

Abstract
Membrane curvature participates in a wide range of cellular processes, and acts as a hotspot for protein interactions and intracellular signalling. Curvature also occurs at the interface between cells and nanotopography of biomaterials and biomedical devices, which could influence the performance of tissue engineering scaffolds and implantable devices. Precisely manipulating membrane curvature is thus of great interest in probing intracellular activities involved with curved membranes. Here we present a detailed protocol to design, fabricate, and characterize nanoscale structures for manipulating membrane curvature and probing curvature-induced phenomena in live cells. This protocol first describes a detailed procedure for the design and fabrication of nanoscale structures using electron-beam lithography. Then, the protocol describes how to use these nanostructures to manipulate local membrane curvature and probe intracellular protein responses. Finally, the protocol describes a procedure to characterize the nanostructure-cell membrane interface using focused ion beam and scanning electron microscopy.
Keywords: membrane curvature, nanoscale, surface topography, nanofabrication, focused ion beam, scanning electron microscopy
INTRODUCTION
Platforms for studying cell membrane curvature
Conformational changes of cell membranes occur in a variety of essential biological processes, including cell division, morphogenesis, migration, cytokinesis, intracellular signalling, and vesicle trafficking. Deformed membranes can have either positive curvature (toward the cytoplasm) or negative curvature (toward the extracellular space). For example, endocytosis, through which cells take in ligands and liquids from the environment, typically involves the formation of positive membrane curvature1,2. In the process of clathrin-mediated endocytosis (CME), clathrin and many other proteins coordinate to bend the membrane and generate clathrin-coated pits. Dynamin is then recruited to the neck region and cleaves the pits from the plasma membrane to yield endosomal vesicles inside the cell. In contrast, negative curvature can be found in cellular protrusions such as filopodia and cilia. Filopodia are involved in cell migration and sensing chemotropic cues3, while cilia can serve as important sensory organelles4. Examining how these morphological features of cell membranes play a role in cell functions is important in our understanding of cell biology, pathology and other biomedical branches5,6.
It is now widely understood that membrane curvature is more than a simple consequence of cellular processes; instead, it is an active topographical hub that promotes protein interactions and intracellular signalling. For example, endocytosis has been found to be intimately linked with cell signalling. As clathrin scaffolds the membrane invagination in CME, activated receptors in the plasma membrane are recruited by interacting with clathrin adaptor proteins, which results in a signalling hub on the plasma membrane2. Endocytic vesicles are subsequently able to transport receptors to the intracellular space to regulate signalling. Similarly, cilia are present in many cells and play critical roles in signal transduction, particularly the Sonic Hedgehog signalling pathway. Membrane curvature is believed to participate in signalling by concentrating specific signalling molecules, thereby increasing their chemical activity.
Membrane curvature may arise from different but often concerted mechanisms such as the accumulation of membrane-shaping proteins, lipid asymmetry with different-sized head groups, and cytoskeletal forces6. An important class of membrane-shaping proteins is the BAR-domain proteins (named after Bin, amphiphysin and Rvs proteins) that feature a “banana-shaped” structure7. Subfamilies of BAR proteins sense and generate both positive membrane curvature (N-BAR and F-BAR proteins) and negative membrane curvature (I-BAR proteins)8,9. Their existence and functions have been reported in important cell activities such as endocytosis10, and neuronal differentiation and outgrowth11. The size of their lipid headgroup and their asymmetric distribution on the two leaflets of the bilayer membrane may favour a particular orientation and degree of membrane curvature. For example, phosphatic acid and lysophosphatidic acid, which are interconvertible, favour opposite orientations of spontaneous curvature12. Importantly, these proteins and lipids not only deform the membrane to certain curvatures, but also are preferentially recruited to curved membranes. Therefore, membrane curvature itself can directly act as a biochemical signal by attracting curvature-sensing proteins and lipids that are involved in intracellular signalling.
The plasma membrane can also be deformed by the topographic features of biomaterials or biomedical devices that are in contact with cells. Therefore, by inducing membrane curvature, surface topography can potentially be used to regulate intracellular signalling and cell behavior.
Implantable devices with nanoscale surface topographies, particularly vertical nanostructures, have been found to induce an intimate contact between the devices and the cell membrane, giving rise to enhanced sensing or modulating performance13,14. In the context of engineering the surface of implants, this cell-topography interaction opens up a variety of solutions for improving the implants’ performance15–17. In particular, titanium (Ti) implants with roughened surfaces are found to promote cell attachment and device integration18. At the cellular level, the presence of nanoscale features affects stem cell differentiation19, proliferation20, and migration21. For example, nanopillars stimulate osteogenic differentiation of mesenchymal stem cells (MSCs)22, nanogratings steer MSCs toward the neuronal lineage23, and Ti nanoroughs modulate macrophage activity24. Therefore, using surface topography to control cell behavior offers a valuable approach for tissue engineering and regenerative medicine.
To understand the functional effects of membrane curvature, in vitro experimental tools and simulations have been developed, aiming to manipulate membrane curvature with precision at the relevant length scales. Giant unilamellar vesicles (GUVs, usually 5–50 μm in diameter) are visible by optical microscopy and micromanipulation with a pipette can generate tubular structures of tens to hundreds of nanometers in diameter from them. These structures have been used in studies of curvature-sensitive proteins, where tubular diameter and membrane tension can be tuned25,26. Small vesicles (usually 50–500 nm in diameter) can be tethered to a substrate and the binding of curvature-sensitive proteins to vesicle surfaces can be quantified by fluorescence microscopy27. Coarse-grained simulations model the movement of molecules in systems much larger than all-atom simulations. They were applied to analyse the effects of membrane tension, protein density, and lipid composition regarding the aggregation of curvature-relevant proteins26–28. The functions of membrane curvature and curvature-related proteins and processes have also been studied extensively in cells. However, precisely controlling the location and the degree of curvature in cells has been challenging. Membrane curvature in cells has been perturbed to some extent by intentional hypertonic osmotic shocks that lead to cell shrinking and curved plasma membrane29, and by overexpression of curvature-generating proteins (e.g., formin-binding protein 17—FBP17) which induces membrane tabulation30. But those global effects fail to pinpoint the location of membrane curvature. The value of membrane curvature (i.e., the diameter of curvature) is also difficult to control, not only due to the limitations of those perturbations to the dynamic and complex system of cell membranes, but also because membrane curvature values in many cellular processes are beyond the diffraction-limited resolution of standard optical microscopy.
Our group and others have recently shown that nanoscale structures can induce stable and well-defined membrane curvatures in live cells. A variety of nanostructures, particularly sparsely-distributed vertical nanostructures, have been fabricated to interface with live cells31–34. It was revealed by fluorescence imaging35 and electron microscopy36 that the plasma membrane tightly wraps around nanostructures to generate stable membrane curvatures 37. The shape and the position of the nanostructure determine the precise value and the location of membrane curvatures on the plasma membrane. These nanostructure-induced membrane curvatures have been shown to recruit curvature-sensing BAR proteins and also significantly enhance curvature-dependent processes such as CME. Beyond the plasma membrane, tall nanopillars are found to deform the nuclear envelope, and potentially contribute to gene expression regulation38. Mounting research demonstrates the use of nanostructures in investigating the phenomena associated with cell membrane deformation.
In this protocol, we describe step-by-step procedures for designing, fabricating, and characterizing nanostructure platforms that can be used to study intracellular processes reliant upon membrane curvature. These nanostructure platforms are transparent and compatible with fluorescence imaging in live cells39. We also describe an electron microscopy method that utilizes focused ion beam and scanning electron microscopy (FIB-SEM) for detailed characterization of the interface between cells and nanostructures36.
Overview of the protocol
The platform technology we developed for manipulating cell membranes involves chips with configurable nanostructures (namely nanochips), cell culture on the nanochips, and the FIB-SEM technique. Accordingly, the method described in this protocol consists of three stages. First, we introduce the design of the nanochip in computer-aided design (CAD) software and fabrication of these nanostructures using the electron beam (E-beam) lithography (Steps 1–47). Second, we describe the surface cleaning and coating procedures for using these nanochips for cell culture and fluorescence imaging. (Steps 48–65). When cleaned properly and handled carefully, these nanochips can be reused for at least 50 times over two years. Third, we present the FIB-SEM technique which can be used to characterize the membrane-nanostructure interface at the nanometer scale (Steps 66–119). We note that the membrane-nanostructure interface is also compatible with other characterization methods such as immunostaining and scanning probe microscopy.
Applications of the protocol
The capability to precisely control membrane curvature in cells is highly desirable for investigating cellular processes that are affected by membrane deformation. Our nanostructure platform affords such a capability, which paves the way for a breadth of applications. E-beam writing used in the nanofabrication procedure generates nanostructures with diameter of curvature from less than 100 nm to micrometers, which covers a wide range of membrane curvatures that are involved in cell activity. Furthermore, the nanostructures on our chips can achieve both positive (into the cytoplasm) and negative (towards the extracellular space) membrane deformations. Although we describe the fabrication procedure for silicon (Si) and quartz chips here, the fabrication method can be adapted to other materials. Therefore, it is possible to combine both nanostructures with other material characteristics (such as the tunable rigidity of polydimethylsiloxane).
Quantitative measurements of the curvature preference of curvature-sensitive proteins
Curvature-sensitive proteins bind to membranes of certain curvature orientation and values, depending on the molecular structures of the proteins. For example, all BAR domain proteins dimerize to form banana-shaped structures as a prerequisite for sensing curvature. However, N-BAR proteins prefer higher curvature values than F-BAR proteins, despite both sensing positive curvature. In contrast, I-BAR proteins prefer negative membrane curvature40. Other proteins are curvature sensitive through mechanisms such as oligomerization or amphipathic helix insertion and will have their distinct curvature preferences41. The binding of such proteins to curvatures also leads to the preferential recruitment of other signalling proteins. Although it is important to quantitatively measure the curvature preference of different curvature-sensitive proteins, it has been challenging to determine the value and the orientation of membrane curvature in live cells.
The protocol described here enables the generation a curvature library (i.e., positive and negative curvatures with varying curvature values) of nanostructures designed with great freedom and fabricated in high precision. A gradient array of membrane curvatures can be generated in the same cell, which provides an ideal system to quantitatively measure the curvature preference of a cellular protein. For example, mammalian cells expressing fluorescently labelled curvature-sensitive proteins can be imaged by live cell imaging. The accumulation of proteins on different curvature values can be measured by their fluorescence intensity. Analysis of the quantitative relationship between protein accumulation (normalized by the membrane area) and curvature value then yields a curvature preference profile for a given curvature-sensitive protein. The same quantification method can also be used for endogenous proteins by immunostaining. With this method, we have reported that amphiphysin 1 strongly prefers 150 nm diameter positive curvature as compared to a flat surface39. We also utilized nanopillars of a range of radii (from 50 nm to 500 nm) to show that dynamin 2 (DMN2) is more likely to accumulate on a curved membrane with a smaller radius (e.g., a 64 nm curvature compared to a 130 nm one). These initial studies demonstrate the quantitative nature of this method. We expect that negative curvatures can also be generated in the plasma membrane by nanohole structures to study I-BAR domain proteins and other negative-curvature sensing proteins.
Studying the effects of membrane curvature on intracellular processes
Many cellular processes involve plasma membrane curvature, such as endocytosis, filopodia, and neurite outgrowth. Curvature-sensitive proteins participating in these processes often contain a curvature-sensing domain and one or more signalling domains that interact with other proteins involved in the process. Therefore, membrane curvatures can actively modulate the dynamics and occurrence frequency of these intracellular processes through curvature-sensitive proteins. Without the ability to precisely manipulate membrane curvature in cells, the generation of membrane curvature is highly dynamic, and neither the curvature value nor the curvature location is controllable. It therefore is difficult to isolate how membrane curvatures actively affect intracellular processes.
The nanostructure platform allows direct manipulation of the plasma membrane curvature, and could be used to investigate how membrane curvature actively affects intracellular processes. For example, we previously demonstrated an application of this method in studying CME38,39,42. By analyzing CME dynamics on the nanochips, we found that the occurrence frequency of CME events per unit area is six times higher on curved membranes with 150 nm radius of curvature than flat membranes. Furthermore, the lifetimes of DNM2 and clathrin are shorter on curved membranes as compared to flat membranes. These observations suggest that nanopillar-induced membrane curvatures are hotspots of CME, and that pre-curved membranes likely reduce the energy barrier to the membrane bending in CME. Beside addressing endocytosis, nanopillars were also shown to induce local actin accumulation in a curvature-dependent manner39. How membrane curvature actively participates in actin dynamics is currently under investigation. Furthermore, many groups have shown that nanostructures directly affect cell behavior such as neurite outgrowth43,44 and stem cell differentiation19,45,46.Membrane curvature and curvature-sensing proteins are likely involved in these nanostructure-induced functional changes. The nanostructure platform described here would afford a new and quantitative approach to investigating these phenomena.
Investigating cellular interactions with topographic features
This third part of this protocol describes a FIB-SEM method that characterizes the interface between cells and vertical nanoscale structures by means of FIB milling and SEM imaging31,47,48. Although utilizing nanostructures to improve the performance of biomedical devices (particularly Ti implants) has led to exciting achievements, our understanding of the interactions between specific cell types and topographies is limited. Transmission electron microscopy (TEM) is currently the primary technique to reveal nanometer scale structures and allows detailed analysis of the cell-to-substrate interface. However, TEM requires sectioning the resin-embedded samples into thin slices with a diamond knife, a procedure not compatible with hard substrates such as Si02 or Ti. Additionally, in a TEM thin section, information about morphological features and their corresponding cellular locations is lost. In order to find sites of interest, e.g. nanostructure locations, one must carry out many rounds of sectioning and imaging.
The FIB-SEM technique described here offers in situ examination of the membrane-to-material interface without blind sectioning and without the loss of cellular context. The ultra-thin plasticization (UTP) procedure allows preservation and visualization with high contrast and resolution of cellular ultrastructures. Taken together with nanostructures, this technique will find many applications in investigating cell-topography interactions. As an example, we previously examined how the cleft distance between the cell membrane and the material surface is affected by nanostructures of different shapes36. In the study, we grew HEK, HL-1 and PC 12 cells on the nanochips, all of which were preserved with the ultra-thin plasticization technique after a few days of culture. Samples were first examined in large areas by SEM. Once a desired location was identified, such as where a cell membrane was in contact with a nanostructure, FIB was used to cut through the sample to reveal the interface, which was then imaged by SEM. Our study showed that nanopillar structures induce a much smaller cleft distance than flat surfaces or nanopores do. Furthermore, we examined nanostructures of both quartz and polystyrenesulfonate (PEDOT). Although the two materials have distinctive Young’s moduli (~80 GPa for quartz, and ~1 GPa for PEDOT), the cleft distances are mainly determined by topography. The FIB-SEM technique can also be used to examine how tissues integrate with Ti implants by allowing direct ultrastructural analysis of the cell to Ti interface of different topographical features and chemical modifications. This will assist in the development new implants with improved tissue integration.
Experimental design
Selection of the nanochip substrate
In this protocol we describe the fabrication of quartz substrates in detail. The same protocol can be adapted to the fabrication of silicon substrates with minor modifications. Nanostructures can also be made from other materials that are compatible with the lithography process. A key consideration is whether the material is compatible with cell culture. Another important consideration is the material’s compatibility with the imaging or testing methods. Some materials such as quartz are transparent, while others such as silicon are not. Other material properties (such as porosity, rigidity, and chemical composition) may also affect cell behavior, so they should be scrutinized according to the research purpose.
Design of nanostructures
The design of nanostructure patterns should also be planned with respect to the intended biological applications. Several key parameters of the nanostructures should be considered: shape, height, spacing, size, and array arrangement (Fig. 1).
Fig. 1 |. Overview of the nanochip-based cell culture platform for manipulating cell membrane curvature.
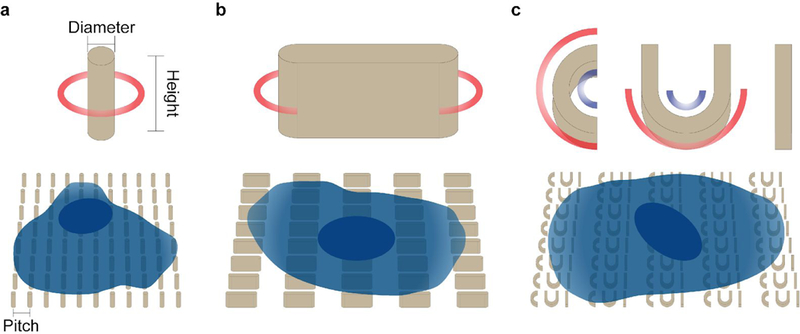
a, Schematics of a single nanopillar and a cell cultured on a nanopillar array. The key configurations include the diameter of circular cross-section of nanopillar, the height of nanopillar, and the pitch between nanopillars, b, Schematics of a single nanobar and a cell cultured on a nanobar array, c, Schematics of a CUI structure and a cell cultured on a CUI array. Red circles and arcs in the schematics indicate positive curvatures, and blue arcs negative curvatures.
The shape of the nanostructures determines whether membrane deformation is positive (invagination toward cell body) or negative (protrusion toward the extracellular space). These two types of curvatures are involved in distinctive cell processes and proteins. Of note, negative curvature is relatively difficult to generate because previous studies have shown the plasma membrane does not protrude far enough to conform to pore-like structures36. The height of nanostructures is usually chosen to be 1–2 μm. which induces a good amount of curved membrane without significantly perturbing cell behaviors. For spacing between nanostructures, we usually choose 2–5 μm The plasma membrane does not fully wrap around dense nanostructures, while very sparse nanostructures will give less curved membrane sites for observation and analysis. The size of nanostructures determines the value of induced membrane curvature. For example, 100-nm-diameter nanopillars will generate a higher curvature value than 900-nm-diameter nanopillars. We have shown that protein accumulation strongly depends on the value of membrane curvature39. The fabrication described here can generate nanopillars with a diameter lower limit at 50–100 nm, and no upper limit. For cellular studies, the 100–1000 nm range would be a good starting point. Large area patterns can be designed with arrays of uniform nanostructures, or arrays of nanostructures with different size parameters. For example, we have used a gradient nanopillar array to quantitatively measure the curvature preference of proteins. Gradient nanopillar arrays are composed of nanopillars with gradually increasing diameters from 100 nm to 1000 nm with a 32 nm incremental step. Gradient arrays of other shapes can be easily designed.
Beside the design of nanostructures, we recommend fabricating fiducial marker (e.g., square patterns or letters) that are in micrometer scale and located at the comers of the nanostructure array used for membrane deformation. Due to the small size of nanostructures, they are often not visible under an optical microscope, making it difficult to locate the positions of curved membranes. Micrometer fiducial markers are easily observable, making much easier to locate nanostructures.
Analytical methods
To observe the effect of membrane curvature on intracellular proteins and cellular processes, we used fluorescence imaging. Both immunostaining after fixation and living cell imaging after transfecting cells with target proteins fused with a fluorescence protein are compatible with our system. Using the nanostructure platforms, both the curvature value and the location of the membrane curvature are precisely defined, which makes it possible to perform meaningful quantitative analysis. We developed a MATLAB graphical user interface, which measures the fluorescence intensity at each nanostructure location and average over thousands of nanostructure pictures to obtain a quantitative value. It is worth noting that the fluorescently tagged proteins may appear to accumulate at nanostructure locations due to a large membrane area wrapping on nanostructures instead of curvature-related protein accumulation. To avoid this effect, we normalize the protein fluorescence intensity by the membrane intensity at the same location. The readout for the plasma membrane can be a fluorescence protein specifically located at the plasma membrane or a lipid dye such as CellMask. Another option is to design a nanostructure that affords two different membrane curvatures and thus has an internal reference on its own. For example, the “bar” structure we designed (Fig. 1b) has two curved ends with high curvature values and two flat side walls with zero curvature for internal reference. The intensity readouts from the two curved ends can be normalized to the intensity readouts from the flat side walls, so that the effects of curvature are convincingly revealed.
MATERIALS
REAGENTS
Nanofabrication
Quartz wafers (525 μm thickness; Silicon Materials Inc., cat. no. 04Q 525–25-1F-SO)
Quartz slips (200 μm thickness; Technical Glass Products)
Si wafers (525 μm thickness, Code-C test; Pure Wafer, cat. no. ST4N05–10TST-C/B)
Acetone (J.T.Baker, cat. no. 9005–05) ! CAUTION It is flammable, and may cause irritation of respiratory tract. Use personal protective equipment.
2-Propanol (Isopropanol, IPA) (J.T.Baker, cat. no. 9079–05) ! CAUTION It is flammable, and may cause irritation of respiratory tract. Use personal protective equipment.
CSAR 62 E-beam photoresist (9% concentration; AR-P 6200, ALLRESIST)
Electra 92 (AR-PC 5091, ALLRESIST)
Deionized (DI) water
Xylene (J.T.Baker, cat. no. 9493–03)
Kapton tape
Diffusion pump oil (Santovac)
Cr etchant (20 % eerie ammonium nitrate, 10 % acetic acid, and 70 % water)
Cell culture and imaging
(3-Aminopropyl)triethoxysilane (APTES) (Sigma-Aldrich, cat. no. 440140)
NIH-3T3 mouse embryonic fibroblast cells (ATCC® CRL-1658™)
U-2 OS human bone osteosarcoma epithelial cells (ATCC® HTB-96™, gifted from David Drubin lab, University of California, Berkeley)
COS-7 Chlorocebus aethiops kidney fibroblast cells (ATCC® CRL-1651™)
HEK 293FT human embryonic kidney cells (Invitrogen, gifted from Velia Siciliano lab, Istituto Italiano di Tecnologia)
Dulbecco’s Modified Eagle’s Medium (DMEM) - high glucose, without glutamine (Sigma-Aldrich, cat. no. D6546)
Fetal bovine serum (FBS) (Sigma-Aldrich, cat. no. F7524)
Penicillin Streptomycin (Pen Strep) 100× (Sigma-Aldrich, cat. no. P4333)
Gibco™ GlutaMAX™ Supplement 100× (Thermo Fisher Scientific, cat. no. 35050061)
MEM non-essential amino acids 100× (EuroClone, cat. no. ECB3054D)
Trypsin-EDTA (0.25%), phenol red (Thermo Fisher Scientific, cat. no. 25200056)
Poly-L-lysine 0.01% (wt/vol) (Sigma-Aldrich, cat. no. P4707)
Gelatin from bovine skin (Sigma-Aldrich, cat. no. G9391)
Trypan blue solution 0.4% (Thermo Fisher Scientific, cat. no.15250061)
Phosphate buffered saline (PBS) 10× (Thermo Fisher Scientific, cat. no. AM9624)
DI water (Thermo Fisher Scientific, cat. no. 10977015)
Hydrogen peroxide 30% (wt/wt) (Thermo Fisher Scientific, cat. no. 10386643)
Sulfuric acid (Thermo Fisher Scientific, cat. no. A300–212)
Lipofectamine™ 2000 Transfection Reagent (Thermo Fisher Scientific, cat. no. 11668027)
Adenosine 5’-triphosphate (ATP) disodium salt hydrate (Sigma-Aldrich, cat. no. A6419)
Magnesium chloride hexahydrate (Sigma-Aldrich, cat. no. M2393)
Potassium phosphate monobasic (Sigma-Aldrich, cat. no. P5655)
Sodium bicarbonate (Sigma-Aldrich, cat. no. S5761)
Ethanol, absolute (200 Proof) (Thermo Fisher Scientific, cat. no. BP2818500)
Paraformaldehyde (PFA) (Sigma-Aldrich, cat. no. P6148)
Plasmids encoding protein of interest (e.g. Dynamin2-EGFP and EGFP-CAAX)
Nail polish (Thermo Fisher Scientific, cat. no. 50–949-071)
HyClone™ Dulbecco’s Modified Eagle’s Medium-high glucose, L-Glutamine, without phenol red (GE Healthcare, cat. no. SH30284.01)
FIB-SEM
Sodium cacodylate (0.2 M, pH 7.4; Electron Microscopy Sciences, cat. no. 11652) ! CAUTION It is irritant to skin and eyes and toxic by inhalation and ingestion. Handle with gloves, goggles and mask and work in a fume hood.
Glutaraldehyde (25% aqueous solution; Sigma-Aldrich, cat. no. G6257)! CAUTION It is hazardous in case of skin contact or inhalation. Handle with gloves and mask and work in a fume hood.
Glycine (BioXtra ≥ 99%, titration; Sigma-Aldrich, cat. no. G7430) ! CAUTION It is irritant to skin and eyes and toxic by inhalation and ingestion. Handle with gloves, goggles and mask and work in a fume hood.
Osmium tetroxide (4% aqueous solution; Electron Microscopy Sciences, cat. no. 19170) ! CAUTION It is very toxic on inhalation and ingestion. Avoid skin and eyes contact. Handle with gloves, goggles and mask and work in a fume hood.
Osmium tetroxide (2% aqueous solution; Electron Microscopy Sciences, cat. no 19172)! CAUTION It is very toxic on inhalation and ingestion. Avoid skin and eyes contact. Handle with gloves, goggles and mask and work in a fume hood.
Potassium ferricyanide (20% aqueous; Electron Microscopy Sciences, cat. no 25102–20) ! CAUTION Avoid eye contact. Handle with gloves, goggles and work in a fume hood.
Thiocarbohydrazide (Sigma-Aldrich, cat. no 21900)! CAUTION It is lethal if inhaled, ingested or in contact with skin and eyes. Handle with gloves, goggles and mask and work in a fume hood.
Uranyl acetate reagent ACS ! CAUTION It is fatal if inhaled and swallowed, causes damage to organs after prolonged and repeated exposition. It has risk of cumulative impact. Handle with gloves, goggles and mask and work in a fume hood. Wear dosimeter.
Tannic acid reagent ACS (Sigma-Aldrich, cat. no. 403040) ! CAUTION It is hazardous in case of skin and eyes contact. Avoid inhalation and ingestion. Handle with gloves, goggles and mask and work in a fume hood.
Ethanol absolute anhydrous (Carlo Erba, cat. no. 414608) ! CAUTION It is slightly hazardous in case of skin and eyes contact. Handle with gloves and work in a fume hood.
Molecular biology reagent (MBR) water (Sigma-Aldrich, cat. no. W4502)
Low viscosity embedding kit (Electron Microscopy Sciences, cat. no. 14300) ! CAUTION Every reagent, included in the kit, is hazardous if inhaled and swallowed. Handle with gloves and mask and work in a fume hood.
EQUIPMENT
Nanofabrication
L-Edit (Tanner)
Sonicator
Hotplate
YES prime oven (Yield Engineering System)
BEAMER (GenlSys)
Spin-coater
E-beam lithography system (JBX-6300FS, JEOL)
AJA E-beam evaporator (AJA International)
PlasmaTherm oxide etcher (PT-OX)
Optical microscope
Scanning electron microscope
Tweezers
Cell culture and imaging
1.5 and 15 ml centrifuge tubes (Eppendorf)
Centrifuge, with adapters for 1.5 and 15 ml centrifuge tubes (Thermo Fisher Scientific)
Tissue culture incubator maintaining 5% CO2. and humidified atmosphere at 37 °C (Thermo Fisher Scientific)
Laminar flow cabinet providing a sterile environment
Tissue culture flasks vented cap (Coming)
TC Flask 75 cm2 (VWR, cat. no. 734–0050)
TC Flask 25 cm2 (VWR, cat. no. 734–2272)
Water bath
Sterile bench for cell culture
Amaxa™ Nucleofector™ II (Lonza)
Plasma cleaner and decontaminator
Fully automated inverted epifluorescence microscope (Leica, DMI6000 B)
High magnification (60× or 100×) objective for epifluorescence microscope (Leica)
Tissue culture dish (35 mm diameter; Coming)
0.2 cm electroporation cuvette (Thermo Fisher Scientific, cat. no. P45050)
Millex-GV Syringe Filter Unit (0.22 μm; Millipore Sigma, cat. no. SLGV033RS)
Nalgene™ Rapid-Flow™ sterile disposable filter units (Thermo Fisher Scientific, cat. no. 09–741-02)
MATLAB (MathWorks)
ImageJ (open source software)
Prism (GraphPad software)
FIB-SEM
Accurate analytical balance
Ampoule cracker
Chemical hood
Dual beam Helios 600 (FEI, Thermo Fisher)
Falcon tubes (15–50 ml)
Foil for alimentary
Laboratory oven
Laboratory fridge
Laboratory scalpel
Molecular biology reagent-grade water (MBR water)
Parafilm M (4 in × 125 ft roll; Fisher Scientific)
Plastic Petri dish (35 mm × 10 mm, non-treated dish; USA Scientific)
Plastic pasteur pipette
Plastic syringe tube (50 ml)
Scissors
Silver conductive paint (RS Pro)
Spatula
Sample supports for SEM
Sputtering machine
Tweezers
Water bath
REAGENT SETUP
Cell culture medium
Prepare 50 ml culture medium by mixing 45 ml DMEM, FBS (10% vol/vol, final concentration), 500 μl of P/S (1% vol/vol, final concentration), 500 μl of MEM non-essential amino acids 100× (1% vol/vol, final concentration), 500 μl of Gibco™ GlutaMAX™ Supplement (1% vol/vol, final concentration).
Sodium cacodylate buffer (0.1 M)
Prepare sodium cacodylate buffer solution (pH 7.2–7.3) by dissolving 50 ml of 0.2 M sodium cacodylate in 50 ml MBR water. This solution can be stored at 4 °C.
Glutaraldehyde primary fixative
Dilute the stock solution of glutaraldehyde from 25% (wt/vol) to 2.5% in 0.1 M sodium cacodylate solution. The solution has to be freshly prepared right before use.
▲ CRITICAL STEP If the required volume is substantial, it is feasible to increase the volume with MBR water and simultaneously dilute stock solutions of sodium cacodylate and glutaraldehyde. Therefore, 50% of the final volume is 0.2 M sodium cacodylate, 40% of the final volume is MBR water and finally 10% of the final volume is glutaraldehyde 25%.
Glycine solution
Mix 30 mg mol−1 of glycine powder in 20 ml of 0.1 M sodium cacodylate buffer until the powder becomes indiscernible.
Secondary fixative
In the chemical hood, take the glass vial containing 5 ml of 4% (wt/vol) tetroxide osmium aqueous solution, carefully break up its upper part using the ampoule cracker and transfer the whole content into a tube with 500 μl of 20% potassium ferrocyanide. Add 4.5 ml of 0.1 M sodium cacodylate buffer to obtain 10 ml secondary fixative. Cover the tube with foil and store it in the dark at room temperature (RT).
Thiocarbohydrazide (1% (wt/vol))
Mix 500 μg thiocarbohydrazide in 50 ml MBR water, preheated to 60 °C. Move the falcon tube upside down several times until salts become indiscernible. Wait until the solution cools down to RT and filter it before use with a 0.22 μm filter.
Tannic acid solution (0.15% (wt/vol))
Dissolve 7.5 μg tannic acid in 50 ml MBR water.
Ethanol solutions for dehydration
Prepare a series of ethanol solutions at ascending concentrations (30, 50, 70, 90, 95% (vol/vol)) by MBR water dilution. Store them in a freezer, so that they are ready for the operations performed on ice. Instead, 100% ethanol should be stored at room temperature.
Spurr’s low viscosity embedding mixture
The mixture is obtained by sequentially adding 10 ml ERL 4221, 8 ml D.E.R. 736 Epoxy Resin and 25 ml Nonenyl Succinic Anhydride Modified (NSA) to a container. After mixing them, add 301 μl Dimethylaminoethanol (DMAE), as the catalyst. All these components are included in the embedding kit. The mixture can be stored in a disposable syringe in a freezer. Remove all the air bubbles. It is recommended to use a vacuum chamber to remove all bubbles otherwise those can create artefact in the cells (Supplementary Fig. 1) and cover the mixture with parafilm in order to avoid air contact. It is highly recommended to prepare it right before use.
▲ CRITICAL STEP If the resin is stored in the freezer, it needs to come to room temperature prior to use, to prevent water condensation.
? TROUBLESHOOTING
Piranha solution
Make a mixture of one part hydrogen peroxide solution (30 % (wt/wt) in water) and three parts of concentrated sulfuric acid by adding hydrogen peroxide to sulfuric acid slowly. Mix gently and expect the container to become hot.
! CAUTION Fast mixing may lead to boiling or splashing. Piranha solution is highly corrosive and an extremely oxidative. Handling piranha requires complete personal protection and with great care. Must prepare and handle it in a functioning fume hood without the presence of organic solvent. Only use glassware or Teflon for piranha solutions. Clearly label the hood and container with a warning sign.
Gelatin in PBS (0.1% (wt/vol))
Prepare 2% (wt/vol) gelatin solution by adding gelatin in DI water. Dissolve gelatin and sterilize by autoclaving at 121 °C, 15 psi for 15 min. Cool the solution to room temperature and prepare 0.1% (wt/vol) gelatin in PBS by mixing 2% (wt/vol) gelatin solution, 10X PBS and DI water.
Electroporation buffer I
Dissolve ATP disodium salt hydrate and magnesium chloride hexahydrate in DI water. The final concentrations of ATP and magnesium chloride are 360mM and 600mM, respectively. Sterilize the solution by a 0.22 μm syringe filter.
Electroporation buffer II
Prepare an aqueous solution containing 88 mM Potassium phosphate monobasic and 14 mM sodium bicarbonate, pH 7.4. Sterilize the solution by a 0.22 μm syringe filter.
PFA solution (4% (wt/vol))
Heat 1 PBS to approximately 60 °C and add PFA powder to it. Slowly add 1 M NaOH dropwise while stirring until the solution clears. Cool the solution to RT. Adjust the pH with diluted HC1 to 6.9. Sterilize the solution by a 0.22 μm filter unit. Aliquot and freeze the solution at −20 °C.
EQUIPMENT SETUP
E-beam lithography system
The JEOL E-beam lithography system utilized in this protocol requires several steps for setting before the sample can be loaded and the job files can be executed. First, electron optical system (EOS) ought to be set to the parameters that are required in the fabrication. It is advised to run test samples and find the EOS setting that has sufficient writing resolution for the designed patterns while taking shorter time. In the E-beam writing with JBX-6300FS equipment and 275 nm thick 9% CSAR 62 E-beam resist, we use 9000 pA current and 130 μm aperture diameter in EOS mode 3 (fast mode). Second, exposure dose should be tuned in tests. It is recommended to fabricate a series of simple patterns (such as circles) with varying sizes, and develop the samples after exposure to see which the doses generate precise nanostructures. Third, after loading samples into the fabrication chamber, calibrations should be executed to confirm that the equipment is running properly prior to E-beam writing.
Besides equipment settings, the JEOL E-beam lithography system we use reads exposure details in jobdeck (.jdf) and schedule (.sdf) files, and it requires preparation. First, the design files generated with computer-aided design (CAD) software should be saved in GDSII format (.gds file). Second, the .gds file is converted to J52 v3.0 (,v30 file) with the BEAMER software (Fig. 2a). The settings in the conversion should be scrutinized to verify the patterns fractured by the software maintain sufficient accuracy (Fig. 2b). Third, .jdf and .sdf files are edited to describe the exposure details. The .jdf file specifies the substrate, arrangement of patterns (.v30 files), the sequence for writing patterns, exposure methods, among others. The .sdf file cites the .jdf, and contains such information as the overall offsets of patterns in the exposure window and dose variations. After the files are edited, they should all be stored in the control computer of the E-beam lithography system and compiled into a single magazine (.mgn) file executable to the system.
Fig. 2 |. Screenshot of pattern design and conversion.
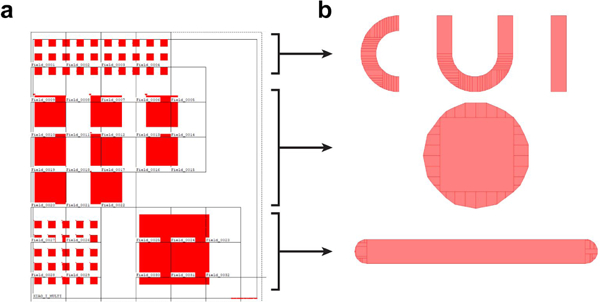
a, Screenshot of overall nanostructure design in the BEAM software. The region of nanostructures is divided into E-beam writing fields. b, Fragmented individual patterns. The individual patterns correspond to the arrays in the overall design: CUI (top), pillar (middle) and bars (bottom).
PROCEDURE
Nanofabrication
Design of nanostructures and fabrication file preparation ● Timing 1–2 h
-
1
Design the nanopattems in L-Edit and save the design as a .gds file.
-
2
The .gds design file is first converted into .v30 files in the BEAMER software (Fig. 2). Edit the jobdeck file (.jdf) and schedule file (.sdf). Store all the files in the control computer of the JEOL E-beam writer and compile them into a single magazine (.mgn) file. (Details are described in EQUIPMENT SETUP.)
Substrate cleaning ● Timing 1 h
-
3
Cut a 100 mm quartz (or Si) wafer into 20 mm × 20 mm square pieces with DISCO wafersaw. Mark the top surface of each piece with a diamond-tipped stylus.
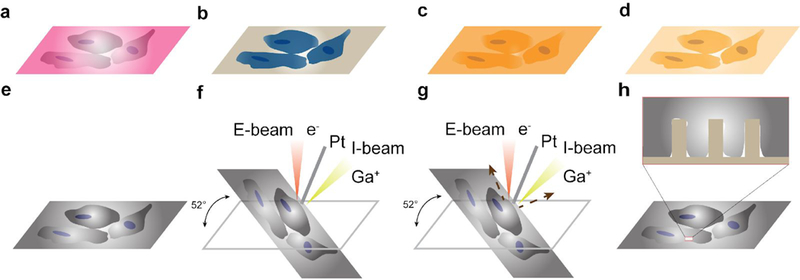
▲ CRITICAL STEP The nanofabrication procedure (Steps 3–47) is applicable to various substrate materials, and most of steps ought to be done in clean rooms to minimize the impurities on chips (Fig. 3).
-
4
Sonicate the substrate pieces in acetone for 5 min. Multiple pieces could be placed in one dish of sufficient acetone to save time, under the premise that they do not overlap during sonication.
-
5
Wash the pieces with a squeeze bottle of IPA to remove acetone, before placing them to a dish of IPA.
-
6
6| Sonicate the substrate piece in IPA for 5 min. Multiple pieces can be placed in one dish of sufficient IPA to save time, under the premise that they do not overlap during sonication.
-
7
Blow dry the piece with nitrogen (N2) gas.
-
8
Heat the substrate piece on a 90 °C hotplate for 10 min to ensure the piece is completely dry.
-
9
Inspect the pieces with an optical microscope to verify the surface is clean.
▲ CRITICAL STEP It is very important to thoroughly clean the substrate piece, which should be done in a clean room. Impurities on the substrate surface may result in a non-uniform E-beam resist coating and detachment of the Cr mask in the lift-off step. Protect the top surface of the substrate piece from scratching.
■ PAUSE POINT The cleaned substrate pieces can be kept in a clean container. The container should only be opened in a clean room to avoid particle contamination of the surface.
Fig. 3 |. Workflow of nanofabrication.
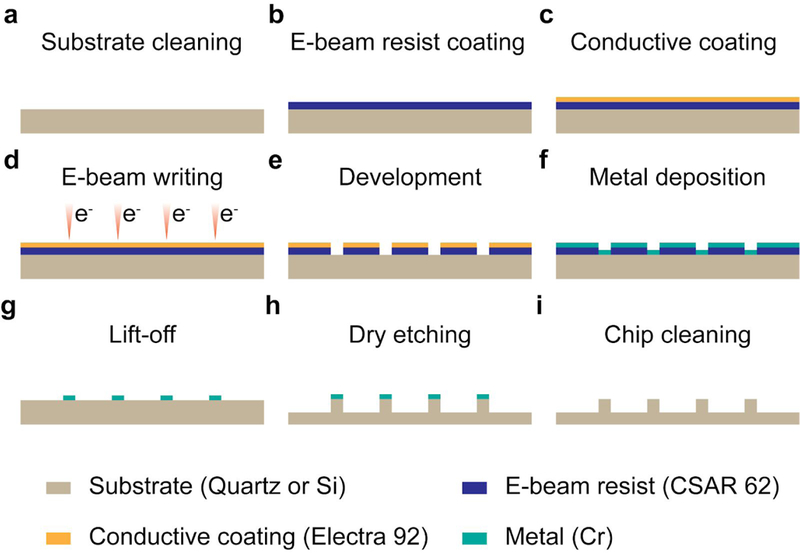
a-i, Steps involved in e-beam writing based nanochip fabrication. A substrate piece is cleaned by sonication in acetone and IPA (a), prior to the E-beam resist coating (CSAR 62) (b) and conductive coating (Electra 92) (c). E-beam writing is done with JEOL JBX-6300FS system (d), followed by the development in xylene and the cleaning in IPA (e). A metal (Cr) is deposited to the substrate piece (f) and then lifted-off along with E-beam resist (g), leaving patterned masks. Dry etching with PT-OX generates nanostructures (h), after which the metal masks are moved with an etchant (i).
E-beam resist and conductive coating ● Timing 30 min
-
10
Use YES prime oven to dehydrate the substrate pieces at 150 °C and coat the wafers with hexamethyldisilazane to induce better coverage of E-beam resists afterwards.
-
11
Spin-coat 9% CSAR 62 at 2000 rpm to generate a 275 nm layer of E-beam resist (in reference to the calibration curve of the manufacturer).
-
12
Bake the substrate piece for 2 min at 80 °C.
-
13
Spin-coat Electra 92 at 1000 rpm to generate a 100 nm conductive coating (in reference to the calibration curve of the manufacturer).
-
14
Bake the substrate piece for 2 min at 80 °C.
-
15
Inspect E-beam resist and conductive coating with an optical microscope. If defects are observed in the regions for exposure, consider cleaning the substrate and redoing the coating. Electra 92 can be removed by deionized water, and CSAR 62 can be removed by sonication in acetone.
■PAUSE POINT The coated substrate pieces can be kept in a clean container for multiple days. The container should only be opened in a clean room.
E-beam writing and development ● Timing 3 h
-
16
Set JEOL JBX-6300FS E-beam writer (Fig. 4a) to EOS mode 3 with 9000 pA current and 130 μm aperture diameter. (Details are described in EQUIPMENT SETUP.)
-
17
Load the substrate pieces to an E-beam writer cassette (Fig. 4b).
▲ CRITICAL STEP Ensure the surface with E-beam resist is on top and the cassette holder has sufficient exposure window for all the patterns to write.
-
18
Load the cassette into the E-beam writer.
-
19
Run calibrations in the E-beam writer user interface to align the beam and confirm the equipment functions properly.
-
20
Run the .mgn file generated in Step 2 to write. The writing progress can be monitored in the E-beam writer user interface.
▲ CRITICAL STEP The writing time depends on the pattern area, dose, current, among other parameters. We normally place two 20 mm × 20 mm square pieces into the cassette in a given writing session (Fig. 4b). Each substrate piece contains four 3 mm × 3 mm nanopattem arrays. To save time, load more samples in the cassette, because multiple loadings require extra chamber pumping and venting, and calibrations.
-
21
After writing, carefully take the substrate pieces off the cassette and avoid scratching the surface.
-
22
Remove the protective coating of Electra 92 with running DI water. Blow dry the substrate with a N2 gun.
-
23
Place the substrate pieces in a dish of xylene for development, and lightly agitate the liquid for 40 s.
-
24
Take the substrate pieces out of xylene, and wash them with IPA from a squeeze bottle.
-
25
Place the substrate pieces in a dish of IPA, and lightly agitate the liquid for 10 s.
-
26
Take the substrate out of IPA and blow it dry with N2 gun.
-
27
Inspect the substrate with an optical microscope. Nanoscale patterns should be visible under an optical microscope (Fig. 5a-c).
■PAUSE POINT The developed substrate pieces can be kept in a clean container. The container should only be opened in a clean room.
Fig. 4 |. E-beam writing system.

a, Photo of JEOL E-beam writing system. The samples are transferred into the machien from the loading chamber, b, Photo of samples in a cassette. The substrate pieces (coated by CSAR 62 and Electra 92) are fixed in a holder which is then placed in a cassette.
Fig. 5 |. Inspections through nanofabrication process.

a-c, Optical micrographs of quartz pieces after E-beam writing and development, including 200 nm diameter pillars with a 2.5 μm pitch (a), bars with varying widths (b), CUI patterns (c). d-f, Optical micrographs of quartz pieces after metal deposition and lift-off, including 200 nm diameter pillars with a 2.5 μm pitch (d), bars with varying widths (e), CUI patterns (f). Scale bars, 5 μm.
Cr deposition, lift-off and cutting ● Timing 2 h
-
28
Mount the developed substrates to a carrier wafer with Kapton tape on the comers. Make sure the surface with E-beam resist faces up. Blow the substrates with a N2 gun.
-
29
Load the carrier wafer into the AJA deposition machine. Deposit a 110 nm thick layer of chromium (Cr) at a 1 Å s−1 speed. In order to achieve a uniform coating, it is recommended to rotate the sample holder during deposition.
■PAUSE POINT The deposited substrate pieces can be kept in a clean container for up to 24 h. The container should only be opened in a clean room. Note that metal might get oxidized after a long storage and become difficult to remove in lift-off.
-
30
Wash a substrate piece with a squeeze bottle of acetone to remove the E-beam resist and the Cr layer. The Cr film detachment should be visible.
-
31
Before the substrate piece dries, place it in a dish of acetone. Sonicate it for 4 min.
-
32
Take the substrate piece out of acetone and wash it with a squeeze bottle of IPA.
-
33
Immerse the substrate piece in IPA and sonicate it for 4 min.
-
34
Inspect the substrate with an optical microscope. Nanopattems should be visible. (Fig. 5d-f)
▲ CRITICAL STEP The lift-off step is crucial and requires careful optimization. Insufficient lift-off fails to remove the Cr layer effectively, which may generate i) abnormal Cr patterns that compromise the pattem-transfer accuracy in etching, ii) residues on the chip (especially among the dense Cr patterns) that interfere with etching or generate a non-smooth substrate surface after etching. Excessive lift-off may damage or remove Cr patterns. Uncleaned surface of substrate may also cause detachment of Cr patterns. (Supplementary Fig. 2) It is prudent to frequently inspect the substrate after a round of acetone and IPA sonication, to decide if further sonication is required. Additionally, it is advised to lift-off one substrate at a time and to use fresh acetone and IPA each time, to minimize the interference from Cr waste in the liquid and to control the lift-off process more closely.
? TROUBLESHOOTING
-
35
Cut each substrate piece into individual 10 mm × 10 mm chips with DISCO wafersaw. Set the parameters appropriately so that the piece is not cut through and can be broken later by bending along the notches (Supplementary Fig. 3).
■PAUSE POINT The chips can be kept in a clean container until etching is required.
? TROUBLESHOOTING
Dry etching ● Timing 1 h
-
36
Clean the substrate pieces in acetone and IPA in sequence, and blow dry them with a N2 gun to remove possible dust and residue generated in the cutting process.
-
37
Load a dummy wafer to PT-OX etcher and run a cleaning recipe to remove the possible residue left from a previous etching session.
-
38
Load a dummy wafer again and execute the etching recipe to season the chamber and verify the equipment runs properly.
-
39
Take a clean wafer as the carrier. Apply small drops of Santovac pump oil to the carrier wafer and place the chips on top of the oil drops. The oil facilitates thermal transfer. Make sure the side with the Cr mask faces up. Press the chips so that the oil spreads well underneath them (Supplementary Fig. 3). Attach the comers of the chips to the carrier wafer with Kapton tape. Blow the chips and carrier wafer with a N2 gun to clean the surface.
-
40
Load the wafer carrier with chips into PT-OX etcher and run the etching procedure. Gas stabilization and ignition steps are normally run prior to the main etching step. We use a C4F8-based etching recipe for quartz and a CF4-based recipe for Si (Tables 2 and 3).
-
41
After etching, remove the substrate pieces from the carrier wafer and carefully remove oil from the bottom of the chip with an acetone-soaked wipe.
-
42
Break the piece into individual chips by flexing along the cut notches made by the wafersaw.
-
43
Inspect the chips with SEM. Si chips can be directly observed with SEM, while quartz chips typically require the sputtering of a conductive layer before imaging (Fig. 6).
▲ CRITICAL STEP Considering etching is an irreversible process, it is highly recommended to carry out this step with caution. The etching rate should be calibrated to decide the etching duration. If the height of the nanostructures is below expected, samples can be etched further as long as the Cr mask is not removed yet.
? TROUBLESHOOTING
■PAUSE POINT The chips can be kept in a clean container until cleaning.
Table 2 |.
Dry-etching recipe for quartz substrate
| Parameter (unit) | Value |
|---|---|
| C4F8 (sccm) | 80 |
| Pressure (mT) | 7 |
| RF1 Bias(W) | 80 |
| RF2 ICP (W) | 1500 |
| Electrode temperature (°C) | 30 |
| Etching rate (nm s−1) | 6.25 |
Table 3 |.
Dry-etching recipe for Si substrate
| Parameter (unit) | Value |
|---|---|
| CF4 (sccm) | 40 |
| o2 (sccm) | 2 |
| Pressure (mT) | 15 |
| RF1 Bias (W) | 50 |
| RF2 ICP (W) | 600 |
| Electrode temperature (Celsius) | 30 |
| Etching rate (nm s−1) | 7 |
Fig. 6 |. SEM images of nanostructures after fabrication.

a-c, SEM images of quartz nanostructure arrays, including 200 nm diameter pillars with a 1 .μm pitch (a), bars with varying widths (b), and CUI patterns (c). d-f, SEM images of individual nanostructures, including a 200 nm diameter pillar (d), a 200 nm wide bar (e), and a set of CUI pattern (f). Scale bars, 5 μm (a-c); 500 nm (d-f). Quartz chips are coated with gold for imaging, and the images are taken with 45° tilting.
Chip cleaning ● Timing 2 h
-
44
Place chips in Cr etchant for 10 min. If the chips were coated with a conductive layer for imaging (e.g., 10 nm Au coating), removal of the conductive layer should be done in advance (e.g., gold etchant for Au coating).
-
45
Clean chips with DI water.
-
46
Clean chips in freshly prepared piranha solution heated at 80 °C for 1 h.
! CAUTION Piranha solution, especially when heated, is highly oxidative and volatile. The cleaning step should be conducted in a fume hood with complete personal protection.
-
47
Thoroughly rinse chip with DI water and blow dry them with air.
■PAUSE POINT The cleaned chips can be kept in a clean container until cell experiment.
Cell culture and imaging
Cell recovery ● Timing variable from days to weeks
-
48
To recover cells for culture, thaw cells (U-2 OS, NIH/3T3, COS-7 and HEK) and then transfer the cryopreservation medium with cells in 10 ml of cell culture medium pre-warmed to 37 °C. Spin the tube with cells at 200 x g (RCF) for 3 min at RT and remove the supernatant. Resuspend the cells in culture medium, transfer cells into a T75 or T25 tissue culture flask, and maintain cells at 37 °C with 5% of CO2 and humidified atmosphere in a tissue culture incubator. Passage cells every 3–4 days (depending on cell type) to prevent overgrowth of adherent cell lines. Maintain the cell culture until the day of plating cells onto nanochips.
Surface treatment ● Timing 2 h
-
49
On the day of cell culture, clean the top surface of the nanochip with a plasma cleaner for 20 min.
-
50
For live cell imaging at high magnification, drill a hole (diameter, less than 10 mm) in 35 mm cell culture dish. Carefully place a nanofabricated thin quartz chip (200 μm thickness) to cover the hole from the outside, with nanopattems facing inside of the dish. Apply nail polish at the comers to anchor the chip and air-dry for 2 min. Seal the chip to the dish by carefully painting the edges with more nail polish and repeat multiple times if necessary. Air-dry for at least 15 min before coating.
▲CRITICAL STEP Imaging setup ought to be chosen in accordance with the research purpose. We recommend three setups (Fig. 7). When imaging cells at low magnification (e.g., 10× and 20×). which normally has a long working distance, a thick nanochip can be placed upright in a regular Petri dish (Fig. 7a). When imaging fixed cells at high magnification (e.g., 60× and 100×). which normally has a short working distance, a thick nanochip can be flipped and placed in a glass-bottomed Petri dish (Fig. 7b). For live cell imaging at high magnification, a thin nanochip can be glued to a Petri dish bottom as described in the above step (Fig. 7c).
-
51
For gelatin coating, drop 10 ul 100% APTES onto the chip. Make sure itspreads to the entire chip surface. Incubate the chip at room temperature for 5 min before removing APTES with DI water.
-
52
Treat the chip with 0.5% glutaraldehyde (in PBS) at RT for 30 min.
-
53
Wash the chip with PBS using a pipette.
-
54
Pre-warm 0.1% (wt/vol) gelatin in PBS solution to 37 °C for complete liquefaction. Treat the chip with the gelatin solution at RT for 30 min.
-
55
Gently wash the chip with PBS using a pipette and keep the chip in PBS solution before cell plating.
▲CRITICAL STEP Cells often require specific surface treatment methods for satisfactory attachment to the nanochips. Steps 49–55 described covalent-bonding-based gelatin surface coating (Supplementary Fig. 4), and this surface is used for U-2 OS and HEK cell culture in this protocol. NIH/3T3 and COS-7 were plated on poly-L-lysine (PLL) treated surface instead. For PLL coating, after Step 49, coat the chip with 0.01% PLL at 37 °C for 1–2 h. Gently wash the chip with PBS.
Fig. 7 |. Optical microscopy setups.
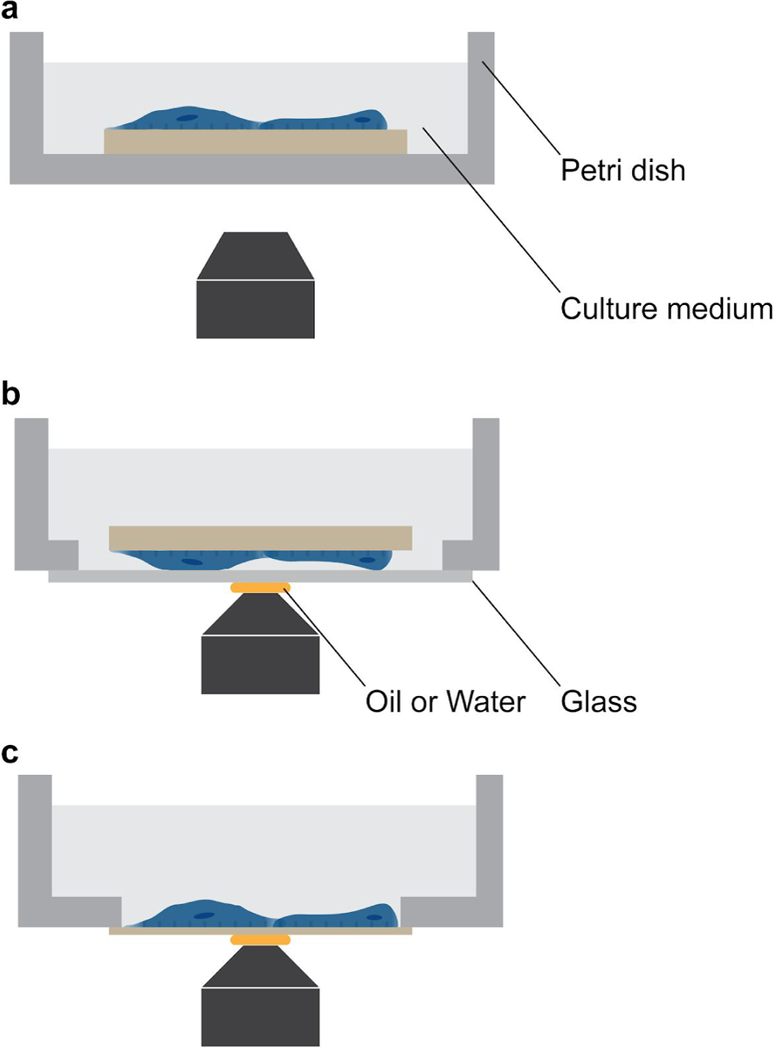
a, Thick nanochip placed in an upright position in a Petri dish for low-magnification imaging. Regular Petri dish can be used for low-magnification objectives whose working distance is relatively long, b, Thick nanochip flipped and placed in a glass-bottom Petri dish for high-magnification imaging. The glass on the bottom is thin, allowing the nanostructures imaged within the relatively short working distance of high-magnification objectives, c, Thin nanochip glued to the bottom of a Petri dish in an upright position. The thin nanochip can be glued with nail polish, which could be dissolved later to clean the nanochip.
Cell plating ● Timing 1 h
-
56
To passage cells, aspirate cell culture media and wash cells once with pre-warmed (37 °C) PBS without Mg2+ and Ca2+. Add 0.25%trypsin-EDTA (0.1 ml cm−2) to cell culture flask and incubate for min at 37 °C to detach cells. Gently pipette up and down to dissociate cells completely into single cells and add equal amount of cell culture medium with 10% (vol/vol) FBS to neutralize trypsin. Transfer the mixture into a 15 ml centrifuge tube, spin it at 200 g (RCF) for 3 min, and remove the supernatant.
▲ CRITICAL STEP Avoid using cells through high passage number, as cell phenotypes may alter after a high number of passages.
-
57
Resuspend cells in 1 ml cell culture medium without Pen Strep. Count the cells using a hemocytometer. Determine their viability by trypan blue exclusion if viability of this cell type is low.
-
58
For transfection with Lipofectamine, dilute 1–2 μg DNA in 100 ul Opti-MEM® I medium without serum. Dilute Lipofectamine™ 2000 in 100 μl Opti-MEM® I medium without serum in a separate tube, stir the tubes gently and incubate at RT for 5 min. Add the diluted Lipofectamine™ 2000 to the tube with the diluted DNA, mix them gently and incubate the solution for 15 min at RT. Dilute about 200,000 cells after counting into 2 ml cell culture medium (with 10% FBS and without Pen Strep) and seed them into a nanochip-mounted 35 mm tissue culture dish. Add DNA-Lipofectamine™ 2000 complex solution into the dish and shake it gently to accelerate the mixing. Maintain the cells at 37 °C in a CO2 incubator for 24–36 h until imaging.
▲ CRITICAL STEP The amount of DNA, Lipofectamine™ 2000 to DNA ratio, and cell type determine the transfection efficiency and final protein expression level in cells. To assess membrane curvature-dependent protein localization, we recommend a relatively low protein expression level in order to minimize protein mislocalization. We used 1–2 μg DNA and a Lipofectamine™ 2000 to DNA ratio of 3 μg−1 to transfect NIH/3T3 cells with Dynamin2-EGFP. When using another DNA construct and cell type, try different conditions for optimized protein expression. Use genome-modified cell lines if necessary.
-
59
Optionally for electroporation, add about 500,000 cells after counting into a centrifuge tube, spin it at 200 × g (RCF) for 3 min and remove supernatant as completely as possible. Resuspend cells in 100 μl electroporation buffer II mixed with 2 μl electroporation buffer I. Add DNA into the tube and mix gently. Transfer cell-DNA suspension into a 0.2 cm electroporation cuvette. Electroporate cells in Amaxa™ Nucleofector™ II with program selected according to the cell type. Immediately add 500 μl pre-warmed culture media without Pen Strep to the cuvette and incubate it 5 min. Resuspend the solution in cuvette and transfer it to a tube. Spin it at 200 × g (RCF) for 3 min, resuspend the cells in pre-warmed culture media and plate them into a nanochip-mounted 35 mm tissue culture dish. After 2 h of incubation at 37 °C in a CO2 incubator, change culture medium to remove dead cells. Maintain transfected cells for 24–36 h until imaging.
▲ CRITICAL STEP The amount of DNA and cell type determine the transfection efficiency and final protein expression level in cells. Tune the amount of DNA in a range of 0.1–2.0 μg to obtain optimal protein expression. We used 0.2 μg Dynamin2-EGFP and 0.5 μg EGFP-CAAX DNA to transfect U-2 OS and COS-7 cells.
? TROUBLESHOOTING
Cell imaging ● Timing variable
-
60
Before live cell imaging, change cell culture media to pre-warmed and 5% CO2 balanced cell culture medium without phenol red.
-
61
Image live cells with an epifluorescence microscope equipped with the appropriate light source, optical filter cubes, high magnification objectives, and objective heater and chambers providing humidified 5% CO2 atmosphere at 37 °C. Use adaptive perfect focus or similar system to avoid focus drifting if time-lapse imaging is required. Focus and sample drifting affect following image analysis. Use light intensity as low as possible, and optimize focus and exposure time as quickly as possible to minimize photobleaching of fluorescent proteins.
▲ CRITICAL STEP Successful membrane curvature generation with nanostructures is the basis for studying curvature related phenomena in cells. So it is advised to label cell membranes with appropriate tags (e.g., EGFP-CAAX for the plasma membrane) in preliminary experiments (Fig. 8).
-
62
Optionally, to image fixed cells, first wash cells with PBS 24–36 h post-transfection and fix cells with 4% PFA in PBS at room temperature for 15 min. Wash fixed cells with PBS three times. If staining is required, treat fixed cells with dye and antibodies following manufacturers’ instructions.
-
63
Nanochips are reusable. After imaging, dissolve dry nail polish by incubating the used culture dish in absolute ethanol for 2 h. Carefully detach the chip from culture dish and let ethanol evaporate completely. Clean chips in freshly prepared piranha solution. Rinse chips with DI water thoroughly.
Fig. 8 |. Micrographs of U2-OS cells cultured on nanochips.
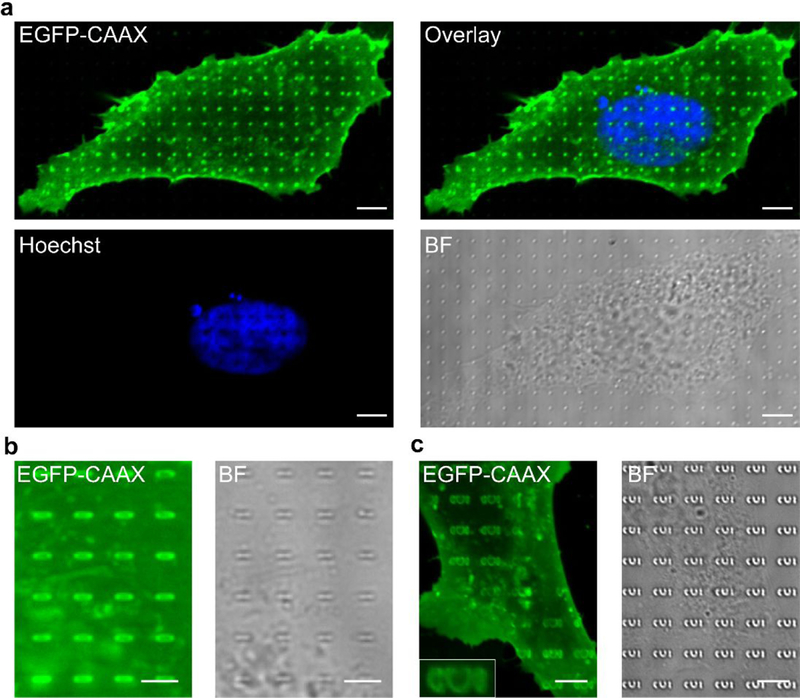
a, Micrographs of a cell cultured on nanopillars of 1 (.un height and 200 nm diameter. The cell is transfected with EGFP-CAAX and stained with Hoechst. b, Micrographs of a part of a cell expressing EGFP-CAAX, cultured on nanobars of 1 μm height, 2 μm length and 200 nm width, c. Micrographs of a part of a cell expressing EGFP-CAAX, cultured on CUI patterns of 1 μm height and 200 nm width. Scale bars, 5 μm. “BF” denotes bright field.
■PAUSE POINT The chips can be kept in a clean container until cleaning.
Image analysis ● Timing variable
-
64
Process and analyse images using ImageJ and MATLAB. We use a custom-written MATLAB code (available upon request) to generate an array of square masks covering the nanobars in the bright field image. These masks are then applied to the corresponding mCherry-CAAX or DNM2-GFP image, and an intensity threshold is used to select only the pillars covered by cells. Finally, the selected nanopillar areas are averaged to create an image of protein accumulation. For time-lapse imaging (for example, images taken every 2 s over a 4 min period), the images are averaged in ImageJ before analysis. For quantitative analysis the images are processed by a rolling ball background subtraction with a radius of 5 μm before analysis. The fluorescence intensity in 500 nm2 squares centred at the end and side of each nanobar are integrated and compared separately to calculate the nanobar end-to-centre ratio.
-
65
Conduct statistical analyses in GraphPad Prism. Set significance level (α) to be 0.05 and no estimated variation within each group of data. Assume equal variances. Report results as P = P value and t (degrees of freedom (df)) = t value.
FIB-SEM
Cell fixation ● Timing 3 h
-
66
Before starting the fixation procedure, it is best to cover the bench with paper towel and foil. In this way, it is possible to confine potential spilling onto the working bench.
▲ CRITICAL STEP The FIB-SEM process (Steps 66–119) requires multiple sample treatments and careful operations with FIB-SEM equipment (Fig. 9).
-
67
Remove the culture medium with a pipette. Be careful not to touch cells and structures.
-
68
Wash the sample by immersion in 0.1 M sodium cacodylate solution for 5 min at RT.
-
69
Next steps need to be carried out on ice. Hence, fill a bowl with ice and cover it with foil.
-
70
Immerse the cell sample in a primary fixative (2.5% glutaraldehyde) for 1–2 h at RT or overnight at 4 °C.
-
71
Remove the fixative and immerse the sample in 0.1 M sodium cacodylate buffer for 5 min at 4 °C. Repeat three times.
■PAUSE POINT It is possible to store the sample in sodium cacodylate buffer for up to 5 days. For longer storage, it is better to proceed to step 73.
▲CRITICAL STEP This step must be carried out on ice. Fill a bucket with ice, place the container of the sample in ice and cover it with foil while waiting.
-
72
Replace sodium cacodylate buffer with 20 mM glycine solution, and keep the sample submerged for 20 min at 4 °C.
-
73
Wash the specimen with sodium cacodylate solution three times at 4 °C, 5 min each time.
Fig. 9 |. Schematics of FIB-SEM workflow.
a, Cells cultured on a nanochip, b, Cells fixed in glutaraldehyde after culture, processed with the RO-T-O procedure and uranyl acetate to increase their contrast, c, Cells on a nanochip embedded in resin, d, resin excess removal and subsequent polymerization. To remove the resin excess, the sample is placed in a vertical position and gently wash them with ethanol, e, Cells on nanochip coated with Au. f, Pt deposition assisted by both E-beam and I-beam, g, I-beam induced cross sectioning, h, Imaging the cell-nanostructure interface after cross-sectioning.
RO-T-O procedure and heavy metals staining ● Timing 21 h
-
74
Replace the sodium cacodylate buffer with a secondary fixative (4% osmium tetroxide and 2% potassium ferrocyanide) and immerse the sample for 1 h at 4 °C. Keep the sample in the dark by covering it with aluminum foil (“RO” step).
▲CRITICAL STEP Osmium tetroxide immersion is a necessary step for FIB-SEM applications because it enhances contrast in FIB-SEM imaging. Osmium tetroxide should penetrate a monolayer of cells in an hour. Very thick tissue samples require a longer immersion time to ensure sufficient penetration.
-
75
Wash the sample with 0.1 M sodium cacodylate solution for 5 min at 4 °C. Repeat two times.
■PAUSE POINT It is possible to store the sample in this solution for several months.
-
76
Leave the sample at room temperature for at least 20 min.
-
77
Wash the sample with MBR water in order to avoid thermal shock and avoid 1% thiocarbohydrazide salt precipitation.
-
78
Remove the water and add fresh filtered 1% thiocarbohydrazide solution to immerse the sample for 20 min at RT (“T” step).
-
79
Wash the sample with MBR water three times at RT, 5 min each time.
-
80
Immerse the sample in a 2% osmium tetroxide aqueous solution for 30 min at RT in the dark (“O” step).
-
81
Wash the sample with MBR water twice at RT, 5 min each time.
-
82
Immerse the sample in filtered (with 0.22 μm filter) 4% uranyl acetate solution at 4 °C overnight.
-
83
Wash the sample with MBR water three times at 4 °C, 5 min each time.
▲CRITICAL STEP Replace the sample container with a new one, after uranyl acetate immersion, to get rid of residues of radioactive materials.
-
84
Remove the water and immerse the sample in 0.15% tannic acid solution for 3 min at 4 °C.
-
85
Wash the sample with MBR water twice at RT, 5 min each time.
Dehydration ● Timing 90 min
-
86
Immerse the sample in 30%, 50%, 70%, and 90% (vol/vol) ethanol in water sequentially at 4 °C, 10 min for each.
▲CRITICAL STEP Be careful not to remove all the liquid when changing solutions, otherwise ethanol could evaporate from cellular compartments causing collapse.
■PAUSE POINT It is possible to leave the sample overnight in ethanol solution of any concentration and place it in the fridge. It is advised to wrap the container of the sample with Parafilm to avoid the evaporation of ethanol.
-
87
Replace the previous solution with 95% (vol/vol) ethanol in water, and wash the sample twice at 4 °C, 10 min each time.
-
88
Wash the sample in 100% ethanol three times, 10 min each time. The first wash should be done at 4 °C, and the other two at RT.
Resin embedding ● Timing 2 days
-
89
When the Spurr’s low viscosity embedding mixture is ready to use (see details in REAGENT SETUP), mix one part resin with three parts 100% ethanol. Immerse the sample in this mixture for 1–2 h at RT.
-
90
Replace the previous mixture with a 1:2 resin to ethanol mixture, and immerse the sample for 1–2 h at RT.
-
91
Leave the sample in a 1:1 resin to ethanol mixture overnight at RT.
▲ CRITICAL STEP Before leaving the sample overnight, wrap its container with parafilm to reduce ethanol evaporation.
-
92
Immerse the sample in a fresh 1:1 resin to ethanol mixture for 2h.
-
93
Immerse the sample in a 1:2 resin to ethanol mixture for 2–3 h.
-
94
Immerse the sample in resin solution (without ethanol) for at least 7 h to provide a complete resin infiltration.
Resin layer thinning and polymerization ● Timing 14 h
-
95
Remove as much resin as possible with a plastic pipette without touching the sample. Carefully remove all the resin excess around and underneath the sample with absorbent paper.
-
96
Place the sample in a vertical position so that excess resin collects at the base due to gravity. Samples can stay vertical for 1–3 h, depending on the desired thickness of the resin layer. The longer the sample stays vertical, the thinner the resin layer will be.
-
97
Hold the sample in a slightly tilted position with tweezers and gently wash the surface by pipetting ethanol.
-
98
Hold the sample in a horizontal position and gently wash it by pipetting ethanol. Wait for 5 s and tilt the substrate to let the ethanol flow away. The volume of ethanol used for the final wash depends on the surface area of the sample.
▲ CRITICAL STEP Removing too much resin may cause artefacts such as cracks in the cell layer or in organelles and intracellular cavities (Supplementary Fig. 5).
? TROUBLESHOOTING
-
99
Lay the sample onto the cleaned and labelled support and place it in the oven preheated to 70 °C. Keep it in the oven for 12 h.
■PAUSE POINT Resin-embedded samples can be stored for a long term without water contact. Water absorption can lead to a sample deterioration.
Sample preparation for imaging ● Timing 100 min
-
100
Place the sample onto a SEM pin stub. Cover the sample edge with conductive silver paste and draw a continuous line from the top of the sample down to the stub, to electrically connect the sample surface to the stub. Leave the silver paste to dry for 1 h before inserting them in the FIB-SEM chamber.
▲ CRITICAL STEP Although the UTP procedure includes heavy metal staining, cells are mostly insulating. Adding a conductive layer on the sample mitigates charging effect in imaging (Supplementary Fig. 6).
? TROUBLESHOOTING
-
101
Place the sample inside the sputter coater and deposit a 10 nm thick layer of gold. The thickness of gold can be adjusted according to the fine structure of the sample to image.
-
102
Mount the sample inside the dual beam chamber of the FIB-SEM equipment.
-
103
Pump the chamber to high vacuum.
-
104
Centre the stage in reference to the E-beam gun.
▲ CRITICAL STEP Be careful when moving the stage inside the chamber and avoid hitting the guns.
FIB-SEM imaging ● Timing variable
-
105
Turn on the E-beam.
-
106
Focus on the sample surface using the secondary electron detector. Apply a voltage between 3 and 15 kV, and a current between 20 pA and 1.2 nA. Magnification, brightness/contrast and stigmation need to be adjusted to yield high-quality images (Supplementary Fig. 7a).
-
107
Select a region of interest (ROI) (Supplementary Fig. 7b).
▲ CRITICAL STEP In case the sample is covered by a large amount of resin which makes the ROI localization challenging, higher voltage and current of the E-beam could be used to improve the visualization of details on sample surface.
-
108
Heat up the platinum (Pt) precursor gas and insert the Pt needle into the chamber before deposition.
-
109
Draw the ROI in the software to cover with a layer of Pt in the FIB-SEM software and proceed with E-beam-induced Pt deposition by fixing a voltage of 3.0 kV and a current in the range of 11–22 nA to generate a thickness between 100 and 500 μm.
▲CRITICAL STEP Both E-beam (in Step 98) and I-beam (in Step 103) induced depositions are applied to cover the ROI with a double layer of platinum and preserve cellular structures (Supplementary Fig. 7c), since the procedure of cross sectioning is very aggressive. Pt gas is injected in close proximity of the sample surface. When it contacts the E-beam, it melts or sublimates due to the high thermal energy produced by the e-beam. The resulting vapor is responsible for the coating. Progressively the E-beam is scanned all over the surface, leading to the Pt deposition layer coating the entire target area. In the same way, during the ion beam-induced deposition, as a consequence of the interaction with the ion beam, Pt gas dissociates into volatile and non-volatile compounds. Here, Pt non-volatile components will be adsorbed onto specimen surfaces, while the volatile ones are evacuated. Because of the high energy of the ion beam, sample milling could take place during deposition. For this reason, it is convenient to run E-beam-induced deposition first and then the I-beam-induced deposition. Deposition time depends on the current value of both I- and E-beams. More specifically, by increasing the current value, a lower deposition time is obtained.
? TROUBLESHOOTING
-
110
Move the Pt needle away from the sample.
-
111
Visualize the Pt-coated area by SEM imaging.
-
112
Find an eucentric point and tilt the stage to 52° and manually adjust the stage height if the stage moves.
▲ CRITICAL STEP Remember to control stage position along the z axis and take great care not to hit the lens or detectors with the sample.
-
113
Turn on the I-beam gun and locate the ROI by imaging with a voltage of 30 kV and a current in the range 1.1–7.7 pA.
-
114
Insert Pt needle again inside the chamber and proceed with ion beam-induced deposition with a voltage of 30 kV and a current from 2.5 nA down to 80 pA, to reach a thickness of 1 μm. The time required for deposition can be adjusted by the current.
▲ CRITICAL STEP Current beyond 2.5 nA can cause the etching of structures and ion contamination around the target area.
-
115
Move the Pt needle away from the sample.
-
116
Visualize the Pt deposition area by SEM imaging.
-
117
Set the area to mill (e.g., 5 μm × 5 μm × 5 μm in x, y and z axes) just in the Pt-coated region and proceed with the ion beam-induced cutting by using a voltage of 30 kV and a current in the range from 65 nA down to 80 pA (Supplementary Fig. 7d).
-
118
Once the cross section is exposed, proceed with I-beam induced polishing to reduce curtaining effects (Supplementary Fig. 8) and remove any contamination due to re-deposition during cutting. Set the polishing area to cover the whole cross section disclosed via the trenching operation and use a voltage of 30 kV and a current in the range of 21 nA down to 80 pA.
▲ CRITICAL STEP It is possible to perform multiple rounds of cross-sectioning along with polishing to rebuild 3D cell structure.
? TROUBLESHOOTING
-
119
Acquire the cell ultrastructures by SEM with the backscattered electron detector. If possible, select dynamic focus, given the stage is tilted to 52°. It is advised to use a dwell time in the range of 30–100 (is, voltage of 3–5 kV, and a current between 0.4 and 1.2 nA. Adjust focus, magnification, brightness/contrast, and stigmation in order to acquire high contrast images.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| Resin preparation (REAGENTS SETUP) |
Micro-nanobubbles formation (Supplementary Fig. 1) | Air trapped in the resin | Remove bubbles by placing the resin container in a vacuum chamber |
| 34 | Missing Cr masks (Supplementary Fig. 2) | Excessive sonication or unclean surface of the substrate pieces that makes Cr attachment to the substrate less firm | The initial cleaning of the substrate must be thorough and the substrate should be carefully checked to make sure there is no stain left in the region to pattern. In the sonication process of lift-off, the chip should be frequently checked |
| 43 | Collapse of nanostructures (Supplementary Fig. 9) | Undercut due to the heat built up at the bottom of the nanostructures | Make the heat transfer from chip to the wafer holder sufficient. Ensure the surface of the holder is clean, and apply vacuum pump oil between chip and wafer holder to facilitate the heat transfer |
| S59 | Poor cell attachment | Inappropriate surface treatment method | First, choose a chemical (such as collagen and gelatin) that effective attaches the cells of interest. Interactions between surface chemistry and cells largely vary, so it is advised to learn from references. Second, utilize a surface treatment approach that effectively immobilize the chemical for attaching cells. Covalent bonding based process is generally strong and reliable, although simple dropping and incubating may also suffice |
| 98 | Damage to cells in resin excess removal (Supplementary Fig. 5) | Placing the sample vertically for too long or washing the sample with too much ethanol can cause detachment of cells and cracks of cellular structures (SI | The optimal resin removal depends on the area of the sample. Before wash the critical samples, test dummy samples of an area comparable to experimental samples |
| 109 | Charging in SEM imaging (Supplementary Fig. 6). | The sample is not conductive enough or well grounded | Properly ground the sample to the sample holder by connecting them with a conductive paste. We suggest to deposit approximately 10 nm of metal on to the specimen. This thickness can be adjusted depending on the sample composition (non-conductive polymer based samples might need 20–25 nm thick metal) |
| 118 | Curtaining effect (i.e., increased surface roughness in the direction of the milling depth) in polishing a cross section (Supplementary Fig. 8) | Underdose of the ion beam | Use low polishing currents (i.e. 80 pA) at a voltage of 30 kV |
● Timing
Nanofabrication
Steps 1 and 2, design of nanostructures and fabrication file preparation: 1–2 h
Steps 3–9, substrate cleaning: 1 h
Steps 10–15, E-beam resist and conductive coating: 30 min
Steps 16–27, E-beam writing and development: 3 h
Steps 28–35, deposition, lift-off and cutting: 2 h
Steps 36–43, dry etching: 1 h
Steps 44–47, chip cleaning: 2 h
Cell culture and imaging
Step 48, cell recovery: variable from days to weeks
Steps 49–55, surface treatment: 2 h
Steps 56–59, cell plating: 1 h
Steps 60–03, cell imaging: variable
Steps 64 and 65, image analysis: variable
FIB-SEM
Steps 66–73, cell fixation: 3 h
Steps 74–85, RO-T-O procedure and heavy metals staining: 21 h
Steps 86–88, dehydration: 90 min
Steps 89–94, resin embedding: 2 days
Steps 95–99, Resin layer thinning and polymerization: 14 h
Steps 100–104, Sample preparation for imaging: variable
Steps 105–119, FIB-SEM imaging: variable
ANTICIPATED RESULTS
Nanostructures obtained with this fabrication method are shown in Figure 6. After proper surface treatment described in Steps 49–55 and cell plating described in Steps 56–59, cells on the nanostructures should grow well and have their plasma membrane conformed to the nanostructures. This cell culture system can be used for various research purposes when combined with imaging, perturbation, and measurement techniques. In this protocol we present results from live cell imaging with fluorescence and from FIB-SEM imaging.
As shown in Figure 8a, cells are transfected with EGFP-CAAX which labels the plasma membrane and are stained with Hoechst to visualize the nucleus. The plasma membrane tightly wraps around the 200 nm diameter pillars, which is seen as fluorescent puncta spots because the nanopillar diameter is beyond the resolution of the epifluorescence microscope. Nuclear staining with Hoechst contains voids, indicating the nucleus is also deformed by nanopillars. Figures 7b and c show membrane wrapping around 200 nm wide nanobars and CUI patterns. Particularly, the EGFP-CAAX renders complete CUI patterns including the negative curvatures in C and U, implying negative plasma membrane curvatures is also successfully induced.
To demonstrate the potential application of our nanostructure platform, we probe the localization of DNM2 in different cell lines cultured on nanobars. DNM2 is a GTP-binding protein responsible for the scission of plasma membrane budding in CME. Cells transfected with either DNM2-GFP or mCherry-CAAX are imaged over time and the obtained images are averaged to register the protein localization. Figure 10 shows representative results obtained from fluorescence imaging. mCherry-CAAX as a plasma membrane labelling shows the outline of the nanobars, confirming that the plasma membrane is deformed by wrapping around nanostructures (Fig. 10a). In contrast, Figures 10b-d illustrate that DMN2-GFP preferred high-curvature membrane (200 nm wide bar ends) in the tested cell lines (e.g., U2-OS, NIH/3T3 and COS-7). This preferential accumulation is more apparent when thefluorescent images of all the 200 nm wide nanobars are averaged (insets of Figure 10e). We used our MATLAB code to analyze the fluorescent intensity values at the ends and centres of 200 nm wide nanobars, and calculated the intensity ratio of each nanobar. The DNM2-GFP intensity ratios in all the cell lines are around 2, whereas the mCherry-CAAX intensity ratio in U2-OS is 1.32. The statistical analysis of the intensity ratios confirms there is a significant difference between mCherry-CAAX and DNM2-GFP in U2-OS cells. Wealso show an epifluorescence image of cells on gradient nanobar array (100 nm to 1000 nm widebars) (Fig. 10f). When the fluorescence images of eachnanobar configuration are averaged (Fig. 10g), it is revealed that DMN2-GFP recruitment is reduced with thicker nanobar.
Fig. 10 |. Probing DNM2 on nanostructures.
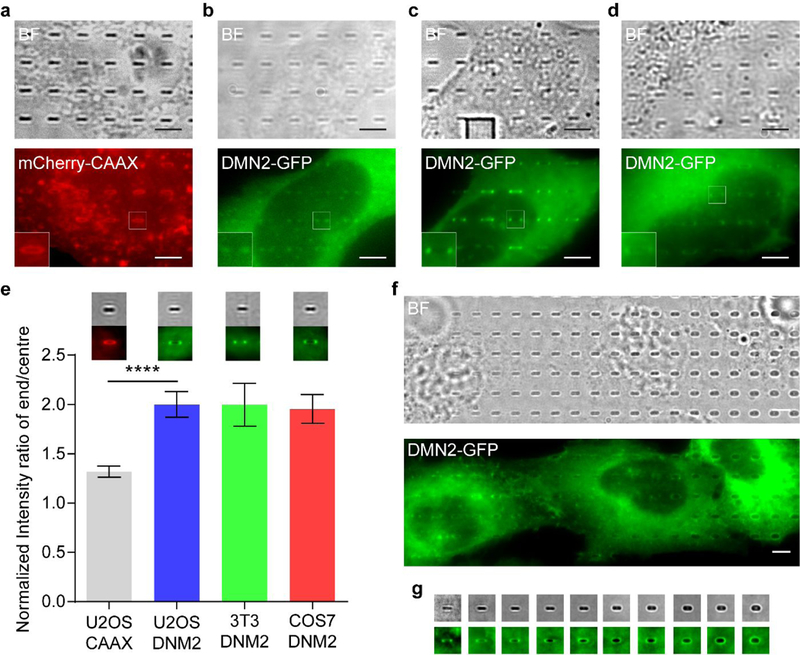
a, Micrographs of a part of U2-OS cell expressing mCherry-CAAX on nanobars, b-d, Micrographs of a part of a cell expressing DMN2-GFP on nanobars. The cells are U2-OS (b), 3T3 (c) and COS7 (d), respectively. Insets are zoom-in views of the corresponding regions in the fluorescence images, e, Quantitative analysis of end/centre ratios of fluorescence intensity values on 200 nm wide nanobars. Insets are the averaged fluorescence images of each cell line according to the bar chart. Error bars represent standard error of mean. Statistical significance of mCherry-CAAX versus DNM2-GFP in U2-OS was evaluated by an unpaired t-test with Welch’s correction. ****p < 0.0001. f, Micrographs of cells expressing DMN2-GFP on nanobars of 100–1000 nm width, g, Micrographs of nanobars of the same width ranging from 100 to 1000 nm, with a 100 nm incremental step. All the micrographs are generated by averaging fluorsecence images taken in live cell imaging. The protein distributions are measured by averaging over 41–181 nanobars. All the nanobars are 1 μm in height and 2 μm in length. Scale bars, 5 μm. “BF” denotes bright field.
Figure 11 displays the images obtained via the FIB–SEM technique. UTP is able to preserve the fine structure of cells on the nanostructures (Fig. 11a). It is sometimes observed that the plasma membrane extends along nanostructures (inset, Fig. 11b). By following the cross-sectioning and polishing steps described in the FIB–SEM section of the protocol and adjusting the imaging settings, the interface between nanostructures and the cell membrane can be clearly revealed (Fig. 11c). Closer look at individual nanostructures allows the measurement of the cleft between plasma membrane and nanostructure surface (Fig. 11d). Thanks to the heavy metal staining, cell organelles can also be observed.
Fig. 11 |. FIB-SEM images of cells cultured on nanostructures.
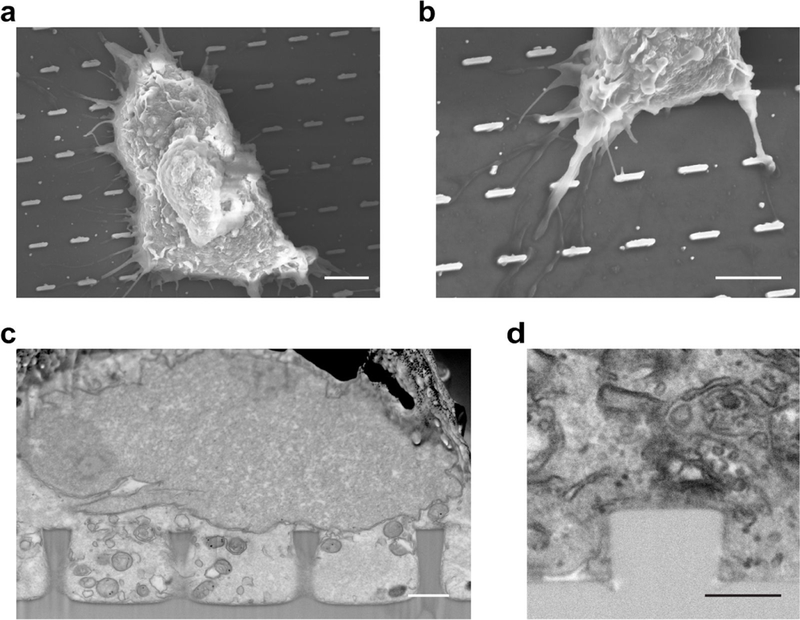
a, SEM image of a cell growing on nanobars, b, SEM image of cell membrane extrusions on nanobars. The nanobars have 200 nm width, 2 μm length, and 1 μm height (a and b). c, Polished cross-sectional micrograph of a part of a cell on nanopillars. The nanopillars have 700 nm diameter and 1.6 μm height, d, Polished cross-sectional micrograph of a part of a cell on a nanopillar. The nanopillar has 700 nm diameter and 500 nm height. Scale bars, 5 μm (a and b), 1 μm (c); 500 nm (d).
Supplementary Material
Acknowledgements
This work was supported by NIH grant 1R01GM125737. B.C. and XL. thank Stanford Nanofabrication Facility and Stanford Nano Shared Facilities for the help with nanofabrication. F.S. and L.M. thank Dr. Valentina Mollo for the help with the embedding procedure and Dr. Antonio Qualtieri for the help with the SEM/FIB in Lecce.
Footnotes
Competing interests
The authors declare no competing interests.
REFERENCES
- 1.Di Fiore PP & von Zastrow M Endocytosis, Signaling, and Beyond. Cold Spring Harb. Perspect. Biol. 6, a016865-a016865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorkin A & von Zastrow M Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 10, 609–622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mattila PK & Lappalainen P Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 9, 446–454 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Motile and non-motile cilia in human pathology: from function to phenotypes. J. Pathol. 241, 564 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Lou H-Y, Zhao W, Zeng Y & Cui B The Role of Membrane Curvature in Nanoscale Topography-Induced Intracellular Signaling. Acc. Chem. Res. 51, 1046–1053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMahon HT & Gallop JL Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438, 590–596 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Habermann B The BAR-domain family of proteins: a case of bending and binding? EMBO Rep. 5, 250–255 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mim C & Unger VM Membrane curvature and its generation by BAR proteins. Trends Biochem. Sci. 37, 526–533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simunovic M, Voth GA, Callan-Jones A & Bassereau P When Physics Takes Over: BAR Proteins and Membrane Curvature. Trends Cell Biol. 25, 780–792 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawson JC, Legg JA & Machesky LM Bar domain proteins: a role in tubulation, scission and actin assembly in clathrin-mediated endocytosis. Trends Cell Biol. 16, 493–498 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Ma Y et al. The inverse F-BAR domain protein srGAP2 acts through srGAP3 to modulate neuronal differentiation and neurite outgrowth of mouse neuroblastoma cells. PLoS One 8, e57865 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kooijman EE et al. Spontaneous curvature of phosphatidic acid and lysophosphatidic acid. Biochemistry 44, 2097–2102 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Abbott J et al. CMOS nanoelectrode array for all-electrical intracellular electrophysiological imaging. Nat. Nanotechnol. 12, 460–466 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Xie C, Lin Z, Hanson L, Cui Y & Cui B Intracellular recording of action potentials by nanopillar electroporation. Nat. Nanotechnol. 7, 185–190 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang M et al. Nanotechnology and Nanomaterials for Improving Neural Interfaces. Adv. Funct. Mater. 28, 1700905 (2017). [Google Scholar]
- 16.Limongi T et al. Fabrication and Applications of Micro/Nanostructured Devices for Tissue Engineering. Nano-Micro Letters 9, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feller L et al. Cellular responses evoked by different surface characteristics of intraosseous titanium implants. Biomed Res. Int. 2015, 171945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wennerberg A & Albrektsson T Effects of titanium surface topography on bone integration: a systematic review. Clin. Oral Implants Res. 20 Suppl 4, 172–184 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Teo BKK et al. Nanotopography modulates mechanotransduction of stem cells and induces differentiation through focal adhesion kinase. ACS Nano 7, 4785–4798 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Rebollar E et al. Proliferation of aligned mammalian cells on laser-nanostructured polystyrene. Biomaterials 29, 1796–1806 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Park J et al. Directed migration of cancer cells guided by the graded texture of the underlying matrix. Nat. Mater. 15, 792–801 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brammer KS, Choi C, Frandsen CJ, Oh S & Jin S Hydrophobic nanopillars initiate mesenchymal stem cell aggregation and osteo-differentiation. Acta Biomater. 7, 683–690 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Teo BKK et al. Nanotopography modulates mechanotransduction of stem cells and induces differentiation through focal adhesion kinase. ACS Nano 7, 4785–4798 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Luu TU, Gott SC, Woo BWK, Rao MP & Liu WF Micro- and Nanopattemed Topographical Cues for Regulating Macrophage Cell Shape and Phenotype. ACSAppl. Mater. Interfaces 7, 28665–28672 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sorre B et al. Nature of curvature coupling of amphiphysin with membranes depends on its bound density. Proc. Natl. Acad. Sci. U. S. A. 109, 173–178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi Z & Baumgart T Membrane tension and peripheral protein density mediate membrane shape transitions. Nat. Commun. 6, 5974 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhatia VK, Hatzakis NS & Stamou D A unifying mechanism accounts for sensing of membrane curvature by BAR domains, amphipathic helices and membrane-anchored proteins. Semin. Cell Dev. Biol. 21, 381–390 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Simunovic M & Voth GA Membrane tension controls the assembly of curvature-generating proteins. Nat. Commun. 6, 7219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsujita K, Takenawa T & Itoh T Feedback regulation between plasma membrane tension and membrane-bending proteins organizes cell polarity during leading edge formation. Nat. Cell Biol. 17, 749–758 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Suetsugu S The proposed functions of membrane curvatures mediated by the BAR domain superfamily proteins. J. Biochem. 148, 1–12 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Kim W, Ng JK, Kunitake ME, Conklin BR & Yang P Interfacing silicon nanowires with mammalian cells. J. Am. Chem. Soc. 129, 7228–7229 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Xie C et al. Noninvasive neuron pinning with nanopillar arrays. Nano Lett. 10, 4020–4024 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu AM et al. Quantification of nanowire penetration into living cells. Nat. Commun. 5, 3613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shalek AK et al. Vertical silicon nanowires as a universal platform for delivering biomolecules into living cells. Proc. Natl. Acad. Sci. U. S. A. 107, 1870–1875 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berthing T et al. Cell membrane conformation at vertical nanowire array interface revealed by fluorescence imaging. Nanotechnology 23, 415102 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Santoro F et al. Revealing the Cell-Material Interface with Nanometer Resolution by Focused Ion Beam/Scanning Electron Microscopy. ACS Nano 11, 8320–8328 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dipalo M et al. Cells Adhering to 3D Vertical Nanostructures: Cell Membrane Reshaping without Stable Internalization. Nano Lett. (2018). doi: 10.1021/acs.nanolett.8b03163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanson L et al. Vertical nanopillars for in situ probing of nuclear mechanics in adherent cells. Nat. Nanotechnol. 10, 554–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao W et al. Nanoscale manipulation of membrane curvature for probing endocytosis in live cells. Nat. Nanotechnol. 12, 750–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H, Pykalainen A & Lappalainen P I-BAR domain proteins: linking actin and plasma membrane dynamics. Curr. Opin. Cell Biol. 23, 14–21 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Antonny B Mechanisms of Membrane Curvature Sensing. Annu. Rev. Biochem. 80, 101–123 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Galic M et al. External push and internal pull forces recruit curvature-sensing N-BAR domain proteins to the plasma membrane. Nat. Cell Biol. 14, 874–881 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bugnicourt G, Brocard J, Nicolas A & Villard C Nanoscale surface topography reshapes neuronal growth in culture. Langmuir 30, 4441–4449 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Kang K et al. Axon-First Neuritogenesis on Vertical Nanowires. Nano Lett. 16, 675–680 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Brammer KS, Choi C, Frandsen CJ, Oh S & Jin S Hydrophobic nanopillars initiate mesenchymal stem cell aggregation and osteo-differentiation. Acta Biomater. 7, 683–690 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Rasmussen CH et al. Enhanced Differentiation of Human Embryonic Stem Cells Toward Definitive Endoderm on Ultrahigh Aspect Ratio Nanopillars. Adv. Funct. Mater. 26, 815–823 (2015). [Google Scholar]
- 47.Wierzbicki R et al. Mapping the complex morphology of cell interactions with nanowire substrates using FIB-SEM. PLoS One 8, e53307 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santoro F et al. Interfacing electrogenic cells with 3D nanoelectrodes: position, shape, and size matter. ACS Nano 8, 6713–6723 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.