

Abstract
Rationale:
Bartter syndrome is an autosomal-recessive inherited disease in which patients present with hypokalemia and metabolic alkalosis. We present 1 case with Bartter syndrome, due to a novel compound heterozygous mutation in the KCNJ1 gene encoding the ATP-sensitive inward rectifier potassium channel in the thick ascending limb of the loop of Henle.
Patient concerns:
A patient was admitted to our hospital because of weakness, polyuria, and polydipsia. At presentation to our hospital, the female Chinese patient was 34 years old and her physical examination was normal. Laboratory studies revealed hypokalemia, metabolic alkalosis, hypercalciuria, hyperparathyroidemia, and hyper-reninemia. In addition, urinary potassium was obviously higher. Computer tomography scan confirmed the patient had the bilateral medullary nephrocalcinosis.
Diagnosis:
Blood samples were received from the patient and her parents, and deoxyribonucleic acid was extracted. The genetic analysis of SLC12A1, SLC12A3, KCNJ1, CLCNKB, BSND, and CASR was performed. The compound heterozygous KCNJ1 gene mutation was validated using conventional Sanger sequencing methods.
Interventions:
The patient was treated with potassium supplementation. Her blood and urine chemistries improved over the next week. Serum potassium normalized with improvement in polyuria and polydipsia over the next month.
Outcomes:
Our patient was compound heterozygous for Thr234Ile and Thr71Met in the KCNJ1 gene. The c.701C>T variant predicted a change from a threonine codon to an isoleucine codon (p.Thr234Ile). The c.212C>T variant predicted a change from a threonine codon to a methionine codon (p.Thr71Met). The unaffected mother was heterozygous for the Thr234Ile mutation, whereas unaffected father was heterozygous for the Thr71Met mutation.
Lessons:
The phenotypes of the patient were similar to other patients with Bartter syndrome. The phenotypes of the patient could eventually be explained by the presence of the novel compound heterozygous p.Thr234Ile/p.Thr71Met variants in the KCNJ1 gene.
Keywords: Bartter syndrome, KCNJ1 gene, late presentation
1. Introduction
Bartter syndrome is an autosomal-recessive disorder, characterized by a set of metabolic abnormalities.[1] Patients with Bartter syndrome present with metabolic alkalosis, hyper-reninemia, hypokalemia, hypercalciuria, polyuria, polydipsia, and hyperaldosteronism,[2] which means that they have problems thriving. Due to impairment of one of the transporters responsible for sodium chloride reabsorption in the thick ascending limb of the loop of Henle, patients with Bartter syndrome develop a mild volume depletion that subsequently activates the renin-angiotensin-aldosterone system. This, in turn, results in secondary hyperaldosteronism, increased distal flow, and sodium delivery, which enhances the excretion of potassium and hydrogen at the secretory sites, leading to metabolic alkalosis and hypokalemia. Without the ability to reabsorb sodium chloride in the thick ascending limb segment of the nephron, the medullary concentration gradient needed to maximally concentrate urine in the presence of arginine vasopressin cannot be obtained and polyuria develops. Patients with Bartter syndrome have lower blood pressure than the general population,[3] due to the pathogenesis of the syndrome, which involves the macula densa and thus the uncoupling of the tubuloglomerular feedback and renin and aldosterone secretion from the volume status.[4]
Bartter syndrome can be divided into the classic form and a more severe antenatal severe type. The antenatal hypercalciuric variant of Bartter syndrome, also known as hyperprostaglandin E syndrome, is a life- threatening disorder, in which both renal tubular hypokalemic alkalosis and profound systemic symptoms manifest. Both classic Bartter syndrome and antenatal Bartter syndrome can be divided into 5 subtypes, according to their underlying genetic defect: types I and II are clinically the most severe subtypes and normally present as antenatal Bartter syndrome and type III is the classic form of Bartter syndrome, presenting later in life with less severe symptoms. Five different genetic defects have been shown to be responsible for either antenatal or classic Bartter syndrome.[5] Type I, with variants in the SLC12A1 gene, encodes bumetanide-sensitive sodium-(potassium)-chloride cotransporter 2. Type II, with variants in the KCNJ1 gene, encodes the ATP-sensitive inward rectifier potassium channel (ROMK). Type III, with variants in the CLCNKB gene, encodes the chloride channel protein ClC-Kb. Type IV is antenatal Bartter syndrome with sensorineural deafness, caused by either variants in the BSND gene encoding Barttin (Bartter syndrome type IVa) or by coexistence of variants in the CLCNKA and CLCNKB genes encoding, respectively, the chloride channel protein ClC-Ka and CLC- Kb (Bartter syndrome type IVb).[6–9]
Here, we report the identification of a novel molecular basis of late-onset Bartter syndrome type II presenting with symptoms.
2. Patient information and clinical findings
A girl with a birth weight of 3050 g and a length of 47.5 cm was born to healthy parents at 38 weeks of gestation. During pregnancy, ultrasound examinations of the fetus were repeatedly performed, and, apart from polyhydramnios, nothing abnormal was noticed. The girl was delivered by a spontaneous vaginal delivery due to polyhydramnios without immediate perinatal/postnatal problems. She was nursed for 2 years, often requiring feeding many times during the night, and she continued to require large amounts of fluid. She had persistent polyuria and polydipsia; however, her weight and height were normal. So the patient was not admitted to the hospital. Two years ago, the patient found she suffered from bilateral medullary nephrocalcinosis by medical examinations. The patient was admitted to our hospital because of the weakness. At presentation to our hospital, the female Chinese patient was 34 years old and her height was 173 cm, weight 68.5 kg, pulse 96/min, respiratory rate 20/min, and blood pressure 110/80 mm Hg. Her physical examination was normal. Laboratory studies revealed hypokalemia, metabolic alkalosis, hypercalciuria, hyperparathyroidemia, and hyper-reninemia. In addition, urinary potassium was obviously higher when the patient suffered from hypokalemia (Table 1). Computer tomography scan confirmed the patient had the bilateral medullary nephrocalcinosis (Fig. 1). The fasting glucose was 5.19 mmol/L and the 2-hour blood glucose after the oral 75 g glucose was 8.69 mmol/L. So the diagnosis was the impaired glucose tolerance. The blood routine examination was normal. The patient did not have any specific drugs and did not have any autoimmune disease. The functions of thyroid, adrenal, and sexual glands were normal. Her father and mother are healthy. In addition, there are not any similar symptoms in all the family. All in all, we need to know the real reasons of the symptoms of the patient. The clinical and paraclinical findings made the diagnosis of desalting nephropathy. In addition, there were no secondary reasons of the disease.
Table 1.
The patient was admitted to the hospital on 30th May and discharged on 9th June; after hospitalization the patient came back to have the examination on 25th June.
Figure 1.
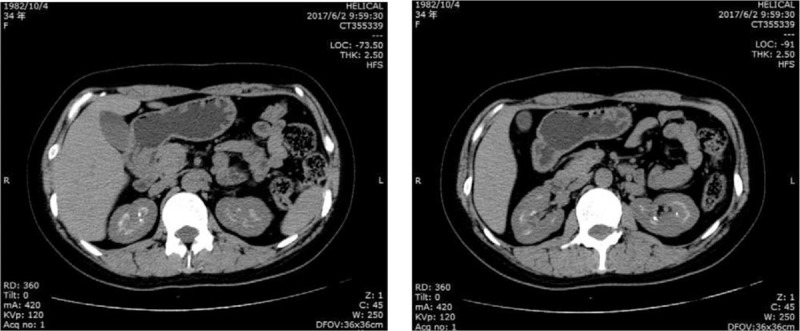
Imaging manifestations of kidney by computer tomography (CT) scan. It showed the patient had the bilateral medullary nephrocalcinosis.
2.1. Diagnostic assessment
To make a precise diagnosis, we then performed targeted sequencing of related Bartter syndrome and Gitelman syndrome genes. The genetic analysis of SLC12A1, SLC12A3, KCNJ1, CLCNKB, BSND, and CASR was performed. Written informed consent of the patent and her parents was obtained before the collection of 5 mL of their peripheral blood for the following experiment. A capture panel (NimbleGen, Madison, WI) of Bartter syndrome genes and Gitelman syndrome was previously designed. Genomic deoxyribonucleic acid (DNA) of all the family members was extracted according to the manufacturer's standard procedure using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). Then the genomic DNA of the family was fragmented by Covaris LE220 (Massachusetts) to generate paired-end library (200–250 bp). The library was enriched by array hybridization according to previously published procedure,[10] followed by elution and postcapture amplification. The products were then subjected to Agilent 2100 Bioanalyzer and ABI StepOne to estimate the magnitude of enrichment. All amplified libraries were subsequently sent to high-throughput sequencing kit for circularization and sequencing on the BGISEQ-500 platform.
To detect the potential variants in the family, we performed bioinformatics processing and data analysis after receiving the primary sequencing data. We used previously published filtering criteria to generate “clean reads” for further analysis.[10] The “clean reads” (with a length of 90 bp) derived from targeted sequencing and filtering were then aligned to the human genome reference using the Burrows-Wheeler Aligner Multi-Vision software package.[11] After alignment, the output files were used to perform sequencing coverage and depth analysis of the target region, single-nucleotide variants (SNVs) and indel calling. We used SOAPsnp software[12] and SAMtools[13] to detect SNVs and indels. All SNVs and indels were filtered and estimated via multiple databases, including NCBI dbSNP, HapMap, 1000 human genome dataset, and database of 100 Chinese healthy adults.
To predict the effect of missense variants, we used dbNSFP, which contains 7 well-established in silico prediction programs (Scale Invariant Feature Transform, PolyPhen-2, LRT, MutationTaster, and PhyloP). Pathogenic variants are assessed under the protocol issued by The American College of Medical Genetics.[14] The Human Gene Mutation Database was used to screen mutations reported in published studies.
All mutations and potential pathogenic variants were validated using conventional Sanger sequencing methods. Segregation analysis was performed if DNA from family members was available.
2.2. Therapeutic intervention
The patient was started on oral potassium chloride 9.5 g/day. Her blood and urine chemistries improved over the next week. Serum potassium normalized with improvement in polyuria and polydipsia over the next month. The weakness and fatigue of the patient has obviously improved.
2.2.1. Follow-up and outcomes
Genes SLC12A1, SLC12A3, KCNJ1, CLCNKB, BSND, and CASR were screened. We detected compound heterozygous missense changes in KCNJ1 gene, encoding the renal outer medullary potassium channel ROMK. The p.Thr234Ile/p.Thr71Met mutation segregated from mother and father, respectively. The c.701C>T variant predicted a change from a threonine codon to an isoleucine codon (p.Thr234Ile). The c.212C>T variant predicted a change from a threonine codon to a methionine codon (p.Thr71Met) (Fig. 2). The compound heterozygous KCNJ1 gene mutation was predicted to be deleterious by >3 bioinformatics software and may give additional potential genetic diagnostic yields. No variants were found in the coding regions of genes SLC12A1, SLC12A3, CLCNKB, BSND, and CASR. The clinical and paraclinical findings with the compound heterozygous KCNJ1 gene mutation made the diagnosis of Bartter syndrome type II.
Figure 2.
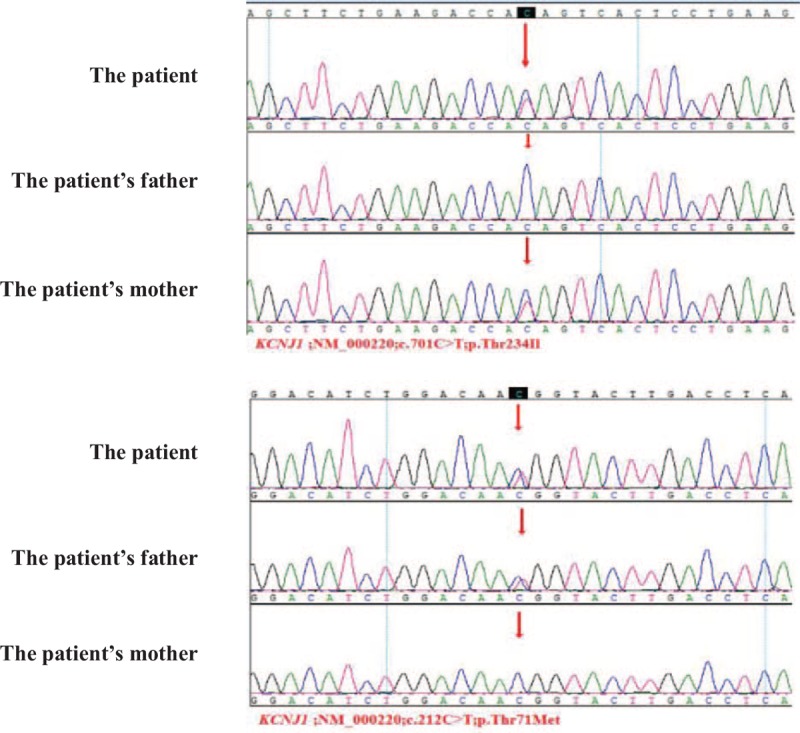
Direct deoxyribonucleic acid (DNA) sequence analysis of the KCNJ1 gene in the patient and her healthy parents. It showed the p.Thr234Ile/p.Thr71Met mutation segregated from mother and father, respectively. The c.701C>T variant predicted a change from a threonine codon to an isoleucine codon (p.Thr234Ile). The c.212C>T variant predicted a change from a threonine codon to a methionine codon (p.Thr71Met).
3. Discussion
Our DNA sequence data showed that the case presented with a novel compound heterozygous gene mutation in the entire coding regions of the KCNJ1 gene. One of the variants (p.Thr71Met) has been reported,[15] and the other variant (p.Thr234Ile) is a new mutation. One heterozygous variant was from her father and the other was from her mother. Only 1 variant cannot induce the disease, so the phenotypes of her father and her mother are normal. Both of the variants could induce the disruption of the function of protein. It will likely deleteriously affect the synthesis of the mRNA and thereby disrupt the function of ROMK. The present study describes the patient with Bartter syndrome with severe hypokalemia, metabolic alkalosis hypercalciuria, hyperparathyroidemia, hyper-reninemia, and nephrocalcinosis due to a novel compound heterozygous KCNJ1 gene mutation. The patient had suffered from polyuria and polydipsia from birth. In addition, the case presented with a low urinary osmolarity of <100 mOsm/kg, hypokalemic alkalosis and urinary potassium and sodium wasting. The treatment seeks to minimize the effects of secondary increased renin, aldosterone, and prostaglandins, and to correct volume depletion and electrolyte shifts. First-line treatment in Bartter syndrome consists of nonsteroidal anti-inflammatory drugs (NSAIDs). A potassium-sparing diuretic,[16,17] combined with potassium and magnesium supplementation and the NSAID, has been shown to have a positive effect.[18] Angiotensin inhibitors can also be given to reduce the production of angiotensin II and aldosterone.[19]
Bartter syndrome type II, with variants in the KCNJ1 gene, encodes the ROMK. Although Bartter syndrome type I and II are clinically the most severe subtypes and normally present as antenatal Bartter syndrome, variants in the SLC12A1 gene have been described as rather benign[20] and a great deal of variance in phenotype can be expected, just as we saw the case in this study. The phenotypic effects of the novel compound heterozygous KCNJ1 gene mutation seem to be comparable to the phenotypes previously described in patients who were homozygous for variants in the KCNJ1 gene; however, the patient was not such severe as those antenatal Bartter syndrome. Our patient was only treated with potassium supplementation. Her serum potassium improved over the next week and normalized with improvement in polyuria and polydipsia over the next month. In addition, in our study the height and weight of the patient were normal at birth and followed by a normal growth and weight gain. Another study reported normalization in height and weight in all 15 patients at a median follow-up of 11 years.[21] The combination of mutations in compound heterozygous state may not be arbitrary, but may follow tertiary or quaternary structure of protein and may be a crucial determinant of the severity of the phenotype.[22] Thus, the compound heterozygous state of p.Thr234Ile and p.Thr71Met may have partial intrinsic transport accounting for the late onset. Similar late onset has been reported in other compound heterozygous mutations of the ROMK and the Na-K-Cl transporter.[20,23]
In conclusion, this case of Bartter syndrome type II is caused by a novel compound heterozygous KCNJ1 gene mutation. Further studies are needed to verify the effect of the novel gene mutation.
Acknowledgments
The authors would like to extend our thanks to all participants of this study.
Author contributions
Data curation: Hongmei Li.
Formal analysis: Shoulong Hu, Rongfeng Wang, Ming Tan, Hongmei Li.
Investigation: Jingyi Li, Yi Nie, Ming Tan.
Methodology: Jingyi Li, Rongfeng Wang, Shuanli Zhu.
Project administration: Jingyi Li.
Resources: Yi Nie.
Supervision: Shoulong Hu.
Writing – original draft: Jingyi Li.
Jingyi Li orcid: 0000-0001-6756-3551.
Footnotes
Abbreviations: DNA = deoxyribonucleic acid, NSAID = nonsteroidal anti-inflammatory drug, ROMK = ATP-sensitive inward rectifier potassium channel, SNV = single-nucleotide variant.
Informed Consent: All the individual participants have written the informed consent in the study. The patient has provided informed consent for publication of the case.
The authors have no conflicts of interest to disclose.
References
- [1].Kurtz I. Molecular pathogenesis of Bartter's and Gitelman's syndromes. Kidney Int 1998;54:1396–410. [DOI] [PubMed] [Google Scholar]
- [2].Stein JH. The pathogenetic spectrum of Bartter's syndrome. Kidney Int 1985;28:85–93. [DOI] [PubMed] [Google Scholar]
- [3].Ji W, Foo JN, O’Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 2008;40:592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol 2011;26:1789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bichet DG, Fujiwara TM. Reabsorption of sodium chloride—lessons from the chloride channels. N Engl J Med 2004;350:1281–3. [DOI] [PubMed] [Google Scholar]
- [6].Simon DB, Karet FE, Hamdan JM, et al. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 1996;13:183–8. [DOI] [PubMed] [Google Scholar]
- [7].Vargas-Poussou R, Feldmann D, Vollmer M, et al. Novel molecular variants of the Na-K-2Cl cotransporter gene are responsible for antenatal Bartter syndrome. Am J Hum Genet 1998;62:1332–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fukuyama S, Okudaira S, Yamazato S, et al. Analysis of renal tubular electrolyte transporter genes in seven patients with hypokalemic metabolic alkalosis. Kidney Int 2003;64:808–16. [DOI] [PubMed] [Google Scholar]
- [9].Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol 2008;4:560–7. [DOI] [PubMed] [Google Scholar]
- [10].Wei X, Ju X, Yi X, et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS One 2011;6:e29500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li R, Li Y, Fang X, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res 2009;19:1124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li H, Handsaker B, Wysoker A, et al. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Peters M, Ermert S, Jeck N, et al. Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int 2003;64:923–32. [DOI] [PubMed] [Google Scholar]
- [16].Vinci JM, Gill JR, Jr, Bowden RE, et al. The kallikrein-kinin system in Bartter's syndrome and its response to prostaglandin synthetase inhibition. J Clin Invest 1978;61:1671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Griffing GT, Komanicky P, Aurecchia SA, et al. Amiloride in Bartter's syndrome. Clin Pharmacol Ther 1982;31:713–8. [DOI] [PubMed] [Google Scholar]
- [18].Vaisbich MH, Fujimura MD, Koch VH. Bartter syndrome: benefits and side effects of long-term treatment. Pediatr Nephrol 2004;19:858–63. [DOI] [PubMed] [Google Scholar]
- [19].Hené RJ, Koomans HA, Dorhout Mees EJ, et al. Correction of hypokalemia in Bartter's syndrome by enalapril. Am J Kidney Dis 1987;9:200–5. [DOI] [PubMed] [Google Scholar]
- [20].Pressler CA, Heinzinger J, Jeck N, et al. Late-onset manifestation of antenatal Bartter syndrome as a result of residual function of the mutated renal Na+-K+-2Cl- co-transporter. J Am Soc Nephrol 2006;17:2136–42. [DOI] [PubMed] [Google Scholar]
- [21].Puricelli E, Bettinelli A, Borsa N, et al. Italian Collaborative Group for Bartter Syndrome. Long-term follow-up of patients with Bartter syndrome type I and II. Nephrol Dial Transplant 2010;25:2976–81. [DOI] [PubMed] [Google Scholar]
- [22].Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol 2009;297:F849–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sharma A, Linshaw MA. A novel compound heterozygous ROMK mutation presenting as late onset Bartter syndrome associated with nephrocalcinosis and elevated 1,25(OH)(2) vitamin D levels. Clin Exp Nephrol 2011;15:572–6. [DOI] [PubMed] [Google Scholar]