

Abstract
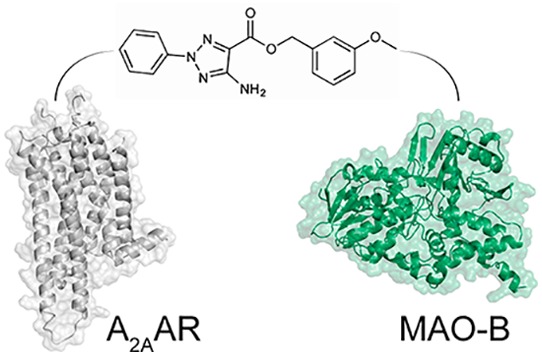
Modulation of multiple biological targets with a single drug can lead to synergistic therapeutic effects and has been demonstrated to be essential for efficient treatment of CNS disorders. However, rational design of compounds that interact with several targets is very challenging. Here, we demonstrate that structure-based virtual screening can guide the discovery of multi-target ligands of unrelated proteins relevant for Parkinson’s disease. A library with 5.4 million molecules was docked to crystal structures of the A2A adenosine receptor (A2AAR) and monoamine oxidase B (MAO-B). Twenty-four compounds that were among the highest ranked for both binding sites were evaluated experimentally, resulting in the discovery of four dual-target ligands. The most potent compound was an A2AAR antagonist with nanomolar affinity (Ki = 19 nM) and inhibited MAO-B with an IC50 of 100 nM. Optimization guided by the predicted binding modes led to the identification of a second potent dual-target scaffold. The two discovered scaffolds were shown to counteract 6-hydroxydopamine-induced neurotoxicity in dopaminergic neuronal-like SH-SY5Y cells. Structure-based screening can hence be used to identify ligands with specific polypharmacological profiles, providing new avenues for drug development against complex diseases.
Introduction
Since the lock-and-key and receptor concepts were introduced more than a century ago,1 drug development has increasingly been focusing on identifying agents that modulate the activity of a single target. The belief that the magic bullet, a compound with high affinity and selectivity for a specific protein, has high potential as a drug candidate is now dominating the pharmaceutical industry. However, the “one target–one drug” paradigm neglects the fact that many diseases are multifactorial and efficient treatment in such cases will require modulation of several proteins. For example, it is now widely accepted that the efficacy of antipsychotic drugs can be ascribed to interactions with multiple members from the G protein-coupled receptor (GPCR) family.2 Compounds that mediate their effects via several targets, which has been coined polypharmacology, show improved efficacy compared to single-target drugs by acting synergistically and avoid side effects associated with combination therapy.3
The realization that the therapeutic effects of several medications rely on polypharmacology2 has sparked interest in applying this strategy to neurological and neuropsychiatric disorders for which traditional drug development approaches have fallen short.4 For example, the recently approved antiparkinson drug safinamide inhibits monoamine oxidase B as well as sodium and calcium channels, which may contribute to its unique properties.5 The vast majority of the known multi-target drugs has been discovered by serendipity, and given the major efforts involved in the generation of single-target lead candidates, development of compounds with dual activity has been anticipated to be very difficult. Rational design of polypharmacology has mainly involved proteins that are either closely related or recognize similar biogenic molecules, e.g., kinases or aminergic GPCRs, but has rarely been accomplished for disparate targets.6−8 Access to atomic resolution information for proteins relevant for the same indication gives the opportunity to design multi-target ligands based on the binding site structures.9,10 A structure-based approach should enable identification of drugs to unrelated targets by taking advantage of common binding site features that are not apparent from protein sequence or ligand similarity.
Two targets relevant for drug development against Parkinson’s disease (PD) were selected to explore the possibility to discover dual-target ligands using structure-based virtual screening. PD is characterized by a progressive loss of dopaminergic neurons, which results in symptoms such as trembling, stiffness, and bradykinesia. To overcome the decreased dopamine levels in the brain, treatment of PD mainly relies on the dopamine precursor levodopa. However, more efficient drugs are urgently needed due to side effects such as dyskinesia and the gradual loss of levodopa efficacy.11 Alternative targets for development of antiparkinson drugs include non-dopaminergic members of the GPCR superfamily and enzymes involved in the degradation of monoamine neurotransmitters.12,13 From these two different target classes, we focused on identifying lead candidates with dual-target activity at the A2A adenosine receptor (A2AAR) and monoamine oxidase B (MAO-B). The idea of developing dual-target A2AAR/MAO-B inhibitors emerged from the serendipitous discovery that the A2A antagonist 8-(3-chlorostyryl)caffeine (CSC) also inhibited MAO-B.14 Although CSC showed promising neuroprotective effects in experimental PD models,14 it has been considered to be undesirable as starting point for drug development due to low solubility and sensitivity to light-induced degradation.15,16 Novel dual-target A2AAR/MAO-B ligands are hence needed to further assess the potential of the multi-target approach in treatment of PD.
A dual A2AAR/MAO-B inhibitor would exert the symptomatic and neuroprotective effects of A2AAR antagonism17 combined with the advantages of sustained dopaminergic signaling due to inhibition of MAO-B.18 As the targets are involved in different biochemical pathways, additive or even synergistic therapeutic effects could be expected for dual-target compounds.3,4 However, considering the dissimilar neurotransmitters recognized by the two proteins and the disparate binding sites revealed by atomic resolution crystal structures (Figure 1), design of dual-target compounds should be very challenging. In the present work, molecular docking screens of a commercial chemical library against crystal structures of the A2AAR and MAO-B binding sites were carried out to identify dual-target ligands. Twenty-four compounds that were top-ranked in the A2AAR and MAO-B screens were assayed experimentally. The screening results enabled us to assess the prospects of designing multi-target ligands using a structure-based approach and to discover starting points for development of a novel class of antiparkinson drugs.
Figure 1.

Targets for development of dual-target antiparkinson agents. (a) Crystal structures of the A2AAR (gray) and MAO-B (green) are shown as cartoons together with 2D representations of adenosine and dopamine. (b) 2D representation of CSC, a dual-target A2AAR and MAO-B inhibitor.24
Results
Docking Screens for Dual-Target Ligands
In order to identify dual-target ligands of the A2AAR and MAO-B, molecular docking screens against the two binding sites were carried out. Sets of known ligands19,20 together with property-matched decoys21 were first docked against available crystal structures to benchmark the binding sites for virtual screening, which was quantified using the adjusted logAUC (Table S1).22 In this step, no particular focus was put on the limited set of known dual-target ligands23−26 to avoid introducing bias in the prospective screen toward these two scaffolds. Crystal structures of the A2AAR in an inactive conformation, which is the state relevant for development of antiparkinson drugs,17 were considered. An A2AAR structure that strongly enriched known ligands over decoys (Figure S1a) and had a relatively open binding site conformation was selected (PDB code 3PWH(27)) to increase the probability of identifying compounds that could be accommodated by several targets. The MAO-B structure (PDB code 2V61(28)) was determined in complex with a noncovalent inhibitor and also showed excellent ligand enrichment (Figure S1b). To assess potential similarities between the A2AAR and MAO-B, the crystal structures and known ligands of the targets were compared. Binding site similarity was assessed using ProBIS,29 an algorithm designed to identify common structural elements of proteins. No statistically significant similarity between the binding sites was detected by this method (Table S2). Ligand 2D similarity was quantified by calculating the Tanimoto similarity coefficient (Tc) for all pairs of A2AAR (3898 compounds) and MAO-B (1671 compounds) ligands from the ChEMBL database19 (Figure S2). 99.9% of the Tc values were <0.30, which suggested that the A2AAR and MAO-B recognize vastly different ligand chemotypes.
Two chemical libraries containing 0.8 million fragment-like compounds (molecular weight of <250 Da) and 4.6 million lead-like compounds (250 Da < molecular weight < 350 Da) from the ZINC database30 were docked separately to the A2AAR and MAO-B structures using DOCK3.6.22 On the basis of previous prospective docking screens against GPCRs,31 the fragment library was likely to give a higher hit rate but at the expense of ligand potency. Conversely, the lead-like compounds were less likely to fit in both binding sites, but hits could be expected to have higher affinity as larger molecules can form more interactions. Several thousand orientations were sampled in the binding sites for each of the 5.4 million molecules with the proteins kept rigid, resulting in billions of predicted complexes that were evaluated using the DOCK3.6 scoring function. The compounds in the fragment- and lead-like libraries were first ranked based on their docking energies22 for the A2AAR and MAO-B. A consensus score was then calculated for each compound as the sum of the ranks from the two screens. In order to identify dual target ligands, the 500 compounds with the lowest consensus scores were inspected visually. In the compound selection step, energy terms neglected by the docking scoring function, commercial availability, and the novelty of the predicted ligands were also considered, as described previously.32 A set of 24 compounds (1–24, Table S3), comprising 13 fragment- and 11 lead-like compounds, was finally prioritized for experimental evaluation. All of the selected compounds had negative docking energies for both targets, indicating favorable interactions between the binding sites and the predicted ligands. The ranks of the selected fragment-like compounds ranged from 34 to 1273 for the A2AAR and from 10 to 2664 for MAO-B. The predicted lead-like compounds had ranks from 465 to 11760 for the A2AAR and from 422 to 14642 for MAO-B. All the selected compounds were hence ranked among the top 0.3% of the screened chemical libraries.
Experimental Evaluation of Docking Predictions
The 24 predicted ligands were evaluated experimentally at both the A2AAR and MAO-B. For the A2AAR, six compounds showed significant (>60%) displacement of radioligand at 30 μM, corresponding to a screening hit rate of 25%. Full dose–response curves were obtained for these ligands, and their Ki values ranged from 19 to 7100 nM (Table 1 and Table S4). In the MAO-B enzyme assay, 12 compounds showed >70% inhibition at 30 μM, corresponding to a hit rate of 50%, and the IC50 values of these inhibitors ranged from 61 to 8700 nM (Table 1 and Table S4). Of the 14 compounds that showed activity for at least one of the targets, seven originated from the fragment-like library and seven were from the set of lead-like compounds. Promising starting points for development of single-target drugs hence emerged from both the fragment and lead-like libraries. As expected, the lead-like compounds were, on average, more potent than the fragment-like, in particular for the A2AAR. On the other hand, the ligand efficiencies (LE, corresponding to the binding energy per atom33) were typically higher for the fragment-like compounds. The virtual screening results are summarized in Figure 2.
Table 1. Experimental Data for Identified Dual-Target Ligands.

Consensus rank from docking screens of a fragment (compound 1) or lead-like (compounds 2–4) library from the ZINC database. The ranks for the individual targets are shown in parentheses (A2AAR/MAO-B).
Ki value expressed as the mean ± SEM from three independent experiments performed in duplicate or triplicate.
IC50 value expressed as the mean ± SEM from three independent experiments performed in duplicate or triplicate.
The maximal Tanimoto coefficient (ECFP4) when compared with all compounds with dual-activity at the A2AAR and MAO-B. The maximal Tanimoto coefficient (ECFP4) for all known ligands is shown in parentheses (A2AAR/MAO-B). 2D structures of the most similar compounds are shown in Table S5.
Figure 2.

Overview of the structure-based screen for dual-target A2AAR/MAO-B ligands. The ZINC lead-like and fragment libraries were screened against A2AAR (gray) and MAO-B (green) crystal structures. Twenty-four top-ranked molecules were selected for experimental evaluation. Twelve and six compounds showed activity at MAO-B and A2AAR, respectively, and four of these displayed dual-activity (Table 1 and Table S4).
Among the 14 experimentally confirmed ligands, four compounds (1–4, Table 1) displayed activity at both the A2AAR and MAO-B, corresponding to a dual-target hit rate of 17%. Compounds 2 and 3 belonged to the same scaffold and the most potent of these (3) had a Ki of 19 nM at the A2AAR and inhibited MAO-B with an IC50 of 100 nM. The two remaining compounds (1 and 4) had micromolar activities at both targets. Of these, compound 1 (Ki = 2700 nM at the A2AAR and IC50 = 5000 nM for MAO-B) was considered to be a more promising starting point for optimization as it was fragment-sized and hence had higher LE values. The predicted binding modes of compounds 1 and 3 are shown in Figure 3. The ligands were deeply buried in the A2AAR binding site and formed hydrogen bonds with Asn2536.55 (superscripts represent Ballesteros–Weinstein residue numbering for GPCRs34). In the MAO-B binding pocket, both compounds 1 and 3 formed hydrogen bonds with Tyr326 and were anchored in a hydrophobic pocket created by Phe103, Ile316, and Phe168. To assess the novelty of the discovered dual-target ligands, we calculated the Tanimoto similarity of the compounds to the two previously identified scaffolds with activity at both targets and all known ligands of the A2AAR or MAO-B (Table 1 and Table S5) from the ChEMBL database.19 Compounds 1–4 had maximal Tanimoto similarity coefficients (Tc) ranging from 0.18 to 0.34 for the dual-target ligands, which is indicative of novel scaffolds.35 The maximal Tc values for all known ligands were higher (0.25–0.50) but remained low for either the A2AAR or MAO-B in each case, demonstrating that dual-target activity would not have been expected from 2D similarity.
Figure 3.

Predicted binding modes of two dual-target ligands: the docking poses of compounds 1 (a, b) and 3 (c, d) in the A2AAR (gray cartoon, PDB code 3PWH(27)) and MAO-B binding sites (green cartoon, PDB code 2V61(28)). Key binding site residues and the ligands are shown in sticks. Hydrogen bonds are shown as black dashed lines.
Structure–Activity Relationships of Dual-Target Ligands
Exploration of structure–activity relationships for the two scaffolds represented by compounds 1 and 3 was guided by docking of commercially available analogs and structure-based design (compounds 1a–i and 3a–k, Table S6). A set of commercially available analogs that were predicted to have the same binding mode as compound 1 was first selected for experimental evaluation, but none of these showed significant affinity for the A2AAR (1c–i, Table S7). Based on the model of compound 1 in complex with the A2AAR, substituents filling the small subpocket in the vicinity of the 7-position of the benzimidazole ring could yield large increases of affinity, as demonstrated by previous structure-based drug design efforts.36,37 Two custom-synthesized compounds were evaluated experimentally based on this prediction (1a,b, Table 2). The chlorine (1a) and methoxy (1b) substituted compounds had 27- and 5-fold better A2AAR binding affinities than compound 1, respectively. These compounds were then evaluated in the MAO-B assay to assess dual-target activity. Whereas compound 1b (IC50 = 7000 nM) showed no improvement over compound 1, compound 1a was a submicromolar inhibitor of both MAO-B (IC50 = 410 nM) and A2AAR (Ki = 99 nM). A representative dose–response curve for compound 1a is shown in Figure S3.
Table 2. Experimental Data for Analogs of Compounds 1 and 3.

Ki value expressed as the mean ± SEM from three independent experiments performed in duplicate or triplicate.
IC50 value expressed as the mean ± SEM from three independent experiments performed in duplicate or triplicate. Inactive compounds (>10 000 nM) were tested in one experiment performed in triplicate.
Compounds 2 and 3 belonged to the same scaffold, and the most potent of these (3) had an affinity of 19 nM for the A2AAR and inhibited MAO-B with an IC50 value of 100 nM. A total of nine analogs (3a–i, Table 2 and Table S8) were assayed. Two commercially available compounds with substituents on the benzyl ring (3a,b, Table 2), which were predicted to occupy the top part of A2AAR binding site and a buried pocket in MAO-B, retained activity at both targets. Commercially available and custom-synthesized analogs devoid of the ester moiety, which may be metabolized in vivo, were also explored by replacing this group with amide groups (3c–i, Table 2 and Table S8). As the ester moiety was facing the solvent in the predicted binding mode for the A2AAR, these analogs were predicted to maintain high activity. Binding assays confirmed that the four compounds were A2AAR ligands, but none of them inhibited MAO-B (Table 2). This result could be explained by the fact that the ester moiety was predicted to bind in a narrow channel connecting the two major subpockets of the MAO-B binding site, which makes it challenging to introduce new substituents in this region (Figure 3). As an additional control, we confirmed that the activity of compound 3 was not mediated by its metabolized form by evaluating the two products of hydrolysis, and these (3j and 3k) were inactive at both targets (Table S8). To summarize, many of the tested analogs retained activity only at one of the targets and compound 3 was still the most potent compound in the series. A representative dose–response curve for compound 3 is shown in Figure S3.
Functional Assays and Selectivity
Functional assays measuring cyclic AMP accumulation mediated by activation of the A2AAR in response to the agonist 5′-N-ethylcarboxamidoadenosine (NECA) in the presence of 1a and 3 (Figure S4) confirmed that these compounds were antagonists, which is the desired efficacy in treatment of PD.17 To evaluate if compounds 1a and 3 were noncovalent inhibitors of MAO-B, a reactivation experiment was carried out. MAO-B was first preincubated in the presence of substrate and either compounds 1a, 3, or the irreversible inhibitor tranylcypromine (trans-(±)-2-phenylcyclopropanamine).18 An excess of substrate was then added, which should displace reversible inhibitors, whereas no effect should be observed for an irreversible mechanism of action. The measured fluorescence increased for compounds 1a and 3, as expected for reversible inhibition, whereas only a small response was observed for the irreversible control inhibitor (Figure S5).
The selectivity profiles of compounds 1a and 3 were further probed by evaluating binding affinities for the A1-, A2B-, and A3AR (Table 3). Compound 1a was 10- and 2-fold selective for the A2AAR over the A1- and A2BAR, respectively, whereas compound 3 showed 12- and 10-fold selectivity over the same subtypes. Both compounds 1a and 3 showed weak affinity for the A3AR subtype (Ki > 10000 nM). Similarly, specificity for MAO-B over MAO-A was also confirmed for compounds 1a and 3. Compound 1a did not show any significant inhibition of MAO-A at 10 μM, whereas compound 3 had an IC50 = 4700 nM, resulting in 47-fold selectivity (Table 3).
Table 3. Activity of Compounds 1a and 3 at A1-, A2B-, and A3ARs and MAO-A.
| ID | A1AR | A2BAR | A3AR | MAO-A |
|---|---|---|---|---|
| (Ki/nM)a | (Ki/nM)a | (%)a | (IC50/nM)b | |
| 1a | 1000 ± 260 | 230 ± 20 | 50 ± 6.0% | >10000 |
| 3 | 220 ± 80 | 190 ± 8.5 | 44 ± 6.5% | 4700 ± 97 |
Percent of displacement at 10 μM or Ki value expressed as the mean ± SEM from three independent experiments performed in duplicate or triplicate.
IC50 value expressed as the mean ± SEM from three independent experiments performed in duplicate or triplicate. Inactive compounds (>10 000 nM) were tested in one experiment performed in triplicate.
Potential concerns in development of multi-target ligands are that the screening hits may be promiscuous scaffolds and that the activity may be due to assay interference or inhibition by colloidal aggregation. However, none of the discovered dual-target ligands contained any of the substructures identified by Baell et al. as common among frequent-hitters (pan-assay interference compounds, PAINS).38 In the case of MAO-B, we also specifically controlled for assay interference by carrying out the experiments in the absence of the target protein with added product, which did not result in significant activity for compound 1a or 3. Furthermore, the MAO-B assays for compounds 1a and 3 were performed in the presence and absence of detergent (Triton X-100, 0.01%) to control for artifactual inhibition due to colloidal aggregation.39 In the case of aggregating compounds, activity will be diminished by Triton X-100, but no significant changes in the IC50 values were observed for 1 or 3a in the presence of detergent. Finally, we found that the commercially available compounds 1 and 3 had been evaluated experimentally in a large number of screens deposited in the PubChem bioassay database and showed no activity in the vast majority of these (Table S9).40 Compound 1 did not show any significant effect in 710 assays and was annotated as active in 39 assays, whereas compound 3 was inactive in in 766 screens and was only active in five cases. A more detailed analysis of the PubChem data revealed that compound 1 showed >50% activity at 10 μM for 16 different targets (Table S10), whereas no targets remained for compound 3 at this concentration.
Evaluation of Compounds 1a and 3 in a Cell Viability Assay
Evaluation of a cytoprotective effect exerted by compounds 1a, 3, and CSC on differentiated dopaminergic neuronal-like SH-SY5Y cells was carried out using the resazurin assay. As shown in Figure 4, 20 μM 6-OHDA resulted in a 37 ± 1.3% reduction in cell viability and was chosen for evaluating compound impact. Pretreatment with 0.15 μM of both compounds 1a and 3 as well as CSC caused statistically significant (F4,115 = 116.4; p < 0.0001) counteraction of the 6-OHDA induced cytotoxicity. Compounds 1a and 3 reduced cytotoxicity by 14 ± 2.1% and 13 ± 2.2%, respectively. The previously discovered dual-target ligand CSC (A2AAR, Ki = 38 nM; MAO-B, IC50 = 18 nM)14,24 resulted in an equally potent protection (13 ± 2.0%). There was no difference in the protective effects of the studied compounds.
Figure 4.

Viability assessment on human dopaminergic neuronal-like cells treated with 6-OHDA toxin. Cells were treated with 0.15 μM 1a, 3, or CSC for 3 h prior to addition of 20 μM 6-OHDA. After 24 h, resazurin was added for another 2 h, whereupon cell viability was assayed. Data are shown as the mean ± SEM and were analyzed using one-way ANOVA with Newman–Keuls multiple comparison test: (###) p < 0.001 versus control and (∗∗∗) p < 0.001 versus 6-OHDA alone.
Discussion and Conclusions
Three main results emerged from our structure-guided screens for dual-target inhibitors of a GPCR and enzyme relevant for development of antiparkinson drugs. First, four dual-target ligands were identified among the 14 screening hits that were confirmed active for either the A2AAR or MAO-B. Second, several of the dual-target compounds were potent. The most promising scaffold had astonishing nanomolar activity at both the A2AAR and MAO-B. The structural models also guided improvement of a weak hit from the virtual screen to yield a second dual-target scaffold with submicromolar potencies. Third, two dual-target compounds showed protective effects in an in vitro PD model.
Structure-based virtual screening has successfully discovered ligands of both GPCRs and enzymes,41,42 but prediction of compounds with multi-target action involving disparate targets has rarely been accomplished. The high docking hit rates (25% and 50%, respectively) are in line with results obtained in previous virtual screening studies against either the A2AAR or MAO-B.20,43−45 The challenges involved in identifying dual-target ligands were reflected by the fact that only 17% of the tested compounds showed activity at both the A2AAR and MAO-B, which is lower than the hit rates obtained previously in docking screens against the A2AAR.43,44 Whereas the hit rate for dual-target ligands was close to 4-fold lower than for the single targets, we were surprised to find that the potencies of the hits were not affected. Remarkably, compound 3 (Ki = 19 nM) not only is among the highest affinity A2AAR ligands to emerge from a virtual screen but also was very potent at MAO-B (IC50 = 100 nM). The fidelity of the predicted binding mode for compound 3 was further supported by a crystal structure of the A2AAR in complex with a related ligand, which was published during the preparation of this manuscript (Figure 5).46 A question that arises is how two proteins can recognize the same compound despite lack of sequence and structural similarity.47 Analysis of the docking results revealed some similarities among the top-ranked compounds and the known dual-target inhibitor CSC (Figure 1). For example, the dual-target scaffolds represented by compounds 1a and 3 both contained two aromatic groups connected by polar moieties. The aromatic groups occupied hydrophobic subpockets at the ends of the extended binding sites, and the polar moieties were stabilized by hydrogen bonds to a side chain positioned centrally in both pockets (Figure 3). Interestingly, CSC (Figure 1), which differs from both compounds 1a and 3 by 2D similarity, has a similar shape and composition of aromatic and polar groups. Despite the differences in sequence and structure, the docking scoring function was thus able to distinguish similarities in the binding site shape and polarity, supporting the use of structure-based virtual screening as a tool for identifying multi-target ligands of disparate targets.
Figure 5.
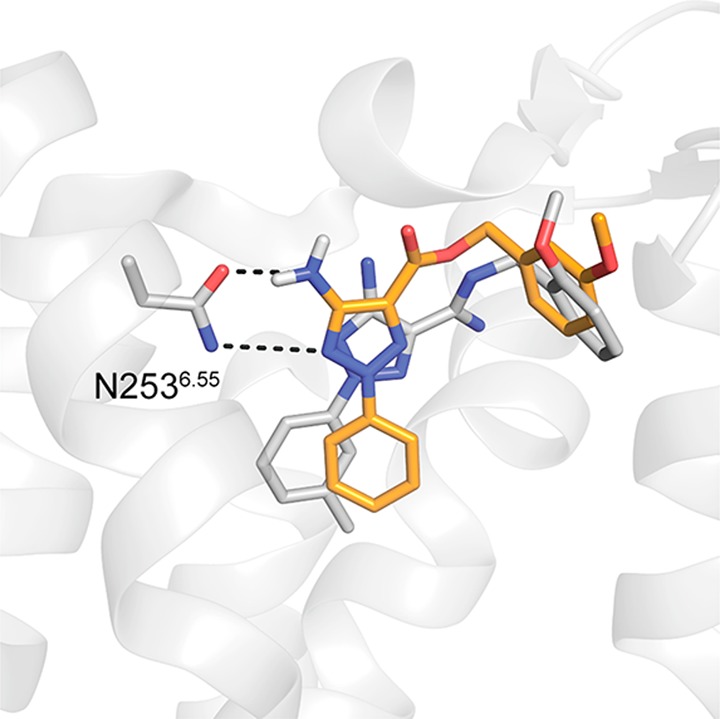
Comparison of the predicted binding mode of compound 3 to a crystal structure of a related antagonist in complex with the A2AAR. The dual-target ligand 3 is shown as sticks with orange carbon atoms. The cocrystallized antagonist (PDB code 5UIG(46)) is depicted in sticks with gray carbon atoms. The A2AAR is depicted as gray cartoons. Hydrogen bonds are shown as black dashed lines.
The potential of polypharmacology in treatment of complex diseases has led to increasing interest in rational design of multi-target ligands.2,3 Such efforts have mainly focused on either optimizing a single-target scaffold for activity at a second target or combining the pharmacophore features of two single-target ligands into one compound.48 The former strategy has mainly been successful for targets that are related or recognize similar biogenic molecules. An elegant example is the computer-aided optimization of an acetylcholinesterase inhibitor for activity at aminergic GPCRs using 2D similarity methods by Besnard et al.7 However, the general applicability of this approach is limited in that it requires access to training sets with known ligands of each target and depends on selecting a suitable starting point for optimization. This is illustrated by the fact that medicinal chemistry efforts to obtain dual-target A2AAR/MAO-B ligands based on an A2AAR scaffold (4H-3,1-benzothiazin-4-one) were successful,24 but optimization of phthalimide inhibitors of MAO-B for activity at the A2AAR failed.49 For unrelated targets, two ligand scaffolds can be linked to obtain a single compound with dual activity. For example, Jörg et al. designed dual-target ligands of the A2AAR and D2 dopamine receptor by connecting the D2 agonist ropinirole to an A2A antagonist via a chemical linker50 whereas De Simone et al. constructed compounds for both the D3 dopamine receptor and fatty acid amide hydrolase by combining two pharmacophores.51 Active compounds were identified in both studies, but the combination of two existing ligands led to high molecular weight compounds with properties that may not be compatible with oral availability and blood–brain barrier penetration. In contrast, the structure-based virtual screening strategy explored in this work was not constrained to the limited chemical space spanned by known scaffolds. High-throughput docking allowed us to carry out an unbiased screen of several million compounds, leading to the discovery of four dual-target ligands. The most potent scaffolds were reversible MAO-B inhibitors and antagonized the A2AAR, which are the desired functional properties in development of PD drugs. The lack of activity of the two scaffolds in high-throughput screens against other proteins and controls made for assay interference and colloidal aggregation suggested that the dual-target activity was unlikely to be due to promiscuous or artifactual inhibition. Moreover, the inhibitors not only did have low molecular weight and good selectivity properties but also showed cytoprotective effects in dopaminergic neuronal-like cells. Compounds 1 and 3a hence provide excellent starting points for development of dual-target A2AAR/MAO-B leads that could be evaluated in vivo for antiparkinson activity.
A few caveats of our approach should be mentioned. First, both the A2AAR and MAO-B have been demonstrated to recognize diverse scaffolds and bind drug-like compounds with high affinity. The high docking hit-rates obtained in this work may not be directly transferrable to less druggable targets. Second, access to multiple high-resolution crystal structures and large sets of known ligands for both targets contributed to the successful selection of compounds for experimental testing, but such wealth of information is not available in all drug discovery projects. Finally, it should be emphasized that the complexity of ligand design increases dramatically when a single scaffold is optimized for multi-target activity. In agreement with this concern and previous studies,24 we found that closely related analogs of our dual-target leads with activity for one target could be completely inactive for the other. Considering the challenges involved in optimization of dual-target compounds, access to atomic resolution information for the target binding sites will be crucial for lead development. In summary, our results demonstrate that molecular docking screening can guide discovery of ligands with specific polypharmacological profiles, which can contribute to development of drugs against complex diseases with improved efficacy and less side effects.
Experimental Section
Molecular Docking Screens
The molecular docking calculations were carried out using DOCK3.622 (http://dock.compbio.ucsf.edu/DOCK3.6/) against a crystal structure of the A2AAR in complex with an antagonist (PDB code 3PWH(27)) and of MAO-B in complex with an inhibitor (PDB code 2V61(28)). The A2AAR structure was prepared by removing non-protein atoms, and thermostabilizing mutations were modified to correspond to the wild type sequence. Side chain rotamers for modified amino acids were selected based on other high-resolution crystal structures of the A2AAR. In the case of MAO-B, all protein residues, the FAD cofactor, and two crystallographic waters (residues 1239 and 1304) were retained in the docking calculations. The protonation states of ionizable side chains of residues Asp, Glu, Lys, and Arg were set according to their most probable state at pH 7. The protonation states of histidines in the binding sites were assigned based on the local hydrogen-bonding network. In the case of the A2AAR, the side chains of His2506.52 and His2787.42 were protonated at the Nε and Nδ positions, respectively.
DOCK3.6 uses a flexible-ligand sampling algorithm that defines the binding site based on a set of matching spheres.52 Forty-five matching spheres were used, and these were derived based on the cocrystallized ligands. The extent of ligand sampling was defined by the bin size, bin size overlap, and distance tolerance, and these parameters were set to 0.4 Å, 0.1 Å, and 1.5 Å, respectively, for both the docked molecules and binding sites matching spheres. Ligand conformations passing a steric filter were scored using the DOCK3.6 physics-based scoring function. The binding energy was estimated as the sum of the van der Waals and electrostatic energies, corrected for ligand desolvation.22 The protein atoms were described using a united atom version of the AMBER force field.53 The dipole moment of the Asn253 side chain in the A2AAR was increased to favor hydrogen bonding to this residue, as described previously.43 Force field parameters for the FAD cofactor in the MAO-B binding site were derived using the generalized AMBER force field.54 The program CHEMGRID55 was used to generate the van der Waals grids,53 and the electrostatic potential maps of the binding sites were obtained using the program Delphi.56 The desolvation energy of a docked compound was obtained by scaling its transfer free energy between solvents with dielectric constants of 78 and 2 with a factor that reflects the degree of burial in the binding site.22 For the best scoring conformation of each docked molecule, 100 steps of rigid-body minimization were carried out.
A chemical library containing 5.4 million compounds was obtained from the ZINC database.30 Known ligands19,20 and DUD-E decoys21 were prepared for docking using the ZINC database protocol.30 All tested compounds were sourced from commercial vendors (Table S3 and S6). Purity (>95%) was confirmed by LC/MS for compounds 1–4, 1a, 1b, 3a, and 3b. The selected molecules were also screened for substructures present in pan-assay interference compounds (PAINS) and did not contain any of these motifs.38
2D Ligand and Binding Site Similarity Calculations
Tanimoto similarity coefficients (Tc) were calculated with ECFP4 fingerprints using Screenmd (JChem version 15.10.12, ChemAxon, 2015). The maximal Tc value between each compound and the previously discovered dual-target A2AAR/MAO-B ligands23−26 or all known ligands of the targets in the ChEMBL database19 (activity <10 μM) was calculated. Binding site similarity was quantified using the ProBis Web server57 based on the A2AAR and MAO-B crystal structures used in the virtual screen. The binding sites were defined as all residues within 7 Å of the cocrystallized ligands.
Binding Assays for the Adenosine Receptors
A2AAR competition binding experiments were carried out in a multiscreen GF/C 96-well plate (Millipore, Madrid, Spain) pretreated with binding buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 10 mM, 2 U/mL adenosine deaminase, pH = 7.4). An amount of 5 μg of membranes from HeLa-A2A cell line was incubated with 3 nM [3H]ZM241385 (4-[2-[[7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-yl]amino]ethyl]-2-tritiophenol) (50 Ci/mmol, 1 mCi/ml, ARC-ITISA 0884) and the studied compound at 25 °C for 30 min and then filtered and washed four times with 250 μL of wash buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 10 mM, pH = 7.4), before measuring in a microplate β scintillation counter (Microbeta Trilux, PerkinElmer, Madrid, Spain). Nonspecific binding was determined in the presence of 50 μM NECA (Sigma E2387).
A1AR competition binding experiments were carried out in a multiscreen GF/C 96-well plate (Millipore, Madrid, Spain) pretreated with binding buffer (Hepes 20 mM, NaCl 100 mM, MgCl2 10 mM, 2 U/mL adenosine deaminase, pH = 7.4). An amount of 5 μg of membranes from the Euroscreen hA1 cell line was incubated with 1 nM [3H]DPCPX (8-cyclopentyl-1,3-bis(1,3-ditritiopropyl)-7H-purine-2,6-dione) (120 Ci/mmol, 1 mCi/mL, PerkinElmer NET974001MC) and the studied compound at 25 °C for 60 min and then filtered and washed four times with 250 μL of wash buffer (Hepes 20 mM, NaCl 100 mM, MgCl2 10 mM, pH = 7.4), before measuring in a microplate β scintillation counter (Microbeta Trilux, PerkinElmer, Madrid, Spain). Nonspecific binding was determined in the presence of 10 μM R-PIA (Sigma P4532).
A2BAR competition binding experiments were carried out in a polypropilene 96-well plate. An amount of 20 μg of membranes from Euroscreen hA2B cell line was incubated with 25 nM [3H]DPCPX (164 Ci/mmol, 1 mCi/ml, PerkinElmer NET974001MC) and the studied compound at 25 °C for 30 min and then filtered and washed four times with 250 μL of wash buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 5 mM, pH = 6.5), before measuring in a microplate β scintillation counter (Microbeta Trilux, PerkinElmer, Madrid, Spain). Nonspecific binding was determined in the presence of 1000 μM NECA (Sigma E2387).
A3AR competition binding experiments were carried out in a multiscreen GF/B 96-well plate (Millipore, Madrid, Spain) pretreated with binding buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 5 mM, 2 U/mL adenosine deaminase, pH = 7.4). An amount of 70 μg of membranes from HeLa-A3 cell line was incubated with 10 nM [3H]NECA (27.6 Ci/mmol, 1 mCi/ml, PerkinElmer NET811250UC) and the studied compound at 25 °C for 180 min and then filtered and washed six times with 250 μL of wash buffer (Tris-HCl, 50 mM, pH = 7.4), before measuring in a microplate β scintillation counter (Microbeta Trilux, PerkinElmer, Madrid, Spain). Nonspecific binding was determined in the presence of 100 μM R-PIA (Sigma P4532).
Nonlinear fitting of the concentration–response curves was carried out by using the GraphPad Prism 4.0 software (San Diego, CA, USA) by applying a four-parameters logistic equation for deriving IC50 values. Ki values were calculated using the Cheng–Prusoff equation. All Ki values are reported as the mean ± SEM.
Functional Assays for the A2AAR
Functional experiments were carried out in a CHO cell line transfected with human A2A receptors by measuring coupling to the Gs signaling pathway. Twenty-four hours before the assay, 104 cells/well were seeded in a 96-well culture plate (Falcon 353072). The cells were washed with wash buffer (Dulbecco’s modified Eagle’s medium (DMEM) nutrient mixture F-12 ham (Sigma D8062), 25 mM Hepes; pH = 7.4). Wash buffer was replaced by incubation buffer (DMEM nutrient mixture F-12 ham (Sigma D8062), 25 mM Hepes, 20 μM Rolipram; pH = 7.4). The tested compounds were added and incubated at 37 °C for 15 min. After incubation, NECA (Sigma E2387) was added in several concentrations and incubated at 37 °C for 15 min. After incubation, the amount of cAMP was determined using cAMP Biotrak enzyme immunoassay (EIA) system kit (GE Healthcare RPN225).
MAO-B and MAO-A Assays
The 24 compounds selected from the from molecular docking screens were initially tested at MAO-B as described previously (IC50 values in Table 1 and Table S4).24 Compounds 1 and 3 and analogs thereof were evaluated using a slightly modified protocol (IC50 values in Table 2 and Table S8). The human recombinant MAO-A and MAO-B enzyme expressed in baculovirus infected BTI insect cells were purchased from Sigma-Aldrich (84 U/mg, catalog no. M7316 and 71 U/mg, catalog no. M 7441, respectively). A volume of 5 μL (2-fold concentration) of human MAO (final 1 U/mL for MAO-B and final 0.5 U/mL for MAO-A) was delivered to the reaction plate (or well) containing the base reaction buffer (50 mM Tris-HCL, pH = 7.5, 0.05% CHAPS, and 1% DMSO). Compounds in concentrations ranging from 10–9 to 10–4 in 100% DMSO were added to the enzyme mixture by acoustic technology (Echo 550; nanoliter range). Samples were preincubated for 30 min at room temperature. A mixture of 5 μL (2-fold concentration) of tyramine (10 μM) with buffer was added to reaction wells, except in the no substrate wells, to initiate the reaction, and samples were incubated for 60 min at room temperature. For the detection step, 10 μL (2-fold concentration) of a premixture of horseradish peroxidase (final 0.1 U/mL) and Amplex Red reagent (final 10 μM) was added to the reaction mixture and fluorescence measurements of hydrogen peroxide and consequently of resorufin were performed using EnVision fluorescence reader (excitation 535 nm and emission 590 nm) over 30 min. In order to control for assay interference, the experiments were carried out in the absence of enzyme but with H2O2 added (mimicking assay product). The MAO-B assays for compounds 1a and 3 were also performed in the presence of Triton X-100 (0.01%) to rule out inhibition due to colloidal aggregation. All data were analyzed using the GraphPad Prism 4.0 software (San Diego, CA, USA). All IC50 values are reported as the mean ± SEM.
Cell Viability Measurement
Reagents for cell culture were acquired through Life Technologies unless otherwise stated. Human SH-SY5Y cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 μg/mL penicillin and streptomycin. Cells were maintained at 37 °C with 95% humidified air and 5% CO2. Cells were grown to a confluency of 70–80% and then seeded into 96-well plates (Corning) at a density of 2.5 × 104 cells/well with phenol-red free DMEM, in 1% FBS, 200 μM l-glutamine, 100 μg/mL penicillin–streptomycin and with 10 μM retinoic acid (Sigma) for 72 h to stimulate differentiation into dopaminergic neuronal-like cells. Cells were then pretreated with the compounds diluted in DMSO (0.1% v/v, Sigma) for 3 h before the addition of 20 μM 6-OHDA (Sigma) for 24 h. 6-OHDA was freshly prepared and diluted in 0.9% sodium chloride (Fresenius-Kabi) and 0.007% ascorbic acid (Sigma). In the cell viability measurements, resazurin (0.02 mg/mL final concentration, Sigma) was added to each well 24 h after 6-OHDA exposure and further incubated for 2 h. Afterward, fluorescence intensity was examined at an excitation of 540 nm and an emission of 590 nm (Spark, Tecan).
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (Grant Agreement 715052). The work was also supported by grants from the Swedish Research Council (Grants 2013-5708 and 2017-4676) and the Science for Life Laboratory to J.C. Computational resources were provided by the Swedish National Infrastructure for Computing (SNIC). We thank OpenEye Scientific Software for the use of OEChem and OMEGA at no cost.
Glossary
Abbreviations Used
- AR
adenosine receptor
- MAO
monoamine oxidase
- logAUC
logarithm of area under the curve
- CSC
8-(3-chlorostyryl)caffeine
- Tc
Tanimoto similarity coefficient
- PAINS
pan-assay interference compounds
- 6-OHDA
6-hydroxydopamine
- ECFP4
extended chemical fingerprints up to four atoms
- FBS
fetal bovine serum
- DMEM
Dulbecco’s modified Eagle medium
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00204.
Author Contributions
∥ M.J. and A.Z. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Rang H. P. The Receptor Concept: Pharmacology’s Big Idea. Br. J. Pharmacol. 2006, 147, S9–S16. 10.1038/sj.bjp.0706457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth B. L.; Sheffler D. J.; Kroeze W. K. Magic Shotguns versus Magic Bullets: Selectively Non-Selective Drugs for Mood Disorders and Schizophrenia. Nat. Rev. Drug Discovery 2004, 3, 353–359. 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J.; Rastelli G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Cavalli A.; Bolognesi M. L.; Mìnarini A.; Rosini M.; Tumiatti V.; Recanatini M.; Melchiorre C. Multi-Target-Directed Ligands to Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- Blair H. A.; Dhillon S. Safinamide: A Review in Parkinson’s Disease. CNS Drugs 2017, 31, 169–176. 10.1007/s40263-017-0408-1. [DOI] [PubMed] [Google Scholar]
- Szabo M.; Lim H. D.; Klein Herenbrink C.; Christopoulos A.; Lane J. R.; Capuano B. Proof of Concept Study for Designed Multiple Ligands Targeting the Dopamine D2, Serotonin 5-HT2A, and Muscarinic M1 Acetylcholine Receptors. J. Med. Chem. 2015, 58, 1550–1555. 10.1021/jm5013243. [DOI] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X.-P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R. C.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated Design of Ligands to Polypharmacological Profiles. Nature 2012, 492, 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Lin H.; Shokat K. M. Targeting the Cancer Kinome through Polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Costanzi S.; Paoletta S. Computational Studies to Predict or Explain G Protein Coupled Receptor Polypharmacology. Trends Pharmacol. Sci. 2014, 35, 658–663. 10.1016/j.tips.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Mason J. S.; Overington J. P. Can We Rationally Design Promiscuous Drugs?. Curr. Opin. Struct. Biol. 2006, 16, 127–136. 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Lees A. J.; Hardy J.; Revesz T. Parkinson’s Disease. Lancet 2009, 373, 2055–2066. 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Meissner W. G.; Frasier M.; Gasser T.; Goetz C. G.; Lozano A.; Piccini P.; Obeso J. A.; Rascol O.; Schapira A.; Voon V.; Weiner D. M.; Tison F.; Bezard E. Priorities in Parkinson’s Disease Research. Nat. Rev. Drug Discovery 2011, 10, 377–393. 10.1038/nrd3430. [DOI] [PubMed] [Google Scholar]
- Schapira A. H. V.; Bezard E.; Brotchie J.; Calon F.; Collingridge G. L.; Ferger B.; Hengerer B.; Hirsch E.; Jenner P.; Le Novère N.; Obeso J. A.; Schwarzschild M. A.; Spampinato U.; Davidai G. Novel Pharmacological Targets for the Treatment of Parkinson’s Disease. Nat. Rev. Drug Discovery 2006, 5, 845–854. 10.1038/nrd2087. [DOI] [PubMed] [Google Scholar]
- Chen J. F.; Steyn S.; Staal R.; Petzer J. P.; Xu K.; Van Der Schyf C. J.; Castagnoli K.; Sonsalla P. K.; Castagnoli N.; Schwarzschild M. A. 8-(3-Chlorostyryl) Caffeine May Attenuate MPTP Neurotoxicity through Dual Actions of Monoamine Oxidase Inhibition and A2A Receptor Antagonism. J. Biol. Chem. 2002, 277, 36040–36044. 10.1074/jbc.M206830200. [DOI] [PubMed] [Google Scholar]
- Hockemeyer J.; Burbiel J. C.; Müller C. E. Multigram-Scale Syntheses, Stability, and Photoreactions of A2A Adenosine Receptor Antagonists with 8-Styrylxanthine Structure: Potential Drugs for Parkinson’s Disease. J. Org. Chem. 2004, 69, 3308–3318. 10.1021/jo0358574. [DOI] [PubMed] [Google Scholar]
- Müller C. E.; Thorand M.; Qurishi R.; Diekmann M.; Jacobson K. A.; Padgett W. L.; Daly J. W. Imidazo[2,1-I ]Purin-5-Ones and Related Tricyclic Water-Soluble Purine Derivatives: Potent A2A- and A3-Adenosine Receptor Antagonists. J. Med. Chem. 2002, 45, 3440–3450. 10.1021/jm011093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armentero M. T.; Pinna A.; Ferré S.; Lanciego J. L.; Müller C. E.; Franco R. Past, Present and Future of A2A Adenosine Receptor Antagonists in the Therapy of Parkinson’s Disease. Pharmacol. Ther. 2011, 132, 280–299. 10.1016/j.pharmthera.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer P.; Laux G. MAO-Inhibitors in Parkinson’s Disease. Exp. Neurobiol. 2011, 20, 1–17. 10.5607/en.2011.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento A. P.; Gaulton A.; Hersey A.; Bellis L. J.; Chambers J.; Davies M.; Krüger F. A.; Light Y.; Mak L.; McGlinchey S.; Nowotka M.; Papadatos G.; Santos R.; Overington J. P. The ChEMBL Bioactivity Database: An Update. Nucleic Acids Res. 2014, 42, D1083–D1090. 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez D.; Gao Z. G.; Moss S. M.; Jacobson K. A.; Carlsson J. Molecular Docking Screening Using Agonist-Bound GPCR Structures: Probing the A2A Adenosine Receptor. J. Chem. Inf. Model. 2015, 55, 550–563. 10.1021/ci500639g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Shoichet B. K. Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J. Chem. Inf. Model. 2010, 50, 1561–1573. 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- Rivara S.; Piersanti G.; Bartoccini F.; Diamantini G.; Pala D.; Riccioni T.; Stasi M. A.; Cabri W.; Borsini F.; Mor M.; Tarzia G.; Minetti P. Synthesis of (E)-8-(3-Chlorostyryl)caffeine Analogues Leading to 9-Deazaxanthine Derivatives as Dual A2A antagonists/MAO-B Inhibitors. J. Med. Chem. 2013, 56, 1247–1261. 10.1021/jm301686s. [DOI] [PubMed] [Google Scholar]
- Stößel A.; Schlenk M.; Hinz S.; Küppers P.; Heer J.; Gütschow M.; Müller C. E. Dual Targeting of Adenosine A2A Receptors and Monoamine Oxidase B by 4 H-3,1-Benzothiazin-4-Ones. J. Med. Chem. 2013, 56, 4580–4596. 10.1021/jm400336x. [DOI] [PubMed] [Google Scholar]
- Koch P.; Akkari R.; Brunschweiger A.; Borrmann T.; Schlenk M.; Küppers P.; Köse M.; Radjainia H.; Hockemeyer J.; Drabczyńska A.; Kieć-Kononowicz K.; Müller C. E. 1,3-Dialkyl-Substituted tetrahydropyrimido[1,2-F]purine-2,4-Diones as Multiple Target Drugs for the Potential Treatment of Neurodegenerative Diseases. Bioorg. Med. Chem. 2013, 21, 7435–7452. 10.1016/j.bmc.2013.09.044. [DOI] [PubMed] [Google Scholar]
- Mikkelsen G. K.; Langgård M.; Schrøder T. J.; Kreilgaard M.; Jørgensen E. B.; Brandt G.; Griffon Y.; Boffey R.; Bang-Andersen B. Synthesis and SAR Studies of Analogues of 4-(3,3-Dimethyl-Butyrylamino)-3,5-Difluoro-N-Thiazol-2-Yl-Benzamide (Lu AA41063) as Adenosine A2A Receptor Ligands with Improved Aqueous Solubility. Bioorg. Med. Chem. Lett. 2015, 25, 1212–1216. 10.1016/j.bmcl.2015.01.062. [DOI] [PubMed] [Google Scholar]
- Doré A. S.; Robertson N.; Errey J. C.; Ng I.; Hollenstein K.; Tehan B.; Hurrell E.; Bennett K.; Congreve M.; Magnani F.; Tate C. G.; Weir M.; Marshall F. H. Structure of the Adenosine A2A Receptor in Complex with ZM241385 and the Xanthines XAC and Caffeine. Structure 2011, 19, 1283–1293. 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda C.; Wang J.; Pisani L.; Caccia C.; Carotti A.; Salvati P.; Edmondson D. E.; Mattevi A. Structures of Human Monoamine Oxidase B Complexes with Selective Noncovalent Inhibitors: Safinamide and Coumarin Analogs. J. Med. Chem. 2007, 50, 5848–5852. 10.1021/jm070677y. [DOI] [PubMed] [Google Scholar]
- Konc J.; Janežič D. ProBiS Algorithm for Detection of Structurally Similar Protein Binding Sites by Local Structural Alignment. Bioinformatics 2010, 26, 1160–1168. 10.1093/bioinformatics/btq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC – A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan A.; Heine P.; Rudling A.; Plückthun A.; Kummer L.; Carlsson J. Ligand Discovery for a Peptide-Binding GPCR by Structure-Based Screening of Fragment- and Lead-Like Chemical Libraries. ACS Chem. Biol. 2017, 12, 735–745. 10.1021/acschembio.6b00646. [DOI] [PubMed] [Google Scholar]
- Carlsson J.; Coleman R. G.; Setola V.; Irwin J. J.; Fan H.; Schlessinger A.; Sali A.; Roth B. L.; Shoichet B. K. Ligand Discovery from a Dopamine D3 Receptor Homology Model and Crystal Structure. Nat. Chem. Biol. 2011, 7, 769–778. 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. Methods Neurosci. 1995, 25, 366–428. 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- Wawer M.; Bajorath J. Similarity-Potency Trees: A Method to Search for SAR Information in Compound Data Sets and Derive SAR Rules. J. Chem. Inf. Model. 2010, 50, 1395–1409. 10.1021/ci100197b. [DOI] [PubMed] [Google Scholar]
- Andrews S. P.; Mason J. S.; Hurrell E.; Congreve M. Structure-Based Drug Design of Chromone Antagonists of the Adenosine A2A Receptor. MedChemComm 2014, 5, 571–575. 10.1039/C3MD00338H. [DOI] [Google Scholar]
- Matricon P.; Ranganathan A.; Warnick E.; Gao Z.-G.; Rudling A.; Lambertucci C.; Marucci G.; Ezzati A.; Jaiteh M.; Dal Ben D.; Jacobson K. A.; Carlsson J. Fragment Optimization for GPCRs by Molecular Dynamics Free Energy Calculations: Probing Druggable Subpockets of the A2A Adenosine Receptor Binding Site. Sci. Rep. 2017, 7, 6398. 10.1038/s41598-017-04905-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Shoichet B. K. A Detergent-Based Assay for the Detection of Promiscuous Inhibitors. Nat. Protoc. 2006, 1, 550–553. 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Bryant S. H.; Cheng T.; Wang J.; Gindulyte A.; Shoemaker B. A.; Thiessen P. A.; He S.; Zhang J. PubChem BioAssay: 2017 Update. Nucleic Acids Res. 2017, 45, D955–D963. 10.1093/nar/gkw1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59, 4103–4120. 10.1021/acs.jmedchem.5b02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez D.; Ranganathan A.; Carlsson J. Discovery of GPCR Ligands by Molecular Docking Screening: Novel Opportunities Provided by Crystal Structures. Curr. Top. Med. Chem. 2015, 15, 2484–2503. 10.2174/1568026615666150701112853. [DOI] [PubMed] [Google Scholar]
- Carlsson J.; Yoo L.; Gao Z.-G.; Irwin J. J.; Shoichet B. K.; Jacobson K. A. Structure-Based Discovery of A2A Adenosine Receptor Ligands. J. Med. Chem. 2010, 53, 3748–3755. 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Jaakola V.-P.; Lane J. R.; Lin J.; IJzerman A. P.; Yeager M.; Kufareva I.; Stevens R. C.; Abagyan R. Structure-Based Discovery of Novel Chemotypes for Adenosine A2A Receptor Antagonists. J. Med. Chem. 2010, 53, 1799–1809. 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldenhuys W. J.; Darvesh A. S.; Funk M. O.; Van der Schyf C. J.; Carroll R. T. Identification of Novel Monoamine Oxidase B Inhibitors by Structure-Based Virtual Screening. Bioorg. Med. Chem. Lett. 2010, 20, 5295–5298. 10.1016/j.bmcl.2010.06.128. [DOI] [PubMed] [Google Scholar]
- Sun B.; Bachhawat P.; Chu M. L.-H.; Wood M.; Ceska T.; Sands Z. A.; Mercier J.; Lebon F.; Kobilka T. S.; Kobilka B. K. Crystal Structure of the Adenosine A2A Receptor Bound to an Antagonist Reveals a Potential Allosteric Pocket. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 2066–2071. 10.1073/pnas.1621423114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barelier S.; Sterling T.; O’Meara M. J.; Shoichet B. K. The Recognition of Identical Ligands by Unrelated Proteins. ACS Chem. Biol. 2015, 10, 2772–2784. 10.1021/acschembio.5b00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morphy R.; Kay C.; Rankovic Z. From Magic Bullets to Designed Multiple Ligands. Drug Discovery Today 2004, 9, 641–651. 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]
- Van Der Walt M. M.; Terre’Blanche G.; Petzer A.; Petzer J. P. The Adenosine Receptor Affinities and Monoamine Oxidase B Inhibitory Properties of Sulfanylphthalimide Analogues. Bioorg. Chem. 2015, 59, 117–123. 10.1016/j.bioorg.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Jörg M.; May L. T.; Mak F. S.; Lee K. C. K.; Miller N. D.; Scammells P. J.; Capuano B. Synthesis and Pharmacological Evaluation of Dual Acting Ligands Targeting the Adenosine A2A and Dopamine D2 Receptors for the Potential Treatment of Parkinson’s Disease. J. Med. Chem. 2015, 58, 718–738. 10.1021/jm501254d. [DOI] [PubMed] [Google Scholar]
- De Simone A.; Ruda G. F.; Albani C.; Tarozzo G.; Bandiera T.; Piomelli D.; Cavalli A.; Bottegoni G. Applying a Multitarget Rational Drug Design Strategy: The First Set of Modulators with Potent and Balanced Activity toward Dopamine D3 Receptor and Fatty Acid Amide Hydrolase. Chem. Commun. 2014, 50, 4904–4907. 10.1039/C4CC00967C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. Hierarchical Docking of Databases of Multiple Ligand Conformations. Curr. Top. Med. Chem. 2005, 5, 739–749. 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner S. J.; Kollman P. A.; Case D. A.; Singh U. C.; Ghio C.; Alagona G.; Profeta S.; Weiner P. A New Force Field for Molecular Mechanical Simulation of Nucleic Acids and Proteins. J. Am. Chem. Soc. 1984, 106, 765–784. 10.1021/ja00315a051. [DOI] [Google Scholar]
- Wang J. M.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Meng E. C.; Shoichet B. K.; Kuntz I. D. Automated Docking with Grid-based Energy Evaluation. J. Comput. Chem. 1992, 13, 505–524. 10.1002/jcc.540130412. [DOI] [Google Scholar]
- Nicholls A.; Honig B. A Rapid Finite Difference Algorithm, Utilizing Successive Over-relaxation to Solve the Poisson–Boltzmann Equation. J. Comput. Chem. 1991, 12, 435–445. 10.1002/jcc.540120405. [DOI] [Google Scholar]
- Konc J.; Janežič D. ProBiS: A Web Server for Detection of Structurally Similar Protein Binding Sites. Nucleic Acids Res. 2010, 38, W436–W440. 10.1093/nar/gkq479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.