

Abstract
PURPOSE
To understand the clinical context of tumor mutational burden (TMB) when comparing a pan-cancer threshold and a cancer-specific threshold.
MATERIALS AND METHODS
Using whole exome sequencing data from primary tumors in The Cancer Genome Atlas (n = 3,534) and advanced and/or metastatic tumors from Weill Cornell Medicine Advanced (n = 696), TMB status was determined using a pan-cancer and cancer-specific threshold. Survival curves, number of samples classified as TMB high, and predicted neoantigens were used to evaluate the differences between thresholds.
RESULTS
The distribution of TMB varied dramatically among cancer types. A cancer-specific threshold was able to adjust for the different TMB distributions, whereas the pan-cancer threshold was often too stringent. The dynamic nature of the cancer-specific threshold resulted in more tumors being classified as TMB high compared with the static pan-cancer threshold. In addition, no significant difference in survival outcomes was found with the cancer-specific threshold compared with the pan-cancer threshold. Furthermore, the cancer-specific threshold maintained higher predicted neoantigen load for the TMB-high samples compared with the TMB-low samples, even when the threshold was lower than the pan-cancer threshold.
CONCLUSION
TMB is determined within the context of cancer type, metastatic state, and disease stage. Compared with a pan-cancer threshold, a cancer-specific threshold classifies more patients as TMB high while maintaining clinical outcomes that are not significantly different. Furthermore, the cancer-specific threshold identifies patients with a high number of predicted neoantigens. Because of the potential impact in the care of patients with cancer, TMB status should be determined in a cancer-specific manner.
INTRODUCTION
Immunotherapy, especially checkpoint inhibition, has been added recently as a successful treatment option for some patients with cancer. It is based on the presence of neoantigens eliciting a response from T cells by recognizing these antigens as foreign and infiltrating the tumor microenvironment.1,2 However, only a subset of patients respond to checkpoint inhibitor immunotherapy; therefore, predictive biomarkers are critical to guide the selection of patients for these therapies. Although high-level programmed death-ligand 1 expression, microsatellite instability, and mismatch-repair deficiency have been deemed clinically relevant, other markers, including tumor mutational burden (TMB), interferon gamma profile, and human leukocyte antigen (HLA) genotype, have also exhibited promising results.3 TMB is commonly defined as the total number of somatic mutations in a tumor genome, divided by the number of bases sequenced in megabases (Mb).4,5 Previously, patients with higher TMB (TMB high) demonstrated increased response to immunotherapy compared with patients with lower TMB (TMB low), especially in non–small-cell lung cancer.6,7 However, no consensus has been established to define a standard threshold for TMB high. In addition, the methods for calculating TMB are inconsistent, especially the inclusion or exclusion of synonymous mutations. Last, previous studies have adopted panel target sequencing with small regions of genomic DNA, possibly excluding neoantigen candidates.8 Therefore, exploring the TMB distribution with more extensive sequencing platforms, such as whole exome sequencing (WES), can provide a more comprehensive view of TMB. In the current study, we compared two TMB thresholds: the Chalmers et al threshold and our Weill Cornell Medicine (WCM) threshold.4 The Chalmers et al threshold is a static cutoff of 20 mutations/Mb applied pan-cancer; it was selected because it was developed using a large data set composed of targeted panel and WES samples. Different cancer types have a wide range of TMB.9 To define more precise TMB thresholds that accurately reflect TMB in each cancer type, the WCM threshold uses interquartile range (IQR): mean (TMB) + 1.25 × IQR (TMB) applied within each cancer, meaning each cancer type will have its own threshold reflective of the distribution of mutations within that cancer type. We sought to improve our understanding of TMB distribution in various cancer types and of how a varying distribution affects a TMB-high threshold.
CONTEXT
Key Objective
Can a cancer-specific tumor mutational burden (TMB) provide more useful information compared with a fixed pan-cancer threshold?
Knowledge Generated
A cancer-specific TMB threshold can dynamically adjust to identify dramatically more patients as TMB high compared with a restrictive pan-cancer threshold. The additional patients do not have a significantly different outcome compared with the other TMB-high patients.
Relevance
When interpreting a patient’s TMB, the overall TMB seen in the specific cancer type should be taken into consideration to give the TMB context.
MATERIALS AND METHODS
Data Acquisition
Data from The Cancer Genome Atlas (TCGA) was obtained from the National Cancer Institute Genomic Data Commons through the R package TCGAbiolinks.10 Clinical and genomic data were obtained for bladder urothelial carcinoma (BLCA; n = 375),11 breast invasive carcinoma (BRCA; n = 598),12 colon adenocarcinoma (COAD; n = 84),13 glioblastoma multiforme (GBM; n = 327),14 kidney chromophobe (KICH; n = 65),15 kidney renal clear cell carcinoma (KIRC; n = 174),16 brain lower grade glioma (LGG; n = 506),14 lung adenocarcinoma (LUAD; n = 90),17 lung squamous cell carcinoma (LUSC; n = 95),18 ovarian serous cystadenocarcinoma (OV; n = 224),19 prostate adenocarcinoma (PRAD; n = 326),20 rectum adenocarcinoma (READ; n = 32),13 thyroid carcinoma (THCA; n = 411),21 and uterine corpus endometrial carcinoma (UCEC; n = 227).22 Detailed characterizations of each cohort can be found in their respective publications. Predicted neoantigens for TCGA samples were obtained through The Cancer Immunome Atlas.23
TMB Calculation
TMB was calculated as the total number of nonsynonymous mutated bases in the tumor genome divided by the Mb of the genome covered. The WCM Advanced cohort was sequenced using our EXaCT-1 WES Haloplex pipeline using tumor-normal pairs,24 which covers 37 Mb of the genome. Samples were collected under a protocol approved by the institutional review board of Weill Cornell Medical College, and written informed consent was obtained. Because TMB is divided by the total size of genome sequenced to help adjust for technical batch effects, the size of sequenced regions was determined for each TCGA cohort according to their respective publications.11-22 Samples with purity of less than 50% according to ABSOLUTE (TCGA) or Clonality Estimate in Tumors (EXaCT-1; Weill Cornell Medine Clinical Genomics Lab, New York, NY) were removed.25,26 Germline variants and variants appearing in the Exome Aggregation Consortium database with a frequency of at least 1% were removed to filter them out.27 Variants with a variant allele frequency (VAF) of less than 10% were filtered out after correcting VAF for tumor purity by dividing VAF by purity. A summary of filtering steps can be seen in the Data Supplement.
Mutational Signatures
Mutational signatures were determined using the deconstructSigs R package.28 For each sample, a matrix containing the frequency a mutation appears within a trinucleotide context was generated. This matrix was compared with the signatures published previously in the Catalogue of Somatic Mutations in Cancer,29 resulting in signature contributions.
Statistical Methods
The Wilcoxon rank sum test was used to compare sample distributions. Statistical work was performed in R (version 3.5.1, R Foundation),30 with plots being generated using the packages ggplot2 and ggsurvminer.31,32 The Survival R package was used for survival curves and Cox regression.33 Regression results were visualized using the forestplot R package.34 The SurvivalROC R package was used to generate receiver operating characteristic (ROC) curves.35 Principal component analysis was performed with the prcomp R function using the output from deconstructSigs.
RESULTS
Distribution of TMB using WES
TMB has been shown previously to vary among cancer types when estimated from targeted-panel sequencing.4 To characterize TMB using WES, we investigated the distribution of TMB within each cancer type in our internal WCM Advanced cohort (n = 696) and the TCGA cohort (n = 3,534; Fig 1; Data Supplement). Only TCGA cohorts with a corresponding publication were included; we excluded cohorts only published through TCGA pan-cancer atlas studies. The WCM Advanced cohort was sequenced using the EXaCT-1 pipeline and contained mostly metastatic samples. The distribution of TMB varied drastically among cancers. For example, the TMB for prostate cancer in TCGA ranged from 0.03 mutations/Mb to 14.13 mutations/Mb, with a mean of 1.23 mutations/Mb (n = 326). However, the TMB for bladder cancer in TCGA ranged from 0.04 mutations/Mb to 99.68 mutations/Mb, with a mean of 6.92 mutations/Mb (n = 375).
FIG 1.

Tumor mutational burden (TMB) varies among cancer types. (A) Distribution of TMB for The Cancer Genome Atlas (TCGA) and (B) Weill Cornell Medicine (WCM) Advanced. Different TMB-high classification thresholds are shown. The Chalmers et al threshold (blue line) is applied pan-cancer, and the WCM threshold (red diamond) is applied per cancer. The bottom of the box represents the 25th percentile, and the top of the box represents the 75th percentile. Each point represents a patient. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; GBM, glioblastoma multiforme; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; LGG, brain lower grade glioma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian serous cystadenocarcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; THCA, thyroid carcinoma; UCEC, uterine corpus endometrial carcinoma.
Because of the higher frequency of metastatic samples in the WCM Advanced cohort, we sought to understand how the distribution of TMB changes between primary and metastatic samples. TMB was compared among 94 primary and metastatic matched samples from 41 different patients in the WCM Advanced cohort (Data Supplement). Overall, metastatic samples had higher TMB than matched primary samples (Wilcoxon one-sided paired P value = .0062; Wilcoxon two-sided paired P value = .012).
Defining the TMB-High Threshold
Recent studies have used a dichotomous TMB measurement to classify patients as TMB high or TMB low.6,36 However, a standard cutoff for TMB has yet to be firmly established. We focused on two TMB thresholds: that of Chalmers et al and that of WCM. The Chalmers et al threshold is a constant cutoff of 20 mutations/Mb for all cancer types,4 whereas our custom WCM threshold is a formula of mean (TMB) + 1.25 × IQR (TMB), adapted from a study by Zehir et al.5 The WCM threshold was applied per cancer to account for the variation in TMB among cancer types.
Data for patients treated with immunotherapy were not available for either cohort; however, TCGA survival outcomes were investigated to understand the differences in survival between TMB high and TMB low using both thresholds. TMB-high patients showed significantly improved survival compared with TMB-low patients for BLCA using both the WCM threshold (log-rank P value = .0014) and the Chalmers et al threshold (log-rank P value = .009; Data Supplement). LUSC showed improved survival for TMB-high patients compared with TMB-low patients when using the WCM cutoff (log-rank P value = .036) but not when using the Chalmers et al cutoff (log-rank P value = .11; Data Supplement). UCEC also showed significantly improved survival for TMB-high patients compared with TMB-low patients when using Chalmers et al (log-rank P value = .023) but did not reach significance when using WCM (log-rank P value = .055; Data Supplement). BRCA, COAD, GBM, LGG, OV, PRAD, READ, and THCA did not have significantly different survival between the TMB-high and TMB-low groups (log-rank P values > .05). To further compare the performances of the two thresholds, time-dependent ROC curves were created at the 5-year time point (Fig 2D; Data Supplement). To explore the difference between the patients selected by the thresholds, we split the samples into three groups: TMB high, WCM-TMB high, and TMB low. The TMB-high group contained the intersection of samples classified as TMB high by both thresholds. The WCM-TMB–high group contained samples classified as TMB high by WCM and TMB low by Chalmers et al (when Chalmers et al had more TMB high classifications [UCEC] than WCM, we used Chalmers-TMB high [CHM-TMB high]). The TMB-low group contained samples classified as TMB low by both methods (Fig 2A). The WCM-TMB–high group did not attain significantly improved survival compared with the TMB-low group for BLCA (log-rank P value = .072; Fig 2B). The TMB-high and WCM-TMB–high groups also did not have significantly different survival (log-rank P value = .60). The WCM threshold selected more patients in the BLCA cohort with better overall survival compared with the TMB-low group. KIRC showed the opposite pattern, with TMB-high patients having poor survival compared with TMB-low patients for the WCM threshold (log-rank P value = .0027 (Fig 2C). Because this trend was specific to renal cancer, there is likely a different relationship between TMB and patients with renal cancer without immunotherapy, such as a high TMB tumor being a more aggressive disease in KIRC. The Chalmers et al threshold did not classify any patients with KIRC as TMB high.
FIG 2.
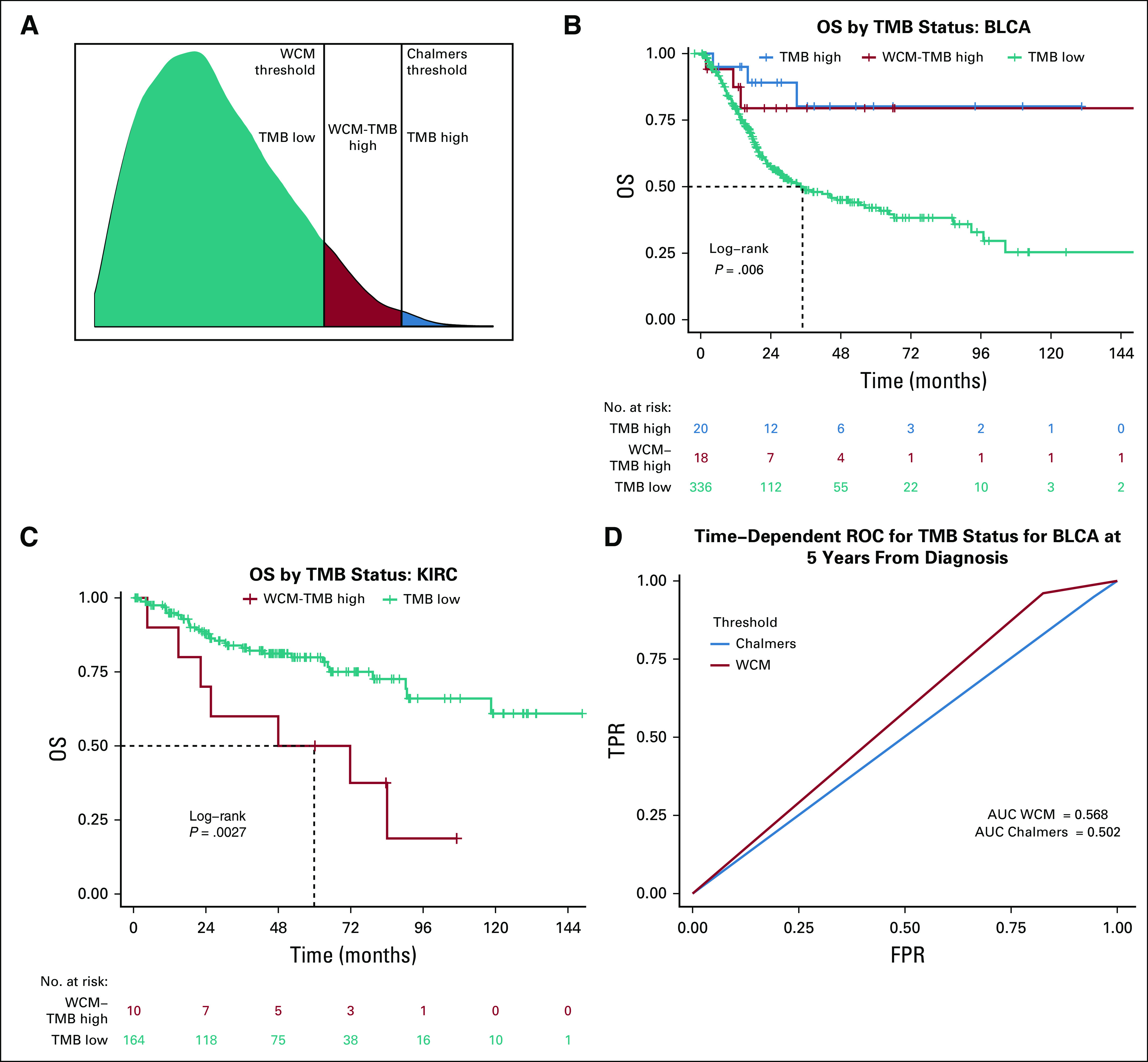
Survival curves for bladder urothelial carcinoma (BLCA) and kidney renal clear cell carcinoma (KIRC) stratified by tumor mutational burden (TMB) status. (A) Visual breakdown of the three different TMB groups. Kaplan-Meier survival curves comparing the TMB classifications between the Weill Cornell Medicine (WCM) threshold and the Chalmers et al threshold for (B) BLCA and (C) KIRC. For BLCA, the difference between TMB high (blue) and TMB low (teal) is significant (log-rank P value = .007). However, the difference between the TMB-high and the WCM-TMB–high (red) curves is not significant (log-rank P value = .60), and the difference between WCM-TMB–high and the TMB-low curves is also not significant (log-rank P value = .072). For KIRC, the Chalmers et al threshold was too high to classify any samples as TMB high, resulting in only two groups: WCM-TMB high and TMB low. The WCM-TMB–high group has significantly worse prognosis than does the TMB-low group (log-rank P value = .0027). (D) Time-dependent receiver operating characteristic (ROC) curve comparing the true positive (TPR) and false positive (FPR) rates for BLCA using both the Chalmers et al threshold and the WCM threshold shows the area under the curve (AUC) for both cutoffs at 5 years from diagnosis. The AUC for WCM is 0.568, whereas the AUC for Chalmers is 0.502. OS, overall survival.
Although the survival trends were not significantly different between the cutoffs, the number of samples classified as TMB high differed between thresholds. Out of 14 TCGA cancer types investigated, eight cancers had a statistically larger TMB-high group for WCM compared with Chalmers et al (BLCA [fold change = 1.9], BRCA [fold change = 6.7], GBM [fold change = 2.7], KIRC [no high by Chalmers], LGG [fold change = 13.0], OV [fold change = 5.67], PRAD [no high by Chalmers], and THCA [no high by Chalmers]); five cancers had no statistically significant change in the number of TMB-high classifications (COAD, KICH, LUAD, LUSC, and READ); and only UCEC had a large number of TMB high classified by Chalmers et al compared with WCM (χ2 test with Yates’s continuity correction; P value < .05; Fig 3A). The group of samples that were classified as high by Chalmers et al but not by WCM was referred to as CHM-TMB high for UCEC. In addition, TMB status was a significant predictor of survival for BLCA, KIRC, and KICH (Fig 3B). Patients with BLCA with high TMB had improved survival compared with those with low TMB (P value = .0028; hazard ratio [HR], 0.29 [95% CI, 0.13 to 0.65]), whereas patients with KIRC (P value = .0047; HR, 3.26 [95% CI, 1.44 to 7.37]) and KICH (P value = .066; HR, 7.16 [95% CI, 0.88 to 58.33]) with high TMB had worse survival compared with those with low TMB.
FIG 3.
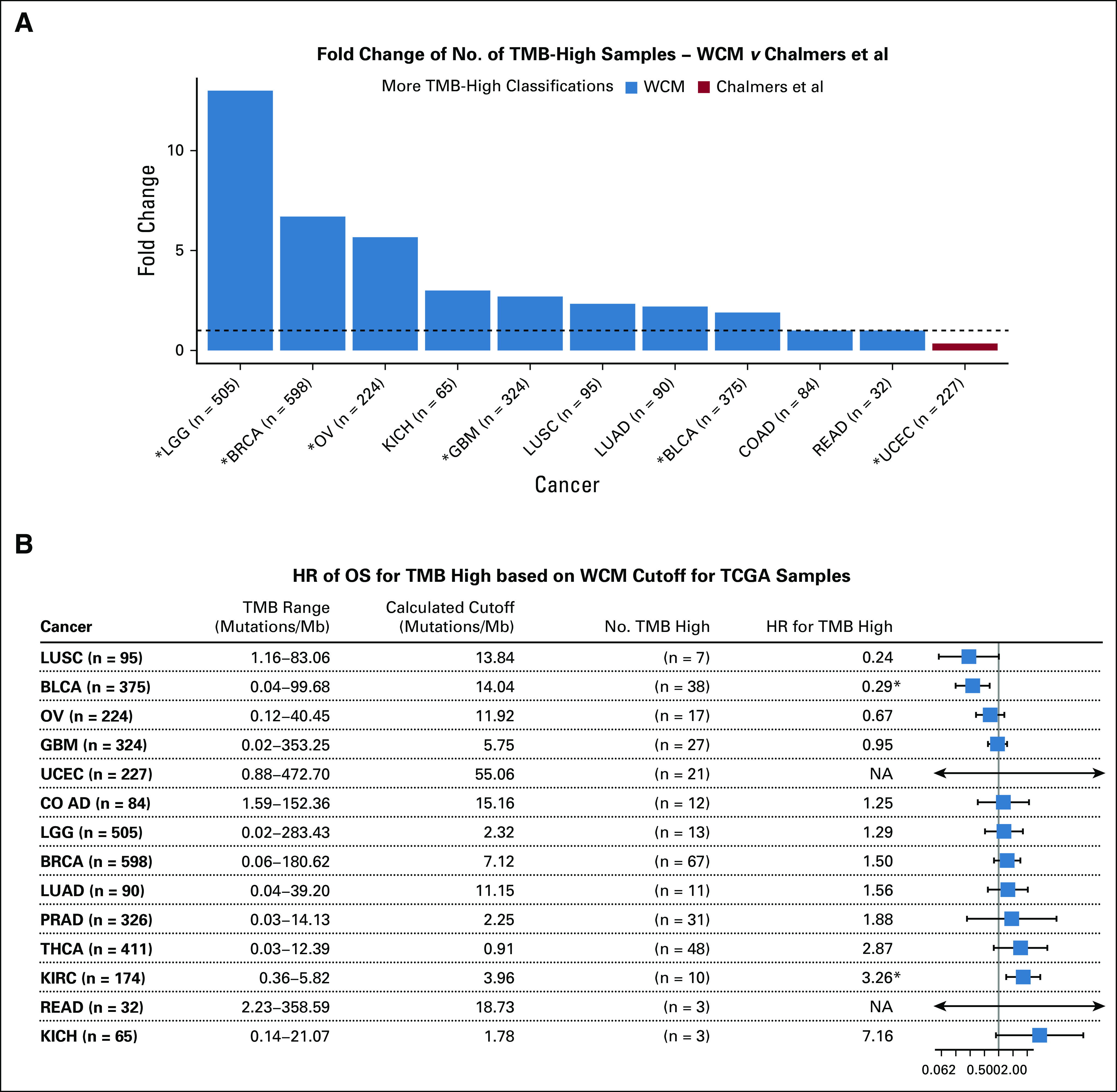
Overview of tumor mutational burden (TMB) classifications for The Cancer Genome Atlas (TCGA). (A) Hazard ratios (HRs) determined by Cox regression for TMB predicting survival for each TCGA cancer type are shown. Significance is determined by a 95% CI and is shown by the boxplot. (*) Significantly increased cancer on the basis of a χ2 test with Yates’ continuity correction (P value < .05). (B) Fold change in TMB-high classifications is shown. The dotted line is at a fold change of 1 (no change). Prostate adenocarcinoma (PRAD) and kidney renal clear cell carcinoma (KIRC) are excluded from the plot because the Chalmers et al threshold did not classify any samples as TMB high. Blue bars represent cancer types with a higher or equal number of TMB-high classifications for the Weill Cornell Medicine (WCM) threshold than the Chalmers et al threshold. Red bar represents a cancer type with a higher number of TMB-high classifications according to Chalmers et al compared with WCM. (†) Statistically significant HRs. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; GBM, glioblastoma multiforme; KICH, kidney chromophobe; LGG, brain lower grade glioma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OS, overall survival; OV, ovarian serous cystadenocarcinoma; READ, rectum adenocarcinoma; THCA, thyroid carcinoma; UCEC, uterine corpus endometrial carcinoma.
Although the WCM TMB cutoff classified more patients as TMB high than did the Chalmers et al cutoff, biologic differences needed to be explored. Predicted neoantigens from The Cancer Immunome Atlas compared the TMB-high, the WCM-TMB–high, and the TMB-low groups for TCGA samples (Fig 4A).23 The TMB-low group had significantly fewer neoantigens than both the TMB-high (Wilcoxon P value < 2.3e-16) and WCM-TMB–high (Wilcoxon P value = 2.1e-14) group. However, the TMB-high and WCM-TMB–high group neoantigen counts were also significantly different (Wilcoxon P value = 7.6e-11; Fig 4A). Because of the observed variation in TMB among cancer types, neoantigens were also compared between the TMB groups for each cancer (Figs 4B and 4C; Data Supplement). TMB high and WCM-TMB–high LUAD cancers did not have a significantly different number of neoantigens (Wilcoxon P value > .05), whereas the TMB-low group had significantly fewer neoantigens than both the TMB-high and the WCM-TMB–high groups (Wilcoxon P value < .05). BLCA showed the number of predicted neoantigens in the TMB-low group to be significantly lower than in the TMB-high (Wilcoxon P value = 9.4e-12) and WCM-TMB–high (Wilcoxon P value = 1.1e-9) groups. However, the TMB-high group also had significantly more neoantigens than the WCM-TMB–high group (Wilcoxon P value > .01). Overall, the TMB-high and WCM-TMB–high groups showed increased neoantigen counts compared with the TMB-low group.
FIG 4.
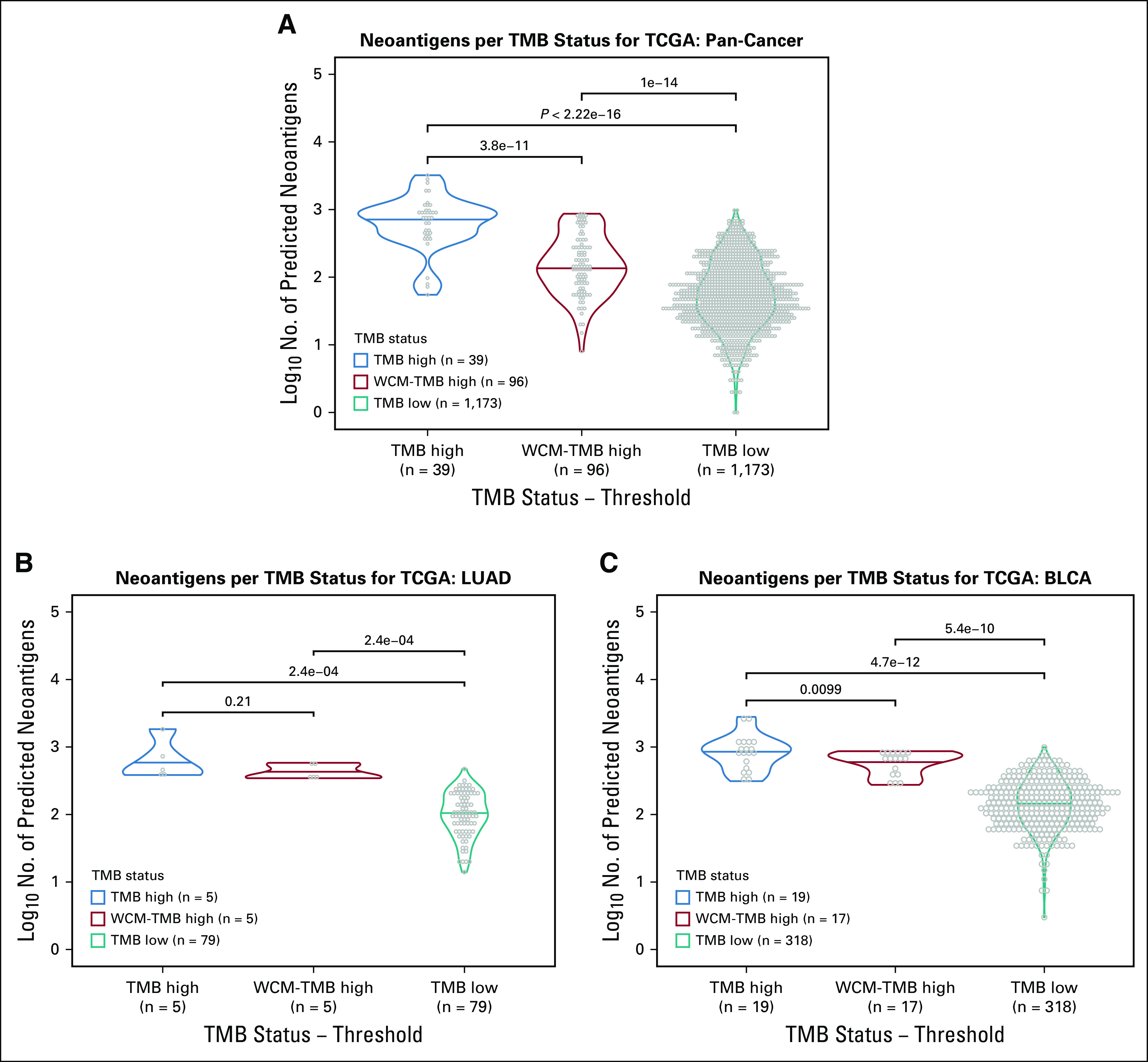
No. of predicted neoantigens differs between tumor mutational burden (TMB) status. Distribution of predicted neoantigens for TMB high (blue), TMB high Weill Cornell Medicine (WCM; red), and TMB low (teal) for The Cancer Genome Atlas (TCGA) shown for (A) all cancers pooled together, (B) lung adenocarcinoma (LUAD), and (C) bladder urothelial carcinoma (BLCA). Values shown above bars are Wilcoxon P values.
Finally, mutational signatures were compared among TMB groups. The weight of contribution for Catalogue of Somatic Mutations in Cancer mutational was calculated for the patients with TCGA in the three different TMB groups and was used to generate principal component analysis plots (Fig 5; Data Supplement).29 BLCA showed an apolipoprotein B mRNA editing enzyme, a catalytic polypeptide-like signature (signature 2), and mismatch repair deficiency (signature 26) that were significantly different for the TMB-low group compared with both the TMB-high (signature 2 Wilcoxon P value = 1.4e-6; signature 26 Wilcoxon P value = .0024) and the WCM-TMB–high (signature 2 Wilcoxon P value = 4.1e-4; signature 26 Wilcoxon P value = .0013) group, whereas there was no significant difference between the TMB-high and the WCM-TMB–high group (P value > .05; Fig 5C). However, UCEC showed the defective polymerase-ε signature (signature 10) contribution to be different in the TMB-high group compared with both the CHM-TMB–high (Wilcoxon P value = 4.4e-9) and the TMB-low (Wilcoxon P value < 2.2e-16) group but not different between the CHM-TMB–high and TMB-low group (Wilcoxon P value = .2).
FIG 5.
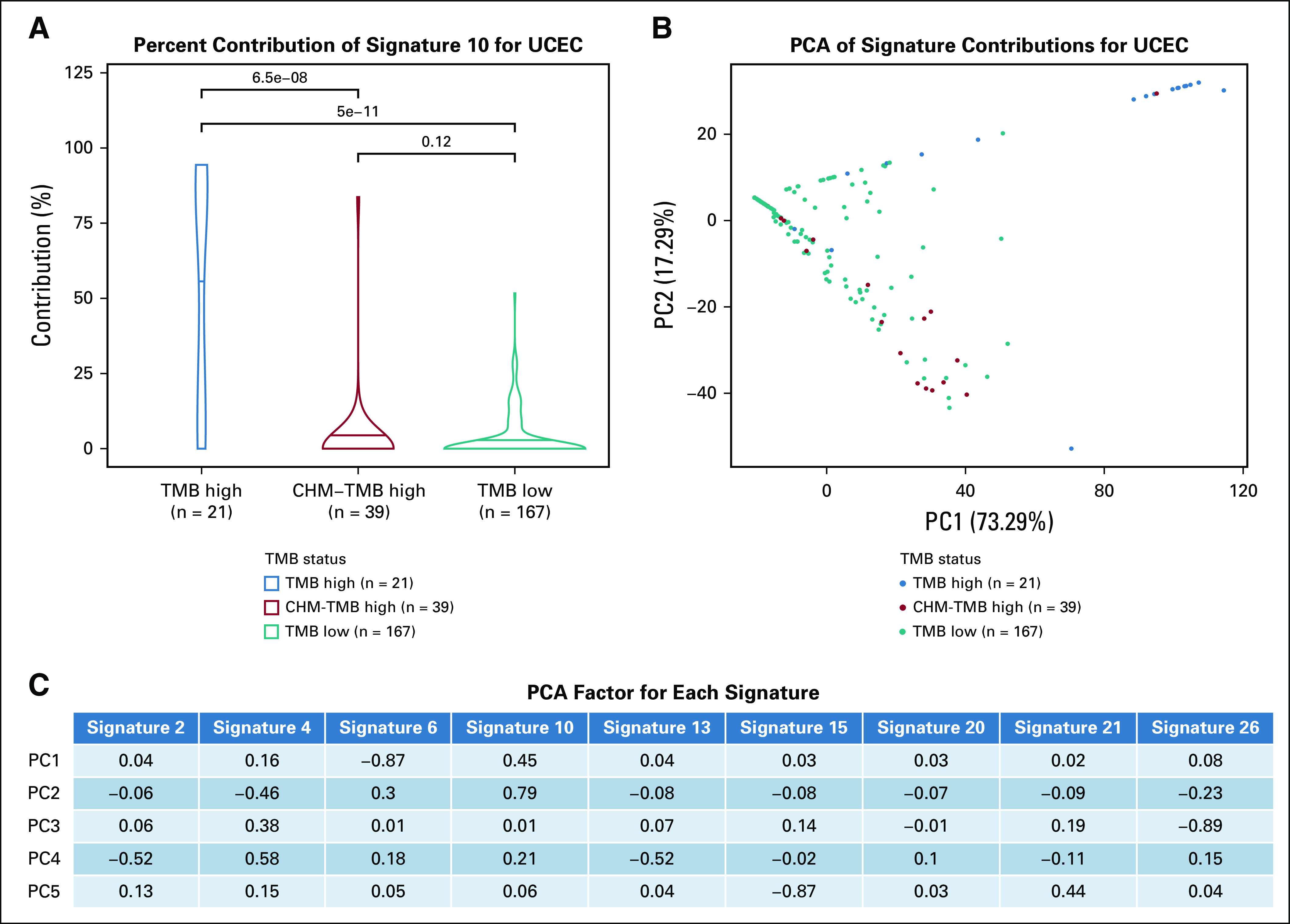
Signature contributions for uterine corpus endometrial carcinoma (UCEC) show that samples cluster by signature 10. (A) Violin plots showing the distribution of the contribution of signature 10 (defective polymerase-ε) in tumor mutational burden (TMB) high, Chalmers (CHM)-TMB high, and TMB low. The mean contribution is significantly different between TMB high and TMB low (Wilcoxon P value = 5e-11), and between TMB high and CHM-TMB high (Wilcoxon P value = 6.5e-8). (B) PCA showing the signature contributions for uterine corpus endemetrial carcinoma (UCEC) with TMB-high samples in blue, WCM-TMB–high samples in red, and TMB-low samples in teal. Only signatures 2, 4, 6, 10, 13, 15, 20, 21, and 26 were considered, to focus on signatures with the most relevant interpretations. (C) The calculated PCA factor loadings are also shown for each signature in the first five principal components. Signature 6 (defective DNA mismatch repair) has the largest factors for principal component 1 (PC1), although it is negative. PCA, principal component analysis; PC2, principal component 2.
Clinical Context of TMB
Cox regression for patients with TCGA was performed to explore additional associations of survival predictors (Data Supplement). For BLCA, disease stage and age were significant predictors of survival, in addition to TMB classified with the WCM threshold (Fig 6). In stage III patients, TMB and age were not significant predictors of survival (TMB P value = .17; age P value = .078). In addition, patients with low TMB and stage III bladder cancer had the lowest survival rate. There was no significant difference in survival between stages I or II and stage III when the patients were TMB high (log-rank P value = .33), suggesting that advanced pathologic stage as a determinant of overall survival may be interpreted in the context of TMB.
FIG 6.
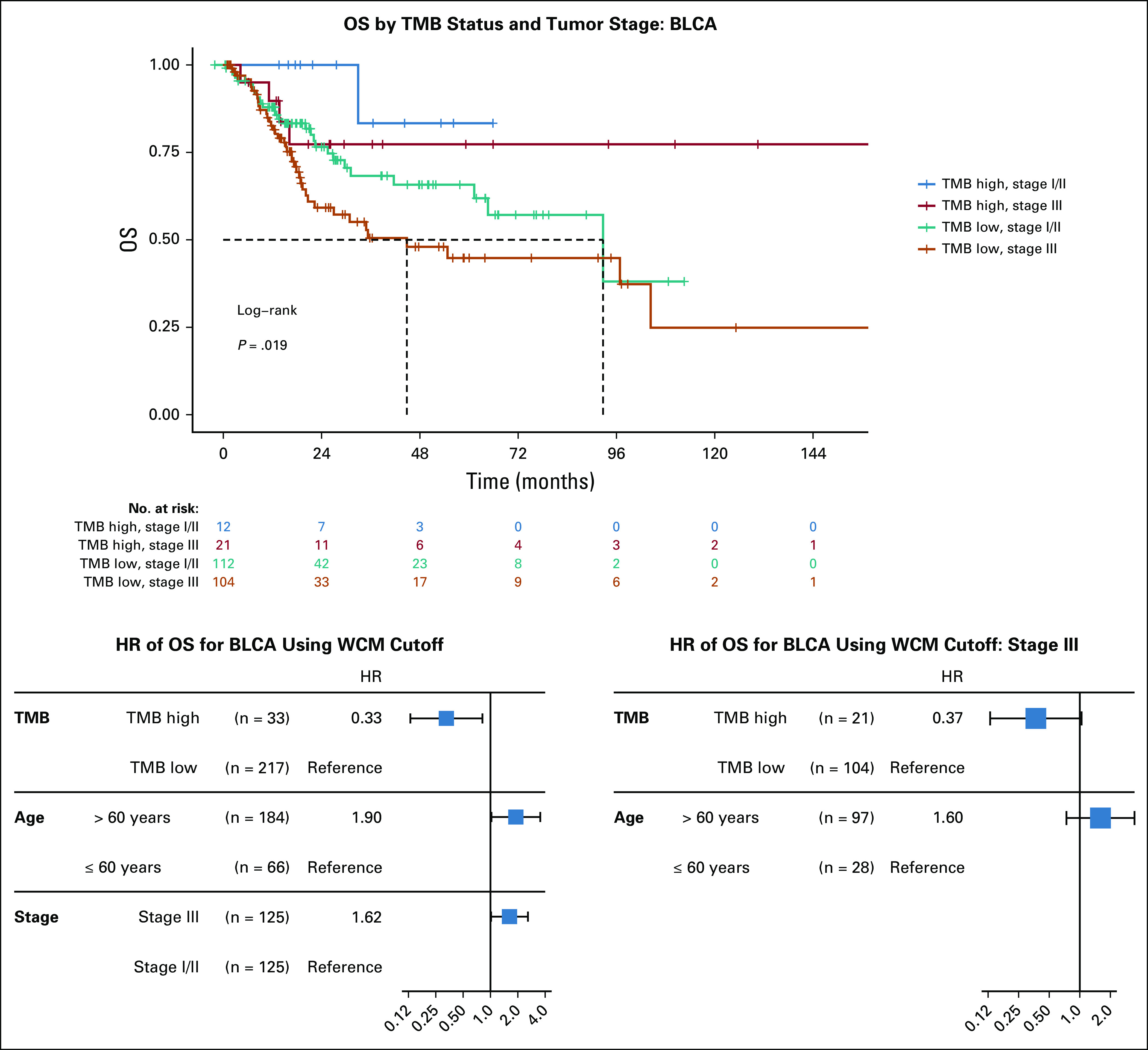
Effect of stage and age with tumor mutational burden (TMB) for bladder urothelial carcinoma (BLCA) in The Cancer Genome Atlas. Kaplan-Meier survival curves for BLCA stratified into both TMB status and disease stage. Cox regression results are also shown for significant predictors, as well as TMB status and age in stage III patients. HR, hazard ratio; OS, overall survival; WCM, Weill Cornell Medicine.
DISCUSSION
TMB varies drastically among cancer types when measured through WES. This variation can make it difficult to classify tumors as having high or low TMB. For example, the Chalmers et al cutoff of 20 mutations/Mb is higher than the maximum TMB seen within TCGA prostate cancer (14.13 mutations/Mb); however, using a lower threshold might be too lenient for TCGA bladder cancer (maximum TMB of 99.68 mutations/Mb). The dramatic difference in TMB among cancer types led us to explore a TMB threshold applied in a cancer-specific manner. When comparing a pan-cancer to a cancer-specific threshold, we found we were able to maintain differences in survival trends between TMB high and TMB low, often while including more patients in the TMB-high group by using the cancer-specific threshold. The WCM cancer-specific threshold classified more patients as TMB high compared with the pan-cancer Chalmers et al threshold in 13 out of the 14 cohorts. Using a TMB threshold that has a larger number of TMB-high classifications while not having significantly different survival trends than a more stringent threshold can potentially open treatment options to patients if they can be shown to respond well. ROC curves showed the two thresholds to have similar performance; however, the performance seen in recent publications suggests that TMB alone is not a critical predictor of survival for nonimmunotherapy treatment response.6,37,38
Differences in TMB distribution between the TCGA and WCM Advanced cohorts could be a result of biologic or technical differences. TCGA contains primary tumor samples, and WCM Advanced contains mostly metastatic tumor samples. When comparing different cohorts of patients, many technical differences can influence TMB, making a static threshold potentially less accurate. The different assays and bioinformatics pipelines for the patient cohorts could have caused batch effects in this study. WCM Advanced was sequenced using Agilent Haloplex, and TCGA used Agilent Sure Select. After sequencing, the EXaCT-1 pipeline and TCGA pipeline use different mutation callers and apply different quality filters. In addition, when comparing TMB among studies, it is important to understand the variants that were considered. We excluded synonymous mutations when calculating TMB, focusing only on variants that were expected to cause an amino acid change. Therefore, our TMB distribution could seem different when compared with studies that include synonymous mutations.
Although this study focused on WES, targeted panels are often used in a clinical setting and have been used for calculating TMB.6,38,39 Targeted panels cover different genes and sizes of the genome and at a higher sequencing depth than WES, influencing the estimated distribution of TMB. The panel must cover at least 2 Mb of the genome to have high sensitivity and specificity when measuring TMB.8 A static threshold would not be able to adjust to the differing levels of TMB resulting from different panels. However, a dynamic threshold would be able to adjust for technical differences.
The cancer-specific threshold adjusted for the variation in TMB; however, the pan-cancer threshold classified more patients as TMB high for UCEC. Mutational signatures for these samples showed a cluster of patients with POLE deficiency, which led to a large number of mutations and a TMB distribution with a large IQR,22 causing the cancer-specific threshold to be strict in this case.
The survival analyses in this study were limited by a lack of patients undergoing immunotherapy treatment. However, the TCGA cohort showed significantly improved survival for TMB-high samples with BLCA compared with TMB-low samples. This difference in prognosis should be further investigated and considered when interpreting the clinical significance of TMB. In addition, the number of predicted neoantigens was compared per cancer, showing that a cancer-specific threshold identifies patients with a higher number of neoantigens compared with TMB-low patients. Moreover, TMB might depict only one factor in the success of immunotherapy. HLA class I molecules play a critical role in T-cell–mediated immunotherapy by presenting tumor antigens to CD8+ T cells. This process is affected by the germline genotype of HLA class I. A recent study has shown that patients with melanoma and non–small-cell lung cancer with heterozygous HLA class I exhibit improved survival compared with patients who are homozygous.40 Because of the limitation of TCGA data, we could not examine the role of HLA class I in the current study. Overall, a prospective study with immunotherapy patients is needed to further elucidate the role of TMB in this context.
ACKNOWLEDGMENT
We thank Juan Sebastian Andrade Martinez for his previous work with the WCM Advanced cohort and Bhavneet Bhinder for her advice on predicted neoantigens.
Footnotes
Presented in part at the Annual Meeting and Expo hosted by the Association of Molecular Pathology, San Antonio, TX, November 1-3, 2018.
Supported by the Caryl and Israel Englander Institute for Precision Medicine, Weill Cornell Medical College (A.S.). Also supported by the National Science Foundation CAREER (O.E.); the Leukemia and Lymphoma Society Specialized Center of Research (O.E.); the Hirschl Trust (O.E.); Starr Cancer Consortium I6-A618 (O.E.) and I8-A8-132 (M.A.S.); National Institutes of Health R01 CA228512 (M.A.S.); and Stand Up to Cancer SU2C-AACR-DT22-17 (M.A.S.) as administered by the American Association for Cancer Research, the scientific partner of SU2C.
A.S., M.A.S., and W.S. are co-senior authors.
AUTHOR CONTRIBUTIONS
Conception and design: Evan M. Fernandez, Kenneth Eng, Bishoy M. Faltas, Mark A. Rubin, Olivier Elemento, Andrea Sboner, Manish A. Shah, Wei Song
Financial support: Mark A. Rubin, Olivier Elemento
Administrative support: Mark A. Rubin, Olivier Elemento, Manish A. Shah
Provision of study materials or patients: Kenneth Eng, Himisha Beltran, Bishoy M. Faltas, Juan Miguel Mosquera, David M. Nanus, Brian D. Robinson, Manish A. Shah
Collection and assembly of data: Evan M. Fernandez, Kenneth Eng, Shaham Beg, Himisha Beltran, Bishoy M. Faltas, Juan Miguel Mosquera, David M. Nanus, David J. Pisapia, Brian D. Robinson, Olivier Elemento, Andrea Sboner, Manish A. Shah
Data analysis and interpretation: Evan M. Fernandez, Himisha Beltran, Bishoy M. Faltas, Juan Miguel Mosquera, David J. Pisapia, Rema A. Rao, Olivier Elemento, Andrea Sboner, Manish A. Shah
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST AND DATA AVAILABILITY STATEMENT
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Evan M. Fernandez
Employment: Celgene
Travel, Accommodations, Expenses: Foundation Medicine
Himisha Beltran
Consulting or Advisory Role: Janssen Oncology, Genzyme, GlaxoSmithKline, AbbVie, Astellas Pharma, AstraZeneca
Research Funding: Janssen (Inst), AbbVie/Stemcentrx (Inst), Eli Lilly (Inst)
Travel, Accommodations, Expenses: Janssen Oncology
Bishoy M. Faltas
Honoraria: Digital Science Press publications
Research Funding: Eli Lilly
Juan Miguel Mosquera
Research Funding: Personal Genome Diagnostics
Travel, Accommodations, Expenses: Personal Genome Diagnostics
David M. Nanus
Consulting or Advisory Role: Roche/Genentech
Research Funding: Novartis (Inst), Boehringer Ingelheim (Inst), Zenith Epigenetics (Inst)
Brian D. Robinson
Patents, Royalties, Other Intellectual Property: Methods for diagnosing and treating prostate cancer
Mark A. Rubin
Honoraria: F. Hoffmann La Roche AG, Novartis, Astellas Pharma
Research Funding: Eli Lilly, Janssen, Millenium Pharmaceuticals, Sanofi
Patents, Royalties, Other Intellectual Property: US Patent (7,767,393 and 7,229,774), Expression Profile of Prostate Cancer, 2007; US Patent (7,332,290), Detection of AMACR Cancer Markers in Urine, 2008; US Patent (7,718,369), Recurrent Gene Fusions in Prostate Cancer, 2010; US Patent (7,803,552 and 7,666,595), Biomarkers for Predicting Prostate Cancer Progression, 2010; US Patent (7,981,609 B2), Methods for Identifying and Using SNP Panels, 2011; US Patent (8,106,037 B2), Identification and Treatment of Estrogen Responsive PCa, 2012; US Patent (9,090,899 B2), Methods of Diagnosing and Treating Prostate Cancer Characterized by NDRG1-ERG Fusion, 2015; US Patent (9,458,213 B2), Compositions and Methods for Diagnosing Prostate Cancer Based on Detection of SLC45A3-ELK4 Fusion Transcript, 2016; US Patent (9,568,483 B2), Molecular Subtyping, Prognosis and Treatment of Prostate Cancer, 2017; US Patent (9,678,077 B2), ERG/TFF3/HMWCK Triple Immunostain for Detection of Prostate Cancer, 2017; US Patent (61,408,341), Exploration of Novel Gene Fusion in Prostate Cancer by RNA-Seq
Travel, Accommodations, Expenses: F. Hoffmann La Roche AG, Novartis, Astellas Pharma
Olivier Elemento
Stock and Other Ownership Interests: Volastra, Owkin, One Three Biotech
Manish A. Shah
Consulting or Advisory Role: Astellas Pharma, Eli Lilly Japan
Research Funding: Gilead Sciences (Inst), Merck (Inst), Boston Biomedical (Inst), Oncolys BioPharma (Inst), Bristol-Myers Squibb (Inst)
Wei Song
Employment: Genentech (I)
Employment: Cytokinetics (I)
Honoraria: Foundation Medicine, Loxo
Consulting or Advisory Role: Foundation Medicine, Loxo
No other potential conflicts of interest were reported.
REFERENCES
- 1.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29:4828–4836. doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tray N, Weber JS, Adams S. Predictive biomarkers for checkpoint immunotherapy: Current status and challenges for clinical application. Cancer Immunol Res. 2018;6:1122–1128. doi: 10.1158/2326-6066.CIR-18-0214. [DOI] [PubMed] [Google Scholar]
- 4.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hellmann MD, Ciuleanu T-E, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–2104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchhalter I, Rempel E, Endris V, et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int J Cancer. doi: 10.1002/ijc.31878. http://doi.wiley.com/10.1002/ijc.31878 [DOI] [PubMed]
- 9.Turajlic S, Litchfield K, Xu H, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017;18:1009–1021. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- 10.Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plimack ER, Dunbrack RL, Brennan TA, et al. Defects in DNA repair genes predict response to neoadjuvant cisplatin-based chemotherapy in muscle-invasive bladder cancer. Eur Urol. 2015;68:959–967. doi: 10.1016/j.eururo.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ceccarelli M, Barthel FP, Malta TM, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis CF, Ricketts CJ, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. doi: 10.1038/nature13385. https://www.nature.com/articles/nature13385 [DOI] [PMC free article] [PubMed]
- 18.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers Nature 489519–525.2012[Erratum: Nature 491:288, 2012] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma Nature 474609–615.2011[Erratum: Nature 490:298, 2012] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abeshouse A, Ahn J, Akbani R, et al. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agrawal N, Akbani R, Aksoy BA, et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–690. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Cancer Genome Atlas Research Network. Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma Nature 49767–73.2013[Erratum: Nature 500:242, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charoentong P, Finotello F, Angelova M, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Reports. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Beltran H, Eng K, Mosquera JM, et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 2015;1:466–474. doi: 10.1001/jamaoncol.2015.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prandi D, Baca SC, Romanel A, et al. Unraveling the clonal hierarchy of somatic genomic aberrations Genome Biol 15439.2014[Erratum: Genome Biol 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenthal R, McGranahan N, Herrero J, et al. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Jones PH, Wedge DC, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. The R project for statistical computing. https://www.R-project.org/
- 31. Wickham H: ggplot2: Elegant Graphics for Data Analysis. Berlin, Germany, Springer-Verlag, 2009. [Google Scholar]
- 32. Kassambara A, Kosinski M: survminer: Drawing Survival Curves using “ggplot2.” http://www.sthda.com/english/rpkgs/survminer/
- 33. Therneau T: A Package for Survival Analysis in S. https://CRAN.R-project.org/package=survival.
- 34. Gordon M, Lumley T: Advanced Forest Plot Using “grid” Graphics. https://cran.r-project.org/web/packages/forestplot.
- 35.Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56:337–344. doi: 10.1111/j.0006-341x.2000.00337.x. [DOI] [PubMed] [Google Scholar]
- 36.Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res. 2016;4:959–967. doi: 10.1158/2326-6066.CIR-16-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samstein RM, Lee C-H, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rizvi H, Sanchez-Vega F, La K, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36:633–641. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chowell D, Morris LGT, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359:582–587. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]