

SUMMARY
Reprogrammed metabolism and cell cycle dysregulation are two cancer hallmarks. p16 is a cell cycle inhibitor and tumor suppressor that is upregulated during oncogene-induced senescence (OIS). Loss of p16 allows for uninhibited cell cycle progression, bypass of OIS, and tumorigenesis. Whether p16 loss affects pro-tumorigenic metabolism is unclear. We report that suppression of p16 plays a central role in reprogramming metabolism by increasing nucleotide synthesis. This occurs by activation of mTORC1 signaling, which directly mediates increased translation of the mRNA encoding ribose-5-phosphate isomerase A (RPIA), a pentose phosphate pathway enzyme. p16 loss correlates with activation of the mTORC1-RPIA axis in multiple cancer types. Suppression of RPIA inhibits proliferation only in p16-low cells by inducing senescence both in vitro and in vivo. These data reveal the molecular basis whereby p16 loss modulates pro-tumorigenic metabolism through mTORC1-mediated upregulation of nucleotide synthesis and reveals a metabolic vulnerability of p16-null cancer cells.
Graphical Abstract
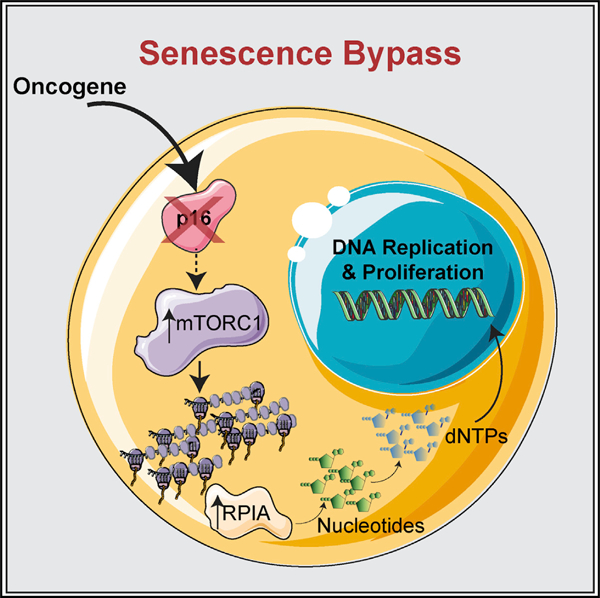
In Brief
Senescence bypass through p16 loss predisposes to transformation and tumorigenesis. Buj et al. found that the loss of p16 upregulates nucleotide metabolism through increased mTORC1-mediated translation of RPIA to bypass senescence in an RB-independent manner. Thus, the mTORC1-RPIA axis is a metabolic vulnerability for p16-null cancers.
INTRODUCTION
Metabolic reprogramming is a hallmark of cancer (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). Transformed and tumorigenic cells require increased deoxyribonucleotide synthesis to fuel the genome replication that sustains their unregulated cell cycle and proliferation. Therefore, it is likely that the cell cycle and nucleotide metabolism are linked. The cell cycle inhibitor p16 is a critical tumor suppressor that is lost as an early event in the progression from senescent benign lesions to cancer (Bennecke et al., 2010; Bennett, 2016; Caldwell et al., 2012; Kriegl et al., 2011; Michaloglou et al., 2005; Shain et al., 2015). Indeed, expression of p16 is low or null in approximately half of all human cancers (Li et al., 2011). Although the loss of p16 is known to play a role in deregulating the cell cycle, whether the loss of p16 expression affects nucleotide metabolism is unknown.
Both increased expression of p16 (Serrano et al., 1997) and decreased levels of deoxyribonucleotide triphosphates (dNTPs) (Aird et al., 2013; Mannava et al., 2013) are characteristics of cellular senescence, a stable cell cycle arrest (Aird and Zhang, 2014, 2015; Dörr et al., 2013; Hernandez-Segura et al., 2018; Wiley and Campisi, 2016). Activation of oncogenes such as BRAFV600E induces senescence to suppress transformation and tumorigenesis (termed oncogene-induced senescence [OIS]) (Pérez-Mancera et al., 2014; Yaswen and Campisi, 2007). Therefore, OIS is considered an important tumor suppressor mechanism in vivo (Braig et al., 2005; Michaloglou et al., 2005). Increased dNTPs or loss of p16 bypasses OIS to allow for transformation and tumorigenesis (Aird et al., 2013, 2015; Damsky et al., 2015; Dankort et al., 2007; Goel et al., 2009; Haferkamp et al., 2008; Sarkisian et al., 2007). Thus, we reasoned that these two processes may be interconnected. Here, we used senescence as a model to study the link between p16 and nucleotide metabolism. We demonstrate that the loss of p16 increases nucleotide synthesis through upregulation of mTORC1 activity.
RESULTS
p16 Knockdown Enhances Nucleotide Synthesis to Bypass Senescence
To determine whether p16 loss affects nucleotide synthesis, we took advantage of our previously published model of dNTP-depletion-induced senescence by knocking down RRM2 (Aird et al., 2013). Knockdown of p16 in shRRM2 cells suppressed senescence markers (Figures 1A–1E and S1A). Data using a second independent hairpin targeting p16 and overexpression of p16 cDNA demonstrate that these results are p16 specific (Figures S1B–S1K). Knockdown of p16 in the pathologically relevant model of BRAFV600E-induced senescence also bypassed senescence (Figures 1F–1J). Knockdown of p16 in both models significantly increased deoxyribonucleotide di-phosphates (dNDPs)/dNTPs even above control levels in some nucleotides (Figures 1K and 1L). Interestingly, we observed an increase in RRM2B in shRRM2/shp16 cells (Figures S1L and S1M), which is likely how these cells reduce nucleoside diphosphates and nucleoside triphosphates (NDPs/NTPs) to dNDPs/ dNTPs. Excitingly, further metabolite analysis demonstrated that nucleotides were also significantly increased upon p16 knockdown in these models (Figures 1M, 1N, and S1N), suggesting that the increase in deoxyribonucleotides is not simply due to increased RRM2B or the proportion of cells in S phase. Together, these data indicate that p16 depletion increases both nucleotide and deoxyribonucleotide synthesis to bypass senescence.
Figure 1. Suppression of p16 Increases Nucleotide Synthesis to Bypass Senescence.

(A–E) IMR90 cells expressing shRNA targeting RRM2 (shRRM2) alone or in combination with an shRNA targeting p16 (shp16). One of 5 experiments is shown.
(A) Immunoblot analysis of the indicated proteins.
(B) Senescence-associated-β-galactosidase (SA-β-Gal) activity, bromodeoxyuridine (BrdU) incorporation, and colony formation (CF). One of 5 experiments is shown. Scale bar, 10 μm.
(C) Quantification of SA-β-Gal activity in (B). n = 3/group; 1 of 5 experiments is shown. Data represent mean ± SD. *p < 0.001.
(D) Quantification of BrdU incorporation in (B). n = 3/group; 1 of 5 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(E) Quantification of colony formation in (B). n = 3/group; 1 of 5 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(F–J) IMR90 cells expressing BRAFV600E alone or in combination with an shRNA targeting p16 (shp16).
(F) Immunoblot analysis of the indicated proteins. One of 3 experiments is shown.
(G) SA-β-Gal activity, BrdU incorporation, and colony formation (CF). One of 5 experiments is shown. Scale bar, 10 µm.
(H) Quantification of SA-β-Gal activity in (G). n = 3/group, 1 of 5 experiments is shown. Data represent mean ± SD. *p < 0.001.
(I) Quantification of BrdU incorporation in (G). n = 3/group; 1 of 5 experiments is shown. Data represent mean ± SEM. *p < 0.002.
(J) Quantification of colony formation in (G). n = 3/group; 1 of 5 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(K–N) Deoxyribonucleotide analysis in the RRM2 (K) and BRAFV600E (L) models and ribonucleotide analysis in the RRM (M) and BRAFV600E (N) models. n > 3/group; 1 of at least 2 experiments is shown. Data represent mean ± SEM. *p < 0.05. NA, not available.
p16 Knockdown Activates mTORC1 to Bypass Senescence and Increase Nucleotide Synthesis
We next aimed to determine the underlying mechanism of nucleotide synthesis upon p16 knockdown. p16 inhibits E2F-mediated transcription in part through regulating the retinoblastoma protein (RB)-E2F interaction (Sherr, 2001). Gene set enrichment analysis (GSEA) of our RNA sequencing (RNA-seq) (GEO: GSE133660) did not show terms related to nucleotide synthesis (Data S1). However, GSEA showed an enrichment in the mTORC1 signaling pathway upon p16 knockdown, which was confirmed by reverse phase protein array (RPPA) (Data S1). Underscoring the pathological relevance of our findings, the mTORC1 signaling pathway was also enriched in melanoma compared with nevi in two independent datasets (Kabbarah et al., 2010; Talantov et al., 2005) (Data S1). Previous studies demonstrated that mTORC1 increases nucleotide synthesis (Ben-Sahra et al., 2013, 2016), suggesting that this may be the mechanism by which the loss of p16 increases nucleotides. We confirmed the activation of mTORC1 signaling by assessing the increased phosphorylation of S6K and 4E-BP1 (Figures 2A and 2B) as well as by mTORC localization at the lysosomal membrane (Figures S2A and S2B). Inhibition of mTORC1 with temsirolimus significantly decreased nucleotides and deoxyribonucleotides and suppressed senescence bypass in both models (Figures 2A–2L and Data S2), whereas it had no effect on parental cell proliferation (Figure S2C). Together, these data demonstrate that activation of mTORC1 downstream of p16 loss drives the observed increase in nucleotides, which is required for senescence bypass.
Figure 2. Suppression of p16 Activates mTORC1 to Increase Nucleotide Synthesis.

(A) IMR90 cells expressing shRNA targeting RRM2 (shRRM2) alone or in combination with an shRNA targeting p16 (shp16) and treated with temsirolimus (Tem). Immunoblot analysis of the indicated proteins. One of 3 experiments is shown.
(B) IMR90 cells expressing BRAFV600E alone or in combination with an shRNA targeting p16 (shp16) and treated with temsirolimus (Tem). Immunoblot analysis of the indicated proteins. One of 3 experiments is shown.
(C and D) Deoxyribonucleotide analysis in the RRM2 (C) and BRAFV600E (D) models. n > 3/group; 1 of at least 3 experiments is shown. Data represent mean ± SEM. *p < 0.05.
(E) Same as (A). SA-β-Gal activity, BrdU incorporation, and colony formation (CF). One of 3 experiments is shown. Scale bar, 10 µm.
(F) Quantification of SA-β-Gal activity in (E). n = 3/group; 1 of 3 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(G) Quantification of BrdU incorporation in (E). n = 3/group; 1 of 3 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(H) Quantification of colony formation in (E). n = 3/group; one of 3 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(I) Same as (B). SA-β-Gal activity, BrdU incorporation, and colony formation (CF). One of 3 experiments is shown. Scale bar, 10 μm.
(J) Quantification of SA-β-Gal activity in (I). n = 3/group; 1 of 3 experiments is shown. Data represent mean ± SEM. *p < 0.02.
(K) Quantification of BrdU incorporation in (I). n = 3/group, 1 of 3 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(L) Quantification of colony formation in (I). n = 3/group, 1 of 3 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(M) Immunoblot analysis of the indicated proteins in cancer cell lines with wild-type p16 expressing shp16. One of at least 2 experiments is shown.
(N) Same as (M) but cells were treated with temsirolimus. n = 3/group; 1 of 2 experiments is shown.
mTORC1 is a master regulator of translation (Ma and Blenis, 2009; Nandagopal and Roux, 2015). Interestingly, increased expression of leading-edge genes associated with the ‘‘translation’’ GSEA term (Data S1) significantly co-occurred with alterations in CDKN2A in multiple cancer types and was associated with worse overall survival (Figures S2D and S2E). Additionally, knockdown of p16 in cancer cell lines with wild-type p16 expression increased phosphorylation of S6K and 4E-BP1 (Figure 2M). Consistently, low p16 expression increased the sensitivity to mTORC1 inhibition (Figures 2N, S2F, and S2G). Together, these data indicate that mTORC1 activation also occurs in cancer cells upon p16 suppression and correlates with increased sensitivity to mTORC1 inhibition.
Finally, we aimed to determine whether increased mTORC1 signaling is dependent on RB. Although RB knockdown suppressed BRAFV600E-induced senescence, it did not increase p-S6K or p-4EBP1 in any of the models tested (Figures S2H–S2N). Consistently, mTOR signaling pathways were not enriched in an independent dataset of RB knockdown in senescence (Data S1) (Chicas et al., 2010). This suggests that the upregulation of mTORC1 activity is not a cell-cycle-dependent phenomenon and that it occurs in an RB-independent pathway. Together, these data demonstrate that activation of mTORC1 signaling upon p16 suppression is critical for nucleotide synthesis and senescence bypass in an RB-independent manner.
mTORC1 Activation by p16 Knockdown Increases Translation of Ribose-5-Phosphate Isomerase A and Promotes Nucleotide Synthesis through the Pentose Phosphate Pathway
Previous reports showed that mTORC1 upregulates purine and pyrimidine metabolism through ATF4-MTHFD2 and CAD, respectively (Ben-Sahra et al., 2013, 2016). We did not observe an increase in MTHFD2 transcription or CAD phosphorylation after p16 knockdown (Figures S3A and S3B). As mTORC1 activity increases translation (Ma and Blenis, 2009), we aimed to determine whether the observed increase in mTORC1-mediated nucleotide synthesis upon p16 suppression increases translation of transcripts involved in nucleotide synthesis. We performed polysome fractionation (Figure S3C) followed by RT-qPCR analysis of transcripts involved in nucleotide synthesis and related anaplerotic pathways. Our results reveal a number of transcripts whose abundance shifted from the light to the heavy polysome fraction upon p16 knockdown (Figure 3A; Table S1), including EEF2, which is known to be translationally regulated by mTORC1 (Thoreen et al., 2012) (Figure S3D). Interestingly, ribose-5-phosphate isomerase A (RPIA), an enzyme that synthesizes the ribose sugar backbone for both purines and pyrimidines (Lane and Fan, 2015) was the top hit (see STAR Methods) (Figure 3A; Table S1). Although MYC increases RPIA transcription (Santana-Codina et al., 2018), we did not observe changes in RPIA gene expression or MYC protein expression in these cells (Figures 3B and S3E). Consistent with the idea that mTORC1 regulates RPIA translation, RPIA protein expression was increased after p16 knockdown and decreased upon mTORC1 inhibition with temsirolimus (Figures 3C and 3D). Increased RPIA protein expression was also observed in 7 isogenic cancer cell lines upon p16 suppression (Figures 3E and 3F) and decreased upon knockdown of RPTOR (Figure S3F). Inhibition of mTORC1 with temsirolimus shifted the RPIA mRNA from the heavy to the light polysome fraction, whereas it did not decrease total RPIA mRNA expression (Figures 3G and S3G). To confirm the direct role of mTORC1 in RPIA translation upon p16 knockdown, we performed polysome fractionation after 3 h of Torin 1 treatment (similar to Thoreen et al., 2012). Torin 1 inhibited mTORC1 activity and RPIA translation (Figures 3H and 3I). Additionally, knockdown of p16 increased 35S-methionine and cysteine incorporation into RPIA, which was decreased by Torin 1 treatment (Figure 3J). Consistent with increased RPIA protein expression, total ribose-5-phosphate (R5P) was increased upon p16 knockdown and decreased by inhibition of mTORC1 (Figures 3K and 3L). Finally, stable isotope labeling using U-13C glucose demonstrated an increase in the M+5 fraction of R5P and multiple nucleotides upon p16 knockdown, which was abrogated by temsirolimus treatment (Figures 3M–3P; Data S2). Taken together, these data demonstrate that knockdown of p16 increases mTORC1-mediated translation of RPIA to fuel R5P and nucleotide synthesis.
Figure 3. Suppression of p16 Increases mTORC1-Mediated RPIA Translation and Nucleotide Synthesis by the Pentose Phosphate Pathway.

(A and B) IMR90 cells expressing shRNA targeting RRM2 (shRRM2) alone or in combination with an shRNA targeting p16 (shp16).
(A) Heatmap of light and heavy fractions from polysome profiling (see Table S1 for raw data). This experiment was performed once with 3 technical replicates.
(B) Total RPIA expression from RNA-seq. Data represent mean ± SEM. ns, not significant.
(C) Same as (A) but cells were treated with temsirolimus (Tem). Immunoblot analysis of the indicated proteins. One of 3 experiments is shown.
(D) IMR90 cells expressing BRAFV600E alone or in combination with shp16 and treated with temsirolimus (Tem). Immunoblot analysis of the indicated proteins. One of 3 experiments is shown.
(E) CDKN2A expression in the indicated cancer cell lines with wild-type p16 expression expressing shp16. *p < 0.006.
(F) Same as (E) but immunoblot analysis of RPIA. One of at least 2 experiments is shown.
(G) Percentage of RPIA mRNA abundance in polysome fractions in the indicated conditions. *p < 0.05.
(H–J) SKMel28 and HT-29 cell expressing shp16 treated with Torin 1.
(H) Immunoblot analysis of the indicated proteins.
(I) Percentage of RPIA mRNA abundance in polysome fractions.
(J) Counts per million (CPM) after [35S]-methionine and cysteine labeling and RPIA immunoprecipitation (IP). Data represent the percentage of CPM normalized by the excised band weight. One of 2 experiments is shown.
(K and L) Ribose-5-phosphate (R5P) abundance in the RRM2 (K) and BRAFV600E (L) models. n > 3/group; 1 of at least 3 experiments is shown. Data represent mean ± SEM. *p < 0.01.
(M and N) R5P M+5 in the RRM2 (M) and BRAFV600E (N) models. n > 3/group; 1 of at least 3 experiments is shown. Data represent mean ± SEM. *p < 0.05.
(O and P) dTTP M+5 and UTP M+5 in the RRM2 (O) and BRAFV600E (P) models. n > 3/group; 1 of at least 3 experiments is shown. Data represent mean ± SEM. *p < 0.03; ND, not detected.
R5P Isomerase A Is a Metabolic Vulnerability of p16-Low Cells In Vitro and In Vivo
To determine whether RPIA is necessary for proliferation of cells with p16 knockdown, we depleted RPIA by using two independent short hairpin RNAs (shRNAs). Our data indicate that RPIA is necessary for proliferation of p16 knockdown cells as it induced senescence in all cell models tested (Figures 4A–4I, S4A–S4F, and S5A–S5F). Knockdown of RPIA alone had no effect on parental cells (Figures S4A–S4F). We did not observe a marked increase in cell death (Figure S5G), suggesting that the observed loss of proliferation is likely due to the senescence-associated cell cycle arrest. Similar results were observed in vivo, where knockdown of RPIA inhibited tumor growth in HT-29 cells with shp16 but not controls (Figures 4J–4L). Consistent with our in vitro data, LMNB1 was decreased only in shp16/shRPIA tumors (Figure 4M). Although there was a decrease in CCNA2 upon RPIA knockdown alone, the difference was significantly larger in shp16/shRPIA compared to shp16 alone tumors (Figure 4N). Together, these data indicate that RPIA-mediated increased nucleotide synthesis is necessary for cancer cell proliferation both in vitro and in vivo and that suppression of RPIA may be a target for cancers with low p16 expression.
Figure 4. Inhibition of RPIA Is a Metabolic Vulnerability for Cells with Low p16.

(A–E) IMR90 cells expressing BRAFV600E alone or in combination with 2 independent shRNAs targeting RPIA (shRPIA).
(A) Immunoblot analysis of the indicated proteins. One of 3 experiments is shown.
(B) SA-β-Gal activity, BrdU incorporation, and colony formation. One of 3 experiments is shown. Scale bar, 10 μm.
(C) Quantification of SA-β-Gal activity in (B). n = 3/group; 1 of 3 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(D) Quantification of BrdU incorporation in (B). n = 3/group; 1 of 3 experiments is shown. Data represent mean ± SEM. *p < 0.02.
(E) Quantification of colony formation in (B). n = 3/group; 1 of 3 experiments is shown. Data represent mean ± SEM. *p < 0.001.
(F–H) ES2 (F), HT-29 (G), and PATU8902 (H) cancer cell lines with wild-type p16 expression expressing shRPIA alone or in combination with shp16. SA-b-Gal activity, BrdU incorporation, and colony formation (CF). n = 3/group; 1 of at least 2 experiments is shown. Scale bar, 10 mm. Data represent mean ± SEM. *p < 0.05 versus shp16 alone.
(I) CCNA2 and LMNB1 fold change in the indicated cells. One of at least 2 experiments is shown. Data represent mean ± SD. *p < 0.05 versus shp16 alone.
(J) Tumor growth curve of the indicated groups.
(K) Representative images of mice from each group. Tumors are outlined in red.
(L) Tumor volume at day 26 post-implantation. *p = 0.0414; ns, not significant.
(M) LMNB1 expression in the indicated tumors. ns, not significant.
(N) CCNA2 expression in the indicated tumors.
See also Figures S4 and S5.
DISCUSSION
The absence of p16 predisposes cells to tumorigenesis (LaPak and Burd, 2014), and its expression is low or null in many human cancers (Cerami et al., 2012; Gao et al., 2013). There is currently no approved targeted therapy for p16-low tumors (Otto and Sicinski, 2017). Therefore, delineating the molecular mechanisms downstream of p16 suppression is critical to identify new therapeutics for these patients. Although the role of p16 loss in deregulating the cell cycle has been known for decades (Sherr, 2001), its role in metabolism is unclear. In this study, we found that mTORC1 signaling activation upon p16 suppression increases nucleotide synthesis. Mechanistically, we found mTORC1 activity leads to increased translation of RPIA and glucose flux through the pentose phosphate pathway to increase nucleotide levels. Suppression of p16 in cancer cells also leads to increased mTORC1 activity and increased RPIA translation and protein expression, and these cells are more sensitive to mTORC1 inhibitors or RPIA suppression than p16 wild-type cells. Together, our results suggest that increased nucleotide metabolism by RPIA upregulation is a metabolic vulnerability of p16-null cancers.
Cancer cells reprogram metabolism to increase biomass needed for growth and proliferation (Pavlova and Thompson, 2016). Modulation of nucleotide and deoxyribonucleotide levels is critical for multiple cancer cell phenotypes, including for the repair of damaged DNA and to ensure rapid proliferation (Kohnken et al., 2015). We previously found that increased dNTPs, either through upregulation of RRM2 expression or loss of ATM, bypass senescence (Aird et al., 2013, 2015). Additionally, a recent paper found that metabolic reprogramming, including increased nucleotide levels, precedes tumor formation in a UVB-induced skin cancer model (Hosseini et al., 2018). Here, we show that the loss of p16 increases nucleotide synthesis through a mechanism mediated by mTORC1. Excitingly, activation of this pathway increased both nucleotides and deoxyribonucleotides. We observed an increase in the other ribonucleotide reductase R2 subunit RRM2B. RRM2B has been shown to play a role in mitochondrial dNTP synthesis and in response to DNA damage (Bourdon et al., 2007; Pontarin et al., 2012). Interestingly, RRM2B was increased in both shRRM2 alone and shRRM2/shp16 cells. This suggests that although RRM2B is likely important for reducing NDPs/NTPs to dNDPs/dNTPs in senescence bypass, its upregulation alone is not sufficient to produce dNDPs/dNTPs. Indeed, these data further support the notion that it is only when upstream nucleotides are also increased, such as when p16 is knocked down, that the expression of RRM2B is critical for senescence bypass.
The canonical function of p16 is upstream of RB to affect E2F and the cell cycle (Sherr, 2001). We found that mTORC1 upregulation downstream of p16 knockdown is independent of RB in multiple cell types. There are an increasing number of studies reporting RB-independent functions of p16 (Al-Khalaf et al., 2013; Jenkins et al., 2011; Lee et al., 2013; Tyagi et al., 2017), suggesting that the non-canonical pathway of p16 loss needs to be explored to identify both mechanistic underpinnings of RB-independent functions and novel therapies for cancer patients with p16-null tumors. As knockdown of p16 increased mTORC at the lysosomal membrane, it is possible that this pathway affects amino acid transporters and/or uptake. Additionally, a previous report in a mouse model of melanomagenesis also found increased mTORC1 signaling upon Cdkn2a knockout due to miR-99/100 expression (Damsky et al., 2015). Finally, it is possible that the effect of p16 knockdown is through cyclin D1/CDK4, which has previously been shown to phosphorylate TSC2, thereby activating mTORC1 signaling (Goel et al., 2016). Further research is needed to understand the connection between the loss of p16 and the activation of mTORC1.
mTORC1 is a master regulator of metabolism by coordinating metabolite availability though translational control of metabolic enzymes (Iurlaro et al., 2014; Zoncu et al., 2011). Recent studies have linked mTORC1 to both purine and pyrimidine synthesis (Ben-Sahra et al., 2013, 2016). Our results indicate that suppression of p16 increases mTORC1-mediated translation of RPIA. Previous studies have shown that RPIA is transcriptionally regulated by mTORC1 signaling (Düvel et al., 2010) or MYC (Santana-Codina et al., 2018). We did not observe a transcriptional increase in RPIA or MYC upregulation in our model, suggesting that RPIA upregulation is context-dependent. Consistent with our results, a recent paper showed that total RPIA mRNA expression is not significantly decreased after 24 h of Torin 1 treatment (Park et al., 2017). Instead, our data demonstrate that RPIA is directly translationally regulated by mTORC1 as inhibition of mTORC1 with a short Torin 1 treatment decreased RPIA transcripts in the heavy polysome fraction as well as RPIA protein expression. Consistent with the idea that our results are MYC-independent, a previous publication demonstrated that MYC mRNA is resistant to Torin 1 inhibition (Thoreen et al., 2012). Taken together, these data demonstrate that the observed increase in RPIA protein upon the loss of p16 is mediated though mTORC1-specific translation.
Cell cycle inhibitors are currently being tested in the clinic for tumors with deletions or mutations in CDKN2A (http://clinicaltrials.gov); however, no US Food and Drug Administration (FDA)-approved therapy currently exists for this subset of patients. Moreover, our data and others demonstrate that p16 may have functions outside of the cell cycle and RB (Al-Khalaf et al., 2013; Jenkins et al., 2011; Lee et al., 2013; Tyagi et al., 2017), suggesting that these inhibitors may not be efficacious in these patients. Excitingly, our results with p16 suppression open up a metabolic vulnerability through activation of mTORC1-mediated nucleotide metabolism. Indeed, we found that isogenic p16-null cells are more sensitive to temsirolimus or suppression of RPIA both in vitro and in vivo. RPIA inhibition has been shown to limit the growth of KrasG12D cell lines and xenografted tumors (Santana-Codina et al., 2018; Ying et al., 2012). Our results demonstrate that RPIA expression could also be exploited as a metabolic target in p16-null cancers.
In conclusion, our study provides a molecular effect of p16 loss whereby mTORC1 signaling is activated to increase nucleotide metabolism. This is different, yet likely linked, to its canonical role in cell cycle regulation. These mechanistic insights have broad implications for understanding pro-tumorigenic metabolism. Moreover, this study provides a metabolic vulnerability for p16-low cancer cells, which may be exploited for therapy.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katherine M. Aird (kaird@psu.edu). There are restrictions to the availability of the shRNA knockdown cell lines generated in this study since they were made with commercial reagents. These will only be distributed with permission of the commercial suppliers.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Normal diploid IMR90 human fibroblasts were cultured according to the ATCC in low oxygen (2% O2) in DMEM (4.5 g/L glucose, Corning cat# 10-017-CV) with 10% FBS supplemented with L-glutamine, non-essential amino acids, sodium pyruvate, and sodium bicarbonate. Experiments were performed on IMR90 between population doubling #25–35. Melanoma (SKMel28), pancreatic (PATU8902), colorectal (HT-29, SW620, and SW480) tumor cells and lentiviral and retroviral packaging cells (293FT and Phoenix, respectively) were cultured in DMEM (Corning, cat# 10–013-CV) with 10% FBS. ES2 ovary tumor cell line was cultured in RPMI 1640 with 10% FBS. TCCSUP bladder cancer cell line was cultured in MEM/EBSS glutamine supplemented with 10% FBS. All cell lines were cultured in MycoZap and were routinely tested for mycoplasma as described in Uphoff and Drexler (2005). All tumor cell lines included in this work express wild-type CDKN2A according to TCGA (Cerami et al., 2012; Gao et al., 2013). According to ATCC, TCCSUP harbors a mutation in RB1. All cell lines were authenticated using STR Profiling (Genetica DNA Laboratories).
Mice
Two-month old male SCID mice were purchased from Charles River Laboratories. All mice were maintained in a HEPA-filtered ventilated rack system at the Penn State College of Medicine animal facility. Mice were housed up to 5 mice per cage and in a 12-hour light/dark cycle. All experiments with animals were performed in accordance with institutional guidelines approved by the Institutional Animal Care and Use Committee (IACUC) at the Penn State College of Medicine.
METHOD DETAILS
Lentiviral and retroviral packaging and infection
Retrovirus production and transduction were performed using the BBS/calcium chloride method (Aird et al., 2013). Phoenix cells (a gift from Dr. Gary Nolan, Stanford University) were used to package the infection viruses. Lentiviral constructs were transfected into 293FT cells using Lipofectamine 2000 (Thermo Fisher). Lentivirus was packaged using the ViraPower Kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions.
The basic IMR90 experiment timeline is delineated in Figure S1A. Briefly, IMR90 cells were infected with pLKO.1 empty vector or pLKO.1-shRRM2, and 24 hours later cells were infected with pLKO.1 empty vector, pLKO.1-shp16 or pLKO.1-shRB. Cells were selected with puromycin (3 µg/mL) for 7 days. Alternatively, IMR90 cells were infected with pBABE control or pBABE BRAFV600E vector and 24 hours later cells were infected with a second round of pBABE control or BRAFV600E vector together with pLKO.1 empty vector, pLKO.1-shp16 or pLKO.1-shRB. Cells were selected with puromycin (3 mg/mL) for 7 days. Where indicated, cells were treated at day 4 with temsirolimus (0.5nM) or infected with pLKO.1-shRPIA. p16 rescue experiment was performed by simultaneous infection with pLKO.1-shp16 and pBABE-p16 overexpression plasmid. For single infections, cells were infected with the corresponding virus and selected in puromycin (1 µg/mL) for 7 days.
Tumor cell lines were infected with pLKO.1 empty vector, pLKO.1-shp16 or pLKO.1-shRB. Cells were selected with puromycin (1 µg/mL) for 4 days. Where indicated, cells were treated at day 4 with increasing concentrations of temsirolimus (0.07–50 µM) in 0.5% FBS for 5 days or infected with pLKO.1 shRPIA or pLKO.1-shRPTOR. For double infections, cells were selected in puromycin (3 µg/mL) for 4 additional days. For Torin 1 experiments, cells were serum starved for 16h and then treated with 250nM Torin 1 for 3h in 0.5% FBS.
RNA-Sequencing and Analysis
Total RNA was extracted from cells at day 7 (Figure S1A) with Trizol (Life Technologies) and DNase treated with RNeasy Mini Kit (QIAGEN, cat#74104) following the manufacturer’s instructions. RNA integrity number (RIN) was measured using BioAnalyzer (Agilent Technologies) RNA 6000 Nano Kit to confirm RIN above 7 for each sample. The cDNA libraries were prepared using KAPA Stranded RNA-Seq Kits with RiboErase (Kapa Biosystems). Next generation sequencing was performed in The Penn State College of Medicine Genome Sciences and Bioinformatics Core facility as previously described in Lynch et al. (2015) using a HiSeq 2500 sequencer (Illumina). Demultiplexed and quality-filtered mRNA-Seq reads were then aligned to human reference genome (GRCh38) using TopHat (v.2.0.9). Differential expression analysis was done using Cuffdiff tool which is available by Cufflinks (v.2.0.2) as described in Lynch et al. (2015). Three technical replicates were used. Raw counts and differential expression analysis generated during this study are available at GEO (GSE133660).
Reverse Phase Protein Array (RPPA)
After 7 days under selection (Figure S1A), cells cultured in 10cm dishes were incubated on ice with 300uL of lysis buffer (1% Triton X-100, 50mM HEPES pH = 7.5, 150mM NaCl, 1.5mM MgCl2, 1mM EGTA, 100mM NaF, 10mM Na pyrophosphate, 1mM Na3VO4 and 10% glycerol) for 20 min with occasional shaking every 5 min. After incubation, cells were scraped off the plate and centrifuged at 18000 g for 10 min at 4°C. Total protein was quantified with Bradford assay and 90ug of protein was diluted 3:1 in SDS sample buffer (40% glycerol, 8% SDS, 0.25M Tris-HCl and 10% B-mercaptoethanol). Lysates were boiled at 95°C for 5 min and stored at −80°C. RPPA data was generated and analyzed by the CCSG-supported RPPA Core Facility at the University of Texas MD Anderson Cancer Center (Akbani et al., 2014). A total of 240 authenticated Antibodies for total protein expression and 64 antibodies for protein phosphorylation were analyzed in this study. The complete antibody list can be found in https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core.html.
Gene set enrichment analysis (GSEA)
Expression values included in the Talantov dataset (18 nevi and 45 primary melanoma tumors) (Talantov et al., 2005) were downloaded from GEO (GSE3189), while the expression values included in the Kabbarah dataset (9 nevi and 31 primary melanoma tumors) (Kabbarah et al., 2010) were downloaded from GEO (GSE46517). Gene Cluster Text files (GTC), as well as Categorical Class files (CLS) were generated independently for, RPPA, Talantov and Kabbarah datasets following the Gene Set Enrichment Analysis (GSEA) documentation indications (http://software.broadinstitute.org/gsea/index.jsp). GTC and CLS files were used to run independent GSEA analysis (javaGSEA desktop application). GSEA for Hallmarks, KEGG and Reactome were run independently under the following parameters: 1000 permutations, weighted enrichment analysis, signal to noise metric for ranking genes, and ‘‘meandiv’’ normalization mode. Following GSEA documentation indications, terms with p value < 0.05 and a q-value < 0.25 were considered significant (Data S1). For both, the RNA-Seq performed in this work (GSE133660) and Chicas dataset (Chicas et al., 2010) (GSE133660), genes were ranked according to the fold-change and p value obtained on the differential gene expression analysis as follows: –log10(p value)*sign(log 2 fold change). Pre-ranked files were built up for the RNA-Seq and Chicas datasets and used to run pre-ranked GSEA (Subramanian et al., 2005) under predefined parameters following GSEA documentation indications.
Polysome fractionation
Eight culture plates per condition (~23 million cells per condition) were incubated with harringtonine (2 µg/mL) for 2 min at 37°C followed by 5 min of cycloheximide (100 µg/mL) treatment at 37°C. Cells were washed twice with PBS after each treatment. Cells were scraped in 600uL of lysis buffer (50mM HEPES, 75mM KCl, 5mM MgCl2, 250mM sucrose, 0.1mg/mL cycloheximide, 2mM DTT, 1% Triton X-100, 1.3% sodium deoxycholate and 5 mL of RNase OUT) on ice. Lysates were rocked for 10 min at 4°C and centrifuged at 3000 g for 15 min at 4°C. 400 mL of lysates supernatant (cytosolic cell extracts) were layered over cold sucrose gradients (10mM HEPES, 75mM KCl, 5mM MgCl2, 0.5mM EDTA and increasing sucrose concentrations from 20% to 47%). Gradients were centrifuged at 34,000 rpms in a Beckman SW41 rotor for 2h and 40 min at 4°C. After centrifugation, light (0 to 2 ribosomes) and heavy (< 2 ribosomes) polysome fractions were collected in Trizol (1:1) using a density gradient fractionation system (Brandel) equipped with a UA-6 absorbance detector and a R1 fraction collector.
RT-qPCR
Total RNA was extracted from cells with Trizol, DNase treated, cleaned, and concentrated using Zymo columns (Zymo Research, Cat# R1013) following the manufacturer’s instructions. Similarly, mRNA from polysome fractions was DNase treated, cleaned, and concentrated using Zymo columns. Total RNA from tumor samples was isolated with NucleoSpin RNA/Protein following manufacturer’s instructions. Optical density values of RNA were measured using NanoDrop One (Thermo Scientific) to confirm an A260 and A280 ratio above 1.9. Relative expression of target genes (listed in Table S2) were analyzed using the QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) with clear 96 well plates (Greiner Bio-One). Primers were designed using the Integrated DNA Technologies (IDT) tool (https://eu.idtdna.com/scitools/Applications/RealTimePCR/) (Table S2). A total of 25ng of RNA was used for One-Step qPCR (Quanta BioSciences) following the manufacturer’s instructions in a final volume of 10ul. Conditions for amplification were: 10 min at 48°C, 5 min at 95°C, 40 cycles of 10 s at 95°C and 7 s at the corresponding annealing temperature (Table S2). The assay ended with a melting curve program: 15 s at 95°C, 1 min at 70°C, then ramping to 95°C while continuously monitoring fluorescence. Alternatively, relative expression of CDKN2A was determined by adapting the method of Zhang et al. (2015). A total of 100ng of RNA was used for One-Step qPCR (Quanta BioSciences) flowing the manufacturer’s instructions in a final volume of 10uL. Conditions for amplification were: 10 min at 48°C, 5 min at 95°C, 4 cycles of 10 s at 95°C and 10 s starting at 66°C and decreasing 2°C per cycle, 50 cycles of 10 s at 95°C and 7 s at 64°C. The assay ended with a melting curve program as above. Each sample was assessed in triplicate. Relative quantification was determined to multiple reference genes (B2M, MRPL9, PSMC4, and PUM1) using the delta-delta Ct method. The percentage of target gene mRNA in each polysome fraction was calculated as in Panda et al. (2017), but Ct values were first normalized to reference genes.
[35S]-methionine/cysteine incorporation followed by IP
After 16h of FBS starvation, cells were treated with 250nM Torin 1 or DMSO in media supplemented with 0.5% FBS for 3h. Thirty minutes prior to the end of Torin 1 treatment, 110uCi [35S]-methionine/cysteine was added to each plate. Cells were wash twice with PBS and pelleted. The pellet was resuspended in 100uL of denaturing Lysis buffer (1mM SDS and 5mM EDTA pH = 8) and boiled at 95°C for 5 min. Lysates were resuspended in 900uL of non-denaturing buffer (20mM Tris-HCl pH = 8, 137mM NaCl, 1mM EDTA pH = 8 and 1% Triton-X), sonicated, and centrifuged (10 min, 13,000 g, 4°C). The supernatant was collected, and the protein concentration was determined using the Bradford assay. 500ug of protein per condition was precleared with 15uL of magnetic beads (rotating 1h, 4°C). 5uL per sample of anti-RPIA or anti-IgG were bound to magnetic beads (rotating 3h, 4°C). Precleared samples were incubated with corresponding magnetic beads with conjugated antibodies (rotating overnight, 4°C). Magnetic beads were washed 3 times for 15 min each at 4°C with cold wash buffer (10mM Tris-HCl pH = 7.4, 1mM EDTA pH = 8, 1mM EGTA pH = 8, 150mM NaCl, 1% Triton-X, 0.2mM Na3VO4, 1mM PMSF and cOmplete Mini EDTA-free 1X). Immunoprecipitates were eluted in 10uL of 1X sample buffer (2% SDS, 10% glycerol, 0.01% bromophenol blue, 62.5mM Tris, pH 6.8, and 0.1M DTT) and boiled 10 min at 65°C and 1000rpm. Immunoprecipitates and 10% of input were separated under a 12% acrylamide gel. Acrylamide gel was stained with Coomassie blue and immunoprecipitated bands corresponding to RPIA (35kDa) were excised from the gel. Excised bands were weighted for normalization purposes before digestion with 1mL of electrode buffer (96mM Tris, 500mM glycine and 0.4% w/s SDS) for 16h at 4°C. Excised band suspensions were used to quantify the counts per minute (CPM) in a Beckman LS6500 scintillation counter. CPMs were normalized to corresponding band weights (mg).
Senescence and proliferation assays
SA-β-Gal staining was performed as previously described (Dimri et al., 1995). Cells were fixed in 2% formaldehyde/0.2% glutaraldehyde in PBS (5 min) and stained (40 mM Na2HPO4, 150 mM NaCl, 2 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, and 1 mg/ml X-gal) overnight at 37°C in a non-CO2 incubator. Images were acquired at room temperature using an inverted microscope (Nikon Eclipse Ts2) with a 20X/0.40 objective (Nikon LWD) equipped with a camera (Nikon DS-Fi3). Each sample was assessed in triplicate and at least 100 cells per well were counted (> 300 cells per experiment).
For BrdU incorporation, cells on coverslips were incubated with 1uM BrdU for 30 min (IMR90, ES2, and SKMel28) or 15 min (SW620, SW480, HT-29, PATU8902, and TCCSUP). Cells were fixed in 4% paraformaldehyde (10min), permeabilized in 0.2% Triton X-100 (5 min), and postfixed in 1% PF 0.01% Tween-20 (30 min). Cells were DNaseI treated (10 min) previous to blocking with 3% BSA/PBS (5 mins). Cells were incubated in anti-BrdU primary antibody in 3% BSA/PBS (1:500) for 1h followed by 3 washes using 1% Triton X-100. Cell were then incubated 1h in FITC anti-Rat secondary antibody in 3% BSA/PBS (1:1000). Finally, cells were incubated with 0.15 µg/ml DAPI in PBS (1 min), mounted, and sealed. Images were acquired at room temperature using a Nikon Eclipse 90i microscope with a 20x/0.17 objective (Nikon DIC N2 Plan Apo) equipped with a CoolSNAP Photometrics camera. Each sample was assessed in triplicate and at least 200 cells per well were counted (> 600 cells per experiment).
For colony formation, an equal number of cells were seeded in 6-well plates and cultured for an additional 2 weeks. Colony formation was visualized by fixing cells in 1% paraformaldehyde (5 min) and staining with 0.05% crystal violet (20 min). Wells were destained in 500 µL 10% acetic acid (5 min). Absorbance (590nm) was measured using a spectrophotometer (Spectra Max 190). Each sample was assessed in triplicate.
Temsirolimus half maximal inhibitory concentration (IC50) assay
Twenty thousand cells per condition were plated in 12-well plates in triplicates. After 24 h regular media was replaced by 0.5% FBS supplemented media with increasing concentrations of temsirolimus (0uM, 0.07uM, 0.21uM, 0.62uM, 1.85uM, 5.56uM, 16.67uM and 50uM). After 5 days cells were fixed in 1% paraformaldehyde (5 min) and stained with 0.05% crystal violet (20 min). Wells were destained in 500uL 10% acetic acid (5 min). Absorbance (590nm) was measured using a spectrophotometer (Spectra Max 190). The half maximal inhibitory concentration (IC50) was defined as the concentration resulting in a 50% reduction in absorbance.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde (10min) and permeabilized in 0.2% Triton X-100 (5 min). Cells were blocked with 3% BSA/ PBS (1h) and incubated in anti-mTORC (1/200) and anti-LAMP2 (1/100) in 3% BSA/PBS (16h) followed by 3 washes using 1% Triton X-100. Cells were then incubated in FITC anti-Rabbit (1/2000) and Cy3 anti-mouse (1/5000) secondary antibodies in 3% BSA/PBS (1 h). Finally, cells were incubated with 0.15 µg/ml DAPI in PBS (1 min), mounted and sealed. Images were acquired at room temperature using a confocal microscope (Leica SP8) with a 64X oil objective. Co-localization analysis was performed using the Leica software in a total of 14 cells per sample.
Western blotting
Cell lysates were collected in 1X sample buffer (2% SDS, 10% glycerol, 0.01% bromophenol blue, 62.5mM Tris, pH 6.8, 0.1M DTT) and boiled (10 min at 95°C). Protein concentration was determined using the Bradford assay. Proteins were resolved using SDS-PAGE gels and transferred to nitrocellulose membranes (Fisher Scientific) (110mA for 2 h at 4°C). Membranes were blocked with 4% BSA in TBS containing 0.1% Tween-20 (TBS-T) for 1 h at room temperature. Membranes were incubated overnight at 4°C in primary antibodies in 4% BSA/TBS 0.025% sodium azide. Membranes were washed 4 times in TBS-T for 5 min at room temperature after which they were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. After washing 4 times in TBS-T for 5 min at room temperature, proteins were visualized on film after incubation with SuperSignal West Pico PLUS Chemiluminescent Substrate (ThermoFisher, Waltham, MA).
Nucleotide Analysis by LC-HRMS
LC-HRMS for nucleotides and other polar metabolites was as previously described (Guo et al., 2016; Kuskovsky et al., 2019). Briefly, an Ultimate 3000 UHPLC equipped with a refrigerated autosampler (at 6°C) and a column heater (at 55°C) with a HSS C18 column (2.1 × 100 mm i.d., 3.5 µm; Waters) was used for separations. Solvent A was 5 mM DIPEA and 200 mM HFIP and solvent B was methanol with 5 mM DIPEA 200 mM HFIP. The gradient was as follows: 100% A for 3 min at 0.18 mL/min, 100% A at 6 min with 0.2 mL/min, 98% A at 8 min with 0.2 mL/min, 86% A at 12 min with 0.2 mL/min, 40% A at 16 min and 1% A at 17.9 min-18.5 min with 0.3 mL/min then increased to 0.4 mL/min until 20 min. Flow was ramped down to 0.18 mL/min back to 100% A over a 5 min re-equilibration. For MS analysis, the UHPLC was coupled to a Q Exactive HF mass spectrometer (Thermo Scientific) equipped with a HESI II source operating in negative mode. The operating conditions were as follows: spray voltage 4000 V; vaporizer temperature 200°C; capillary temperature 350°C; S-lens 60; in-source CID 1.0 eV, resolution 60,000. The sheath gas (nitrogen) and auxiliary gas (nitrogen) pressures were 45 and 10 (arbitrary units), respectively. Single ion monitoring (SIM) windows were acquired around the [M-H]- of each analyte with a 20 m/z isolation window, 4 m/z isolation window offset, 1e6 ACG target and 80 ms IT, alternating in a Full MS scan from 70–950 m/z with 1e6 ACG, and 100 ms IT. Data was analyzed in XCalibur v4.0 and/or Tracefinder v4.1 (Thermo Scientific) using a 5 ppm window for integration of the peak area of all analytes. Used standards for nucleotides and deoxinucleotides both isotope labeled and non-labeled are indicated on the table. No suitable source of stable isotope labeled ADP, dADP, dTDP, CDP, or dCDP was found, thus the mono-phosphate was used as a surrogate internal standard. Guanine nucleotides were not quantified due to spectral overlap with the highly abundant adenine nucleotides.
Glucose labeling and analysis
Cells were seeded in 10 cm culture plates and at the end of the indicated treatment (Figure S1A) cells were washed twice with PBS and incubated 8h in DMEM (Cat# D5030) supplemented with 5mM of 13C6-D-glucose, 0.5% of charcoal stripped FBS and 20mM of HEPES. Isotopologue patterns for dNDPs, dNTPs, and ribose-5-phosphate were analyzed by LC-HRMS as indicated above. Adjustment for natural isotopic abundance was conducted through open source and publicly available FluxFix (Trefely et al., 2016).
Flow Cytometry
For 7AAD staining both cells and media were collected and centrifuged (1000 rpm for 5 min) followed by resuspension in 500uL of 7AAD staining solution (5uL of 7AAD solution and 38mM NaCitrate). Stained cells were run on a 10-color FACSCanto flow cytometer (BD biosciences). Data were analyzed with FlowJo Software. Each sample was assessed in triplicate.
Murine tumor model
HT-29 colorectal carcinoma cells were infected with shRNA targeting p16 and RPIA alone or in combination. After 2 days of puromycin selection (3 µg/mL), 3 million cells were resuspended in 200 µL of PBS and injected subcutaneously into the left flank of SCID mice. Mice were monitored daily to identify palpable tumors. Mice weight and tumor length (L) and width (W) (L > W) were measured every 3 days after a tumor volume of 200mm3. Tumor volume was calculated as 1/2 (L x W2). All animals were sacrificed at day 26 post injection and tumor tissues collected for following experiments. A total of 22 animals were used in this study (6 control, 6 shRPIA, 4 shp16 and 6 shp16/shRPIA).
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism version 7.0 was used to perform statistical analysis. The level of significance between two groups was assessed with unpaired t test. For dataset with more than two groups, one-way ANOVA followed by Tukey’s post hoc test was applied. P values < 0.05 were considered significant. The IC50 dose-response curves were plotted, and IC50 values calculated using the log(inhibitor) versus normalized response function in GraphPad Prism Version 7.0. Kaplan-Meier survival plots differences between the two groups were analyzed with log-rank (Mantel-Cox) test using GraphPad Prism version 7.0. Data for the indicated tumors was obtained from cBioportal (Cerami et al., 2012; Gao et al., 2013). Unpaired t test between shRRM2/shp16 and shRRM2 alone was assessed with GraphPad Prism version 7.0 for each transcript included in the polysome fractionation experiment (Figure 3A; Table S1). P values were adjusted according to Benjamini and Hochberg’s false discovery rate (FDR) and the percentage of shift between the light and the heavy fraction in shRRM2/shp16 was calculated (% transcript in heavy fraction - % transcript in light fraction) for each transcript. Hits were defined as those transcripts that were significantly (FDR P-value < 0.05) decreased in the light fraction and significantly (FDR P-value < 0.05) increased in the heavy fraction in shRRM2/shp16 when compared with shRRM2 alone. With these criteria we narrowed the list to three transcripts: ribose-5-phosphate isomerase A (RPIA), nucleoside diphosphate kinase A (NME1), and nucleoside diphosphate kinase 3 (NME3). NME1/NME3 are known metastasis suppressors (Boissan et al., 2018); therefore, we focused on RPIA. Longitudinal and cross-sectional analysis of tumor volume where calculated using TumorGrowth tool with default parameters (Enot et al., 2018).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RRM2 | Santa Cruz Biotechnology | Cat # sc-398294 |
| p16 | Abcam | Cat# ab108349; RRID:AB_10858268 |
| Vinculin | Sigma-Aldrich | Cat# V9131; RRID:AB_477629 |
| S6K | Cell Signaling Technology | Cat# 2708; RRID:AB_390722 |
| Phospho S6K (Thr389) | Cell Signaling Technology | Cat# 9234; RRID:AB_2269803 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9644; RRID: AB_2097841 |
| Phospho 4E-BP1 (Ser65) | Cell Signaling Technology | Cat# 9451; RRID: AB_330947 |
| RPTOR | Cell Signaling Technology | Cat# 2280; RRID: AB_561245 |
| β-Actin | Sigma-Aldrich | Cat# A1978; RRID:AB_476692 |
| RPIA | Abcam | Cat# ab181235 |
| BRAF | Santa Cruz Biotechnology | Cat# sc-5284; RRID:AB_2721130 |
| RB | BD Biosciences | Cat# 554136; RRID:AB_39525 |
| CAD | Cell Signaling Technology | Cat# 11933; RRID: AB_2797772 |
| Phospho CAD (Ser1859) | Cell Signaling Technology | Cat # 70307; RRID: AB_2799782 |
| BrdU | Abcam | Cat# ab6326; RRID:AB_305426 |
| mTOR | Cell Signaling Technology | Cat# 2983; RRID:AB_2105622 |
| LAMP2 | Santa Cruz Biotechnology | Cat# sc-18822; RRID:AB_626858 |
| Anti-mouse HRP | Cell Signaling Technology | Cat# 7076; RRID:AB_330924 |
| Anti-rabbit HRP | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Anti-rat FITC | Jackson ImmunoResearch Labs | Cat# 712–095-150; RRID:AB_2340651 |
| Anti-rabbit FITC | Jackson ImmunoResearch Labs | Cat# 711–095-152; RRID:AB_2315776 |
| Anti-mouse Cy3 | Jackson ImmunoResearch Labs | Cat# 715–165-150; RRID:AB_2340813 |
| Bacterial and Virus Strains | ||
| Stbl3 Chemically Competent E. | Fisher Scientific | Cat# C737303 |
| DH5α Competent Cells | Fisher Scientific | Cat# 18265–017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BrdU | Alfa Aesar | Cat # H27260 |
| [35S]-methionine/cysteine | Perkin Elmer | Cat # NEG772 |
| X-Gal | Sigma-Aldrich | Cat # B4252 |
| D-Glucose-13C6 | Sigma-Aldrich | Cat # 389374 |
| Puromycin | GIBCO | Cat # A11138–02 |
| Polybrene | Sigma-Aldrich | Cat # H9268 |
| Propidium Iodide | Sigma-Aldrich | Cat# P4170 |
| PureProteome™ Protein G Magnetic Beads | EMD Millipore | Cat # LSKMAGG10 |
| cOmplete Mini EDTA-free | Roche | Cat # 11836170001 |
| Crystal violet | Harleco | Cat # 192–12 |
| DMEM 17 | Corning | Cat# 10–017-CV |
| DMEM 13 | Corning | Cat# 10–013-CV |
| RPMI | GIBCO | Cat# 11875093 |
| DMEM w/o glucose or glutamine | Sigma-Aldrich | Cat# D5030 |
| MEM/EBSS glutamine | HyClone | Cat# SH30024.01 |
| MEM Nonessential Amino Acids | Corning | Cat# 25025CL |
| Glutagro | Corning | Cat# 25015CL |
| Sodium Bicarbonate | Corning | Cat# 25035CL |
| Sodium Pyruvate | Corning | Cat# 25000CL |
| FBS | VWR | Cat# 16000–044 |
| Charcoal stripped FBS | Sigma-Aldrich | Cat# F6765 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Trizol | Ambion | Cat# 15596018 |
| Glutaraldehyde | Polysciences, Inc. | Cat# 01909 |
| RNase Out | Invitrogen | Cat# 10777019 |
| Formaldehyde | VWR | Cat# 0493 |
| Paraformaldehyde | Sigma-Aldrich | Cat# 158127 |
| Temsirolimus | Selleckchem | Cat# S1044 |
| Torin 1 | Med Chem Express | Cat# HY-13003 |
| 7AAD | Tonbo | Cat# 13–6993-T500 |
| Optima LC-MS grade water | Thermo Fisher Scientific | Cat# W6–4 |
| Optima LC-MS grade acetonitrile | Thermo Fisher Scientific | Cat# A955–4 |
| Optima LC-MS grade methanol | Thermo Fisher Scientific | Cat# A456–4 |
| 1,1,1,3,3,3-hexafluoro 2-propanol (HFIP) | Sigma-Aldrich | Cat# 105228 |
| Diisopropylethylamine (DIPEA) | Sigma-Aldrich | Cat# D125806 |
| AMP standard | Sigma-Aldrich | Cat# 01930 |
| dAMP standard | Sigma-Aldrich | Cat# D6375 |
| ATP standard | Sigma-Aldrich | Cat# A26209 |
| dATP standard | Sigma-Aldrich | Cat# D6500 |
| dTMP standard | Sigma-Aldrich | Cat# T7004 |
| dTTP standard | Sigma-Aldrich | Cat# T0251 |
| CMP standard | Sigma-Aldrich | Cat# C1006 |
| dCMP standard | Sigma-Aldrich | Cat# D7625 |
| CTP standard | Sigma-Aldrich | Cat# C1506 |
| dCTP standard | Sigma-Aldrich | Cat# D4635 |
| UMP standard | Sigma-Aldrich | Cat# U6375 |
| dUMP standard | Sigma-Aldrich | Cat# D3876 |
| UDP standard | Sigma-Aldrich | Cat# 94330 |
| UTP standard | Sigma-Aldrich | Cat# U6625 |
| dUTP standard | Sigma-Aldrich | Cat# D4001 |
| AMP-13C,15N standard | Sigma-Aldrich | Cat# 650676 |
| dAMP-13C,15N standard | Sigma-Aldrich | Cat# 648620 |
| ATP-13C, 15N standard | Sigma-Aldrich | Cat# 645702 |
| dATP-13C, 15N standard | Sigma-Aldrich | Cat# 646237 |
| TTP-13C, 15N standard | Sigma-Aldrich | Cat# 646202 |
| CTP-13C, 15N standard | Sigma-Aldrich | Cat# 645699 |
| dTPC-13C, 15N standard | Sigma-Aldrich | Cat# 646229 |
| UTP-13C, 15N standard | Sigma-Aldrich | Cat# 645672 |
| dTMP-13C, 15N standard | Sigma-Aldrich | Cat# 648590 |
| CMP-13C, 15N standard | Sigma-Aldrich | Cat# 650692 |
| UMP-13C, 15N standard | Sigma-Aldrich | Cat# 651370 |
| Critical Commercial Assays | ||
| NucleoSpin RNA/Protein | Macherey-Nagel | Cat#740933 |
| Deposited Data | ||
| RNA-Seq | This paper | GEO: GSE133660 |
| Experimental Models: Cell Lines | ||
| Fibroblasts: IMR90 | ATCC | CCL-186 |
| Fibroblasts: IMR90 shControl | This paper | N/A |
| Fibroblasts: IMR90 shRRM2 | This paper | N/A |
| Fibroblasts: IMR90 shp16 | This paper | N/A |
| Fibroblasts: IMR90 shRRM2/shp16 | This paper | N/A |
| Fibroblasts: IMR90 shRRM2/shRB | This paper | N/A |
| Fibroblasts: IMR90 shRPIA | This paper | N/A |
| Fibroblasts: IMR90 shRRM2/shp16/shRPIA | This paper | N/A |
| Fibroblasts: IMR90 shRRM2/shp16/p16 OE | This paper | N/A |
| Fibroblasts: IMR90 Control | This paper | N/A |
| Fibroblasts: IMR90 BRAFV600E | This paper | N/A |
| Fibroblasts: IMR90 BRAFV600E/shp16 | This paper | N/A |
| Fibroblasts: IMR90 BRAFV600E/shRB | This paper | N/A |
| BRAFV600E/shp16/shRPIA | This paper | N/A |
| Embryonic kidney: 293FT | Thermo Fisher | R70007 |
| Embryonic kidney: Phoenix (QNX) | Dr. Gary Nolan | N/A |
| Skin: SKMel28 | ATCC | HTB-72 |
| Skin: SKMel28 shControl | This paper | N/A |
| Skin: SKMel28 shRPIA | This paper | N/A |
| Skin: SKMel28 shp16 | This paper | N/A |
| Skin: SKMel28 shp16/shRPIA | This paper | N/A |
| Skin: SKMel28 shRB | This paper | N/A |
| Skin: SKMel28: shp16/shRPTOR #1 | This paper | N/A |
| Skin: SKMel28: shp16/shRPTOR #2 | This paper | N/A |
| Pancreas: PATU8902 | G. DeNicola Laboratory | N/A |
| Pancreas: PATU8902 shControl | This paper | N/A |
| Pancreas: PATU8902 shRPIA | This paper | N/A |
| Pancreas: PATU8902 shp16 | This paper | N/A |
| Pancreas: PATU8902 shp16/shRPIA | This paper | N/A |
| Ovary: ES-2 | ATCC | Crl-1978 |
| Ovary: ES-2 shControl | This paper | N/A |
| Ovary: ES-2 shRPIA | This paper | N/A |
| Ovary: ES-2 shp16 | This paper | N/A |
| Ovary: ES-2 shp16/shRPIA | This paper | N/A |
| Bladder: TCC-SUP | ATCC | HTB-5 |
| Bladder: TCC-SUP shControl | This paper | N/A |
| Bladder: TCC-SUP shRPIA | This paper | N/A |
| Bladder: TCC-SUP shp16 | This paper | N/A |
| Bladder: TCC-SUP shp16/shRPIA | This paper | N/A |
| Colon: SW620 | ATCC | CCL-227 |
| Colon: SW620 shControl | This paper | N/A |
| Colon: SW620 shRPIA | This paper | N/A |
| Colon: SW620 shp16 | This paper | N/A |
| Colon: SW620 shp16/shRPIA | This paper | N/A |
| Colon: SW480 | ATCC | CCL-228 |
| Colon: SW480 shControl | This paper | N/A |
| Colon: SW480 shRPIA | This paper | N/A |
| Colon: SW480 shp16 | This paper | N/A |
| Colon: SW480 shp16/shRPIA | This paper | N/A |
| Colon: HT-29 | ATCC | HTB-38 |
| Colon: HT-29 shControl | This paper | N/A |
| Colon: HT-29 shRPIA | This paper | N/A |
| Colon: HT-29 shp16 | This paper | N/A |
| Colon: HT-29 shp16/shRPIA | This paper | N/A |
| Skin: SKMel28 shRB | This paper | N/A |
| Skin: SKMel28: shp16/shRPTOR #1 | This paper | N/A |
| Skin: SKMel28: shp16/shRPTOR #2 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| C.B-17/ICRHsd-Prkdc scid mice | Envigo | 182 |
| Oligonucleotides | ||
| RT-qPCR Primers (Listed in Table S2) | This paper | N/A |
| Software and Algorithms | ||
| GSEA | Broad Institute | N/A |
| GraphPad Prism 7 | N/A | N/A |
| IDT tool for primer design | Integrated DNA Technologies | N/A |
| Cufflinks | Version v.2.0.2 | N/A |
Highlights.
p16 knockdown activates mTORC1 to increase nucleotide synthesis and bypass senescence
mTORC1 directly increases translation of RPIA to increase ribose-5-phosphate
Activation of the mTORC1 pathway downstream of p16 suppression is independent of RB
RPIA suppression induces senescence only in cells and tumors with low p16
ACKNOWLEDGMENTS
We would like to acknowledge Dr. Alice Soragni (UCLA), Dr. Kristin Eckert and Dr. Nadine Hempel (Penn State College of Medicine), Dr. Gina DeNicola (Moffitt Cancer Center), and Dr. Rugang Zhang (The Wistar Institute) for providing cell lines. We would like to thank Drs. Juan Andres Melendez and Robert P. Feehan for providing helpful discussion. This work was supported by grants from the NIH (F31CA236372 to E.S.D., P50CA174523 and U54CA224070 to M.H., R01DK13499 and R01K156548 to S.R.K., K22ES026235 and R01GM132261 to N.W.S., and R00CA194309 to K.M.A.); the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to M.H.); the W.W. Smith Charitable Trust (to K.M.A.); and the Penn State Cancer Institute Postdoctoral Fellowship (to R.B.). The RPPA Core Facility is funded by NCI P30CA16672.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.084.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
The RNA-Seq generated during this study is available at GEO (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE133660.
REFERENCES
- Aird KM, and Zhang R (2014). Metabolic alterations accompanying oncogene-induced senescence. Mol. Cell. Oncol 1, e963481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird KM, and Zhang R (2015). Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett 356 (2 Pt A), 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, Wu H, Wei Z, Wagner SN, Herlyn M, and Zhang R (2013). Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep 3, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird KM, Worth AJ, Snyder NW, Lee JV, Sivanand S, Liu Q, Blair IA, Wellen KE, and Zhang R (2015). ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep 11, 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, et al. (2014). A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun 5, 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khalaf HH, Mohideen P, Nallar SC, Kalvakolanu DV, and Aboussekhra A (2013). The cyclin-dependent kinase inhibitor p16INK4a physically interacts with transcription factor Sp1 and cyclin-dependent kinase 4 to transactivate microRNA-141 and microRNA-146b-5p spontaneously and in response to ultraviolet light-induced DNA damage. J. Biol. Chem 288, 35511–35525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, and Manning BD (2013). Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, and Manning BD (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, Robine S, Jung A, Arkan MC, Kirchner T, and Greten FR (2010). Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell 18, 135–146. [DOI] [PubMed] [Google Scholar]
- Bennett DC (2016). Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res 29, 122–140. [DOI] [PubMed] [Google Scholar]
- Boissan M, Schlattner U, and Lacombe ML (2018). The NDPK/NME super-family: state of the art. Lab. Invest 98, 164–174. [DOI] [PubMed] [Google Scholar]
- Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chrétien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, et al. (2007). Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet 39, 776–780. [DOI] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dörken B, Jenuwein T, and Schmitt CA (2005). Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436, 660–665. [DOI] [PubMed] [Google Scholar]
- Caldwell ME, DeNicola GM, Martins CP, Jacobetz MA, Maitra A, Hruban RH, and Tuveson DA (2012). Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene 31, 1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, and Lowe SW (2010). Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17, 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky W, Micevic G, Meeth K, Muthusamy V, Curley DP, Santhanakrishnan M, Erdelyi I, Platt JT, Huang L, Theodosakis N, et al. (2015). mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell 27, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Filenova E, Collado M, Serrano M, Jones K, and McMahon M (2007). A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev 21, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Däbritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, et al. (2013). Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425. [DOI] [PubMed] [Google Scholar]
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enot DP, Vacchelli E, Jacquelot N, Zitvogel L, and Kroemer G (2018). TumGrowth: An open-access web tool for the statistical analysis of tumor growth curves. OncoImmunology 7, e1462431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel VK, Ibrahim N, Jiang G, Singhal M, Fee S, Flotte T, Westmoreland S, Haluska FS, Hinds PW, and Haluska FG (2009). Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 28, 2289–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, Ramm S, Palmer AC, Yuzugullu H, Varadan V, et al. (2016). Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 29, 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Worth AJ, Mesaros C, Snyder NW, Glickson JD, and Blair IA (2016). Diisopropylethylamine/hexafluoroisopropanol-mediated ion-pairing ultra-high-performance liquid chromatography/mass spectrometry for phosphate and carboxylate metabolite analysis: utility for studying cellular metabolism. Rapid Commun. Mass Spectrom 30, 1835–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferkamp S, Becker TM, Scurr LL, Kefford RF, and Rizos H (2008). p16INK4a-induced senescence is disabled by melanoma-associated mutations. Aging Cell 7, 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hernandez-Segura A, Nehme J, and Demaria M (2018). Hallmarks of Cellular Senescence. Trends Cell Biol 28, 436–453. [DOI] [PubMed] [Google Scholar]
- Hosseini M, Dousset L, Mahfouf W, Serrano-Sanchez M, Redonnet-Vernhet I, Mesli S, Kasraian Z, Obre E, Bonneu M, Claverol S, et al. (2018). Energy Metabolism Rewiring Precedes UVB-Induced Primary Skin Tumor Formation. Cell Rep 23, 3621–3634. [DOI] [PubMed] [Google Scholar]
- Iurlaro R, León-Annicchiarico CL, and Muñoz-Pinedo C (2014). Regulation of cancer metabolism by oncogenes and tumor suppressors. Methods Enzymol 542, 59–80. [DOI] [PubMed] [Google Scholar]
- Jenkins NC, Liu T, Cassidy P, Leachman SA, Boucher KM, Goodson AG, Samadashwily G, and Grossman D (2011). The p16(INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene 30, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabbarah O, Nogueira C, Feng B, Nazarian RM, Bosenberg M, Wu M, Scott KL, Kwong LN, Xiao Y, Cordon-Cardo C, et al. (2010). Integrative genome comparison of primary and metastatic melanomas. PLoS One 5, e10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohnken R, Kodigepalli KM, and Wu L (2015). Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol. Cancer 14, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegl L, Neumann J, Vieth M, Greten FR, Reu S, Jung A, and Kirchner T (2011). Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Mod. Pathol 24, 1015–1022. [DOI] [PubMed] [Google Scholar]
- Kuskovsky R, Buj R, Xu P, Hofbauer S, Doan MT, Jiang H, Bostwick A, Mesaros C, Aird KM, and Snyder NW (2019). Simultaneous isotope dilution quantification and metabolic tracing of deoxyribonucleotides by liquid chromatography high resolution mass spectrometry. Anal. Biochem 568, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AN, and Fan TWM (2015). Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43, 2466–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPak KM, and Burd CE (2014). The molecular balancing act of p16(INK4a) in cancer and aging. Mol. Cancer Res 12, 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Choi BY, Cho YY, Lee SY, Huang Z, Kundu JK, Kim MO, Kim DJ, Bode AM, Surh YJ, and Dong Z (2013). Tumor suppressor p16(INK4a) inhibits cancer cell growth by downregulating eEF1A2 through a direct interaction. J. Cell Sci 126, 1744–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Poi MJ, and Tsai MD (2011). Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 50, 5566–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch CJ, Kimball SR, Xu Y, Salzberg AC, and Kawasawa YI (2015). Global deletion of BCATm increases expression of skeletal muscle genes associated with protein turnover. Physiol. Genomics 47, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, and Blenis J (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol 10, 307–318. [DOI] [PubMed] [Google Scholar]
- Mannava S, Moparthy KC, Wheeler LJ, Natarajan V, Zucker SN, Fink EE, Im M, Flanagan S, Burhans WC, Zeitouni NC, et al. (2013). Depletion of Deoxyribonucleotide Pools Is an Endogenous Source of DNA Damage in Cells Undergoing Oncogene-Induced Senescence. Am. J. Path 182, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, and Peeper DS (2005). BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724. [DOI] [PubMed] [Google Scholar]
- Nandagopal N, and Roux PP (2015). Regulation of global and specific mRNA translation by the mTOR signaling pathway. Translation (Austin) 3, e983402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda AC, Martindale JL, and Gorospe M (2017). Polysome Fractionation to Analyze mRNA Distribution Profiles. Bio. Protoc 7, e2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Reyna-Neyra A, Philippe L, and Thoreen CC (2017). mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep 19, 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Mancera PA, Young AR, and Narita M (2014). Inside and out: the activities of senescence in cancer. Nat. Rev. Cancer 14, 547–558. [DOI] [PubMed] [Google Scholar]
- Pontarin G, Ferraro P, Bee L, Reichard P, and Bianchi V (2012). Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. USA 109, 13302–13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, Wang X, Bronson RT, Lyssiotis CA, Ying H, and Kimmelman AC (2018). Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun 9, 4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, and Chodosh LA (2007). Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol 9, 493–505. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, et al. (2015). The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med 373, 1926–1936. [DOI] [PubMed] [Google Scholar]
- Sherr CJ (2001). The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol 2, 731–737. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, and Wang Y (2005). Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin. Cancer Res 11, 7234–7242. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefely S, Ashwell P, and Snyder NW (2016). FluxFix: automatic isotopologue normalization for metabolic tracer analysis. BMC Bioinformatics 17, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi E, Liu B, Li C, Liu T, Rutter J, and Grossman D (2017). Loss of p16INK4A stimulates aberrant mitochondrial biogenesis through a CDK4/Rb-independent pathway. Oncotarget 8, 55848–55862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff CC, and Drexler HG (2005). Detection of mycoplasma contaminations. Methods Mol. Biol 290, 13–23. [DOI] [PubMed] [Google Scholar]
- Wiley CD, and Campisi J (2016). From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab 23, 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen P, and Campisi J (2007). Oncogene-induced senescence pathways weave an intricate tapestry. Cell 128, 233–234. [DOI] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wang J, Deng F, Yan Z, Xia Y, Wang Z, Ye J, Deng Y, Zhang Z, Qiao M, et al. (2015). TqPCR: A Touchdown qPCR Assay with Significantly Improved Detection Sensitivity and Amplification Efficiency of SYBR Green qPCR. PLoS One 10, e0132666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, and Sabatini DM (2011). mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol 12, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.