

Abstract
Purpose
Previously we showed that AAV5-mediated expression of either human M- or L-opsin promoted regrowth of cone outer segments and rescued M-cone function in the treated M-opsin knockout (Opn1mw−/−) dorsal retina. In this study, we determined cone viability and window of treatability in aged Opn1mw−/− mice.
Methods
Cone viability was assessed with antibody against cone arrestin and peanut agglutinin (PNA) staining. The rate of cone degeneration in Opn1mw−/− mice was quantified by PNA staining. AAV5 vector expressing human L-opsin was injected subretinally into one eye of Opn1mw−/− mice at 1, 7, and 15 months old, while the contralateral eyes served as controls. M-cone–mediated retinal function was analyzed 2 and 13 months postinjection by full-field ERG. L-opsin transgene expression and cone outer segment structure were examined by immunohistochemistry.
Results
We showed that dorsal M-opsin dominant cones exhibit outer segment degeneration at an early age in Opn1mw−/− mice, whereas ventral S-opsin dominant cones were normal. The remaining M-opsin dominant cones remained viable for at least 15 months, albeit having shortened or no outer segments. We also showed that AAV5-mediated expression of human L-opsin was still able to rescue function and outer segment structure in the remaining M-opsin dominant cones when treatment was initiated at 15 months of age.
Conclusions
Our results showing that the remaining M-opsin dominant cones in aged Opn1mw−/− mice can still be rescued by gene therapy is helpful for establishing the window of treatability in future blue cone monochromacy clinical trials.
Keywords: blue cone monochromacy, gene therapy, opsins, X-linked genetic disease, AAV
Blue cone monochromacy (BCM) is an X-linked visual disorder characterized by impaired function of both red (L, long wavelength) and green (M, middle wavelength) cone photoreceptors due to mutations in the OPN1LW/OPN1MW gene cluster on the X-chromosome.1–5 The prevalence of BCM is approximately 1 in 100,000. Because only rods and S-cones subserve visual function in these patients, affected males typically present at birth/early infancy with severely impaired color discrimination, and reduced visual acuity that may progress to 20/200, myopia, pendular nystagmus, and photoaversion.6,7
Studies of the molecular basis of BCM have revealed two major mechanisms that lead to L- and M-cone photoreceptor dysfunction3–5: (1) partial or complete deletion of the locus control region (LCR) abolishes transcription of the OPN1LW/OPN1MW gene array (the LCR is located upstream of the OPN1LW/OPN1MW gene array and controls their transcription, ensuring only one opsin gene is expressed in any one cone photoreceptor5,8,9); or (2) presence of a deleterious C203R missense mutation either in a single OPN1LW/MW hybrid gene or in multiple OPN1LW/MW genes. Studies using adaptive optics scanning laser ophthalmoscopy showed that both genotypes of BCM patients demonstrate a disrupted cone mosaic with a reduced number of cones in the fovea early in life.10–13 Both genotypes also show a trend toward increased thinning of the foveal outer nuclear layer in older patients. Patients with the C203R mutation, however, appear to have a less aggressive disease progression than patients with the LCR deletion. They have decades-longer retention of the foveal outer nuclear layer and a slower rate of developing inner segment/outer segment defects, suggesting that there is a much wider window of opportunity for applying treatment to this BCM subset.13 These studies also provide convincing evidence that although cone photoreceptor cells are degenerated from early life in both genotypes, there are sufficient numbers of remaining cones in the central retina that retain sufficient residual structure and presumably viability to serve as feasible targets for gene therapy.
In mice, most cones coexpress M- and S-opsin, but in a dorsal-ventral gradient, with M-opsin dominant in the dorsal retina and S-opsin dominant in the ventral.14–18 For simplicity, in this article we used the terminology M-cones for dorsal cones predominately expressing M-opsin and no S-opsin was detected by immunohistochemistry, and S-cones for ventral cones predominately expressing S-opsin and no M-opsin was detected by immunohistochemistry. Previously we showed that the dorsal M-cones of Opn1mw−/− mice do not form cone outer segments, very much like the remaining cones in human BCM fovea with shortened outer segments; therefore, Opn1mw−/− mice can be used as a model for BCM. We also showed that AAV-mediated expression of either human OPN1LW or OPN1MW in Opn1mw−/− cones rescued M-cone function and promoted regeneration of cone outer segments in the dorsal retina.19,20 In the present study, we characterized the numbers of viable cones in dorsal and ventral retinas in young Opn1mw−/− mice and their rate of degeneration with age. We also assessed whether AAV-mediated delivery of human L-opsin can still rescue dorsal M-cones in aged Opn1mw−/− mice and the longevity of that rescue effect. These results provide important information on the age window for gene therapy in future clinical trials. We have used human L-opsin in this study because we believe this is a better choice of vector for future BCM clinical studies. We think it is best to treat BCM patients with only one opsin to minimize biologic variables and simplify the interpretation of study results, at least initially. L-opsin might be a better choice than M-opsin because when added to the endogenous S-cone frequencies, L-opsin response spectrum to light frequencies would give rise to a wider spectrum of perceivable frequencies than an M-opsin plus S-opsin combination.
Methods
Animals
All mice used in this study were maintained in the University of Florida Health Science Center Animal Care Service Facilities on a 12-hour/12-hour light/dark cycle. All animals were maintained under standard laboratory conditions (18°C–23°C, 40%–65% humidity) with food and water available ad libitum. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Florida and conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and National Institutes of Health regulations. The generation and characterization of Opn1mw−/− mice has been described previously.20
AAV Vector
The AAV vector expressing human OPN1LW driven by PR2.1 promoter (PR2.1-hOPN1LW) was described previously.21 This vector was packaged in serotype 5 AAV and was purified according to previously published method.22
Subretinal Injection
Eyes of Opn1mw−/− mice were dilated with Tropi-Phen (phenylephrine HCL 2.5%, Tropicamide 1% Ophthalmic Solution; Pine Pharmaceuticals, Tonawanda, NY, USA) 15 minutes before injection. Transcorneal subretinal injections were performed with a 33-gauge blunt-end needle attached on a 5-mL Hamilton syringe. First, an entering puncture was introduced at the edge of the cornea with a 30-gauge disposable needle, then 1 μL of viral vector mixed with fluorescein dye (0.01% final concentration) was injected through the corneal opening and delivered into the subretinal space as described before.23,24 An injection was considered successful if it detached at least 80% of the retina visualized by the fluorescence bleb that was monitored by video camera attached to the injection scope allowing real-time assessment of surgical procedures. Atropine eye drops (Akorn, Inc., Lake Forest, IL, USA) and neomycin/polymyxin B/dexamethasone ophthalmic ointment (Bausch & Lomb, Inc., Tampa, FL, USA) were applied after injection. One eye was injected, whereas the contralateral eye was uninjected and served as a control; 5 × 109 vector genomes (vg) in 1 μL was injected. Antisedan (Orion Corporation, Espoo, Finland) at 1 mg/kg was given intramuscularly following injection as an anesthetic reversal. We performed AAV5-GFP injections in Opn1mw−/− mice in our previous study20 and observed no adverse effect.
Electroretinography
M-cone ERG responses were analyzed using a UTAS Visual Diagnostic System with a Big Shot Ganzfeld dome (LKC Technologies, Gaithersburg, MD, USA). All mice were anesthetized with an intraperitoneal injection of ketamine (72 mg/kg)/xylazine (4 mg/kg). The pupils were dilated with Tropi-Phen. Mice were first exposed to white light for 5 minutes at 30 cd/m2 to suppress rod function. Then M-cone ERGs were recorded by stimulation of the green channel middle-wavelength light (510 nm) at intensities of −0.6, 0.4, and 1.4 log cd.s/m2. Twenty-five recordings were averaged for each of the three light intensities. ERG data were presented as average ± SEM. Seven mice were recorded for each treatment group. Statistical analysis was performed by 1-way ANOVA with the Bonferroni post hoc test to compare each treatment group with untreated control group. Significance was defined as a P value <0.01.
Our ERG machine has a 510-nm filter for detection of mouse M-cone. From our previous study,19 we found that the 510-nm filter can still detect human L-opsin delivered by AAV in Opn1mw−/− eyes because of the spectrum overlap between human L-opsin and mouse M-opsin. All animals were light-adapted before ERG to suppress any rod responses and we never detected any responses from contralateral untreated Opn1mw−/− eyes under photopic conditions when we performed M-cone ERG.
Preparation of Arrestin4 Monoclonal Antibody
Poly(A)+ RNA was isolated from C57/BL6J mouse retinas (QuickPrep mRNA isolation kit; GE Life Sciences, Chicago, IL, USA) and reverse transcribed, priming with oligo(dT) (First-strand cDNA synthesis kit; GE Life Sciences). Arrestin4 cDNA was amplified from this cDNA using oligonucleotides specific to the 5′ and 3′ ends of the open reading frame murine arrestin4 cDNA, incorporating EcoRI restriction sites before the initiating methionine codon and after the stop codon. The cDNA was cloned into pET-28a (Novagen, Madison, WI, USA) to incorporate a His tag on the amino terminus. Arrestin4 protein was expressed in BL21(DE3) Escherichia coli in the presence of 30 μM IPTG. The protein was purified by nickel affinity chromatography (His GraviTrap; GE Life Sciences) in 50 mM sodium phosphate (pH 8.0), 300 mM NaCl, 10 mM imidazole, and 6 M guanidine hydrochloride and then dialyzed in PBS. The arrestin4 protein was used to immunize BALB/c mice (Jackson Laboratory, Bar Harbor, ME, USA), as previously described.25 Hybridoma cells lines prepared from the splenocytes from the immunized mice were characterized by immunoblot and immunohistochemistry to identify monoclonal antibodies that were specific for arrestin4.
Preparation of Retinal Whole Mounts and PNA Staining
Mice were humanely euthanized, the eyes were marked at 12 o'clock on the cornea with a burn marker and then enucleated and fixed with 4% paraformaldehyde in PBS for 1 hour. The cornea and lens were then removed. To make flat mounts, the entire retina was carefully dissected from the eyecup and radial cuts were made from the edges to the equator of the retina. Retinal flat mounts were blocked in 3% BSA for 2 hours, then labeled with Biotinylated Peanut Agglutinin (PNA) (Vector Laboratories, Burlingame, CA, USA) and S-opsin antibody (MilliporeSigma, Burlington, MA, USA) in PBS overnight. The flat mounts were then washed with PBS three times, followed by incubating with Fluorescein Avidin D (Vector Laboratories) and immunoglobulin (Ig)G secondary antibody tagged with Alexa-594 (Molecular Probes, Eugene OR, USA) diluted 1:500 in PBS at room temperature for 2 hours, then washed with PBS. The flat mounts were flattened on slides with a fine brush, one drop of Vectashield Mounting Medium for Fluorescence (H-1400; Vector Laboratories) was applied and then covered with a coverslip. The flat mounts were imaged with a Leica (Wetzlar, Germany) Fluorescence Microscope LAS X Widefield System. Four images were taken from each flat mount, two from dorsal area and two from the ventral area, marked by a burn mark. PNA-positive cells from each image were counted in an area that is equivalent to 0.01 mm2 of retina using the counting tool in Adobe Photoshop (Adobe Systems, Inc., San Jose, CA, USA). Counts of 12 images from six different mice (three males, three females) were average for dorsal and ventral regions, and the SD was calculated. Statistical analysis was performed by one-way ANOVA with Bonferroni posttests to compare the difference among each age group for dorsal or ventral regions. Significance was defined as P < 0.05.
Frozen Retinal Section Preparation and Immunohistochemistry
Mice were light-adapted at 2000 lux under white fluorescent light for 1 hour to facilitate cone arrestin (CAR) translocation to cone outer segments.26 They were then killed immediately and their eyes marked at 12 o'clock on the cornea with a burn marker and enucleated. The eyes were fixed in 4% paraformaldehyde for 30 minutes then the cornea and lens were removed without disturbing the retina. The retinas were further fixed for additional 2 to 3 hours at room temperature. The eyecups were then rinsed with PBS and cryoprotected with 30% sucrose/PBS for 3 hours at room temperature, then embedded in cryostat compound (Tissue TEK OCT; Sakura Finetek USA, Inc., Torrance, CA, USA) and frozen at –80°C. Retinas were cut perpendicularly from dorsal to ventral at 12-μm thickness. For immunohistochemistry, retinal sections were rinsed in PBS and blocked in 3% BSA, 0.3% Triton X-100 in PBS for 1 hour at room temperature. Sections were then incubated with primary antibodies at room temperature overnight. The following primary antibodies were used: anti-L/M-opsin, anti-S-opsin (both from MilliporeSigma), cone arrestin, PDE6γ' (kindly provided by Bernd Wissinger, PhD). After incubation with first antibody, the slides were washed with PBS three times, followed by incubating with IgG secondary antibody tagged with Alexa-594 or Alexa-488 (Molecular Probes) diluted 1:500 in PBS at room temperature for 2 hours, then washed with PBS. Sections were mounted with Vectashield Mounting Medium for Fluorescence (H-1400; Vector Laboratories) and cover slipped. Sections were imaged with a Leica Fluorescence Microscope LAS X Widefield System.
CAR staining was performed in two 15-month-old, and two 20-month-old Opn1mw−/− mice.
Results
Rate of Cone Degeneration in Opn1mw−/− Mice
Previously we showed that the dorsal M-cones of 2-month-old Opn1mw−/− mice had significantly shortened outer segments; however, their cone sheaths were detectible by PNA staining, indicating that these cones remain viable.19 We also showed that ventral retinas (dominated by S-cones) of Opn1mw−/− mice had normal cone structure. Here we stained retinal sections of 15-month-old Opn1mw−/− mice with an antibody against cone arrestin. Cone arrestin staining was defuse throughout the cone photoreceptors from the synaptic terminals to the cone outer segments,26 and is another marker for assessing cone viability. We found that in aged Opn1mw−/− dorsal retinas, cone arrestin staining was still present in synaptic terminals, axons, cell bodies, and inner segments, an indication that these cones are viable. Similar to what we observed before, these dorsal cones had shortened outer segments compared with their ventral counterparts (Fig. 1A). An image taken from dorsal-ventral transition zone showed that cone arrestin staining extended from synaptic terminals into outer segments in ventral S-opsin dominant cones, whereas staining stopped at inner segments in the dorsal region where S-opsin was not detected (Fig. 1B).
Figure 1.

(A) Fluorescent images of cryosections from dorsal and ventral retinas in a 15-month-old Opn1mw−/− mouse stained for cone arrestin antibody. Staining in the cone synapses, axons, cell bodies, and inner segments in the dorsal retina was still present, indicating dorsal cones were viable. (B) An image taken from dorsal-ventral transition zone of a 15-month-old Opn1mw−/− mouse showed that cone arrestin staining (red) in the dorsal area (left side) was similar to its counterparts in the ventral area (right side), except in the dorsal area staining stopped at the distal end of inner segments, whereas staining extended into outer segments in the ventral area colocalizing with S-opsin staining (green). Left and right panels were the same images except the right shows S-opsin staining. Four mice were stained for CAR antibody. ONL, outer nuclear layer; OPL, outer plexiform layer; IS, inner segments; OS, outer segments. Scale bar: 20 μm.
Next we characterized the number of viable cones in dorsal and ventral retinas in Opn1mw−/− mice of different ages by labeling retinal flat mounts with PNA at 21 days, 3 months, 11 months, and 15 months (Fig. 2A). In the wild-type controls, we counted 100 to 140 cones per 0.01 mm−2 of retina, similar to a previous reported cone density of 1.2 ± 0.2 × 104 mm−2.14,16,27 We found no statistical difference in cone densities between young and aged wild-type mice (Fig. 2C) (n = 6, P > 0.05 for both dorsal and ventral), also consistent with a previous report.28 We show here that at postnatal day 21 (P21), Opn1mw−/− mice had similar numbers of viable cones in both dorsal and ventral retinas as in the 3-month-old wild-type controls (n = 6, P > 0.05 for both dorsal and ventral) (Fig. 2C). However, dorsal M-cones in Opn1mw−/− mice degenerated rapidly. At 3 months of age, Opn1mw−/− dorsal retinas had retained only ∼70% viable M-cones compared with age-matched wild-type controls (n = 6, P < 0.01), whereas the density of ventral S-cones was not significantly different from wild-type controls (P > 0.05). Dorsal cones further degenerated with age, with 11- and 15-month-old Opn1mw−/− dorsal retinas containing only ∼50% of viable M-cones compared with the age-matched wild-type controls (n = 6, P < 0.0001), whereas the numbers of ventral S-cones were similar to their age-matched wild-type controls (P > 0.05).
Figure 2.

The time course of cone degeneration in Opn1mw−/− mice. (A) Representative images of retinal flat mounts taken from dorsal and ventral retinas of Opn1mw−/− mice and wild-type controls at different ages. Retinal whole mounts were stained with S-opsin antibody (red) and PNA (green). S-opsin is primarily detected in the ventral retina. (B) A representative image of a flat-mounted 3-month-old Opn1mw−/− retina with overlaid squares indicating regions analyzed for cone densities. (C) Quantification of PNA staining from dorsal and ventral retinas of Opn1mw−/− mice at different ages and wild-type controls. Each bar represents PNA-positive signals counted in 0.01 mm2 of surface area of retina from 12 images taken from six different mice. Data show average ± SD. *P < 0.01. ns, not significant.
Expression of Human L-opsin Restores M-cone Function in Aged Opn1mw−/− Mice
Consistent with above observation, AAV-mediated expression of human L-opsin rescued M-cone function in aged Opn1mw−/− mice. Previously we showed that AAV5-mediated expression of either human M- or L-opsin promoted regrowth of cone outer segments and rescued M-cone function when delivered in 1-month-old M-opsin knockout (Opn1mw−/−) mice.19 Here we show that subretinal injection of AAV vector expressing human OPN1LW driven by the cone-specific promoter PR2.1 rescued ERG responses to M-cone preferred middle-wavelength light stimuli in 15-month-old Opn1mw−/− mice (Fig. 3). ERG responses in treated aged mice were lower than those in treated younger mice, consistent with the lesser number of viable dorsal cones in aged mice; however, significant ERG function was preserved in the aged mice, even in mice treated at 15 months. The averaged maximum M-cone b-wave amplitude in Opn1mw−/− mice treated at 1 month of age and examined for ERG function at 2 months postinjection (P1M +2M) was 96 ± 4.3 μV; b-wave amplitudes in Opn1mw−/− mice at P7M + 2M, P7M +13M, and P15M + 2M were 72 ± 7.6 μV, 43 ± 4.2 μV, and 53 ± 4.3 μV, respectively (average ± SEM, n = 7). ERG rescue in all cases was significantly higher than the unrecordable ERGs from untreated contralateral control eyes (P < 0.001).
Figure 3.
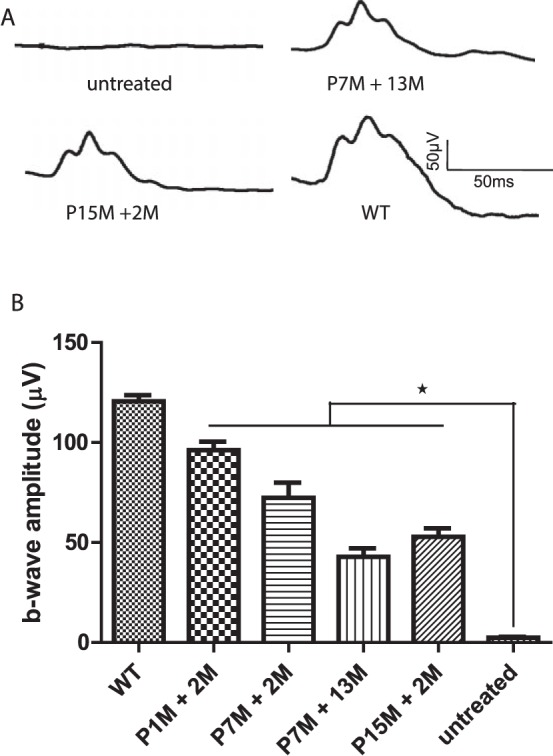
(A) Representative ERG traces from an untreated Opn1mw−/− mouse (17 months old), Opn1mw−/− mouse treated at 7 months of age, and receiving ERG at 13 months postinjection (P7M +13M), Opn1mw−/− mouse treated at 15 months of age and receiving ERG at 2 months postinjection (P15M +2M), and wild-type control (WT, 17-month-old). (B) Averaged M-cone ERG responses in Opn1mw−/− mice treated at different ages. Untreated Opn1mw−/− mice (17 months old). Each bar represents the mean ± SEM of M-cone b-wave amplitudes recorded at 1.4 log cd.s/m2 (n = 7 for each group, *P < 0.001). P1M + 2M, treated at 1 month of age and receiving ERG at 2 months postinjection; P7M + 2M, treated at 7 months of age and receiving ERG at 2 months postinjection; P7M + 13M, treated at 7 months of age and receiving ERG at 13 months postinjection; P15M + 2M, treated at 15 months of age and receiving ERG at 2 months postinjection; WT, wild-type controls (17-month-old).
Treatment Restores Outer Segments in Dorsal Cones of Aged Opn1mw−/− Mice
AAV-mediated expression of OPN1LW in treated Opn1mw−/− mice is expressed in both dorsal and ventral retinas. To confirm that the cone outer segment structure was regenerated in treated dorsal cones where M-opsin normally dominates, but is missing in Opn1mw−/− mice, we performed immunohistochemistry using antibodies against proteins specifically expressed in cone outer segments. First we showed that the AAV-delivered OPN1LW was expressed in the treated Opn1mw−/− dorsal retinas. A representative image of a retinal whole mount from an Opn1mw−/− mouse injected at 15 months of age and killed 2 months postinjection showed that OPN1LW protein was detected in the dorsal retina and localized to the distal tip of PNA staining, whereas mouse OPN1MW was not expressed in the untreated Opn1mw−/− dorsal retina (Fig. 4). Low-resolution retinal cross sections from these mice showed that vectored OPN1LW was expressed across the entire retina in both dorsal and ventral hemispheres. (Fig. 5A). In the ventral retina, OPN1LW was colocalized with endogenous S-opsin. In the dorsal retina, OPN1LW was localized in the cone outer segments, confirming regeneration of cone outer segments (Fig. 5B).
Figure 4.
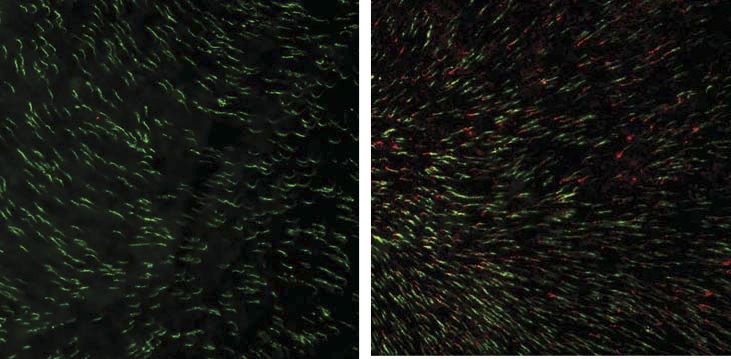
AAV-delivered OPN1LW is expressed in Opn1mw−/− dorsal cones when treated at 15 months of age and tested at 2 months postinjection. A representative image of an Opn1mw−/− retinal flat mount immunostained with OPN1LW (red) and PNA (green) showing that OPN1LW is expressed in dorsal retinal cones and localized to the tip of PNA staining in the treated eye (right). In contrast, no OPN1LW expression was detected in the untreated contralateral eye (left).
Figure 5.

Cryosections of AAV-mediated human OPN1LW expression in Opn1mw−/− mice treated at 15 months of age and tested at 2 months postinjection. (A) Retinal sections show that M-opsin is not expressed in the untreated retina, and was expressed across the entire retina in the treated eye. (B) Images taken from dorsal retinas of untreated and treated eyes showed that the OPN1LW is localized correctly in the outer segments after treatment. Scale bar: 20 μm.
We also stained treated Opn1mw−/− sections (PM15 + 2M) with antibody against cone phosphodiesterase γ' subunit (PDE6γ'), which is normally localized to cone outer segments. In untreated eyes, PDE6γ' staining was mislocalized to the inner segments of dorsal cones (Fig. 6A), whereas in the ventral retina it was colocalized with endogenous S-opsin in cone outer segments (Fig. 6B). After treatment, a significant portion of PDE6γ' staining was detected in cone outer segments in the dorsal retina (Fig. 6), suggesting reloading of PDE6γ' on cone outer segment regeneration.
Figure 6.

Cone outer segments are regenerated in treated aged Opn1mw−/− dorsal retinas. (A) In dorsal retinas, PDE6γ' staining (green) is primarily detected in cone inner segments in untreated Opn1mw−/− retinas. After treatment, PDE6γ' staining is primarily detected in cone outer segments. (B) In ventral retinas, PDE6γ' staining is colocalized with endogenous S-opsin (red) in cone outer segments in both untreated and treated Opn1mw−/− retinas. Scale bar: 20 μm.
We performed immunohistochemistry from three treated mice for each treatment group shown in Figure 3 and confirmed regeneration of cone outer segments in all cases. We noticed that it appeared that fewer numbers of dorsal cones were rescued in animals treated at older age than those treated at younger age, corresponding to reduced numbers of viable dorsal cones in aged Opn1mw−/− mice. We also performed immunohistochemistry from mice injected at 15 months of age and killed at 12 months postinjection, only a few dorsal cones were found to contain outer segments, suggesting that treatment effect was gradually lost over time when mice were treated at this advanced age.
Discussion
In this study, we characterized the degeneration rate of mutant M-cones in Opn1mw−/− mice and explored the window of treatability. We found that mutant M-cones already degenerated at early life and they continued to degenerate with age. Aged Opn1mw−/− mice (15 months old) had ∼50% of the viable dorsal M-cones compared with age-matched wild-type controls. These mutant M-cones, although having significantly shortened or no outer segments, remained viable and continued to express other essential cone outer segment proteins. We further demonstrated that delivery of AAV5 expressing human OPN1LW resulted in regenerated cone outer segments in the remaining M-cones in treated aged Opn1mw−/− mice. Moreover, other cone phototransduction proteins localized normally to the outer segments on regeneration, and treated M-cones responded appropriately to middle-wavelength light.
In previous studies on retinal structural changes in BCM human patients, it was found that there are a reduced number of cones early in life. Some patients showed evidence of progressive central retinal cone loss and macular atrophy at later stages. High-resolution retinal imaging in BCM patients confirmed the existence of inner segments as part of the remaining cone cells, albeit with significantly shortened but detectable outer segments. This evidence indicates that remaining mutant cones in human patients remain viable, suggesting potential value of gene therapy for BCM. We also detected degeneration of mutant dorsal cones early in young Opn1mw−/− mice, consistent with observations in BCM human patients. Our results confirm that gene therapy is still possible in older patients as long as they retain sufficient residual foveal cones.
There are two major mechanisms causing BCM: LCR deletion and C203R missense mutation. The Opn1mw−/− mouse model represents patients with LCR deletion. However, the C203R mutation might have a different disease mechanism because its corresponding mutation in rhodopsin (C187Y) causes early and severe autosomal dominant retinitis pigmentosa.29 Interestingly, a recent study in BCM patients comparing the phenotype caused by large deletion mutations with those having the C203R mutation showed that disease progression in patients with the C203R genotype appeared less aggressive than in their cohort of patients with deletion mutations.13 We are in the process of characterizing a mouse model representing the C203R mutation and performing proof-of-concept experiments.
In wild-type mice, the formation of the outer segments is complete at P21.30,31 We showed that at P21, Opn1mw−/− mice had a similar density of viable dorsal cones as in the 3-month old wild-type controls suggesting that differentiation and formation of M-cones was not affected initially in the absence of M-opsin. Mutant M-cones degenerated with age; however, the remaining cones, although having shortened or no outer segments, remained alive for a long time. This persistence is in sharp contrast to the fate of rod photoreceptors in rhodopsin knockout mice in which rods undergo rapid cell death.32,33 The hardiness of cones maybe explained by the fact that most cones coexpress M-opsin and S-opsin.14,16–18 It was shown that in the S-opsin knockout mouse, cones in the distal ventral can survive for at least 1.5 years.16 In addition, knocking-out S-opsin led to a higher level expression of M-opsin.16 Consistent with this, we also noticed higher S-opsin expression in the Opn1mw−/− mice.20 Therefore, it is likely that some S-opsin expression in the dorsal cones of Opn1mw−/− mice contributes to their survival.
In conclusion, our results show that the remaining M-cones in aged Opn1mw−/− mice can be rescued by cone opsin gene therapy and this has important implications for defining the time window for possibly similar therapies in humans. Our results also provide important information for patient selection criteria for future BCM clinical studies.
Acknowledgments
The authors thank Bernd Wissinger (professor of Molecular Genetics of Sensory Systems, Institute for Ophthalmic Research, Centre for Ophthalmology, Tuebingen, Germany) for kindly providing us with PDE6γ' antibody.
Supported by National Institutes of Health Grants EY021721 (WWH), EY08123 (WB), EY019298 (WB), and EY014800-039003 (National Eye Institute core grant to the Department of Ophthalmology, University of Utah); unrestricted grants to the Departments of Ophthalmology at the University of Florida and the University of Utah from Research to Prevent Blindness (RPB; New York); and an RPB Nelson Trust Award (WB); and by grants from BCM Families Foundation, Macula Vision Research Foundation, and AGTC Inc.
Disclosure: W.-T. Deng, AGTC Inc. (I); J. Li, None; P. Zhu, None; B. Freedman, None; W.C. Smith, None; W. Baehr, None; W.W. Hauswirth, AGTC Inc. (F, I, C, R), P
References
- 1.Buena-Atienza E, Ruther K, Baumann B, et al. De novo intrachromosomal gene conversion from OPN1MW to OPN1LW in the male germline results in Blue Cone Monochromacy. Sci Rep. 2016;6:28253. doi: 10.1038/srep28253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner JC, Michaelides M, Holder GE, et al. Blue cone monochromacy: causative mutations and associated phenotypes. Mol Vis. 2009;15:876–884. [PMC free article] [PubMed] [Google Scholar]
- 3.Kazmi MA, Sakmar TP, Ostrer H. Mutation of a conserved cysteine in the X-linked cone opsins causes color vision deficiencies by disrupting protein folding and stability. Invest Ophthalmol Vis Sci. 1997;38:1074–1081. [PubMed] [Google Scholar]
- 4.Nathans J, Davenport CM, Maumenee IH, et al. Molecular genetics of human blue cone monochromacy. Science. 1989;245:831–838. doi: 10.1126/science.2788922. [DOI] [PubMed] [Google Scholar]
- 5.Smallwood PM, Wang Y, Nathans J. Role of a locus control region in the mutually exclusive expression of human red and green cone pigment genes. Proc Natl Acad Sci U S A. 2002;99:1008–1011. doi: 10.1073/pnas.022629799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michaelides M, Johnson S, Simunovic MP, et al. Blue cone monochromatism: a phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye (Lond) 2005;19:2–10. doi: 10.1038/sj.eye.6701391. [DOI] [PubMed] [Google Scholar]
- 7.Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol. 2004;88:291–297. doi: 10.1136/bjo.2003.027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Macke JP, Merbs SL, et al. A locus control region adjacent to the human red and green visual pigment genes. Neuron. 1992;9:429–440. doi: 10.1016/0896-6273(92)90181-c. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi T, Motulsky AG, Deeb SS. Visual pigment gene structure and expression in human retinae. Hum Mol Genet. 1997;6:981–990. doi: 10.1093/hmg/6.7.981. [DOI] [PubMed] [Google Scholar]
- 10.Carroll J, Dubra A, Gardner JC, et al. The effect of cone opsin mutations on retinal structure and the integrity of the photoreceptor mosaic. Invest Ophthalmol Vis Sci. 2012;53:8006–8015. doi: 10.1167/iovs.12-11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll J, Rossi EA, Porter J, et al. Deletion of the X-linked opsin gene array locus control region (LCR) results in disruption of the cone mosaic. Vision Res. 2010;50:1989–1999. doi: 10.1016/j.visres.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cideciyan AV, Hufnagel RB, Carroll J, et al. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum Gene Ther. 2013;24:993–1006. doi: 10.1089/hum.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sumaroka A, Garafalo AV, Cideciyan AV, et al. Blue cone monochromacy caused by the C203R missense mutation or large deletion mutations. Invest Ophthalmol Vis Sci. 2018;59:5762–5772. doi: 10.1167/iovs.18-25280. [DOI] [PubMed] [Google Scholar]
- 14.Applebury ML, Antoch MP, Baxter LC, et al. The murine cone photoreceptor: a single cone type expresses both S and M opsins with retinal spatial patterning. Neuron. 2000;27:513–523. doi: 10.1016/s0896-6273(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 15.Haverkamp S, Wassle H, Duebel J, et al. The primordial, blue-cone color system of the mouse retina. J Neurosci. 2005;25:5438–5445. doi: 10.1523/JNEUROSCI.1117-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daniele LL, Insinna C, Chance R, et al. A mouse M-opsin monochromat: retinal cone photoreceptors have increased M-opsin expression when S-opsin is knocked out. Vision Res. 2011;51:447–458. doi: 10.1016/j.visres.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyubarsky AL, Falsini B, Pennesi ME, Valentini P, Pugh EN., Jr. UV- and midwave-sensitive cone-driven retinal responses of the mouse: a possible phenotype for coexpression of cone photopigments. J Neurosci. 1999;19:442–455. doi: 10.1523/JNEUROSCI.19-01-00442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikonov SS, Kholodenko R, Lem J, Pugh EN., Jr. Physiological features of the S- and M-cone photoreceptors of wild-type mice from single-cell recordings. J Gen Physiol. 2006;127:359–374. doi: 10.1085/jgp.200609490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng WT, Li J, Zhu P, et al. Human L- and M-opsins restore M-cone function in a mouse model for human blue cone monochromacy. Mol Vis. 2018;24:17–28. [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Deng WT, Du W, et al. Gene-based therapy in a mouse model of blue cone monochromacy. Sci Rep. 2017;7:6690. doi: 10.1038/s41598-017-06982-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mancuso K, Hauswirth WW, Li Q, et al. Gene therapy for red-green colour blindness in adult primates. Nature. 2009;461:784–787. doi: 10.1038/nature08401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zolotukhin S, Potter M, Zolotukhin I, et al. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 23.Pang JJ, Boye SL, Kumar A, et al. AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEbeta mutation. Invest Ophthalmol Vis Sci. 2008;49:4278–4283. doi: 10.1167/iovs.07-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pang JJ, Chang B, Kumar A, et al. Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther. 2006;13:565–572. doi: 10.1016/j.ymthe.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Smith TS, Spitzbarth B, Li J, et al. Light-dependent phosphorylation of Bardet-Biedl syndrome 5 in photoreceptor cells modulates its interaction with arrestin1. Cell Mol Life Sci. 2013;70:4603–4616. doi: 10.1007/s00018-013-1403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu X, Li A, Brown B, et al. Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol Vis. 2002;8:462–471. [PubMed] [Google Scholar]
- 27.Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams GA, Jacobs GH. Cone-based vision in the aging mouse. Vision Res. 2007;47:2037–2046. doi: 10.1016/j.visres.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards JE, Scott KM, Sieving PA. Disruption of conserved rhodopsin disulfide bond by Cys187Tyr mutation causes early and severe autosomal dominant retinitis pigmentosa. Ophthalmology. 1995;102:669–677. doi: 10.1016/s0161-6420(95)30972-4. [DOI] [PubMed] [Google Scholar]
- 30.May-Simera H, Nagel-Wolfrum K, Wolfrum U. Cilia—the sensory antennae in the eye. Prog Retin Eye Res. 2017;60:144–180. doi: 10.1016/j.preteyeres.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Sedmak T, Wolfrum U. Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol Cell. 2011;103:449–466. doi: 10.1042/BC20110034. [DOI] [PubMed] [Google Scholar]
- 32.Humphries MM, Rancourt D, Farrar GJ, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 33.Lem J, Krasnoperova NV, Calvert PD, et al. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A. 1999;96:736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]