

Abstract
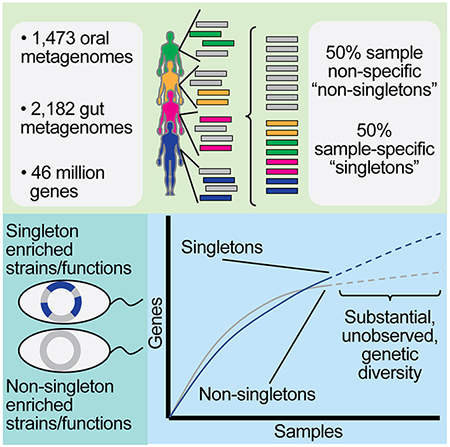
Despite substantial interest in the species diversity of the human microbiome and its role in disease, the scale of its genetic diversity, which is fundamental to deciphering human-microbe interactions, has not been quantified. Here, we conducted a cross-study meta-analysis of metagenomes from two human body niches, the mouth and gut, covering 3,655 samples from 13 studies. We found staggering genetic heterogeneity in the dataset, identifying a total of 45,666,334 non-redundant genes (23,961,508 oral, 22,254,436 gut) at the 95% identity level. 50% of all genes were “singletons”, or unique to a single metagenomic sample. Singletons were enriched for different functions (compared to non-singletons) and arose from sub-population specific microbial strains. Overall, these results provide potential bases for the unexplained heterogeneity observed in microbiome-derived human phenotypes. Based on these data we built a resource, which can be accessed at https://microbial-genes.bio.
Graphical Abstract
eTOC blurb:
Tierney et al. present a meta-analysis of metagenomes covering 3,655 samples from two body sites. They identify 45,666,334 non-redundant genes in the human oral and gut microbiome, with half of every person’s microbial gene content being completely unique. These rare genes, denoted singletons, predominantly arise from extremely rare microbial strains.
Introduction
Recent studies have made great strides in deepening our understanding of the strain-level diversity within the human gut microbiome, with the identification of 150,000 and 92,143 distinct microbial strains in two large meta-analyses since the beginning of 2019 alone (Almeida et al., 2019; Pasolli et al., 2019). Additionally, others have demonstrated the importance of minute gene-level variation across strains in human health and disease (Zeevi et al., 2019). However, the implications of these discoveries for the overall microbial gene content of the human microbiota remains unexplored. The field still does not have a grasp on the scope of the microbiome’s genetic content – in the gut and otherwise – a question crucial for understanding microbial function in the context of host disease (Sandoval-Motta et al., 2017).
The total number of distinct genetic elements within all prokaryotes is currently unknown, and theoretical estimates start at one billion genes(Wolf et al., 2016); (Lapierre and Gogarten, 2009) and range to maxima defined by permutations of nucleotide arrangements or thermodynamic stability in the context of protein folding(Lapierre and Gogarten, 2009). Specifically, in the human microbiome, most metagenomic analyses and methods that consider genes focus on core gene families(Lloyd-Price et al. 2017; Truong et al. 2015), where a core gene is defined as being present once, not a paralog, and more similar to its orthologs than any other gene in any other species(Young et al. 2006; Tettelin et al. 2005). Others have addressed metagenomic gene content by producing “gene catalogs,” the set of all genes identified via assembly across a large number of samples. Within the human gut microbiome, up to 10 million non-redundant genes have been identified by major sequencing consortiums using de novo approaches(Dusko Ehrlich and The MetaHIT Consortium, 2011; Forster et al., 2016; Li et al., 2014; Nielsen et al., 2014; Qin et al., 2010). These efforts have been almost exclusively associated with the gut microbiome, are relatively limited in terms of sample sizes, and do not focus on the overall rarity of genes across a population.
Moreover, there is a need to link our understanding of metagenomics back to that of traditional microbial genetics. Microbial genetic elements can be grouped into “pan-genomes,” which describe the set of all genes found in all strains of a particular species (Tettelin et al., 2005). The size of a pan-genome is most influenced by its effective population size and ability to migrate to new niches (McInerney et al., 2017). However, other intermittently present genes contribute significantly to the size and function of the pan-genome. In newly sequenced prokaryotic isolate genomes, up to a third of these genes have no detectable homologs in other species (Daubin and Ochman, 2004; Yin and Fischer, 2006). These “ORFans” are distinct from all open reading frames (ORFs) in the genome and are hypothesized to be neutral to selection pressure (i.e. ORFans are replaced at the natural rate of DNA uptake, recombination and loss) (Wolf et al., 2016). With an increasing emphasis in the field on the importance of strain-level variation in the gut microbiome,(Zhao et al. 2019), there is a need to identify the contribution of ORFan-like genes to overall metagenome gene content. We hypothesized, especially given the recent discoveries of massive strain diversity in the gut, that these genes would increase variation in gene content of the human microbiome.
Here, we sought to build a multi-body site microbiome gene catalog as a publically available resource for the scientific community. We further aimed to use this catalog to identify, and taxonomically and functionally document, the metagenomic analogs of ORFan genes. Then, with ORFans in mind, we attempted to determine the scale of sequencing that would be required to sufficiently sample the total genomic content – the universe of genes – of each niche, therefore building a “complete” gene catalog of the human microbiome.
Results
A pan-microbiome genetic database
Like prior gene catalog analyses, we utilized a de novo approach (as, by design, reference-based approaches only detect genes present in a reference database) to construct non-redundant microbiome gene-catalogs from publicly available short read data. We aggregated 2,183 samples from 6 gut microbiome studies. For the oral microbiome dataset, we retrieved 1,473 oral microbiome metagenomic samples from 7 studies, a cohort ~2x larger than the largest consortium effort to study this niche (Lloyd-Price et al., 2017). For a table of definitions used in this paper, please see Table 1.
Table 1:
Table of definitions used in the paper.
| Term | Definition |
|---|---|
| metagenome | total genomic potential of a microbial community (in this work, we use this term interchangeably with “sample” and “metagenomic sample”) |
| singleton gene | a gene detected in only one metagenomic sample across a defined collection of samples |
| non-singleton gene | a gene detected in more than one metagenomic sample across a defined collection of samples |
| ORFan gene | genes that have no detectable homologs in other species and are distinct from all open reading frames (ORFs) in the genome |
| universe of genes | the set of all non-redundant genetic elements across all communities of organisms in a given niche |
| gene rarefaction curve | a curve tracking the accumulation of new genes as samples are incrementally added |
| gene discovery curve | the derivative of the rarefaction curve (It estimates the rate at which new genes are added to the catalog when samples are added incrementally, and it can be used to estimate the size and burden of sampling of the universe of genes.) |
| singleton fraction curve | a curve estimating the fraction of a gene catalog that consists of singletons vs. non-singletons as samples are added incrementally (It is used to estimate the total number of samples that would be required for all singletons to be seen twice and thus no longer be singletons.) |
| mixture contig | A contig from de novo assembly consisting of both singletons and non-singletons |
| singleton contig | A contig from de novo assembly consisting of only singletons |
| non-singleton contig | A contig from de novo assembly consisting of only non-singletons |
We performed a meta-analysis of this aggregated metagenomic data, de novo assembling each metagenome (Fig. 1A–D, Supp Table 1). This analysis uncovered a universe of prokaryotic genes massive in scale. Extending existing approaches,(Li et al. 2014; Nielsen et al. 2014; Qin et al. 2012) we initially defined a unique gene as being distinct from all other ORFs at the 95%-identity level. Overall, we predicted 157,241,550 ORFs from the assembled oral data, compared to 136,672,846 from the gut data. Clustering at the 95% identity threshold, the initial oral and gut catalogs contained 23,961,508 and 22,254,436 consensus genes, respectively. When these oral and gut catalogs were clustered together at 95% identity, the resultant, non-redundant, catalog had 45,666,334 genes, as at this percent identity cutoff 549,610 ORFs overlapped (Fig 2A).
Figure 1: Meta-analysis of the oral and gut microbiomes.

A-B) We aggregated publically available oral and gut short read data and assembled it into contigs (in this example, each contig comes from a single sample). C) Gene open-reading-frames (ORFs) are identified on assembled contigs D) ORFs are clustered at 95% identity to identify a non-redundant gene catalog E) Database content, description of backend, description of UI F-K) Downstream singleton analytical pipeline. F) We identify singletons and non-singletons in our dataset and G) compare their functional annotations. H) We then map genes to contigs, which we group into 3 categories: singleton-contigs (those consisting of only singletons), non-singleton contigs (those consisting of only non-singletons) and mixture contigs (those consisting of both singletons and non-singletons). i) We filter short contigs and bin the remainder according to the taxonomic classification of their gene content. We then attempt to identify the source of singletons as either J) horizontal gene transfer (HGT) and/or K) rare, singleton-rich microbial strains.
Figure 2: The genetic diversity of the oral and gut microbiomes.

A) The overlap in genetic content (95% identity level) between the oral and gut microbiomes. B) Distribution of ORF cluster sizes at 95% identity in our oral (blue) and gut (red) gene catalogs. C) Iterative clustering of our amino acid gene catalogs. D) Distribution of gene cluster sizes for amino acid gene catalogs generated at the 50% identity level. E) Sorensen-Dice index measuring dissimilarity in gene content between all pairs of individuals. F) Sorensen-Dice dissimilarity of individuals in terms of MetaPhlAn2-derived species content.
Using this final catalog, which is replete with functional and taxonomic annotations, we built a publicly available and searchable PostgresQL database with an associated front-end that contains summary data (i.e. gene counts per body site, average gene length, number of genes in each consensus gene cluster, etc.) as well as information on our pipelines (Fig 1E). Our database has 2,418 different gene EC Numbers(Bairoch, 2000), 222,308 unique gene annotations and 15,746 NCBI taxonomies annotated within it. We additionally report consensus gene sequences and the number of genes in each 95% identity cluster. Finally, we also have made available for download MetaPhlAn2(Truong et al. 2015) output for each sample and all of the gene catalogs generated in the latter sections of this study.
The oral and gut microbiomes contain vast and individual-specific genetic content
We explored the reasons behind the substantial size of these gene catalogs. We hypothesized this effect was driven by the metagenomic equivalent of ORFan genes. As such, we sought to determine the frequency of occurrence of each gene on a sample-by-sample basis. Some genes assembled in multiple metagenomic samples (non-singletons), while other genes were found in exactly one sequencing sample (singletons). The oral gene catalog contained 11,891,670 (49.6%) singletons and 12,069,838 (50.4%) non-singletons, whereas the gut gene catalog contained 12,621,933 (56.7%) singletons and 9,632,503 (43.2%) non-singletons (Fig 2B). On average, 2.9% of the genes in each sample were singletons (standard deviation ∓3.5%).
We carried out substantial analysis on synthetic and real data with different assemblers and parameters to determine if singleton genes were artifacts of our analytic pipeline or false positive/short/low coverage genes. We found that singletons had modest associations with false positive genes, low coverage genes/contigs, short genes/contigs, or particular assemblers/assembly parameters relative to non-singletons. (Supp Table 2, Figs 1–3). We additionally sought to determine whether prior gene catalog analyses contained singletons, and found that the Metahit Integrated Gene Catalog,(Li et al., 2014) contained is 46% singletons (out of a total of 9.9 million genes [Supp Fig 3F]). Second, we tested whether singleton identification could be explained by low depth of sequencing. If that were universally true, singletons could be present in many samples just below the threshold of detection by assembly. We were unable to identify a strong correlation (Spearman correlation: 0.22, p<.05) between total read count and singleton gene count within a sample (Supp Fig 3G–J), implying depth alone is not driving singleton presence. Finally, to confirm the parameters for our choice of assembler, MEGAHIT, was supported by the literature, we reviewed every study (N = 99, 67 of which we had access to and were not dissertations/books) currently citing the MEGAHIT publication and determined similar projects used the same assembly settings (Supp Table 3).
We next relaxed the gene catalog clustering identity threshold to determine if ORFans/singletons were artifacts of high percent identities (Fig 2C). To circumvent computational limitations of clustering in nucleic acid space, we first translated the nucleic acid catalog to amino acids and lowered the clustering threshold from 100% amino acid identity to the limit of reasonable computational feasibility, 50% identity. While the catalog size shrank with the lower identity thresholds as expected, the fraction of singleton genes in the catalog remained approximately constant, particularly at lower percent identities, reflecting that the high proportion of singleton genes was not influenced by clustering thresholds. At 50% identity, the oral microbiome gene catalog contained 7,842,539 consensus genes, 3,255,115 (41.5%) of which were singletons (compared to 10,465,169 genes, 49.9% singletons, in the gut, Fig 2D).
Notably, while the oral gene catalog was larger at 95% nucleotide identity and contained more singletons, it was smaller than the gut catalog (and contained fewer singletons) at 50% identity, implying overall lesser overall sequence variation at low percent identities in the former than the latter. For the remainder of this manuscript, singleton and non-singleton genes will refer to those generated at the 50% clustering level, unless otherwise specified.
We next sought to determine if subjects (human hosts) with similar reference-based species content, which we identified using MetaPhlAn2, also had similar genetic content. We found this not to be the case. Using Sorensen-Dice dissimilarity (where 0 is identical and 1 is most dissimilar), we found that the human microbiome exhibits more inter-individual similarity of overall species content (mean Sorensen-Dice oral=0.43, mean Sorensen-Dice gut=0.60, Fig 2E) versus that of genes (mean Sorensen-Dice oral=0.85, mean Sorensen-Dice gut=0.95, Fig. 2F). Moreover, we found that most samples were equally dissimilar from each other, and the presence of singletons could not be explained by a few completely distinct samples in our dataset. Last, while genetic content varied between samples, singleton genes were evenly distributed throughout the sample population (Supp Fig 3G–H, Supp Table 1).
Singletons are functionally and taxonomically distinct from non-singletons
Further, we collapsed each gene annotated with EC numbers (Bairoch, 2000) from Prokka into Minpath (Ye and Doak, 2009) annotations. Overall, 12.8% of singletons in the mouth and 12.9% in the gut were functionally annotated by Prokka, compared to 36.7% of oral non-singletons and 34.6% of gut non-singletons (Fig 3A–B). While we were limited by relatively scant functional annotation information, we sought to test, using Sorensen-Dice dissimilarity, whether individual samples had, on average, the same pathways. We found this as well not to be the case (mean oral=0.43, mean gut=0.29, Supp Fig 4A).
Figure 3: The known and unknown functional diversity of the oral and gut microbiomes.

A-B) Fractions of genes functionally annotated in the oral and gut microbiomes. Genes labeled with pathway annotations were used in the Minpath analyses. C) Sorensen-dice dissimilarity of individuals in terms of overall pathway content.
We sought to taxonomically and functionally characterize the singletons that remained in the 50% identity amino acid catalog. We compared the enrichment of functional annotations across singleton and non-singleton genes (Fig 1F–G, 4, Supp Table 4). We found non-singletons and singletons to have little overlap in their functional diversity. In the top 50 most enriched minpath classes for gut and oral non-singletons, 27 overlapped, whereas only 9 of the top 50 oral and gut singleton enriched pathways did. Overall, non-singletons were enriched for primary metabolic processes, such as the Citric Acid Cycle and amino acid biosynthesis, whereas singletons were enriched for a wide range of diverse biosynthesis and degradation pathways.
Figure 4: Enrichment of functions in gut/oral niches for singletons/non-singletons.

Here we display the top 50 most enriched pathways for oral singletons (A), oral non-singletons (B), gut singletons (C), and gut non-singletons (D). Blue bars are pathways enriched in both oral and gut non-singletons, red bars are pathways enriched in both oral and gut singletons, and the green bar is a pathway enriched in both oral singletons and gut non-singletons.
In addition to a subset of singletons arising from genes with divergence greater than 50% identity, we hypothesized that singletons may arise from (1) horizontal gene transfer (HGT) or (2) extremely rare microbial species/strains, or some combination of the two. To test these hypotheses, we mapped genes back to their original contigs and classified contigs that arose exclusively from non-singletons (73M) (ie, only containing non-singletons), exclusively from singletons (2.5M), and contigs arising from both (1M, Fig 1H). 78.7% of singletons and 90.1% of non-singletons could be taxonomically annotated using NCBI’s refseq database (Supp Fig 4B–C). We grouped contigs (Fig 1I) using these gene-level annotations and searched the resulting groups for evidence of horizontal gene transfer and taxonomic variation between singleton and non-singleton contigs (Fig. 1J–K).
To test hypothesis (1) and screen for potential HGT, we searched for contigs consisting of both non-singletons and singletons where the non-singletons were annotated as coming from one species/genus and singletons were annotated to a different taxa/genus. We found that HGT did not contribute substantially to singleton presence. The genes on the contigs that were a mixture of singletons and non-singletons tended to emerge from the same species or genus. Only 8,557 (0.8%) of all mixture contigs in the oral microbiome contained potential cross-genus HGT. In the gut, there were 33,224 of these cross-genus, mixture contigs, a total of 1.8%.
Singletons arise from rare, sub-population specific bacterial strains
In testing our second hypothesis (highly uncommon microbial strains as the source of singletons), we identified differences in the taxa from which singleton-contigs and non-singleton-contigs originated. For each taxa, the singleton and non-singleton counts were in some cases modest, and we observed some rare taxa had more singleton than non-singleton contigs. The Pearson correlation between singleton-contig and non-singleton-contig counts for each taxa was 0.27 in the oral microbiome and 0.34 in the gut (Supp Table 5, Supp Fig. 4D–L). We sought to identify if the bias towards particular taxonomies was being driven by singletons arising from shorter contigs or contigs with fewer ORF’s. We found this not to be the case (Supp Fig 5, Supp Table 6). In total, we found that contigs with greater than one gene mapped to 2,071 and 2,476 species-level taxonomic annotations in the oral and gut microbiomes, respectively. Of these, 1,155 (55%) species in the mouth and 1,648 (67%) in the gut had more singleton than non-singleton contigs. We refer to these contigs as arising from “rare strains,” and from their presence concluded that hypothesis (2) was more likely than (1).
Having found that singletons were enriched in different taxa than non-singletons, we sought to test if singleton-only contigs came from subpopulation-specific strains. The alternative would be that species that contained singleton contigs were evenly distributed across the population. To test this, we compared the number of samples in which singleton and non-singleton contigs with given taxonomic annotations appeared. On average, in both the oral and gut microbiomes (Fig 5A), we found singletons-contig-derived taxonomic annotations in fewer samples (oral_mean=6.7, gut_mean=8.3, Wilcoxon test p<.05) than non-singletons (oral_mean=22.0, gut_mean=25.0, Wilcoxon test p<.05), demonstrating that singleton-enriched taxa are uncommon with respect to the entire population. We further tested to see if even when singleton and non-singleton contigs mapped to the same taxonomies, singleton-rich strains still arose from specific individuals or subpopulations (Fig 5B). We found this to be the case; for example, 28 singleton and 42 non-singleton contigs map to Eubacterium rectale, however the singleton contigs come from 1 individual, whereas the non-singleton contigs are from 39 different individuals (Fig 5C).
Figure 5: Singleton taxa as sub-population specific, rare strains.

A) Counts of taxonomic annotations for singleton and non-singleton contigs in the oral and gut microbiomes. B) Number of metagenomes singleton contigs and non-singleton contigs are present in for different taxonomies. Each point represents a different taxonomic annotation. C) Examples of strain-specific “fingerprints.” Each pair of rows corresponds to singleton and non-singleton contigs containing at least two genes that were binned into the same taxonomic annotation. Columns are different metagenomic samples (each corresponding to a different individual). Green boxes correspond to singleton contigs. Red boxes correspond to non-singleton contigs.
Estimating the burden of sequencing the human microbiome
Given the size and heterogeneity of our gene catalog, we sought to identify the amount of sequencing that would be required to capture the entire “universe of genes” in the oral and gut microbiomes. We used 10 rarefaction methods to estimate the rate of accumulation of unique genes at the 50% identity level (Fig 6, Supp Fig 6). We found imprecise estimates of the total gene content of the human microbiome. We claim this catalog is sampling between 8%-72% and 4%-50% of the total potential genetic richness of the gut and oral microbiomes, respectively. Assuming a constant rate of singleton accumulation with sampling, we estimate that to achieve a point where only 1% of genes per sample sequenced had not been seen before, we would need to sequence on the order of 5,698 samples in the oral microbiome and 23,530 samples in the gut. However, given the high variation associated with our extrapolations (which ranged from on the order of 40 million to 200 million) in Supplemental Figure 17, we emphasize that these estimates could be off by up to an order of magnitude. Furthermore, they are dependent on parameters that are challenging to optimize, such as gene sequence percent identity threshold. Therefore, we can only conclude that gene variation within the human microbiome is vast and deeply uncertain in scope despite the relatively large sample sizes of our meta-analysis.
Figure 6: Extrapolating the gene content of the human microbiome.

A-B) Extrapolation of the universe of genes using curves fit to our oral microbiome data (A) and gut microbiome data (B). Yellow dashed lines demarcate sampling required to observe certain percentages of new singletons per sample. Purpose dashed line marks size of this study. Green dashed line is the asymptotic number of genes in the oral microbiome. C-D) Alternative, more conservative extrapolation methods for estimating total gene content in the oral/gut niches.
Discussion
We have built a large, microbiome-gene database that incorporates multiple human body sites clustered at a range of percent identities. We built this resource with a focus on the variation of genomic content across the human microbiome, identifying an order of magnitude more genes in both the oral and gut microbiomes than ever before. We also identified singletons, which we propose are the metagenomic equivalent of ORFan genes. On analysis of our catalog, we find that the genetic richness in the human microbiome has been underestimated and undersampled, though estimating the degree and uncertainty of undersampling was nontrivial, despite this large collection of data.
In line with other recently published work (Almeida et al., 2019; Pasolli et al., 2019), our results indicate substantial strain-level diversity. For context, consider the following: suppose the average prokaryote has 5,000 genes(Land et al. 2015) and that 90% of genetic content is shared between genomes of a single species(Zhu et al. 2015). To explain the size of the 95% identity oral gene catalog (24 million genes), each of the 2,000 species we identified would require on the order of 20 sub-species/strains. If we were to only consider the 788 species identified by reference-based methods, each species would require on the order of 50 strains. Finally, outside of only showing diversity in strains, we were additionally able to show that strains rich in singletons can act as microbial fingerprints, tending to be unique to sub-populations within this dataset, and in some cases even individuals.
Questions remain regarding best analytic practices for de novo metagenomic studies, and, in the future, meta-analyses of metagenomic studies. Reference-genome-based approaches are superior to gene catalog analyses in terms of computational feasibility and interpretability; however, given the lack of observed correlation between taxonomy and genetic content, databases derived from primarily cultured isolates may lack many functionally important genes. As such, the successful biological interpretation of metagenomic findings is contingent upon building resources and databases with microbial genetic diversity in mind, considering both ORFans and otherwise.
We found that singleton genes are enriched in functionality for a variety of unrelated metabolic functions compared to non-singletons, which were enriched in more conserved bacterial processes. However, functions encoded by singletons are not irrelevant. We identified a number of pathways (e.g., antibiotic resistance, cell wall biosynthesis), that may impact both the structure of the microbiome and host health. The limited overlap in top enriched singleton functions between the oral and gut, compared to non-singletons, implies that singletons encode more niche-specific functions than non-singletons. As such, given the functional variety encoded within singleton genes, we propose that singletons form an evolutionary organ within the microbiome, one that can be leveraged by microbes to adapt readily to environmental conditions. It is possible that, analogously to recent work done in the field of human genetics (Wainschtein et al., 2019), ORFan genes may explain a large portion of the currently unexplained variation in microbiome-associated human disease states.(Sandoval-Motta et al., 2017) Recent work has demonstrated that this may be the case. For example, sub-population specific and intransient strains are associated with human disease and colonization (Zeevi et al., 2019).
The definition of a singleton gene states that it was only observed in a single sample Therefore, a gene could feasibly still be present in other samples in such low abundance that it cannot be identified via assembly. While computing relative abundance is fraught with the challenge of spurious alignments, especially in the case of low abundance genes, it could partially address this issue and is a reasonable future direction for this work. However, given the low correlation identified between sample depth and singleton presence (Supp Fig 3), we posit that forces other than undersampling are, at least in part, driving singleton presence in our data.
Overall, we have built a resource intended for studying gene-level variation across multiple human body sites and samples. We also showed that the gene landscape of the human microbiome is immense and that its heterogeneity across people is staggering. Moreover, we have quantified the need to increase sequencing efforts to fully explore both the oral and gut niches, as well as other body sites. Our findings imply that an order of magnitude more sequencing data (than currently exists) is necessary to sufficiently sample (with only 1% of genes being novel per metagenome) human microbiome sequence diversity and function at even the 50% identity level. That being said, clearly this estimation is immensely challenging due to both the variation in available modeling methods as well as the difficulty of sequence-identity-based microbial gene definitions (i.e. 95% vs 50% would yield vastly different results). It is also worth noting that despite large samples sizes, our cohorts are geographically constrained, with most of our data coming from European and American subjects. As such, future estimates human microbiome gene content will likely be further improved by capturing even greater geographic heterogeneity.
These results make a comprehensive genetic understanding of the human microbiome, or even a compilation of its non-redundant gene catalog, seem very challenging. However, with greater focus on de novo assembled genes, we can avoid oversimplified analytical approaches, such as those based exclusively on taxonomy. Additionally, using extrapolated data, we see a clear need to increase sample sizes in metagenomic studies to the orders of tens of thousands if we are to adequately sequence the “genome” of the human microbiome. Incorporation of these large-scale gene level analyses into currently existing technologies can add a genetic context to the meaning of microbial species, allowing for more meaningful studies rooted in microbial genetics. We hope the scientific community will be able to use the set of resources provided here to deepen the field’s understanding of the relationship between taxonomy and microbial genetic variation.
Star Methods
Contact for resource and reagent sharing
Further information and requests for resources, software, and data sharing should be directed to and will be fulfilled by the Lead Contact, Aleksandar Kostic (Aleksandar.Kostic@joslin.harvard.edu).
Method details
Overview of the approach
We aggregated 3,655 publically available oral and gut microbiome metagenomes used de novo assembly, Open-Reading-Frame (ORF) calling, and sequence-based clustering via CD-HIT(Li and Godzik, 2006) to identify a set of 45,666,334 non-redundant genes within them. We found 23,961,508 and 22,254,436 non-redundant genes in the oral and gut cavities, respectively. To enable access to our data by the broader research community, we built a public-facing interface, queryable PostgresQL database, and data repository hosted on Figshare.
To validate our gene-calling pipeline, we performed extensive analysis on synthetic and real data. Synthetic read data were generated with Art(Huang et al, 2012) from complete oral microbe isolate genomes downloaded from the Human Oral Microbiome Database. We assembled our synthetic metagenomes with metaSPAdes(Forouzan et al., 2018) and MEGAHIT running a variety of settings.(Li et al., 2015) We called ORF’s and checked the false discovery rate of each assembler as well as the correlation with genes of different prevalence (i.e. incidence in multiple samples or just one) with coverage and length of genes/contigs.
We quantified the distribution of genes across samples within our dataset, undertaking an in-depth analysis as to the number, frequency, taxonomic, and functional classifications of these genes. We quantified taxonomy by alignment to NCBI’s NR database with Diamond that had been indexed with NCBI’s taxon mapping files (available at ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/). Functional classifications were carried out as part of our ORF calling process, which leverages information from UniProt, Pfam, TIGRFAMs, and NCBI’s RefSeq to classify genes ab initio. We were able to identify 2,418 discrete pathway ECiDs (out of 222,308 unique gene annotations) as well as confidently map to 15,746 microbial NCBI ID’s. We have additionally made these results available as part of our resource, which can be searched by gene name, gene annotation, or taxonomy, if need be.
Further, we clustered our gene catalog at a variety of percent identities (down to 50% amino acid identity) to study the rate of clustering of different genes. We have made these available as downloadable links in our dataset as well.
We provide example scripts for each phase in our pipeline at https://github.com/kosticlab/universe_of_genes_scripts.
Synthetic data benchmarking of gene catalog pipeline
An outline of our methodological pipeline can be viewed in Supplementary Figure 1.
We attempted to address if singletons are likely to be false positive genes. Due to the gene-level focus in our data we do not analyze misassemblies, as contigs are an intermediate stage in our analysis. We felt it more prudent to search for predictors of success in our primary endpoints: genes. Our confidence in this analytic decision was further increased by our literature-review-based analysis yielding that MEGAHIT and metaSPAdes have been shown in other publications to yield equivalent numbers of misassemblies when compared head-to-head (see “Literature search for comparisons between MegaHit and other assemblers, including metaSPAdes” in the Star Methods).
This in mind, we carried out an extensive analysis of the performance of different assemblers at different levels of coverage on synthetic metagenomic data in order to answer four questions:
Is there a best assembler in terms of false discovery rate and singleton discovery at varying coverages?
Are low coverage contigs/genes usually false positives?
Are short contigs/genes usually false positives?
Can we identify optimal quality filtering parameters such that we minimize false positive genes and maximize true positive genes– a minimum contig length/coverage or gene length/coverage?
Aggregation of complete microbial genomes
We downloaded all 467 complete, circularized genomes, as well as their corresponding Open-Reading-Frame predictions, from the Human Oral Microbiome Database (www.homd.org).
Relevant code: synthetic_data_benchmarking/download_homd_data.py
Construction of synthetic read data
We ran Art(Huang et al., 2012) to create synthetic read data at 1X coverage for each genome (parameters: art_illumina -ss HS25 -sam -i input_genome -p -l 150 -f 1 -m 200 -s 10 -o paired_dat, where input_genome is a fasta file containing the complete genome assembly). These are the recommended parameters in Art’s README for generating synthetic Illumina sequencing data.
Relevant code, for example parameters: synthetic_data_benchmarking/art_parameters_example.sh
Construction of synthetic metagenomes
We found from our MetaPhlAn2 output, on average, there were 95 species per sample in our oral microbiome data. As such, we randomly picked 95 of the 467 genomes to be combined into a synthetic metagenome at varying levels of coverage. We randomly selected a value X (where X > 0) between a specific coverage range for each of the 95 metagenomes. If the value were greater than 1, we combined X copies of the 1X coverage synthetic read file for that genome into the metagenome. If it were less than 1, we subsampled that fraction of reads from each of the fastq files using seqtk (parameters: -s X)
Relevant code: synthetic_data_benchmarking/run_synthetic_data_modeling.py.
Assembly and gene calling parameters
We assembled with the following parameters:
-
MEGAHIT (parameter descriptions taken from http://www.metagenomics.wiki/tools/assembly/megahit):
meta ‘--min-count 2 --k-list 21,41,61,81,99’ (generic metagenomes, default)
meta-sensitive ‘--min-count 2 --k-list 21,31,41,51,61,71,81,91,99’ (more sensitive but slower)
meta-large ‘--min-count 2 --k-list 27,37,47,57,67,77,87’ (large & complex metagenomes, like soil)
-
metaSPAdes (used default parameters):
metaSPAdes.py -1 synthetic_metagenome_1.fq.gz -2 synthetic_metagenome_2.fq.gz --only-assembler -o output
-
Prokka
prokka --outdir prokka_output --addgenes --metagenome --cpus 0 --mincontiglen 1 assembly_output
Relevant code: synthetic_data_benchmarking/run_synthetic_data_modeling.py.
Coverage ranges
We ran our pipeline at three coverage ranges. Each of the 95 organisms in each metagenome was added to said metagenome at a level of coverage within a specific range. In the recent Pasolli et al. used a minimum coverage cutoff of 10X for genome extraction from metagenomic data, they – S s such, we chose to test coverage ranges centered around this value. We performed 10 iterations (i.e. generated 10 synthetic metagenomes) for each range. The ranges we chose were low coverage (0-1X), low-medium coverage (0-10X) and medium-high coverage (10-20X).
Identification of false positive genes
We identified false positive predicted genes by aligning our predicted genes back to the Open-Reading-Frames found in the 95 complete genomes we initially put into the synthetic metagenome. We aligned with Diamond(Buchfink et al., 2015) (additional parameters: --max-target-seqs 1 --id .95). Genes in the predicted gene set that did not align to the “ground truth” genes were marked as false positives.
Relevant code: synthetic_data_benchmarking/run_synthetic_data_modeling.py.
Computing coverage for each gene/contig
We computed coverage of each gene/contig by aligning raw reads back to the predicted gene/contig output files, respectively, using BBMap (parameters: bbmap/bbwrap.sh ref=$reference in=$f1 in2=$f2 out=output kfilter=22 subfilter=15 maxindel=80). We based these parameters based on those recommended in the MEGAHIT wiki for computing contig coverage (https://github.com/voutcn/megahit/wiki/An-example-of-real-assembly). We computed average coverage per contig/gene, as well as average percent of contig/gene covered.
Relevant code: synthetic_data_benchmarking/run_synthetic_data_modeling.py, synthetic_data_benchmarking/compute_gene_contig_coverage.sh.
Summary-level analysis
We carried out linear regression and correlational analyses on summary-level data, which consisted of averaged statistics across all the genes/contigs . We computed average false discovery rate for a given iteration/coverage level, average contig/gene coverage (total percent covered as well as fold coverage), and average contig/gene length. Using base-R’s glm function, we ran the regression
False discovery rate ~ Assembler type, where Assembler Type is a categorical variable consisting of the four different assembly parameters we used. Further, we used the stat_comp function from the ggpubr package to compute correlations false discovery rate and gene/contig fold coverage/length.
Relevant code: statistical_analysis_and_figures/summary_data_analysis.R
Gene-by-gene false positive analysis
We used base-R’s glm function to run the following two logistic regressions, using contig-level and gene-level summary statistics, respectively:
False positive gene-Gene length (per 1sd) + Gene avg fold coverage (per 1sd) + Assembler type + Genome coverage range
False positive gene ~ Contig length (per 1sd) + Contig avg fold coverage (per 1sd) + Assembler type + Genome coverage range
We computed two different regressions to avoid including highly correlated variables (gene length/coverage and contig length/coverage) in the same model.
false_positive = a given gene is a false positive (1) or a true positive (0)
fold_coverage_contig = the fold coverage for the contig a particular gene arose from
fold_coverage_gene = the fold of coverage for a gen
length_contig = the length of a contig a particular gene arose from
length_gene = the length of a gene
assembler_type = which of the 4 assembly parameters were used (megahit large, megahit sensitive, megahit default, metaSPAdes)
coverage_range = which coverage range a given gene came from (0 to 1, 0 to 10, 10 to 20)
We plotted the distributions of gene/contig length/coverage by false positive/singleton status in Supplementary Figure 2. Judging from the relationships displayed in these distributions, we hypothesized that shorter genes would have a higher probability of being a false positive.
Total number of false positive genes assembled by MEGAHIT default was 514,446 (15.3%). Gene length ranged from 61 to 2,448 bases with a mean of 267.2 and a median of 243.0. Contig length ranged from 64 to 5,905 bases with a mean of 480.4 and a median of 430.4. Gene average fold coverage ranged from 0 to 3722.385 reads with a mean of 23.458 and a median of 19.609. Contig average fold coverage ranged from 0 to 3646.935 reads with a mean of 23.185 and a median of 20.397. In order to aid in interpretability of our analysis, we normalized each of these variables by their standard deviations for each regression they were used in (gene length SD: 123.22 bases, contig length SD: 202.70 bases, gene average fold coverage SD: 21.16 reads, contig average fold coverage SD: 19.03 reads). By doing this, our odds ratios could be interpreted as change in odds for a gene being a false positive given a 1 standard deviation change in length/coverage.
Relevant code: statistical_analysis_and_figures/gene_by_gene_synthetic_analysis.R
Clustering and identification of singleton genes
We additionally clustered all the metagenomes within each assembler parameter/type group (so across all coverage ranges) into four separate non-redundant gene catalogs, so we could identify how singleton status of a given gene associated with coverage statistics and assembly method. To do so, we grouped all 10 iterations within a given coverage range and used CD-HIT to cluster the genes therein (parameters: cdhit/cd-hit -n 3 -i all_genes_for_cdhit -T 0 -M 0 -s .9 -aS .9 -c .5 -o cdhit_output_50perc) at the 50% identity level. We chose 50% identity to mimic the analysis that we had done in much of the paper.
Relevant code: synthetic_data_benchmarking/run_synthetic_cdhit_analysis.py
Gene-by-gene singleton analysis
We used base-R’s glm function to run the following two logistic regressions, using contig-level and gene-level summary statistics, respectively:
Singleton gene ~ False positive gene + Gene length (per 1sd) + Gene avg fold coverage (per 1sd) + Assembler type + Genome coverage range
Singleton gene ~ False positive gene + Contig length (per 1sd) + Contig avg fold coverage (per 1sd) + Assembler type + Genome coverage range
The parameter definitions are the same as above with the addition of singleton_status, which refers to if, after clustering, a gene was a singleton (1) or a non-singleton (0). We computed area under the curve (AUC) estimates using using the roc function in the pROC package.(Robin et al. 2011)
Relevant code:statistical_analysis_and_figures/gene_by_gene_synthetic_singleton_analysis.R
Benchmarking of gene catalog pipeline on real data
Modeling singleton gene status and oral microbiome contig coverage/length, gene length, and read counts
We used the following regressions to find associations between contig coverage/gene length/contig length/depth of sequencing and singleton status in our oral microbiome data. We computed AUC estimates using using the roc function in the pROC package.(Robin et al. 2011)
Singleton gene ~ Gene length (per 1sd) + Total reads (per 1sd)
Singleton gene ~ Contig length (per 1sd) + Contig avg fold coverage (per 1sd) + Total reads (per 1sd)
One drawback of the synthetic data analysis is that due to small sample size compared to our actual study, the singleton gene fraction was higher than we would have expected (i.e. some non-singletons may have been classified as singletons). As such, we modeled our real data as well. In this case, we lack information on true/false positive genes, but we have larger sample sizes and a lower overall fraction of singleton genes.
We used bbMap once again to compute contig-by-contig coverage for each predicted element that was identified by PROKKA. For this analysis, we opted initially not to compute gene-by-gene coverage (or contig-by-contig coverage for the gut microbiome) due to the 1) additional time and monetary cost that would be required and 2) the similarity between the gene/contig results in the synthetic data analysis.
Given the similar distribution of singleton/non-singleton genes in association with our independent variables of interest (SuppSupp. Fig. 3), we hypothesized our regressions would yield small effect sizes and minimal changes in the probability of a gene being a singleton compared to baseline. We found modest in effect size but statistically significant associations between singleton genes and coverage of the contig from which a gene came, gene length, and contig length (SuppSupp. Table 2). The total number of singletons was 3,183,181 (2.0%). Contig length ranged from 200 to 1,041,740 base pairs with a mean of 1,343 and a median of 2,390. Contig coverage, in terms of average number of reads aligning to each base of a contig, ranged from 0 to 98,048.39 reads with a mean of 16.58 and a median of 5,910. Gene length ranged from 66 to 31,656 with a mean of 671 and a median of 513. As with our synthetic data analysis, in our regressions we normalized each continuous variable by its standard deviation (Gene length SD: 547.15 bases, Contig length SD: 40,924.06 bases, Contig average fold coverage SD: 59.10 reads, Total reads SD: 83,030,393 reads).
Relevant code: statistical_analysis_and_figures/gene_by_gene_singleton_analysis_real_data_oral.R
Comparison between MEGAHIT and metaSPAdes in identification of singleton genes
We ran our assembly, gene calling, and gene catalog construction pipeline on a subset of 10 randomly selected samples, computing gene-by-gene and contig-by-contig coverage (as above) and identifying singletons. We computed AUC estimates using using the roc function in the pROC package.(Robin et al. 2011)We ran the following regressions for our analysis:
Singleton gene ~ Assembler type
Relevant code: statistical_analysis_and_figures/gene_by_gene_metaspades_megahit_real.R
Meta-analytic data collection
We identified 13 publications (Supplementary Table 1) with shotgun sequencing metagenomic data taken from any human oral and gut microbiomes. We used 2,182 gut samples and 1,473 oral samples. We downloaded relevant study data from either the National Center for Biotechnology Information (NCBI), the European Bioinformatics Institute (EBI), the metagenomics RAST server (MG-RAST), or the Human Oral Microbiome Database (HOMD).
Raw read filtering and quality control
If reads had not been trimmed or had human sequences filtered out in their respective studies, we used KneadData(https://bitbucket.org/biobakery/humann2/wiki/Home) to do so prior to assembly. This pipeline involves two primary steps. 1) Aligning raw reads back to the human genome reference (GRCh37/hg19) to filter out human contaminants (settings: --very-sensitive). 2) Using Trimmomatic to remove adapter contamination (settings: SLIDINGWINDOW 4:20, MINLEN 50).
Open-reading-frame prediction and initial functional annotation
We ran Prokka(Seemann, 2014) (version: 1.12, settings: --metagenome --addgenes --mincontiglen 1) on the raw contigs from our de novo assembly to predict genes.
Assembly, gene calling, and construction of non-redundant gene catalogue
We assembled raw reads into contiguous sequences (or, contigs) using MEGAHIT(Li et al., 2016) V1.1.2 (parameters: --default --mem-flag 2). We removed contigs under 200 base pairs in length. We used Prokka(Seemann, 2014) V1.12 to annotate genes from the MEGAHIT output (settings: --cpus 0 --addgenes -metagenome --mincontiglen 1). We used the default databases installed with Prokka (UniProt, Pfam, TIGRFAMs, and NCBI’s RefSeq) for functional annotation. We then ran CD-HIT-EST(Li and Godzik, 2006) V4.6.8 with a 95% identity cutoff (--n 10 --c .95 -aS .9 -S .9 -M 0 -T 0). We removed genes under 100 bases in length that did not align to any sequence NR reference database at 95% identity. For any other gene catalogs we made we either used CD-HIT or CD-HIT-EST with varying percent identity and word length (according to the instructions in the CD-HIT user’s manual https://github.com/weizhongli/cdhit/blob/master/doc/cdhit-user-guide.pdf)
Iterative gene catalogue construction
We translated our nucleic acid gene catalog into amino acids with Python’s Biopython(Cock et al., 2009) package. We ran CD-HIT V4.6.8 with the same parameters as above on the translated gene catalog with progressively lower percent identities, starting at 100% and decreasing in increments of 5 down to 50%. For example, we fed the output of CD-HIT run at 100% identity into another CD-HIT run with the -c flag changed to .95, the output of which was run through CD-HIT again at -c .9, and so on.
Relevant code: gene_catalog_construction/iterative_cdhit.sh, gene_catalog_construction/parse_iterative_cdhit.py
Reference-based species identification
We ran MetaPhlAn2(Truong et al., 2015) V2.1.0 with the default settings to identify the species content in each sample. To create incidence data from the MetaPhlAn output, which we used to in our cross-sample dissimilarity calculations, we collapsed the raw output into a relative abundance matrix, where the columns were samples and the rows were species. We then created an incidence matrix by recoding non-zero cells as having values of 1.
Calculation of cross-sample dissimilarity
Similarity metrics were calculated using Sorenson-Dice(Sørensen, 1948) similarity, which is simply Bray-Curtis Dissimilarity applied to prevalence rather than abundance data. To speed up data processing, we used a custom, parallelized, c++ implementation.
Relevant code: gene_catalog_construction/sorensen.cpp
MinPath annotation
We ran MinPath(Ye and Doak, 2009) V1.4 (command: python ../MinPath1.2.py -any ecid_mapping -map ec2path -report ec.report -details ec.details) on the set of all EcID’s captured in each gene catalog to identify a mapping between gene, EcID, and parsimonious pathway annotation.
Functional enrichment analysis
We identified pathways enriched in singletons or non-singletons for the gut and oral microbiomes using a Fisher’s Exact test, where we compared the ratio of counts of singletons and non-singletons of any given pathway to the overall ratio of singletons to non-singletons across all populations. We adjusted for False-Discovery Rate using Benjamini-Yekutieli(Benjamini and Yekutieli, 2001) correction. For the plots in Figure 4, we reported the top 50 most enriched pathways in the gut microbiome and oral microbiome for singletons and non-singletons. For the plot in Supplementary Figures 13-14, we show the top 25 most enriched species/genera using the same methods.
Relevant code: statistical_analysis_and_figures/orfleton_figures_both.Rmd
Gene-level taxonomic annotation
We used Diamond’s(Buchfink et al., 2015) taxonomic annotation configuration (which uses NCBI’s taxon nodes and taxonmap files in conjunction with the Lowest Common Ancestor algorithm) to align against NCBI’s RefSeq non-redundant protein database, which we downloaded from ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz. After combining the separate files, we configured the diamond database with the command “diamond makedb --in nr.gz --db nr --taxonnodes nodes.dmp --taxonmap prot.accession2taxid.gz ” We used Diamond’s default cutoff, a minimum e-value of 0.0001, to identify confident hits.
Relevant code: gene_catalog_construction/full_contig_parsing_and_singleton_hunting_pipeline.py
Gene-based taxon contig binning
We mapped genes onto the contigs from which they originated, and we binned contigs into a particular taxonomic group if at least 75% of the genes on a given contig had the same taxonomic annotation. To increase our confidence in our annotations, we filtered out contigs with fewer than two genes on them, as well as those that were binned at a taxonomic level above genus (kingdom, phylum, class, order, family levels).
Relevant code: contig_analysis/build_contig_database.py, contig_analysis/bin_contigs_species.py
Identification of horizontal gene transfer
We tested of horizontal gene transfer was noticeably giving rise to singletons by examining in mixture-contigs, those consisting of both non-singletons and singletons. We searched for where the discordant species or genus taxonomic annotations of singletons(s) and non-singleton(s), excluding those annotations that were unable to be ascribed to any specific microbe. Out of concern of being biased by low resolution annotations, we only classified HGT as possibly having occurred when the taxonomic bins of the singleton(s) and the non-singleton(s) were of different genera.
Relevant code: contig_analysis/bin_contigs_species.py
Preparing data for functional and taxonomic enrichment scatterplots
In order to display enrichment within singletons and non-singletons (Supplementary Figure 10B,F) while accounting for sample size, prior to plotting we normalized the counts of number of genes/contigs in particular pathway/taxa by dividing by the total number of singletons or non-singletons for a given niche. For example, if 10 singletons and 10 non-singletons aligned to a pathway and a total of 100 singletons and 10000 non-singletons were found to align to any pathway at all, we divided 10/100 and 10/10000 to get normalized values of .1 and .001, respectively, for said pathway.
Construction of disaggregated sample-based rarefaction curves
We create an R-by-S disaggregated sample-based rarefaction matrix D (where drs is the expected number of r-tons when s samples are drawn from a set of S samples), starting from a binary G-by-S incidence matrix W (where wgs = 1 if the gene g was found in sample s). The rarefaction curve can be solved analytically using hypergeometric distributions, and depends only on the frequency in which each gene is found(Ugland et al., 2003).
First, we calculate the frequency yg that gene g appears in all the samples, as the row sum of the incidence matrix W(1), then calculate the incidence frequency count qk(2) where qk is the number of times that genes appear k times in the sample S, and l() = 1 if its argument is true.
| 1) |
| 2) |
Second, let R denote the maximum value of k where qk ≠ 0. Then, the expected number of r-tons accumulated after s random samples collected without replacement dr,s is calculated as follows:
| 3) |
where the hypergeometric function (4), h(s, S, r, k) returns the probability of drawing exactly r out of k possible units when sampling s times without replacement out of a set S. For example, suppose we have a collection of 20 samples. The probability of finding exactly 3 incidences in a 10-ton set if we choose at random 12 of the 20 samples, is h(12, 20, 3, 10) and is calculated using binomial coefficients as follows:
| 4) |
Finally, the r-ton sample-based rarefaction curves are plotted from the rth line of D using the Python matplotlib(Hunter, 2007) package. The aggregated sample-based rarefaction curve is the expected number of unique genes when s samples are collected without replacement:
| 5) |
Creation of the gene discovery curve from the rarefaction curve
The gene discovery curve Q is the derivative of the rarefaction curve, where qs is the number of new genes discovered on sample s:
| 6) |
Determining a fitness function for the gene discovery curve
The gene discovery curve qs from (6) is used to extrapolate gene richness in the microbiome pangenome by polynomially fitting qs.
We used the function curve_fit from the Python package scipy.optimize to fit the discovery curve to a function. Because the discovery curve is derived from a rarefaction curve, the chosen function must, on the positive x-axis, be continuous, non-increasing, and convex. Further, as there are combinatorial limits on genes, the function must asymptotically reach 0. Finally, it must fit qs with high-fidelity, especially at the right tail, in order to get the best estimator. As none of the usual eligible candidate functions (e.g. negative exponential, negative power curves) adequately fit the discovery curve at the right tail, it was necessary to select our own.
As the qs appeared curved in logarithmic space, we applied a 2nd degree polynomial regression on logarithm-transformed data (i.e. fitting a curve after remapping the axes to and x =log log s :
| 7) |
where a, b, and c are the regression parameters. Note that if a = 0, the fitting function becomes a power curve.
Determining the marginal sample s that yields a maximum number or percentage of new genes
We determined the marginal sample s required to contain less than a fraction of new genes zfrac. As f(s, a, b, c) is not trivial to invert in logarithmic space, we use the root function from scipy.optimize in Python to solve for s:
| 8) |
Similarly, the same root finding algorithm is used to find the marginal sample s that yields less than znum new genes, by solving:
| 9) |
Relevant code: extrapolation/RollingSpeciesEstimator.r, extrapolation/species_estimator.py
Gene richness estimation of oral/gut microbiome pangenomes
As the area under the gene discovery curve function is finite for a< 0 and s > 0, we integrate to extrapolate the rarefaction curve for an arbitrary value of s, by using the scipy.integrate function from Python. The richness of oral and gut genes asymptotically reaches 91,439,476 and 238,585,237 genes, respectively, when s = infinity.
Relevant code: extrapolation/RollingSpeciesEstimator.r, extrapolation/species_estimator.py
Estimating the number of singletons in the extrapolated rarefaction curve
A function that extrapolates the number of singletons as a function of samples collected must meet certain properties: the function must be continuous for s > 0, represent a non-increasing fraction of the rarefaction curve, ideally be the same value as the rarefaction curve at s = 1, asymptotically reach zero, and fit d1,s with high-fidelity, especially at the right tail, in order to get the best estimator. As none of the functions that we attempted fit the above criteria, we decided to regress the fraction of singletons in logarithmic space to the fitting function (7) while setting a = 0, then multiply it with the extrapolated rarefaction curve.
Relevant code: extrapolation/RollingSpeciesEstimator.r, extrapolation/species_estimator.py
Gene richness estimation of oral microbiome pangenome via the Chao2 and Chao-Bunge estimator
From the disaggregated rarefaction curve D, we estimate the gene richness of the oral microbiome by using estimators available in the SPECIES R package. Rolling estimates using Chao2, Chao-Bunge, Jackknife, and Chao-Lee were produced as samples were collected.
Relevant code: extrapolation/RollingSpeciesEstimator.r, extrapolation/species_estimator.py
Cloud Computing
All analyses were carried out entirely in the cloud on a combination of Amazon Web Services (AWS) and Microsoft Azure resources. We ran our initial assemblies on AWS spot instances using Aether(Luber and Tierney et al., 2017) and stored the resulting data on Azure’s cloud storage. We used Azure, Linux-based virtual machines running Ubuntu 16.04 for the remainder of our analyses.
Figure generation
All plotting, except for that done for the rarefaction curves, was done in R using the packages “ggplot2” and “cowplot” (https://cran.r-project.org/web/packages/cowplot/index.html). Rarefaction analysis and extrapolation was done using Python’s “Matplotlib”(Hunter, 2007) package. Figures were assembled in Adobe Illustrator (https://www.adobe.com/products/illustrator.html).
Quantification and statistical analysis
We used Fisher’s exact tests, linear, and logistic regression to quantify associations between various covariates across the manuscript. We controlled for multiple hypothesis testing with Benjamini-Yekutieli p-value adjustment.
Data and software availability
Example scripts for each step of this analytical pipeline are publicly accessible at https://github.com/kosticlab/universe_of_genes_scripts of genes scripts. When relevant, each section of the methods section below refers to script in this repository used for that particular analysis. The post-assembly pipeline, which includes non-redundant gene catalog construction, gene catalog quality control, gene-level taxonomy mapping, iterative gene catalog construction, binary gene incidence matrix generation, and Sorenson-Dice dissimilarity calculation, is run by “gene_catalog_construction/full_contig_parsing_and_singleton_hunting_pipeline.py”. We built our public facing database using Microsoft Azure’s Database for PostgreSQL service. We built our website with a Flask API..
Additional resources
We additionally present a database of our results at https://microbial-genes.bio.
Supplementary Material
Supplementary Table 1, related to Figures 1 and 2: Summaries of data used for meta-analysis, download links for all datasets, and sample-by-sample summary statistics/assembly quality metrics.
Supplementary Table 2, related to STAR methods: Regression output from synthetic and real data analysis intended to determine the validity of singleton genes.
Supplementary Table 3, related to STAR methods: Literature review of Megahit parameters. At the time of writing searched the methods sections of 110 (99 distinct, 67 that we had access to and were not books or dissertations) publications that cited Megahit on Google Scholar and, when possible, recorded the parameters they used. Each publication is fully referenced in the works cited list.
Supplementary Table 4, related to Figure 4: Functional enrichment, related to Figure 4. Contains raw data regarding functions enriched in oral singletons/non-singletons and gut singletons/non-singletons.
Supplementary Table 5, related to Figure 5: Taxonomic enrichment of singletons/non-singletons, related to Figure 5 and Supplementary Figure 5. Contains raw output regarding strains enriched in oral singletons/non-singletons and gut singletons/non-singletons.
Supplementary Table 6, related to Figure 5: Relationship between contig length/gene content and taxonomic enrichment across singletons/non-singletons, related to Figure 3–5. Pearson correlations between normalized counts of taxonomies assigned to non-singleton contigs and singleton contigs as a function of contig length and number of genes per contig.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and Algorithms | ||
| MEGAHIT (v1.1.2) | (Li et al. 2015) | https://github.com/voutcn/megahit |
| metaSPAdes (v3.13.0) | (Nurk et al. 2017) | https://github.com/ablab/spades |
| Prokka (v1.12) | (Seemann 2014) | https://github.com/tseemann/prokka |
| CD-HIT (v4.6.8) | (Li and Godzik 2006) | https://github.com/weizhongli/cdhit |
| Diamond (v0.9.24) | (Buchfink et al. 2015) | https://github.com/bbuchfink/diamond |
| Art (MountRainier) | (Huang et al. 2012) | https://www.niehs.nih.gov/research/resources/software/biostatistics/art/index.cfm |
| Data links and publicly available resource information | Supplementary Table 1 | |
| Database of results | https://microbial-genes.bio | |
Highlights:
Cross-study meta-analysis of metagenomes covering 3,655 samples from two body sites
Meta-analysis uncovers staggering microbial gene diversity
50% of all genes in a metagenomic sample are individual-specific or ‘singletons’
Individual’s microbiomes’ can be fingerprinted via rare microbial strains
Acknowledgements
We thank Debora Marks for her advice at the outset of our project, as well as Microsoft Azure and Amazon Web Services for providing compute resources for this work. This research was additionally supported by the National Institutes of Health NIEHS (T32 DK110919, R00ES23504 and R21ES205052), the National Science Foundation (1636870), the National Institute of Allergy and Infectious Disease (R01AI127250), the American Diabetes Association (ADA) Pathway to Stop Diabetes Initiator Award #1-17-INI-13, and a Smith Family Foundation Award for Excellence in Biomedical Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors have no competing interests to declare.
References
- Almeida Alexandre, Mitchell Alex L., Boland Miguel, Forster Samuel C., Gloor Gregory B., Tarkowska Aleksandra, Lawley Trevor D., and Finn Robert D.. 2019. “A New Genomic Blueprint of the Human Gut Microbiota.” Nature, February 10.1038/s41586-019-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei Adrian-Ştefan, Salcher Michaela M., Mehrshad Maliheh, Rychtecký Pavel, Znachor Petr, and Ghai Rohit. 2019. “Niche-Directed Evolution Modulates Genome Architecture in Freshwater Planctomycetes.” The ISME Journal, January 10.1038/s41396-018-0332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayling Martin, Clark Matthew D., and Leggett Richard M.. 2018. “New Approaches for Assembly of Short-Read Metagenomic Data.” e27332v1 PeerJ Preprints. 10.7287/peerj.preprints.27332v1. [DOI] [Google Scholar]
- Bairoch A 2000. “The ENZYME Database in 2000.” Nucleic Acids Research 28 (1): 304–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Yoav, and Yekutieli Daniel. 2001. “The Control of the False Discovery Rate in Multiple Testing under Dependency.” Annals of Statistics 29 (4): 1165–88. [Google Scholar]
- Bredon Marius, Dittmer Jessica, Noël Cyril, Moumen Bouziane, and Bouchon Didier. 2018. “Lignocellulose Degradation at the Holobiont Level: Teamwork in a Keystone Soil Invertebrate.” Microbiome 6 (1): 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink Benjamin, Xie Chao, and Huson Daniel H.. 2015. “Fast and Sensitive Protein Alignment Using DIAMOND.” Nature Methods 12 (1): 59–60. [DOI] [PubMed] [Google Scholar]
- Carlos Camila, Fan Huan, and Currie Cameron R.. 2018. “Substrate Shift Reveals Roles for Members of Bacterial Consortia in Degradation of Plant Cell Wall Polymers.” Frontiers in Microbiology 9 (March): 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Zijian. 2018. “PPAD, Porphyromonas Gingivalis and the Subgingival Microbiome in Periodontitis and Autoantibody-Positive Individuals at Risk of Rheumatoid Arthritis.” Phd, University of Leeds. http://etheses.whiterose.ac.uk/22247/. [Google Scholar]
- Chen Zigui, Rob DeSalle Mark Schiffman, Herrero Rolando, Wood Charles E., Ruiz Julio C., Clifford Gary M., Chan Paul K. S., and Burk Robert D.. 2018. “Niche Adaptation and Viral Transmission of Human Papillomaviruses from Archaic Hominins to Modern Humans.” PLoS Pathogens 14 (11): e1007352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock Peter J. A., Antao Tiago, Chang Jeffrey T., Chapman Brad A., Cox Cymon J., Dalke Andrew, Friedberg Iddo, et al. 2009. “Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics.” Bioinformatics 25 (11): 1422–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubin Vincent, and Ochman Howard. 2004. “Bacterial Genomes as New Gene Homes: The Genealogy of ORFans in E. Coli.” Genome Research 14 (6): 1036–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado Beatriz, Bach Alex, Guasch Isabel, González Carmen, Elcoso Guillermo, Pryce Jennie E., and Gonzalez-Recio Oscar. 2019. “Whole Rumen Metagenome Sequencing Allows Classifying and Predicting Feed Efficiency and Intake Levels in Cattle.” Scientific Reports 9 (1): 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc Frédéric, Kuch Melanie, Gibb Gillian C., Hughes Jonathan, Szpak Paul, Southon John, Enk Jacob, Duggan Ana T., and Poinar Hendrik N.. 2018. “Resolving the Phylogenetic Position of Darwin’s Extinct Ground Sloth (Mylodon Darwinii) Using Mitogenomic and Nuclear Exon Data.” Proceedings. Biological Sciences / The Royal Society 285 (1878). 10.1098/rspb.2018.0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Bo, Yi Yunhai, Liang Lifeng, and Shi Qiong. 2017. “High Throughput Identification of Antimicrobial Peptides from Fish Gastrointestinal Microbiota.” Toxins 9 (9). 10.3390/toxins9090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusko Ehrlich S, and The MetaHIT Consortium. 2011. “MetaHIT: The European Union Project on Metagenomics of the Human Intestinal Tract” In Metagenomics of the Human Body, 307–16. Springer, New York, NY. [Google Scholar]
- Flota José Jaime Maussan. n.d. “CONSULTING SERVICES REPORT.” The-Alien-Project.com. https://www.the-alien-project.com/wp-content/uploads/2018/12/ABRAXAS-EN.pdf.
- Forouzan Esmaeil, Shariati Parvin, Sadat Mousavi Maleki Masoumeh, Asghar Karkhane Ali, and Yakhchali Bagher. 2018. “Practical Evaluation of 11 de Novo Assemblers in Metagenome Assembly.” Journal of Microbiological Methods 151 (June): 99–105. [DOI] [PubMed] [Google Scholar]
- Forster Samuel C., Browne Hilary P., Kumar Nitin, Hunt Martin, Denise Hubert, Mitchell Alex, Finn Robert D., and Lawley Trevor D.. 2016. “HPMCD: The Database of Human Microbial Communities from Metagenomic Datasets and Microbial Reference Genomes.” Nucleic Acids Research 44 (D1): D604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georganas Evangelos, Egan Rob, Hofmeyr Steven, Goltsman Eugene, Arndt Bill, Tritt Andrew, Buluc Aydin, Oliker Leonid, and Yelick Katherine. 2018. “Extreme Scale De Novo Metagenome Assembly.” arXiv [cs.DC]. arXiv. http://arxiv.org/abs/1809.07014. [Google Scholar]
- Gerner Samuel M., Rattei Thomas, and Graf Alexandra B.. 2018. “Assessment of Urban Microbiome Assemblies with the Help of Targeted in Silico Gold Standards.” Biology Direct 13 (1): 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanás Gómez-Lama, Carmen David Ruano-Rosa, Legarda Garikoitz, Paloma Pizarro-Tobias Antonio Valverde-Corredor, Juan Carlos Triviño Amalia Roca, and Mercado-Blanco Jesus. 2018. “Bacillales Members from the Olive Rhizosphere Are Effective Biological Control Agents against the Defoliating Pathotype of Verticillium Dahliae.” Collection FAO: Agriculture 8 (7): 90. [Google Scholar]
- Graham E, Heidelberg JF, and Tully B. 2017. “Undocumented Potential For Primary Productivity In A Globally-Distributed Bacterial Photoautotroph.” bioRxiv. https://www.biorxiv.org/content/10.1101/140715v2.abstract. [DOI] [PMC free article] [PubMed]
- Graham Emily B., Crump Alex R., Kennedy David W., Arntzen Evan, Fansler Sarah, Purvine Samuel O., Nicora Carrie D., Nelson William, Tfaily Malak M., and Stegen James C.. 2018. “Multi ‘Omics Comparison Reveals Metabolome Biochemistry, Not Microbiome Composition or Gene Expression, Corresponds to Elevated Biogeochemical Function in the Hyporheic Zone.” The Science of the Total Environment 642 (November): 742–53. [DOI] [PubMed] [Google Scholar]
- Han Maozhen, Yang Pengshuo, Zhong Chaofang, and Ning Kang. 2018. “The Human Gut Virome in Hypertension.” Frontiers in Microbiology 9 (December): 3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan Geoffrey D., Duhaime Melissa B., Koutra Danai, and Schloss Patrick D.. 2018. “Biogeography and Environmental Conditions Shape Bacteriophage-Bacteria Networks across the Human Microbiome.” PLoS Computational Biology 14 (4): e1006099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan Geoffrey D., Duhaime Melissa B., Ruffin Mack T. 4th, Koumpouras Charlie C., and Schloss Patrick D.. 2018. “Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome.” mBio 9 (6). 10.1128/mBio.02248-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Peng, Zhang Yan, Xiao Kangpeng, Jiang Fan, Wang Hengchao, Tang Dazhi, Liu Dan, et al. 2018. “The Chicken Gut Metagenome and the Modulatory Effects of Plant-Derived Benzylisoquinoline Alkaloids.” Microbiome 6 (1): 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Weichun, Li Leping, Myers Jason R., and Marth Gabor T.. 2012. “ART: A next-Generation Sequencing Read Simulator.” Bioinformatics 28 (4): 593–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD 2007. “Matplotlib: A 2D Graphics Environment.” Computing in Science Engineering 9 (3): 90–95. [Google Scholar]
- Jackman Shaun D., Vandervalk Benjamin P., Mohamadi Hamid, Chu Justin, Yeo Sarah, Austin Hammond S, Jahesh Golnaz, et al. 2017. “ABySS 2.0: Resource-Efficient Assembly of Large Genomes Using a Bloom Filter.” Genome Research 27 (5): 768–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner Manuel, Thorson Erin, Sharp Christine E., Dong Xiaoli, Liu Dan, Li Carmen, and Strous Marc. 2017. “Assessing Species Biomass Contributions in Microbial Communities via Metaproteomics.” Nature Communications 8 (1): 1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger Marie E., Delmont Tom O., Eren AM, Meyer Kyle M., Guo Jiarong, Khan Kiran, Rodrigues Jorge L. M., et al. 2018. “New Biological Insights Into How Deforestation in Amazonia Affects Soil Microbial Communities Using Metagenomics and Metagenome-Assembled Genomes.” Frontiers in Microbiology 9 (July): 1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusy Dominik, Motyka Michal, Bocek Matej, Vogler Alfried P., and Bocak Ladislav. 2018. “Genome Sequences Identify Three Families of Coleoptera as Morphologically Derived Click Beetles (Elateridae).” Scientific Reports 8 (1): 17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land Miriam, Hauser Loren, Jun Se-Ran, Nookaew Intawat, Leuze Michael R., Ahn Tae-Hyuk, Karpinets Tatiana, et al. 2015. “Insights from 20 Years of Bacterial Genome Sequencing.” Functional & Integrative Genomics 15 (2): 141–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre Pascal, and Gogarten J. Peter. 2009. “Estimating the Size of the Bacterial Pan-Genome.” Trends in Genetics: TIG 25 (3): 107–10. [DOI] [PubMed] [Google Scholar]
- Learman Deric R., Ahmad Zahra, Brookshier Allison, Henson Michael W., Hewitt Victoria, Lis Amanda, Morrison Cody, et al. 2019. “Comparative Genomics of 16 Microbacterium Spp. That Tolerate Multiple Heavy Metals and Antibiotics.” PeerJ 6 (January): e6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Dinghua, Liu Chi-Man, Luo Ruibang, Sadakane Kunihiko, and Lam Tak-Wah. 2015. “MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph.” Bioinformatics 31 (10): 1674–76. [DOI] [PubMed] [Google Scholar]
- Li Dinghua, Luo Ruibang, Liu Chi-Man, Leung Chi-Ming, Ting Hing-Fung, Sadakane Kunihiko, Yamashita Hiroshi, and Lam Tak-Wah. 2016. “MEGAHIT v1.0: A Fast and Scalable Metagenome Assembler Driven by Advanced Methodologies and Community Practices.” Methods 102 (June): 3–11. [DOI] [PubMed] [Google Scholar]
- Li Hong-Yi, Wang Hang, Wang Hai-Tiao, Xin Pei-Yong, Xu Xin-Hua, Ma Yun, Liu Wei-Ping, et al. 2018. “The Chemodiversity of Paddy Soil Dissolved Organic Matter Correlates with Microbial Community at Continental Scales.” Microbiome 6 (1): 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Junhua, Jia Huijue, Cai Xianghang, Zhong Huanzi, Feng Qiang, Sunagawa Shinichi, Arumugam Manimozhiyan, et al. 2014. “An Integrated Catalog of Reference Genes in the Human Gut Microbiome.” Nature Biotechnology 32 (8): 834–41. [DOI] [PubMed] [Google Scholar]
- Li Weizhong, and Godzik Adam. 2006. “Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences.” Bioinformatics 22 (13): 1658–59. [DOI] [PubMed] [Google Scholar]
- Lloyd-Price Jason, Mahurkar Anup, Rahnavard Gholamali, Crabtree Jonathan, Orvis Joshua, Brantley Hall A, Brady Arthur, et al. 2017. “Strains, Functions and Dynamics in the Expanded Human Microbiome Project.” Nature 550 (7674): 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luber Jacob M., Tierney Braden T., Cofer Evan M., Patel Chirag J., and Kostic Aleksandar D.. 2017. “Aether: Leveraging Linear Programming For Optimal Cloud Computing In Genomics.” Bioinformatics , December 10.1093/bioinformatics/btx787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Robbie M., Moniruzzaman Mohammad, Mucci Nicholas C., Willis Anusuya, Woodhouse Jason N., Xian Yuejiao, Xiao Chuan, Brussaard Corina P. D., and Wilhelm Steven W.. 2019. “Cylindrospermopsis Raciborskii Virus and Host: Genomic Characterization and Ecological Relevance.” Environmental Microbiology 21 (6): 1942–56. [DOI] [PubMed] [Google Scholar]
- Maus Irena, Rumming Madis, Bergmann Ingo, Heeg Kathrin, Pohl Marcel, Nettmann Edith, Jaenicke Sebastian, et al. 2018. “Characterization of Bathyarchaeota Genomes Assembled from Metagenomes of Biofilms Residing in Mesophilic and Thermophilic Biogas Reactors.” Biotechnology for Biofuels 11 (June): 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney James O., McNally Alan, and O’Connell Mary J.. 2017. “Why Prokaryotes Have Pangenomes.” Nature Microbiology 2 (March): 17040. [DOI] [PubMed] [Google Scholar]
- Mizzi Jessica E., Lounsberry Zachary T., Titus Brown C, and Sacks Benjamin N. 2017. “Draft Genome of Tule Elk Cervus Canadensis Nannodes.” F1000Research 6 (September): 1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H. Bjørn, Almeida Mathieu, Juncker Agnieszka Sierakowska, Rasmussen Simon, Li Junhua, Sunagawa Shinichi, Plichta Damian R., et al. 2014. “Identification and Assembly of Genomes and Genetic Elements in Complex Metagenomic Samples without Using Reference Genomes.” Nature Biotechnology 32 (8): 822–28. [DOI] [PubMed] [Google Scholar]
- Nurk Sergey, Meleshko Dmitry, Korobeynikov Anton, and Pevzner Pavel A.. 2017. “metaSPAdes: A New Versatile Metagenomic Assembler.” Genome Research 27 (5): 824–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary Nuala A., Wright Mathew W., Brister J. Rodney, Ciufo Stacy, Haddad Diana, McVeigh Rich, Rajput Bhanu, et al. 2016. “Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation.” Nucleic Acids Research 44 (D1): D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pärnänen Katariina, Karkman Antti, Tamminen Manu, Lyra Christina, Hultman Jenni, Paulin Lars, and Virta Marko. 2016. “Evaluating the Mobility Potential of Antibiotic Resistance Genes in Environmental Resistomes without Metagenomics.” Scientific Reports 6 (October): 35790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasolli Edoardo, Asnicar Francesco, Manara Serena, Zolfo Moreno, Karcher Nicolai, Armanini Federica, Beghini Francesco, et al. 2019. “Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle.” Cell 176 (3): 649–62. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin Nastassia V., Pratte Zoe A., Regensburger Matthew, Hall Eric, Gilde Kailen, Dove Alistair D. M., and Stewart Frank J.. 2018. “Microbiome Dynamics in a Large Artificial Seawater Aquarium.” Applied and Environmental Microbiology 84 (10). 10.1128/AEM.00179-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedron Renato, Esposito Alfonso, Bianconi Irene, Pasolli Edoardo, Tett Adrian, Asnicar Francesco, Cristofolini Mario, Segata Nicola, and Jousson Olivier. 2019. “Genomic and Metagenomic Insights into the Microbial Community of a Thermal Spring.” Microbiome 7 (1): 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Junjie, Li Ruiqiang, Raes Jeroen, Arumugam Manimozhiyan, Kristoffer Solvsten Burgdorf Chaysavanh Manichanh, Nielsen Trine, et al. 2010. “A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing.” Nature 464 (7285): 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Junjie, Li Yingrui, Cai Zhiming, Li Shenghui, Zhu Jianfeng, Zhang Fan, Liang Suisha, et al. 2012. “A Metagenome-Wide Association Study of Gut Microbiota in Type 2 Diabetes.” Nature 490 (7418): 55–60. [DOI] [PubMed] [Google Scholar]
- Rebollar Eria A., Ana Gutiérrez-Preciado Cecilia Noecker, Eng Alexander, Hughey Myra C., Medina Daniel, Walke Jenifer B., et al. 2018. “The Skin Microbiome of the Neotropical Frog Craugastor Fitzingeri: Inferring Potential Bacterial-Host-Pathogen Interactions From Metagenomic Data.” Frontiers in Microbiology 9 (March): 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengasamy Vasudevan. 2018. Engineering High Performance Workflows for End-to-End Acceleration of Genomic Applications. The Pennsylvania State University. [Google Scholar]
- Rengasamy V, Medvedev P, and Madduri K. 2017. “Parallel and Memory-Efficient Preprocessing for Metagenome Assembly.” In 2017 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW), 283–92. ieeexplore.ieee.org. [Google Scholar]
- Robin Xavier, Turck Natacha, Hainard Alexandre, Tiberti Natalia, Lisacek Frederique, Sanchez Jean-Charles, and Muller Markus. 2011. “pROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves.” BMC Bioinformatics 12 (March): 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux Simon, Emerson Joanne B., Eloe-Fadrosh Emiley A, and Sullivan Matthew B. 2017. “Benchmarking Viromics: An in Silico Evaluation of Metagenome-Enabled Estimates of Viral Community Composition and Diversity.” Peer J 5 (September): e3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royalty Taylor, and Steen Andrew D.. 2018. “Simulation-Based Approaches to Characterize Metagenome Coverage as a Function of Sequencing Effort and Microbial Community Structure.” bioRxiv. 10.1101/356840. [DOI] [Google Scholar]
- Sandoval-Motta Santiago, Aldana Maximino, Martinez-Romero Esperanza, and Frank Alejandro. 2017. “The Human Microbiome and the Missing Heritability Problem.” Frontiers in Genetics 8 (June): 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz Frederik, Alteio Lauren, Goudeau Danielle, Ryan Elizabeth M., Yu Feiqiao B., Malmstrom Rex R., Blanchard Jeffrey, and Woyke Tanja. 2018. “Hidden Diversity of Soil Giant Viruses.” Nature Communications 9 (1): 4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann Torsten. 2014. “Prokka: Rapid Prokaryotic Genome Annotation.” Bioinformatics 30 (14): 2068–69.24642063 [Google Scholar]
- Shiller AM, Chan EW, Joung DJ, Redmond MC, and Kessler JD. 2017. “Light Rare Earth Element Depletion during Deepwater Horizon Blowout Methanotrophy.” Scientific Reports 7 (1): 10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen T. 1948. “{A Method of Establishing Groups of Equal Amplitude in Plant Sociology Based on Similarity of Species and Its Application to Analyses of the Vegetation on Danish Commons}.” Biol. Skr. 5: 1–34. [Google Scholar]
- Souvorov Alexandre, Agarwala Richa, and Lipman David J.. 2018. “SKESA: Strategic K-Mer Extension for Scrupulous Assemblies.” Genome Biology 19 (1): 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven Blaire, and Kuske Cheryl Rae. 101AD “Resuscitation of Intact and Disturbed Biological Soil Crusts in Response to a Wetting Event Characterized By Metatranscriptomic Sequencing.” Frontiers in Microbiology 9 (LA-UR-17–31418). https://www.osti.gov/servlets/purl/1479950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton Thomas D. S., Clooney Adam G., Ryan Feargal J., Ross R. Paul, and Hill Colin. 2019. “Choice of Assembly Software Has a Critical Impact on Virome Characterisation.” Microbiome 7 (1): 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin Hervé, Masignani Vega, Cieslewicz Michael J., Donati Claudio, Medini Duccio, Ward Naomi L., Angiuoli Samuel V., et al. 2005. “Genome Analysis of Multiple Pathogenic Isolates of Streptococcus Agalactiae: Implications for the Microbial ‘pan-Genome.’” Proceedings of the National Academy of Sciences of the United States of America 102 (39): 13950–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus Brown C, Moritz Dominik, O’Brien Michael P, Reidl Felix, Reiter Taylor, and Sullivan Blair D.. 2019. “Exploring Neighborhoods in Large Metagenome Assembly Graphs Reveals Hidden Sequence Diversity.” bioRxiv. 10.1101/462788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong Duy Tin, Franzosa Eric A., Tickle Timothy L., Scholz Matthias, Weingart George, Pasolli Edoardo, Tett Adrian, Huttenhower Curtis, and Segata Nicola. 2015. “MetaPhlAn2 for Enhanced Metagenomic Taxonomic Profiling.” Nature Methods 12 (10): 902–3. [DOI] [PubMed] [Google Scholar]
- Tschitschko Bernhard, Erdmann Susanne, DeMaere Matthew Z., Roux Simon, Panwar Pratibha, Allen Michelle A., Williams Timothy J., et al. 2018. “Genomic Variation and Biogeography of Antarctic Haloarchaea.” Microbiome 6 (1): 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully Benjamin J., Graham Elaina D., and Heidelberg John F.. 2018. “The Reconstruction of 2,631 Draft Metagenome-Assembled Genomes from the Global Oceans.” Scientific Data 5 (January): 170203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully Benjamin J., Sachdeva Rohan, Graham Elaina D., and Heidelberg John F.. 2017. “290 Metagenome-Assembled Genomes from the Mediterranean Sea: A Resource for Marine Microbiology.” PeerJ 5 (July): e3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi Anuj, Singh Balwinder, Billekallu Thammegowda Naveen K., and Singh Niraj K.. 2019. “Shotgun Metagenomics Offers Novel Insights into Taxonomic Compositions, Metabolic Pathways and Antibiotic Resistance Genes in Fish Gut Microbiome.” Archives of Microbiology, January 10.1007/s00203-018-1615-y. [DOI] [PubMed] [Google Scholar]
- Ugland Karl I., Gray John S., and Ellingsen Kari E.. 2003. “The Species-Accumulation Curve and Estimation of Species Richness.” The Journal of Animal Ecology 72 (5): 888–97. [Google Scholar]
- Vasconcellos AF, Silva JMF, de Oliveira AS, Prado PS, Nagata T, and Resende RO. 2019. “Genome Sequences of Chikungunya Virus Isolates Circulating in Midwestern Brazil.” Archives of Virology 164 (4): 1205–8. [DOI] [PubMed] [Google Scholar]
- Verbruggen Heroen, Marcelino Vanessa R., Guiry Michael D., Cremen Ma Chiela M, and Jackson Christopher J.. 2017. “Phylogenetic Position of the Coral Symbiont Ostreobium (Ulvophyceae) Inferred from Chloroplast Genome Data.” Journal of Phycology 53 (4): 790–803. [DOI] [PubMed] [Google Scholar]
- Wainschtein Pierrick, Jain Deepti P., Yengo Loic, Zheng Zhili, TOPMed Anthropometry Working Group, Trans-Omics for Precision Medicine Consortium, L. Adrienne Cupples, et al. 2019. “Recovery of Trait Heritability from Whole Genome Sequence Data.” bioRxiv. 10.1101/588020. [DOI] [Google Scholar]
- Wang Jincheng, Tang Lili, Zhou Hongyuan, Zhou Jun, Glenn Travis C., Shen Chwan-Li, and Wang Jia-Sheng. 2018. “Long-Term Treatment with Green Tea Polyphenols Modifies the Gut Microbiome of Female Sprague-Dawley Rats.” The Journal of Nutritional Biochemistry 56 (June): 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xiong X, Cao W, Zhang C, Werren J, and Wang X. 2018. “Genome Assembly of the A-Group Wolbachia in Nasonia Oneida and Phylogenomic Analysis of Wolbachia Strains Revealed Genome Evolution and Lateral Gene Transfer.” bioRxiv. https://www.biorxiv.org/content/10.1101/508408v1.abstract.
- Ward Lewis M., Hemp James, Shih Patrick M., McGlynn Shawn E., and Fischer Woodward W.. 2018. “Evolution of Phototrophy in the Chloroflexi Phylum Driven by Horizontal Gene Transfer.” Frontiers in Microbiology 9 (February): 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Lewis M., McGlynn Shawn E., and Fischer Woodward W.. 2017a. “Draft Genome Sequence of Chloracidobacterium Sp. CP2_5A, a Phototrophic Member of the Phylum Acidobacteria Recovered from a Japanese Hot Spring.” Genome Announcements 5 (40). 10.1128/genomeA.00821-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —. 2017b. “Draft Genome Sequences of a Novel Lineage of Armatimonadetes Recovered from Japanese Hot Springs.” Genome Announcements 5 (40). 10.1128/genomeA.00820-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LM, McGlynn SE, and Fischer WW. 2018. “Draft Genome Sequences of Two Basal Members of the Anaerolineae Class of Chloroflexi from a Sulfidic Hot Spring.” Genome Announcements 6 (25). 10.1128/genomeA.00570-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Yuri I., Makarova Kira S., Lobkovsky Alexander E., and Koonin Eugene V.. 2016. “Two Fundamentally Different Classes of Microbial Genes.” Nature Microbiology 2 (November): 16208. [DOI] [PubMed] [Google Scholar]
- Xing Xin, Liu Jun S., and Zhong Wenxuan. 2017. “MetaGen: Reference-Free Learning with Multiple Metagenomic Samples.” Genome Biology 18 (1): 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Yuzhen, and Doak Thomas G.. 2009. “A Parsimony Approach to Biological Pathway Reconstruction/inference for Genomes and Metagenomes.” PLoS Computational Biology 5 (8): e1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Yanbin, and Fischer Daniel. 2006. “On the Origin of Microbial ORFans: Quantifying the Strength of the Evidence for Viral Lateral Transfer.” BMC Evolutionary Biology 6 (August): 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younge Noelle E., Araújo-Pérez Félix, Brandon Debra, and Seed Patrick C.. 2018. “Early-Life Skin Microbiota in Hospitalized Preterm and Full-Term Infants.” Microbiome 6 (1): 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J Peter W., Crossman Lisa C., Johnston Andrew W. B., Thomson Nicholas R., Ghazoui Zara F., Hull Katherine H., Wexler Margaret, et al. 2006. “The Genome of Rhizobium Leguminosarum Has Recognizable Core and Accessory Components.” Genome Biology 7 (4): R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaikova Elena, Goerlitz David S., Tighe Scott W., Wagner Nicole Y., Bai Yu, Hall Brenda L., Bevilacqua Julie G., Weng Margaret M., Samuels-Fair Maya D., and Johnson Sarah Stewart. 2019. “Antarctic Relic Microbial Mat Community Revealed by Metagenomics and Metatranscriptomics.” Frontiers in Ecology and Evolution 7: 1. [Google Scholar]
- Zeevi David, Korem Tal, Godneva Anastasia, Bar Noam, Kurilshikov Alexander, Lotan-Pompan Maya, Weinberger Adina, et al. 2019. “Structural Variation in the Gut Microbiome Associates with Host Health.” Nature, March, 1. [DOI] [PubMed] [Google Scholar]
- Zhao Shijie, Lieberman Tami D., Poyet Mathilde, Kauffman Kathryn M., Gibbons Sean M., Groussin Mathieu, Xavier Ramnik J., and Alm Eric J.. 2019. “Adaptive Evolution within Gut Microbiomes of Healthy People.” Cell Host & Microbe, April https://doi.org/10.1016Zj.chom.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Renjun, Zeng Shenzheng, Hou Dongwei, Liu Jian, Weng Shaoping, He Jianguo, and Huang Zhijian. 2019. “Occurrence of Human Pathogenic Bacteria Carrying Antibiotic Resistance Genes Revealed by Metagenomic Approach: A Case Study from an Aquatic Environment.” Journal of Environmental Sciences 80 (June): 248–56. [DOI] [PubMed] [Google Scholar]
- Zhou Youlian, Zhenjiang Zech Xu Yan He, Yang Yunsheng, Liu Le, Lin Qianyun, Nie Yuqiang, et al. 2018. “Gut Microbiota Offers Universal Biomarkers across Ethnicity in Inflammatory Bowel Disease Diagnosis and Infliximab Response Prediction.” mSystems 3 (1). 10.1128/mSystems.00188-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Ana, Sunagawa Shinichi, Mende Daniel R., and Bork Peer. 2015. “Inter-Individual Differences in the Gene Content of Human Gut Bacterial Species.” Genome Biology 16 (April): 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinke Laura A., Glombitza Clemens, Bird Jordan T., Roy Hans, Jorgensen Bo Barker, Lloyd Karen G., Amend Jan P., and Reese Brandi Kiel. 2019. “Microbial Organic Matter Degradation Potential in Baltic Sea Sediments Is Influenced by Depositional Conditions and In Situ Geochemistry.” Applied and Environmental Microbiology 85 (4). 10.1128/AEM.02164-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1, related to Figures 1 and 2: Summaries of data used for meta-analysis, download links for all datasets, and sample-by-sample summary statistics/assembly quality metrics.
Supplementary Table 2, related to STAR methods: Regression output from synthetic and real data analysis intended to determine the validity of singleton genes.
Supplementary Table 3, related to STAR methods: Literature review of Megahit parameters. At the time of writing searched the methods sections of 110 (99 distinct, 67 that we had access to and were not books or dissertations) publications that cited Megahit on Google Scholar and, when possible, recorded the parameters they used. Each publication is fully referenced in the works cited list.
Supplementary Table 4, related to Figure 4: Functional enrichment, related to Figure 4. Contains raw data regarding functions enriched in oral singletons/non-singletons and gut singletons/non-singletons.
Supplementary Table 5, related to Figure 5: Taxonomic enrichment of singletons/non-singletons, related to Figure 5 and Supplementary Figure 5. Contains raw output regarding strains enriched in oral singletons/non-singletons and gut singletons/non-singletons.
Supplementary Table 6, related to Figure 5: Relationship between contig length/gene content and taxonomic enrichment across singletons/non-singletons, related to Figure 3–5. Pearson correlations between normalized counts of taxonomies assigned to non-singleton contigs and singleton contigs as a function of contig length and number of genes per contig.
Data Availability Statement
Example scripts for each step of this analytical pipeline are publicly accessible at https://github.com/kosticlab/universe_of_genes_scripts of genes scripts. When relevant, each section of the methods section below refers to script in this repository used for that particular analysis. The post-assembly pipeline, which includes non-redundant gene catalog construction, gene catalog quality control, gene-level taxonomy mapping, iterative gene catalog construction, binary gene incidence matrix generation, and Sorenson-Dice dissimilarity calculation, is run by “gene_catalog_construction/full_contig_parsing_and_singleton_hunting_pipeline.py”. We built our public facing database using Microsoft Azure’s Database for PostgreSQL service. We built our website with a Flask API..