

Abstract

The functionalization of semiconducting single-walled carbon nanotubes (SWNTs) with sp3 defects that act as luminescent exciton traps is a powerful means to enhance their photoluminescence quantum yield (PLQY) and to add optical properties. However, the synthetic methods employed to introduce these defects are currently limited to aqueous dispersions of surfactant-coated SWNTs, often with short tube lengths, residual metallic nanotubes, and poor film-formation properties. In contrast to that, dispersions of polymer-wrapped SWNTs in organic solvents feature unrivaled purity, higher PLQY, and are easily processed into thin films for device applications. Here, we introduce a simple and scalable phase-transfer method to solubilize diazonium salts in organic nonhalogenated solvents for the controlled reaction with polymer-wrapped SWNTs to create luminescent aryl defects. Absolute PLQY measurements are applied to reliably quantify the defect-induced brightening. The optimization of defect density and trap depth results in PLQYs of up to 4% with 90% of photons emitted through the defect channel. We further reveal the strong impact of initial SWNT quality and length on the relative brightening by sp3 defects. The efficient and simple production of large quantities of defect-tailored polymer-sorted SWNTs enables aerosol-jet printing and spin-coating of thin films with bright and nearly reabsorption-free defect emission, which are desired for carbon nanotube-based near-infrared light-emitting devices.
Keywords: single-walled carbon nanotubes, sp3 defects, functionalization, photoluminescence, diazonium salts
The combination of narrow-band emission in the near-infrared (NIR) and high charge carrier mobilities make semiconducting single-walled carbon nanotubes (SWNTs) highly desirable materials for optoelectronic devices, such as light-emitting diodes1,2 and field-effect transistors.3−5 The fabrication of such devices based on homogeneous thin films requires concentrated SWNT inks without any metallic nanotubes6 that can be processed easily and reproducibly. While photoluminescence (PL) from SWNTs was first observed in aqueous dispersions stabilized by surfactants,7 nowadays selective wrapping with conjugated polymers in organic solvents is often applied for the extraction of semiconducting SWNTs with the highest purity and photoluminescence quantum yields (PLQYs).8 In conjunction with mild and scalable exfoliation methods such as shear force mixing,9 large volumes of dispersions with high concentrations of long SWNTs with high quality can be produced by polymer wrapping. These dispersions are ideal for the deposition of homogeneous films by spin-coating,10 aerosol-jet11 or inkjet printing,12 owing to the beneficial properties of organic solvents (surface tension, viscosity, vapor pressure, etc.). However, despite the substantial progress in terms of purification and processing, light-emitting devices based on carbon nanotubes are still limited by their low absolute PLQYs (∼1% in dispersion and ∼0.1% in films) resulting from the presence of low-lying dark exciton states13 and the fast diffusion of mobile excitons to nonradiative quenching sites.14,15
The most promising approach to enhance the PLQY of SWNTs is their functionalization with a limited number of oxygen16,17 or sp3 defects18−20 that act as luminescent exciton traps with depths on the order of 100 meV. Excitons trapped at these defect sites are not subject to otherwise dominant diffusion-limited contact-quenching,14 and the modification of the local electronic structure by the defects enables radiative relaxation and red-shifted emission (labeled as E11*, or E11*– for different binding configurations).21−24 As demonstrated by Piao et al.,18 the emission from sp3 defects that are introduced to the SWNT lattice via aryldiazonium chemistry can be much brighter than that of pristine SWNTs. This report motivated a series of studies on the population mechanism25 and relaxation dynamics26−29 of these emissive trap states and the impact of the different binding configurations on their optical properties.22,30−32 Moreover, it was shown that sp3 defects in SWNTs can be employed for room-temperature single-photon emission in the NIR,29−31,33 PL imaging within the second biological window34 and sensing.35 However, the synthetic methods for the creation of such sp3 defects usually involve the use of highly polar reagents,18,36 such as diazonium salts. They are thus limited to dispersions of SWNTs in water. While it is possible to sort semiconducting SWNTs by chromatography or two-phase extraction of aqueous dispersions,37,38 the superior purity of polymer-sorted SWNTs for a minimum of purification steps makes them preferable for optoelectronic applications.6,9,39,40
Given the advantages of polymer-wrapped SWNTs in organic solvents it is highly desirable to develop an easy and reproducible sp3 functionalization method for these systems. The main challenge is the conflict between the solubility of polar diazonium salts and the colloidal stability of SWNTs wrapped with nonpolar conjugated polymers. Although the in situ generation of diazonium salts may be used, this approach suffers from limited scalability, as it requires elevated temperatures and inert conditions.41,42 Other attempts, for example, by dip-doping, were limited to predeposited polymer-sorted nanotubes and only successful with very reactive diazonium salts.31
Here, we present a simple and scalable method to create luminescent sp3 defects in polymer-wrapped (6,5) SWNTs in organic nonhalogenated solvents. The addition of a polar cosolvent and a phase-transfer agent allows the preformed diazonium salts to be solubilized and to react with the SWNTs at room temperature. Because of the high concentration and purity of the resulting dispersions of functionalized SWNTs, the absorption features arising from the defects can be analyzed in detail for the first time. These data give insights into reorganization processes upon exciton trapping and site-to-site interactions. To reliably quantify the functionalization-induced brightening, we determine the PLQY by absolute measurements in an integrating sphere and demonstrate the influence of defect density and trap depth on the PLQY enhancement. We reveal the strong impact of initial SWNT quality and length on the relative brightening by sp3 defects. This simple functionalization method provides access to large quantities of polymer-wrapped and sp3-functionalized SWNTs that can be easily processed into thin or thick films with enhanced PLQY that may enable brighter NIR light-emitting devices.
Results and Discussion
Our method for creating luminescent sp3 defects in polymer-wrapped SWNTs relies on the solubilization of preformed aryldiazonium salts by an ether crown43 (here, 18-crown-6) in a moderately polar medium (see Figure 1). This reaction approach avoids chlorinated solvents while facilitating large-scale functionalization of polymer-wrapped carbon nanotubes with a minimum of processing steps. As a model system we use (6,5) SWNTs, which are extracted with high purity by selective dispersion with the polyfluorene copolymer poly-[(9,9-dioctylfluorenyl-2,7-diyl)-alt-co-(6,6′)-(2,2′-bipyridine)] (PFO-BPy) in toluene (see Methods). A mixture of toluene and acetonitrile (MeCN) in an 80:20 vol % ratio is sufficiently polar to solubilize aryldiazonium salts at concentrations on the order of a few grams per liter in the presence of 18-crown-6 as a phase-transfer agent, yet it does not lead to substantial aggregation of (6,5) SWNTs or free PFO-BPy as corroborated by absorption and PL spectra (see the Supporting Information, Figure S1). A low concentration (1 × 10–9 mol L–1) of potassium acetate (KOAc) is included to optimize the selectivity of the functionalization process (for details, see the Supporting Information, Figure S2). After a typical reaction time of 16 h at room temperature and in the dark, the SWNTs are collected by vacuum filtration, and unreacted diazonium salt is washed off with acetonitrile and toluene. This washing step is crucial, as we found the SWNT PL emission of the reaction mixture to be strongly quenched by diazonium salts adsorbed on the SWNT surface. In aqueous media less than 10% of the added diazonium salt actually reacts with the SWNT lattice to form sp3 defects,44 and the reactivity in organic solvents turns out to be even lower. This drop in reactivity might result from differences in the reaction mechanism (see the Supporting Information), the surface accessibility provided by the wrapping polymer or steric hindrance due to ether crown complexation. Hence, it is likely that a large number of unreacted aryldiazonium cations are adsorbed on the SWNTs and quench the PL. After they are washed, the sp3-functionalized (6,5) SWNTs are easily redispersed in pure solvent (here, toluene) by bath sonication, and their intrinsic spectroscopic properties in dispersion can be studied. Note that the filtration and washing step does not constitute an additional workload, as it also removes most of the excess polymer, which is often a necessary step in film and device fabrication. A detailed experimental protocol for sp3 functionalization of PFO-BPy-wrapped (6,5) SWNTs is provided in the Supporting Information.
Figure 1.

Schematic functionalization of PFO-BPy-wrapped (6,5) SWNTs by aryldiazonium salts in a toluene/acetonitrile mixture containing 18-crown-6 as a phase-transfer agent.
The overall procedure is scalable to large amounts of functionalized (6,5) SWNTs that can be redispersed at different low or high concentrations thus enabling reliable absorption measurements, determination of the absolute PLQY, and creation of thick luminescent films. To establish the robustness and applicability of our procedure for polymer-wrapped SWNTs, we employ the outlined routine to introduce aryl defects from low to high defect densities and with substituents ranging from electron-withdrawing to electron-donating character and then characterize their optical properties in detail.
First, we discuss the spectral characteristics of sp3-functionalized PFO-BPy-wrapped (6,5) SWNTs using the example of 4-bromophenyl defects as shown in Figure 2. In addition to the strong E11 transition at ∼994 nm, the absorption spectra of the dispersions in Figure 2a show an absorption band at ∼1142 nm that grows systematically with the concentration of reagent used. Since unreacted diazonium salt and adsorbed byproducts were removed by washing, and the untreated reference sample does not show any absorption features in this spectral range, we can unambiguously assign this band to sp3 defects. Previous assignments of this transition in the literature were complicated by the presence of minority chiralities and low concentrations of functionalized SWNTs.18 These issues are absent from dispersions prepared by polymer-wrapping. The highest E11*/E11 absorbance ratio in this study was 0.13; however, without knowledge of the extinction coefficient of the defect absorption this value still does not enable an absolute quantification of the number of defects in the lattice (vide infra).
Figure 2.

(a) Normalized absorption spectra and (b) PL spectra recorded under pulsed excitation (575 nm, ∼0.5 mJ cm–2) of PFO-BPy-wrapped (6,5) SWNTs in toluene functionalized using different concentrations of 4-bromobenzenediazonium tetrafluoroborate in the reaction mixture to adjust the defect density. (c) Stokes shift of E11 (black) and E11* (red) transition as a function of diazonium salt concentration. Solid lines are guides.
Emission Spectra and Stokes Shift
Photoluminescence excitation at the E22 transition (575 nm) of (6,5) SWNTs gave the emission spectra presented in Figure 2b showing PL arising from mobile (E11) and trapped (E11*) excitons at ∼999 and ∼1161 nm, respectively. At the highest shown defect density, there is an additional red tail that stretches to 1450 nm and has been attributed to deeper trap states (E11*–) resulting from different arrangements of aryl moieties on the SWNT lattice.22,30−32 Note that all emission features are red-shifted compared to functionalized (6,5) SWNTs dispersed in aqueous surfactant solutions owing to interaction with the conjugated wrapping polymer and differences in the dielectric environment.45 Since the PL was collected through an objective, the NIR absorption of toluene does not affect the spectra as a result of the extremely short path length within the liquid. As expected, the E11* PL intensity increases relative to the E11 with rising diazonium concentration and, thus, defect density. Below a diazonium salt concentration of ∼0.037 mmol L–1, the E11 and E11* transition energies depend only weakly on defect density, supporting the picture of well-isolated sp3 defects. However, above this level, the E11 transition blue-shifts both in absorption and in emission with further increasing defect density, whereas the E11* transition red-shifts. The origin of these opposing spectral shifts is currently not well-understood, but it may provide an interesting starting point for future computational studies. The spectral shifts are more pronounced in absorption than in emission, and this difference is reflected clearly in the Stokes shifts (see Figure 2c). The untreated (6,5) SWNTs exhibit an E11 Stokes shift of 6 meV that reaches 11 meV for highly functionalized samples. Conversely, the E11* Stokes shift drops from 18 to 14 meV as the defect density increases.
The small E11 Stokes shift originates from the rigidity of the SWNT structure (i.e., small reorganization energy) in combination with the delocalized nature of the E11 exciton.46 In contrast to that, quantum chemical calculations predict a significant distortion in the vicinity of an sp3 defect upon exciton trapping.21,26,47,48 At low defect densities the observed E11* Stokes shift is ∼3 times larger than that associated with the E11 exciton. Generally, we find Stokes shifts from 18 to 21 meV depending on the substituent on the aryl group (see Supporting Information, Figure S3). Following the assignment of the E11* transition in (6,5) SWNTs to the ortho++ defect binding configuration30 (equivalent to ortho L90 in ref (22)), the experimentally measured E11* Stokes shift for 4-Br-phenyl defects is almost an order of magnitude smaller than predicted by calculations (18 vs ∼140 meV).22 Even though part of this discrepancy might result from imperfect energy scaling in the quantum chemical calculations,49 it is unlikely to be the sole origin. Further, the range of reorganization energies (20 to 120 meV) extracted from temperature-dependent PL measurements via a model involving vibrational reorganization upon exciton trapping is well-above our measured E11* Stokes shifts.47 This mismatch suggests that the relationship between the optical trap depth given by the energy difference between the E11 and E11* transition and the thermal detrapping energy might be more complex than previously assumed. Nevertheless, the general trend in E11* Stokes shifts reflects the reported behavior of reorganization energies.47 Both quantities decrease as a function of defect density, most likely due to site-to-site interactions and associated electronic state delocalization.47Figure S4 (Supporting Information) summarizes the PL spectra obtained for (6,5) SWNTs functionalized with other aryl defects bearing more electron-withdrawing (4-nitro and 3,5-dichloro) or electron-donating (4-methoxy) substituents. Note that all PL spectra are normalized to the E11 intensity, and thus no conclusions about the relative PLQYs can be drawn from these spectra alone.
Defect Density Metrics
An important experimental consideration is whether the density of the created defects scales linearly with the reagent concentration. Hence, we recorded Raman spectra of the functionalized SWNTs and determined the intensity of the D mode, which is expected to be proportional to the density of symmetry-breaking sp3 defects,50 relative to the G+ mode, which is a measure of the density of sp2 carbon atoms in the probed spot (Supporting Information, Figure S5). Similar to the functionalization process in an aqueous environment,18 we find a roughly linear relationship between the D/G+ ratio and the diazonium concentration (Figure 3a). In addition to this linearity, we observe that the reagent concentration window across which defect luminescence is detectable in organic solvent spans 3 orders of magnitude. In water this range is only 1 order of magnitude and occurs at much lower concentrations.18 While this aspect is simply the result of the lower reactivity of the diazonium species in organic media, it is highly beneficial in practical terms, as it facilitates precise tuning of the defect density.
Figure 3.

Raman, absorption, and PL properties of functionalized (6,5) SWNTs using different concentrations of 4-bromobenzenediazonium tetrafluoroborate. (a) Integrated Raman D/G+ ratio and integrated E11*/E11 absorbance ratio vs diazonium salt concentration. Solid lines are guides. (b) Integrated E11*/E11 PL ratio vs integrated E11*/E11 absorbance ratio for different excitation densities and linear fits to the data.
In addition to the D/G+ ratio of the Raman signals,
the ratio of E11* to E11 absorption was evaluated after background subtraction51,52 and plotted as a function of the diazonium concentration (Figure 3a). The correlation
is roughly linear at low defect levels, but it becomes sublinear at
high levels. This dependence might be explained by assuming the oscillator
strengths (per carbon atom) of pristine and functionalized regions
in a one-dimensional (1D) lattice to be constant at low defect densities.
In this case, defect creation reduces the number of carbon atoms contributing
to E11 absorption, whereas E11* absorption increases by an increment. Then, the E11*/E11 absorbance
ratio is given by 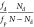 , with fd and fp as the oscillator
strengths of defective and
pristine lattice regions, respectively; Nd denotes the number of sp3 hybridized carbon atoms, and N indicates the total number of carbon atoms in the lattice.
For low defect densities, that is, Nd ≪ N, this geometric model predicts an approximately linear
dependence in agreement with the experimental data. However, at high
defect densities, a superlinear increase would be expected, which
contradicts the measured sublinear behavior. This difference is likely
caused by a drop in oscillator strength of the defects once they start
clustering. Computational studies have already revealed the strong
impact of the addition pattern around the aryl moiety on the oscillator
strength of optical transitions.21,22 Furthermore,
this trend is in agreement with the limiting case of a fully functionalized
SWNT, for which all transitions vanish, and only a scattering background
remains.53
, with fd and fp as the oscillator
strengths of defective and
pristine lattice regions, respectively; Nd denotes the number of sp3 hybridized carbon atoms, and N indicates the total number of carbon atoms in the lattice.
For low defect densities, that is, Nd ≪ N, this geometric model predicts an approximately linear
dependence in agreement with the experimental data. However, at high
defect densities, a superlinear increase would be expected, which
contradicts the measured sublinear behavior. This difference is likely
caused by a drop in oscillator strength of the defects once they start
clustering. Computational studies have already revealed the strong
impact of the addition pattern around the aryl moiety on the oscillator
strength of optical transitions.21,22 Furthermore,
this trend is in agreement with the limiting case of a fully functionalized
SWNT, for which all transitions vanish, and only a scattering background
remains.53
On the basis of these different metrics for the sp3 functionalization level, it is possible to test their relation to the emission properties of (6,5) SWNTs, in particular, the integrated E11*/E11 PL ratio. This ratio is plotted as a function of the diazonium concentration, the Raman D/G+ ratio, and the E11*/E11 absorbance ratio in the Supporting Information, Figure S6. All graphs show a roughly linear correlation with the best fit found for the E11*/E11 PL ratio versus the E11*/E11 absorbance ratio. Importantly, even though the E11*/E11 PL ratio is frequently used itself as a measure of defect density,27,47 it is only meaningful for a given excitation power density owing to the nonlinear behavior of both E11 and E11* emissions. While PL from mobile excitons saturates at high excitation power due to exciton–exciton annihilation,54,55 the saturation of defect emission has been attributed to state-filling due to the fast exciton diffusion to trap sites (∼10 ps) and the long lifetimes of localized excitons (∼200 ps).56 Since defect emission generally saturates at lower excitation power than E11 emission, the E11*/E11 PL ratio decreases as a function of pump power (see Supporting Information, Figure S7a,b). As shown in Figure S7c, PL spectra collected under lamp excitation (∼1 mW cm–2) are strongly dominated by defect emission. The impact of the power dependence on the correlation between E11*/E11 PL ratio and E11*/E11 absorbance ratio can be seen in Figure 3b, where the slope of the linear fit changes with excitation power.
In summary, the Raman D/G+ ratio and the E11*/E11 absorbance ratio can be used as metrics for the sp3 defect density of functionalized polymer-wrapped SWNTs. The E11*/E11 PL ratio should only be used as a measure of functionalization for a known and fixed excitation power, and care should be take when comparing data from different experiments.
Photoluminescence Quantum Yields and Lifetimes
One of the main reasons to create luminescent sp3 defects in SWNTs on a large scale is the goal to substantially increase their typically low PL quantum yields, that is, the ratio of emitted to absorbed photons, for practical applications. In the following we investigate the effect of sp3 defects on the PL quantum yield of polymer-wrapped (6,5) SWNTs. At this point, it must be emphasized that the functionalization level that maximizes the PLQY of the ensemble of functionalized SWNTs is not necessarily the one that leads to the most efficient radiative relaxation at an individual trap site. The efficiency of exciton trapping and competition with diffusive contact-quenching are equally important. In other words, even if very low defect densities on the order of ∼1 defect per nanotube support radiative relaxation of trapped excitons with the highest efficiency (i.e., emitted photons per trapped excitons),28,31 they will not be able to compete with diffusive contact-quenching given the estimated quenching site densities on SWNTs of ∼9 μm–1.57
Here, we aim to understand how the PLQY of an ensemble of functionalized SWNTs is affected by the defect density, the substituent on the aryl group influencing the trap depth and the dispersion method, that is, quality and length of the SWNTs. To address these points without the uncertainties associated with the use of a reference fluorophore,58 the PLQY was determined directly by measuring the laser absorption at the E22 transition and PL emission of the sample in an integrating sphere (absolute method as demonstrated previously).9 Since the PLQY, especially of the defect state, is a function of the excitation power density due to the nonlinear behavior discussed above, we note that all measurements were performed under pulsed excitation (pulse width ∼6 ps) with an energy density of ∼1.5 nJ cm–2. As discussed in the Supporting Information, interaction between multiple excitons can be safely neglected at these extremely low pulse energies.
Figure 4a shows the evolution of the total PLQY as well as the spectral contributions of the main peaks, namely, E11 and E11*, for shear-mixed (6,5) SWNTs functionalized with 4-nitrophenyl defects. Corresponding graphs for other substituents can be found in the Supporting Information, Figure S8. Note that, unlike SWNTs in aqueous dispersions, polymer-wrapped (6,5) SWNTs already exhibit quite good PLQY values of up to 2.4%.9 After functionalization, even low defect densities lead to a sharp drop of E11 PLQY and the emergence of strong E11* emission. At 0.037 mmol L–1, a local maximum in PLQY versus diazonium concentration is observed, at which the E11 still has an efficiency of 1.1%, but the E11* already contributes an additional 2.5%. This range of defect density could be favorable when elevated PLQYs are needed, but trap sites should be well-isolated. We note that this maximum might be less pronounced for SWNTs with higher initial quenching site density and, thus, lower starting levels of E11 PLQY.
Figure 4.

(a) Spectral contributions to the PLQY vs concentration of 4-nitrobenzenediazonium tetrafluoroborate as a measure of defect density. Solid lines are guides. (b) Optimum PLQYs found for different substituents as a function of their optical trap depth. The corresponding substituents—in order of increasing optical trap depth—are 4-OMe (blue), 3,5-Cl2 (green), 4-Br (orange), and 4-NO2 (red). The gray shaded areas represent the typical ranges of E11 and total PLQY of pristine (6,5) SWNTs dispersed by shear force mixing. (c) Lifetime components (τ) extracted from a biexponential fit to the time-resolved PL decay as a function of the optical trap depth. Note that PLQY and lifetime measurements were performed on the same samples.
At a diazonium salt concentration of 0.37 mmol L–1, the PLQY of the defect emission peaks at 3.5% with a total PLQY of 3.8%. Hence, more than 90% of photons are emitted through the defect channel. This scenario leads to the highest E11* and total PLQY observed in this concentration series. As the number of defects rises further, the E11 contribution converges to zero, but the E11* emission efficiency decreases as well. This observation is in agreement with the negligible PL found for highly functionalized SWNTs.59 The outlined PLQY evolution of functionalized, polymer-wrapped (6,5) SWNTs agrees qualitatively with the trends for PL intensities of surfactant-dispersed (6,5) SWNTs in water decorated with the same type of aryl defects.18 In analogy to the 4-nitrophenyl (4-NO2) case, the optimum reagent concentrations yielding the maximum PLQY were identified for the other substituents (Supporting Information, Figure S8). As expected from the reactivity pattern, only 0.037 mmol L–1 of the electron-poor and very reactive 3,5-dichloro (3,5-Cl2) reagent were required, as opposed to 3.7 mmol L–1 of the electron-rich 4-methoxy (4-OMe) compound. The 4-bromo (4-Br) reagent has a medium reactivity, and 0.37 mmol L–1 was found to be optimal for maximum PL.
In Figure 4b the maximum E11* and total PLQYs are plotted as a function of the optical trap depth of the substituted aryl defect, that is, the difference between the E11 and E11* PL emission energies calculated from Figures 2b and S4 and listed in Table 1. Even though the substituent on the aryl group modulates the optical trap depth by no more than ∼10%, it has a strong impact on the maximum PLQY. Evidently, both E11* and total PLQY increase with the optical trap depth. Furthermore, the deeper the trap, the smaller the residual E11 contribution (i.e., difference between total and E11* PLQY).
Table 1. Correlation of Defect Type,a Optical Trap Depth, and Emission Properties.
| defect type | optical trap depth (meV) | maximum total PLQY (%) | long lifetime component (ps) |
|---|---|---|---|
| 4-OMe | 167.5 | 3.0 | 205 |
| 3,5-Cl2 | 167.8 | 3.1 | 212 |
| 4-Br | 175.3 | 3.6 | 240 |
| 4-NO2 | 183.9 | 3.8 | 257 |
Substituent on the aryl group.
This trend is best discussed in combination with an analysis of the PL decay dynamics. We recorded the PL decay at the respective peak wavelengths of the E11* emission using time-correlated single-photon counting (TCSPC) and fitted a biexponential decay to the transients (refer to Supporting Information, Figures S9 and S10 for a representative histogram and results for all substituents and defect densities). In agreement with literature,26,27 these E11* PL decays feature a long-lived and a short-lived decay component (τlong and τshort, respectively) with comparable amplitudes. The short time constant has been interpreted as the time scale for redistribution of exciton population among bright and dark states localized at the defect.26,27 This redistribution period is followed by a slower decay of the trapped excitons through radiative and nonradiative channels.26,27 Consequently, the longer time constant is critical for the emission efficiency. Quantum chemical calculations suggest radiative lifetimes of ∼2 ns for bright trap states,29 but measured lifetimes are a few hundred picoseconds. Hence, recombination of trapped excitons is still dominated by nonradiative pathways. Specifically, localization-enhanced multiphonon decay (MPD) and electronic-to-vibrational energy transfer (EVET) in the liquid phase were proposed as key mechanisms.26,27 The lifetime components found for the samples discussed in Figure 4b are shown in Figure 4c as a function of the optical trap depth. Note that, as the PLQY is maximized at moderate defect densities, the lifetimes in this regime should also be the most reliable, since there is neither a significant residual contribution from the E11 phonon sideband nor significant site-to-site interaction.
Similar to the trend in PLQY, τlong is larger for deeper traps. Recently, it was observed that τlong increases for a given SWNT chirality as longer-wavelength defect emission is probed.26 It was concluded that thermal detrapping of excitons was responsible for this dependence, because longer wavelengths are associated with deeper traps. In addition, the corresponding time scales on the order of ∼100 ps can compete with MPD and EVET in most solvents.26 In our case, the variation of the substituent on the aryl group causes a variation of the trap depth and, thus, exciton lifetime. The values for τlong range from 205 ps for 4-OMe- to 257 ps for 4-NO2-substituted defects and agree well with those found for similar systems.26,27
Concurrently to the long lifetime component, the total PLQY rises from 3.0% to 3.8% with the fraction of photons emitted viaE11* continuously increasing from 81% to 92%. If we assume that we compensated the differences in reactivity of the various diazonium salts by adjusting their concentrations, all samples compared in Figure 4b,c should have similar defect densities. With this assumption we can attribute the substituent dependence of PLQY to the changes in trap depth, which affects the rate of thermal detrapping. The loss of defect state population suppresses the E11* PLQY, and the detrapped mobile E11 excitons are either quenched or decay radiatively, thereby increasing the E11 PLQY, in accordance with the trend in Figure 4b. As the trap depth appears to be one of the factors limiting the PLQY in this study, the design of divalent dopants that could bind to the SWNT in an arrangement creating deeper traps is a promising route to even brighter SWNTs.23,36
Length Dependence of PL Brightening
Apart from the parameters discussed above, the defect density and trap depth, we find the PLQY to depend strongly on the quality of the initially dispersed SWNTs. To illustrate the variation of the PLQY in the starting material (shear-mixed, PFO-BPy-wrapped (6,5) SWNTs), the typical range is indicated in Figure 4b. A similar degree of batch-to-batch variation is observed in sp3-functionalized (6,5) SWNTs with individual samples reaching total PLQYs up to 4.3% and others less than 3%. The general variation in SWNT dispersion quality is governed by many factors including the degree of mechanical damage of the SWNTs, coverage by dispersant, humidity, and temperature. In the following, we discuss the role of tube length and initial exciton quenching site density on the PL brightening upon sp3 functionalization. We are particularly interested in this aspect with regard to the discrepancy between the twofold PLQY enhancement observed here and previous reports of 5- to 10-fold brightening of (6,5) SWNTs that were dispersed in aqueous surfactant solution by tip sonication and sorted by gel chromatography.18 Because of the harshness of tip sonication, the dispersed SWNTs are much shorter (average lengths of 350–700 nm)26,60 than shear-mixed SWNTs with average lengths of ∼1.7 μm.9
In addition to nanotube ends many types of sidewall defects also act as exciton quenching sites, thus promoting nonradiative relaxation and decreasing the PLQY depending on the dispersion method.9,14 To investigate the effect of higher quenching site densities (before any diazonium treatment) on the optical properties of the SWNTs after sp3 functionalization without compromising the chiral purity of the sample, we shortened presorted, shear-mixed (6,5) SWNTs by tip sonication. The degree of nanotube shortening was controlled by the duration of ultrasonication. The length distributions of the shear-mixed stock and tip-sonicated nanotube batches are provided in the Supporting Information, Figure S11. Atomic force microscopy statistics reveal that the average length drops from 1.7 μm in the shear-mixed stock to 0.5 μm after the longest sonication. As shown in Figure 5a, the reduction in SWNT length leads to a significant decrease in PLQY from 1.6% to 0.7%. We find that short sonication times that result in marginal reductions of tube length still strongly affect the PLQY, which is most likely due to the creation of sidewall defects. After the shortening step, each batch was treated with the optimized concentration of 4-bromobenzenediazonium tetrafluoroborate (0.37 mmol L–1) to produce (4-bromophenyl) sp3 defects. Note that the shortening and functionalization steps were performed in this order to account for reactivity differences in SWNTs with more or less damaged conjugated lattices. In this way, we mimic the scenario of starting materials dispersed under harsh conditions.
Figure 5.

(a) PLQY of PFO-BPy-wrapped (6,5) SWNTs in toluene as a function of the average nanotube length. (b) PLQY (black ●) after functionalization (final) with 4-bromo-benzenediazonium tetrafluoroborate vs PLQY before functionalization (initial). The brightening factor (red ●) is defined as the ratio of final PLQY/initial PLQY. Solid lines are guides.
In Figure 5b, the spectrally integrated PLQY of sp3 -functionalized (6,5) SWNTs is plotted versus the initial PLQY of the corresponding batch of pristine polymer-wrapped SWNTs. It is evident that higher initial PLQYs also lead to higher postfunctionalization PLQYs, which may be seen as the result of more efficient channeling of excitons to defect sites owing to a reduced probability of contact-quenching. However, the relative brightening, which we define as the ratio of final PLQY/initial PLQY, consistently follows an inverse trend. While the shortest SWNTs with low initial PLQY exhibit a ∼2.5-fold brightening, the longest SWNTs are only brightened by a factor of ∼1.5.
In summary, while high-quality functionalized SWNTs feature the highest absolute PLQYs, the degree of brightening is higher for lower-quality SWNTs. This observation is in agreement with the recent demonstration of photoluminescence from ultrashort and usually dark SWNTs by emissive trap sites.34 As SWNTs with large numbers of nonradiative quenching sites will never display efficient E11 emission due to fast exciton diffusion and contact quenching,14,61 sp3 defects allow a fraction of the exciton population to be harvested that otherwise would decay nonradiatively. The remaining discrepancy between our observed 2.5-fold brightening and the 7-fold brightening previously reported18 for aqueous dispersions of (6,5) SWNTs with the same defect type likely stems from two factors. First, the surfactant-dispersed SWNTs were even shorter on average60 than the shortest batch studied in this work. Second, polymer-wrapped SWNTs usually have higher PLQYs than surfactant-stabilized SWNTs due to a higher degree of debundling and stronger dielectric shielding.62 Untreated SWNTs dispersed in water suffer from faster nonradiative decay, which is alleviated by defect-induced exciton localization, as this precludes diffusive sampling of the dielectric environment.29 In polymer-wrapped SWNTs, however, the contrast between both cases should be smaller; thus, functionalization results in a lower PLQY enhancement.
PLQY and Brightening of SWNT Films
If sp3 functionalization is to be used to improve the quantum efficiency of SWNTs in light-emitting devices, it is crucial that the PLQY enhancement (relative to pristine SWNTs) is retained in a film with intertube and substrate interactions. The localization of excitons might even reduce the large PLQY drop from SWNT dispersions to thin films, which would be highly desirable. However, suitable deposition techniques for homogeneous films with defined thickness over large areas are required to reliably investigate and compare the thin-film photoluminescence and PLQY of functionalized versus pristine polymer-wrapped SWNTs. To achieve this, we scaled up the functionalization process to reaction volumes of several hundred milliliters containing a total mass of ∼0.5 mg of highly enriched, polymer-wrapped (6,5) SWNTs. Following our standard procedure, we tuned the defect density to the desired level corresponding to the maximum PLQY as determined above. Next, concentrated inks of functionalized and pristine SWNTs were prepared and used for aerosol-jet printing, which is an efficient method to deposit thick films in a controlled and spatially confined way.63,64 The low to moderate numbers of sp3 defects introduced to the (6,5) SWNTs do not affect the colloidal stability or viscosity of the nanotube ink to an extent that would require adjustment of printing conditions. Hence, both pristine and functionalized (6,5) SWNTs were deposited under identical conditions. We printed four pairs of stripes (for comparison of pristine and functionalized SWNTs) with increasing thickness, all of them being visible with the bare eye. Currently, there is no deposition technique for aqueous SWNT dispersions that enables the formation of comparably homogeneous, dense, and large-area films, except filtration.65 For a comparison of PL intensities, the respective stripes must be equally thick. Mapping of the Raman G+ mode intensity confirmed that the stripes of each pair indeed have very similar thicknesses. With similar numbers of emitters, the PL intensity under identical excitation conditions can be used as a relative measure of PLQY. To achieve a direct comparison, the laser excitation spot was expanded to homogeneously illuminate a pair of printed stripes, and the PL was imaged onto a two-dimensional (2D) InGaAs camera. The resulting PL images of all printed stripes are shown in the Supporting Information, Figure S12, together with the integrated Raman intensity and brightfield optical microscope images. The stripes of functionalized SWNTs are consistently brighter than the reference stripes. Careful analysis of the PL intensity as a function of film thickness shows that the films of functionalized SWNTs are on average a factor of 1.7 brighter than films of pristine SWNTs (see the Supporting Information, Figure S13). The PL micrograph of the thickest pair of stripes is shown in Figure 6. Clearly, the emission intensity of defect-functionalized SWNTs is higher. Subsequently, a grating was inserted to spectrally disperse the PL emission along one coordinate. The corresponding hyperspectral image is given next to the real-space image in Figure 6. The strong PL of the functionalized stripe originates from E11* emission. Further, the asymmetry of the E11 feature in contrast to the peak shape of the E11* illustrates that, apart from differences in the PLQY, the E11 emission suffers from significant self-absorption due to the small Stokes shift, whereas the E11* is nearly reabsorption-free.60
Figure 6.

PL micrograph and hyperspectral image of printed stripes of pristine and 4-bromophenyl-functionalized PFO-BPy-wrapped (6,5) SWNTs on glass recorded under continuous wave excitation at 640 nm.
As a complementary test of enhanced emission efficiencies in films, we also spin-coated thin films of both samples with areas of ∼1 cm2 and measured the absolute PLQY in an integrating sphere in analogy to the liquid samples. Despite low emission intensities, the measurement was reproducible, and PLQYs of 0.18 ± 0.05% for the reference and 0.31 ± 0.06% for the sp3-functionalized sample were found. The corresponding brightening factor of 1.7 matches the enhancement of PL intensity from the printed stripes exactly. Although the PL enhancement is not larger than in dispersion, it is at least retained for thin and thick functionalized SWNT films.
Conclusions
We have presented a simple and scalable phase-transfer reaction scheme for the functionalization of polymer-wrapped SWNTs with luminescent sp3 defects in organic nonhalogenated solvents based on aryldiazonium chemistry. As a result of the high monochiral purity and concentration of the functionalized nanotube dispersions, we were able to unambiguously identify the E11* absorption of the sp3 defects and found E11* Stokes shifts of ∼20 meV at low defect densities. We determined the absolute PLQY of the sp3-functionalized (6,5) SWNTs in dispersion and probed its dependence on defect density and trap depth. Careful tuning of the defect density gave PLQYs of up to 4% with ∼90% of photons emitted through the defect channel. The length and quality of the as-dispersed SWNTs strongly affected both the absolute PLQY and the relative brightening factor upon introduction of luminescent defects. While long SWNTs with high initial PLQY also gave the highest postfunctionalization PLQY, the greatest relative brightening was found for the shortest nanotubes with the lowest initial PLQY. The presented method to functionalize polymer-wrapped SWNTs with sp3 defects on a large scale in organic solvents further enabled the fabrication of homogeneous thin and thick nanotube films with bright and nearly reabsorption-free emission. While we only employed PFO-BPy-wrapped (6,5) nanotubes in this study, we presume that the general method can be applied likewise to other nanotube species with different wrapping polymers. The reproducible and large-scale sp3 functionalization of highly purified polymer-sorted semiconducting SWNTs will enable the fabrication of brighter carbon nanotube-based NIR emitting devices such as light-emitting diodes and field-effect transistors.
Methods
Selective Dispersion of (6,5) SWNTs
As described previously,9 (6,5) SWNTs were selectively extracted from CoMoCAT raw material (Chasm Advanced Materials, SG65i-L58, 0.38 g L–1) by shear force mixing (Silverson L2/Air, 10 230 rpm, 72 h) and polymer-wrapping with PFO-BPy (American Dye Source, Mw = 40 kg mol–1, 0.5 g L–1) in toluene. Aggregates were removed by centrifugation at 60 000g (Beckman Coulter Avanti J26XP centrifuge) for 90 min and subsequent filtration (poly(tetrafluoroethylene) (PTFE) syringe filter, 5 μm pore size).
Shortening of (6,5) SWNTs
Dispersions in toluene obtained from shear force mixing were tip-sonicated (Sonics, Vibracell VXC-500) using a tapered microtip at 35% amplitude with 8 s on, 2 s off pulses at 5 °C for 4.5–23 h to produce different degrees of nanotube shortening. The sonicated dispersions were centrifuged at 60 000g for 45 min, and the supernatant was used for analysis.
sp3 Functionalization
PFO-BPy-wrapped (6,5) SWNTs were covalently functionalized with a series of commercially available diazonium salts (4-bromo-, 4-methoxy-, 4-nitro-, and 3,5-dichlorobenzenediazonium tetrafluoroborate, Sigma-Aldrich). For a detailed protocol, refer to the Supporting Information. Reactions were performed at a (6,5) SWNT concentration of 0.36 mg L–1 (corresponding to an E11 absorbance of 0.2 for 1 cm path length)66 in an 80:20 vol-% toluene/acetonitrile mixture. Briefly, a toluene solution of 18-crown-6 (18-crown-6, 99%, Sigma-Aldrich) was added to the as-prepared SWNT dispersion such that the 18-crown-6 concentration after addition of all other components was 7.6 mmol L–1. The diazonium salt was dissolved in acetonitrile, and an appropriate volume of this solution was added to the reaction vessel. After thorough mixing and a waiting period of 5 min, a solution of potassium acetate (KOAc) in 80:20 vol-% toluene/acetonitrile and 7.6 mmol L–1 18-crown-6 was added to the mixture to yield a KOAc concentration of ∼1 × 10–9 mol L–1. The reaction proceeded at room temperature in the dark; stirring was not required. After typically 16 h, the mixture was passed through a PTFE membrane filter (Merck Millipore, JVWP, 0.1 μm pore size) to collect the SWNTs. The filter cake was washed with acetonitrile and toluene to remove unreacted diazonium salt and excess polymer. Finally, the filter cake was redispersed in a small volume of pure toluene by bath sonication for 30 min.
Printing of SWNTs
Pristine and sp3-functionalized (6,5) SWNTs were printed under identical conditions. Toluene and terpineol (mixture of isomers, Sigma-Aldrich) were added to the dispersion to adjust the SWNT concentration to 5.4 mg L–1 (E11 absorbance of 3 cm–1) and the terpineol concentration to 2 vol-%. This ink was used for aerosol jet printing (Aerosol Jet 200 printer, Optomec) with an ultrasonic atomizer.63,64 A 200 μm inner diameter nozzle was used at a sheath gas flow of 30 sccm and carrier gas flow of 25 sccm. The film thickness was tuned by the number of printing cycles. The sample stage was at 100 °C to facilitate fast evaporation of toluene. Residual terpineol was rinsed off with tetrahydrofuran and isopropyl alcohol.
Optical Measurements
Absorption spectra were recorded with a Cary 6000i absorption spectrometer (Varian). Liquid-phase PL spectra in the low excitation power density regime were collected with a HORIBA Jobin Yvon Fluorolog spectrofluorometer equipped with a 450 W xenon arc lamp and a liquid nitrogen cooled InGaAs line camera. For acquisition of PL spectra at higher excitation densities, a home-built setup was used. The sample was excited by the spectrally filtered output of a picosecond-pulsed supercontinuum laser source (Fianium WhiteLase SC400) focused by a 50× NIR-optimized objective (N.A. 0.65, Olympus). Scattered laser light was blocked by a dichroic long-pass filter (875 nm cutoff). The PL emission from the sample was dispersed by an Acton SpectraPro SP2358 spectrograph (grating 150 lines mm–1) and detected with a liquid nitrogen cooled InGaAs line camera (Princeton Instruments OMA V). For PL lifetime measurements using a time-correlated single photon counting scheme, the spectrally selected PL emission was focused onto a gated InGaAs/InP avalanche photodiode (Micro Photon Devices) via a 20× NIR-optimized objective (Mitutoyo). The PL quantum yield of dispersions and thin films was determined by an absolute method as reported earlier.9 Samples were placed inside an integrating sphere (Labsphere), and the absorption of laser light at 575 nm (E22 transition) as well as the PL emission were measured by transmitting the light through an optical fiber and coupling into the spectrometer. Photoluminescence images were acquired using an imaging spectrograph (Princeton Instruments IsoPlane SCT 320) and a thermoelectrically cooled 2D InGaAs camera (Princeton Instruments NIRvana). The sample was homogeneously illuminated with a 640 nm laser diode (Coherent OBIS, 40 mW continuous wave), and scattered laser light was blocked by a dichroic long-pass filter (850 nm cutoff).
Acknowledgments
This project received funding from the European Research Council under the European Union’s Horizon 2020 research and innovation programme (Grant No. 817494). J.L. acknowledges support by the Volkswagenstiftung (Grant No. 93404). V.L. thanks the DAAD-RISE programme. The authors thank B. S. Flavel and H. Li for helpful discussions and A. Graf and F. Grün for initial input.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b03792.
Dispersion stability in reaction medium, mechanistic considerations and impact of KOAc on functionalization, detailed sp3 functionalization protocol, E11* absorption and emission maxima and Stokes shifts, PL spectra of (6,5) SWNTs functionalized with different aryl defects, Raman spectra of functionalized (6,5) SWNTs, different measures of defect densities, power-dependence of photoluminescence, PLQY vs defect density for all substituents, defect state decay dynamics, AFM statistics of SWNT length distributions, characterization of printed (6,5) SWNT films (PDF)
Author Present Address
∥ Department of Chemistry, Purdue University, West Lafayette, IN 47907, United States.
Author Present Address
⊥ Institute of Semiconductor and Solid State Physics, Johannes Kepler University Linz, A-4040 Linz, Austria.
The authors declare no competing financial interest.
Supplementary Material
References
- Mueller T.; Kinoshita M.; Steiner M.; Perebeinos V.; Bol A. A.; Farmer D. B.; Avouris P. Efficient Narrow-Band Light Emission from a Single Carbon Nanotube p-n Diode. Nat. Nanotechnol. 2010, 5, 27–31. 10.1038/nnano.2009.319. [DOI] [PubMed] [Google Scholar]
- Graf A.; Murawski C.; Zakharko Y.; Zaumseil J.; Gather M. C. Infrared Organic Light-Emitting Diodes with Carbon Nanotube Emitters. Adv. Mater. 2018, 30, 1706711. 10.1002/adma.201706711. [DOI] [PubMed] [Google Scholar]
- Graf A.; Held M.; Zakharko Y.; Tropf L.; Gather M. C.; Zaumseil J. Electrical Pumping and Tuning of Exciton-Polaritons in Carbon Nanotube Microcavities. Nat. Mater. 2017, 16, 911–917. 10.1038/nmat4940. [DOI] [PubMed] [Google Scholar]
- Jakubka F.; Grimm S. B.; Zakharko Y.; Gannott F.; Zaumseil J. Trion Electroluminescence from Semiconducting Carbon Nanotubes. ACS Nano 2014, 8, 8477–8486. 10.1021/nn503046y. [DOI] [PubMed] [Google Scholar]
- Avouris P.; Freitag M.; Perebeinos V. Carbon-Nanotube Photonics and Optoelectronics. Nat. Photonics 2008, 2, 341–350. 10.1038/nphoton.2008.94. [DOI] [Google Scholar]
- Wei L.; Flavel B. S.; Li W.; Krupke R.; Chen Y. Exploring the Upper Limit of Single-Walled Carbon Nanotube Purity by Multiple-Cycle Aqueous Two-Phase Separation. Nanoscale 2017, 9, 11640–11646. 10.1039/C7NR03302H. [DOI] [PubMed] [Google Scholar]
- O’Connell M. J.; Bachilo S. M.; Huffman C. B.; Moore V. C.; Strano M. S.; Haroz E. H.; Rialon K. L.; Boul P. J.; Noon W. H.; Kittrell C.; Ma J.; Hauge R. H.; Weisman R. B.; Smalley R. E. Band Gap Fluorescence from Individual Single-Walled Carbon Nanotubes. Science 2002, 297, 593–596. 10.1126/science.1072631. [DOI] [PubMed] [Google Scholar]
- Samanta S. K.; Fritsch M.; Scherf U.; Gomulya W.; Bisri S. Z.; Loi M. A. Conjugated Polymer-Assisted Dispersion of Single-Wall Carbon Nanotubes: The Power of Polymer Wrapping. Acc. Chem. Res. 2014, 47, 2446–2456. 10.1021/ar500141j. [DOI] [PubMed] [Google Scholar]
- Graf A.; Zakharko Y.; Schießl S. P.; Backes C.; Pfohl M.; Flavel B. S.; Zaumseil J. Large Scale, Selective Dispersion of Long Single-Walled Carbon Nanotubes with High Photoluminescence Quantum Yield by Shear Force Mixing. Carbon 2016, 105, 593–599. 10.1016/j.carbon.2016.05.002. [DOI] [Google Scholar]
- Schneider S.; Brohmann M.; Lorenz R.; Hofstetter Y. J.; Rother M.; Sauter E.; Zharnikov M.; Vaynzof Y.; Himmel H.-J.; Zaumseil J. Efficient n-Doping and Hole Blocking in Single-Walled Carbon Nanotube Transistors with 1,2,4,5-Tetrakis(Tetramethylguanidino)Benzene. ACS Nano 2018, 12, 5895–5902. 10.1021/acsnano.8b02061. [DOI] [PubMed] [Google Scholar]
- Rother M.; Brohmann M.; Yang S.; Grimm S. B.; Schießl S. P.; Graf A.; Zaumseil J. Aerosol-Jet Printing of Polymer-Sorted (6,5) Carbon Nanotubes for Field-Effect Transistors with High Reproducibility. Adv. Electron. Mater. 2017, 3, 1700080. 10.1002/aelm.201700080. [DOI] [Google Scholar]
- Bucella S. G.; Salazar-Rios J. M.; Derenskyi V.; Fritsch M.; Scherf U.; Loi M. A.; Caironi M. Inkjet Printed Single-Walled Carbon Nanotube Based Ambipolar and Unipolar Transistors for High-Performance Complementary Logic Circuits. Adv. Electron. Mater. 2016, 2, 1600094. 10.1002/aelm.201600094. [DOI] [Google Scholar]
- Mortimer I. B.; Nicholas R. J. Role of Bright and Dark Excitons in the Temperature-Dependent Photoluminescence of Carbon Nanotubes. Phys. Rev. Lett. 2007, 98, 027404. 10.1103/PhysRevLett.98.027404. [DOI] [PubMed] [Google Scholar]
- Hertel T.; Himmelein S.; Ackermann T.; Stich D.; Crochet J. Diffusion Limited Photoluminescence Quantum Yields in 1-D Semiconductors: Single-Wall Carbon Nanotubes. ACS Nano 2010, 4, 7161–7168. 10.1021/nn101612b. [DOI] [PubMed] [Google Scholar]
- Amori A. R.; Hou Z.; Krauss T. D. Excitons in Single-Walled Carbon Nanotubes and Their Dynamics. Annu. Rev. Phys. Chem. 2018, 69, 81–99. 10.1146/annurev-physchem-050317-014241. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Bachilo S. M.; Simonette R. A.; Beckingham K. M.; Weisman R. B. Oxygen Doping Modifies Near-Infrared Band Gaps in Fluorescent Single-Walled Carbon Nanotubes. Science 2010, 330, 1656–1660. 10.1126/science.1196382. [DOI] [PubMed] [Google Scholar]
- Miyauchi Y.; Iwamura M.; Mouri S.; Kawazoe T.; Ohtsu M.; Matsuda K. Brightening of Excitons in Carbon Nanotubes on Dimensionality Modification. Nat. Photonics 2013, 7, 715–719. 10.1038/nphoton.2013.179. [DOI] [Google Scholar]
- Piao Y.; Meany B.; Powell L. R.; Valley N.; Kwon H.; Schatz G. C.; Wang Y. Brightening of Carbon Nanotube Photoluminescence through the Incorporation of sp3 Defects. Nat. Chem. 2013, 5, 840–845. 10.1038/nchem.1711. [DOI] [PubMed] [Google Scholar]
- Brozena A. H.; Kim M.; Powell L. R.; Wang Y. Controlling the Optical Properties of Carbon Nanotubes with Organic Colour-Centre Quantum Defects. Nat. Rev. Chem. 2019, 3, 375–392. 10.1038/s41570-019-0103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H. B.; Wang P.; Wu X.; Qu H.; Ren X.; Wang Y. One-Pot, Large-Scale Synthesis of Organic Color Center-Tailored Semiconducting Carbon Nanotubes. ACS Nano 2019, 13, 8417–8424. 10.1021/acsnano.9b04087. [DOI] [PubMed] [Google Scholar]
- Kilina S.; Ramirez J.; Tretiak S. Brightening of the Lowest Exciton in Carbon Nanotubes via Chemical Functionalization. Nano Lett. 2012, 12, 2306–2312. 10.1021/nl300165w. [DOI] [PubMed] [Google Scholar]
- Gifford B. J.; Kilina S.; Htoon H.; Doorn S. K.; Tretiak S. Exciton Localization and Optical Emission in Aryl-Functionalized Carbon Nanotubes. J. Phys. Chem. C 2018, 122, 1828–1838. 10.1021/acs.jpcc.7b09558. [DOI] [Google Scholar]
- Shiraki T.; Shiraishi T.; Juhasz G.; Nakashima N. Emergence of New Red-Shifted Carbon Nanotube Photoluminescence Based on Proximal Doped-Site Design. Sci. Rep. 2016, 6, 28393. 10.1038/srep28393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y.; Minami S.; Takehana Y.; Dang J. S.; Aota S.; Matsuda K.; Miyauchi Y.; Yamada M.; Suzuki M.; Zhao R. S.; Zhao X.; Nagase S. Tuning of the Photoluminescence and Up-Conversion Photoluminescence Properties of Single-Walled Carbon Nanotubes by Chemical Functionalization. Nanoscale 2016, 8, 16916–16921. 10.1039/C6NR04214G. [DOI] [PubMed] [Google Scholar]
- Hartmann N. F.; Yalcin S. E.; Adamska L.; Haroz E. H.; Ma X.; Tretiak S.; Htoon H.; Doorn S. K. Photoluminescence Imaging of Solitary Dopant Sites in Covalently Doped Single-Wall Carbon Nanotubes. Nanoscale 2015, 7, 20521–20530. 10.1039/C5NR06343D. [DOI] [PubMed] [Google Scholar]
- He X.; Velizhanin K. A.; Bullard G.; Bai Y.; Olivier J. H.; Hartmann N. F.; Gifford B. J.; Kilina S.; Tretiak S.; Htoon H.; Therien M. J.; Doorn S. K. Solvent- and Wavelength-Dependent Photoluminescence Relaxation Dynamics of Carbon Nanotube sp3 Defect States. ACS Nano 2018, 12, 8060–8070. 10.1021/acsnano.8b02909. [DOI] [PubMed] [Google Scholar]
- Hartmann N. F.; Velizhanin K. A.; Haroz E. H.; Kim M.; Ma X.; Wang Y.; Htoon H.; Doorn S. K. Photoluminescence Dynamics of Aryl sp3 Defect States in Single-Walled Carbon Nanotubes. ACS Nano 2016, 10, 8355–8365. 10.1021/acsnano.6b02986. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Velizhanin K. A.; He X.; Sarpkaya I.; Yomogida Y.; Tanaka T.; Kataura H.; Doorn S. K.; Htoon H. Photoluminescence Intensity Fluctuations and Temperature-Dependent Decay Dynamics of Individual Carbon Nanotube sp3 Defects. J. Phys. Chem. Lett. 2019, 10, 1423–1430. 10.1021/acs.jpclett.8b03732. [DOI] [PubMed] [Google Scholar]
- He X.; Sun L.; Gifford B. J.; Tretiak S.; Piryatinski A.; Li X.; Htoon H.; Doorn S. K. Intrinsic Limits of Defect-State Photoluminescence Dynamics in Functionalized Carbon Nanotubes. Nanoscale 2019, 11, 9125–9132. 10.1039/C9NR02175B. [DOI] [PubMed] [Google Scholar]
- Saha A.; Gifford B. J.; He X.; Ao G.; Zheng M.; Kataura H.; Htoon H.; Kilina S.; Tretiak S.; Doorn S. K. Narrow-Band Single-Photon Emission through Selective Aryl Functionalization of Zigzag Carbon Nanotubes. Nat. Chem. 2018, 10, 1089–1095. 10.1038/s41557-018-0126-4. [DOI] [PubMed] [Google Scholar]
- He X.; Hartmann N. F.; Ma X.; Kim Y.; Ihly R.; Blackburn J. L.; Gao W.; Kono J.; Yomogida Y.; Hirano A.; Tanaka T.; Kataura H.; Htoon H.; Doorn S. K. Tunable Room-Temperature Single-Photon Emission at Telecom Wavelengths from sp3 Defects in Carbon Nanotubes. Nat. Photonics 2017, 11, 577–582. 10.1038/nphoton.2017.119. [DOI] [Google Scholar]
- He X.; Gifford B. J.; Hartmann N. F.; Ihly R.; Ma X.; Kilina S. V.; Luo Y.; Shayan K.; Strauf S.; Blackburn J. L.; Tretiak S.; Doorn S. K.; Htoon H. Low-Temperature Single Carbon Nanotube Spectroscopy of sp3 Quantum Defects. ACS Nano 2017, 11, 10785–10796. 10.1021/acsnano.7b03022. [DOI] [PubMed] [Google Scholar]
- He X.; Htoon H.; Doorn S. K.; Pernice W. H. P.; Pyatkov F.; Krupke R.; Jeantet A.; Chassagneux Y.; Voisin C. Carbon Nanotubes as Emerging Quantum-Light Sources. Nat. Mater. 2018, 17, 663–670. 10.1038/s41563-018-0109-2. [DOI] [PubMed] [Google Scholar]
- Danné N.; Kim M.; Godin A. G.; Kwon H.; Gao Z.; Wu X.; Hartmann N. F.; Doorn S. K.; Lounis B.; Wang Y.; Cognet L. Ultrashort Carbon Nanotubes That Fluoresce Brightly in the Near-Infrared. ACS Nano 2018, 12, 6059–6065. 10.1021/acsnano.8b02307. [DOI] [PubMed] [Google Scholar]
- Kwon H.; Kim M.; Meany B.; Piao Y.; Powell L. R.; Wang Y. Optical Probing of Local pH and Temperature in Complex Fluids with Covalently Functionalized, Semiconducting Carbon Nanotubes. J. Phys. Chem. C 2015, 119, 3733–3739. 10.1021/jp509546d. [DOI] [Google Scholar]
- Kwon H.; Furmanchuk A.; Kim M.; Meany B.; Guo Y.; Schatz G. C.; Wang Y. Molecularly Tunable Fluorescent Quantum Defects. J. Am. Chem. Soc. 2016, 138, 6878–6885. 10.1021/jacs.6b03618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Gordeev G.; Garrity O.; Reich S.; Flavel B. S. Separation of Small-Diameter Single-Walled Carbon Nanotubes in One to Three Steps with Aqueous Two-Phase Extraction. ACS Nano 2019, 13, 2567–2578. 10.1021/acsnano.8b09579. [DOI] [PubMed] [Google Scholar]
- Cui J.; Su W.; Yang D.; Li S.; Wei X.; Zhou N.; Zhou W.; Xie S.; Kataura H.; Liu H. Mass Production of High-Purity Semiconducting Carbon Nanotubes by Hydrochloric Acid Assisted Gel Chromatography. ACS Appl. Nano Mater. 2019, 2, 343–350. 10.1021/acsanm.8b01942. [DOI] [Google Scholar]
- Derenskyi V.; Gomulya W.; Gao J.; Bisri S. Z.; Pasini M.; Loo Y.-L.; Loi M. A. Semiconducting SWNTs Sorted by Polymer Wrapping: How Pure Are They?. Appl. Phys. Lett. 2018, 112, 072106. 10.1063/1.5011388. [DOI] [Google Scholar]
- Hennrich F.; Li W.; Fischer R.; Lebedkin S.; Krupke R.; Kappes M. M. Length-Sorted, Large-Diameter, Polyfluorene-Wrapped Semiconducting Single-Walled Carbon Nanotubes for High-Density, Short-Channel Transistors. ACS Nano 2016, 10, 1888–1895. 10.1021/acsnano.5b05572. [DOI] [PubMed] [Google Scholar]
- Bahr J. L.; Tour J. M. Highly Functionalized Carbon Nanotubes Using in situ Generated Diazonium Compounds. Chem. Mater. 2001, 13, 3823–3824. 10.1021/cm0109903. [DOI] [Google Scholar]
- Wang J.; Shea M. J.; Flach J. T.; McDonough T. J.; Way A. J.; Zanni M. T.; Arnold M. S. Role of Defects as Exciton Quenching Sites in Carbon Nanotube Photovoltaics. J. Phys. Chem. C 2017, 121, 8310–8318. 10.1021/acs.jpcc.7b01005. [DOI] [Google Scholar]
- Beadle J. R.; Korzeniowski S. H.; Rosenberg D. E.; Garcia-Slanga B. J.; Gokel G. W. Phase-Transfer-Catalyzed Gomberg-Bachmann Synthesis of Unsymmetrical Biarenes: A Survey of Catalysts and Substrates. J. Org. Chem. 1984, 49, 1594–1603. 10.1021/jo00183a021. [DOI] [Google Scholar]
- Usrey M. L.; Lippmann E. S.; Strano M. S. Evidence for a Two-Step Mechanism in Electronically Selective Single-Walled Carbon Nanotube Reactions. J. Am. Chem. Soc. 2005, 127, 16129–16135. 10.1021/ja0537530. [DOI] [PubMed] [Google Scholar]
- Shiraki T.; Niidome Y.; Toshimitsu F.; Shiraishi T.; Shiga T.; Yu B.; Fujigaya T. Solvatochromism of near Infrared Photoluminescence from Doped Sites of Locally Functionalized Single-Walled Carbon Nanotubes. Chem. Commun. 2019, 55, 3662–3665. 10.1039/C9CC00829B. [DOI] [PubMed] [Google Scholar]
- Jones M.; Engtrakul C.; Metzger W. K.; Ellingson R. J.; Nozik A. J.; Heben M. J.; Rumbles G. Analysis of Photoluminescence from Solubilized Single-Walled Carbon Nanotubes. Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 71, 115426. 10.1103/PhysRevB.71.115426. [DOI] [Google Scholar]
- Kim M.; Adamska L.; Hartmann N. F.; Kwon H.; Liu J.; Velizhanin K. A.; Piao Y.; Powell L. R.; Meany B.; Doorn S. K.; Tretiak S.; Wang Y. Fluorescent Carbon Nanotube Defects Manifest Substantial Vibrational Reorganization. J. Phys. Chem. C 2016, 120, 11268–11276. 10.1021/acs.jpcc.6b02538. [DOI] [Google Scholar]
- Ma X.; Adamska L.; Yamaguchi H.; Yalcin S. E.; Tretiak S.; Doorn S. K.; Htoon H. Electronic Structure and Chemical Nature of Oxygen Dopant States in Carbon Nanotubes. ACS Nano 2014, 8, 10782–10789. 10.1021/nn504553y. [DOI] [PubMed] [Google Scholar]
- Gifford B. J.; Sifain A. E.; Htoon H.; Doorn S. K.; Kilina S.; Tretiak S. Correction Scheme for Comparison of Computed and Experimental Optical Transition Energies in Functionalized Single-Walled Carbon Nanotubes. J. Phys. Chem. Lett. 2018, 9, 2460–2468. 10.1021/acs.jpclett.8b00653. [DOI] [PubMed] [Google Scholar]
- Dresselhaus M. S.; Jorio A.; Souza Filho A. G.; Saito R. Defect Characterization in Graphene and Carbon Nanotubes Using Raman Spectroscopy. Philos. Trans. R. Soc., A 2010, 368, 5355–5377. 10.1098/rsta.2010.0213. [DOI] [PubMed] [Google Scholar]
- Pfohl M.; Tune D. D.; Graf A.; Zaumseil J.; Krupke R.; Flavel B. S. Fitting Single-Walled Carbon Nanotube Optical Spectra. ACS Omega 2017, 2, 1163–1171. 10.1021/acsomega.6b00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov A. V.; Ghosh S.; Tsyboulski D. A.; Bachilo S. M.; Weisman R. B. Analyzing Absorption Backgrounds in Single-Walled Carbon Nanotube Spectra. ACS Nano 2011, 5, 1639–1648. 10.1021/nn1035922. [DOI] [PubMed] [Google Scholar]
- Nair N.; Usrey M. L.; Kim W.-J.; Braatz R. D.; Strano M. S. Estimation of the (n, m) Concentration Distribution of Single-Walled Carbon Nanotubes from Photoabsorption Spectra. Anal. Chem. 2006, 78, 7689–7696. 10.1021/ac0610917. [DOI] [PubMed] [Google Scholar]
- Wang F.; Dukovic G.; Knoesel E.; Brus L. E.; Heinz T. F. Observation of Rapid Auger Recombination in Optically Excited Semiconducting Carbon Nanotubes. Phys. Rev. B: Condens. Matter Mater. Phys. 2004, 70, 241403. 10.1103/PhysRevB.70.241403. [DOI] [Google Scholar]
- Wang F.; Wu Y.; Hybertsen M. S.; Heinz T. F. Auger Recombination of Excitons in One-Dimensional Systems. Phys. Rev. B: Condens. Matter Mater. Phys. 2006, 73, 245424. 10.1103/PhysRevB.73.245424. [DOI] [Google Scholar]
- Iwamura M.; Akizuki N.; Miyauchi Y.; Mouri S.; Shaver J.; Gao Z.; Cognet L.; Lounis B.; Matsuda K. Nonlinear Photoluminescence Spectroscopy of Carbon Nanotubes with Localized Exciton States. ACS Nano 2014, 8, 11254–11260. 10.1021/nn503803b. [DOI] [PubMed] [Google Scholar]
- Hartleb H.; Späth F.; Hertel T. Evidence for Strong Electronic Correlations in the Spectra of Gate-Doped Single-Wall Carbon Nanotubes. ACS Nano 2015, 9, 10461–10470. 10.1021/acsnano.5b04707. [DOI] [PubMed] [Google Scholar]
- Stürzl N.; Lebedkin S.; Kappes M. M. Revisiting the Laser Dye Styryl-13 as a Reference Near-Infrared Fluorophore: Implications for the Photoluminescence Quantum Yields of Semiconducting Single-Walled Carbon Nanotubes. J. Phys. Chem. A 2009, 113, 10238–10240. 10.1021/jp905166s. [DOI] [PubMed] [Google Scholar]
- Doyle C. D.; Rocha J.-D. R.; Weisman R. B.; Tour J. M. Structure-Dependent Reactivity of Semiconducting Single-Walled Carbon Nanotubes with Benzenediazonium Salts. J. Am. Chem. Soc. 2008, 130, 6795–6800. 10.1021/ja800198t. [DOI] [PubMed] [Google Scholar]
- Powell L. R.; Piao Y.; Ng A. L.; Wang Y. Channeling Excitons to Emissive Defect Sites in Carbon Nanotube Semiconductors Beyond the Dilute Regime. J. Phys. Chem. Lett. 2018, 9, 2803–2807. 10.1021/acs.jpclett.8b00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrah D. M.; Swan A. K. The Role of Length and Defects on Optical Quantum Efficiency and Exciton Decay Dynamics in Single-Walled Carbon Nanotubes. ACS Nano 2011, 5, 647–655. 10.1021/nn1031214. [DOI] [PubMed] [Google Scholar]
- Nish A.; Hwang J. Y.; Doig J.; Nicholas R. J. Highly Selective Dispersion of Single-Walled Carbon Nanotubes Using Aromatic Polymers. Nat. Nanotechnol. 2007, 2, 640–646. 10.1038/nnano.2007.290. [DOI] [PubMed] [Google Scholar]
- Berger F. J.; Higgins T. M.; Rother M.; Graf A.; Zakharko Y.; Allard S.; Matthiesen M.; Gotthardt J. M.; Scherf U.; Zaumseil J. From Broadband to Electrochromic Notch Filters with Printed Monochiral Carbon Nanotubes. ACS Appl. Mater. Interfaces 2018, 10, 11135–11142. 10.1021/acsami.8b00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother M.; Brohmann M.; Yang S.; Grimm S. B.; Schießl S. P.; Graf A.; Zaumseil J. Aerosol-Jet Printing of Polymer-Sorted (6,5) Carbon Nanotubes for Field-Effect Transistors with High Reproducibility. Adv. Electron. Mater. 2017, 3, 1700080. 10.1002/aelm.201700080. [DOI] [Google Scholar]
- He X.; Gao W.; Xie L.; Li B.; Zhang Q.; Lei S.; Robinson J. M.; Hároz E. H.; Doorn S. K.; Wang W.; Vajtai R.; Ajayan P. M.; Adams W. W.; Hauge R. H.; Kono J. Wafer-Scale Monodomain Films of Spontaneously Aligned Single-Walled Carbon Nanotubes. Nat. Nanotechnol. 2016, 11, 633–638. 10.1038/nnano.2016.44. [DOI] [PubMed] [Google Scholar]
- Streit J. K.; Bachilo S. M.; Ghosh S.; Lin C. W.; Weisman R. B. Directly Measured Optical Absorption Cross Sections for Structure-Selected Single-Walled Carbon Nanotubes. Nano Lett. 2014, 14, 1530–1536. 10.1021/nl404791y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.