

Abstract
Polymerization-induced self-assembly (PISA) is a powerful platform technology for the rational and efficient synthesis of a wide range of block copolymer nano-objects (e.g., spheres, worms or vesicles) in various media. In situ small-angle X-ray scattering (SAXS) studies of reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization have previously provided detailed structural information during self-assembly (see M. J. Derry et al., Chem. Sci. 2016, 7, 5078–5090). However, conducting the analogous in situ SAXS studies during RAFT aqueous emulsion polymerizations poses a formidable technical challenge because the inherently heterogeneous nature of such PISA formulations requires efficient stirring to generate sufficiently small monomer droplets. In the present study, the RAFT aqueous emulsion polymerization of 2-methoxyethyl methacrylate (MOEMA) has been explored for the first time. Chain extension of a relatively short non-ionic poly(glycerol monomethacrylate) (PGMA) precursor block leads to the formation of sterically-stabilized PGMA-PMOEMA spheres, worms or vesicles, depending on the precise reaction conditions. Construction of a suitable phase diagram enables each of these three morphologies to be reproducibly targeted at copolymer concentrations ranging from 10 to 30% w/w solids. High MOEMA conversions are achieved within 2 h at 70 °C, which makes this new PISA formulation well-suited for in situ SAXS studies using a new reaction cell. This bespoke cell enables efficient stirring and hence allows in situ monitoring during RAFT emulsion polymerization for the first time. For example, the onset of micellization and subsequent evolution in particle size can be studied when preparing PGMA29-PMOEMA30 spheres at 10% w/w solids. When targeting PGMA29-PMOEMA70 vesicles under the same conditions, both the micellar nucleation event and the subsequent evolution in the diblock copolymer morphology from spheres to worms to vesicles are observed. These new insights significantly enhance our understanding of the PISA mechanism during RAFT aqueous emulsion polymerization.
Introduction
Emulsion polymerization is a remarkably efficient and environmentally-friendly process that is applicable to many water-immiscible vinyl monomers, including styrene, methacrylates, acrylates, vinyl acetate, vinyl chloride etc.1,2 It is employed on a global scale by many chemical companies to prepare tens of millions of tons of copolymer latexes every year. Such particles are widely used for paints, coatings, varnishes, adhesives, as additives for concrete and as the mobile phase for various immunodiagnostic assays.3 It is well-known that microcompartmentalization facilitates fast polymerization kinetics for such heterogeneous formulations, which enables the efficient formation of high molecular weight polymer chains in a highly convenient low-viscosity latex form.3−8
The inherently heterogeneous nature of emulsion polymerization makes reliable sampling of such reactions somewhat problematic. Moreover, it is essential that such formulations are efficiently stirred to ensure that micrometer-sized monomer droplets are generated, otherwise there is insufficient interfacial area between this water-immiscible reagent and the aqueous continuous phase to enable the polymerization to proceed. The kinetics of emulsion polymerization have been monitored in situ using 1H nuclear magnetic resonance (NMR) spectroscopy coupled with a flow cell9 or by utilizing Raman10 or near-IR spectroscopy.11 However, such techniques do not provide any information regarding the evolution in particle morphology during the polymerization.
Over the past decade or so, there has been considerable interest in conducting aqueous emulsion polymerizations using pseudo-living radical polymerization.12−16 In particular, reversible addition–fragmentation chain transfer (RAFT) polymerization enables the controlled polymerization of a wide range of functional monomers to form well-defined amphiphilic diblock copolymers.17−20 In principle, this surfactant-free approach should enable access to nanoparticles with various copolymer morphologies using a technique known as polymerization-induced self-assembly (PISA).21 However, in practice there are many literature examples of RAFT aqueous emulsion polymerization formulations that only produce kinetically-trapped spheres.22−34 Exceptions to this restrictive paradigm usually involve the use of statistical copolymers as the water-soluble precursor block to form spheres, worms/fibers or vesicles.35−43 However, the use of ionizable monomers in such examples means that the copolymer morphology also depends on parameters such as the stabilizer block composition,35−38 solution pH35,37,39 and salt concentration.35,36
In contrast, it is well-known that RAFT aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA), which has an aqueous solubility of 100 g dm–3 at 70 °C, invariably allows convenient access to spheres, worms or vesicles provided that a sufficiently short non-ionic steric stabilizer block is utilized.44,45 In view of such observations, we hypothesized that the aqueous solubility of the vinyl monomer might be an important parameter when attempting to prepare worms or vesicles via RAFT aqueous emulsion polymerization. This is because the mass transport of sparingly soluble monomers from the emulsion droplets to the growing diblock copolymer nanoparticles is expected to be rather limited on the time scale of the polymerization. This retarded rate of diffusion reduces the extent of solvation of the growing hydrophobic polymer chains by the unreacted monomer, which is believed to be important for achieving the desired evolution in copolymer morphology during PISA.21,45 In this context, it is widely recognized that other parameters such as the chemical structure35−39,41,42 and copolymer architecture40−42 of the steric stabilizer can also play a decisive role in avoiding the formation of kinetically-trapped spheres. Nevertheless, Cockram et al.46 investigated the RAFT aqueous emulsion polymerization of 4-hydroxybutyl methacrylate (HBMA), which has an aqueous solubility of 20 g dm–3 at 70 °C. A new non-spherical “monkey nut” morphology was obtained for this PISA formulation when using a partially ionized poly(methacrylic acid) stabilizer block at pH 5.46 This encouraging result led us to evaluate glycidyl methacrylate (GlyMA), which has a comparable aqueous solubility of 24–25 g dm–3 at 80 °C.47 Initially, only kinetically-trapped spheres were obtained when using a non-ionic poly(glycerol monomethacrylate) (PGMA) stabilizer.32 However, reducing the mean degree of polymerization (DP) of this hydrophilic block to just 25 enabled the synthesis of well-defined worms.48 In the present study, we introduce 2-methoxyethyl methacrylate (MOEMA) as a third example of a methacrylic monomer exhibiting moderate aqueous solubility (19.6 g dm–3 at 70 °C) that again enables the restrictive paradigm of kinetically-trapped spheres to be avoided without recourse to statistical copolymer and/or ionizable stabilizer blocks. As far as we are aware, the RAFT aqueous emulsion polymerization of MOEMA has not yet been reported.
Recently, we reported the in situ study of a PISA formulation using small-angle X-ray scattering (SAXS).49 This is a powerful characterization technique that can provide precise structural information over multiple length scales.50,51 Indeed, in situ SAXS studies of various types of polymerizations have been reported in the literature.49,52,53 For example, this technique has provided compelling evidence for a recently postulated vesicle growth mechanism during PISA, which places an unexpected constraint on the thermodynamic stability of the vesicle phase.52 However, these prior experiments involved RAFT dispersion polymerization. Such homogeneous formulations require no mechanical agitation, so these PISA syntheses can be conducted within glass capillaries (∼125 μL) that are well-suited for in situ SAXS studies. In contrast, efficient stirring is usually essential to generate sufficiently small monomer droplets to ensure efficient mass transport in the case of RAFT aqueous emulsion polymerization.36,46 This makes in situ SAXS studies much more problematic for such heterogeneous formulations. In an attempt to overcome this technical problem, Paulis et al. developed a thermostated cell for in situ monitoring of the miniemulsion terpolymerization of methyl methacrylate (MMA), n-butyl acrylate and stearyl acrylate using conventional free radical polymerization in the presence of an organically modified clay.54 However, this work focused on monitoring changes in the interlayer distance between clay platelets during the polymerization, rather than examining the nucleation and particle growth events. As far as we are aware, there are no other reports of in situ SAXS studies conducted during any type of heterogeneous polymerization.
Herein we introduce a bespoke stirrable reaction cell (Figure 1) to conduct the first in situ SAXS studies during RAFT aqueous emulsion polymerization.55 We focus on using MOEMA because the relatively high aqueous solubility of this monomer allows the evolution of diblock copolymer nanoparticle morphology from spheres to vesicles via worms when using a non-ionic PGMA stabilizer block (Figure 2). Systematic variation of the copolymer concentration and the mean target DP of the PMOEMA block enables construction of a phase diagram (or morphology map) for a series of PGMA29-PMOEMAy diblock copolymer nano-objects. Moreover, the volumetric capacity of this new reaction cell is sufficiently large (2.0 mL) to enable postmortem analysis of the final nanoparticle dispersion by transmission electron microscopy (TEM), 1H NMR spectroscopy, dynamic light scattering (DLS) and gel permeation chromatography (GPC). Such detailed studies enhance our understanding of the true nature of these heterogeneous polymerizations.52
Figure 1.
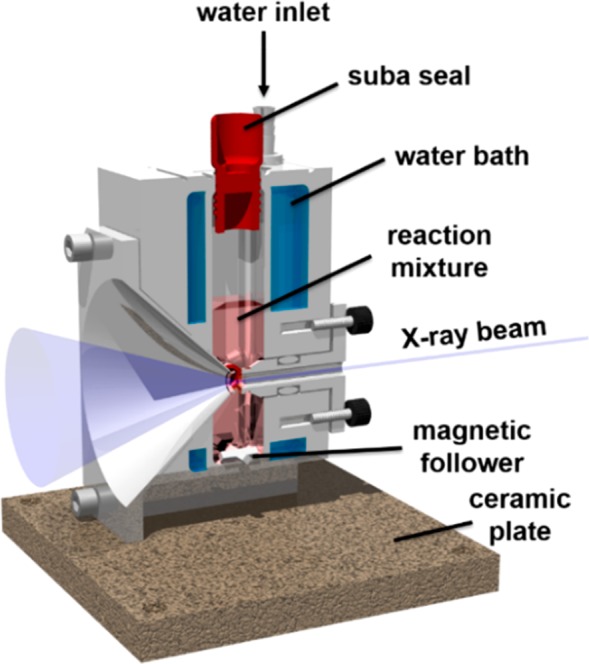
Schematic representation of a cross-section of the bespoke stirrable reaction cell used for in situ SAXS experiments performed during RAFT aqueous emulsion polymerization of MOEMA at 70 °C. The volume of reaction solution within this cell is approximately 2.0 mL, which provides sufficient diblock copolymer (∼200 mg) for postmortem analysis using multiple characterization techniques (see main text for further details).
Figure 2.

Synthesis of PGMA29-PMOEMAy diblock copolymer nano-objects via RAFT aqueous emulsion polymerization of MOEMA using a water-soluble PGMA29 precursor block at 70 °C, where y ranges from 30 to 110. Phase diagram constructed for PGMA29-PMOEMAy diblock copolymer nano-objects synthesized via RAFT aqueous emulsion polymerization of MOEMA at copolymer concentrations ranging from 10 to 30% w/w. Representative TEM images recorded for: (a) PGMA29-PMOEMA36 spheres prepared at 30% w/w; (b) PGMA29-PMOEMA45 worms prepared at 20% w/w; (c) a PGMA29-PMOEMA58 mixed phase comprising worms, jellyfish and vesicles prepared at 10% w/w; (d) PGMA29-PMOEMA78 vesicles prepared at 10% w/w; (e) a PGMA29-PMOEMA85 mixed phase (comprising spheres, worms and vesicles) prepared at 30% w/w; (f) PGMA29-PMOEMA95 vesicles prepared at 20% w/w.
Results and Discussion
The importance of efficient stirring during RAFT aqueous emulsion polymerization is well-known in the PISA literature.36,56 In the absence of any stirring, little or no polymerization occurs because there is simply insufficient interfacial area between the water-immiscible monomer phase and the water-soluble initiator located in the aqueous continuous phase.
A PGMA macromolecular chain transfer agent (macro-CTA) with a mean degree of polymerization (DP) of 29 was synthesized via RAFT ethanolic solution polymerization of GMA at 70 °C, as previously described.32,44 This PGMA29 macro-CTA (Mn = 9600 g mol–1; Đ = 1.12)57 was then chain-extended via RAFT aqueous emulsion polymerization of MOEMA at 70 °C in mildly acidic aqueous solution using 4,4′-azobis(4-cyanovaleric acid) (ACVA) as a well-known water-soluble radical initiator;27,30,35−39,46,56,58 see reaction scheme in Figure 2. The mean target DP of the core-forming PMOEMA block was systematically varied between 30 and 110 while the copolymer concentration was adjusted between 10 and 30% w/w. For well-stirred reaction mixtures, the PGMA macro-CTA acts as an emulsifier: laser diffraction and optical microscopy studies indicated the formation of polydisperse MOEMA monomer droplets with a volume-average diameter of around 14 μm at 20 °C (data not shown). On heating to 70 °C, high MOEMA conversions (≥97%) were achieved for all PISA syntheses, as confirmed by 1H NMR spectroscopy studies (Table S1). GPC studies indicated monomodal curves and relatively narrow molecular weight distributions (Đ ≤ 1.17) for all diblock copolymers synthesized at copolymer concentrations of 10, 15 or 20% w/w (Figure S1 and Table S1). For diblock copolymers prepared at 30% w/w, somewhat higher dispersities (1.11 ≤ Đ ≤ 1.49) were observed owing to the appearance of a high molecular weight peak (Figure S1b). The origin of this feature is not currently known but it appears to be associated with mixed phase copolymer morphologies. As expected, a linear evolution in molecular weight with increasing PMOEMA DP was observed (Figure S2). PGMA29-PMOEMAy diblock copolymer nano-objects were analyzed by DLS and TEM in order to construct a phase diagram (Figure 2). Spherical nanoparticles were obtained when targeting relatively short PMOEMA DPs (y = 30–41) (Figure 2a). A pure worm phase spans the whole range of copolymer concentrations and is relatively narrow (less than 10 MOEMA units for any given reaction concentration) (Figure 2b). Similar observations have been reported for other aqueous PISA formulations.44,59,60
Vesicles can be obtained at all copolymer concentrations investigated, but shorter hydrophobic PMOEMA blocks are required to access this morphology if such PISA syntheses are conducted at lower concentrations. For example, PGMA29-PMOEMA70 at 10% w/w corresponds to a pure vesicle phase (Figure 2d), whereas the same composition at 30% w/w only produces a mixed phase comprising spheres, worms and vesicles. As previously discussed, it is usually fairly straightforward to produce pure worms or vesicles via RAFT aqueous dispersion polymerization.44 In contrast, the precise design rules for RAFT aqueous emulsion polymerization are still being elucidated. Here, we demonstrate that well-defined spheres, worms or vesicles can be obtained by selecting a suitable monomer (e.g., MOEMA) that is mainly water-immiscible but nevertheless exhibits appreciable aqueous solubility at the reaction temperature of 70 °C. Similar findings have been recently reported when using HBMA46 or GlyMA,48 although the full range of copolymer morphologies were not observed in these prior studies. In contrast, other research groups have reported the formation of non-spherical morphologies when polymerizing sparingly soluble monomers such as styrene.35−42 However, these prior PISA syntheses involve using statistical copolymer precursors as the stabilizer block. Notwithstanding these important contributions to the literature, we believe that, for at least some formulations, the aqueous monomer solubility can be an important parameter for understanding the scope and limitations of RAFT aqueous emulsion polymerization.
Two formulations were selected in order to assess the kinetics of the RAFT aqueous emulsion polymerization of MOEMA at 10% w/w solids (Figure 3): PGMA29-PMOEMA30 spheres and PGMA29-PMOEMA70 vesicles. In each case a MOEMA conversion of more than 95% was achieved within 2 h at 70 °C. The kinetic data for the synthesis of the PGMA29-PMOEMA30 spheres were acquired via in situ1H NMR studies, using a set-up recently reported for a RAFT dispersion polymerization formulation in non-polar media.61 In contrast, the kinetic data obtained for the PGMA29-PMOEMA70 diblock copolymer vesicles were obtained by withdrawing small aliquots periodically from the reaction mixture, quenching the polymerization in each case and then utilizing ex situ1H NMR spectroscopy to determine the intermediate monomer conversion.
Figure 3.
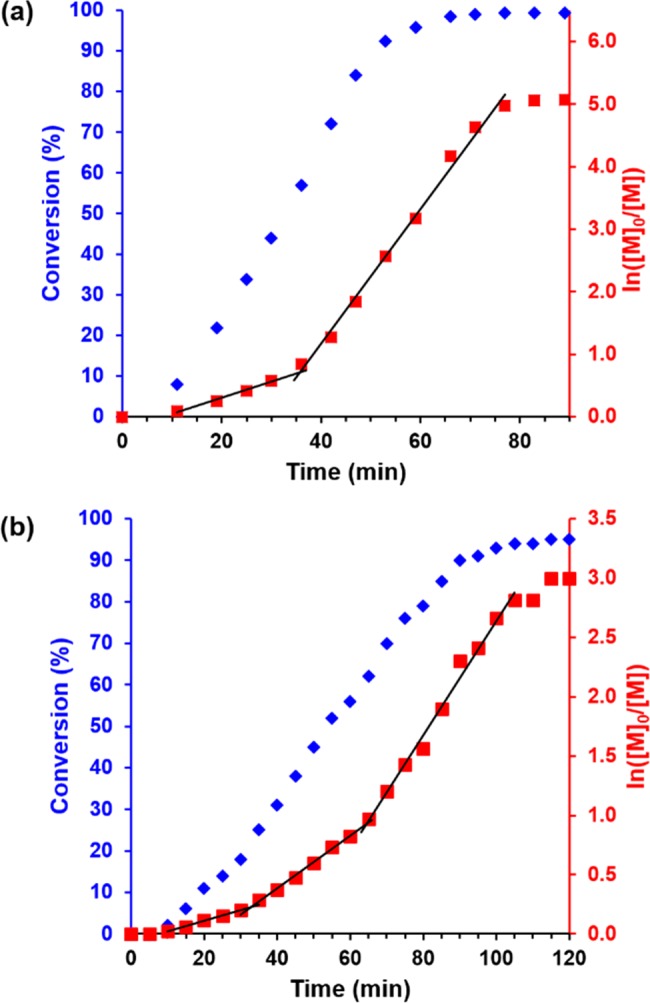
Kinetic studies of the RAFT aqueous emulsion polymerization of MOEMA at 70 °C targeting (a) PGMA29-PMOEMA30 diblock copolymer spheres at 10% w/w and (b) PGMA29-PMOEMA70 diblock copolymer vesicles at 10% w/w. Conversion vs time curves are indicated by blue diamonds and corresponding semilogarithmic plots are shown as red squares. MOEMA conversions were calculated either from (a) in situ1H NMR studies performed in D2O or (b) ex situ1H NMR spectra recorded for quenched aliquots of the reaction solution diluted in d6-DMSO. See experimental section in the Supporting Information for further details.
For the synthesis of PGMA29-PMOEMA30 spheres, a 4-fold rate enhancement was observed after 35 min, which corresponds to approximately 55% conversion and a theoretical DP of 17 (Figure 3a). This is attributed to micellar nucleation of the growing diblock copolymer chains. Similar rate acceleration effects have been reported for RAFT aqueous dispersion polymerization45,62 and also other RAFT aqueous emulsion polymerization formulations.25 Comparable observations were also made during the synthesis of PGMA29-PMOEMA70 vesicles (Figure 3b). Thus, a 2.5-fold increase in the rate of polymerization occurred at around 32 min; this corresponds to 20% conversion and the critical PMOEMA DP required for micellar nucleation is 14. However, in this case a further 2-fold rate enhancement was observed at 65 min (61% conversion). The PMOEMA DP is calculated to be 43 for this higher conversion, which is consistent with the diblock copolymer composition required to form worms as indicated in the phase diagram (Figure 2). This suggests that the second rate enhancement is most likely associated with a sphere-to-worm transition. A similar increase in the rate of polymerization was reported by Charleux and co-workers for the RAFT aqueous emulsion polymerization of MMA, which led to kinetically-trapped spheres.39 However, the same group observed a retardation effect during the RAFT aqueous emulsion polymerization of styrene,38 which was attributed to the reduced number of particles per unit volume associated with the sphere-to-worm transition. In this context, it is perhaps worth noting that similar two-stage rate enhancements have been observed for RAFT dispersion polymerizations conducted in non-polar media,61,63 so this phenomenon is not necessarily a consequence of the heterogeneous nature of RAFT aqueous emulsion polymerization. Such subtle changes in polymerization kinetics during PISA are not properly understood and clearly warrant further studies.
SAXS Analysis of PGMA29-PMOEMAy Nano-Objects
SAXS patterns were recorded for 1.0% w/w aqueous dispersions of four examples of PGMA29-PMOEMAy nano-objects synthesized at 10% w/w, see Figure 4 and Table S2. The y values targeted were 38, 43, 70 and 84. According to the phase diagram shown in Figure 2, such diblock compositions should correspond to spheres, worms, vesicles and vesicles, respectively.
Figure 4.
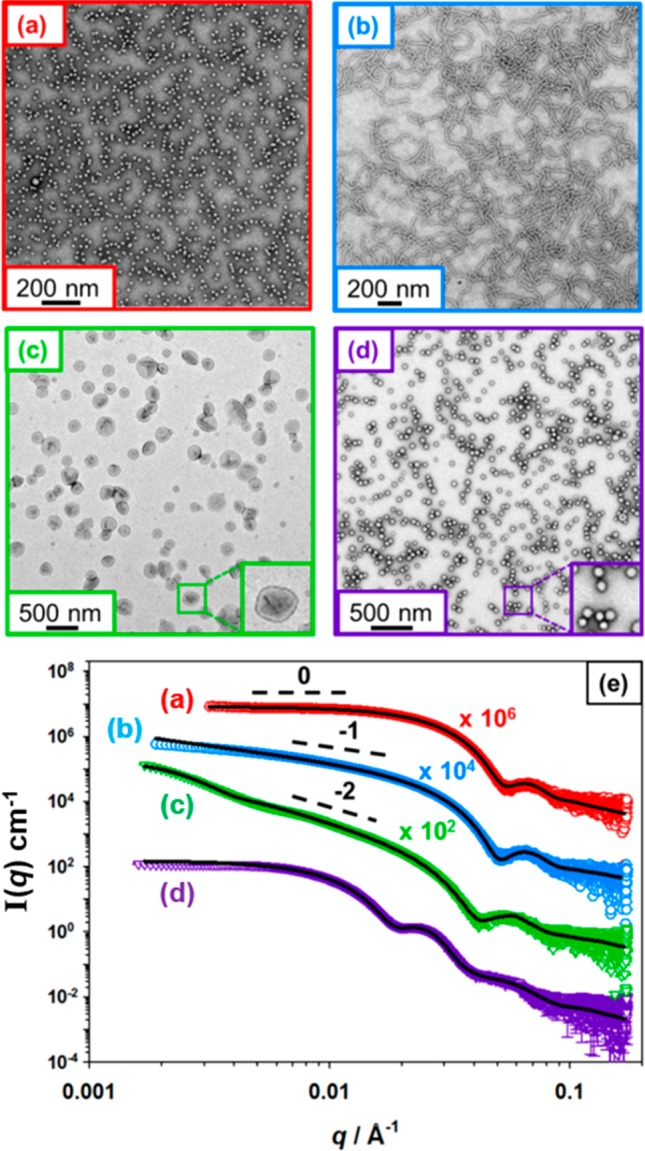
TEM images obtained for (a) PGMA29-PMOEMA38 spheres, (b) PGMA29-PMOEMA43 worms, (c) relatively large PGMA29-PMOEMA70 vesicles and (d) relatively small PGMA29-PMOEMA84 vesicles. (e) Corresponding SAXS patterns recorded at 1.0% w/w for the same four copolymer dispersions, which were each originally prepared at 10% w/w. Black solid lines show the data fits obtained for each SAXS pattern using an appropriate spherical micelle, worm-like micelle or vesicle model. Each low q gradient is consistent with the TEM images.
The radially integrated patterns obtained for these four PGMA29-PMOEMAy dispersions are plotted as the X-ray scattering intensity, I(q), vs the scattering vector, q. It is well-known that the gradient in the low q region of a SAXS pattern can be used to assign the predominant copolymer morphology.64 Spherical micelles exhibit a low q gradient of zero, rigid rods (for which worm-like micelles are a reasonable approximation) have a gradient of −1, and a gradient close to −2 indicates the presence of relatively flat bilayers (or vesicles with thin membranes). Inspecting Figure 4, the SAXS pattern for PGMA29-PMOEMA38 can be satisfactorily fitted using a spherical micelle model65 (Figure 4a,e), which indicated a sphere volume-average diameter (Ds) of 19 nm. This is consistent with the z-average diameter (Dz) of 26 nm obtained from DLS studies. As expected, the SAXS pattern recorded for PGMA29-PMOEMA43 could be fitted using a worm-like micelle model62 (Figure 4b,e), which reported a worm cross-sectional volume-average diameter (Dw) of 16 nm. Patterns obtained for PGMA29-PMOEMA70 and PGMA29-PMOEMA84 were each satisfactorily fitted using a vesicle model66 (Figure 4c,e and Figure 4d,e, respectively) with vesicle volume-average diameters (Dv) of 144 and 49 nm, respectively. As expected, the corresponding Dz indicated by DLS were somewhat larger at 178 and 51 nm, respectively. It is perhaps worth emphasizing that the PGMA29-PMOEMA84 vesicles are unusually small. Based on TEM analysis alone, these nanoparticles were initially incorrectly assigned as spheres, not least because there was little or no evidence for vesicle deformation under the ultrahigh vacuum conditions required for electron microscopy. In retrospect, the lack of collapse on drying is not unexpected for such relatively small vesicles, for which the mean thickness of the hydrophobic part of the membrane (Tm) of approximately 9.3 nm indicated by SAXS analysis is comparable to that of the overall vesicle radius of ∼25 nm.
Like the PGMA29-PMOEMA84 vesicles synthesized at 10% w/w, morphology assignments also proved to be somewhat problematic for PGMA29-PMOEMA89 synthesized at 10% w/w solids, PGMA29-PMOEMA95 and PGMA29-PMOEMA100 prepared at 20% w/w solids, and PGMA29-PMOEMA110 prepared at 30% w/w solids. Initially, TEM images were tentatively interpreted as spheres in each case. However, there was also some evidence for vesicular morphologies by TEM, with at least some of the dried nano-objects exhibiting somewhat darker central areas (see Figure S3). Indeed, the corresponding SAXS patterns could be satisfactorily fitted using a vesicle model (see Figure S4 and Table S2), whereas attempted data fits using a spherical micelle model were unsuccessful (see Figure S5). The mean vesicle dimensions determined by SAXS, TEM and DLS are summarized in Table S3. These vesicles are relatively small (Dv = 50–91 nm by SAXS), which most likely explains why they resist collapse under ultrahigh vacuum conditions and hence were originally judged to be spheres in our initial TEM studies.
In Situ SAXS Studies During RAFT Aqueous Emulsion Polymerization Using the Bespoke Stirrable Cell
The bespoke stirrable reaction cell (Figure 1) is machined from aluminum: it allows efficient magnetic stirring of the reaction solution and controlled heating by means of a water jacket connected to a circulating water bath. Moreover, this cell can be hermetically sealed, enabling air-sensitive polymerizations to be conducted under an inert atmosphere. Thus, it is particularly useful for conducting RAFT aqueous emulsion polymerizations, which are inherently heterogeneous in nature. Reaction solution volumes of ∼2.0 mL were utilized, which provides sufficient material to enable postmortem characterization of the resulting diblock copolymer nanoparticles after the in situ SAXS experiments. This offers an important advantage compared to the ∼125 μL glass capillaries used by Derry et al. for their in situ SAXS studies of RAFT dispersion polymerization.49 Given the relatively fast kinetics (Figure 3), a synchrotron X-ray source is essential to achieve the high temporal resolution required for in situ SAXS studies of the RAFT aqueous emulsion polymerization of MOEMA at 70 °C. This enables acquisition of many SAXS patterns even within the relatively short reaction time scale of 80–120 min.
To evaluate the feasibility of conducting the RAFT aqueous emulsion polymerization of MOEMA using the stirrable reaction cell, two PGMA29-PMOEMAy formulations were targeted where the final morphology was either spheres or vesicles, respectively. In the former case, a progressive increase in particle diameter was anticipated after micellar nucleation. In the latter case, the in situ evolution in copolymer morphology from spheres to worms to vesicles should be observed.62 Data were collected until no further change in the scattering pattern was observed, which was taken to indicate the end of the polymerization. It is emphasized that, as far as we are aware, these experiments represent the first in situ SAXS studies attempted for any aqueous emulsion polymerization formulation.
PGMA29-PMOEMA30 Spheres
PGMA29-PMOEMA30 synthesized at 10% w/w yielded well-defined spherical nano-objects in laboratory-based experiments (Figure 2). Therefore, this PISA formulation was selected for in situ experiments. The postmortem characterization of this aqueous dispersion of PGMA29-PMOEMA30 nanoparticles is summarized in Table S4 and Figure S6. GPC analysis of PGMA29-PMOEMA30 diblock copolymer chains prepared in the equivalent laboratory-based synthesis indicated very similar molecular weight data compared to that obtained after the in situ SAXS experiment (Mn = 10 500 and Đ = 1.21 vs Mn = 10 600 g mol–1 and Đ = 1.21, respectively). TEM images recorded for both dispersions confirmed the formation of a well-defined spherical morphology in each case (Figure 5). DLS studies (see Table S4) indicated a Dz of 19 nm (PDI = 0.09) for the laboratory-based synthesis of PGMA29-PMOEMA30 spheres, whereas the nanoparticles obtained after the in situ SAXS experiment using the same PISA formulation had an almost identical Dz of 18 nm (PDI = 0.10).
Figure 5.

Representative TEM images recorded for the dried PGMA29-PMOEMA30 spheres prepared via RAFT aqueous emulsion polymerization of MOEMA at 10% w/w: (a) after in situ SAXS experiments using the stirrable reaction cell shown in Figure 1 and (b) after a laboratory-based synthesis using precisely the same PISA formulation.
SAXS patterns were collected every 5 min for 90 min (Figure 6a). Two aspects of this RAFT aqueous emulsion polymerization were used to compare the progress of the polymerization during the in situ SAXS experiments with the kinetic data obtained from the laboratory-based synthesis. These features were: (i) the characteristic time required for the onset of micellar nucleation and (ii) the time at which the MOEMA polymerization was judged to have reached completion. Initially, the growing PMOEMA chains remained fully soluble in the reaction mixture and the rate of the ensuing RAFT solution polymerization was relatively slow.62 However, once this block had become sufficiently hydrophobic to induce micellar nucleation, a significant rate enhancement was observed.37,45 This phenomenon is readily apparent in the kinetic data obtained for the laboratory-based synthesis (Figure 3a).
Figure 6.
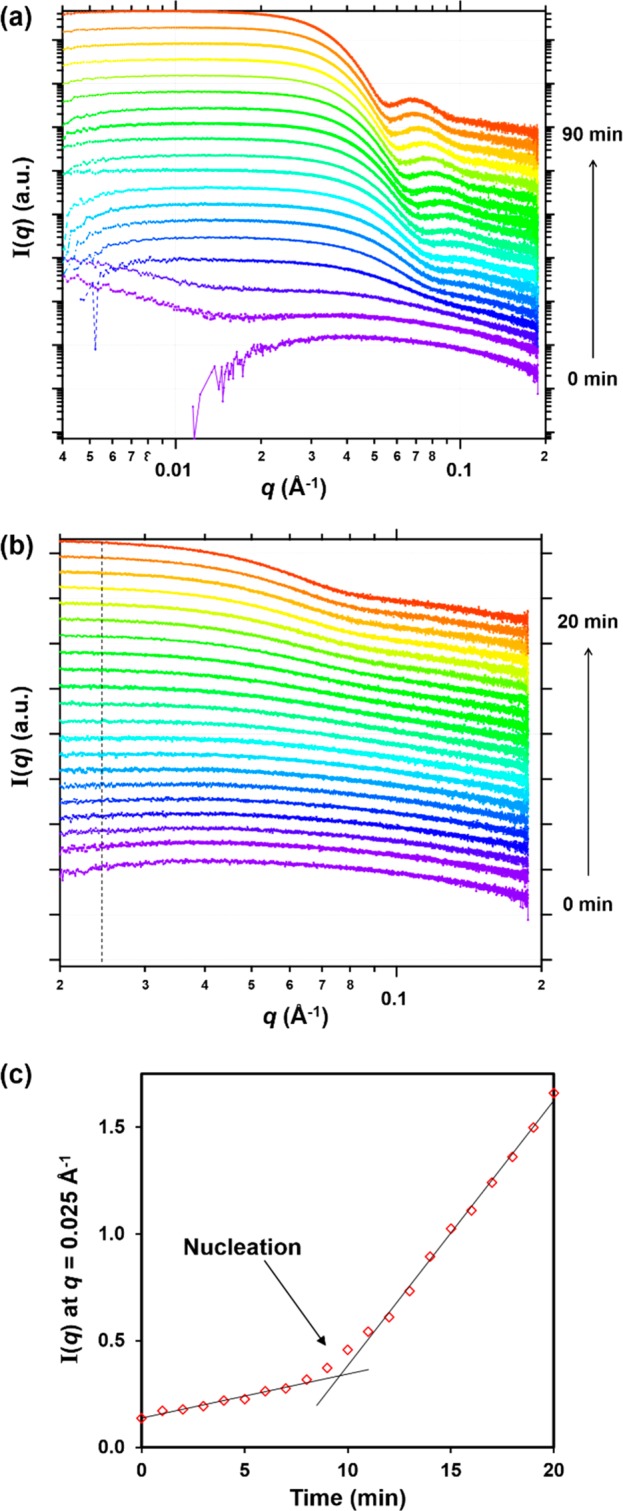
SAXS patterns recorded during the RAFT aqueous emulsion polymerization of MOEMA targeting PGMA29-PMOEMA30 at 10% w/w solids using the stirrable reaction cell (Figure 1). (a) Patterns recorded every 5 min from 0 to 90 min. (b) Patterns recorded every 1 min from 0 to 20 min and (c) a plot of I(q) at q = 0.025 Å–1 vs time. The onset of micellar nucleation is indicated by an arrow. Patterns are offset by an arbitrary factor to aid clarity in panels a and b.
To recap, micellar nucleation occurred at ∼35 min (Figure 3a), which corresponded to a MOEMA conversion of 55% and a critical DP of 17 for the structure-directing PMOEMA block. In situ NMR experiments indicated that a MOEMA conversion of 99% was achieved within 90 min at 70 °C for this PISA formulation. In the literature, visual inspection has been used to identify the onset of micellar nucleation for RAFT aqueous dispersion polymerizations because there is an associated increase in turbidity for such initially homogeneous PISA formulations.62 However, in the case of RAFT aqueous emulsion polymerization, the inherently heterogeneous nature of the reaction mixture precludes this approach (i.e., the initial aqueous monomer emulsion is already highly turbid). Fortunately, SAXS can readily distinguish between molecularly dissolved copolymer chains and the formation of nascent micelles, since the latter species lead to more intense X-ray scattering at low q (Guinier regime). To determine the onset of micellization during the in situ SAXS experiment, the scattering intensity, I(q), at an arbitrary q value of 0.025 Å–1 was plotted as a function of time for the first 20 min of the polymerization (Figure 6c). The increase in scattering intensity observed after 9–10 min indicates the formation of larger scattering objects and hence corresponds to micelle formation. One of the reviewers of this manuscript suggested that we calculate the Porod invariant (Q)67,68 for our SAXS data and plot this parameter against time. This alternative approach yielded essentially the same time for the micellar nucleation event (data not shown). It is noteworthy that the onset of micellar nucleation occurs on a somewhat shorter time scale for this in situ SAXS experiment compared to the equivalent laboratory-based PISA synthesis. This is not unexpected, as a significant rate enhancement was observed for the RAFT dispersion polymerization of benzyl methacrylate in mineral oil.49 The faster rate of polymerization in the presence of the high-energy X-rays was attributed to the ionizing nature of such radiation, which can generate an additional radical flux.69,70
The MOEMA polymerization was judged to have reached completion within 90 min, because no discernible change in the scattering pattern was observed after this time point. This was supported by postmortem analysis of the quenched reaction mixture by 1H NMR spectroscopy, which indicated a final MOEMA conversion of 98%. Such kinetics are comparable to those observed for laboratory-based syntheses, although the nucleation event occurs earlier during the in situ SAXS studies. Micelle formation is indicated by the appearance of a local minimum at q = 0.091 Å–1. This distinctive signature is first observed after 15 min and enables estimation of the mean core radius (Rs) of the nascent spherical micelles using the well-known relationship, d = 4.49/q, where d is a real-space distance corresponding to Rs. During the MOEMA polymerization, the local minimum gradually shifts to lower q at longer reaction times, indicating the expected progressive increase in nanoparticle radius (Figure S7).49 A sphere radius, Rs, of 8.3 nm was calculated from the final SAXS pattern, corresponding to a mean sphere diameter, Ds, of 16.6 nm; this is comparable to the postmortem z-average diameter of 18 nm indicated by DLS studies. After the MOEMA polymerization was complete, a SAXS pattern was also recorded for a 1.0% w/w aqueous dispersion of spheres after a 10-fold dilution of the final reaction solution. This approach enabled a satisfactory data fit to be obtained using the spherical micelle model without requiring any prior knowledge of the structure factor that is associated with interparticle interactions (Figure S8a and Table S5). A Ds value of 18.8 nm was determined from this data fit. By incorporating an appropriate structure factor, the final scattering pattern recorded for the undiluted 10% w/w aqueous dispersion could also be fitted to a spherical micelle model, giving a comparable Ds value of 19.1 nm (Figure S8b and Table S5).
PGMA29-PMOEMA70 Vesicles
For a RAFT dispersion polymerization, targeting a suitably asymmetric diblock copolymer composition under appropriate conditions normally leads to the consecutive formation of spheres, worms and ultimately vesicles during the course of the reaction.49,62 To investigate whether the stirrable reaction cell allowed access to such higher order copolymer morphologies, the RAFT aqueous emulsion polymerization of MOEMA was conducted targeting PGMA29-PMOEMA70 at a copolymer concentration of 10% w/w. According to the phase diagram (Figure 2), this PISA formulation should produce vesicles as the final copolymer morphology at full MOEMA conversion. Postmortem GPC characterization of the PGMA29-PMOEMA70 diblock copolymer chains indicated Mn values of 16 100 and 16 600 g mol–1 for the in situ SAXS experiment and corresponding laboratory-based synthesis, respectively, with relatively low dispersities (Đ < 1.15) being obtained in each case (Table S4 and Figure S9). Representative TEM images for these PGMA29-PMOEMA70 diblock copolymer nano-objects are shown in Figure 7.
Figure 7.

Representative TEM images recorded for the dried PGMA29-PMOEMA70 diblock copolymer nano-objects obtained by RAFT aqueous emulsion polymerization of MOEMA at 10% w/w: (a) using the stirrable reaction cell shown in Figure 1 and (b) via the corresponding laboratory-based synthesis.
A predominantly vesicular morphology is clearly present in both cases, but some short worms and spheres are also discernible for the MOEMA polymerization conducted in the stirrable reaction cell (Figure 7a). DLS studies indicated that somewhat smaller vesicles were obtained during the in situ SAXS experiment compared to those produced in the laboratory-based synthesis (Dz were 124 and 178 nm, respectively), with relatively narrow size distributions (PDI ≤ 0.08) being obtained in each case. This size difference is consistent with the minor populations of short worms and spheres observed by TEM.
It is not currently clear whether this subtle difference in copolymer morphology is the result of a difference in mechanical agitation efficiency or perhaps the rate enhancement induced by X-ray irradiation. SAXS patterns were recorded regularly during the MOEMA polymerization at 70 °C, with no further change in such data being discernible after 90 min (Figure 8). Assuming that the polymerization was complete on this time scale, this suggests a modest rate enhancement compared to that observed for the corresponding laboratory-based synthesis, which required 2 h at 70 °C to achieve more than 95% conversion. A local minimum at q = 0.065 Å–1 became evident after around 15 min, suggesting that the transformation from dissolved copolymer chains to nascent spherical micelles occurred within this time frame. Close inspection of scattering patterns recorded at 1 min intervals within the first 15 min indicated that micellar nucleation appears to occur between 6 and 8 min (Figure 8c). As observed for the PGMA29-PMOEMA30 spheres, micellar nucleation clearly occurs on a significantly shorter time scale than the 32 min indicated by 1H NMR analysis of the equivalent laboratory-based experiment (Figure 3b). As discussed above, a similar rate enhancement has been reported for the in situ dispersion polymerization of benzyl methacrylate in mineral oil49 by Derry and co-workers, who attributed this phenomenon to the additional radical flux generated by the high-energy X-rays.69,70 A plot of I(q) at an arbitrary q value of 0.025 Å–1 vs time indicated that the onset of micellar nucleation actually occurs at a reaction time of approximately 6 min (Figure 8d). The expected evolution in copolymer morphology from spheres to worms to vesicles at longer polymerization times was confirmed by the change in gradient at low q (for 0.005 ≤ q ≤ 0.015 Å–1) (Figure 8b). The initial gradient is close to zero, as expected for spherical micelles. This gradient becomes significantly more negative after 27 min, at which the mean PMOEMA DP is estimated to be 37. According to the phase diagram (Figure 2), this DP corresponds to the onset of the sphere-to-worm transition. Furthermore, the plot of I(q) at q = 0.01 Å–1 vs time (Figure 8b) indicates a sharp increase in the scattering intensity between 25 and 30 min, further supporting such a morphological transition. For the time period from 27 to 53 min, the low q gradient is close to −1, which indicates the presence of linear worms. The monotonic increase in I(q) during this period suggests the growth of initially short worms to form longer worms via 1D fusion with multiple spheres. The low q gradient becomes approximately −2 after 70 min, which indicates the formation of vesicles. At the end of the polymerization (∼90 min) the scattering intensity becomes constant and the gradient tends to −2.4. Approximately the same gradient (−2.3) was observed over the same q range (0.005 ≤ q ≤ 0.015 Å–1) for the final diblock copolymer vesicles after dilution to 1.0% w/w (Figure S10). Thus, this more negative gradient indicates the formation of relatively thick-walled vesicles at the end of the reaction. The final scattering pattern acquired for the in situ synthesis conducted at 10% w/w was also fitted to a vesicle model using a restricted q range of 0.005–0.14 Å–1 to avoid any influence of the structure factor (Figure S11 and Table S5).49 This approach gave a Dv of 115 nm, which is consistent with the Dz value of 124 nm reported by DLS.
Figure 8.

SAXS patterns recorded during the PISA synthesis of PGMA29-PMOEMA70 via RAFT aqueous emulsion polymerization of MOEMA at 10% w/w solids using the stirrable reaction cell (Figure 1). (a) Patterns recorded every 5 min from 0 to 120 min. (b) The corresponding plot of I(q) at q = 0.01 Å–1 and the gradient at low q (0.005 ≤ q ≤ 0.015 Å–1) vs time. (c) Patterns recorded each minute from 0 to 15 min. (d) The corresponding plot of I(q) at q = 0.025 Å–1 vs time. The onset of micellar nucleation at 5–6 min is indicated by an arrow. Patterns are offset by an arbitrary factor to aid clarity in panels a and c.
Depending on the precise time interval, the local minima observed in these SAXS patterns (for which q values range from 0.074 to 0.040 Å–1) can provide approximate dimensions for the spheres, worms and vesicles. More specifically, real-space distances corresponding to the sphere core diameter (Ds), worm cross-sectional diameter, (Dw), or the overall vesicle membrane thickness (Tv) were calculated (Figure S12). After micellar nucleation, Ds increased monotonically from 12.2 to 16.5 nm as the MOEMA polymerization progressed. Subsequently, during the sphere-to-worm transition, the Dw initially dropped slightly to 15.1 nm as multiple spheres fuse to form short worms, then increased monotonically up to 18.6 nm. Following the worm-to-vesicle transition, Tv is around 15.3 nm, which suggests some degree of interdigitation by the hydrophobic PMOEMA chains within the vesicle membrane. It should be recognized that the above data is best used as a relative guide to changes in the nano-object dimensions during the MOEMA polymerization. Nevertheless, fitting the SAXS pattern obtained for the pure vesicles at full monomer conversion using the appropriate scattering model indicated essentially the same volume-average membrane thickness (Tm + 4Rg = 15.3 nm), where Tm is the thickness of the hydrophobic part of the membrane alone, once the radius of gyration of the stabilizer chains is taken into account. In principle, if a higher PMOEMA DP was targeted the Tv should increase further with increasing monomer conversion52 but in this particular case the target core-forming block DP was only just sufficient to form vesicles (see phase diagram in Figure 2).
Finally, we examined whether the stirrable reaction cell could provide sufficient mechanical agitation to enable the RAFT aqueous emulsion polymerization of a sparingly soluble monomer using a closely related PGMA48 macro-CTA.30 Accordingly, 2,2,2-trifluoroethyl methacrylate (TFEMA; aqueous solubility ∼2.9 g dm–3 at 25 °C)71 was utilized instead of MOEMA. This PISA synthesis was conducted at 10% w/w solids and proceeded to 99% conversion within 170 min at 70 °C (see Figure S13 in the Supporting Information). Postmortem TEM studies confirmed the formation of well-defined kinetically-trapped spheres, while DLS analysis indicated an intensity-average diameter of 60 nm (PDI = 0.08). The in situ SAXS data indicated an upturn in scattering intensity after approximately 1 h and a gradual evolution in particle size over the course of the polymerization (final PTFEMA core diameter, Ds = 47 nm). This experiment is important because it suggests that the new stirrable reaction cell should allow in situ SAXS studies of the RAFT aqueous emulsion polymerization of most water-immiscible vinyl monomers, regardless of their aqueous solubility.
Conclusions
In summary, the RAFT aqueous emulsion polymerization of 2-methoxyethyl methacrylate (MOEMA) at 70 °C using a water-soluble poly(glycerol monomethacrylate) (PGMA) macromolecular chain transfer agent can produce diblock copolymer worms and vesicles as well as spherical nanoparticles. Systematic variation of the diblock copolymer composition and the copolymer concentration enabled construction of a phase diagram, which is essential for reproducible targeting of each of these three morphologies. A bespoke reaction cell provides sufficient mechanical agitation to form micrometer-sized monomer droplets and hence enables the first in situ SAXS studies to be conducted for any RAFT aqueous emulsion polymerization. Moreover, this new cell has a reaction volume of ∼2.0 mL, which is sufficient to allow postmortem analysis of the final diblock copolymer nano-objects using 1H NMR spectroscopy, DLS and TEM, as well as GPC analysis of the diblock copolymer chains.
A modest rate enhancement was observed during these in situ SAXS experiments compared to the equivalent laboratory-based syntheses. This kinetic effect is attributed to the ionizing nature of the high-energy X-ray radiation and is much less than that previously reported for RAFT dispersion polymerizations conducted in mineral oil using capillary cells.48 Analysis of the in situ SAXS patterns indicated that a significant increase in the rate of polymerization occurs immediately after micellar nucleation, which suggests the formation of monomer-swollen nascent micelles. This is consistent with the rate enhancement observed by 1H NMR spectroscopy studies. It is believed that the relatively high aqueous solubility of MOEMA leads to enhanced PMOEMA chain mobility within the growing monomer-swollen nanoparticle cores at intermediate conversion. Such core solvation is most likely important for the observed evolution in copolymer morphology under certain conditions. Thus, provided that the water-soluble PGMA steric stabilizer block is relatively short (DP = 29), sphere–sphere fusion occurs efficiently on the time scale of the polymerization, which enables the formation of well-defined worms or vesicles as well as spheres. Determination of the low q gradient during these in situ SAXS studies has enabled the direct observation of the in situ evolution in diblock copolymer morphology from spheres to worms to vesicles during RAFT aqueous emulsion polymerization for the first time. This is important, because such PISA formulations are applicable to a much wider range of vinyl monomers than RAFT aqueous dispersion polymerization.45 Moreover, such worms may be useful as new aqueous thickeners21 while the vesicles may offer potential applications for microencapsulation21 or as opacifiers.41
In preliminary experiments, we have recently confirmed that the same experimental set-up also enables in situ studies of (i) the RAFT aqueous emulsion polymerization of sparingly soluble monomers such as 2,2,2-trifluoroethyl methacrylate,30 (ii) charge-stabilized latexes via conventional aqueous emulsion polymerization72 and (iii) the formation of colloidal polymer/silica nanocomposite particles.73 Thus, this new approach is of broader significance and can be expected to provide important new insights regarding the evolution of structure for various heterogeneous colloidal soft matter systems in the future.
Acknowledgments
S.P.A. thanks the ERC for an Advanced Investigator grant (PISA 320372) to support F.L.H. and the EPSRC for an Established Career Fellowship in Particle Technology (EP/R003009). The Leverhulme Trust is also thanked for postdoctoral funding of M.J.D. (RPG-2016-330). The authors thank the European Synchrotron Radiation Facility for providing beam time at the ID02 beamline (sc4601 and sc4864) and Diamond Light Source for providing beam time at the I22 beamline (sm19852). Dr. Sandra van Meurs is thanked for her assistance with the in situ NMR experiments. Dr. Svetomir Tzokov is thanked for carbon coating TEM grids.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b06788.
Full experimental section and further characterization details of the PGMA29-PMOEMAy and PGMA48-PTFEMA300 diblock copolymer nano-objects prepared at 10–30% w/w solids (including monomer conversions, molecular weight data (Mn, Mw and Đ), dynamic light scattering results, DMF GPC chromatograms, additional TEM images and small-angle X-ray scattering patterns; further details of the SAXS models and analytical approach (PDF)
Author Present Address
§ Department of Materials, Loughborough University, Loughborough LE11 3TU, United Kingdom
The authors declare no competing financial interest.
Supplementary Material
References
- Lovell P. A.; El-Aasser M. S.. Emulsion Polymerization and Emulsion Polymers; Wiley, 1997. [Google Scholar]
- Gilbert R. G.Emulsion polymerization: a mechanistic approach; Academic Press, 1995. [Google Scholar]
- Chern C. S. Emulsion polymerization mechanisms and kinetics. Prog. Polym. Sci. 2006, 31 (5), 443–486. 10.1016/j.progpolymsci.2006.02.001. [DOI] [Google Scholar]
- Tauer K.; Hernandez H.; Kozempel S.; Lazareva O.; Nazaran P. Towards a consistent mechanism of emulsion polymerization - new experimental details. Colloid Polym. Sci. 2008, 286 (5), 499–515. 10.1007/s00396-007-1797-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith W. V.; Ewart R. H. Kinetics of Emulsion Polymerization. J. Chem. Phys. 1948, 16 (6), 592–599. 10.1063/1.1746951. [DOI] [Google Scholar]
- Harkins W. D. General Theory of Mechanism of Emulsion Polymerization. 2. J. Polym. Sci. 1950, 5 (2), 217–251. 10.1002/pol.1950.120050208. [DOI] [Google Scholar]
- Harkins W. D. A General Theory of the Mechanism of Emulsion Polymerization. J. Am. Chem. Soc. 1947, 69 (6), 1428–1444. 10.1021/ja01198a053. [DOI] [PubMed] [Google Scholar]
- Thickett S. C.; Gilbert R. G. Emulsion polymerization: State of the art in kinetics and mechanisms. Polymer 2007, 48 (24), 6965–6991. 10.1016/j.polymer.2007.09.031. [DOI] [Google Scholar]
- Vargas M. A.; Cudaj M.; Hailu K.; Sachsenheimer K.; Guthausen G. Online Low-Field H-1 NMR Spectroscopy: Monitoring of Emulsion Polymerization of Butyl Acrylate. Macromolecules 2010, 43 (13), 5561–5568. 10.1021/ma1006599. [DOI] [Google Scholar]
- Chen X. Y.; Laughlin K.; Sparks J. R.; Linder L.; Farozic V.; Masser H.; Petr M. In Situ Monitoring of Emulsion Polymerization by Raman Spectroscopy: A Robust and Versatile Chemometric Analysis Method. Org. Process Res. Dev. 2015, 19 (8), 995–1003. 10.1021/acs.oprd.5b00045. [DOI] [Google Scholar]
- Bao H.; Chen Z.; Xu W.; Wu P.; Wang Y.; Gao B.; Liu J. Synthesis of nano-sized magnetic colloidal particles from ferrous salts and hydroxylated poly(butyl methacrylate-b-glycidyl methacrylate). Colloid J. 2006, 68 (5), 644–647. 10.1134/S1061933X06050176. [DOI] [Google Scholar]
- Monteiro M. J.; Cunningham M. F. Polymer Nanoparticles via Living Radical Polymerization in Aqueous Dispersions: Design and Applications. Macromolecules 2012, 45 (12), 4939–4957. 10.1021/ma300170c. [DOI] [Google Scholar]
- Zetterlund P. B.; Thickett S. C.; Perrier S.; Bourgeat-Lami E.; Lansalot M. Controlled/Living Radical Polymerization in Dispersed Systems: An Update. Chem. Rev. 2015, 115 (18), 9745–9800. 10.1021/cr500625k. [DOI] [PubMed] [Google Scholar]
- Prescott S. W.; Ballard M. J.; Rizzardo E.; Gilbert R. G. Rate optimization in controlled radical emulsion polymerization using RAFT. Macromol. Theory Simul. 2006, 15 (1), 70–86. 10.1002/mats.200500052. [DOI] [Google Scholar]
- Ferguson C. J.; Hughes R. J.; Nguyen D.; Pham B. T. T.; Gilbert R. G.; Serelis A. K.; Such C. H.; Hawkett B. S. Ab initio emulsion polymerization by RAFT-controlled self-assembly. Macromolecules 2005, 38 (6), 2191–2204. 10.1021/ma048787r. [DOI] [Google Scholar]
- Ferguson C. J.; Hughes R. J.; Pham B. T. T.; Hawkett B. S.; Gilbert R. G.; Serelis A. K.; Such C. H. Effective ab initio emulsion polymerization under RAFT control. Macromolecules 2002, 35 (25), 9243–9245. 10.1021/ma025626j. [DOI] [Google Scholar]
- Keddie D. J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43 (2), 496–505. 10.1039/C3CS60290G. [DOI] [PubMed] [Google Scholar]
- Chiefari J.; Chong Y. K.; Ercole F.; Krstina J.; Jeffery J.; Le T. P. T.; Mayadunne R. T. A.; Meijs G. F.; Moad C. L.; Moad G.; Rizzardo E.; Thang S. H. Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31 (16), 5559–5562. 10.1021/ma9804951. [DOI] [Google Scholar]
- Chong Y. K.; Le T. P. T.; Moad G.; Rizzardo E.; Thang S. H. A more versatile route to block copolymers and other polymers of complex architecture by living radical polymerization: The RAFT process. Macromolecules 1999, 32 (6), 2071–2074. 10.1021/ma981472p. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process - A Third Update. Aust. J. Chem. 2012, 65 (8), 985–1076. 10.1071/CH12295. [DOI] [Google Scholar]
- Canning S. L.; Smith G. N.; Armes S. P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self Assembly. Macromolecules 2016, 49 (6), 1985–2001. 10.1021/acs.macromol.5b02602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger J.; Stoffelbach F.; Bui C.; Alaimo D.; Jerome C.; Charleux B. Amphiphilic poly(ethylene oxide) macromolecular RAFT agent as a stabilizer and control agent in ab initio batch emulsion polymerization. Macromolecules 2008, 41 (12), 4065–4068. 10.1021/ma800544v. [DOI] [Google Scholar]
- Rieger J.; Osterwinter G.; Bui C. O.; Stoffelbach F.; Charleux B. Surfactant-Free Controlled/Living Radical Emulsion (Co)polymerization of n-Butyl Acrylate and Methyl Methacrylate via RAFT Using Amphiphilic Poly(ethylene oxide)-Based Trithiocarbonate Chain Transfer Agents. Macromolecules 2009, 42 (15), 5518–5525. 10.1021/ma9008803. [DOI] [Google Scholar]
- Rieger J.; Zhang W. J.; Stoffelbach F.; Charleux B. Surfactant-Free RAFT Emulsion Polymerization Using Poly(N,N-dimethylacrylamide) Trithiocarbonate Macromolecular Chain Transfer Agents. Macromolecules 2010, 43 (15), 6302–6310. 10.1021/ma1009269. [DOI] [Google Scholar]
- Zhang W. J.; D’Agosto F.; Boyron O.; Rieger J.; Charleux B. One-Pot Synthesis of Poly(methacrylic acid-co-poly(ethylene oxide) methyl ether methacrylate)-b-polystyrene Amphiphilic Block Copolymers and Their Self-Assemblies in Water via RAFT-Mediated Radical Emulsion Polymerization. A Kinetic Study. Macromolecules 2011, 44 (19), 7584–7593. 10.1021/ma201515n. [DOI] [Google Scholar]
- Chaduc I.; Girod M.; Antoine R.; Charleux B.; D’Agosto F.; Lansalot M. Batch Emulsion Polymerization Mediated by Poly(methacrylic acid) MacroRAFT Agents: One-Pot Synthesis of Self-Stabilized Particles. Macromolecules 2012, 45 (15), 5881–5893. 10.1021/ma300875y. [DOI] [Google Scholar]
- Cunningham V. J.; Alswieleh A. M.; Thompson K. L.; Williams M.; Leggett G. J.; Armes S. P.; Musa O. M. Poly(glycerol monomethacrylate)-Poly(benzyl methacrylate) Diblock Copolymer Nanoparticles via RAFT Emulsion Polymerization: Synthesis, Characterization, and Interfacial Activity. Macromolecules 2014, 47 (16), 5613–5623. 10.1021/ma501140h. [DOI] [Google Scholar]
- Carlsson L.; Fall A.; Chaduc I.; Wågberg L.; Charleux B.; Malmström E.; D’Agosto F.; Lansalot M.; Carlmark A. Modification of cellulose model surfaces by cationic polymer latexes prepared by RAFT-mediated surfactant-free emulsion polymerization. Polym. Chem. 2014, 5 (20), 6076–6086. 10.1039/C4PY00675E. [DOI] [Google Scholar]
- Truong N. P.; Dussert M. V.; Whittaker M. R.; Quinn J. F.; Davis T. P. Rapid synthesis of ultrahigh molecular weight and low polydispersity polystyrene diblock copolymers by RAFT-mediated emulsion polymerization. Polym. Chem. 2015, 6 (20), 3865–3874. 10.1039/C5PY00166H. [DOI] [Google Scholar]
- Akpinar B.; Fielding L. A.; Cunningham V. J.; Ning Y.; Mykhaylyk O. O.; Fowler P. W.; Armes S. P. Determining the Effective Density and Stabilizer Layer Thickness of Sterically Stabilized Nanoparticles. Macromolecules 2016, 49 (14), 5160–5171. 10.1021/acs.macromol.6b00987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton F. L.; Ruda M.; Lansalot M.; D’Agosto F.; Malmström E.; Carlmark A. Xyloglucan-Functional Latex Particles via RAFT-Mediated Emulsion Polymerization for the Biomimetic Modification of Cellulose. Biomacromolecules 2016, 17 (4), 1414–1424. 10.1021/acs.biomac.6b00036. [DOI] [PubMed] [Google Scholar]
- Hatton F. L.; Lovett J. R.; Armes S. P. Synthesis of well-defined epoxy-functional spherical nanoparticles by RAFT aqueous emulsion polymerization. Polym. Chem. 2017, 8 (33), 4856–4868. 10.1039/C7PY01107E. [DOI] [Google Scholar]
- Engström J.; Hatton F. L.; Wågberg L.; D’Agosto F.; Lansalot M.; Malmström E.; Carlmark A. Soft and rigid core latex nanoparticles prepared by RAFT-mediated surfactant-free emulsion polymerization for cellulose modification - a comparative study. Polym. Chem. 2017, 8 (6), 1061–1073. 10.1039/C6PY01904H. [DOI] [Google Scholar]
- Gibson R. R.; Armes S. P.; Musa O. M.; Fernyhough A. End-group ionisation enables the use of poly(N-(2-methacryloyloxy)ethyl pyrrolidone) as an electrosteric stabiliser block for polymerisation-induced self-assembly in aqueous media. Polym. Chem. 2019, 10 (11), 1312–1323. 10.1039/C8PY01619D. [DOI] [Google Scholar]
- Boisse S.; Rieger J.; Belal K.; Di-Cicco A.; Beaunier P.; Li M. H.; Charleux B. Amphiphilic block copolymer nano-fibers via RAFT-mediated polymerization in aqueous dispersed system. Chem. Commun. 2010, 46 (11), 1950–1952. 10.1039/b923667h. [DOI] [PubMed] [Google Scholar]
- Boisse S.; Rieger J.; Pembouong G.; Beaunier P.; Charleux B. Influence of the Stirring Speed and CaCl2 Concentration on the Nano-Object Morphologies Obtained via RAFT-Mediated Aqueous Emulsion Polymerization in the Presence of a Water-Soluble macroRAFT Agent. J. Polym. Sci., Part A: Polym. Chem. 2011, 49 (15), 3346–3354. 10.1002/pola.24771. [DOI] [Google Scholar]
- Zhang X. W.; Boisse S.; Zhang W. J.; Beaunier P.; D’Agosto F.; Rieger J.; Charleux B. Well-Defined Amphiphilic Block Copolymers and Nano-objects Formed in Situ via RAFT-Mediated Aqueous Emulsion Polymerization. Macromolecules 2011, 44 (11), 4149–4158. 10.1021/ma2005926. [DOI] [Google Scholar]
- Zhang W. J.; D’Agosto F.; Boyron O.; Rieger J.; Charleux B. Toward Better Understanding of the Parameters that Lead to the Formation of Nonspherical Polystyrene Particles via RAFT-Mediated One-Pot Aqueous Emulsion Polymerization. Macromolecules 2012, 45 (10), 4075–4084. 10.1021/ma300596f. [DOI] [Google Scholar]
- Zhang W. J.; D’Agosto F.; Dugas P. Y.; Rieger J.; Charleux B. RAFT-mediated one-pot aqueous emulsion polymerization of methyl methacrylate in presence of poly(methacrylic acid-co-poly(ethylene oxide) methacrylate) trithiocarbonate macromolecular chain transfer agent. Polymer 2013, 54 (8), 2011–2019. 10.1016/j.polymer.2012.12.028. [DOI] [Google Scholar]
- Lesage de la Haye J.; Zhang X. W.; Chaduc I.; Brunel F.; Lansalot M.; D’Agosto F. The Effect of Hydrophile Topology in RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem., Int. Ed. 2016, 55 (11), 3739–3743. 10.1002/anie.201511159. [DOI] [PubMed] [Google Scholar]
- Pham B. T. T.; Nguyen D.; Huynh V. T.; Pan E. H.; Shirodkar-Robinson B.; Carey M.; Serelis A. K.; Warr G. G.; Davey T.; Such C. H.; Hawkett B. S. Aqueous Polymeric Hollow Particles as an Opacifier by Emulsion Polymerization Using Macro-RAFT Amphiphiles. Langmuir 2018, 34 (14), 4255–4263. 10.1021/acs.langmuir.7b03410. [DOI] [PubMed] [Google Scholar]
- Nguyen D.; Huynh V.; Pham N.; Pham B.; Serelis A.; Davey T.; Such C.; Hawkett B. SPION-Decorated Nanofibers by RAFT-Mediated Free Radical Emulsion Polymerization-Induced Self Assembly. Macromol. Rapid Commun. 2019, 40 (2), 1800402. 10.1002/marc.201800402. [DOI] [PubMed] [Google Scholar]
- Tan J.; Dai X.; Zhang Y.; Yu L.; Sun H.; Zhang L. Photoinitiated Polymerization-Induced Self-Assembly via Visible Light-Induced RAFT-Mediated Emulsion Polymerization. ACS Macro Lett. 2019, 8 (2), 205–212. 10.1021/acsmacrolett.9b00007. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Ryan A. J.; Armes S. P. Predictive Phase Diagrams for RAFT Aqueous Dispersion Polymerization: Effect of Block Copolymer Composition, Molecular Weight, and Copolymer Concentration. Macromolecules 2012, 45 (12), 5099–5107. 10.1021/ma301059r. [DOI] [Google Scholar]
- Warren N. J.; Armes S. P. Polymerization-Induced Self-Assembly of Block Copolymer Nano-objects via RAFT Aqueous Dispersion Polymerization. J. Am. Chem. Soc. 2014, 136 (29), 10174–10185. 10.1021/ja502843f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockram A. A.; Neal T. J.; Derry M. J.; Mykhaylyk O. O.; Williams N. S. J.; Murray M. W.; Emmett S. N.; Armes S. P. Effect of Monomer Solubility on the Evolution of Copolymer Morphology during Polymerization-Induced Self-Assembly in Aqueous Solution. Macromolecules 2017, 50 (3), 796–802. 10.1021/acs.macromol.6b02309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe L. P. D.; Ryan A. J.; Armes S. P. From a Water-Immiscible Monomer to Block Copolymer Nano-Objects via a One-Pot RAFT Aqueous Dispersion Polymerization Formulation. Macromolecules 2013, 46 (3), 769–777. 10.1021/ma301909w. [DOI] [Google Scholar]
- Hatton F. L.; Park A. M.; Zhang Y.; Fuchs G. D.; Ober C. K.; Armes S. P. Aqueous one-pot synthesis of epoxy-functional diblock copolymer worms from a single monomer: new anisotropic scaffolds for potential charge storage applications. Polym. Chem. 2019, 10 (2), 194–200. 10.1039/C8PY01427B. [DOI] [Google Scholar]
- Derry M. J.; Fielding L. A.; Warren N. J.; Mable C. J.; Smith A. J.; Mykhaylyk O. O.; Armes S. P. In situ small-angle X-ray scattering studies of sterically-stabilized diblock copolymer nanoparticles formed during polymerization-induced self-assembly in non-polar media. Chem. Sci. 2016, 7 (8), 5078–5090. 10.1039/C6SC01243D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan T.; Wacklin H.; Konovalov O.; Lund R. Recent applications of synchrotron radiation and neutrons in the study of soft matter. Crystallogr. Rev. 2017, 23 (3), 160–226. 10.1080/0889311X.2016.1277212. [DOI] [Google Scholar]
- Fürst C.; Zhang P.; Roth S. V.; Drechsler M.; Förster S. Self-assembly of block copolymers via micellar intermediate states into vesicles on time scales from milliseconds to days. Polymer 2016, 107, 434–444. 10.1016/j.polymer.2016.09.087. [DOI] [Google Scholar]
- Warren N. J.; Mykhaylyk O. O.; Ryan A. J.; Williams M.; Doussineau T.; Dugourd P.; Antoine R.; Portale G.; Armes S. P. Testing the Vesicular Morphology to Destruction: Birth and Death of Diblock Copolymer Vesicles Prepared via Polymerization-Induced Self-Assembly. J. Am. Chem. Soc. 2015, 137 (5), 1929–1937. 10.1021/ja511423m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alauhdin M.; Bennett T. M.; He G. P.; Bassett S. P.; Portale G.; Bras W.; Hermida-Merino D.; Howdle S. M. Monitoring morphology evolution within block copolymer microparticles during dispersion polymerisation in supercritical carbon dioxide: a high pressure SAXS study. Polym. Chem. 2019, 10 (7), 860–871. 10.1039/C8PY01578C. [DOI] [Google Scholar]
- Paulis M.; Bonnefond A.; Micusik M.; Leiza J. R. New agitated and thermostatized cell for in situ monitoring of fast reactions by synchrotron SAXS. J. Synchrotron Radiat. 2009, 16, 869–871. 10.1107/S0909049509036310. [DOI] [PubMed] [Google Scholar]
- A stirrable reaction cell has been recently utilized by Howdle and co-workers to perform SAXS studies during RAFT dispersion polymerizations conducted in supercriti-cal carbon dioxide in order to the evolution of internal nanostructure in diblock copolymer microparticles. See reference (53).
- Cockram A. A.; Bradley R. D.; Lynch S. A.; Fleming P. C. D.; Williams N. S. J.; Murray M. W.; Emmett S. N.; Armes S. P. Optimization of the high-throughput synthesis of multiblock copolymer nanoparticles in aqueous media via polymerization-induced self-assembly. React. Chem. Eng. 2018, 3 (5), 645–657. 10.1039/C8RE00066B. [DOI] [Google Scholar]
- The GPC trace obtained for this precursor block exhibits low molecular weight tailing. This feature is postulated to be a GPC artifact for this particular polymer/solvent combination. We emphasize that chain extension of this precursor block proceeds with high blocking efficiency to produce well-defined diblock copolymer chains that do not exhibit any low molecular weight tailing.
- One reviewer of this paper suggested that we should also examine the use of 2,2′-azobisisobutyronitrile (AIBN) for the RAFT aqueous emulsion polymerization of MOEMA. This nonionic azo initiator has a much lower aqueous solubility than ACVA, but it still enabled the synthesis of PGMA29-PMOEMA30 spheres (final MOEMA conversion = 95% after 2 h at 70 °C; Mn = 12 000 g mol–1, Mw/Mn = 1.14; DLS diameter = 15 nm (polydispersity = 0.11).
- Figg C. A.; Carmean R. N.; Bentz K. C.; Mukherjee S.; Savin D. A.; Sumerlin B. S. Tuning Hydrophobicity To Program Block Copolymer Assemblies from the Inside Out. Macromolecules 2017, 50 (3), 935–943. 10.1021/acs.macromol.6b02754. [DOI] [Google Scholar]
- Byard S. J.; Williams M.; McKenzie B. E.; Blanazs A.; Armes S. P. Preparation and Cross-Linking of All-Acrylamide Diblock Copolymer Nano-Objects via Polymerization-Induced Self-Assembly in Aqueous Solution. Macromolecules 2017, 50 (4), 1482–1493. 10.1021/acs.macromol.6b02643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornel E. J.; van Meurs S.; Smith T.; O’Hora P. S.; Armes S. P. In Situ Spectroscopic Studies of Highly Transparent Nanoparticle Dispersions Enable Assessment of Trithiocarbonate Chain-End Fidelity during RAFT Dispersion Polymerization in Nonpolar Media. J. Am. Chem. Soc. 2018, 140 (40), 12980–12988. 10.1021/jacs.8b07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanazs A.; Madsen J.; Battaglia G.; Ryan A. J.; Armes S. P. Mechanistic Insights for Block Copolymer Morphologies: How Do Worms Form Vesicles?. J. Am. Chem. Soc. 2011, 133 (41), 16581–16587. 10.1021/ja206301a. [DOI] [PubMed] [Google Scholar]
- Lopez-Oliva A. P.; Warren N. J.; Rajkumar A.; Mykhaylyk O. O.; Derry M. J.; Doncom K. E. B.; Rymaruk M. J.; Armes S. P. Polydimethylsiloxane-Based Diblock Copolymer Nano-objects Prepared in Nonpolar Media via RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2015, 48 (11), 3547–3555. 10.1021/acs.macromol.5b00576. [DOI] [Google Scholar]
- Glatter O.; Kratky O.. Small-angle X-ray Scattering; Academic Press: London, 1982. [Google Scholar]
- Pedersen J. Form factors of block copolymer micelles with spherical, ellipsoidal and cylindrical cores. J. Appl. Crystallogr. 2000, 33 (3), 637–640. 10.1107/S0021889899012248. [DOI] [Google Scholar]
- Bang J.; Jain S. M.; Li Z. B.; Lodge T. P.; Pedersen J. S.; Kesselman E.; Talmon Y. Sphere, cylinder, and vesicle nanoaggregates in poly (styrene-b-isoprene) diblock copolymer solutions. Macromolecules 2006, 39 (3), 1199–1208. 10.1021/ma052023+. [DOI] [Google Scholar]
- Porod G. Die Röntgenkleinwinkelstreuung von dichtgepackten kolloiden Systemen. Colloid Polym. Sci. 1951, 124 (2), 83–114. 10.1007/BF01512792. [DOI] [Google Scholar]
- Lindner P.; Zemb T.. Neutrons, X-rays and Light: Scattering Methods Applied to Soft Condensed Matter; Elsevier, 2002; pp 49–71. [Google Scholar]
- Tseng S. J.; Chien C. C.; Liao Z. X.; Chen H. H.; Kang Y. D.; Wang C. L.; Hwu Y.; Margaritondo G. Controlled hydrogel photopolymerization inside live systems by X-ray irradiation. Soft Matter 2012, 8 (5), 1420–1427. 10.1039/C1SM06682J. [DOI] [Google Scholar]
- Qiao M. H.; Yan F. Q.; Sim W. S.; Deng J. F.; Xu G. Q. X-ray-induced polymerization of furan overlayers on Ru(001). Surf. Sci. 2000, 460 (1–3), 67–73. 10.1016/S0039-6028(00)00492-1. [DOI] [Google Scholar]
- https://chem.nlm.nih.gov/chemidplus/rn/352-87-4.
- Goodwin J. W.; Hearn J.; Ho C. C.; Ottewill R. H. Studies on Preparation and Characterization of Monodisperse Polystyrene Lattices. 3. Preparation without Added Surface-Active Agents. Colloid Polym. Sci. 1974, 252 (6), 464–471. 10.1007/BF01554752. [DOI] [Google Scholar]
- Balmer J. A.; Mykhaylyk O. O.; Armes S. P.; Fairclough J. P. A.; Ryan A. J.; Gummel J.; Murray M. W.; Murray K. A.; Williams N. S. J. Time-Resolved Small-Angle X-ray Scattering Studies of Polymer-Silica Nanocomposite Particles: Initial Formation and Subsequent Silica Redistribution. J. Am. Chem. Soc. 2011, 133 (4), 826–837. 10.1021/ja106924t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.