

Abstract
Background/Purpose
Rheumatoid arthritis (RA) patients with the lowest circulating low-density lipoprotein concentrations (LDL-C) are at heightened risk for cardiovascular disease (CVD) events. However, atherosclerotic burden within this subgroup is unknown.
Methods
RA patients pooled from 4 cohort studies of CVD (n=546) were compared with non-RA controls from the Multi-Ethnic Study of Atherosclerosis (MESA; n=5,279). Those on lipid lowering medications were excluded. Differences in cardiac computed tomography-derived coronary arterial calcium (CAC) Agatston scores between the RA and control groups were compared across strata of LDL-C.
Results
Among those with low LDL-C (LDL-C<70 mg/dL), mean adjusted CAC scores were 3-fold higher for RA patients compared with controls (18.6 vs. 4.6 Agatston units, respectively; p<0.001), a difference significantly greater than that of any other LDL-C stratum except LDL-C>160 mg/dL. Similarly, 32% of the RA patients with low LDL-C had a CAC score ≥100 Agatston units compared with only 7% of controls in the same LDL-C stratum (OR=5.97; p<0.001), a difference significantly greater than all of the other LDL-C strata. Low LDL-C was most strongly associated with higher CAC among RA patients who were white race, ever smokers, and the non-obese. Other than a higher frequency of current smokers, RA patients with low LDL-C did not have more CVD risk factors or higher measures of RA disease activity or severity when compared with RA patients with higher LDL-C.
Conclusions
RA patients with low LDL-C may represent a group appropriate for heightened screening and prevention of atherosclerotic CVD.
Keywords: cardiovascular, risk prediction, lipids, inflammation
INTRODUCTION
Among individuals with rheumatoid arthritis (RA), rates of myocardial infarction (MI) and overall cardiovascular disease (CVD) mortality are 50% higher than in non-RA controls(1, 2), rates that are comparable to those in individuals with diabetes(3). Accordingly, RA patients have a greater burden of atherosclerosis, with coronary artery calcium (CAC) scores markedly higher than in non-RA controls(4–7). Although the majority of prior studies of accelerated atherogenesis in RA have focused on the contribution of chronic systemic inflammation, traditional CVD risk factors are also important, but may differ from the non-RA population(8). In particular, several observational studies have identified RA patients with the lowest circulating low-density lipoprotein concentration (LDL-C) levels (i.e. LDL-C<70 mg/dL) as those with unexpectedly high risk for CVD events, with risk comparable to, or exceeding that observed in RA patients with the highest LDL-C levels(9, 10). The etiologic mechanism underlying this observation, now commonly referred to as the “lipid paradox”, is unclear, although inflammation-induced reduction in lipid levels has been postulated. In contrast, high HDL-C and low triglyceride levels appear to be associated with decreased risk of CVD events in both RA and non-RA population. Whether RA patients with very low LDL-C levels have a greater burden of atherosclerosis is unknown, but its clarification has important implications for CVD prevention strategies.
For the present study, we compared CAC scores between RA patients and non-RA controls within and between strata of circulating fasting lipids. We hypothesized that RA patients with very low LDL-C levels who were not treated with lipid lowering therapy would demonstrate subclinical CAC scores higher than those of non-RA controls. Further, we postulated that the subgroup of RA patients with very low LDL-C would have more severe and active RA disease as a potential mediator of higher CAC scores in this sub-group.
METHODS
Participants and Recruitment
RA patients were pooled from 4 cohort studies of CVD in RA in which cardiac computed tomography (CT) was obtained(4–6, 11). The 4 cohort studies enrolled patients from and around Nashville, Tennessee (n=169); Pittsburgh, Pennsylvania (n=195); Baltimore, Maryland (n=197); and New York, New York (n=101). Detailed methods and the findings of each study have been previously reported(4–6, 11). RA patients were enrolled between 2001 and 2005 for the Nashville cohort, between 2000 and 2004 for the Pittsburgh cohort, between 2004 and 2006 for the Baltimore cohort, and between 2011 and 2015 for the New York City cohort. Each of the cohorts included patients who fulfilled American College of Rheumatology (ACR) 1987 classification criteria(12). The RA sample for the analyses reported here was restricted to those without prior CVD events or procedures and those who were not treated with lipid lowering medications, for a final cohort total of 546 RA patients (Nashville n=137; Pittsburgh n=165; Baltimore n=161; New York n=83). Each study was approved by the Institutional Review Board (IRB) of the associated university, and all subjects provided written informed consent prior to enrollment. The IRB of Columbia University Medical Center approved the pooled analyses.
Non-RA controls were enrollees in the Multi-Ethnic Study of Atherosclerosis (MESA). A description of the MESA study design and methods has been published(13). In brief, MESA enrolled a multi-ethnic cohort of 6,814 participants from six US communities between 2000 and 2002, all of whom had a cardiac CT performed at baseline for quantification of CAC according to the Agatston method. MESA participants with RA were excluded(14) along with those treated with lipid lowering medications. In total, 1,535 controls were excluded, leaving 5,279 MESA controls.
Assessments
RA patients in the Baltimore and New York cohorts underwent 64-slice cardiac multidetector row CT (MDCT). RA patients in the Nashville and Pittsburgh cohorts underwent cardiac electron-beam CT (EBCT). In MESA, both MDCT and EBCT were used. The comparability of both methods has been validated(15). CAC was quantified using the Agatston method(16) in each cohort. In all cohorts, demographics, smoking history, and current medications were assessed by participant self-report. Resting blood pressure and anthropometrics were assessed similarly for all cohorts, and a fasting blood sample was stored from which circulating lipid concentrations and glucose were measured. For the pooled analyses, RA cases and controls were classified as having hypertension and diabetes based on the same definitions. Hypertension was defined by systolic blood pressure ≥ 140 mmHg, diastolic blood pressure ≥ 90 mmHg, or antihypertensive medication use. Diabetes was defined as a fasting serum glucose ≥ 126 mg/dl or use of antidiabetic medications. Circulating C-reactive protein was measured in all RA cases and controls except enrollees in the Pittsburgh cohort.
Duration of RA from diagnosis and duration of morning stiffness were assessed by self-report. Joints were examined for swelling and tenderness for the Nashville, Baltimore, and New York City cohorts, but not the Pittsburgh cohort.
Statistical Analysis
Participant characteristics were compared between the RA and control groups using t-tests for normally distributed continuous variables, the Kruskal-Wallis test for non-normally distributed continuous variables, and the chi-square goodness-of-fit or Fisher’s exact test, as appropriate, for categorical variables. Due to demographic imbalances between the RA and control groups, we additionally compared non-demographic characteristics using linear or ordinary logistic regression, according to the characteristic, in models that included variables for RA status, age, sex, and race. Non-normally distributed continuous variables were transformed as required. Demographically-adjusted means and percentages and their 95% confidence intervals (CIs) were calculated and transformed variables were back-transformed for ease of interpretation.
Next, we compared CAC scores between the RA and control groups by strata of LDL-C, defined (in mg/dL) as LDL-C<70, 70–99, 100–129, 130–159, and ≥160, using linear regression with CAC, transformed as log CAC+1 to meet the normality requirements for linear regression, modeled as the dependent variable and RA x LDL strata modeled as an interaction term. Back-transformed mean CAC scores, and their corresponding 95% CIs were calculated and plotted for the RA and control groups within each stratum. Between-LDL-C strata differences in the magnitude of the within-stratum RA vs. control difference in CAC score were compared by calculating p-values for the multiplicative interaction terms for each LDL-C stratum referent to the stratum of LDL-C<70 mg/dl. Additional models included adjustment for relevant shared characteristics unbalanced by RA status and associated with CAC in univariate models at the p<0.20 level (age, sex, race, waist circumference, ever and current smoking, diabetes, hypertension, HDL-C, and aspirin use).
Ordinal logistic regression was used to model CAC≥100 and ≥300 units with covariates modeled as described above for linear regression. Similar models were constructed to model non-HDL-C (using published cut-points from current guidelines(17)) and HDL-C (modeled in quintiles). Sensitivity analyses explored differences in the patterns of association of LDL-C strata with CAC restricted to strata of patient characteristics (age>60 years, sex, white vs. non-white race, ever smoking, hypertension, diabetes, BMI above and below 30 kg/m2) and by RA cohort.
Finally, we compared patient characteristics according to LDL-C below and above 70 mg/dL using univariate tests described above for the RA and control groups separately. Differences in the associations of characteristics with LDL-C≤70 mg/dL between the RA and control groups were compared by modeling LDL-C<70 mg/dL as the dependent variable in ordinary logistic regression models that that included RA x characteristic interaction terms as the primary covariates of interest. Throughout, a two-tailed alpha of 0.05 was utilized. STATA SE Version 14 (StataCorp LLC, College Station, TX) was used for all analyses.
RESULTS
Characteristics of the 546 RA patients and 5,279 controls are summarized in Table 1. Compared with controls, RA patients were significantly younger and more likely to be female and white race. Adjusting for these demographics, RA patients had a significantly higher frequency of underweight by BMI (i.e. BMI<18.5 kg/m2), a significantly lower waist circumference, and a higher prevalence of hypertension, which included higher mean adjusted SBP, DBP, and more frequent use of anti-hypertensives, compared with controls. RA patients were more frequent users of NSAIDs, including COX-2 inhibitors, and less frequent users of aspirin than controls. While total cholesterol did not significantly differ between the groups, RA patients had a lower mean demographically adjusted LDL-C compared with controls, and the demographically adjusted frequency of LDL-C<70 mg/dL in the RA group was double that of the control group (12% vs. 6%, respectively). As expected, mean adjusted CRP was higher in the RA group compared with controls. As previously established, the average demographically adjusted CAC score was twice as high in the RA group compared with controls, as were the frequencies of CAC>100 and >300 units.
Table 1.
Participant Characteristics According to Rheumatoid Arthritis Status
| Demographically Adjusted† |
||||||
|---|---|---|---|---|---|---|
| Control (n=5,279) |
RA (n=546) |
p-value | Control mean (95% CI) |
RA mean (95% CI) |
p-value | |
| Age, years | 61 ± 10 | 56 ± 11 | <0.001 | |||
| Male, n (%) | 2514 (48) | 112 (21) | <0.001 | |||
| White, n (%) | 2010 (38) | 443 (81) | <0.001 | |||
| BMI; kg/m2 | 28.1 ± 5.5 | 28.3 ± 5.9 | 0.58 | 28.1 (28.0, 28.3) | 28.2 (27.7, 28.7) | 0.72 |
| BMI<18.50 kg/m2, n(%) | 54 (1) | 10 (2) | 0.082 | 0.9 (0.7, 0.1) | 2.1 (1.1, 3.9) | 0.030 |
| BMI=18.50–24.99 kg/m2, n(%) | 1575 (30) | 171 (31) | 0.44 | 30 (29, 31) | 28 (25, 32) | 0.42 |
| BMI=25.00-29.99 kg/m2, n(%) | 2036 (39) | 177 (33) | 0.006 | 38 (37, 39) | 35 (31, 40) | 0.27 |
| BMI≥30.00 kg/m2, n(%) | 1614 (31) | 186 (34) | 0.082 | 30 (29, 31) | 33 (29, 38) | 0.14 |
| Waist circumference | 97 ± 14 | 93± 16 | <0.001 | 97 (97, 98) | 94 (93, 95) | <0.001 |
| Diabetes, n (%) | 557 (11) | 27 (5) | <0.001 | 8 (8, 9) | 8 (6, 12) | 0.85 |
| Ever smoking, n (%) | 2589 (49) | 261 (48) | 0.58 | 49 (48, 50) | 49 (44, 53) | 0.91 |
| Current smoking, n (%) | 709 (13) | 74 (14) | 0.94 | 13 (12, 14) | 12 (10, 16) | 0.90 |
| Hypertension, n (%) | 2144 (41) | 249 (46) | 0.024 | 38 (37, 40) | 56 (51, 61) | <0.001 |
| SBP, mm Hg | 126 ± 21 | 126 ± 19 | 0.35 | 125 (125, 126) | 131 (130, 133) | <0.001 |
| DBP, mm Hg | 72 ± 10 | 75 ± 10 | <0.001 | 72 (71, 72) | 77 (76, 78) | <0.001 |
| Anti-hypertensive use; n (%) | 1695 (32) | 184 (34) | 0.43 | 30 (29, 31) | 41 (36, 45) | <0.001 |
| Current NSAIDs, n (%) | 1164 (22) | 301 (55) | <0.001 | 22 (21, 23) | 45 (41, 50) | <0.001 |
| COX-2 inhibitors*, n (%) | 299 (6) | 129 (28) | <0.001 | 5 (4, 6) | 27 (22, 32) | <0.001 |
| Current aspirin use, n (%) | 1157 (22) | 70 (13) | <0.001 | 20 (19, 21) | 11 (9, 14) | <0.001 |
| Total cholesterol, mg/dL | 196 ± 34 | 199 ± 39 | 0.057 | 196 (195, 197) | 195 (192, 198) | 0.47 |
| LDL-C, mg/dL | 120 ± 31 | 118 ± 33 | 0.21 | 120 (119, 121) | 117 (114, 120) | 0.070 |
| LDL<70, n (%) | 244 (5) | 47 (9) | <0.001 | 6 (6, 7) | 12 (9, 15) | <0.001 |
| LDL ≥ 130, n (%) | 1859 (35) | 195 (36) | 0.82 | 35 (34, 37) | 34 (30, 38) | 0.60 |
| HDL-C, mg/dL | 51 ± 15 | 56 ± 17 | <0.001 | 52 (51, 52) | 53 (52, 54) | 0.041 |
| Triglycerides*, mg/dL | 111 (78-161) | 112 (79-153) | 0.76 | 110 (108, 111) | 115 (109, 121) | 0.13 |
| Non-HDL-C, mg/dL | 145 ± 35 | 143 ± 38 | 0.26 | 145 (144, 146) | 142 (139, 145) | 0.13 |
| CRP**, mg/L | 1.9 (0.8-4.3) | 4.0 (1.4-9.7) | <0.001 | 1.9 (1.9, 2.0) | 3.7 (3.3, 4.2) | <0.001 |
| CAC Score, units | 0 (0-65) | 0 (0-90) | 0.35 | 7 (7, 7) | 14 (12, 17) | <0.001 |
| CAC ≥1 unit, n (%) | 2443 (46) | 266 (49) | 0.28 | 44 (43, 46) | 61 (56, 66) | <0.001 |
| CAC ≥100 units, n (%) | 1097 (21) | 132 (24) | 0.064 | 14 (13, 15) | 27 (23, 31) | <0.001 |
| CAC ≥300 units, n (%) | 556 (11) | 66 (12) | 0.26 | 6 (5, 6) | 12 (9, 15) | <0.001 |
Demographically adjusted means and 95% CIs are derived from linear or logistic regression, as appropriate to the characteristic of interest, in models adjusting for age, sex, and race/ethnicity. Where required, characteristics requiring normal transformation for modeling were back-transformed.
Available in all controls and 463 RA patients
Available in all controls and 439 RA patients
Available in 5,251 controls and 436 RA patients
In the non-adjusted columns, mean ± SD or median (IQR) depicted, unless otherwise noted. In the adjusted columns, means and 95% CIs are depicted for continuous and categorical variables
BMI=body mass index; SBP=systolic blood pressure; DBP=diastolic blood pressure; NSAID=non-steroidal anti-inflammatory medication; COX=cyclooxygenase; LDL-C=low density lipoprotein concentration; HDL-C=high density lipoprotein concentration; CRP=C-reactive protein; CAC=coronary artery calcification; RA=rheumatoid arthritis
Having a LDL-C Less than 70 mg/dL was Associated with a Markedly Higher CAC Score in RA Patients Compared with non-RA Controls
Adjusted CAC scores according to strata of LDL-C, non-HDL-C, and HDL-C are depicted in panels A-C of Figure 1. Mean CAC scores were significantly higher for the RA vs. control groups across all LDL-C strata after adjusting for demographics and relevant CVD risk factors (Figure 1A) and demonstrated a U-shaped pattern in the RA group compared with a linear increase, on average, in the control group. The greatest difference in mean adjusted CAC scores between the RA and control groups was observed in those with an LDL-C<70 mg/dL in which the mean adjusted CAC score was more than 3-fold higher for the RA group compared with the control group (18.6 vs. 4.6 Agatston units, respectively; p<0.001). This magnitude of difference in adjusted CAC scores between the RA and control groups was significantly larger for the LDL-C<70 mg/dL stratum compared with the three next highest strata (i.e. p-value for interaction all<0.05). Mean adjusted CAC scores were also higher for the RA group across all strata of non-HDL-C (Fig 1B) and HDL-C (Fig 1C); however, the magnitude of the difference in mean adjusted CAC scores between the RA and control groups within the lowest stratum did not differ significantly from that of the other strata. Similar patterns were observed across each of the 4 RA cohorts (data not shown)
Figure 1. Adjusted Coronary Artery Calcium Levels According to LDL-C Strata: RA vs. Control.
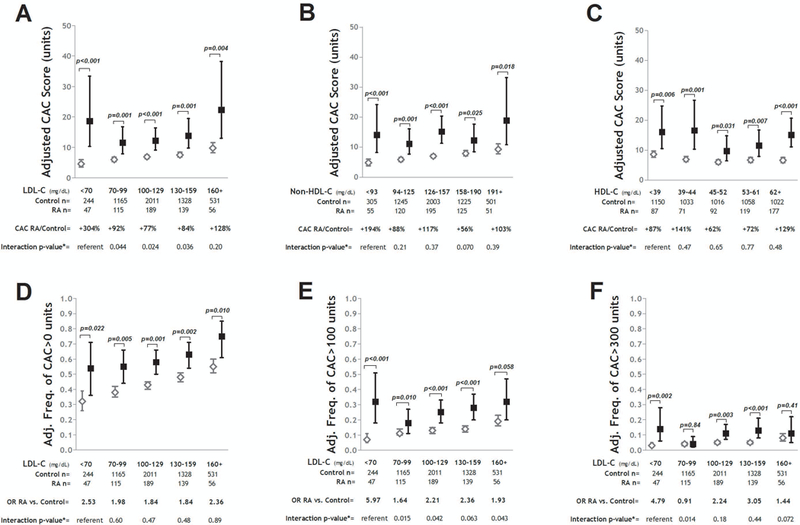
Graphs depict adjusted mean CAC scores and 95% confidence intervals (CIs) for strata of LDL-C (Panel A); non-HDL-C (Panel B); and HDL-C (Panel C). Relative differences in CAC scores for the RA vs. control groups are indicated per stratum. Interaction p-values compare the relative difference in CAC between the RA and control groups for the given stratum compared with the lowest (referent) stratum. Panels D-F depict the differences in the adjusted frequencies of any CAC (CAC>0 units) (Panel D), CAC>100 units (Panel E), and CAC>300 units (Panel F) with their associated 95% CIs and odds ratios (ORs) for the RA vs. control groups for each LDL-C stratum. Interaction p-values compare the magnitude of the OR between the RA and control groups for the given stratum compared with the lowest (referent) stratum. For all panels, models were adjusted for age, sex, race, waist circumference, smoking, diabetes, hypertension, HDL-C (where appropriate), and aspirin use.
Adjusted frequencies of any CAC (i.e. CAC>0), CAC>100 units, and CAC>300 units according to LDL-C strata are depicted in panels D-F of Figure 1. For any CAC (Fig 1D), the greatest relative difference in the adjusted frequency was observed in the lowest LDL-C stratum; however, the magnitude of difference was not statistically significant compared with the differences within the other strata. For CAC>100 units (Fig 1E), the adjusted odds of CAC>100 units was nearly 6-fold higher for RA patients with a LDL-C<70 mg/dL compared with controls in the same stratum. The magnitude of this difference was significantly larger for this stratum compared with the RA vs. control differences of the other strata (i.e. interaction p-values all<0.05). A similar pattern was observed for CAC>300 units (Fig 1F); although the magnitude of difference for the lowest LDL-C stratum was only significantly different from the difference in the LDL-C 70–99 mg/dL stratum. These patterns were similar across each of the 4 RA cohorts (data not shown).
The Association of Low LDL-C with Higher CAC Scores was Observed Only Among RA Patients of White Race, Ever Smokers, and the non-Obese
We explored whether the association of low LDL-C with higher CAC scores differed according to patient characteristics. There were three subgroups of RA patients, non-White race, never smokers, and those with a BMI>30 kg/m2, for which the difference in mean adjusted CAC score between the RA and control groups was not the greatest in the LDL-C<70 mg/dL stratum relative to other LDL strata (depicted in Figure 2). Accordingly, the associations of low LDL-C with CAC were stronger when the White, ever smoker, and BMI<30 kg/m2 subgroups were analyzed separately (also depicted in Figure 2). The pattern was similar for both former and current smokers, so ever smokers were modeled as a single group (data not shown). Similarly, patterns were similar for the normal BMI and overweight BMI groups, which were combined and modeled together.
Figure 2. Adjusted Coronary Artery Calcium Levels According to LDL-C Strata Stratified by Race, Smoking, and Body Mass index: RA vs. Control.
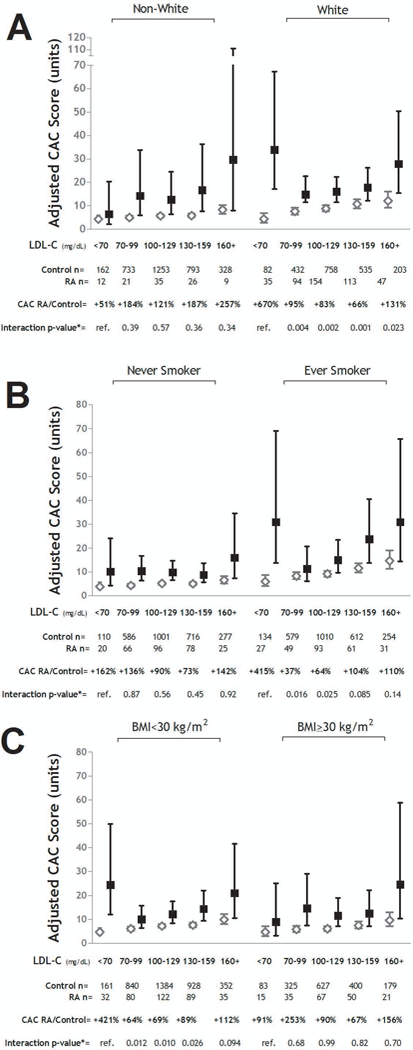
Graphs depict adjusted mean CAC scores and 95% confidence intervals (CIs) for strata of LDL-C for the RA vs. control groups stratified by White Race (Panel A), ever smoking (Panel B), and obesity (i.e. BMI≥30 mg/m2)(Panel C). Relative differences in CAC scores for the RA vs. control groups are indicated per stratum. Interaction p-values compare the relative difference in CAC between the RA and control groups for the given stratum compared with the lowest (referent) stratum. For all panels, models were adjusted for age, sex, race (where appropriate), waist circumference, smoking (where appropriate), diabetes, hypertension, HDL-C, and aspirin use
In analyses restricted to the subgroup of white ever-smokers, RA patients with an LDL-C<70 mg/dL had a mean adjusted CAC score nearly 10-fold higher than controls in the same stratum (61.2 vs 5.7 units, respectively; p<0.001), a difference that was significantly greater than the differences between the RA and control groups within each of the other LDL strata (Figure 3A). A similar pattern was observed for CAC>100 units, with more than two-thirds of the white, ever-smoking RA patients with LDL-C<70 mg/dL demonstrating CAC>100 units compared with only 8% of the similar controls, after adjustment (OR=23.85; p<0.001: Figure 3C). This difference was, as before, significantly greater than the differences observed in the other LDL strata. Differences within the lowest LDL-C stratum were even greater when restricting to participants of white race who were ever-smokers with a BMI<30 kg/m2 (Figure 3B and D).
Figure 3. Adjusted Coronary Artery Calcium Levels According to LDL-C Strata Restricted to High Impact Subgroups: RA vs. Control.
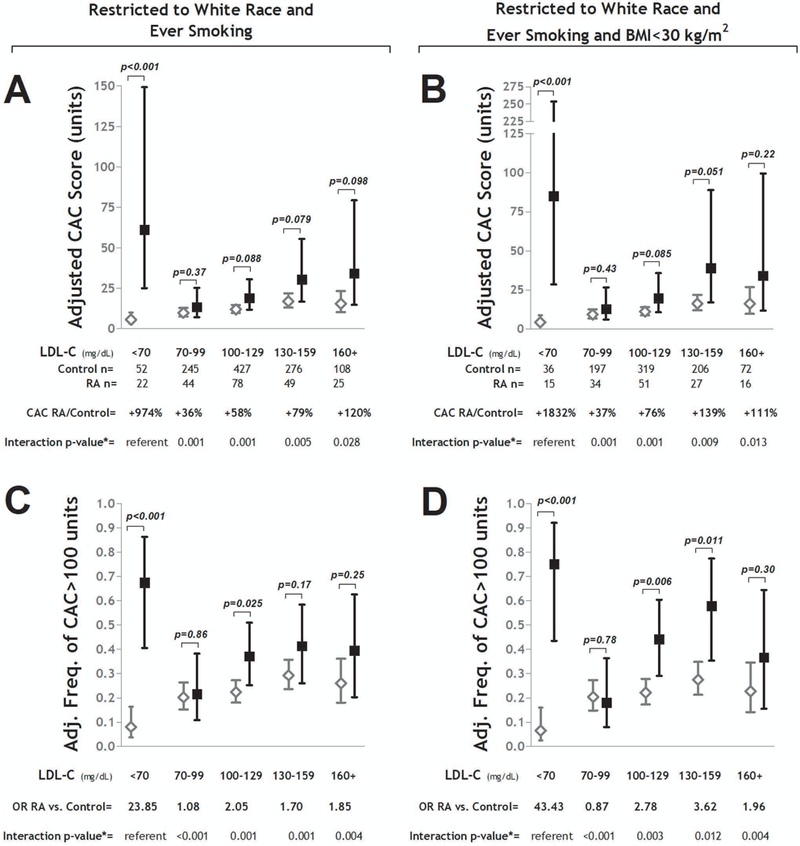
Panels A and B depict adjusted mean CAC scores and 95% confidence intervals (CIs) for strata of LDL-C for the RA vs. control groups restricted to those of White race who are ever smokers (Panel A), and those of White race who are both ever smokers and non-obese (Panel B). Relative differences in CAC scores for the RA vs. control groups are indicated per stratum. Interaction p-values compare the relative difference in CAC between the RA and control groups for the given stratum compared with the lowest (referent) stratum. Panels C and D depict the differences in the adjusted frequencies of any CAC≥100 units with their associated 95% CIs and odds ratios (ORs) for the RA vs. control groups for each LDL-C stratum restricted to those of White race who are ever smokers (Panel C), and those of White race who are both ever smokers and non-obese (Panel D). Interaction p-values compare the magnitude of the OR between the RA and control groups for the given stratum compared with the lowest (referent) stratum. For all panels, models were adjusted for age, sex, race, waist circumference, diabetes, hypertension, HDL-C, and aspirin use
CVD Risk Factors and RA Disease Activity/Severity Measures were not Highly Prevalent among RA Patients with Low LDL-C
We explored whether RA patients with low LDL-C demonstrated a risk factor profile that could explain their markedly higher CAC scores (summarized in Table 2). With the exception of having a significantly higher prevalence of current smokers, the frequencies of other CVD risk factors were not higher among RA patients with low LDL-C compared with RA patients with higher LDL-C. Several CVD risk factors (BMI, waist circumference, and triglycerides) were lower in RA patients with low LDL-C compared with those with higher LDL-C. Importantly, the lower average BMI in the low LDL-C RA groups was not driven by a higher proportion of those in the underweight (i.e. BMI<18.5 kg/m2) category. Likewise, RA disease and treatment characteristics were not higher in those with low LDL-C, and the presence of shared epitope alleles was significantly lower among those with low LDL-C. Associations of demographics, lifestyle characteristics, and CVD risk factors with low LDL-C were generally similar for non-RA controls compared with the RA group, with the exceptions of BMI and waist circumference, which were not lower in the controls with low LDL-C.
Table 2.
Participant Characteristics According to Rheumatoid Arthritis Status and Low Density Lipoprotein Concentration Strata
| RA |
Control |
||||||
|---|---|---|---|---|---|---|---|
| LDL ≤ 70 mg/dL (n=47) |
LDL > 70 mg/dL (n=499) |
p-value | LDL ≤ 70 mg/dL (n=244) |
LDL > 70 mg/dL (n=5035) |
p-value | Interaction p-value |
|
| Age, years | 55 ± 15 | 57 ± 11 | 0.26 | 62 ± 11 | 61 ± 10 | 0.67 | 0.20 |
| Male, n (%) | 13 (28) | 99 (20) | 0.20 | 113 (46) | 2401 (48) | 0.68 | 0.17 |
| White, n (%) | 35 (74) | 408 (82) | 0.22 | 82 (34) | 1928 (38) | 0.14 | 0.60 |
| BMI; kg/m2 | 26.5 ± 5.8 | 28.4 ± 5.9 | 0.030 | 28.2 ± 5.9 | 28.1 ± 5.5 | 0.78 | 0.020 |
| BMI<18.50 kg/m2, n(%) | 2 (4) | 8 (2) | 0.21 | 4 (2) | 50 (1) | 0.32 | 0.61 |
| BMI=18.50–24.99 kg/m2, n(%) | 21 (45) | 150 (30) | 0.041 | 77 (32) | 1498 (30) | 0.57 | 0.11 |
| BMI=25.00–29.99 kg/m2, n(%) | 9 (19) | 168 (34) | 0.040 | 80 (33) | 1956 (39) | 0.059 | 0.22 |
| BMI≥30.00 kg/m2, n(%) | 15 (32) | 171 (34) | 0.73 | 83 (34) | 1531 (30) | 0.26 | 0.43 |
| Waist circumference | 87 ± 16 | 93 ± 16 | 0.011 | 98 ± 16 | 97 ± 14 | 0.61 | 0.005 |
| Diabetes, n (%) | 3 (6) | 24 (5) | 0.50 | 42 (17) | 515 (10) | 0.001 | 0.57 |
| Ever smoking, n (%) | 27 (57) | 234 (47) | 0.17 | 134 (55) | 2455 (49) | 0.060 | 0.73 |
| Current smoking, n (%) | 11 (23) | 63 (13) | 0.039 | 54 (22) | 655 (13) | <0.001 | 0.90 |
| Hypertension, n (%) | 20 (43) | 229 (46) | 0.66 | 125 (51) | 2019 (40) | 0.001 | 0.14 |
| SBP, mm Hg | 123 ± 21 | 127 ± 19 | 0.21 | 126 ± 21 | 126 ± 21 | 0.82 | 0.44 |
| DBP, mm Hg | 75 ± 10 | 75 ± 10 | 0.72 | 71 ± 11 | 72 ± 10 | 0.38 | 0.68 |
| Anti-hypertensive use; n (%) | 17 (37) | 167 (33) | 0.63 | 113 (46) | 1582 (31) | <0.001 | 0.23 |
| Total cholesterol, mg/dL | 139 ± 24 | 205 ± 35 | <0.001 | 136 ± 22 | 199 ± 32 | <0.001 | 0.77 |
| HDL-C, mg/dL | 56 ± 21 | 56 ± 17 | 0.83 | 52 ± 20 | 51 ± 15 | 0.22 | 0.88 |
| Triglycerides*, mg/dL | 87 (67–127) | 112 (81–152) | 0.005 | 97 (63–174) | 109 (77–156) | 0.075 | 0.092 |
| CRP**, mg/L | 3.4 (0.5–8.0) | 4.0 (1.4–10.0) | 0.23 | 2.2 (0.8–5.1) | 1.9 (0.8–4.2) | 0.20 | 0.10 |
| CRP≥5mg/L, n(%) | 15 (41) | 161 (47) | 0.48 | 62 (25) | 998 (20) | 0.037 | 0.14 |
| Current NSAIDs, n (%) | 27 (57) | 274 (55) | 0.74 | 57 (23) | 1107 (22) | 0.61 | 0.82 |
| COX-2 inhibitors***, n (%) | 7 (20) | 122 (28) | 0.28 | 17 (7) | 282 (6) | 0.37 | 0.17 |
| Current aspirin use, n (%) | 8 (17) | 62 (12) | 0.37 | 53 (22) | 1104 (22) | 0.94 | 0.37 |
| RA duration, years | 10 (3–20) | 9 (3–19) | 0.76 | -- | -- | -- | |
| RF seropositive, n (%) | 31 (66) | 363 (75) | 0.18 | -- | -- | -- | |
| DAS28-CRP** | 3.6 (2.5 – 4.7) | 3.7 (2.8–4.5) | 0.42 | -- | -- | -- | |
| Morning stiffness, minutes | 20 (5–60) | 20 (5–60) | 0.77 | -- | -- | -- | |
| Any shared epitope alleles*, n (%) | 15 (52) | 256 (70) | 0.039 | -- | -- | -- | |
| Any non-biologics, n (%) | 39 (83) | 425 (85) | 0.66 | -- | -- | -- | |
| Current methotrexate, n (%) | 29 (62) | 319 (64) | 0.76 | -- | -- | -- | |
| Current hydroxychloroquine, n (%) | 14 (30) | 103 (21) | 0.14 | -- | -- | -- | |
| Current biologics, n (%) | 15 (32) | 166 (33) | 0.84 | -- | -- | -- | |
| Current prednisone, n (%) | 21 (45) | 215 (43) | 0.83 | -- | -- | -- | |
Mean ± SD or median (IQR) depicted, unless otherwise noted
BMI=body mass index; SBP=systolic blood pressure; DBP=diastolic blood pressure; NSAID=non-steroidal anti-inflammatory medication; LDL-C=low density lipoprotein concentration; HDL-C=high density lipoprotein concentration; CRP=C-reactive protein; RF=rheumatoid factor; RA=rheumatoid arthritis
available in n=385 RA patients
available in n=382 RA patients
available in n=463 RA patients
DISCUSSION
In this study, the first to explore coronary atherosclerotic burden among RA patients with very low LDL-C not treated with lipid-lowering medications, we observed a U-shaped association of LDL-C with CAC score among RA patients that was not present in non-RA controls; the largest relative difference in CAC score between the RA and control groups was observed for those with an LDL-C less than 70 mg/dL. The magnitude of this association was larger among those of white race, ever smokers, and the non-obese. However, other than a higher proportion of current smokers among those with very low LDL-C, traditional CVD risk factors and RA characteristics did not account for the findings. The study also confirmed, in the largest sample to date, higher overall CAC scores in RA across the entire range of LDL-C.
Observational studies reporting lower levels of circulating total cholesterol and LDL-C among RA patients compared with non-RA controls date back decades(18). Recognition that the magnitude of association of LDL-C with CVD events was lower among RA patients compared with the general population derive from more recent studies(8). However, the identification of RA patients with very low LDL-C levels as being at heightened CVD event risk was reported only in 2010 by Myasoedova et al., who named the association the “lipid paradox”(9). Since then, the association has been reported in additional cohorts(10); however, whether the association truly differs between RA and non-RA populations has been questioned(19).
Our findings lend credence to heightened CVD risk for RA patients with very low LDL-C, particularly since more than 30% of the RA patients in this group had a CAC score ≥100 units, an established threshold predictive of future atherosclerotic CVD events(20). Moreover, 75% of those with very low LDL-C in the highest risk group that we identified (white race, ever smoker, and non-obese) had a CAC score ≥100 units. Such individuals would not be considered high risk based on risk algorithms validated in the general population that are weighted heavily toward CVD risk driven by hyperlipidemia, such as the current ACC/AHA guideline. These algorithms have consistently been shown to underperform in RA patients(21, 22), suggesting that additional predictive factors for RA patients should be identified. However, efforts to improve prediction by factoring in systemic inflammatory markers have been unsuccessful(21, 22).
In RA, systemic inflammatory markers vary with time and treatment, and current levels are likely not reflective of levels from the past that may have contributed to atherogenesis. Low LDL-C in the setting of no treatment with lipid lowering medications may represent a more consistent and stable marker of an RA-driven atherogenic propensity, and RA patients with this phenotype may be appropriate for more aggressive CVD screening and primary prevention measures, including targeting of non-lipid risk factors. Using cardiac CT for secondary screening for atherosclerosis is already advocated for those at uncertain or intermediate risk in the general population(23); however, the utility of any such strategy has not been evaluated in RA patients with very low LDL-C. Our data provide support that studies evaluating the utility of secondary screening with an imaging assessment of atherosclerosis among RA patients with very low LDL-C are warranted.
Mechanistically, it is unclear what factor(s) may be mediating the disconnect between circulating LDL-C and atherogenesis in this subgroup of RA patients. Inflammatory cytokines associated with RA, such as IL-6, upregulate LDL receptors and scavenger receptors for modified LDL particles on hepatocytes and macrophages, potentially leading to lower circulating LDL levels while also being pro-atherogenic(24, 25). However, we did not observe an association of higher CRP or DAS levels with low LDL-C, making these unlikely mediators of our observed associations. Another mechanism potentially leading to reduced circulating LDL is oxidation, as oxidized LDL particles are more readily taken up by macrophages and removed from circulation(26). RA patients, on average, have higher levels of oxidized LDL(27). HDL protects against such LDL oxidation, largely through the activity of its paraoxonase cargo. In RA, HDL particles are deficient of paraoxonase(28) and paraoxonase function is diminished(29), an effect that is potentially reversible with treatment(30). However, whether these, or other mechanisms mediate the low LDL phenotype to be pro-atherogenic warrants further investigation. Very low LDL-C, along with lower HDL-C and triglycerides, has also been linked to higher mortality in patients with moderate to severe heart failure(31). Whether this phenomenon is due to an increase in atherosclerosis, consistent with our findings, or a consequence of malnutrition and/or the cachectic hypermetabolic state of advanced heart failure is unclear.
The fact that a larger effect of very low LDL-C on CAC was seen among RA patients of white race who had smoked was in keeping with the expected contribution of these risk factors; however, the protective effect of higher BMI was unexpected. Interestingly, higher BMI has also been associated with lower all-cause and CVD mortality in RA patients(32, 33). It has been postulated that this is due to the presence of sarcopenia and frailty induced by prolonged disease activity and severity that characterizes RA patients with lower BMIs; however, in the study by del Rincon et al(32), the protective effect of BMI on all-cause mortality was incremental, even when moving from the normal weight to overweight to obese BMI categories. Nevertheless, because the number of patients with low BMI and very low LDL-C is a relatively small subset of the RA population, it seems unlikely that this is the primary mechanism whereby BMI appears to be protective against all-cause and CVD mortality in RA.
Our study has notable strengths and limitations. Among strengths, the RA patient sample captured the participants of four of the largest North American cohort studies of CAC in RA and is sufficiently large to explore associations within subsets of patients. Likewise, the ability to leverage the size of the MESA cohort for non-RA controls allowed additional precision to detect differences within subsets. Among limitations, there were differences in the four RA cohorts in inclusion/exclusion criteria, dates of enrollment, data captured, and geographic location. However, the primary exposures and outcomes were collected in a similar way between cohorts and, in sensitivity analyses, there were no differences in the associations of very low LDL-C with CAC between the cohorts. There were also differences in demographics between the pooled RA sample and the MESA control group. However, we chose not to sacrifice precision by attempting to match or restrict inclusion on demographic variables; rather, we used multivariable regression to adjust for demographic differences and we conducted sensitivity analyses restricted to age groups, sex, and race, with notable differences noted only for race, as discussed above. Because of the smaller sample size of the subgroup analyses and the inherent reduction in statistical power, the magnitude and significance of the associations should be interpreted as less reliable than that of the main effects identified in the full cohort. However, these subgroup dichotomies are potentially hypothesis generating and warrant validation in subsequent studies. Finally, our comparisons are cross-sectional only with no ability to determine temporality in the associations. In particular, future studies exploring atherosclerosis progression according to LDL strata are warranted. We did not find an interaction of age or RA duration on the association of very low LDL-C with CAC, suggesting that the association is not related solely to the cumulative effects of RA disease.
In summary, RA patients not treated with lipid lowering medications with the lowest circulating LDL-C (i.e.<70 mg/dL) had markedly higher CAC scores relative to non-RA controls, including high level of CAC scores potentially associated with CVD events (i.e. CAC>100 units), even after adjusting for relevant confounders. The association was not observed for HDL-C or non-HDL-C, suggesting an effect specific to LDL-C. The association was stronger in subsets of RA patients, particularly those of White race, ever smokers, and the non-obese. However, these patients did not appear to be at such high risk based either on their traditional CVD risk profile or on RA disease or treatment characteristics. Our data support the so-called “lipid paradox” in which RA patients with similarly low LDL-C levels have been noted to be at unexpectedly high risk for CVD events and suggest a susceptible subgroup of RA patients that may be appropriate for additional CVD screening and/or preemptory aggressive primary prevention efforts targeting non-lipid risk factors.
ACKNOWLEDGMENTS
We would like to thank the Johns Hopkins Bayview Medical Center General Clinical Research Center and staff, the field center of the Baltimore MESA cohort, and the MESA Coordinating Center at the University of Washington, Seattle.
The authors thank the other investigators, staff, and participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
FUNDING
Supported by the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases grants AR-056116 for Dr. Stein, AR-050026 for Dr. Bathon) and NIH/NCRR/GCRC Grant M01-RR000056 for Dr. Wasko. The MESA was supported by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168 and N01-HC-95169 from the National Heart, Lung, and Blood Institute and by grants UL1-TR-000040 and UL1-TR-001079 from NCRR.
Footnotes
COMPETING INTERESTS
None of the authors report conflicts of interest related to the content of this manuscript
REFERENCES
- 1.Avina-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis and Rheumatism 2008;59:1690–1697. [DOI] [PubMed] [Google Scholar]
- 2.Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis 2012;71:1524–1529. [DOI] [PubMed] [Google Scholar]
- 3.Peters MJ, van Halm VP, Voskuyl AE, Smulders YM, Boers M, Lems WF, et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum 2009;61:1571–1579. [DOI] [PubMed] [Google Scholar]
- 4.Chung CP, Oeser A, Raggi P, Gebretsadik T, Shintani AK, Sokka T, et al. Increased coronary-artery atherosclerosis in rheumatoid arthritis: relationship to disease duration and cardiovascular risk factors. Arthritis and Rheumatism 2005;52:3045–3053. [DOI] [PubMed] [Google Scholar]
- 5.Kao AH, Krishnaswami S, Cunningham A, Edmundowicz D, Morel PA, Kuller LH, et al. Subclinical coronary artery calcification and relationship to disease duration in women with rheumatoid arthritis. The Journal of rheumatology 2008;35:61–69. [PubMed] [Google Scholar]
- 6.Giles JT, Szklo M, Post W, Petri M, Blumenthal RS, Lam G, et al. Coronary arterial calcification in rheumatoid arthritis: comparison to the multi-ethnic study of atherosclerosis. Arthritis research & therapy 2009;11:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karpouzas GA, Malpeso J, Choi TY, Li D, Munoz S, Budoff MJ. Prevalence, extent and composition of coronary plaque in patients with rheumatoid arthritis without symptoms or prior diagnosis of coronary artery disease. Ann Rheum Dis 2014;73:1797–1804. [DOI] [PubMed] [Google Scholar]
- 8.Crowson CS, Rollefstad S, Ikdahl E, Kitas GD, van Riel P, Gabriel SE, et al. Impact of risk factors associated with cardiovascular outcomes in patients with rheumatoid arthritis. Ann Rheum Dis 2018;77:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Myasoedova E, Crowson CS, Kremers HM, Roger VL, Fitz-Gibbon PD, Therneau TM, et al. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann Rheum Dis 2011;70:482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navarro-Millan I, Yang S, DuVall SL, Chen L, Baddley J, Cannon GW, et al. Association of hyperlipidaemia, inflammation and serological status and coronary heart disease among patients with rheumatoid arthritis: data from the National Veterans Health Administration. Ann Rheum Dis 2016;75:341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winchester R, Giles JT, Nativ S, Downer K, Zhang HZ, Bag-Ozbek A, et al. Association of Elevations of Specific T Cell and Monocyte Subpopulations in Rheumatoid Arthritis With Subclinical Coronary Artery Atherosclerosis. Arthritis Rheumatol 2016;68:92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis and Rheumatism 1988;31:315–324. [DOI] [PubMed] [Google Scholar]
- 13.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, et al. Multi-ethnic study of atherosclerosis: objectives and design. American Journal of Epidemiology 2002;156:871–881. [DOI] [PubMed] [Google Scholar]
- 14.Majka DS, Vu TT, Pope RM, Teodorescu M, Karlson EW, Liu K, et al. Association of Rheumatoid Factors With Subclinical and Clinical Atherosclerosis in African American Women: The Multiethnic Study of Atherosclerosis. Arthritis Care Res (Hoboken) 2017;69:166–174. [DOI] [PubMed] [Google Scholar]
- 15.Mao SS, Pal RS, McKay CR, Gao YG, Gopal A, Ahmadi N, et al. Comparison of coronary artery calcium scores between electron beam computed tomography and 64-multidetector computed tomographic scanner. J Comput Assist Tomogr 2009;33:175–178. [DOI] [PubMed] [Google Scholar]
- 16.Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M, Detrano R. Quantification of Coronary-Artery Calcium Using Ultrafast Computed-Tomography. Journal of the American College of Cardiology 1990;15:827–832. [DOI] [PubMed] [Google Scholar]
- 17.Elshazly MB, Martin SS, Blaha MJ, Joshi PH, Toth PP, McEvoy JW, et al. Non-high-density lipoprotein cholesterol, guideline targets, and population percentiles for secondary prevention in 1.3 million adults: the VLDL-2 study (very large database of lipids). J Am Coll Cardiol 2013;62:1960–1965. [DOI] [PubMed] [Google Scholar]
- 18.Svenson KL, Lithell H, Hallgren R, Vessby B. Serum lipoprotein in active rheumatoid arthritis and other chronic inflammatory arthritides. II. Effects of anti-inflammatory and disease-modifying drug treatment. Arch Intern Med 1987;147:1917–1920. [PubMed] [Google Scholar]
- 19.Liao KP, Liu J, Lu B, Solomon DH, Kim SC. Association between lipid levels and major adverse cardiovascular events in rheumatoid arthritis compared to non-rheumatoid arthritis patients. Arthritis Rheumatol 2015;67:2004–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Budoff MJ, Nasir K, McClelland RL, Detrano R, Wong N, Blumenthal RS, et al. Coronary calcium predicts events better with absolute calcium scores than age-sex-race/ethnicity percentiles: MESA (Multi-Ethnic Study of Atherosclerosis). Journal of the American College of Cardiology 2009;53:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crowson CS, Matteson EL, Roger VL, Therneau TM, Gabriel SE. Usefulness of risk scores to estimate the risk of cardiovascular disease in patients with rheumatoid arthritis. Am J Cardiol 2012;110:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arts EE, Popa CD, Den Broeder AA, Donders R, Sandoo A, Toms T, et al. Prediction of cardiovascular risk in rheumatoid arthritis: performance of original and adapted SCORE algorithms. Ann Rheum Dis 2015. [DOI] [PubMed] [Google Scholar]
- 23.Goff DC Jr., Lloyd-Jones DM, Bennett G, Coady S D’Agostino RB, Gibbons R, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129:S49–73. [DOI] [PubMed] [Google Scholar]
- 24.Lubrano V, Gabriele M, Puntoni MR, Longo V, Pucci L. Relationship among IL-6, LDL cholesterol and lipid peroxidation. Cell Mol Biol Lett 2015;20:310–322. [DOI] [PubMed] [Google Scholar]
- 25.Hashizume M, Mihara M. Atherogenic effects of TNF-alpha and IL-6 via up-regulation of scavenger receptors. Cytokine 2012;58:424–430. [DOI] [PubMed] [Google Scholar]
- 26.Chistiakov DA, Bobryshev YV, Orekhov AN. Macrophage-mediated cholesterol handling in atherosclerosis. J Cell Mol Med 2016;20:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMahon M, Grossman J, FitzGerald J, Dahlin-Lee E, Wallace DJ, Thong BY, et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum 2006;54:2541–2549. [DOI] [PubMed] [Google Scholar]
- 28.Isik A, Koca SS, Ustundag B, Celik H, Yildirim A. Paraoxonase and arylesterase levels in rheumatoid arthritis. Clin Rheumatol 2007;26:342–348. [DOI] [PubMed] [Google Scholar]
- 29.Tanimoto N, Kumon Y, Suehiro T, Ohkubo S, Ikeda Y, Nishiya K, et al. Serum paraoxonase activity decreases in rheumatoid arthritis. Life Sci 2003;72:2877–2885. [DOI] [PubMed] [Google Scholar]
- 30.McInnes IB, Thompson L, Giles JT, Bathon JM, Salmon JE, Beaulieu AD, et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann Rheum Dis 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charach G, Rabinovich A, Ori A, Weksler D, Sheps D, Charach L, et al. Low levels of low-density lipoprotein cholesterol: a negative predictor of survival in elderly patients with advanced heart failure. Cardiology 2014;127:45–50. [DOI] [PubMed] [Google Scholar]
- 32.Escalante A, Haas RW, del Rincon I. Paradoxical effect of body mass index on survival in rheumatoid arthritis: role of comorbidity and systemic inflammation. Archives of Internal Medicine 2005;165:1624–1629. [DOI] [PubMed] [Google Scholar]
- 33.Kremers HM, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE. Prognostic importance of low body mass index in relation to cardiovascular mortality in rheumatoid arthritis. Arthritis and Rheumatism 2004;50:3450–3457. [DOI] [PubMed] [Google Scholar]