

Abstract
In vitro to in vivo extrapolation (IVIVE) is a critical component of the efforts to prioritize and assess environmental chemicals using high-throughput in vitro assays. The plasma unbound fraction (Fub) is a key toxicokinetic parameter in IVIVE, and is usually measured via the Rapid Equilibrium Dialysis (RED) assay widely used for pharmaceuticals. However, pharmaceuticals have a narrower range of physicochemical properties than environmental chemicals. Motivated by the observation that high LogKOW compounds appeared to have disproportionately low Fub measurements using RED, we added a protein-free control in order to verify equilibration to 100% unbound in the absence of proteins. We found that many high LogKOW non-pharmaceuticals fail to equilibrate in RED in protein-free controls, and thus had apparent values of Fub = 0 in plasma. In these cases, Solid Phase Microextraction (SPME) as an alternative method provided an accurate, though more time-consuming, alternative to accurately determine Fub. We propose an updated IVIVE workflow that adds a protein-free control to the RED protocol, with the use of alternative approaches, such as SPME, in cases where compounds fail to adequately equilibrate. These refinements will provide additional confidence in the use of IVIVE as part of high-throughput screening programs of chemicals.
Keywords: In vitro-in vivo extrapolation, high-throughput screening, protein binding, environmental chemicals, toxicokinetics
Introduction
The National Academies’ seminal report Toxicity Testing in the 21st Century: A Vision and Strategy (National Research Council, 2007), ushered in a new era in vitro-based toxicology aimed at prioritizing and assessing the tens of thousands of chemicals in commerce. The report envisioned that pharmacokinetic modeling would be needed to relate effective concentrations in vitro and environmental exposure levels, generally referred to know as “in vitro-in vivo extrapolation,” or IVIVE (Yoon et al., 2012). Indeed, IVIVE is now recognized as essential to enabling prioritization and decision-making based on in vitro testing of chemicals (Bell et al., 2018; Wetmore, 2015).
The need to conduct IVIVE on a large scale across many chemicals has in turn led to the development of “high throughput toxicokinetic” (HTTK) models that only require a small number of experimental measurements in order to parameterize (Pearce et al., 2017; Wambaugh et al., 2015). By far the most widely used HTTK-based approach to IVIVE utilizes a simple steady state model that relies on two measurements that can be done in vitro: hepatic clearance and plasma protein binding (Rotroff et al., 2010; Wetmore et al., 2012). Specifically, for a given oral dose, the steady state plasma concentration (Css) in this case is given by
| (1) |
Because linear kinetics is assumed, the dose is usually set to a unit value of 1 mg/kg·day. Fub equals the unbound fraction of parent chemical, measured through in vitro plasma protein binding. Clint equals intrinsic hepatic clearance, measured through in vitro hepatic clearance and scaled up to human physiological values. GFR is glomerular filtration rate and Q1 is liver blood flow, set at human physiological values. The calculation of Css then assumes that elimination is solely due to hepatic metabolism and renal filtration, with only the unbound chemical fraction available for metabolism and elimination. The standard approach is then to measure Clint using cryopreserved hepatocytes, and to measure Fub using Rapid Equilibrium Dialysis (RED) (Rotroff et al., 2010; Wetmore et al., 2012). This approach has been used for over 400 chemicals that had been tested in the U.S. EPA ToxCast screening battery (Wetmore, 2015). An “Oral Equivalent Dose” (OED) is determined by applying reverse dosimetry to determined exposure levels needed to reach steady state blood concentrations equal to the effective in vitro concentrations (e.g., AC50):
| (2) |
Overall, these assumptions are thought to be conservative in the sense of not underestimating the dose at which bioactivity would be observed.
Much of the emphasis on improving the accuracy of IVIVE has focused on the prediction of metabolic clearance (Bell et al., 2018). However, in many cases, protein binding may have equal or greater impact on overall kinetics. Because of the success and broad applicability of RED method for predicting freely available concentrations of pharmaceuticals (Bohnert and Gan, 2013), use of RED to determine Fub has become part of the standard protocol for IVIVE analyses (Wetmore, 2015). However, due to the Lipinski “rule of 5” (Lipinski et al., 2001), pharmaceutical compounds commonly have a much narrower range of chemical properties as compared to the broader universe of chemicals in commerce.
For instance, one of Lipinski rules is that the octanol-water partition coefficient (LogKow) be no greater than five, while a large number of industrial chemicals have values greater than five (e.g., many polychlorinated byphenyl (PCB) and polycyclic aromatic hydrocarbon (PAH) compounds). High LogKow is also associated with non-specific binding to polymer-based plates used in in vitro experiments (Auner et al., 2019), raising the question as to whether apparently high values of binding derived from RED assays may be confounded by non-specific binding. For instance, a substantial fraction of chemical compounds tested using RED have been reported to have very small (<5%) or negligible values for Fub (Wetmore, 2015), in which case a nominal value of Fub = 5% has been assigned for the purposes of IVIVE. However, these data (Figure 1A) show that there appears to be strong correlation between higher LogKow and smaller values of the Fub. Among the 51 chemicals with LogKow≥5, only three had measured Fub > 5%. Additionally, among the 97 chemicals with non-detectable Fub, only 9 had a LowKow <3 (Figure 1B). This correlation raises the concern that some of the small reported values for Fub may be due to limitations in the domain of applicability of RED assay, and not reflect the true degree of plasma protein binding. For instance, the reported Fub based on RED for the pesticide permethrin was 0% (Wetmore et al., 2012), while a radiolabel-based study reported much higher values of Fub = 11%-80% (Sethi et al., 2014). Therefore, there is a critical need to routinely verify whether and when RED may be giving spurious results, as well as to testing alternative methods for measuring plasma protein binding in such cases.
Figure 1.
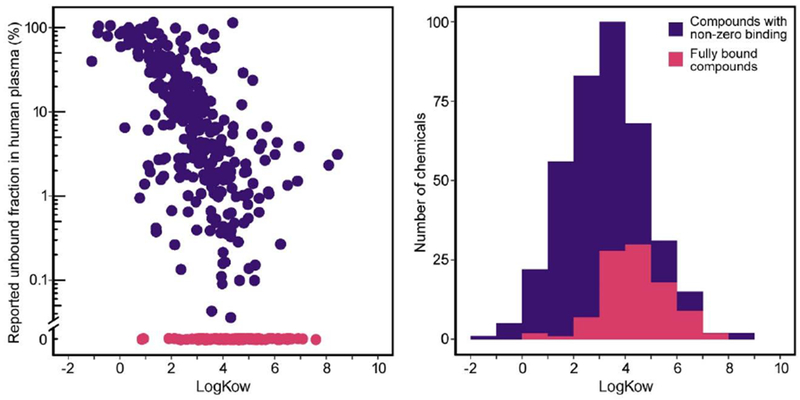
Relationship between logKow and unbound fraction in RED by Wetmore et al. (Wetmore, 2015; Wetmore et al., 2012). having non-zero unbound fractions in purple and those with zero human plasma measured using A. Scatter-plot with chemicals unbound fractions in magenta and shown along the lower x-axis. B. Histogram distribution of logKow values from panel A overall (purple), and for the subset with zero measured unbound fractions shown in magenta.
To address this need, we first selected a subset of chemicals previously tested with RED assay that had a range of values for LogKow. We then augmented the standard RED protocol to include an additional protein-free control, so as to verify equilibration reflecting Fub=100% in the absence of protein binding. Finally, we used Solid Phase Microextraction (SPME) as an alternative method for determining Fub. We found that it is not uncommon for nonpharmaceuticals that RED fails to equilibrate in the absence of proteins, leading to an apparent value of Fub = 0, and thus suggesting that Fub in plasma determined by RED assay may not be accurate. Additionally, we found that in these cases, SPME provides an accurate, though more time-consuming, alternative to determining Fub.
Materials and Methods
Chemicals
Phosphate buffer saline (PBS), LC-MS grade acetonitrile, dimethyl sulfoxide (DMSO), LC/MS grade water with 0.1% formic acid, and LC-MS grade methanol were purchased from Fisher Scientific (Waltham, MA). Pharmaceuticals: propranolol, sotalol, and isoproterenol were purchased from Molecular Devices (Sunnyvale, CA). Cisapride monohydrate was purchased from Sigma-Aldrich (St. Louis, MO). These pharmaceuticals were selected because their toxicokinetics has been extensively studied and they cover a range of plasma protein binding values as well as octanol/water partition coefficient’s (logKow). Environmental chemicals: carbaryl, pirimicarb, permethrin, acenaphthene, benzo(k)fluoranthene, chrysene, dibenz(a,h)anthracene, and phenanthrene were purchased from Sigma-Aldrich. Prometon was purchased from Accustandard (New Haven, CT). Acenaphthene, carbaryl, permethrin, pirimicarb, and prometon have been evaluated with the RED device in other studies and were selected to assess reproducibility of the RED assay data (Wetmore et al., 2012). Remaining chemicals were selected to evaluate logKow effects within the RED assay. In total, all selected chemicals cover a broad spectrum of logKow values, ranging from 0.1 (isoproterenol) to 6.75 (dibenz(a,h)anthracene) (Table 1). All chemicals and reagents were stored according to manufacturer’s guidelines. Pharmaceuticals and environmental chemicals were purchased in neat form and diluted in 100% DMSO to working stock concentration of 2 mM and stored at < −70°C until use.
Table 1.
Chemicals Evaluated
| Chemical | CASRN | Chemical Class | LogKOW | Literature % unbound (equilibrium dialysis) | Literature % unbound (SPME) |
|---|---|---|---|---|---|
| Isoproterenol | 51-30-9 | drug | 0.1 | 35%a | NA |
| Propranolol | 318-98-9 | drug | 3.48 | 7% – 39%d | 25%e |
| Cisapride | 260779-88-2 | drug | 3.18 | 2-3%c | NA |
| Pirimicarb | 23103-98-2 | pesticide | 1.7 | 16%b | NA |
| Carbaryl | 63-25-2 | pesticide | 2.36 | 69%b | NA |
| Prometon | 1610-18-0 | pesticide | 2.99 | 0%b | NA |
| Permethrin | 52645-53-1 | pesticide | 6.5 | 0%b | NAf |
| Naphthalene | 91-20-3 | industrial | 3.3 | 1,6%b | NA |
| Acenaphthene | 83-32-9 | industrial | 3.92 | 4.5%b | NA |
| Biphenyl | 92-52-4 | industrial | 4.01 | 1.6%b | NA |
| Dib enzot hiop he ne | 132-65-0 | industrial | 4.38 | NA | NA |
| Phenanthrene | 85-01-8 | industrial | 4.46 | NA | NA |
| Pyrene | 129-00-0 | industrial | 4.88 | 2.4%b | NA |
| Chrysene | 218-01-9 | industrial | 5.81 | NA | NA |
| Benzo(k) fluoranthene | 207-08-9 | industrial | 6.11 | NA | NA |
| Dibenz(a,h) anthracene | 53-70-3 | industrial | 6.75 | NA | NA |
FDA package insert
(Sethi et al., 2014) reported fraction unbound values from 11%-80% depending on concentration, using radiolabeled permethrin and organic solvent exfraction.
Rapid Equilibrium Dialysis (RED) Assay
Plasma protein binding was evaluated for each chemical utilizing the rapid equilibrium dialysis (RED) method as described in other publications (Figure 2A) (Rotroff et al., 2010; Wetmore et al., 2012), but modified to incorporate no protein equilibrium controls (Figure 2B). Human plasma was recovered from whole blood donations using anti-coagulant (K2EDTA) and pooled from healthy donors at a U.S. Food and Drug Administration-licensed donor center (HMPLEDTA2; Bioreclamation, Westbury, NY). All donors tested negative for HIV V2 AB and HCV AB and non-reactive for HBSAG, HIV-1 RNA, HCV RNA, HBV DNA and STS. Prior to analysis, human plasma, stored at <−70°C, was thawed to room temperature and centrifuged at 2000×g for 10 minutes to remove particulates (Waters et al., 2008; Wetmore et al., 2012). The assay was conducted using RED inserts (catalog no. 90006, Pierce Biotechnology, Rockford, IL) according to manufacturer’s instructions, with protocol modification to incorporate protein free equilibrium controls. The RED membrane 8K MWCO was used in all experiments. The only deviation from previously published protocols was addition of equilibrium controls comprising of PBS buffer in both sample and buffer chambers, which are designed to verify that chemicals frilly equilibrated within the device in the absence of proteins (Figure 2B).
Figure 2. Overview of experimental design.
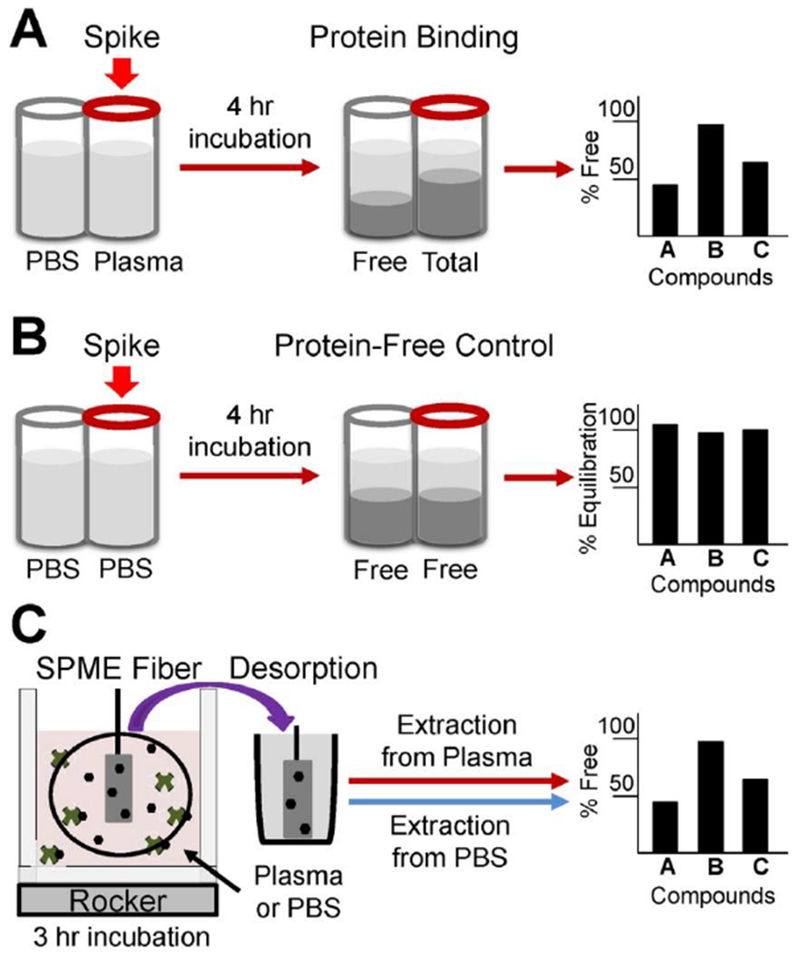
(A) Rapid Equilibrium Dialysis (RED) assay following manufacturer protocol to measure percent unbound in plasma; (B) additional protein-free control for RED assay to measure percent equilibration in absence of proteins; (C) alternative method to measure percent unbound in plasma using Solid Phase Micro-Extraction (SPME), adapted from (Musteata et al., 2006).
DMSO-dissolved chemical stock solutions were diluted 200-fold in human plasma to test concentration of 10 μM. This concentration was used by previous investigators for generating data for IVIVE (Rotroff et al., 2010; Wetmore et al., 2012), and corresponds to a typical in vitro test concentration. Preliminary experiments with the three drugs conducted at both 1 and 10 μM gave similar results, so subsequent experiments were only conducted at 10 μM. Moreover, because the aim of IVIVE to estimate oral equivalent doses from in vitro assay results, which are then compared to human exposure levels for the purposes of prioritization, it is only necessary to measure Fub in the range of nominal test concentrations. Specifically, if the margin of exposure is large, then a smaller Fub at lower environmental concentrations (where there is less saturation of binding sites) would only make the margin of exposure larger. On the other hand, if the margin of exposure is small, then the test concentrations are already in the range of human exposures.
Final DMSO concentration in each assay was 0.5% (v/v). Sealing tape was placed on each RED device and it was incubated at 370C for 4 hours at 100 oscillations per minute on an orbital rocker (Waters et al., 2008; Wetmore et al., 2012). Upon completion of incubation, 50 μL aliquots were removed from each chamber and matrix matched with equal volumes of plasma, or buffer. Samples were diluted with 300μL 100% acetonitrile and frozen at −80°C until analysis (Waters et al., 2008; Wetmore et al., 2012). Aliquots of spiked human plasma, and PBS working stock solutions were removed to measure percent recovery. These percent recovery samples followed the same matrix match and acetonitrile dilution pattern. All RED assays were completed in triplicate.
The percentage of a chemical that remains unbound was calculated by measuring the concentration within both chambers, sample and buffer. The concentration in the buffer chamber was then divided by the concentration detected in the sample chamber and multiplied by 100. Experiments were performed in triplicate and percent unbound values were averaged to determine the final unbound value. No testing concentrations were below the analytical detection limits.
Solid Phase Micro-Extraction (SPME) Assay
SPME techniques present a possible alternative to accurately measure protein binding for chemicals not suitable for the RED assay. The SPME device consists of small rods covered in a material that absorbs a fraction of the chemical in equilibrium with the sample’s unbound concentration. This technique has been utilized in a variety of applications such as ecological contamination monitoring, in vitro protein binding modeling, and analysis of a variety of chemicals (Blaauboer, 2010; Musteata et al., 2006; Peltenburg et al., 2015).
Protein binding analysis followed previously described methods (Musteata et al., 2006; Peltenburg et al., 2015) with some modifications at room temperature (Figure 2C). C18 SPME fibers were preconditioned in methanol/Milli-Q water solution (50:50). Samples were placed into 2 ml amber glass vials containing 200 μL glass inserts. Total sample volume was 100 μL and analyses were performed in triplicate. Prior to SPME fiber extraction, samples were allowed to equilibrate on an orbital shaker (500 rpm) for lhr. After equilibration, SPME fibers were inserted through the vial cap septa and placed in the incubator on an orbital shaker (500 rpm) for 3 hrs. After that, SPME fibers were removed, rinsed briefly with Milli-Q water and placed in 100 μL of 100% acetonitrile. Fibers were placed on an orbital shaker (500 rpm) and desorbed for 30 min. Standard solutions were prepared in PBS, following the same dilution patterns and fiber extraction, desorption procedures as previously mentioned. SPME protein binding controls (propranolol, acenaphthene, and permethrin) were tested at 10 μM concentrations in pooled human plasma, prepared in the same manner as in the RED assay. These control chemicals were incorporated in order to validate the SPME method’s ability to produce accurate and precise protein binding data.
Determination of unbound chemical concentrations using SPME followed procedures outlined elsewhere (Musteata et al., 2006). Briefly, the fiber constant (fc), representing the partition coefficient between unbound chemical in solution and the amount of absorbed to the fiber, was determined by analyzing standard solutions of chemical in PBS.
| (3) |
Where C0,s is the initial concentration prior to fiber extraction and Ce,s represents the concentration of the chemical extracted by the fiber. When SPME procedure was performed in a sample containing proteins and a chemical is extracted by the fiber (Ce), the unbound concentration (Cfree) in the sample is determined using the following equation.
| (4) |
The final total concentration (Ct) of a chemical in the sample was determined using the following equation, where Co represents the initial chemical concentration prior to fiber extraction.
| (5) |
Ultimately, the percentage unbound (% Unbound) was calculated from the total and free concentration of the chemical as displayed below.
| (6) |
Analytical Chemistry
All analytical measurements were performed using Agilient (Santa Clara, CA) 6470 triple quadrupole mass spectrometer operating in positive ion mode with a Waters Acquity H class HPLC (Milford, MA). Chromatography separation was performed on a C18 column (Agilent Zorbex Eclipse Plus C18 3.0×50mm, 1.8 micron) with a C18 guard column. Complete HPLC/MS and GC/MS conditions for all chemicals are listed in Supplemental Tables 1–2.
Analysis of pharmaceuticals:
Aqueous mobile phase consisted of 0.1% formic acid and acetonitrile for organic mobile phase. Sample (10 μL) injections were separated using the following a solvent gradient: (1) 2% organic for 1 min; (2) linear gradient ramp to 95% organic over 1.5min; (3) 95% organic maintained for 1.5 min; (4) linear gradient ramp to 2% organic over 0.2 min; (5) 2% organic condition held for 3.8 min until next injection. Total analysis time was 8 minutes at a flow rate of 400 μL per minute.
Analysis of environmental chemicals:
Chromatography conditions followed a previously described method with slight modification (Wetmore et al., 2012). Aqueous mobile phase consisted of 0.1 % formic acid and methanol for organic mobile phase. Sample (5 μL) injections were separated using the following a solvent gradient: (1) 20% organic for 0.5 min; (2) linear gradient ramp to 100% organic over 4.5 min; (3) maintain 100% organic for 1 min; (4) linear gradient ramp to 20% organic over 0.5 min; and (5) maintain 20% organic for 2 min prior to the next injection. Total analysis time was 8.5 min per sample at a flow rate of 400 μL per minute. All samples (environmental chemicals, and pharmaceuticals) were introduced to the mass spectrometer in splitless mode with an AJS ESI ion source.
Results
Rapid Equilibrium Dialysis with Additional Controls
As shown in Table 1, a library of 13 chemicals, comprising of pharmaceuticals, pesticides, and industrial chemicals, with logKow values ranging from less than 1 to greater than 5 were selected to test the reproducibility and applicability of RED.
Evaluated pharmaceuticals (cisapride, propranolol, and isoproterenol) all had LogKOW <5, and the resulting Fub measurements were consistent with drug label references or literature-based values (Figure 3A). Cisapride was the most highly bound and isoproterenol was mostly free. Additionally, all pharmaceutical successfully equilibrated within the RED device in PBS controls (Figure 3B), with protein-free free fractions of >80%. We also measured mass balance for these compounds in both protein binding experiments and equilibrium controls, with recovery compared to stock solutions >70% for cisapride and isoproterenol, but only 43% for propranolol.
Figure 3. Rapid equilibrium dialysis results.
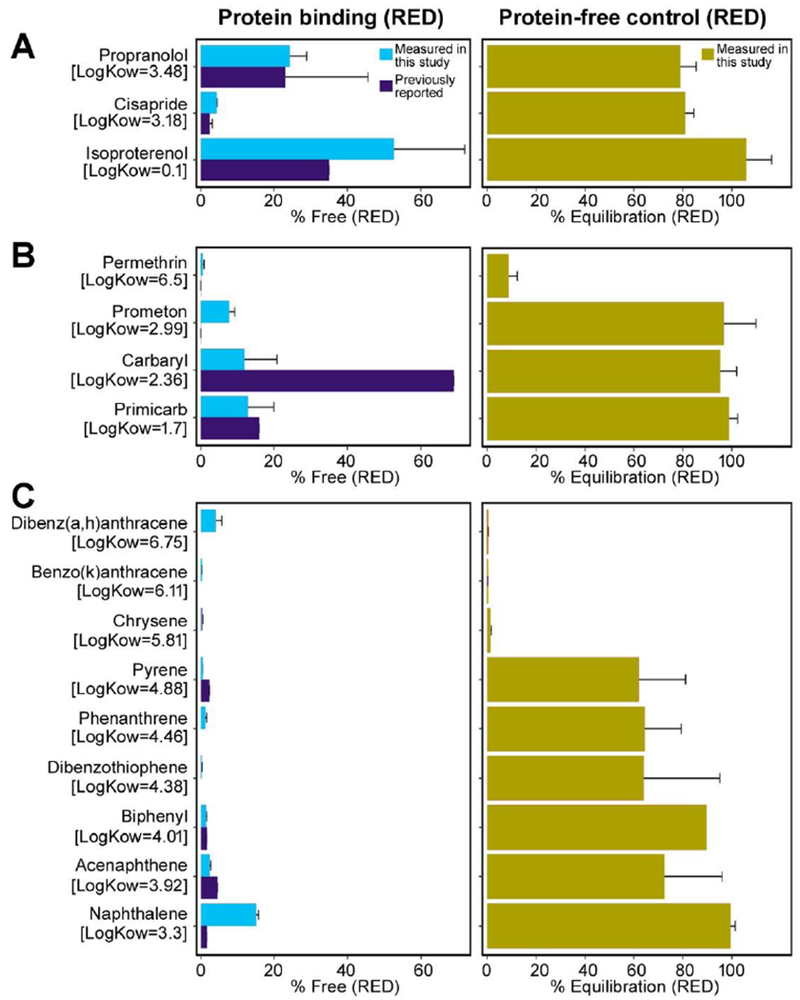
A, C, and E: Measured unbound fraction in pooled human plasma compared to reported values in the literature using equilibrium dialysis methods for drugs (A), pesticides (C), and industrial chemicals (E). B, D, and F: Corresponding measured equilibration percentage using equilibrium dialysis with PBS in both chambers, for drugs (B), pesticides (D), and industrial chemicals (F).
For pesticides, measured values for pirimicarb and permethrin were very similar to those previously reported using RED, whereas values for prometon and carbaryl were substantially different (Figure 3C). For instance, carbaryl yielded a lower value of 12% unbound, as compared to a reported value of 70% unbound (Wetmore et al., 2012), displaying a more highly bound characteristic similar to pirimicarb and other carbamate insecticides (Alden, 1991). The three pesticides with logKow <3 (prometon, carbaryl, and pirimicarb) all successfully equilibrated with PBS controls, with protein-lfee free fractions >95%; however, permethrin was poorly equilibrated, with a protein-free free fraction of <10% (Figure 3D).
For the industrial chemicals other than naphthalene, Fub<5% was measured, consistent with previous reports (Figure 3E). For naphthalene, a value of Fub=15% was measured, whereas it was also previously reported to be ~2% (Wetmore et al., 2012) (Figure 3E). However, there is a clear trend of lower equilibration with higher logKow, with the lowest logKow compound (naphthalene) completed equilibrated with a protein-free free fraction of 100%, and the compounds with logKow >5 completely unequilibrated (Figure 3F). Only biphenyl and naphthalene had equilibration of >80%. All completely unequilibrated compounds have an “apparent” unbound fraction of 0%.
No volume shifts in liquid across the two sides of the membrane were observed. Additionally, for the three pharmaceuticals, 4 PAHs, and permethrin, similar results (not shown) were observed using the 12K MWCO membrane, extending the incubation time from 4 to 5 hr, or using deproteinated plasma instead of PBS for equilibrium controls. For the larger pore size membrane, mass balance was also measured compared to stock solutions, with recoveries between 47% and 103%.
Solid Phase MicroExtraction
Because low logKow chemicals appeared to consistently equilibrate in the RED device under protein-free conditions, only the subset of chemicals with logKow >3 were tested using SPME. Only for propranolol has Fub been previously measured using SPME, and our results for this compound are consistent with the reported value (Figure 4). For compounds with logKow >3.5, SPME results for Fub were consistently higher than those from RED (Figure 4). In particular, all of the compounds with “apparent” unbound fraction of 0% via RED had measurable values for Fub based on SPME. For instance, for permethrin, SPME yielded Fub of near to 70%, while the RED device resulted in a value <1%. Additionally, the SPME value was within the range of ll%-80% reported by (Sethi et al., 2014), who used 14C- labeled permethrin analyzed through a 3-phase organic solvent extraction procedure to isolate bound and unbound concentrations.
Figure 4. Comparison of RED and SPME results.
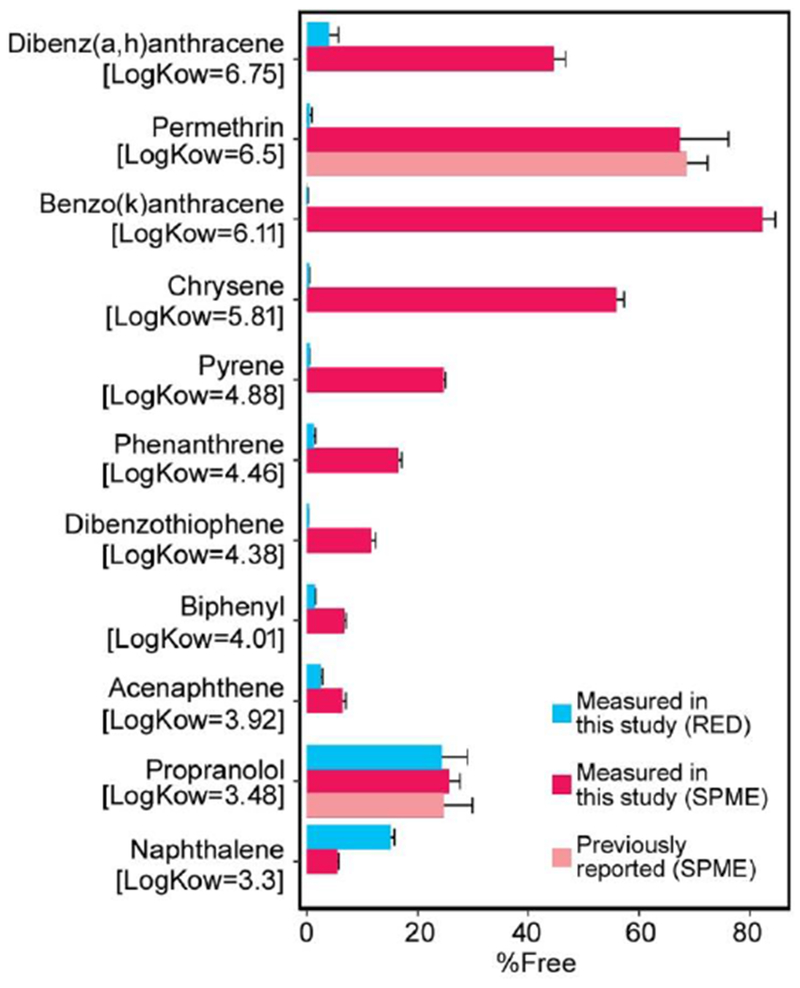
Unbound fraction in pooled human plasma measured using rapid equilibrium dialysis, measured using solid phase microextraction, and reported in the literature using solid phase microextraction.
Conclusions
High-throughput in vitro assays offer the promise of more humane, human-relevant, and efficient testing of chemical toxicity as compared to animal bioassays. However, in this new paradigm, the challenge of extrapolating from experimental animals to humans is replaced by an equally daunting challenge of extrapolating from in vitro bioactivity to in vivo toxicity. A critical component to addressing this challenge is IVIVE, which combines in vitro and in silico approaches to convert in vitro concentrations to equivalent in vivo exposure levels. IVIVE requires determination of a chemical’s unbound fraction, as this parameter plays a significant role in assessing its distribution throughout the body, and is also important for determination of its rate of elimination via metabolism and excretion. This parameter is also used widely to estimate bioavailability and safe dosing levels (Bohnert and Gan, 2013). In the pharmaceutical industry, equilibrium dialysis is recognized as the standard, validated approach to determining free fraction of a drug in plasma. The incorporation of RED into pharmaceutical evaluation dramatically reduced time, labor, and data uncertainty common with other equilibrium dialysis methods (Waters et al., 2008). Thus, RED assay was adapted to screen hundreds of environmental chemicals with a hope to provide data necessary for informing in vitro to in vivo extrapolation (IVIVE) of the dose in chemical risk assessments (Wetmore, 2015).
However, because the range of chemical properties is much wider among environmental chemicals than it is among pharmaceuticals, it is necessary to better understand the domain of applicability of RED so as to ensure accurate prediction of chemical toxicokinetics. This study confirms that chemical water solubility represents a critical factor in determining suitability of the RED approach toward accurate protein binding evaluation. The implementation of protein-free controls through the evaluation of environmental chemicals spanning a range of water solubility revealed that hydrophobic chemicals with larger values of logKow > 5 failed to equilibrate fully within the device, thus yielding inaccurate data. Moreover, the trend towards poor equilibration appears to be apparent at even lower values of logKow of 3 or 4. Interestingly, the degree of equilibration does not seem to be influenced by mass balance recovery, as both equilibrating pharmaceuticals and non-equilibrating high logKow chemicals had similar ranges of recovery. Thus, significant non-specific binding or loss does not appear to be the reason for the lack of equilibrium, suggesting that the RED membrane is for some reason ill-suited for high logKow compounds. However, we could not definitively determine whether that attachment to the membrane was the specific cause of these problems.
Whatever their source(s), the resultant inaccuracies can result in erroneous IVIVE and pharmacokinetic modeling by altering derived OEDs. The IVIVE models used to extrapolate from in vitro concentration to in vivo dose (Bell et al., 2018; Wambaugh et al., 2015; Wetmore, 2015), take a health-protective approach by matching in vitro active concentrations to Css based on total blood concentration, as opposed to free concentration. As a result, increasing the unbound chemical concentration contributes to a greater metabolic clearance, constituting to a higher OED, so the challenges with using data from RED assay as identified in this study may result in “conservative” estimates that tend to overestimate risks.
A recent comparison of IVIVE methods with in vivo toxicokinetic data in the rat confirms that IVIVE predictions using these methods tend to be “conservative” in the sense that they overestimate the steady state Css (Wambaugh et al., 2018), although only 3 of the 45 compounds investigated had logKow > 5. In addition, Casey et al. (2018) recently suggested improvements to IVIVE by incorporating Fub into the calculation of the estimated dose by matching in vitro active concentrations to Css based free concentration:
| (7) |
This equation is the same as equation (1) but multiplied by Fub. In this case, underestimating Fub can result in an underestimate of the Css, leading to equivalent in vivo doses that may not be adequately protective. Thus, accurate estimation of Fub remains a critical concern for implementing IVIVE in high throughput screening and prioritization of chemicals.
SPME is a widely available technique for chromatographic-spectrometric analysis that relies on solvent-free sample preparation whereby the analytes are extracted from a gaseous or liquid sample by absorption in, or adsorption on, a fiber that is coated with various polymers and placed inside an injection needle or inside a capillary (Pragst, 2007). SPME is applied in the analysis of various biological fluids and specimens in both clinical and forensic toxicology. In addition, SPME is used as sampling tool for freely dissolved concentrations, including for pharmaceuticals, especially for highly protein-bound compounds (Peltenburg et al., 2015). The SPME technique implemented in this study demonstrates its suitability as an alternative method to RED assays to evaluate in vitro protein binding of more hydrophobic environmental chemicals. Because of the low concentration levels of chemicals in plasma, microextraction sample preparation methods also allow for less consumption of solvent, reagents, and packing materials, and small sample volumes can be used (Moein et al., 2014). Moreover, SPME has additional advantages, such as increasing detection signal during chemical analysis by reducing instrumental noise commonly attributed to matrix effects (Maciel et al., 2019).
Overall, our results suggest that IVIVE approaches that rely on RED for estimating the extent of protein binding may be inaccurate for more hydrophobic compounds. Fortunately, the accuracy of RED for any given compound can be verified through use of a protein-free control, which measure how well the compound equilibrates within the RED device in the absence of proteins. Thus, we propose an updated IVIVE workflow that adds a protein-free control to the RED protocol. Then, in cases where compounds fail to adequately equilibrate, alternative approaches to measuring protein binding, such as SPME, would be used. Additional optimization of alternative methods would be beneficial in order to better define a comprehensive workflow for protein binding measurements for using in IVIVE. Furthermore, additional studies comparing IVIVE predictions with in vivo methods to measure free and bound chemicals in plasma would be useful for validation, particularly for high logKOW compounds for which there is greater uncertainty in the measurement of Fub. These refinements will provide additional confidence in the use of IVIVE as part of high-throughput screening programs of chemicals, further advancing the National Academies’ (2007) vision for Toxicity Testing in the 21st Century.
Supplementary Material
HIGHLIGHTS.
Fraction unbound (Fub) is a key parameter for in vitro-in vivo extrapolation
Rapid equilibrium dialysis (RED) may lead to incorrect Fub for lipophilic compounds
Checking equilibration of protein-free controls can verify the validity of RED
Solid phase microextraction is a recommended alternative for measuring Fub
Acknowledgments
The authors with to that B.A. Wetmore, J.F. Wambaugh, and R.S. Thomas for useful discussions during the design of this study. This work was supported, in part, by grants from the U.S. EPA (STAR RD83561202) and National Institutes of Health (P42 ES027704), and institutional support from Texas A&M University. K.C. Ferguson was supported by the U.S. Army Advanced Civil Schooling Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors declare they have no competing interest.
References
- Alden CL, 1991. Toxicologic pathology of the kidney, In: Haschek-Hock WM, Rousseaux CG (Eds.), Fundamentals of Toxicologic Pathology. Academic Press, New York. [Google Scholar]
- Auner AW, Tasneem KM, Markov DA, McCawley LJ, Hutson MS, 2019. Chemical-PDMS binding kinetics and implications for bioavailability in microfluidic devices. Lab Chip. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SM, Chang X, Wambaugh JF, Allen DG, Bartels M, Brouwer KLR, Casey WM, Choksi N, Ferguson SS, Fraczkiewicz G, Jarabek AM, Ke A, Lumen A, Lynn SG, Paini A, Price PS, Ring C, Simon TW, Sipes NS, Sprankle CS, Strickland J, Troutman J, Wetmore BA, Kleinstreuer NC, 2018. In vitro to in vivo extrapolation for high throughput prioritization and decision making. Toxicol In Vitro 47, 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaauboer BJ, 2010. Biokinetic modeling and in vitro-in vivo extrapolations. J Toxicol Environ Health B Crit Rev 13, 242–252. [DOI] [PubMed] [Google Scholar]
- Bohnert T, Gan LS, 2013. Plasma protein binding: from discovery to development. J Pharm Sci 102, 2953–2994. [DOI] [PubMed] [Google Scholar]
- Casey WM, Chang X, Allen DG, Ceger PC, Choksi NY, Hsieh JH, Wetmore BA, Ferguson SS, DeVito MJ, Sprankle CS, Kleinstreuer NC, 2018. Evaluation and Optimization of Pharmacokinetic Models for in Vitro to in Vivo Extrapolation of Estrogenic Activity for Environmental Chemicals. Environ Health Perspect 126, 97001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung EN, Chen YH, Lau YY, 2003. Semi-automatic high-throughput determination of plasma protein binding using a 96-well plate filtrate assembly and fast liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 795, 187–194. [DOI] [PubMed] [Google Scholar]
- Kariv I, Cao H, Oldenburg KR, 2001. Development of a high throughput equilibrium dialysis method. J Pharm Sci 90, 580–587. [DOI] [PubMed] [Google Scholar]
- Kelly JG, McDevitt DG, 1978. Plasma protein binding of propranolol and isoprenaline in hyperthyroidism and hypothyroidism. Br J Clin Pharmacol 6, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug deliv Rev 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Maciel EVS, de Toffoli AL, Lancas FM, 2019. Current status and future trends on automated multidimensional separation techniques employing sorbent-based extraction columns. J Sep Sci 42, 258–272. [DOI] [PubMed] [Google Scholar]
- Moein MM, Said R, Bassyouni F, Abdel-Rehim M, 2014. Solid phase microextraction and related techniques for drugs in biological samples. J Anal Methods Chem 2014, 921350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musteata FM, Pawliszyn J, Qian MG, Wu JT, Miwa GT, 2006. Determination of drug plasma protein binding by solid phase microextraction. J Pharm Sci 95, 1712–1722. [DOI] [PubMed] [Google Scholar]
- National Research Council, 2007. Toxicity testing in the 21st century: A vision and a strategy. The National Academies Press, Washington, DC. [Google Scholar]
- Pearce RG, Setzer RW, Strope CL, Wambaugh JF, Sipes NS, 2017. httk: R Package for High-Throughput Toxicokinetics. J Stat Softw 79, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltenburg H, Bosman IJ, Hermens JL, 2015. Sensitive determination of plasma protein binding of cationic drugs using mixed-mode solid-phase microextraction. J Pharm Biomed Anal 115, 534–542. [DOI] [PubMed] [Google Scholar]
- Pragst F, 2007. Application of solid-phase microextraction in analytical toxicology. Anal Bioanal Chem 388, 1393–1414. [DOI] [PubMed] [Google Scholar]
- Rotroff DM, Wetmore BA, Dix DJ, Ferguson SS, Clewell HJ, Houck KA, LeCluyse EL, Andersen ME, Judson RS, Smith CM, Sochaski MA, Kavlock RJ, Boellmann F, Martin MT, Reif DM, Wambaugh JF, Thomas RS, 2010. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol Sci 117, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi PK, Muralidhara S, Bruckner JV, White CA, 2014. Measurement of plasma protein and lipoprotein binding of pyrethroids. J Pharmacol Toxicol Methods 70, 106–111. [DOI] [PubMed] [Google Scholar]
- Wambaugh JF, Hughes MF, Ring CL, MacMillan DK, Ford J, Fennell TR, Black SR, Snyder RW, Sipes NS, Wetmore BA, Westerhout J, Setzer RW, Pearce RG, Simmons JE, Thomas RS, 2018. Evaluating In Vitro-In Vivo Extrapolation of Toxicokinetics. Toxicol Sci 163, 152–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambaugh JF, Wetmore BA, Pearce R, Strope C, Goldsmith R, Sluka JP, Sedykh A, Tropsha A, Bosgra S, Shah I, Judson R, Thomas RS, Woodrow Setzer R, 2015. Toxicokinetic Triage for Environmental Chemicals. Toxicol Sci 147, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters NJ, Jones R, Williams G, Sohal B, 2008. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J Pharm Sci 97, 4586–4595. [DOI] [PubMed] [Google Scholar]
- Wetmore BA, 2015. Quantitative in vitro-to-in vivo extrapolation in a high-throughput environment. Toxicology 332, 94–101. [DOI] [PubMed] [Google Scholar]
- Wetmore BA, Wambaugh JF, Ferguson SS, Sochaski MA, Rotrofi; D.M., Freeman, K., Clewell, H.J., 3rd, Dix, D.J., Andersen, M.E., Houck, K.A., Allen, B., Judson, R.S., Singh, R, Kavlock, R.J., Richard, A.M., Thomas, R.S., 2012. Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol Sci 125, 157–174. [DOI] [PubMed] [Google Scholar]
- Yoon M, Campbell JL, Andersen ME, Clewell HJ, 2012. Quantitative in vitro to in vivo extrapolation of cell-based toxicity assay results. Crit Rev Toxicol 42, 633–652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.