

Abstract
Background
The SLC29A3 gene, encoding a nucleoside transporter protein, is found in intracellular membranes. Based on the literatures, mutations in this gene cause a wide range of clinical manifestations including H syndrome, pigmented hypertrichosis with insulin dependent diabetes, Faisalabad histiocytosis, and dysosteosclerosis. However, all these disorders with their different names and terminologies are actually the same entity termed H syndrome.
Case presentation
We report four GJB2 and GJB6 negative deaf patients from two Iranian related families who present the associated symptoms of SLC29A3-disorder. Whole Exome Sequencing (WES) using Next Generation Illumina Sequencing was used to enrich all exons of protein-coding genes as well as some other important genomic regions in one of studied patients. A novel homozygous frame-shift mutation c.307-308delTT (p.Phe103fs) in exon 3 of SLC29A3 gene was identified in a 35 years old man with profound hearing loss, camptodactyly, rheumatoid arthritis and delayed puberty without any skin changes, short stature and insulin dependent diabetes mellitus. The mutation found was also confirmed by Sanger sequencing in other studied patients and their healthy parents. In compared to proband, however the clinical manifestations of these patients were different, indicating variable expressivity of mutant SLC29A3 gene as well as possible involvement of other modifier genes.
Conclusion
The present study uncovered a rare novel homozygous frame-shift mutation c.307-308delTT in SLC29A3 gene of four related patients with various manifestation of SLC29A3-disorder. Such studies can help to conduct genetic counseling and subsequently, prenatal diagnosis more accurately for individuals at the high risk of these types of genetic disorders.
Keywords: SLC29A3, Frame-shift mutation, SLC29A3-disorder, Iran
Background
The SLC29A3 gene, also called ENT3 (equilibrative nucleoside transporter 3), encodes a nucleoside transporter protein. The ENT3 protein is found in intracellular membranes, especially in lysosomal and mitochondrial membranes [1, 2]. Because of wide spread biochemical roles of nucleoside molecules, any disruption in metabolism and trafficking of nucleosides could result in various abnormal phenotypes.
Based on the literatures, the mutations in the SLC29A3 gene cause a wide range of different clinical manifestations including H syndrome (defined by cardiac anomalies, camptodactyly, short stature, hypergonadotropic hypogonadism, hepatosplenomegaly, scrotal masses, growth retardation, and sensorineural hearing loss [3–7]), pigmented hypertrichosis with insulin dependent diabetes (PHID) (characterized by hyperpigmented and hypertrichotic skin, hepatosplenomegaly, hypogonadism, diabetes mellitus, comptodactyly, clinodactyly, chronic inflammatory syndrome, and growth retardation [8]), Faisalabad histiocytosis (FHC) (defined by deafness, growth retardation, hypogonadism, camptodactyly, and Rosai Dorfman disease [9, 10]), and dysosteosclerosis (DSS) [8, 11, 12] (characterized by hyperpigmented skin, frontal bossing, mid-face hypoplasia, short stature, sclerotic platyspondyly, otosclerosis, and compression of CNS and cranial nerves [11]). However, it is worth mentioning that all these disorders, with their different names and terminologies, are actually the same entity termed H syndrome. For this reason, their description as different entities is misleading. As outlined above, a number of phenotypes including patches and plaques of hyperpigmented and hypertrichotic skin, short stature, camptodactyly, hearing problems, myelofibrosis, hepatosplenomegaly, pulmonary stenosis, pericarditis, diabetes, and hypogonadism [7, 11, 13] are common among H syndrome affected patients but not all phenotypes are seen in all patients. In this report, we describe a novel homozygous frame-shift mutation in SLC29A3 gene and its related phenotypes in two Iranian related families.
Case presentation
The studied families are of Iranian origin located in Semnan province (central Iran). As shown in Fig. 1, four affected individuals are included in two related families (a brother and a sister from each family), with two common ancestors. All four patients were born at term, with normal health indicators and metrics, following an uneventful pregnancy. Their ages ranged between 35 and 46 years. The clinical and demographic data of the patients are summarized in Table 1. In addition, camptodactyly phenotype in patient III-5 is depicted in Fig. 2.
Fig. 1.

Pedigree and segregation analysis of the SLC29A3 gene mutation (c.307-308delTT). All four patients (III.2, III.5, III.8, and III.11) are homozygote mutants, whereas parents of proband (II.1 and II.2) are heterozygote. Furthermore, sequence analysis of the three normal siblings of the patients in two families revealed that two of them (III.1 and III.6) are heterozygote and one (III-9) is normal
Table 1.
Summary of clinical and demographic findings
| Family 1 | Family 2 | |||
|---|---|---|---|---|
| Patient III-2 | Patient III-5 | Patient III-8 | Patient III-11a | |
| Sex | Female | Male | Female | Male |
| Age (years) | 46 | 35 | 37 | 41 |
| Height (cm) | 170 | 178 | 167 | 180 |
| Age of onset (years)b | 15 | 6 | 7–8 | 5 |
| Hearing loss | Mild | Profound | Profound | Profound |
| Camptodactyly | + | + | + | + |
| ESR (mm/h) | 70–110 | 45 | No data | No data |
| Polyclonal gammopathy | Severe | Mild | Mild | – |
| Skin changes | No | No | No | No |
| IDDM | – | – | + | + |
| Arthropathy | + | + | + | + |
| Delayed puberty | Yes | Yes | No | No |
| CRP mg/dL | 30–90 | 34 | No data | No data |
| Rheumatoid arthritis | + | + | + | + |
| Lymph nodes | Generalized Lymphoadenopathy | No | No | No |
ESR erythrocyte sedimentation rate, IDDM Insulin dependent diabetes mellitus, CRP C reactive protein
a The patient III-11 died due to the diabetes
b For clinical manifestations
Fig. 2.
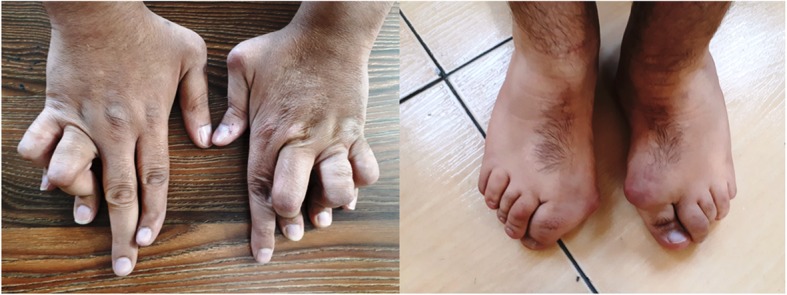
The hands and feet of patient III.5 which show camptodactyly phenotype
After obtaining informed consent, the peripheral blood samples were collected from patients and their family members. Genomic DNA was extracted from blood samples by QIAamp DNA Blood Mini Kit (Germany) according to the manufacturer’s instructions. At first, the patients were investigated for mutations of GJB2 gene using ASPCR (Allele Specific PCR) and sequencing followed by screening of two known deletions del(D13S1830) and del(D13S1854) of GJB6 gene. Since the studied patients were negative for GJB2 and GJB6 genes mutations, the patient III-5 was more investigated by Next Generation Sequencing. For this purpose, Whole Exome Sequencing (WES) was used to enrich all exons of protein-coding genes as well as some other important genomic regions. The WES was performed to sequence close to 100 million reads on Illumina HiSeq2000 Sequencer. Generally, the test platform examined > 95% of the targeted regions with sensitivity of above 99%. In this test, point mutations and micro-insertions/deletions as well as duplications (< 20 bp) can be simultaneously detected. Analysis of the sequencing results was performed using BWA aligner [14], annovar [15] and GATK [16] open access software as well as public databases ExAC, gnomAD, Kaviar (~Known VARiants) and GME. Additionally, ACMG guidelines and local population database with more than 1500 unrelated individuals (BayanGene) were used. Standard bioinformatics tools used were as follows: CADD_phred, REVEL, MCAP, SIFT, Polyphen, LRT, MutationTaster, and MutationAssessor. Furthermore, to confirm the novel mutation found in the SLC29A3 gene, PCR and sequencing were performed for patients and their normal family members using following primers: F-5′ CAGTCCATGGGCAGAAGTGT 3′ and R- 5′ TCGCCTACCTGTTGACAAGC 3′ (PCR product: 401 bp). Finally, the Sanger sequencing data was analyzed by Chromas software.
Based on the results, all four analyzed patients were negative for GJB2 mutations and two common large deletions of GJB6 gene. Subsequently, the NGS analysis of proband (patient III-5) revealed one deleterious homozygous mutation in SLC29A3 gene. This homozygous deletion includes two thymidine nucleotides (c.307-308delTT) of exon 3 causing a frame-shift mutation that immediately, makes a stop codon (TGA) instead of Phe103 codon (TTT) (SLC29A3:NM_001174098:exon3:c.307-308delTT:p.Phe103fs) (Fig. 3). The mutation found, validated by Sanger sequencing.
Fig. 3.

Schematic illustration for loss of 10 out of 11 transmembrane domains of ENT3 protein as a consequence of c.307-308delTT (p.Phen103fs) frame-shift mutation in SLC29A3 gene. a The normal sequence of the gene and the corresponding amino acid sequence have shown on the left. Two deleted thymidines are underlined. The mutated sequence and the corresponding truncated protein are depicted on the right. b Ten out of 11 transmembrane domains of ENT3 protein are deleted due to frame-shift mutation c.307-308delTT (p.Phen103fs)
At the next step, segregation analysis of novel homozygous mutation found was performed for other family members including another three patients (patients III-2, III-8, and III-11). As expected, parents of proband were heterozygote for reported mutation. A similar result was achieved from the parents of patient III-8 (data not shown). Furthermore, the obtained results showed that patients III-2, III-8, and III-11 have the found deleterious mutation. Furthermore, three normal siblings of the patients were also checked for the deleterious mutation 307-308delTT of SLC29A3 gene. The results clearly revealed that two of them (III-1 and III-6) are heterozygote and one (III-9) is normal homozygote. The corresponding family pedigree with the results of sequence analyses are illustrated in Fig. 1.
Discussion and conclusion
As previously mentioned, the mutation in SLC29A3 gene causes a wide range of mild to severe different medical manifestations which are known as the same entity termed H syndrome. To date, more than 22 pathogenic or likely pathogenic mutations have been reported in the SLC29A3 gene (Table 2). As shown in Table 2, the preliminary report for 308-309delTT (OMIM: 612373.0009) frame-shift mutation, which is resulted in truncated protein has been published in 2010 [10]. However, they found a heterozygous form of this mutation in combination with heterozygous missense mutation Gly437Arg as compound heterozygote, which is led to H syndrome. Here, we have explained the first report of novel homozygous form of this deleterious mutation from two Iranian related families. All four reported patients were results of consanguineous marriages. As previously pointed out, this frame-shift mutation is resulted in premature protein truncation. The truncated ENT3 protein loses its 10 out of 11 transmembrane domains (Fig. 3). Therefore, it seems that this homozygous mutation, as a loss of function mutation, leads to elimination of ENT3 protein function.
Table 2.
A summary of the reported pathogenic/likely pathogenic mutations of SLC29A3 gene
| Nucleotide change | Genotype | Consequence | (Clinical significance) Main Phenotypes | Family origin | Reference/ (variation ID in ClinVar) |
|---|---|---|---|---|---|
| c.243delA | Homo | p.Lys81Asnfs |
Pathogenic Nasal infiltration, Pancreatic exocrine deficiency, Insulin-dependent diabetes, Contractures of the fingers, Contractures of the toes, Retroperitoneal fibrosis |
Moroccan | [17] |
| c.308-309delTT | Hetero | p.Phe103Terfs |
(Pathogenic) Histiocytosis, Rosai-Dorfman disease |
Turkish and Palestinian | [10] |
| c.300 + 1G > A (IVS2 + 1G > A)a | Homo | Splice site | Pakistani | ||
| c.73C > T | – | p.Arg25Ter |
(Likely pathogenic) Histiocytosis-lymphadenopathy plus syndrome |
– | (ID: 212200) |
| c.300 + 1G > C | – | Splice site |
(Pathogenic) not provided |
(ID: 427021) | |
| c.347 T > G | Homo | p.Met116Arg |
(Pathogenic) insulin-dependent diabetes mellitus and pigmented hypertrichotic skin lesions |
Australian Lebanese | [8] |
| c.940delT | p.Tyr314ThrfsTer91 | Indian | |||
| c.1309G > A | p.Gly437Arg | Pakistani | |||
| c.1330G > T | p.Glu444Ter | North American Caucasian | |||
| c.1346C > G | p.Thr449Arg | Australian Lebanese | |||
| c.479G > A | – | p.Trp160Ter |
(Pathogenic) Histiocytosis-lymphadenopathy plus syndrome |
– | (ID: 573984) |
| c.607 T > C | Hetero | p.Ser203Pro |
(Pathogenic) Dysosteosclerosis |
– | [11] |
| c.1157G > A | p.Arg386Gln | ||||
| c.1346C > G | Homo | p.Thr449Arg | |||
| c.714_715invTG | – | p.Val239Ile |
(Likely pathogenic) Histiocytosis-lymphadenopathy plus syndrome |
– | (ID: 300363) |
| c.1001A > G | – | p.Asn334Ser |
(Likely pathogenic) Histiocytosis-lymphadenopathy plus syndrome |
– | (ID: 300368) |
| c.1045delC | Homo | p.Leu349Serfs |
(Pathogenic) Hyperpigmentation, Fixed flexion contractures of proximal interphalangeal joints, Hallux valgus, Prominent gynecomastia, histiocytic and dendritic infiltrate |
Bulgarian | [18] |
| c.1087C > T | p.Arg363Trp | Spanish | [4] | ||
| c.1088G > A | p.Arg363Gln | Arab | |||
| c.1228C > T | – | p.Gln410Ter |
(Pathogenic) Histiocytosis-lymphadenopathy plus syndrome |
– | (ID: 130338) |
| c.1279G > A | Hetero | p.Gly427Ser |
(Pathogenic) seronegative polyarthritis, hypogonadotropic hypogonadism, hearing loss, Proptosis, Arthropathy, Camptodactyly, Delayed puberty, Polyclonal gammopathy |
Arab | [7] |
| c.307-308delTT | Homo | p.Phe103Terfs |
(Pathogenic) Hearing loss, Camptodactyly, Polyclonal gammopathy, Arthropathy, Delayed puberty, Rheumatoid arthritis |
Iranian | This report |
aIVS InterVening Sequence (i.e. an intron)
As shown in Table 1, some of the common manifestations have been seen in our patients. Nonetheless, some of the phenotypes have not seen or were different among patients of two studied families. For example, although the short stature is reported in mostly reports for H syndrome [7, 11, 13], all four patients in this report have normal height (167–180 cm), in accordance with two previously reported cases from Morocco [17]. Similarly, and in accordance with some reports [8, 12], all patients in this study have moderate (patient III-2) to profound deafness (patients III-5, III-8, and III-11). Deafness has also been reported in other previous reports [4, 18], which is in accordance with our observations. Although these reported patients showed other manifestations including, hyperpigmentation, hypertrichosis, fixed flexion contractures of proximal interphalangeal joints, Hallux valgus and fixed flexion contractures of toe joints. However, the hearing loss has not been reported in originally reported PHID (other form of H syndrome) patients [19, 20]. Delayed puberty was observed in patients of family 1 (Patients III-2 and III-5). This observation is in line with those reported by Cliffe et al. (2009) and Hussain et al. (2009) [8, 19].
In accordance with other reports [7, 11], all four studied patients were suffered from camptodactyly. While the normal ESR range is 0–22 mm/h for men and 0–29 mm/h for women, a high ESR was observed in two patients III-2 and III-5. On the other words, it seems that the high levels of CRP in patients III-2 and III-5 could because of rheumatoid arthritis in these patients (normal CRP level is below 3.0 mg/L). Although, there was no data for ESR and CRP levels of patients III-8 and III-11, clinical symptoms of the rheumatoid arthritis were also confirmed by a rheumatologist in these patients. This result is in agreement with clinical symptoms of a previously reported patient [7]. While, the severe polyclonal gammopathy was observed in patient III-2, a mild representation was observed in patients III-5 and III-8 indicating variable expressivity of mutant SLC29A3 gene. Polyclonal gammopathy could be due to the elevated production of immunoglobulins by plasma cells [21]. Unlike the monoclonal gammopathy, polyclonal gammopathy is associated with a nonmalignant condition. In the present study, patient III-2 has been shown generalized lymphadenopathy. This observation is similar to previously reported lymphadenopathy in mutated SLC29A3 patients [5, 7, 8, 10, 11, 20]. Corresponding to the previously report from Morocco [17], despite the hyperpigmented/hypertrichotic skin is the common feature in SLC29A3 mutants [11], none of the patients reported in this paper have skin changes. Similarly, although mutation in SLC29A3 gene mostly leads to diabetes [7, 11, 22], we observed this phenotype only in the patients of the family 2 (patients III-8 and III-11) showing the variable expressivity and eventually involvement of other genes. Similar observation has been conducted by Spiegel et al. (2010) [7]. All four patients were suffered from arthropathy and rheumatoid arthritis. This is in accordance with several previously reported cases [7, 23].
In conclusion, this report shows that a homozygous frame-shift mutation c.307-308delTT results in a complex of clinical manifestations of H syndrome. In addition to allelic heterogeneity, it seems that these various clinical symptoms clearly confirm the pleiotropic effects and variable expressivity of SLC29A3 gene as well as possible involvement of other genes. On the other words, the obtained results showed that whole-exome sequencing is a useful method discovering genes involved in human genetic diseases.
Acknowledgments
We are grateful to patients and their families who kindly consented to join the study. We thank Dr. Masoumeh Heshmati and Dr. Nooshin Barikrow (from Pharmaceutical Sciences Research Center, Tehran medical Sciences, Islamic Azad University, Tehran, Iran) for technical collaborations, as well.
Abbreviations
- ASPCR
Allele Specific PCR
- CRP
C reactive protein
- DSS
Dysosteosclerosis
- ENT3
Equilibrative nucleoside transporter 3
- ESR
Erythrocyte sedimentation rate
- FHC
Faisalabad histiocytosis
- GJB2
Gap junction beta-2 protein
- GJB6
Gap Junction Beta 6 protein
- IDDM
Insulin dependent diabetes mellitus
- NGS
Next generation sequencing
- PHID
Pigmented hypertrichosis with insulin dependent diabetes
- SHML
Histiocytosis with massive lymphadenopathy
- SLC29A3
Solute Carrier Family 29 Member 3
- WES
Whole Exome Sequencing
Authors’ contributions
SN, SB, and TT, performed experimental assays; FP, organized this study, reviewed clinical and laboratory data, and finalized this manuscript. MF, performed patient record management; HF, performed data analysis, literature review and drafted the manuscript. All authors read and approved the final manuscript.
Funding
No funding was obtained for this study.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Ethics approval and consent to participate
This study had been approved by the ethics committee of the pharmaceutical sciences branch of Islamic Azad University, Tehran, Iran. Written informed consent was obtained from the parents to take part in this study and for possible publication. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sadaf Noavar, Email: noavarsadaf@yahoo.com.
Samira Behroozi, Email: behroozisamira@yahoo.com.
Taraneh Tatarcheh, Email: taraneh.tatarcheh@gmail.com.
Farshid Parvini, Phone: +98-2331533197, Phone: +98-2333655410, Email: f.parvini@semnan.ac.ir.
Majid Foroutan, Email: dr_forotan@semums.ac.ir.
Hossein Fahimi, Phone: +98-2122248282, Email: h.fahimi@iaups.ac.ir.
References
- 1.Endo Y, Obata T, Murata D, Ito M, Sakamoto K, Fukushima M, et al. Cellular localization and functional characterization of the equilibrative nucleoside transporters of antitumor nucleosides. Cancer Sci. 2007;98(10):1633–1637. doi: 10.1111/j.1349-7006.2007.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elwi AN, Damaraju VL, Baldwin SA, Young JD, Sawyer MB, Cass CE. Renal nucleoside transporters: physiological and clinical implications. Biochem Cell Biol. 2006;84(6):844–858. doi: 10.1139/o06-198. [DOI] [PubMed] [Google Scholar]
- 3.Molho-Pessach V, Agha Z, Aamar S, Glaser B, Doviner V, Hiller N, et al. The H syndrome: a genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol. 2008;59(1):79–85. doi: 10.1016/j.jaad.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 4.Molho-Pessach V, Suarez J, Perrin C, Chiaverini C, Doviner V, Tristan-Clavijo E, et al. The H syndrome: two novel mutations affecting the same amino acid residue of hENT3. J Dermatol Sci. 2010;57(1):59–61. doi: 10.1016/j.jdermsci.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Kismet E, KöseoĞlu V, Atay AA, Devecİ S, Demİrkaya E, Tuncer K. Sinus histiocytosis with massive lymphadenopathy in three brothers. Pediatr Int. 2005;47(4):473–476. doi: 10.1111/j.1442-200x.2005.02096.x. [DOI] [PubMed] [Google Scholar]
- 6.Broshtilova V, Ramot Y, Molho-Pessach V, Zlotogorski A. Diabetes mellitus may be the earliest and sole manifestation of the H syndrome. Diabet Med. 2009;26(11):1179–1180. doi: 10.1111/j.1464-5491.2009.02843.x. [DOI] [PubMed] [Google Scholar]
- 7.Spiegel R, Cliffe ST, Buckley MF, Crow YJ, Urquhart J, Horovitz Y, et al. Expanding the clinical spectrum of SLC29A3 gene defects. Eur J Med Genetics. 2010;53(5):309–313. doi: 10.1016/j.ejmg.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 8.Cliffe ST, Kramer JM, Hussain K, Robben JH, de Jong EK, de Brouwer AP, et al. SLC29A3 gene is mutated in pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome and interacts with the insulin signaling pathway. Hum Mol Genet. 2009;18(12):2257–2265. doi: 10.1093/hmg/ddp161. [DOI] [PubMed] [Google Scholar]
- 9.Rossbach HC, Dalence C, Wynn T, Tebbi C. Faisalabad histiocytosis mimics Rosai-Dorfman disease: brothers with lymphadenopathy, intrauterine fractures, short stature, and sensorineural deafness. Pediatr Blood Cancer. 2006;47(5):629–632. doi: 10.1002/pbc.20605. [DOI] [PubMed] [Google Scholar]
- 10.Morgan NV, Morris MR, Cangul H, Gleeson D, Straatman-Iwanowska A, Davies N, et al. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 2010;6(2):e1000833. doi: 10.1371/journal.pgen.1000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campeau PM, Lu JT, Sule G, Jiang M-M, Bae Y, Madan S, et al. Whole-exome sequencing identifies mutations in the nucleoside transporter gene SLC29A3 in dysosteosclerosis, a form of osteopetrosis. Hum Mol Genet. 2012;21(22):4904–4909. doi: 10.1093/hmg/dds326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonard L, Couloigner V, Pierrot S, Louha M, Gherbi S, Denoyelle F, et al. Progressive hearing loss associated with a unique cervical node due to a homozygous SLC29A3 mutation: a very mild phenotype. Eur J Med Genetics. 2012;55(1):56–58. doi: 10.1016/j.ejmg.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Melki I., Lambot K., Jonard L., Couloigner V., Quartier P., Neven B., Bader-Meunier B. Mutation in the SLC29A3 Gene: A New Cause of a Monogenic, Autoinflammatory Condition. PEDIATRICS. 2013;131(4):e1308–e1313. doi: 10.1542/peds.2012-2255. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with burrows–wheeler transform. bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164–e16e. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolze A, Abhyankar A, Grant AV, Patel B, Yadav R, Byun M, et al. A mild form of SLC29A3 disorder: a frameshift deletion leads to the paradoxical translation of an otherwise noncoding mRNA splice variant. PLoS One. 2012;7(1):e29708. doi: 10.1371/journal.pone.0029708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Libdeh AA, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet. 2008;83(4):529–534. doi: 10.1016/j.ajhg.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussain K, Padidela R, Kapoor RR, James C, Banerjee K, Harper J, et al. Diabetes mellitus, exocrine pancreatic deficiency, hypertrichosis, hyperpigmentation, and chronic inflammation: confirmation of a syndrome. Pediatr Diabetes. 2009;10(3):193–197. doi: 10.1111/j.1399-5448.2008.00470.x. [DOI] [PubMed] [Google Scholar]
- 20.Prendiville J, Rogers M, Kan A, Fd C, Wong M, Junker A, et al. Pigmented hypertrichotic dermatosis and insulin dependent diabetes: manifestations of a unique genetic disorder? Pediatr Dermatol. 2007;24(2):101–107. doi: 10.1111/j.1525-1470.2007.00352.x. [DOI] [PubMed] [Google Scholar]
- 21.Dispenzieri A, Gertz MA, Therneau TM, Kyle RA, editors. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc. 2001;76(5):476–87. [DOI] [PubMed]
- 22.Molho-Pessach V, Ramot Y, Camille F, Doviner V, Babay S, Luis SJ, et al. H syndrome: the first 79 patients. J Am Acad Dermatol. 2014;70(1):80–88. doi: 10.1016/j.jaad.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 23.Bloom JL, Lin C, Imundo L, Guthery S, Stepenaskie S, Galambos C, et al. H syndrome: 5 new cases from the United States with novel features and responses to therapy. Pediatr Rheumatol. 2017;15(1):76. doi: 10.1186/s12969-017-0204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.