associative and predictive biomarkers of dementia in HIV-1-infected patients. Neurology 68:1481–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan L, Qiao L, Wei F, Yin J, Liu L, Ji Y, Smith D, Li N, Chen D (2013) Cytokines in CSF correlate with HIV-associated neurocognitive disorders in the post-HAART era in China. J Neurovirol 19:144–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beckhauser TF, Francis-Oliveira J, De Pasquale R (2016) Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. Journal of experimental neuroscience 10:23–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louboutin JP, Agrawal L, Reyes BA, van Bockstaele EJ, Strayer DS (2012) Gene delivery of antioxidant enzymes inhibits human immunodeficiency virus type 1 gp120-induced expression of caspases. Neurosci 214:68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hui L, Chen X, Bhatt D, Geiger NH, Rosenberger TA, Haughey NJ, Masino SA, Geiger JD (2012) Ketone bodies protection against HIV-1 Tat-induced neurotoxicity. J Neurochem 122:382–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akay C, Cooper M, Odeleye A, Jensen BK, White MG, Vassoler F, Gannon PJ, Mankowski J, Dorsey JL, Buch AM, Cross SA, Cook DR, Pena MM, Andersen ES, Christofidou-Solomidou M, Lindl KA, Zink MC, Clements J, Pierce RC, Kolson DL, Jordan-Sciutto KL (2014) Antiretroviral drugs induce oxidative stress and neuronal damage in the central nervous system. J Neurovirol 20:39–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agrawal L, Louboutin JP, Reyes BA, Van Bockstaele EJ, Strayer DS (2012) HIV-1 Tat neurotoxicity: a model of acute and chronic exposure, and neuroprotection by gene delivery of antioxidant enzymes. Neurobiol Dis 45:657–670 [DOI] [PubMed] [Google Scholar]
- 19.Viviani B, Corsini E, Binaglia M, Galli CL, Marinovich M (2001) Reactive oxygen species generated by glia are responsible for neuron death induced by human immunodeficiency virus-glycoprotein 120 in vitro. Neurosci 107:51–58 [DOI] [PubMed] [Google Scholar]
- 20.Kim SH, Smith AJ, Tan J, Shytle RD, Giunta B (2015) MSM ameliorates HIV-1 Tat induced neuronal oxidative stress via rebalance of the glutathione cycle. American journal of translational research 7:328–338 [PMC free article] [PubMed] [Google Scholar]
- 21.Kim HJ, Martemyanov KA, Thayer SA (2008) Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci 28:12604–12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF (2012) Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 tat transgenic mice. Biol Psychiatry 73:443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L (1994) Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 367:188–193 [DOI] [PubMed] [Google Scholar]
- 24.Kim HJ, Shin AH, Thayer SA (2011) Activation of cannabinoid type 2 receptors inhibits HIV-1 envelope glycoprotein gp120-induced synapse loss. Mol Pharmacol 80:357–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Festa L, Gutoskey CJ, Graziano A, Waterhouse BD, Meucci O (2015) Induction of Interleukin-1beta by Human Immunodeficiency Virus-1 Viral Proteins Leads to Increased Levels of Neuronal Ferritin Heavy Chain, Synaptic Injury, and Deficits in Flexible Attention. J Neurosci 35:10550–10561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertrand SJ, Aksenova MV, Mactutus CF, Booze RM (2013) HIV-1 Tat protein variants: critical role for the cysteine region in synaptodendritic injury. Exp Neurol 248:228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertrand SJ, Mactutus CF, Aksenova MV, Espensen-Sturges TD, Booze RM (2014) Synaptodendritic recovery following HIV Tat exposure: neurorestoration by phytoestrogens. J Neurochem 128:140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hahn YK, Podhaizer EM, Farris SP, Miles MF, Hauser KF, Knapp PE (2015) Effects of chronic HIV-1 Tat exposure in the CNS: heightened vulnerability of males versus females to changes in cell numbers, synaptic integrity, and behavior. Brain Struct Funct 220:605–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michaud J, Fajardo R, Charron G, Sauvageau A, Berrada F, Ramla D, Dilhuydy H, Robitaille Y, Kessous-Elbaz A (2001) Neuropathology of NFHgp160 transgenic mice expressing HIV-1 env protein in neurons. J Neuropathol Exp Neurol 60:574–587 [DOI] [PubMed] [Google Scholar]
- 30.Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF (2013) Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry 73:443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin AH, Kim HJ, Thayer SA (2012) Subtype selective NMDA receptor antagonists induce recovery of synapses lost following exposure to HIV-1 Tat. Br J Pharmacol 166:1002–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raybuck JD, Hargus NJ, Thayer SA (2017) A GluN2B-Selective NMDAR Antagonist Reverses Synapse Loss and Cognitive Impairment Produced by the HIV-1 Protein Tat. J Neurosci 37:7837–7847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu XT (2015) HIV-1 Tat-mediated calcium dysregulation and neuronal dysfunction in vulnerable brain regions. Curr Drug Targets [DOI] [PMC free article] [PubMed]
- 34.Krogh KA, Wydeven N, Wickman K, Thayer SA (2014) HIV-1 protein Tat produces biphasic changes in NMDA-evoked increases in intracellular Ca concentration via activation of Src kinase and nitric oxide signaling pathways. J Neurochem 130:642–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD (2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78:457–467 [DOI] [PubMed] [Google Scholar]
- 36.Viviani B, Gardoni F, Bartesaghi S, Corsini E, Facchi A, Galli CL, Di Luca M, Marinovich M (2006) Interleukin-1beta Released by gp120 Drives Neural Death through Tyrosine Phosphorylation and Trafficking of NMDA Receptors. J Biol Chem 281:30212–30222 [DOI] [PubMed] [Google Scholar]
- 37.Jara JH, Singh BB, Floden AM, Combs CK (2007) Tumor necrosis factor alpha stimulates NMDA receptor activity in mouse cortical neurons resulting in ERK-dependent death. J Neurochem 100:1407–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prendergast MA, Rogers DT, Mulholland PJ, Littleton JM, Wilkins LH, Self RL, Nath A (2002) Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-D-aspartate receptor. Brain Res 954:300–307 [DOI] [PubMed] [Google Scholar]
- 39.King JE, Eugenin EA, Hazleton JE, Morgello S, Berman JW (2010) Mechanisms of HIV-tat-induced phosphorylation of N-methyl-D-aspartate receptor subunit 2A in human primary neurons: implications for neuroAIDS pathogenesis. Am J Pathol 176:2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu H, Bae M, Tovar-y-Romo LB, Patel N, Bandaru VV, Pomerantz D, Steiner JP, Haughey NJ The human immunodeficiency virus coat protein gp120 promotes forward trafficking and surface clustering of NMDA receptors in membrane microdomains. J Neurosci 31:17074–17090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mishra A, Kim HJ, Shin AH, Thayer SA (2012) Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J Neuroimmune Pharmacol 7:571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Thayer SA (2018) Monoacylglycerol lipase inhibitor JZL184 prevents HIV-1 gp120-induced synapse loss by altering endocannabinoid signaling. Neuropharmacol 128:269–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green MV, Thayer SA (2016) NMDARs Adapt to Neurotoxic HIV Protein Tat downstream of a GluN2A-Ubiquitin Ligase Signaling Pathway. J Neurosci 36:12640–12649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ru W, Tang SJ (2015) HIV-1 gp120Bal down-Regulates Phosphorylated NMDA Receptor Subunit 1 in Cortical Neurons via Activation of Glutamate and Chemokine Receptors. J Neuroimmune Pharmacol [DOI] [PMC free article] [PubMed]
- 45.Masliah E, Roberts ES, Langford D, Everall I, Crews L, Adame A, Rockenstein E, Fox HS (2004) Patterns of gene dysregulation in the frontal cortex of patients with HIV encephalitis. J Neuroimmunol 157:163–175 [DOI] [PubMed] [Google Scholar]
- 46.Shipton OA, Paulsen O (2014) GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos Trans R Soc Lond B Biol Sci 369:20130163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci 27:2846–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, Xu L, Duan WH, Xiong ZQ (2008) Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 39:3042–3048 [DOI] [PubMed] [Google Scholar]
- 49.Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414. [DOI] [PubMed] [Google Scholar]
- 50.Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci [DOI] [PMC free article] [PubMed]
- 51.Rozzi SJ, Avdoshina V, Fields JA, Trejo M, Ton HT, Ahern GP, Mocchetti I (2017) Human Immunodeficiency Virus Promotes Mitochondrial Toxicity. Neurotox Res [DOI] [PMC free article] [PubMed]
- 52.Fitting S, Knapp PE, Zou S, Marks WD, Bowers MS, Akbarali HI, Hauser KF (2014) Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na(+) influx, mitochondrial instability, and Ca(2)(+) overload. J Neurosci 34:12850–12864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haughey NJ, Mattson TP (2002) Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins tat and gp120. JAIDS Journal of Acquired Immune Deficiency Syndromes 31:S55–S61 [DOI] [PubMed] [Google Scholar]
- 54.Malik S, Eugenin EA (2017) Role of Connexin and Pannexin containing channels in HIV infection and NeuroAIDS. Neurosci Lett [DOI] [PMC free article] [PubMed]
- 55.Weilinger NL, Lohman AW, Rakai BD, Ma EM, Bialecki J, Maslieieva V, Rilea T, Bandet MV, Ikuta NT, Scott L, Colicos MA, Teskey GC, Winship IR, Thompson RJ (2016) Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat Neurosci 19:432–442 [DOI] [PubMed] [Google Scholar]
- 56.Hu R, Chen J, Lujan B, Lei R, Zhang M, Wang Z, Liao M, Li Z, Wan Y, Liu F, Feng H, Wan Q (2016) Glycine triggers a non-ionotropic activity of GluN2A-containing NMDA receptors to confer neuroprotection. Sci Rep 6:34459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R (2013) Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci U S A 110:4027–4032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gelman BB, Nguyen TP (2010) Synaptic proteins linked to HIV-1 infection and immunoproteasome induction: proteomic analysis of human synaptosomes. J Neuroimmune Pharmacol 5:92–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin AH, Thayer SA (2013) Human immunodeficiency virus-1 protein Tat induces excitotoxic loss of presynaptic terminals in hippocampal cultures. Mol Cell Neurosci 54:22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hargus NJ, Thayer SA (2013) Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J Neurosci 33:17908–17920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44:5–21 [DOI] [PubMed] [Google Scholar]
- 62.Strack S, Colbran RJ (1998) Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J Biol Chem 273:20689–20692 [DOI] [PubMed] [Google Scholar]
- 63.Ding JD, Kennedy MB, Weinberg RJ (2013) Subcellular organization of camkii in rat hippocampal pyramidal neurons. J Comp Neurol 521:3570–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swulius MT, Waxham MN (2008) Ca(2+)/calmodulin-dependent protein kinases. Cell Mol Life Sci 65:2637–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3:175–190 [DOI] [PubMed] [Google Scholar]
- 66.Hook SS, Means AR (2001) Ca(2+)/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol 41:471–505 [DOI] [PubMed] [Google Scholar]
- 67.Tian L, Stefanidakis M, Ning L, Van Lint P, Nyman-Huttunen H, Libert C, Itohara S, Mishina M, Rauvala H, Gahmberg CG (2007) Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J Cell Biol 178:687–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Louboutin JP, Reyes BA, Agrawal L, Van Bockstaele EJ, Strayer DS (2011) HIV-1 gp120 upregulates matrix metalloproteinases and their inhibitors in a rat model of HIV encephalopathy. Eur J Neurosci 34:2015–2023 [DOI] [PubMed] [Google Scholar]
- 69.Sanderson JL, Gorski JA, Dell’Acqua ML (2016) NMDA Receptor-Dependent LTD Requires Transient Synaptic Incorporation of Ca(2)(+)-Permeable AMPARs Mediated by AKAP150-Anchored PKA and Calcineurin. Neuron 89:1000–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188 [DOI] [PubMed] [Google Scholar]
- 71.Rutherford LC, Nelson SB, Turrigiano GG (1998) BDNF Has Opposite Effects on the Quantal Amplitude of Pyramidal Neuron and Interneuron Excitatory Synapses. Neuron 21:521–530 [DOI] [PubMed] [Google Scholar]
- 72.Desai NS, Rutherford LC, Turrigiano GG (1999) BDNF regulates the intrinsic excitability of cortical neurons. Learning and Memory 6:284–291 [PMC free article] [PubMed] [Google Scholar]
- 73.Bachis A, Avdoshina V, Zecca L, Parsadanian M, Mocchetti I (2012) Human immunodeficiency virus type 1 alters brain-derived neurotrophic factor processing in neurons. J Neurosci 32:9477–9484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nosheny RL, Bachis A, Acquas E, Mocchetti I (2004) Human immunodeficiency virus type 1 glycoprotein gp120 reduces the levels of brain-derived neurotrophic factor in vivo: potential implication for neuronal cell death. Eur J Neurosci 20:2857–2864 [DOI] [PubMed] [Google Scholar]
- 75.Rahimian P, He JJ (2016) HIV-1 Tat-shortened neurite outgrowth through regulation of microRNA-132 and its target gene expression. J Neuroinflammation 13:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Albrecht D, Garcia L, Cartier L, Kettlun AM, Vergara C, Collados L, Valenzuela MA (2006) Trophic factors in cerebrospinal fluid and spinal cord of patients with tropical spastic paraparesis, HIV, and Creutzfeldt-Jakob disease. AIDS Res Hum Retroviruses 22:248–254 [DOI] [PubMed] [Google Scholar]
- 77.Meeker RB, Poulton W, Markovic-Plese S, Hall C, Robertson K (2011) Protein changes in CSF of HIV-infected patients: evidence for loss of neuroprotection. J Neurovirol 17:258–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Banerjee S, Liao L, Russo R, Nakamura T, McKercher SR, Okamoto S, Haun F, Nikzad R, Zaidi R, Holland E, Eroshkin A, Yates JR 3rd, Lipton SA (2012) Isobaric tagging-based quantification by mass spectrometry of differentially regulated proteins in synaptosomes of HIV/gp120 transgenic mice: implications for HIV-associated neurodegeneration. Exp Neurol 236:298–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raja R, Ronsard L, Lata S, Trivedi S, Banerjea AC (2017) HIV-1 Tat potently stabilises Mdm2 and enhances viral replication. Biochem J 474:2449–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perez-Otano I, Ehlers MD (2005) Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci 28:229–238 [DOI] [PubMed] [Google Scholar]
- 81.Nguyen TP, Soukup VM, Gelman BB (2010) Persistent hijacking of brain proteasomes in HIV-associated dementia. Am J Pathol 176:893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD (2003) Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40:595–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fuchs SY, Adler V, Buschmann T, Wu X, Ronai Z (1998) Mdm2 association with p53 targets its ubiquitination. Oncogene 17:2543–2547 [DOI] [PubMed] [Google Scholar]
- 84.Aprea S, Del Valle L, Mameli G, Sawaya BE, Khalili K, Peruzzi F (2006) Tubulin-Mediated Binding of Human Immunodeficiency Virus-1 Tat to the Cytoskeleton Causes Proteasomal-Dependent Degradation of Microtubule-Associated Protein 2 and Neuronal Damage. J Neurosci 26:4054–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rechsteiner M, Rogers SW (1996) PEST sequences and regulation by proteolysis. Trends Biochem Sci 21:267–271 [PubMed] [Google Scholar]
- 86.Levy JM, Chen X, Reese TS, Nicoll RA (2015) Synaptic Consolidation Normalizes AMPAR Quantal Size following MAGUK Loss. Neuron 87:534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bissel SJ, Wang G, Ghosh M, Reinhart TA, Capuano S 3rd, Stefano Cole K, Murphey-Corb M, Piatak M Jr, Jr., Lifson JD, Wiley CA (2002) Macrophages relate presynaptic and postsynaptic damage in simian immunodeficiency virus encephalitis. Am J Pathol 160:927–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kovalevich J, Langford D (2012) Neuronal toxicity in HIV CNS disease. Future virology 7:687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lam S, Wiercinska E, Teunisse AF, Lodder K, ten Dijke P, Jochemsen AG (2014) Wild-type p53 inhibits pro-invasive properties of TGF-beta3 in breast cancer, in part through regulation of EPHB2, a new TGF-beta target gene. Breast cancer research and treatment 148:7–18 [DOI] [PubMed] [Google Scholar]
- 90.Yuferov V, Ho A, Morgello S, Yang Y, Ott J, Kreek MJ (2013) Expression of ephrin receptors and ligands in postmortem brains of HIV-infected subjects with and without cognitive impairment. J Neuroimmune Pharmacol 8:333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brandimarti R, Khan MZ, Fatatis A, Meucci O (2004) Regulation of cell cycle proteins by chemokine receptors: A novel pathway in human immunodeficiency virus neuropathogenesis? J Neurovirol 10 Suppl 1:108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ferrarese C, Aliprandi A, Tremolizzo L, Stanzani L, De Micheli A, Dolara A, Frattola L (2001) Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 57:671–675 [DOI] [PubMed] [Google Scholar]
- 93.Meisner F, Neuen-Jacob E, Sopper S, Schmidt M, Schlammes S, Scheller C, Vosswinkel D, Ter Meulen V, Riederer P, Koutsilieri E (2008) Disruption of excitatory amino acid transporters in brains of SIV-infected rhesus macaques is associated with microglia activation. J Neurochem 104:202–209 [DOI] [PubMed] [Google Scholar]
- 94.Wang Z, Trillo-Pazos G, Kim SY, Canki M, Morgello S, Sharer LR, Gelbard HA, Su ZZ, Kang DC, Brooks AI, Fisher PB, Volsky DJ (2004) Effects of human immunodeficiency virus type 1 on astrocyte gene expression and function: potential role in neuropathogenesis. J Neurovirol 10 Suppl 1:25–32 [DOI] [PubMed] [Google Scholar]
- 95.Potter MC, Figuera-Losada M, Rojas C, Slusher BS (2013) Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol 8:594–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Melendez RI, Roman C, Capo-Velez CM, Lasalde-Dominicci JA (2016) Decreased glial and synaptic glutamate uptake in the striatum of HIV-1 gp120 transgenic mice. J Neurovirol 22:358–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dreyer EB, Lipton SA (1995) The coat protein gp120 of HIV-1 inhibits astrocyte uptake of excitatory amino acids via macrophage arachidonic acid. Eur J Neurosci 7:2502–2507 [DOI] [PubMed] [Google Scholar]
- 98.Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA (1996) Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem 271:15303–15306 [DOI] [PubMed] [Google Scholar]
- 99.Marty V, El Hachmane M, Amedee T (2008) Dual modulation of synaptic transmission in the nucleus tractus solitarius by prostaglandin E2 synthesized downstream of IL-1beta. Eur J Neurosci 27:3132–3150 [DOI] [PubMed] [Google Scholar]
- 100.Bellizzi MJ, Lu SM, Masliah E, Gelbard HA (2005) Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor. J Clin Invest 115:3185–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu SM, Tong N, Gelbard HA (2007) The phospholipid mediator platelet-activating factor mediates striatal synaptic facilitation. J Neuroimmune Pharmacol 2:194–201 [DOI] [PubMed] [Google Scholar]
- 102.Bazan NG (1998) The neuromessenger platelet-activating factor in plasticity and neurodegeneration. Prog Brain Res 118:281–291 [DOI] [PubMed] [Google Scholar]
- 103.Musante V, Summa M, Neri E, Puliti A, Godowicz TT, Severi P, Battaglia G, Raiteri M, Pittaluga A (2010) The HIV-1 Viral Protein Tat Increases Glutamate and Decreases GABA exocytosis from Human and Mouse Neocortical Nerve Endings. Cerebral Cortex 20:1974–1984 [DOI] [PubMed] [Google Scholar]
- 104.Perry SW, Norman JP, Litzburg A, Zhang D, Dewhurst S, Gelbard HA (2005) HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol 174:4333–4344 [DOI] [PubMed] [Google Scholar]
- 105.Salter MW, Dong Y, Kalia LV, Liu XJ, Pitcher G (2009) Regulation of NMDA Receptors by Kinases and Phosphatases Biology of the NMDA Receptor Taylor & Francis Group, LLC., Boca Raton FL: [PubMed] [Google Scholar]
- 106.Krogh KA, Lyddon E, Thayer SA (2014) HIV-1 Tat activates a RhoA signaling pathway to reduce NMDA-evoked calcium responses in hippocampal neurons via an actin-dependent mechanism. J Neurochem [DOI] [PMC free article] [PubMed]
- 107.Babiloni C, Buffo P, Vecchio F, Onorati P, Muratori C, Ferracuti S, Roma P, Battuello M, Donato N, Noce G, Di Campli F, Gianserra L, Teti E, Aceti A, Soricelli A, Viscione M, Andreoni M, Rossini PM, Pennica A (2014) Cortical sources of resting-state EEG rhythms in “experienced” HIV subjects under antiretroviral therapy. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology 125:1792–1802 [DOI] [PubMed] [Google Scholar]
- 108.Modi M, Mochan A, Modi G (2009) New onset seizures in HIV--seizure semiology, CD4 counts, and viral loads. Epilepsia 50:1266–1269 [DOI] [PubMed] [Google Scholar]
- 109.Kasyanov A, Tamamura H, Fujii N, Xiong H (2006) HIV-1 gp120 enhances giant depolarizing potentials via chemokine receptor CXCR4 in neonatal rat hippocampus. Eur J Neurosci 23:1120–1128 [DOI] [PubMed] [Google Scholar]
- 110.Gelman BB, Spencer JA, Holzer CE 3rd, Soukup VM (2006) Abnormal striatal dopaminergic synapses in National NeuroAIDS Tissue Consortium subjects with HIV encephalitis. J Neuroimmune Pharmacol 1:410–420 [DOI] [PubMed] [Google Scholar]
- 111.Zhu J, Ananthan S, Mactutus CF, Booze RM (2011) Recombinant human immunodeficiency virus-1 transactivator of transcription1–86 allosterically modulates dopamine transporter activity. Synapse 65:1251–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM (2009) In vivo microdialysis in awake, freely moving rats demonstrates HIV-1 Tat-induced alterations in dopamine transmission. Synapse 63:181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM (2009) The human immunodeficiency virus-1-associated protein, Tat1–86, impairs dopamine transporters and interacts with cocaine to reduce nerve terminal function: a no-net-flux microdialysis study. Neurosci 159:1292–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Midde NM, Gomez AM, Zhu J (2012) HIV-1 Tat protein decreases dopamine transporter cell surface expression and vesicular monoamine transporter-2 function in rat striatal synaptosomes. J Neuroimmune Pharmacol 7:629–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scheller C, Arendt G, Nolting T, Antke C, Sopper S, Maschke M, Obermann M, Angerer A, Husstedt IW, Meisner F, Neuen-Jacob E, Muller HW, Carey P, Ter Meulen V, Riederer P, Koutsilieri E (2010) Increased dopaminergic neurotransmission in therapy-naive asymptomatic HIV patients is not associated with adaptive changes at the dopaminergic synapses. Journal of neural transmission (Vienna, Austria : 1996) 117:699–705 [DOI] [PubMed] [Google Scholar]
- 116.Purohit V, Rapaka R, Frankenheim J, Avila A, Sorensen R, Rutter J (2013) National Institute on Drug Abuse symposium report: drugs of abuse, dopamine, and HIV-associated neurocognitive disorders/HIV-associated dementia. J Neurovirol 19:119–122 [DOI] [PubMed] [Google Scholar]
- 117.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK (2004) Role of microglia in central nervous system infections. Clin Microbiol Rev 17:942–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tremblay ME, Marker DF, Puccini JM, Muly EC, Lu SM, Gelbard HA (2013) Ultrastructure of microglia-synapse interactions in the HIV-1 Tat-injected murine central nervous system. Commun Integr Biol 6:e27670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lu SM, Tremblay ME, King IL, Qi J, Reynolds HM, Marker DF, Varrone JJ, Majewska AK, Dewhurst S, Gelbard HA (2011) HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One 6:e23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mosser CA, Baptista S, Arnoux I, Audinat E (2017) Microglia in CNS development: Shaping the brain for the future. Prog Neurobiol 149–150:1–20 [DOI] [PubMed] [Google Scholar]
- 121.Hong S, Dissing-Olesen L, Stevens B (2016) New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36:128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Aarts MM, Tymianski M (2003) Novel treatment of excitotoxicity: targeted disruption of intracellular signalling from glutamate receptors. Biochem Pharmacol 66:877–886 [DOI] [PubMed] [Google Scholar]
- 123.McGuire JL, Gill AJ, Douglas SD, Kolson DL (2016) The complement system, neuronal injury, and cognitive function in horizontally-acquired HIV-infected youth. J Neurovirol 22:823–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Collini P, Noursadeghi M, Sabroe I, Miller RF, Dockrell DH (2010) Monocyte and macrophage dysfunction as a cause of HIV-1 induced dysfunction of innate immunity. Curr Mol Med 10:727–740 [DOI] [PubMed] [Google Scholar]
- 125.Kedzierska K, Azzam R, Ellery P, Mak J, Jaworowski A, Crowe SM (2003) Defective phagocytosis by human monocyte/macrophages following HIV-1 infection: underlying mechanisms and modulation by adjunctive cytokine therapy. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology 26:247–263 [DOI] [PubMed] [Google Scholar]
- 126.Debaisieux S, Lachambre S, Gross A, Mettling C, Besteiro S, Yezid H, Henaff D, Chopard C, Mesnard JM, Beaumelle B (2015) HIV-1 Tat inhibits phagocytosis by preventing the recruitment of Cdc42 to the phagocytic cup. Nat Commun 6:6211. [DOI] [PubMed] [Google Scholar]
- 127.Krogh KA, Lyddon E, Thayer SA (2015) HIV-1 Tat activates a RhoA signaling pathway to reduce NMDA-evoked calcium responses in hippocampal neurons via an actin-dependent mechanism. J Neurochem 132:354–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME (2008) Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455:1198–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu C, Hermes DJ, Mackie K, Lichtman AH, Ignatowska-Jankowska BM, Fitting S (2016) Cannabinoids Occlude the HIV-1 Tat-Induced Decrease in GABAergic Neurotransmission in Prefrontal Cortex Slices. J Neuroimmune Pharmacol 11:316–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xu C, Fitting S (2016) Inhibition of GABAergic Neurotransmission by HIV-1 Tat and Opioid Treatment in the Striatum Involves mu-Opioid Receptors. Front Neurosci 10:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gelman BB, Chen T, Lisinicchia JG, Soukup VM, Carmical JR, Starkey JM, Masliah E, Commins DL, Brandt D, Grant I, Singer EJ, Levine AJ, Miller J, Winkler JM, Fox HS, Luxon BA (2012) The National NeuroAIDS Tissue Consortium Brain Gene Array: Two Types of HIV-Associated Neurocognitive Impairment. PLoS One 7:e46178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.de San Martin JZ, Delabar JM, Bacci A, Potier MC (2017) GABAergic over-inhibition, a promising hypothesis for cognitive deficits in Down syndrome. Free Radic Biol Med [DOI] [PubMed]
- 133.Contestabile A, Magara S, Cancedda L (2017) The GABAergic Hypothesis for Cognitive Disabilities in Down Syndrome. Front Cell Neurosci 11:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Souchet B, Guedj F, Penke-Verdier Z, Daubigney F, Duchon A, Herault Y, Bizot JC, Janel N, Creau N, Delatour B, Delabar JM (2015) Pharmacological correction of excitation/inhibition imbalance in Down syndrome mouse models. Frontiers in behavioral neuroscience 9:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu Z, Guo Z, Gearing M, Chen G (2014) Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat Commun 5:4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cavanna AE, Ali F, Rickards HE, McCorry D (2010) Behavioral and cognitive effects of anti-epileptic drugs. Discovery medicine 9:138–144 [PubMed] [Google Scholar]
- 137.Xia P, Chen H-sV, Zhang D, Lipton SA (2010) Memantine Preferentially Blocks Extrasynaptic over Synaptic NMDA Receptor Currents in Hippocampal Autapses. J Neurosci 30:11246–11250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lipton SA (1992) Memantine prevents HIV coat protein-induced neuronal injury in vitro [see comments]. Neurology 42:1403–1405 [DOI] [PubMed] [Google Scholar]
- 139.Toggas SM, Masliah E, Mucke L (1996) Prevention of HIV-1 gp120-induced neuronal damage in the central nervous system of transgenic mice by the NMDA receptor antagonist memantine. Brain Res 706:303–307 [DOI] [PubMed] [Google Scholar]
- 140.Muller WE, Pergande G, Ushijima H, Schleger C, Kelve M, Perovic S (1996) Neurotoxicity in rat cortical cells caused by N-methyl-D-aspartate (NMDA) and gp120 of HIV-1: induction and pharmacological intervention. Progress in molecular and subcellular biology 16:44–57 [DOI] [PubMed] [Google Scholar]
- 141.Albers GW, Atkinson RP, Kelley RE, Rosenbaum DM (1995) Safety, tolerability, and pharmacokinetics of the N-methyl-D-aspartate antagonist dextrorphan in patients with acute stroke. Dextrorphan Study Group. Stroke 26:254–258 [DOI] [PubMed] [Google Scholar]
- 142.Grotta J, Clark W, Coull B, Pettigrew LC, Mackay B, Goldstein LB, Meissner I, Murphy D, LaRue L (1995) Safety and tolerability of the glutamate antagonist CGS 19755 (Selfotel) in patients with acute ischemic stroke. Results of a phase IIa randomized trial. Stroke 26:602–605 [DOI] [PubMed] [Google Scholar]
- 143.Garwood CJ, Cooper JD, Hanger DP, Noble W (2010) Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Frontiers in psychiatry 1:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cheng S, Hou J, Zhang C, Xu C, Wang L, Zou X, Yu H, Shi Y, Yin Z, Chen G (2015) Minocycline reduces neuroinflammation but does not ameliorate neuron loss in a mouse model of neurodegeneration. Sci Rep 5:10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Garcez ML, Mina F, Bellettini-Santos T, Carneiro FG, Luz AP, Schiavo GL, Andrighetti MS, Scheid MG, Bolfe RP, Budni J (2017) Minocycline reduces inflammatory parameters in the brain structures and serum and reverses memory impairment caused by the administration of amyloid beta (1–42) in mice. Prog Neuropsychopharmacol Biol Psychiatry 77:23–31 [DOI] [PubMed] [Google Scholar]
- 146.Si Q, Cosenza M, Kim MO, Zhao ML, Brownlee M, Goldstein H, Lee S (2004) A novel action of minocycline: inhibition of human immunodeficiency virus type 1 infection in microglia. J Neurovirol 10:284–292 [DOI] [PubMed] [Google Scholar]
- 147.Zink MC, Uhrlaub J, DeWitt J, Voelker T, Bullock B, Mankowski J, Tarwater P, Clements J, Barber S (2005) Neuroprotective and anti-human immunodeficiency virus activity of minocycline. JAMA 293:2003–2011 [DOI] [PubMed] [Google Scholar]
- 148.Sacktor N, Miyahara S, Deng L, Evans S, Schifitto G, Cohen BA, Paul R, Robertson K, Jarocki B, Scarsi K, Coombs RW, Zink MC, Nath A, Smith E, Ellis RJ, Singer E, Weihe J, McCarthy S, Hosey L, Clifford DB (2011) Minocycline treatment for HIV-associated cognitive impairment: results from a randomized trial. Neurology 77:1135–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nakasujja N, Miyahara S, Evans S, Lee A, Musisi S, Katabira E, Robertson K, Ronald A, Clifford DB, Sacktor N (2013) Randomized trial of minocycline in the treatment of HIV-associated cognitive impairment. Neurology 80:196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mocchetti I, Bachis A, Campbell LA, Avdoshina V (2014) Implementing neuronal plasticity in NeuroAIDS: the experience of brain-derived neurotrophic factor and other neurotrophic factors. J Neuroimmune Pharmacol 9:80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Everall IP, Trillo-Pazos G, Bell C, Mallory M, Sanders V, Masliah E (2001) Amelioration of neurotoxic effects of HIV envelope protein gp120 by fibroblast growth factor: a strategy for neuroprotection. J Neuropathol Exp Neurol 60:293–301 [DOI] [PubMed] [Google Scholar]
- 152.Fields J, Dumaop W, Langford TD, Rockenstein E, Masliah E (2014) Role of neurotrophic factor alterations in the neurodegenerative process in HIV associated neurocognitive disorders. J Neuroimmune Pharmacol 9:102–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bachis A, Major EO, Mocchetti I (2003) Brain-derived neurotrophic factor inhibits human immunodeficiency virus-1/gp120-mediated cerebellar granule cell death by preventing gp120 internalization. J Neurosci 23:5715–5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B (2003) CB1 Cannabinoid Receptors and On-Demand Defense Against Excitotoxicity. Science 302:84–88 [DOI] [PubMed] [Google Scholar]
- 155.Shen M, Thayer SA (1998) Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol 54:459–462 [DOI] [PubMed] [Google Scholar]
- 156.Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA (1999) Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 19:2987–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim HJ, Waataja JJ, Thayer SA (2008) Cannabinoids inhibit network-driven synapse loss between hippocampal neurons in culture. J Pharmacol Exp Ther 325:850–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Purohit V, Rapaka RS, Rutter J (2014) Cannabinoid receptor-2 and HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol 9:447–453 [DOI] [PubMed] [Google Scholar]
- 159.Avraham HK, Jiang S, Fu Y, Rockenstein E, Makriyannis A, Zvonok A, Masliah E, Avraham S (2014) The cannabinoid CB(2) receptor agonist AM1241 enhances neurogenesis in GFAP/Gp120 transgenic mice displaying deficits in neurogenesis. Br J Pharmacol 171:468–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gorantla S, Makarov E, Roy D, Finke-Dwyer J, Murrin LC, Gendelman HE, Poluektova L (2012) Immunoregulation of a CB2 receptor agonist in a murine model of neuroAIDS. J Neuroimmune Pharmacol 5:456–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF (2011) Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334:809–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kinsey SG, Wise LE, Ramesh D, Abdullah R, Selley DE, Cravatt BF, Lichtman AH (2013) Repeated Low Dose Administration of the Monoacylglycerol Lipase Inhibitor JZL184 Retains CB1 Receptor Mediated Antinociceptive and Gastroprotective Effects. J Pharmacol Exp Ther 345:492–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Scheiman JM (2016) NSAID-induced Gastrointestinal Injury: A Focused Update for Clinicians. Journal of clinical gastroenterology 50:5–10 [DOI] [PubMed] [Google Scholar]
- 164.Ferrando SJ, Rabkin JG, van Gorp W, Lin SH, McElhiney M (2003) Longitudinal improvement in psychomotor processing speed is associated with potent combination antiretroviral therapy in HIV-1 infection. The Journal of neuropsychiatry and clinical neurosciences 15:208–214 [DOI] [PubMed] [Google Scholar]
- 165.Thurnher MM, Schindler EG, Thurnher SA, Pernerstorfer-Schon H, Kleibl-Popov C, Rieger A (2000) Highly active antiretroviral therapy for patients with AIDS dementia complex: effect on MR imaging findings and clinical course. AJNR American journal of neuroradiology 21:670–678 [PMC free article] [PubMed] [Google Scholar]
- 166.Schellenberg GD, D’Souza I, Poorkaj P (2000) The genetics of Alzheimer’s disease. Current psychiatry reports 2:158–164 [DOI] [PubMed] [Google Scholar]
- 167.Corder EH, Robertson K, Lannfelt L, Bogdanovic N, Eggertsen G, Wilkins J, Hall C (1998) HIV-infected subjects with the E4 allele for APOE have excess dementia and peripheral neuropathy. Nat Med 4:1182–1184 [DOI] [PubMed] [Google Scholar]
- 168.Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW (2007) HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A 104:3438–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Keblesh JP, Dou H, Gendelman HE, Xiong H (2009) 4-Aminopyridine improves spatial memory in a murine model of HIV-1 encephalitis. J Neuroimmune Pharmacol 4:317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nakanishi N, Kang YJ, Tu S, McKercher SR, Masliah E, Lipton SA (2016) Differential Effects of Pharmacologic and Genetic Modulation of NMDA Receptor Activity on HIV/gp120-Induced Neuronal Damage in an In Vivo Mouse Model. J Mol Neurosci 58:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Meisner F, Scheller C, Kneitz S, Sopper S, Neuen-Jacob E, Riederer P, ter Meulen V, Koutsilieri E (2008) Memantine upregulates BDNF and prevents dopamine deficits in SIV-infected macaques: a novel pharmacological action of memantine. Neuropsychopharmacol 33:2228–2236 [DOI] [PubMed] [Google Scholar]
- 172.Navia BA, Dafni U, Simpson D, Tucker T, Singer E, McArthur JC, Yiannoutsos C, Zaborski L, Lipton SA (1998) A phase I/II trial of nimodipine for HIV-related neurologic complications. Neurology 51:221–228 [DOI] [PubMed] [Google Scholar]
- 173.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I (2011) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17:3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Schifitto G, Yiannoutsos CT, Simpson DM, Marra CM, Singer EJ, Kolson DL, Nath A, Berger JR, Navia B, Group Actg 301 Team AA (2006) A placebo-controlled study of memantine for the treatment of human immunodeficiency virus-associated sensory neuropathy. J Neurovirol 12:328–331 [DOI] [PubMed] [Google Scholar]
- 175.Schifitto G, Navia BA, Yiannoutsos CT, Marra CM, Chang L, Ernst T, Jarvik JG, Miller EN, Singer EJ, Ellis RJ, Kolson DL, Simpson D, Nath A, Berger J, Shriver SL, Millar LL, Colquhoun D, Lenkinski R, Gonzalez RG, Lipton SA (2007) Memantine and HIV-associated cognitive impairment: a neuropsychological and proton magnetic resonance spectroscopy study. Aids 21:1877–1886 [DOI] [PubMed] [Google Scholar]