

Abstract
NFU1 deficiency is a rare metabolic disorder affecting iron–sulfur cluster synthesis, an essential pathway for lipoic acid-dependent enzymatic activities and mitochondrial respiratory chain complexes. It is a little-known cause of pulmonary arterial hypertension (PAH), while PAH is a prominent feature of the disease. We herein report on a female infant diagnosed as having idiopathic PAH since 1 month of age, who did not respond to bosentan plus sildenafil. NFU1 deficiency was only suggested and confirmed at 10 months of age when she demonstrated neurological deterioration along with high glycine levels in body fluids. Unexplained PAH in early infancy should prompt clinicians to perform amino acid chromatography searching for high glycine levels. Early recognition will avoid further invasive procedures and enable appropriate genetic counseling to be offered. No effective treatment is currently able to prevent the fatal course of this metabolic condition.
Keywords: Hyperglycinemia, lipoic acid, neurological regression, NFU1, pulmonary hypertension
INTRODUCTION
Idiopathic pulmonary arterial hypertension (PAH) in early infancy is a severe condition, diagnosed when no other pediatric disease is found accounting for high pulmonary pressures. We herein report on an infant presenting with severe idiopathic PAH since the 1st month of life. The clinical evolution eventually prompted the clinicians to search for a rare genetic cause of PAH.
CLINICAL SUMMARY
This female infant was the first child of second-degree consanguineous Portuguese parents. She was born full term and had an uneventful neonatal period. At 1 month of age, she presented with respiratory deterioration, rapidly requiring intensive care. Chest X-ray revealed diffuse pulmonary infiltrates with consolidation of the right lung. She exhibited acute respiratory distress syndrome (ARDS), with Bordetella pertussis detected in the respiratory secretions by polymerase chain reaction (PCR) analysis. Echocardiogram identified suprasystemic pulmonary artery pressures. The right ventricular systolic pressure was estimated at 70 mmHg based on tricuspid valve insufficiency. Concomitant systemic blood pressure was 64/29 mmHg. The right ventricle (RV) was dilated, and the pulmonary artery (PA) was severely enlarged (15 mm = Z score + 6.6). Prostaglandin E1 was administered with unfavorable outcome, after which nitric oxide was applied, improving the pulmonary pressures, which normalized within 1 week. RV size returned to normal values, and the nitric oxide was stopped. PAH was considered to be secondary to pertussis-related ARDS.
At 3 months of age, she presented with a new acute deterioration, including metabolic acidosis, hypoxemia, and tachycardia. She exhibited failure to thrive. Echocardiogram again revealed severe pulmonary hypertension with a dilated and hypertrophied RV [Figure 1], squeezing the left ventricle [Figure 2]. Pulmonary pressure was evaluated on the pulmonary valve insufficiency in the absence of tricuspid regurgitation. Mean PA pressure was estimated around 60 mmHg [Figure 3], while systemic blood pressure was 90/55 mmHg. Cardiac catheterization in FiO2 0.3 demonstrated a mean pulmonary pressure of 22 mmHg and pulmonary vascular resistance of 10 Wood unit/m2, which represented 50% of the systemic vascular resistance. Acute vasoreactivity testing showed no change under oxygen FiO2 1.00 and nitric oxide 20 ppm [Table 1].
Figure 1.
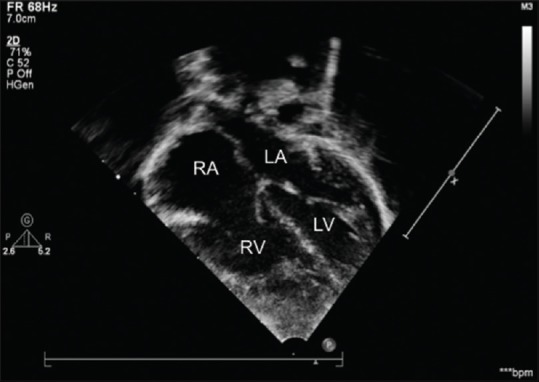
Apical four-chamber view showing a dilated heart and hypertrophied right ventricle. LA: Left atrium; RA: Right atrium; LV: Left ventricle; RV: Right ventricle
Figure 2.
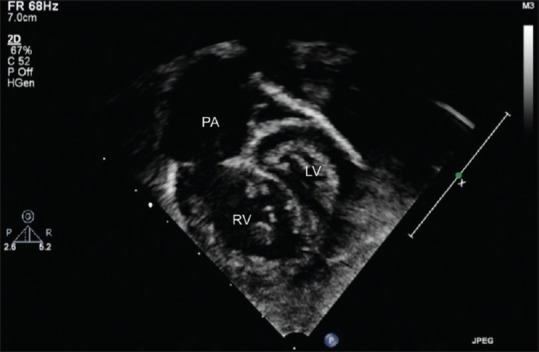
Subcostal view showing a dilated pulmonary artery and right ventricle, squeezing the left ventricle with systolic septal bowing into the left ventricle
Figure 3.
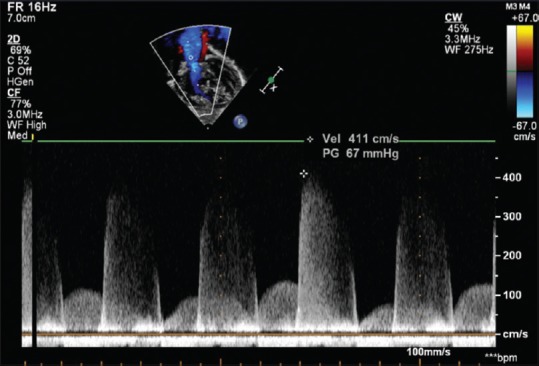
Continuous wave Doppler recording the high-velocity pulmonary regurgitation related to high pulmonary artery pressure
Table 1.
Catheterization parameters at baseline and with vasoreactivity testing
| FiO2 0.3 | FiO2 1.00 + NO 20 ppm | |
|---|---|---|
| PAP (mmHg) | 37/11 (22) | 36/10 (21) |
| SAP | 50/28 (42) | 50/29 (42) |
| PVR (Wood units ×m2) | 10 | 7.4 |
| SVR (Wood units ×m2) | 22 | 17 |
| PVR/SVR | 0.47 | 0.43 |
| Qp (L/min/m2) | 1.6 | 2.2 |
| Qp/Qs | 1 | 1 |
PAP: Pulmonary arterial pressure, SAP: Systemic arterial pressure, PVR: Pulmonary vascular resistances, SVR: Systemic vascular resistances, Qp: Pulmonary flow, Qs: Systemic flow, NO: Nitric oxide
Given the presence of transient lactic acidosis upon this episode, urine organic acids and the plasma acylcarnitine profile were assessed, yet with unremarkable results, while plasma amino acids were not investigated. The girl's condition improved in the next 2 days after fluid infusion and bicarbonate therapy were administered. Diagnostic workup ruled out associated congenital heart disease, chronic lung or liver disorders, infections (HIV), and thromboembolic PAH. Causes of inheritable PAH were investigated. Sequencing of BMPR2, ACVRL1, ENG, and KCNK3 genes identified no causal mutations. Array comparative genomic hybridization proved normal. The diagnosis of idiopathic PAH was retained, given that no underlying causal condition was found. Due to disease severity, specific PAH therapy was initiated, using a combination of sildenafil and bosentan, progressively increased to 30 mg/day (3 × 10 mg) and 20 mg/day (2 × 10 mg), respectively. In the following weeks, serial echocardiograms showed persistent pulmonary hypertension of systemic level, without significant ventricular dysfunction. Bosentan had to be stopped due to liver dysfunction. Treatment with intravenous epoprostenol was considered yet not administered.
At 9 months of age, the child suffered her first episode of refractory seizures. Clinical examination revealed neurological regression, along with axial hypotonia, spastic quadriplegia, and poor visual contact. Brain imaging revealed symmetric lesions in the white matter with necrotic areas [Figure 4]. Metabolic screening revealed high glycine levels in both the plasma (337 mmol/L; normal: 74–290) and urine (1198 mmol/mol creatinine; normal: 114–445), without lactic acidosis. In the following weeks, her neurological condition progressively deteriorated. She died at 10 months of age from neurological and metabolic decompensation. PCR amplification and sequencing of the 8 exons and parts of the flanking introns of the NFU1 gene identified the repeatedly reported[1] pathogenic homozygous mutation c. 622G>T (p. Gly208Cys) (RefSeq: NM_001002755.2). Both parents proved to be heterozygous carriers of the pathogenic variant.
Figure 4.
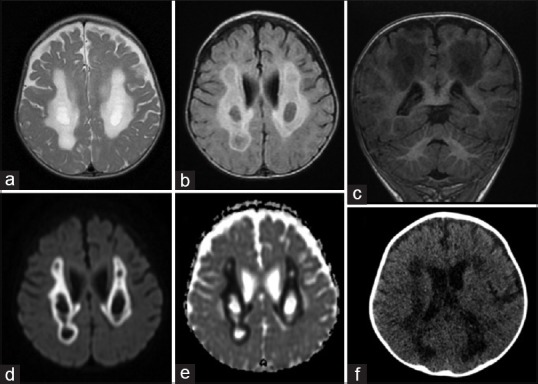
Brain MRI, Axial T2 (a) and Flair (b), coronal T1 (c), diffusion (d) and apparent diffusion coefficient (e) sequences; Computed tomography (CT) scanner (f). Images showing diffuse abnormal intensity of the white matter involving periventricular regions
DISCUSSION
Biogenesis of the iron–sulfur (Fe–S) clusters (ISCs) is a complex process that requires several proteins for their synthesis, assembly, maturation, and delivery to their target apoproteins.[2] Among the ISC machinery components currently associated with human diseases, the NFU1 gene (OMIM 60810) is particularly essential to targeting the (4Fe–4S) clusters into lipoic acid synthetase and succinate dehydrogenase (complex II of the mitochondrial respiratory chain), in a late-acting function of the protein maturation (Fe–S) pathway.[1] NFU1 deficiency (OMIM #605711), an autosomal recessive inherited disorder, is therefore responsible for decreased lipoic acid synthesis and impaired respiratory chain function. Lipoic acid is an essential cofactor for the lipoylation of the E2 subunit of four mitochondrial dehydrogenases: pyruvate dehydrogenase, alpha-ketoglutarate dehydrogenase, 2-oxoadipate dehydrogenase, and Branched chain keto acid dehydrogenase, as well as the H protein of the glycine cleavage system.[3] All these explain the biochemical hallmarks of NFU1-deficient patients,[1,2,3,4] namely high glycine levels in body fluids, variable lactic acidosis, and urinary excretion of Krebs cycle intermediates. Approximately 40 NFU1-deficient patients have previously been described, presenting a relatively homogeneous clinical outcome. Symptoms appear in the 1st month of life, with death generally followed before 2 years of age. Failure to thrive, PAH, psychomotor retardation, and neurological regression are the main clinical features. No patients with antenatal symptoms have been described. Brain magnetic resonance imaging typically reveals diffuse cavitating leukoencephalopathy. More than 50% of infant patients were found to display significant PAH,[2] which could be an isolated and prominent finding initially, as was the case for our patient. Interestingly, pulmonary samples from the NFU1-deficient individuals with PAH showed obstructive vasculopathy with proximal and acinar arterial involvement.[1] Pulmonary vascular remodeling is thought to be secondary to dysregulated endothelial energy metabolism,[5] though the pathophysiological mechanisms are not yet fully understood. It should be noted that the clinical spectrum of NFU1 deficiency is probably not fully known since Tonduti et al.[6] described a milder phenotype in an adult patient with spastic paraplegia.
While glycogen storage disease and Gaucher disease are metabolic disorders commonly associated with PAH, NFU1 deficiency is not mentioned among the metabolic disorders listed in the last updated clinical classification of pediatric pulmonary hypertension.[7] NFU1 deficiency should be considered in the differential diagnosis of pulmonary hypertension during infancy since its diagnosis can be rapidly established by amino and organic acid chromatography. Combined features of neurological deterioration, cavitating leukoencephalopathy, unexplained PAH, and hyperglycinemia are key findings that should prompt the clinician to perform NFU1 gene sequencing. Early recognition of this fatal multiple mitochondrial dysfunction syndrome should avoid a long diagnostic odyssey with useless molecular investigations and invasive procedures, while enabling adequate genetic counseling and prenatal testing to be offered.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, Del Toro M, et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–67. doi: 10.1016/j.ajhg.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tort F, Ferrer-Cortes X, Ribes A. Differential diagnosis of lipoic acid synthesis defects. J Inherit Metab Dis. 2016;39:781–93. doi: 10.1007/s10545-016-9975-4. [DOI] [PubMed] [Google Scholar]
- 3.Mayr JA, Feichtinger RG, Tort F, Ribes A, Sperl W. Lipoic acid biosynthesis defects. J Inherit Metab Dis. 2014;37:553–63. doi: 10.1007/s10545-014-9705-8. [DOI] [PubMed] [Google Scholar]
- 4.Ahting U, Mayr JA, Vanlander AV, Hardy SA, Santra S, Makowski C, et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front Genet. 2015;6:123. doi: 10.3389/fgene.2015.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Q, Chan SY. Mitochondrial and metabolic drivers of pulmonary vascular endothelial dysfunction in pulmonary hypertension. Adv Exp Med Biol. 2017;967:373–83. doi: 10.1007/978-3-319-63245-2_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tonduti D, Dorboz I, Imbard A, Slama A, Boutron A, Pichard S, et al. New spastic paraplegia phenotype associated to mutation of NFU1. Orphanet J Rare Dis. 2015;10:13. doi: 10.1186/s13023-015-0237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansmann G. Pulmonary hypertension in infants, children, and young adults. J Am Coll Cardiol. 2017;69:2551–69. doi: 10.1016/j.jacc.2017.03.575. [DOI] [PubMed] [Google Scholar]