

Abstract
High-grade gliomas, particularly glioblastomas (grade IV), are devastating diseases with dismal prognoses; afflicted patients seldom live longer than 15 months, and their quality of life suffers immensely. Our current standard of-care therapy has remained essentially unchanged for almost 15 years, with little new therapeutic progress. We desperately need a better biologic under standing of these complicated tumors in acomplicated organ. One area of rejuvenated study relates to extracellular vesicles (EVs)—membrane-enclosed nano- or microsized particles that originat from the endosomal system or are shed from the plasma membrane. EVs contribute to tumor heterogeneity (including the maintenance of glioma stem cells or their differentiation), the impacts of hypoxia (angiogenesis and coagulopathies), interactions amid the tumor microenvironment (concerning the survival of astrocytes, neurons, endothelial cells, blood vessels, the blood–brain barrier, and the ensuing inflammation), and influences on the immune system (both stimulatory and suppressive). This article reviews glioma EVs and the ways that EVs manifest themselves as autocrine, paracrine, and endocrine factors in proximal and distal intra- and intercellular communications. The reader should note that there is much controversy, and indeed confusion, in the field over the exact roles for EVs in many biological processes, and we will engage some of these difficulties herein.
Keywords: exosomes, microvesicles, extracellular vesicles, brain tumor, glioma, immune response, astrocytes, inflammation, microenvironment
1. INTRODUCTION
High-grade gliomas (HGGs), such as glioblastomas (which are grade IV gliomas) and other grade III glial tumors, are devastating diseases of the brain and central nervous system; because of their locations in the brain, treatment of these diseases may lead to debilitating collateral damage in an organ that can suffer little of that. The disease and treatments inflict staggering financial burdens on patients and their families and on the health-care system (114), with a degenerating quality of life that can literally change the very nature of self.
Current standard-of-care therapy for primary gliomas—maximal surgical resection followed by chemoradiation and then more chemotherapy—has remained largely unchanged since 2005 and results in a median survival of less than 15 months (131). Tumor recurrence is almost universal, and few salvage strategies are available for those patients. For the age groups with the highest incidence (over age 40 and over age 75), five-year survival rates range from 7.7% to 1%. Five year survival rates for other high-grade glial tumors [grade III anaplastic astrocytomas, oligoastrocytomas, and oligodendrogliomas (86)] in those age groups range from approximately 30% to 1% (102). Despite advancements in our knowledge of the molecular pathways active in gliomas, targeted therapy successes have been modest at best (94), and the prognosis for patients with HGGs remains abysmal. Thus, new avenues of therapy are essential, including immunotherapy. Our knowledge of HGG biology also desperately needs to expand.
Therapeutic resistance is at the heart (or brain?) of HGG malignancy (6) and has been recognized for decades (9, 91). While our understanding of these defense mechanisms has surely improved, our efforts to counter the resistance have proven essentially futile. HGGs use both defensive and offensive strategies to overcome the challenges of interventions, and extracellular agents of the tumors, such as extracellular vesicles (EVs), are gaining acceptance as manipulators of the tumor milieu (66). HGG EVs affect both the host structures (e.g., the brain) and the cells that form the microenvironment, including invading immune cells.
Immunotherapy in the form of immune checkpoint inhibitors has shown tremendous advancements in other cancers, notably with US Food and Drug Administration approval of the use of such drugs (i.e., antibodies) in the treatment of various cancers (52). These achievements led to cancer immunotherapy’s designation as the breakthrough of the year in 2013 (19). Despite those successes, checkpoint inhibitors and other forms of immunotherapy have been far less beneficial in the treatment of HGGs (93). Glioma-induced immune suppression, both local and systemic, is a profound barrier to immunotherapy, with multiple soluble, enzymatic, and cell-associated factors blamed for immune resistance (87, 147). In addition to the generation of a protumor environment, one common denominator among these immunosuppressive mechanisms is the presence of EVs derived from the tumor, from the cells of the microenvironment, and from the immune cells themselves. These vesicles weave a tapestry of interaction of both pro- and antitumor activities that almost always benefits the tumor (108). In this context, this article reviews a spectrum of research that has established EVs as prominent actors in the pathophysiology and aberrant biology of HGGs, focusing on EV influences on tumor heterogeneity, on cells of the tumor microenvironment, and on the immune responses against HGGs.
2. EXTRACELLULAR VESICLES: A BRIEF AND INCOMPLETE HISTORY OF NOMENCLATURE
We have known for decades that cells release various forms of extracellular particles, ranging from “platelet dust” (53, 155) to “roundish bodies, having amorphous structure” (10, p. 33) likely to be the matrix vesicles of calcification (4) to tissue-specific derivatives called prostasomes (130). The relevance of such membranous vesicles has perhaps been more recently appreciated with the coining of the term exosome by Rose Johnstone (63) [referring to “vesicles…literally jettisoned from maturing blood reticulocytes” (51, p. 367)], based on work both from her own group (105) and from Cliff Harding in Phil Stahl’s laboratory (50). In these cases, the term exosome described vesicles that originated in the endosomal system but were released extracellularly, in a sort of reverse endocytosis (62). We now employ this term for vesicles that form in maturing endosomes, which produce lumenal invaginations that bud off inside the endosome as intralumenal vesicles or intravesicular bodies; the endosomal compartment where this budding occurs is now called a multivesicular body. Canonically, multivesicular bodies merge into the lysosomal pathway for content degradation or recycling. However, if the multivesicular body instead fuses with the cellular plasma membrane, the interior vesicles could be released extracellularly and would then be termed exosomes (160).
However, such nomenclature preceded Johnstone’s definition, and not always in reference to EVs. The term exosome has been used in several scientific contexts as far back as 1970 by Allen Fox and Sei Byung Yoon (33–35), who described in Drosophila “DNA segments [that] enter the cells of treated individuals and become firmly associated with their homologous chromosome segments, but are never integrated into the linear structure of the chromosome” (34, p. 1608). To further complicate matters, the term exosome complex refers to a multiprotein conglomerate RNA-processing machine required for the maturation of certain rRNAs and the degradation of some mRNAs (97). Trams et al.(140) had previously proposed the term exosome (in a brain tumor cell line, curiously enough) for exfoliated membrane vesicles that had similar sizes and biochemical qualities to those of the shed membrane particles that today we likely would call ectosomes or microvesicles (even as they did then). In a 2005 review, Johnstone (62) admitted some regret over neglecting to check for prior use of the word. Thus, one designation for a subset of EVs historically has covered much ground, and not necessarily in relationship to EVs at all.
The term microvesicle, denoting lipid-membrane-enclosed extracellular particles, goes back (debatably) at least as far as the original EVs, the neuronal synaptic vesicles (22). There has been a trend in the EV field to reserve the term microvesicle for membrane particles released (described variously as shed, blebbed, exfoliated, or exocytosed) directly from the cell’s plasma membrane (39); early publications noted the plasma membrane as the source of these vesiculations (11, 165). The term ectosome, originally used to describe the cortical portion of sponges (12), is also used in reference to vesicles that are directly assembled and released at the plasma membrane (18) and provides another source of confusion (18). The term oncosome would thus mean vesicles [originally considered to be microvesicles (1); the coining of this term is credited to Brian Meehan (92)] released from tumor cells carrying oncogenic cargo, perhaps extended to the entire cancer vesiculome (79). In EV usage, oncosomes are increasingly seen in the context of so-called large oncosomes (25, 26), 1–10 μm in diameter, which were studied and popularized by the Di Vizio laboratory (95, 99). However, even some nontransformed cells release large vesicles (71), and suggestions have been made that such non-oncosomes could be called megavesicles or shed cytoplasts (92). Meehan et al. (92) maintained that the distinction between oncosomes and large oncosomes is important.
The ultimate definition of an exosome (as opposed to a microvesicle, ectosome, shed vesicle, etc.) is the vesicle’s cellular or compartmental origin—that is, release through the endosomal pathway via the multivesicular body, at the plasma membrane for exosomes, or via direct pinching off from the plasma membrane for microvesicles (160). Experimentally, we seldom witness such events, and instead attempt to define or categorize EVs based on biophysical and biochemical parameters. Differences in vesicle sizes and sedimentation coefficients (i.e., differences in the g-forces at which vesicles will pellet during centrifugation) have become surrogate means of defining vesicle types (39). Those size ranges tend to be 30–150 nm for exosomes, 100–2,000 nm for microvesicles (etc.), and up to 10 μm for apoptotic bodies [a third EV type not discussed in depth here, along with (deoxy)ribonuclear particles and lipoprotein particles] (41, 42). Zhang et al. (164) recently described small EVs of approximately 30 nm called exomeres, and cryo-electron microscopy has revealed many types of single-to-multiple membranous EVs of sundry shapes and sizes (162). Other differences at the molecular level have also been noted between lower and higher sedimentation rates (60, 158), and those differences even extend to what we would call the exosome (or small EV) level (71, 153).
It is little wonder that the EV nomenclature can be confusing (for a fascinating compilation of EV names in the literature, see 144). This complexity is one reason that the field tends to use the all-encompassing term EV to refer to almost any such membrane-enclosed content released from essentially any cell type (39; see also 125). I use the term EV in the remainder of the review while being fully cognizant that the vesicular definitions may need refinement in some situations where vesicle generation is actually visualized, in order to distinguish between exosomes and other extracellularly released vesicles.
EVs have been referred to as “tiny cellular surrogates” (40), reflecting the cellular content and identity in the EVs released from parental cells, including, to some extent, the topography of membrane components. EV cargo repositories are available online for proteins, some RNAs, and some lipids (e.g., from ExoCarta, EVpedia, and Vesiclepedia) (65) and for various RNAs as part of the exRNA Atlas (http://exrna-atlas.org). Lipidomics remains an area of development (127), and work on the metabolomics of EVs is slowly gaining ground (2, 104, 110). Thus, EV molecular contents are likely critical players in the impacts of EVs on recipient cells in terms of activities delivered, induced, or generated. Because these contents might reflect the status of the original cell in sickness and in health, they may also be sources of biomarkers in disease pathologies (114). Groups are working to develop guidelines for terms, definitions, and reproducibility (154). Figure 1 depicts key aspects of the biogenesis and contents of EVs. Note that cargo sorting of the varied components of EVs is likely a critical aspect of EV identity and functionality, even if it is not well understood (3). Furthermore, alternate localizations of proteins that are generally considered cytosolic [e.g., heat shock proteins (HSPs)] and lipids that ordinarily face inside the cell (e.g., phosphatidylserine) are examples of noncanonical presentations that can affect EV–target cell interactions and recipient cell impact (1, 29, 43). Molecular entities such as various RNAs, proteins, and lipids may also be transferred from EVs to recipient cells, or the molecules on EVs may have impacts on recipient cells. Some examples of this with references found within this review are shown in Table 1.
Figure 1.
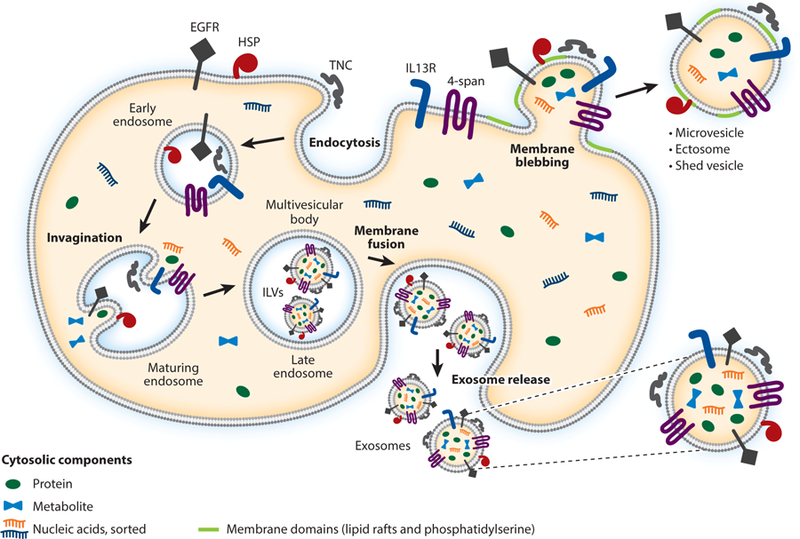
Types of extracellular vesicles (EVs) and their formation. EVs consist of three classes that are defined by their biogenesis but have frequent overlap of molecular content: exosomes, microvesicles, and apoptotic bodies (the last of which are not discussed here). The figure depicts a highly diagrammatic cell with various types of membrane and extracellular proteins that are of interest in high-grade gliomas: epidermal growth factor receptor (EGFR), heat shock proteins (HSPs), interleukin 13R (IL13R), tenascin C (TNC), and a generic tetraspanin protein (4-span). (Note that the cells, organelles, and vesicles are not drawn to scale.) Other cytosolic proteins or biomolecules (nucleic acids and metabolites) are also represented. Endocytosis (e.g., phagocytic, fluid-phase pinocytic, receptor mediated, and caveolar endocytosis) routes membrane and membrane-associated proteins into the endocytic pathways and early endosomes. Endosomal trafficking drops the lumenal pH, promoting alterations in lipids and RAB GTPases, which bestow identities on the intracellular vesicles. The endosomes mature and progress toward late endosomes and multivesicular bodies (MVBs). Here, lumenal pH changes, membrane lipid reorganization, and ESCRT complex activities (not shown) drive invaginations or reverse budding of the endosome limiting membrane. Cytosolic components (proteins, metabolites, and nucleic acids) are sorted into these bubbles. Note that some intracellular components (e.g., some RNAs) are not sorted into EVs, indicating a selective process. When they bud off into the lumenal interior, the newly formed interior vesicles are called intralumenal vesicles (ILVs), or sometimes intravesicular bodies. Note that these vesicles somewhat replicate the cellular topography, with lipids and membrane proteins facing outward and cytosolic components contained within. Canonically, MVBs fuse with lysosomes (not shown); alternatively, the MVBs fuse with the plasma membrane to release the ILVs outside the cell, referred to as exosomes. Other EVs derive from membrane blebbing, or outward protrusion of the cellular membrane, which may be facilitated by membrane domain (lipid raft) formation, sometimes characterized by the flipping of phosphatidylserine to the external surface of the plasma membrane. Cytosolic components may enter into the membrane extensions, which bleb, pinch off, or exocytose, releasing extracellular membrane vesicles. These EVs are termed microvesicles, ectosomes, or shed vesicles, among other monikers. Again, there is a recapitulation of the cellular topography; however, a variety of sorting mechanisms for both proteins and lipids result in an inexact replication.
Table 1.
Examples of extracellular vesicle RNA, protein, and lipid transfer or impact on recipient cells
3. AN OVERVIEW OF RESEARCH ON EXTRACELLULAR VESICLES IN BRAIN TUMORS
Brain tumors, particularly HGGs, have numerous features that make them difficult to treat. Tumor EVs contribute to essentially all of these detrimental features, including tumor heterogeneity, aspects of the tumor microenvironment and constituent cells, and the effects of HGG EVs on the immune system.
3.1. Tumor Heterogeneity
HGGs are notoriously heterogeneous conglomerates of cell types. While large-scale DNA sequencing allowed for classification into four glioblastoma subtypes [proneural, neural, classical, and mesenchymal (148)], single-cell RNA-sequencing technologies have revealed further heterogeneity even within a given tumor subclass (61). Stem cell–like populations, more differentiated cells, cells close to the hypoxic core, and proliferative and invasive cells at the tumor margin may all be of tumor origin and differ both spatially and temporally, particularly in response to treatment. Invading immune cell types are of nontumor origin, while endothelial cells may be brain or tumor derived, and other brain-resident cells (of glial origin, neuronal, etc.) are all affected by the tumor (111). EVs from both the tumor and the microenvironmental cells shape that heterogeneity.
Gene expansion of receptor tyrosine kinases, particularly epidermal growth factor receptor (EGFR) and its mutant version, EGFR variant III (EGFRvIII), are signatures of the classical glioblastoma subtype (148). Heterogeneous expression of intratumoral receptor tyrosine kinases is commonplace, with crosstalk among such cells mediated by cytokines (58). However, as glioma EVs reemerged in 2008–2009 as potential players in tumor biology, Al-Nedawi et al. (1) demonstrated horizontal transfer of EGFRvIII via EVs from cells expressing it to cells that did not express it. The recipient cells activated downstream signaling pathways and generated a more aggressive phenotype. This provides a new mechanism for receptor tyrosine kinase heterogeneity within a tumor, and further supported vesicular release of EGFRvIII into the peripheral circulation, which has biomarker potential, known at both the protein (43) and mRNA (126) levels. Tumor EGFRvIII expression also alters the EV proteome from such cells, with particular emphasis on EV-bound or EV-contained proinvasive entities, such as extracellular matrix proteins and extracellular matrix–binding proteins, proteases, and adhesion molecules (17). There is also a likely relationship between signal peptide peptidase and the secretome of EGFRvIII-expressing cells, especially of various tumorigenic cytokines, such as transforming growth factor beta (TGFB) (151). As TGFB is a known component of glioblastoma EVs (43), this work suggests a relationship between EGFRvIII and TGFB, either or both of which are transferable in or on tumor EVs and are thus capable of downstream impacts on recipient cells proximally and distally.
Because cancer stem cells—or, specifically, glioma stem cells (GSCs)—are tumor initiators and drivers of recurrence, they are often seen as the apex cells of the cancer cell hierarchy, with heterogeneity developing as different tumor niches are occupied (159). These concepts are not without controversy (90), but the general precepts persist. GSC subclassification also follows the defined genetics, but as noted (61), there can be subtype heterogeneity within a given tumor. Ricklefs et al. (118) demonstrated that EVs from GSCs of different subtypes (proneural and mesenchymal, in this study) are proteomically distinguishable from each other and can transfer EVs to other cells, with potent influences on tumor growth. The mixed populations of GSCs, with accompanying EV cross-transfer, led to shorter survival in tumor-implanted murine models, and EV proteome signatures could predict adverse patient outcomes (118).
As mentioned above, EVs harbor many RNA types, and the demonstration that functional RNAs could be transported to cells by EVs has had enormous influence on the field (143). It is also clear that mRNAs and microRNAs (miRs) are differentially loaded as EV cargo relative to their presence and concentrations in cells (80, 126). There are several described mechanisms for such sorting, and the implications reach into both the biology of EVs and their use as biomarker carriers (112). As both markers and influencers, miRs from both GSCs and their EVs could be used to distinguish intratumor subtypes both genetically and spatially (mesenchymal, nodular, and perinecrotic zone versus proneural, migratory, and infiltrative edge). In mixed-cell populations, EVs from nodular cells could promote migration of the infiltrative cells in vitro and in vivo, including passage of miRs. This suggests that EVs contribute to the heterogeneity of tumors, with EV cargo influencing cell behavior (37). For instance, brain-specific miR-128 is associated with better patient outcome and is often downregulated in glioblastomas. In studies of differential miR-128 expression in GSCs, less aggressive proneural GSCs expressing a miR-128 anti-miR (antisense molecule) became more aggressive when coinoculated with a mesenchymal GSC line that overexpressed miR-128. Rooj et al. (119) postulated that intercellular EV communication abrogated the tumor-suppressing effect of miR-128 overexpression by the mesenchymal GSCs. Thus, EVs may contribute to intratumoral heterogeneity, which generally leads to worse outcomes.
A key feature of GSCs is their association with tumor vascular niches (16), which contribute to the maintenance of stem cell–like properties. However, GSC EVs may also influence the endothelial cells that make up the tumor blood vessels. Curiously, EVs from GSCs of different genetic subtypes are differentially taken up by endothelial cells, with those from mesenchymal subtypes taken up more proficiently than those from proneural subtypes. Furthermore, differentiation of either subtype of GSC yields EVs that have less avid endothelial cell uptake but nonetheless results in increased endothelial cell proliferation (again, more so from mesenchymal GSCs) (129). These findings add a further complicating layer to the role of EVs in a heterogeneous environment, wherein the effects of EVs from GSCs can change depending not only on the GSC subtype but also on the differentiated outcome of the GSCs.
HGG heterogeneity also includes cells not of tumor origin, such as brain endothelial cells, microglia, astrocytes, and other cells of the central nervous system. Some cells will migrate in from outside the central nervous system, including immune cells and some mesenchymal stem cells (MSCs), perhaps of bone marrow origin, that become brain residents (57). As one might expect, these MSCs interact with the tumor through EVs. Using in vitro settings, Del Fattore et al. (24) incubated EVs—exosome-like or small EVs—of MSCs from different sources (bone marrow, umbilical cord or Wharton’s jelly, and adipose tissue) with a glioblastoma cell line, which led to distinctive outcomes. MSC EVs from all sources were internalized by glioblastoma cells; bone marrow and umbilical cord MSC EVs reduced glioblastoma cell proliferation, Nwhile adipose tissue MSC EVs actually enhanced it with a concurrent cell cycle shift to the S and G2/M phases. Bone marrow and umbilical cord EVs also induced apoptosis in the glioblastoma cells; umbilical cord EVs could be loaded with the chemotherapy agent vincristine for delivery to the tumor cells to further promote cell death. Such results are significant because MSC EVs are approaching therapeutic use in a variety of diseases (67). These data suggest that the origin of the EVs is important, and (as noted below) EVs from some sources may promote tumor growth. Another use of umbilical cord (Wharton’s jelly) MSCs transfected with a miR-124 construct (with miR-124 acting as a tumor suppressor) implied that the miR could be delivered to glioblastoma cells by EVs, inducing enhanced chemosensitivity and reduced tumor cell migration(123). This study prepared EVs from MSCs by precipitation, which would result in a combination of EV types for delivery; it also used transwell assays, which would allow larger EVs to passage to the glioma cells. However, another study with umbilical cord MSCs demonstrated their chemotaxis into brain-ensconced GSCs via CCL2 and CXCL12 secreted by GSCs and the receptors CCR2 and CXCR4 on the MSCs (106). MSCs in the GSCs led to increased tumor growth with invasive phenotypes, which the authors attributed, at least in part, to MSC EVs. These were identified by immunohistochemical stain for EV or exosome markers (CD9 and CD63, which are present on several different EV subtypes), although the level of resolution would be insufficient for such staining to be definitive. Obviously, these very different results suggest that further study is necessary to promote the use of MSCs or their EVs in therapeutic settings against gliomas.
A recent study isolated MSCs directly from patient glioblastomas; the cells were genetically similar to bone marrow MSCs and did not show the genetic signatures of the tumors, thus indicating their origin in normal tissue (32). EVs (exosome-like small EVs) from MSCs could promote the proliferation of GSCs in vitro and in vivo, with MSC EV miR-1587 playing a role, presumably by downregulating expression of the tumor suppressor mRNA of NCOR1 (32). Normal cells within tumors are frequently co-opted to serve the tumor, and EVs from those normal cells may be a primary means of interaction. Glioma heterogeneity may be strongly influenced by these forms of communication.
3.2. Angiogenesis, Hypoxia, and Coagulopathies
A glioma characteristic closely related to heterogeneity is that of tumor-driven angiogenesis (156), which has been unsuccessfully targeted in numerous clinical trials (85). EVs have long been implicated in blood vessel biology (139) in normal and pathologic states. This section discusses the impacts of glioma hypoxia on its EVs and on coagulopathies, which are frequently associated with gliomas.
In one of the seminal papers on glioblastoma EVs, Skog et al. (126) demonstrated the presence of angiogenic components contained in glioblastoma EVs (in addition to the above-mentioned RNA content) that could stimulate in vitro endothelial cell tubule formation. The EVs were described as microvesicles, although the EV preparation was similar to exosome/small-EV methodologies. Curiously, later reports indeed showed roles for glioblastoma EV RNA cargo, such as the long intervening/intergenic noncoding RNA POU3F3 (linc-POU3F3). linc-POU3F3 was found at increased levels in HGGs, cell lines, and cell line EVs. EVs (described as exosomes) from glioblastoma cell line A172 containing linc-POU3F3 could promote endothelial cell proliferation, migration, tubule formation, and expression of angiogenic factors. Such EVs increased arteriole formation in a chick chorioallantoic membrane assay in vivo, and linc-POU3F3 knockdown inhibited all of these phenomena (75). In a very similar set of experiments, the same group used EVs (also described as exosomes) from glioblastoma cell line U87, which expressed high levels of another linc-RNA, linc-CCAT2 (76). EV transfer of linc-CCAT2 to endothelial cells drove all of the above-mentioned angiogenic features (which were reversed by silencing of linc-CCAT2) but also inhibited endothelial cell apoptosis under hypoxic conditions (76). Another linc-RNA, HOTAIR, was linked to VEGFA expression and release in glioma cells, with concomitant activation of angiogenic programs in endothelial cells. Silencing HOTAIR mitigated angiogenic potential, but overexpression of VEGFA could somewhat recover the effects. The authors suggested that HOTAIR could be transferred to endothelial cells by glioblastoma EVs, but those conclusions were not thoroughly pursued (76). When overexpressed in glioblastoma GSCs, the tumor-ubiquitous miR-21 (42) correlated with increased VEGF mRNA in the GSC EVs; those EVs (described as exosomes) promoted angiogenic phenotypes in endothelial cells (proliferation, tubule formation, and activation of the pFLK/VEGFR2 pathway). Knockdown of miR-21 or VEGF abrogated those phenotypes. It should be noted that GSC EVs from control cells could significantly drive the same angiogenic behaviors, but at lower levels (133). These reports show that the RNA content of glioblastoma EVs, ranging from mRNAs to long noncoding RNAs, lincRNAs, and miRs, can play a significant role in fostering angiogenic phenotypes in endothelial cells and could thus be a mechanism for HGG angiogenesis. It should also be noted that the EV preparations across the various studies were not identical, and these differences could have effects on the study outcomes.
Glioblastoma EV protein cargo could presumably also affect tumor angiogenesis. Treps et al. (142) reported the presence of VEGFA in or on glioblastoma GSC EVs (associated with EV membranes shown in superresolution microscopy); those EVs (called the cleaned EV fraction, with mostly small-EV characteristics) stimulated endothelial cell permeability, tubule formation, and sprouting, and such events were reduced by silencing VEGFA in the GSCs. Furthermore, using the Cancer Genome Atlas database, they showed that higher levels of VEGFA RNA expression correlated with shorter overall survival in patients, and they demonstrated the presence of increased levels of VEGFA on EVs from glioblastoma patient sera. The presence of VEGFA on serum EVs raises the possibility of potential titration of blocking antibodies (e.g., bevacizumab in blood) before the drug can reach target sites in the brain, although another study indicated that a unique form of VEGF on breast cancer EVs was ineffective at binding the antibody (30). On the other hand, earlier results suggested that EVs from the plasma of glioblastoma patients were ineffective at driving endothelial cell viability and proliferation, tubule formation, or phosphoactivation of the AKT/CTNNB1 (β-catenin) pathway, all of which were promoted by EVs from glioblastoma patient cerebrospinal fluid (82). Admittedly, the EVs (described as microvesicles) were not purified on the basis of known cancer markers, and their preparation would capture all categories of EVs, but these results nonetheless raise the question of whether circulating EVs in blood ever return to the brain, and if so, whether they have any angiogenic impact. Still, as mentioned, such EVs could theoretically sponge up antibodies against tumor targets if the antibodies were administered intravenously. Glioblastoma cells themselves can internalize bevacizumab and subsequently release it on EVs, possibly as an escape mechanism (124), but the further sequelae are unknown.
Hypoxia and angiogenesis are closely tied in the natural history of HGGs, as are their mechanisms of therapy resistance (88). Also related are the role of tissue factor (TF) in the rather high frequency of coagulopathies in patients with central nervous system tumors (78) and the fact that TF is often a component of blood EVs in various pathologies (146). As far back as 1984, EVs (described as microvesicles) from U87 glioblastoma cells were shown to exhibit coagulation and platelet aggregation activities that required thrombin and blood factors, but these activities could be inhibited by antibodies against TF (7). The activity was higher on a per-protein basis in the EVs than the isolated cellular plasma membranes. Thus, TF presence, enrichment, and activity on glioblastoma EVs have been known at the cellular level for some time.
Svensson et al.(135) examined the relationships between hypoxia and TF in cellular and molecular studies, including the action of hypoxic glioblastoma EVs on hypoxic endothelial cells. The EVs were described as microvesicles but could contain largely small-EV populations. Endothelial cells under hypoxia upregulated PAR2, an important signaling regulator of angiogenesis, along with heparin-binding EGF-like growth factor (HB-EGF), which stimulated ERK½ signaling and drove angiogenic features in endothelial cells. TF and factor VIIa were upregulated on EVs from hypoxic glioblastoma cells, and this protease complex (with factor X) further triggered ERK phosphorylation and endothelial cell proliferation and migration, thus potentially promoting angiogenesis in a hypoxic environment. Using two cell lines derived from rat C6 glioma, Monteiro et al. (98) showed that an aggressive variant (P7) expressed more TF than the nontumorigenic ST1, with the former line able to generate active protease factor Xa via TF and factor VIIa and increasing the expression of both PAR1 and PAR2. All of these features were upregulated during hypoxic stress, particularly for P7 cells. Both cell types generated EVs (described as, and bearing characteristics of, microvesicles), with P7 EVs displaying TF activity, but this was not examined under hypoxia. These results imply that, at least in the tumor microenvironment, TF activity may be passaged by tumor EVs, and those EVs may interact with malleable endothelial cells in angiogenesis under hypoxia.
The regulatory effects of miRs on TF expression can be complicated and possibly dependent on tumor type. D’Asti et al. (21) showed that, in a rare but deadly pediatric brain tumor (embryonal tumor with multilayered rosettes), miR-520g is upregulated and targets the expression of TF mRNA. This leads to downregulation of TF in both cells and EVs, with concomitant reduction of procoagulant activity and coagulation-driven signaling. Furthermore, forced expression of miR-520g in medulloblastoma (another pediatric brain tumor) showed similar results, along with enhanced EV production, with vesicles containing more miR-520g and less TF (the EVs were accurately referred to as the total EV fraction). Growth of medulloblastoma cells under stem cell–like conditions also upregulated miR-520g and downregulated TF, suggesting that the stem cell phenotype may be less stimulatory to endothelial cells than more differentiated cells, as the same group has shown (129). Hypoxia can also change the miR content of GSCs, in this case more quantitatively than qualitatively (163). GSC EVs (exosome/small-EV fraction) from hypoxic cells induced slightly but significantly higher cell proliferation of both the parent U87 cells and endothelial cells. While these results are in some contrast to those found by Spinelli et al. (129) (using exosome-like EVs), there are too many differences between the experimental systems to draw solid conclusions, such as the use of primary glioma stem cell cultures versus converting the long-passaged U87 line to a stem-like phenotype. Interestingly, even though this topic was not pursued, two of the upregulated miRs from the study by Zhang et al. (163), miR-6876 and miR-1910, likely have affinity for the 3 untranslated region of TF mRNA, as found in TargetScan (M.W. Graner, personal observation). The roles of stem cell–like tumor cells and their EVs in the various microenvironment settings will likely remain an area of active study.
As mentioned, glioblastoma hypoxic environments are well known, but the molecular effects on cells and their ensuing interactions require further study. EVs (exosome-like) from hypoxic glioblastoma cells and those (likely large and small EVs) found in the blood of glioblastoma patients have distinct proteomic and transcriptomic signatures (compared with normoxic cell– derived EVs or control blood EVs), and several of the features are related to worse survival outcomes, as determined by mining the National Cancer Institute’s Repository of Brain Neoplasia Data (REMBRANDT) database (72). Hypoxic glioblastoma EVs could enhance aorta-explant sprouting, endothelial cell tubule formation, proliferation (and reduced cell death), and migration compared with normoxic EVs; the conditioned media from the treated endothelial cells could also stimulate glioblastoma cell proliferation and migration. Hypoxic glioblastoma EVs are also better promoters of in vivo xenograft glioblastomas (in a nonorthotopic setting) in terms of growth kinetics, vascularity, and mesenchymal phenotypes (72). Another study of hypoxic glioblastoma EVs (mostly exosome-like small EVs) found upregulation of LOX, TSP1, and VEGF proteins in or on those EVs compared with EVs from normoxic cells, and the hypoxic EVs preserved endothelial cell viability in the face of hypoxia while promoting tubule length and branching (69). Hypoxic glioblastoma EVs thus contained proteins involved in aberrant vascularity in tumors, and also prompted transcriptomic changes in recipient glioblastoma cells, including pathways of metabolic interruption—signaling that leads to ischemic preconditioning, intercellular communication, and neurologic functioning. In this report (69), the U87 cells were not grown under stem cell conditions, which is important to remember when comparing results and analyses, especially in light of previous work with stem cell–like versus differentiated tumor cells (129).
In addition to the angiogenic features of hypoxic glioblastoma EVs, the vesicles can alter endothelial cell barrier capabilities that affect the blood–brain barrier. Confirming previous work (142), Zhao et al. (166) found that VEGFA is a component of hypoxic glioblastoma EVs (somewhat characterized as small EVs) that is involved in the downregulation of occludin and claudin-5 proteins as components of blood–brain barrier tight junctions. VEGFA and EVs from hypoxic glioblastoma cells increased endothelial cell proliferation, disrupted endothelial cell permeability barriers, and reduced trans-endothelial electrical resistance in vitro. In vivo, injections of hypoxic EVs promoted Evans blue dye extravasation across the blood–brain barrier and into the brain parenchyma. Another report implicated SEMA3A from GSC EVs (shown to contain both exosomes/small EVs as well as microvesicles) as an important player in vascularization in vitro, in situ, and in vivo, also showing that differentiated cells expressed far less SEMA3A (141). Blocking its receptor, NRP1, could abrogate the effects (even though, curiously, blocking VEGFA showed only limited reduction), as could knockdown of SEMA3A. EVs from glioblastoma patient blood (EVs from sera—all other mentions of blood EVs in this review were obtained from plasma) also had potent permeability activities that could be mitigated by blocking NRP1. These studies raise important issues because blood vessel normalization is a complicated phenomenon in the roles and effects of antiangiogenic therapies for gliomas (138). Single-agent targeting may be missing other critical effectors in angiogenesis and endothelial cell barrier characteristics.
Most studies have mainly pursued basic mechanisms at the molecular and cellular levels, but other investigations have been directly patient oriented. Looking at studies from patients’ blood, one report of TF activity in blood EVs (called microparticles, i.e., microvesicles) suggested that this activity was not prognostic for the coagulopathic complication venous thromboembolism in brain cancer patients (137). However, the specific nature of the patients in that population was unclear, making a proper assessment difficult. A study of 61 patients diagnosed with glioblastoma looked at the patients’ blood EVs for procoagulant activity over time (via clotting and fibrinolytic assays; TF was not directly assessed) and in relation to the extent of surgical resection [all patients received prophylactic antithrombic treatments and underwent Stupp protocol therapy (131)] (122). The EVs, also referred to as microparticles, would have been very dense EVs given the g-force and short centrifugation time for collection. On the other hand, the blood collections themselves were carefully controlled to mitigate extraneous factors. In the early stages (before surgery and one week after surgery), all patients had higher EV procoagulant activity, but this activity subsequently declined over seven months if the patients received a gross total resection. The EV procoagulant activities were higher if the resection was subtotal and if there were signs of tumor progression. Venous thromboembolism occurred in approximately 64% of the patients with higher EV activity. It should be noted that the particles obtained were largely of the microvesicle category, and no attempt was made to use putative tumor markers to distinguish the vesicles’ origins. Expanding on those studies with the same cohort, the same group did utilize a potential tumor marker [glial fibrillary acidic protein (GFAP), which may certainly be relevant as a circulating glioblastoma marker (64)] both with and without TF. The patients had higher levels of GFAP+ EVs, TF+ EVs, and GFAP+/TF+ EVs than controls did, with GFAP+ EVs and GFAP+/TF+ EVs increasing after resection. After seven months, patients with subtotal resections and/or those showing disease progression showed further increases in GFAP+/TF+ EVs. GFAP−/TF+ EVs (which could come from several cellular sources) were higher in patients who developed venous thromboembolism or those at further risk of venous thromboembolism (121). These results further suggest the potential for coagulopathies related to EVs that may be derived from tumor and other cellular origins.
Circulating endothelial cells, and sometimes their EVs, are characteristic of vascular perturbation, such as might occur in cancers (38). Circulating endothelial cell numbers (defined as CD45−/CD31+ cells) are known to be higher in pretreatment glioblastoma patients, but in a study by Bennett et al. (8), they declined in the brief postoperative measure (five days), with no tested markers correlating with patient outcome. The patient population in this study was small and somewhat mixed with primary and recurrent cases, and the overall survival was less than that of standard of care for primary cases, potentially confounding the results. Another study quantified blood EVs (phosphatidylserine+) and circulating endothelial cells (defined as CD146+/lectin UAE-1+) using EV clotting time and thrombin generation as activity assays (115). Glioblastoma patients underwent surgical biopsy or complete or subtotal resection and received Stupp protocol treatment (131). Blood and biomarker B sampling was done soon after surgery and at the end of radiochemotherapy (presumably six weeks after the start of treatment). The authors found that circulating endothelial cells were higher before and after treatment; EVs (referred to only as microparticles and defined by ability to bind annexin V) and thrombin generation decreased, while clotting time was higher at both time points. A high level (99th percentile) of pretreatment circulating endothelial cells correlated with worse overall survival, even though, in this small sample, progression-free survival and overall survival were seemingly low compared with those of most glioblastoma populations. These studies raise questions about the utility of EVs, circulating endothelial cells, and coagulant activities as biomarkers, but clearly these are small studies with dissimilar time courses and marker evaluations. Additionally, these studies would likely have excluded the exosome/small-EV fraction, which is known to transport TF, for instance (70). Unified, agreed-upon markers or activities, patient populations, and time courses of evaluation would aid in making such studies more coherent and therefore actionable.
3.3. The High-Grade Glioma Microenvironment: Life, Death, and Inflammation
Tumor microenvironments are interplays between the tumor cells themselves and the normal cells in which they find themselves. For HGGs, at a very simplistic level, cells of the microenvironment would include neurons and cells of glial origin: astrocytes, oligodendrocytes, ependymocytes, and microglia. As noted above, vasculature components such as endothelial cells and pericytes could also be part of the microenvironment. This section focuses mainly on the highly abundant astrocytes, with special emphasis on inflammation.
An earlier work analyzing the effects of EVs from a derived murine low-grade tumor [characterized as an oligodendroglioma (134)] showed that such vesicles could induce apoptosis when incubated with fetal rat primary cortical neurons via FASL presenton the EVs (20). While the vesicle preparation methods were not definitive by more current standards (e.g., it is unclear whether the serum used in the media had been depleted of bovine EVs, and the EVs likely included all classes), EVs from NIH3T3 cells presumably grown in the same type of medium did not have the proapoptotic effects. The same group followed up with studies that indicated that some level of apoptosis (40%) was induced in rat astrocytes treated with the same murine oligodendroglioma vesicles, but this time suggesting TRAIL [also called tumor necrosis factor ligand superfamily member 10 (TNFSF10)] as the EV component driving apoptosis (84). While the presence of FASL on brain tumor EVs has not been widely scrutinized, Hellwinkel et al. (56) did not find significant FASL on exosome-like EVs from an anaplastic oligodendroglioma line, but did note it on two glioblastoma lines.
Unlike the papers described above, a recent study of human glioblastoma exosome-like EVs incubated with human astrocytes showed a dramatic proinflammatory response from the astrocytes—whose conditioned medium drove glioblastoma cell proliferation—and also prosurvival tumor-like signaling pathways that promoted astrocyte growth in soft agar (103). Using the rat C6 glioma line, Taheri et al.(136) also found that C6 EV (microvesicles 200–300 nm in diameter) treatment of rat astrocytes stimulated astrocyte proliferation and GFAP mRNA expression and also upregulated mRNA expression of the matrix metalloproteinases MMP2 and MMP14 (with no effect on MMP9 mRNA, and downregulation of TIMP2). Further studies of glioblastoma EVs (exosome-like EVs) and astrocytes indeed demonstrated gelatinase activity from astrocytes treated with glioblastoma EVs (47), which is consistent with the astrocyte MMP9 release seen by Oushy et al. (103). There were also extensive proteomic analyses performed on astrocytes treated with GSC EVs compared with glioblastoma EVs from the differentiated GSCs, with signaling pathways involving EIF2, eIF4, p70S6K, and mTOR being prominent (47). In concordance with this work, Oushy et al. (103) also saw activation of p70S6K (phosphorylation); release of interleukin 3 (IL3), IL5, and IL15 [identified as upstream regulators by Hallal et al. (47)]; and immune cell activation as a top pathway analysis biofunction (with CD3 acting as an upstream regulator). These combined results suggest that glioblastoma EVs profoundly affect recipient astrocytes, which can then alter the tumor microenvironment, which again is generally beneficial to the tumor. The extracellular matrix alterations when glioblastomas engage astrocytes have precedence, particularly regarding the activation of matrix metalloproteinases and PLAUR [a urokinase plasminogen activator receptor (77)], as noted by Oushy et al. (103). Determining whether such astrocytes can actually form tumors [i.e., as seen following relatively minimal genetic manipulation (116)] will require further study.
Curiously, the inflammatory cytokine TNFA also promotes release of EVs (90–200 nm in diamter) from astrocytes themselves, which requires the action of glutaminase and the influence of reactive oxygen species (150). This suggests that proinflammatory cytokine release from astrocytes treated with glioblastoma EVs (e.g., 103) may result in astrocyte autocrine stimulation to maintain the inflammatory milieu in a brain tumor setting; it also suggests that glutaminase may be a therapeutic target. On the other hand, the inflammatory environment may also change the glioblastoma EV (exosome-like) output in terms of protein content; treatment of glioblastoma cells with IL1B or TNFA generates differential proteomes in glioblastoma EVs, including increases in various chaperones, such as CRYAB/αB-crystallin, GRP78, and HSP90 and HSP60 family members, all of which are known tumorigenic drivers (68).
The inflammatory microenvironment may have differential effects on HGG cell types—in this case, another oligodendroglioma, ostensibly being used as an oligodendrocyte model. Podbielska et al. (109) treated oligodendroglioma cells with TNFA and/or interferon gamma (IFNG), which resulted in some cell death but was synergistic in combination. EVs (called exosomes) collected from the treated cells had no cytotoxic effects when applied to naive cells but caused significant cell death when the cells were pretreated with IFNG, suggesting that the synergy in the cytokine combination’s cytotoxicity may involve EVs. The results support the idea that ceramide species enriched in EVs from the treated donor cells acted as cell death effectors when those EVs were applied to recipient cells in conjunction with T helper 1 cytokines. It should be noted that the EVs usedherewouldnotbeapoptoticbodiesintheory, butthatpossibilitywasnotformallyruledout. It is curious that EVs reported to be cell death mediators are those derived from oligodendroglioma lines (20, 84), suggesting that not all gliomas are created equal in terms of their EV effects on themselves and other cells as recipients.
Microglia, the macrophages of the brain, along with their bone marrow–derived monocyte cousins, make up a significant proportion of the cellular content of glioblastomas and have potent implications for glioma progression (48). However, there have been relatively few investigations of microglia or glioblastoma EV involvement in the glioblastoma microenvironment. Yang et al. (161) studied changes in microglia EV proteomic outputs following lipopolysaccharide stimulation of the cells. EV heterogeneity increased (i.e., from exosome-like vesicles to a variety of larger-diameter species, 200–350 nm), along with proinflammatory cytokines (TNFA and IL6) and proteins involved in RNA binding, ribosome function, transduction, and translation; EV production and these inflammatory changes were closely tied to TNFA in vitro and in vivo. In another setting, glioblastoma EVs (likely an exosome/small-EV and microvesicle fraction) also promoted release of TNFA and IL6 (among other cytochemokines) from microglia (145). In that study, the authors observed glioblastoma EV transfer to microglia in vitro and in vivo (the latter with intravital two-photon microscopy); highly upregulated miRs from glioblastomas that were transferred to microglia included miR-451 and miR-21, which correlated with a dramatic decrease in target MYC/c-Myc mRNA. The downstream implications of the transcription factor downregulation were not pursued here but are undoubtedly important.
4. HIGH-GRADE GLIOMAS, EXTRACELLULAR VESICLES, AND THE IMMUNE SYSTEM
As noted above, immune suppression by HGGs can be profound (107) and provides formidable barriers to immunotherapy (93). The role of brain tumor EVs in immune suppression and stimulation has not received the same level of attention given to other tumor types, but there is research that deserves mention.
4.1. High-Grade Glioma Extracellular Vesicles in Immune Stimulation
There have been an overwhelming number of accounts of tumor EVs as purveyors of immunosuppression (e.g., 45, 74); however, there are also instances of tumor and other EVs acting as immune stimulators, including their utility as vaccines or vaccine components (73). The rationale for their use in vaccines was that EVs can act as virus-sized surrogates for their parental cells, containing putative antigens as well as danger signals such as HSPs (44) to provide an antigen–adjuvant combination. This appeared to be the case in an early murine study of glioma EVs (exosome-like) that used SMA-560vIII cell culture–derived EVs as prophylactic vaccines, which had potent T and B cell stimulatory properties that resulted in 90% tumor rejection (43). Surprisingly, the vaccine showed no efficacy in a therapeutic setting; the authors noted that this lack of effectiveness may have resulted from built-in immune suppression when the tumors were preestablished. Alternatively, it is formally possible that EVs are insufficient as vaccines in more stringent settings, but there are numerous reports of successful EV vaccine responses (73).
Dendritic cells are the premier antigen-presenting cell of the immune system and are central to the paradigms of cell-based cancer vaccines (120). They are used as anticancer vaccines in clinical settings following incubation of (patient-derived) dendritic cells with tumor antigens. The antigens are processed for display on major histocompatibility complex (MHC) class I and class II molecules, along with other forms of dendritic cell stimulation. In these cases, the dendritic cells themselves are injected into patients; in other cases, EVs derived from dendritic cells [sometimes called dendritic cell exosomes (DEXs)] are harvested from the culture supernatants and utilized themselves as the anticancer vaccines (described in 73). The highly simplified concept is that the antigen-loaded or antigen-stimulated dendritic cells will migrate to lymph nodes to activate cognate T cells that recognize the presented antigens. The activated T cells then move into the periphery to locate and destroy tumors displaying the provoking antigens. Dendritic cell EVs are believed to stimulate endogenous dendritic cells for activation of T cells by delivering processed antigens and powerful costimulatory molecules. The perceived advantage of dendritic cell EVs in these settings is that the EVs may be harvested over extended periods and then stored, thawed, and utilized without facing the procedural or regulatory hurdles encountered with live cells (73).
While there have been many vaccine clinical trials for gliomas (152), none have involved dendritic cell EVs. However, Bu et al. (13) performed a human ex vivo study in which they cultured patient tumors and harvested exosome-like EVs. Those autologous EVs promoted significant dendritic cell stimulation and activation or cytotoxicity of patient T cells against their own tumors. In preclinical models, the same group employed a CELLine 1000 culture system to enhance dendritic cell growth and harvest conditioned media for exosome-like EV collection (14). They cultured murine dendritic cells and incubated them with chaperone-rich cell lysate (CRCL) (46) as an antigen source from mouse glioma cells (GL261). Dendritic cell EVs from the CRCL treated dendritic cells had quantitatively more stimulatory features (MHC, ICAM, HSP, CD80, and CD86 expression) than did dendritic cell EVs from cells treated with GL261 lysate alone. This led to a complicated study in which dendritic cells were treated with GL261 CRCL, GL261 lysate, or nonglioma cell lysate; dendritic cell EVs were harvested; and those EVs, which were largely uncharacterized, were incubated as antigen sources on naive dendritic cells (15). Those newly treated dendritic cells were then used as cellular vaccines against preestablished intracranial GL261, where the dendritic cells pulsed with dendritic cell EVs from previous dendritic cells treated with GL261 CRCL drove increased survival, smaller tumor size, greater tumor cytotoxicity, and a far more activated immune response compared with other antigen sources. These studies demonstrate that glioblastoma EVs or other forms of tumor antigen can provide stimulus and specific antigens to dendritic cells, and those dendritic cells or their EVs may then be used as cancer vaccines.
Other uses of glioblastoma EVs as immunogens for dendritic cells include a unique combination of rat (C6) glioblastoma EVs (purified with a commercial affinity column) and α-galactosylceramide (α-GalCer, an analog of a marine sponge glycolipid) that serves as a potent stimulator of invariant natural killer T cells (81). α-GalCer is presented to this specialized class of natural killer cells on dendritic cell–expressed CD1d, which can lead to activation of the dendritic cells as well as natural killer, T, and B cells but may also anergize invariant natural killer T cells (132). In this report (81), injection of dendritic cells loaded with α-GalCer and glioblastoma EVs promoted immune responses in conjunction with prolonged survival for rats with intracranial C6 tumors (compared with treatments of dendritic cells loaded with lipid/C6-lysate). There have been numerous clinical trials with dendritic cells pulsed with α-GalCer, so the safety is reasonably well established (149). The question remains of what a clinically feasible source of glioblastoma EVs for human trials would be. It is not certain that a sufficient number of autologous tumor cells could be grown in cell culture to allow harvesting of EVs from those media supernatants in a timely fashion, given that patients with glioblastomas have a 15-month median survival time. One might suggest that mixtures of various glioblastoma EVs from several cell types may be scalable to obtain quantities for dendritic cell pulsing; such EVs, almost assuredly with different MHC backgrounds, might be especially good stimulators of dendritic cells but may also be subject to substantial regulatory hurdles.
A recent clinical trial (ClinicalTrials.gov ID P5">) was initiated following a previous trial (5)in which autologous tumor cultures under went apoptosis using an antisense oligodeoxynucleotide targeting the mRNA of IGF1R. The apoptosing cells were encapsulated in a lucite biodiffusion chamber that was implanted into a surgically fashioned subcutaneous pocket in the rectus sheath. There is a presumed release of tumor EVs (54) (possibly apoptotic bodies) that promote immune responses (reviewed in 42). Curiously, in the earlier trial, several patients suffered deep vein thrombosis (with other patients then treated with enoxaparin), suggesting either enhanced release of EVs or exacerbation of related coagulopathies. While immune studies are presumably forthcoming, this study seemingly relies on the concept of self-vaccination via EV release following immunogenic cell death (reviewed in 73).
4.2. Extracellular Vesicles as Immune Response Biomarkers
Immunotherapy, particularly anticancer vaccination, is regarded as a bright hope for treating HGGs (152). However, we still lack useful biomarkers of immune response or failure in these treatment settings. The use of blood EVs in liquid biopsy may provide information regarding the effects of treatment, bearing in mind that the EVs may derive from numerous cell types, including immune cells (114). Blood EV profiling has shown elements of promise in two anti-HGG vaccine trials.
A phase I/II trial employing a specialized dendritic cell subset pulsed with four different glioma-associated antigens demonstrated efficacy and relationships between blood IL12 levels and time to progression in patients with recurrent HGGs (101). Exosome-like EVs obtained from patient blood samples were probed for total EV protein concentration and for levels of immune- and tumor-related mRNAs (100); the total EV protein levels correlated with the presence of tumor versus healthy donor controls and with the grade of the tumor (III or IV), and showed reduced quantity following vaccination.mRNAs coding for IL8, TIMP1, TGFB, and ZAP70 generally decreased following vaccination, with the decreasing amounts of IL8 and TGFB mRNA correlating with immune responses, suggesting that decreases in tumor-related gene expression may relate to antitumor immune response.
In another study, patients with recurrent HGGs were immunized with an emulsified, modified survivin peptide plus granulocyte-macrophage colony stimulating factor as an adjuvant (31). The investigators used imaging flow cytometry to analyze exosome-like EVs from the sera (note : not plasma) of immunized patients and controls and found that increases in CD9+/survivin+ or CD9+/survivin+/GFAP+ EVs following vaccination were associated with tumor progression (36). One might suspect that increased tumor volume relates to survivin release, but there was no clear correlation between magnetic resonance imaging (MRI)–measurable disease and EV numbers. The localization of GFAP and survivin on EV surfaces implies tumor extracellular release of the proteins, but these biologic mechanisms remain mysterious.
4.3. High-Grade Glioma Extracellular Vesicles in Immunosuppression
As noted above, cancer-related EVs are culprits of immunosuppression, as has been shown in several instances in HGG EVs. Using one of the few syngeneic (immune-competent) murine models, Liu et al. (83) found that treatment of mice with EVs (called exosomes but not characterized) from GL26 tumor cells increased intracranial tumor growth and had immune-suppressive effects on CD8+ T cells, which they determined to be the primary effectors in terms of cytotoxicity and IFNGoutput. This is almost the opposite of what Graner et al.(43)hadfoundinadifferentmurine model, where prophylactic vaccination dramatically inhibited tumor growth and drove B and T cell activation. However, in the study by Liu et al. (83), it is unclear how the EVs were introduced (i.e., into the brain or subcutaneously) and whether the tumor inoculation took place before or during treatment. Such details matter in terms of the context of EV exposure to dendritic cells (e.g., Langerhans cells in the skin) and the potential for already-present tumor-induced immune suppression.
Other reports of glioma EV immunosuppression have come from human settings. Myeloid cells are known to be markedly affected by gliomas (157), particularly with the immune suppressive monocyte-derived suppressor cells, and glioma EV (likely exosomes and microvesicles, based on their sizes) impacts on monocytes have been documented by de Vrij et al. (23). In that study, the authors showed that glioblastoma EVs could promote M2-type (considered tumor-supportive) monocyte-to-macrophage differentiation, but with lesser effects on microglia except for increased MMP14 release. Harshyne et al. (55) confirmed this result using glioblastoma patient blood EVs; they found M2-like monocyte phenotypes in blood, with the EVs promoting a T helper 2–type/M2-type cytokine release from EV-exposed (and soluble factor–exposed) monocytes. The EVs here were determined to be exosomes and microvesicles by particular enzyme-linked immunosorbent assay (ELISA) and flow cytometry techniques. Another report implicated changes in monocyte phenotypes toward monocyte-derived suppressor cells in populations of peripheral blood mononuclear cells treated with glioblastoma patient plasma EVs or GSC EVs (27); the induced monocyte-derived suppressor cells were responsible for the resulting T cell suppression (decreased proinflammatory cytokine release and reduced proliferation of some T cell populations). The EVs used here were prepared by precipitation, but the activities were verified against EVs prepared by differential centrifugation.
An often-overlooked immune regulatory type is the regulatory B cell population. glioblastoma EVs (described as exosomes but minimally characterized) bearing placental growth factor (PGF) could induce TGFB+ regulatory B cells from naive peripheral blood mononuclear cells; the regulatory B cells suppressed antigen-induced proliferation of T cells and led to reduced amounts of granzyme B and perforin, the effector molecules of T cell–mediated cytotoxicity (49). Glioma-specific regulatory B cells could also be isolated from glioblastoma tissue, implicating another infiltrating cell type that glioblastoma EVs can convert to a regulatory or suppressor phenotype.
Because HGG EVs are arguably the most important immune effectors controlling tumors, their influence on T cells is an area of ongoing study and has led to some incongruent conclusions. Work from the late Andrew Parsa’s group (59) indicated that glioma EVs (described as exosomes and characterized only by Western blot) are rather ineffective at inducing relevant phenotypic changes in peripheral monocytes and T cells other than reductions in HLA-DR on monocytes. On the other hand, Hellwinkel et al. (56) had contrasting results in terms of the stimulation or suppression of peripheral blood mononuclear cells and T cells, depending on the quantities of HGG EVs (exosome-like EVs) used to treat the lymphocytes. Up to a certain concentration of EVs from a grade III anaplastic oligodendroglioma, Jurkat T cells and healthy donor peripheral blood mononuclear cells, stimulated with phytohemagglutinin, increased IL2 and IFNG output [also seen with medulloblastoma EVs (29)], only to have those cytokines drop precipitously at higher concentrations of EV incubation. The proliferation of peripheral blood mononuclear cells actually increased with increased EV concentrations, and the signaling pathways and cytokine and chemokine profiles were also dose dependent, as was the loss of T cell activation status (IFNG+/CD69+ cells). Curiously, both low and high HGG EV concentrations reduced T cell motility as a possible alternative form of immune suppression. As mentioned above, the authors found minimal FASL on the EVs and, as in the study by the Parsa group (59), little or no PD-L1 (J. Hellwinkel, unpublished results).
In a very recent study, Mirzaei et al. (96) identified tenascin C (TNC), a known glioma antigen (113), as a GSC EV protein (EVs characterized to have exosome-like sizes) that reduced T cell proliferation and activation status (reduced CD69+/IFNG+/IL2+ cells) and led to signaling changes via integrin interactions with TNC. In contrast to the studies by Hellwinkel et al.(56) and the Parsa group (59), Ricklefs et al. (117) found varying levels of PD-L1 on some glioblastomas and GSCs and their EVs (with exosome-like and microvesicle-like sizes). Regardless of high or low PD-L1 levels, the EVs could suppress activation of antigen- or TCR-stimulated T cells, but for PD-L1-high EVs, the deactivation could be somewhat mitigated with anti-PD1 antibodies (blocking the PD1 receptor on T cells). IFNG stimulation of PD-L1-low glioblastoma cells could upregulate the ligand on EVs, making them more susceptible to the effects of the PD1-blocking antibody. These results are important in the context of the use of such checkpoint inhibitors in current cancer therapy paradigms.
Situational contexts involving HGG EV immunosuppression may affect peripheral circulating T cells as opposed to tumor-infiltrating T cells. One presumes that the density of EVs in the tumor microenvironment would be quite high, but with the caveat that we know little about the kinetics and dynamics of cellular release, recipient cell uptake, or reprocessing and release of potentially hybrid EVs from the recipient cells, or even what is meant by extracellular space in a solid tumor (pointed out in 56). The implications that HGG EVs can promote an inflammatory environment, including outputs from astrocytes, and yet T cell cytotoxicity is generally minimal within tumors bear witness to the “wounds that do not heal” (28). HGG EV roles may be quite different as circulating entities, where perhaps they more closely mimic liposome liposome biodistribution (128) but at least could encounter numerous classes of immune cells, with potentially vastly different consequences. We must keep in mind that our research settings may not best replicate those environments.
5. CONCLUSIONS
The relationships among glioma EVs, their autocrine impacts on the tumor cells themselves, and their paracrine and endocrine impacts on cells of the microenvironment and periphery are obviously extraordinarily complicated. The tightly entwined interactions of HGGs and their host brain cells lead to both offensive and defensive postures from the tumors, with a co-opting of almost every system that ultimately leads to a protumorigenic situation benefiting the HGG. The roles of HGG EVs in these nefarious processes are only just being realized despite decades of work, with few means to redirect or eliminate these particles. Figure 2 highlights some of the impacts HGG EVs may have on surrounding brain cells and infiltrating endothelial and immune cells.
Figure 2.
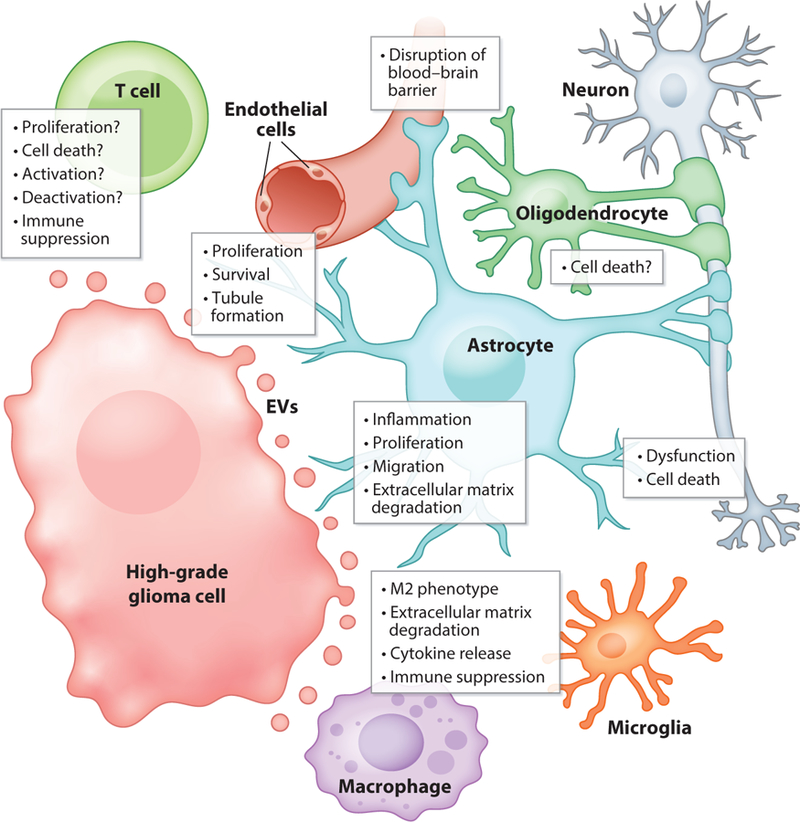
Effects of high-grade glioma (HGG) extracellular vesicles (EVs) on resident brain cells (neurons, astrocytes, oligodendrocytes, and microglia) and some infiltrating cells (endothelial cells or blood vessels, T cells, and macrophages). Astrocytes make contact with blood vessels, where HGG EVs can disrupt the blood–brain barrier; astrocytes and oligodendrocytes also contact neurons.
From a therapeutic perspective, we have not yet developed sufficiently specific targets or targeting modalities to remove EVs from the extensively heterogeneous system, although such a device may exist for EV trapping in blood (89). As noted above, it is not obvious how important such peripheral elimination might be in terms of an immune response, for example. Preventing EV release would likely be extremely valuable, but gross banishment of EVs from an organism would surely be detrimental; thus, tumor-specific targeting would presumably be necessary, but we currently lack any such technology or deliverable accuracy. One implication from the works spotlighted here is the recognition that particular signaling pathways often prevalent in tumors may be value-added targets there appears to be little overall clinical awareness of EV-driven tumor processes; such tumor-promoting EV effects need to be included in the therapeutic mind-set, with the hope that an encompassing gestalt will encourage cross-disciplinary thinking and action. This remains true not only for our unmet needs in treating patients with brain tumors but also across an entire spectrum of disease and health, where EVs are not simply cellular debris but are active participants in biology and physiology.
ACKNOWLEDGMENTS
The author is supported by grants from the National Institutes of Health (1R01 GM129046-01, 1R21MH118174-01) and by the Department of Neurosurgery at the University of Colorado.
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, et al. 2008. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol 10:619–24 [DOI] [PubMed] [Google Scholar]
- 2.Altadill T, Campoy I, Lanau L, Gill K, Rigau M, et al. 2016. Enabling metabolomics based biomarker discovery studies using molecular phenotyping of exosome-like vesicles. PLOS ONE 11:e0151339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anand S, Samuel M, Kumar S, Mathivanan S. 2019. Ticket to a bubble ride: cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta Proteins Proteom In press. 10.1016/j.bbapap.2019.02.005 [DOI] [PubMed]
- 4.Anderson HC. 1969. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell Biol 41:59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrews DW, Resnicoff M, Flanders AE, Kenyon L, Curtis M, et al. 2001. Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin-like growth factor type I receptor in malignant astrocytomas. J. Clin. Oncol 19:2189–200 [DOI] [PubMed] [Google Scholar]
- 6.Balca-Silva J, Matias D, Carmo AD, Sarmento-Ribeiro AB, Lopes MC, Moura-Neto V. 2019. Cellular and molecular mechanisms of glioblastoma malignancy: implications in resistance and therapeutic strategies. Semin. Cancer Biol In press 10.1016/j.semcancer.2018.09.007 [DOI] [PubMed]
- 7.Bastida E, Ordinas A, Escolar G, Jamieson GA. 1984. Tissue factor in microvesicles shed from U87MG human glioblastoma cells induces coagulation, platelet aggregation, and thrombogenesis. Blood 64:177–84 [PubMed] [Google Scholar]
- 8.Bennett IE, Guo H, Kountouri N, D’Abaco GM, Hovens CM, et al. 2015. Preoperative biomarkers of tumour vascularity are elevated in patients with glioblastoma multiforme. J. Clin. Neurosci 22:1802–8 [DOI] [PubMed] [Google Scholar]
- 9.Bloom HJ. 1982. Intracranial tumors: response and resistance to therapeutic endeavors, 1970–1980. Int. J. Radiat. Oncol. Biol. Phys 8:1083–113 [DOI] [PubMed] [Google Scholar]
- 10.Bonucci E 1967. Fine structure of early cartilage calcification. J. Ultrastruct. Res 20:33–50 [DOI] [PubMed] [Google Scholar]
- 11.Boudot R, Miletic D, Dziuban P, Affolderbach C, Knapkiewicz P, et al. 2011. First-order cancellation of the Cs clock frequency temperature-dependence in Ne-Ar buffer gas mixture. Opt. Express 19:3106–14 [DOI] [PubMed] [Google Scholar]
- 12.Bretting H, Konigsmann K. 1979. Investigations on the lectin-producing cells in the sponge Axinella polypoides (Schmidt). Cell Tissue Res 201:487–97 [DOI] [PubMed] [Google Scholar]
- 13.Bu N, Wu H, Sun B, Zhang G, Zhan S, et al. 2011. Exosome-loaded dendritic cells elicit tumor-specific CD8+ cytotoxic T cells in patients with glioma. J. Neurooncol 104:659–67 [DOI] [PubMed] [Google Scholar]
- 14.Bu N, Wu H, Zhang G, Ma X, Zhao P, et al. 2015. Exosome from chaperone-rich cell lysates-loaded dendritic cells produced by CELLine 1000 culture system exhibits potent immune activity. Biochem. Biophys. Res. Commun 456:513–18 [DOI] [PubMed] [Google Scholar]
- 15.Bu N, Wu H, Zhang G, Zhan S, Zhang R, et al. 2015. Exosomes from dendritic cells loaded with chaperone-rich cell lysates elicit a potent T cell immune response against intracranial glioma in mice. Mol. Neurosci 56:631–43 [DOI] [PubMed] [Google Scholar]
- 16.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. 2012. The brain tumor microenvironment. Glia 60:502–14 [DOI] [PubMed] [Google Scholar]
- 17.Choi D, Montermini L, Kim DK, Meehan B, Roth FP, Rak J. 2018. The impact of oncogenic EGFRvIII on the proteome of extracellular vesicles released from glioblastoma cells. Mol. Cell. Proteom 17:1948–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cocucci E, Meldolesi J. 2015. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 25:364–72 [DOI] [PubMed] [Google Scholar]
- 19.Couzin-Frankel J 2013. Breakthrough of the year 2013. Cancer immunotherapy. Science 342:1432–33 [DOI] [PubMed] [Google Scholar]
- 20.D’Agostino S, Salamone M, Di Liegro I, Vittorelli ML. 2006. Membrane vesicles shed by oligodendroglioma cells induce neuronal apoptosis. Int. J. Oncol 29:1075–85 [PubMed] [Google Scholar]
- 21.D’Asti E, Huang A, Kool M, Meehan B, Chan JA, et al. 2016. Tissue factor regulation by miR-520g in primitive neuronal brain tumor cells: a possible link between oncomirs and the vascular tumor microenvironment. Am. J. Pathol 186:446–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Robertis ED, Bennett HS. 1955. Some features of the submicroscopic morphology of synapses in frog and earthworm. J. Biophys. Biochem. Cytol 1:47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Vrij J, Maas SL, Kwappenberg KM, Schnoor R, Kleijn A, et al. 2015. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int. J. Cancer 137:1630–42 [DOI] [PubMed] [Google Scholar]
- 24.Del Fattore A, Luciano R, Saracino R, Battafarano G, Rizzo C, et al. 2015. Differential effects of extracellular vesicles secreted by mesenchymal stem cells from different sources on glioblastoma cells. ExpertOpin. Biol. Ther 15:495–504 [DOI] [PubMed] [Google Scholar]
- 25.Di Vizio D, Kim J, Hager MH, Morello M, Yang W, et al. 2009. Oncosome formation in prostate cancer:association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res 69:5601–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, et al. 2012. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am. J. Pathol 181:1573–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domenis R, Cesselli D, Toffoletto B, Bourkoula E, Caponnetto F, et al. 2017. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLOS ONE 12:e0169932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dvorak HF. 2015. Tumors: wounds that do not heal—redux. Cancer Immunol. Res 3:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Epple LM, Griffiths SG, Dechkovskaia AM, Dusto NL, White J, et al. 2012. Medulloblastoma exosome proteomics yield functional roles for extracellular vesicles. PLOS ONE 7:e42064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng Q, Zhang C, Lum D, Druso JE, Blank B, et al. 2017. A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumour angiogenesis. Nat. Commun 8:14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, et al. 2016. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother 65:1339–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figueroa J, Phillips LM, Shahar T, Hossain A, Gumin J, et al. 2017. Exosomes from glioma-associated mesenchymal stem cells increase the tumorigenicity of glioma stem-like cells via transfer of miR-1587. Cancer Res 77:5808–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fox AS, Duggleby WF, Gelbart WM, Yoon SB. 1970. DNA-induced transformation in Drosophila: evidence for transmission without integration. PNAS 67:1834–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox AS, Yoon SB. 1970. DNA-induced transformation in Drosophila: locus-specificity and the establishment of transformed stocks. PNAS 67:1608–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fox AS, Yoon SB, Gelbart WM. 1971. DNA-induced transformation in Drosophila: genetic analysis of transformed stocks. PNAS 68:342–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galbo PM Jr., Ciesielski MJ, Figel S, Maguire O, Qiu J, et al. 2017. Circulating CD9+/GFAP+/survivin+ exosomes in malignant glioma patients following survivin vaccination. Oncotarget 8:114722–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Godlewski J, Ferrer-Luna R, Rooj AK, Mineo M, Ricklefs F, et al. 2017. MicroRNA signatures and molecular subtypes of glioblastoma: the role of extracellular transfer. Stem Cell Rep 8:1497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goon PK, Boos CJ, Stonelake PS, Lip GY. 2005. Circulating endothelial cells in malignant disease. Future Oncol 1:813–20 [DOI] [PubMed] [Google Scholar]
- 39.Gould SJ, Raposo G. 2013. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell. Vesicles 2:20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graner MW. 2012. Brain tumor exosomes and microvesicles: pleiotropic effects from tiny cellular surrogates. In Molecular Targets of CNS Tumors, ed. Garami M, pp. 43–78. London: InTechOpen [Google Scholar]
- 41.Graner MW. 2018. B cell exosomes/extracellular vesicles as vehicles of B cell antigen presentation: implications for cancer vaccine therapies. In Diagnostic and Therapeutic Applications of Exosomes in Cancer, ed. Amiji MM, Ramesh R, pp. 325–64. London: Academic [Google Scholar]
- 42.Graner MW. 2018. Extracellular vesicles in cancer immune responses: roles of purinergic receptors. Semin. Immunopathol 40:465–75 [DOI] [PubMed] [Google Scholar]
- 43.Graner MW, Alzate O, Dechkovskaia AM, Keene JD, Sampson JH, et al. 2009. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J 23:1541–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graner MW, Bigner DD. 2006. Therapeutic aspects of chaperones/heat-shock proteins in neurooncology. Expert Rev. Anticancer Ther 6:679–95 [DOI] [PubMed] [Google Scholar]
- 45.Graner MW, Schnell S, Olin MR. 2018. Tumor-derived exosomes, microRNAs, and cancer immune suppression. Semin. Immunopathol 40:505–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graner MW, Zeng Y, Feng H, Katsanis E. 2003. Tumor-derived chaperone-rich cell lysates are effective therapeutic vaccines against a variety of cancers. Cancer Immunol. Immunother 52:226–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hallal S, Mallawaaratchy DM, Wei H, Ebrahimkhani S, Stringer BW, et al. 2018. Extracellular vesicles released by glioblastoma cells stimulate normal astrocytes to acquire a tumor-supportive phenotype via p53 and MYC signaling pathways. Mol. Neurobiol In press. 10.1007/s12035-0181385-1 [DOI] [PMC free article] [PubMed]
- 48.Hambardzumyan D, Gutmann DH, Kettenmann H. 2016. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci 19:20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han S, Feng S, Ren M, Ma E, Wang X, et al. 2014. Glioma cell-derived placental growth factor induces regulatory B cells. Int. J. Biochem. Cell Biol 57:63–68 [DOI] [PubMed] [Google Scholar]
- 50.Harding CV, Heuser JE, Stahl PD. 1983. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol 97:329–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harding CV, Heuser JE, Stahl PD. 2013. Exosomes: looking back three decades and into the future. J. Cell Biol 200:367–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hargadon KM, Johnson CE, Williams CJ. 2018. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol 62:29–39 [DOI] [PubMed] [Google Scholar]
- 53.Hargett LA, Bauer NN. 2013. On the origin of microparticles: from “platelet dust” to mediators of intercellular communication. Pulm. Circ 3:329–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harshyne LA, Hooper KM, Andrews EG, Nasca BJ, Kenyon LC, et al. 2015. Glioblastoma exosomes and IGF-1R/AS-ODN are immunogenic stimuli in a translational research immunotherapy paradigm. Cancer Immunol. Immunother 64:299–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harshyne LA, Nasca BJ, Kenyon LC, Andrews DW, Hooper DC. 2016. Serum exosomes and cytokines promote a T-helper cell type 2 environment in the peripheral blood of glioblastoma patients. Neuro-Oncology 18:206–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hellwinkel JE, Redzic JS, Harland TA, Gunaydin D, Anchordoquy TJ, Graner MW. 2015. Gliomaderived extracellular vesicles selectively suppress immune responses. Neuro-Oncology 18:497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hossain A, Gumin J, Gao F, Figueroa J, Shinojima N, et al. 2015. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells 33:2400–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, et al. 2010. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 24:1731–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iorgulescu JB, Ivan ME, Safaee M, Parsa AT. 2016. The limited capacity of malignant glioma-derived exosomes to suppress peripheral immune effectors. J. Neuroimmunol 290:103–8 [DOI] [PubMed] [Google Scholar]
- 60.Jeppesen DK, Hvam ML, Primdahl-Bengtson B, Boysen AT, Whitehead B, et al. 2014. Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J. Extracell. Vesicles 3:25011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson E, Dickerson KL, Connolly ID, Hayden Gephart M. 2018. Single-cell RNA-sequencing in glioma. Curr. Oncol. Rep 20:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnstone RM. 2005. Revisiting the road to the discovery of exosomes. Blood Cells Mol. Dis 34:214–19 [DOI] [PubMed] [Google Scholar]
- 63.Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. 1987. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem 262:9412–20 [PubMed] [Google Scholar]
- 64.Jung CS, Foerch C, Schanzer A, Heck A, Plate KH, et al. 2007. Serum GFAP is a diagnostic marker for glioblastoma multiforme. Brain 130:3336–41 [DOI] [PubMed] [Google Scholar]
- 65.Keerthikumar S, Gangoda L, Gho YS, Mathivanan S. 2017. Bioinformatics tools for extracellular vesicles research. Methods Mol. Biol 1545:189–96 [DOI] [PubMed] [Google Scholar]
- 66.Keklikoglou I, Cianciaruso C, Güç E, Squadrito ML, Spring LM, et al. 2019. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol 21:190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keshtkar S, Azarpira N, Ghahremani MH. 2018. Mesenchymal stem cell-derived extracellular vesicles: novel frontiers in regenerative medicine. Stem Cell Res. Ther 9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kore RA, Abraham EC. 2014. Inflammatory cytokines, interleukin-1 beta and tumor necrosis factoralpha, upregulated in glioblastoma multiforme, raise the levels of CRYAB in exosomes secreted by U373 glioma cells. Biochem. Biophys. Res. Commun 453:326–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kore RA, Edmondson JL, Jenkins SV, Jamshidi-Parsian A, Dings RPM, et al. 2018. Hypoxia-derived exosomes induce putative altered pathways in biosynthesis and ion regulatory channels in glioblastoma cells. Biochem. Biophys. Rep 14:104–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kourembanas S 2015. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu. Rev. Physiol 77:13–27 [DOI] [PubMed] [Google Scholar]
- 71.Kowal J, Arras G, Colombo M, Jouve M, Morath JP, et al. 2016. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. PNAS 113:E968–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, et al. 2013. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. PNAS 110:7312–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kunigelis KE, Graner MW. 2015. The dichotomy of tumor exosomes (TEX) in cancer immunity: Is it all in the ConTEXt? Vaccines 3:1019–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurywchak P, Tavormina J, Kalluri R. 2018. The emerging roles of exosomes in the modulation of immune responses in cancer. Genome Med 10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lang HL, Hu GW, Chen Y, Liu Y, Tu W, et al. 2017. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci 21:959–72 [PubMed] [Google Scholar]
- 76.Lang HL, Hu GW, Zhang B, Kuang W, Chen Y, et al. 2017. Glioma cells enhance angiogenesis and inhibit endothelial cell apoptosis through the release of exosomes that contain long non-coding RNA CCAT2. Oncol. Rep 38:785–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, et al. 2003. Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J. Neurosci 23:4034–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Le Rhun E, Perry JR. 2016. Vascular complications in glioma patients. Handb. Clin. Neurol 134:251–66 [DOI] [PubMed] [Google Scholar]
- 79.Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. 2011. Microvesicles as mediators of intercellular communication in cancer—the emerging science of cellular ‘debris.’ Semin. Immunopathol 33:455–67 [DOI] [PubMed] [Google Scholar]
- 80.Li CC, Eaton SA, Young PE, Lee M, Shuttleworth R, et al. 2013. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol 10:1333–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu H, Chen L, Liu J, Meng H, Zhang R, et al. 2017. Co-delivery of tumor-derived exosomes with alphagalactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett 411:182–90 [DOI] [PubMed] [Google Scholar]
- 82.Liu S, Sun J, Lan Q. 2014. Glioblastoma microvesicles promote endothelial cell proliferation through Akt/beta-catenin pathway. Int. J. Clin. Exp. Pathol 7:4857–66 [PMC free article] [PubMed] [Google Scholar]
- 83.Liu ZM, Wang YB, Yuan XH. 2013. Exosomes from murine-derived GL26 cells promote glioblastoma tumor growth by reducing number and function of CD8+ T cells. Asian Pac. J. Cancer Prev 14:309–14 [DOI] [PubMed] [Google Scholar]
- 84.Lo Cicero A, Schiera G, Proia P, Saladino P, Savettieri G, et al. 2011. Oligodendroglioma cells shed microvesicles which contain TRAIL as well as molecular chaperones and induce cell death in astrocytes. Int. J. Oncol 39:1353–57 [DOI] [PubMed] [Google Scholar]
- 85.Lombardi G, Pambuku A, Bellu L, Farina M, Della Puppa A, et al. 2017. Effectiveness of antiangiogenic drugs in glioblastoma patients: a systematic review and meta-analysis of randomized clinical trials. Crit. Rev. Oncol. Hematol 111:94–102 [DOI] [PubMed] [Google Scholar]
- 86.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, et al. 2016. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131:803–20 [DOI] [PubMed] [Google Scholar]
- 87.Magana-Maldonado R, Chavez-Cortez EG, Olascoaga-Arellano NK, Lopez-Mejia M, Maldonado-Leal FM, et al. 2016. Immunological evasion in glioblastoma. BioMed. Res. Int 2016:7487313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mahase S, Rattenni RN, Wesseling P, Leenders W, Baldotto C, et al. 2017. Hypoxia-mediated mechanisms associated with antiangiogenic treatment resistance in glioblastomas. Am. J. Pathol 187:940–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marleau AM, Chen CS, Joyce JA, Tullis RH. 2012. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med 10:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mazzoleni S, Galli R. 2012. Gliomagenesis: a game played by few players or a team effort? Front. Biosci. (Elite Ed.) 4:205–13 [DOI] [PubMed] [Google Scholar]
- 91.McComb RD, Bigner DD. 1984. The biology of malignant gliomas—a comprehensive survey. Clin. Neuropathol 3:93–106 [PubMed] [Google Scholar]
- 92.Meehan B, Rak J, Di Vizio D. 2016. Oncosomes – large and small: what are they, where they came from? Extracell. Vesicles 5:33109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mildenberger I, Bunse L, Ochs K, Platten M. 2017. The promises of immunotherapy in gliomas. Curr. Opin. Neurol 30:650–58 [DOI] [PubMed] [Google Scholar]
- 94.Miller JJ, Wen PY. 2016. Emerging targeted therapies for glioma. Expert Opin. Emerg. Drugs 21:441–52 [DOI] [PubMed] [Google Scholar]
- 95.Minciacchi VR, Freeman MR, Di Vizio D. 2015. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol 40:41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mirzaei R, Sarkar S, Dzikowski L, Rawji KS, Khan L, et al. 2018. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology 7:e1478647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. 1997. The exosome: a conserved eukaryotic RNA processing complex containing multiple 3 exoribonucleases. Cell 91:457–66 [DOI] [PubMed] [Google Scholar]
- 98.Monteiro RQ, Lima LG, Gonçalves NP, Arruda de Souza MR, Leal AC, et al. 2016. Hypoxia regulates the expression of tissue factor pathway signaling elements in a rat glioma model. Oncol. Lett 12:315–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morello M, Minciacchi VR, de Candia P, Yang J, Posadas E, et al. 2013. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle 12:3526–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Muller L, Muller-Haegele S, Mitsuhashi M, Gooding W, Okada H, Whiteside TL. 2015. Exosomes isolated from plasma of glioma patients enrolled in a vaccination trial reflect antitumor immune activity and might predict survival. Oncoimmunology 4:e1008347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, et al. 2011. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with α-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol 29:330–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, et al. 2017. CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology 19:v1–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oushy S, Hellwinkel JE, Wang M, Nguyen GJ, Gunaydin D, et al. 2018. Glioblastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Philos. Trans. R. Soc. Lond. B 373:20160477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Palomo L, Casal E, Royo F, Cabrera D, van-Liempd S, Falcon-Perez JM. 2014. Considerations for applying metabolomics to the analysis of extracellular vesicles. Front. Immunol 5:651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pan BT, Johnstone RM. 1983. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: selective externalization of the receptor. Cell 33:967–78 [DOI] [PubMed] [Google Scholar]
- 106.Pavon LF, Sibov TT, de Souza AV, da Cruz EF, Malheiros SMF, et al. 2018. Tropism of mesenchymal stem cell toward CD133+ stem cell of glioblastoma in vitro and promote tumor proliferation in vivo. Stem Cell Res. Ther 9:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Perng P, Lim M. 2015. Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front. Oncol 5:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pink RC, Elmusrati AA, Lambert D, Carter DRF. 2018. Royal Society Scientific Meeting: extracellular vesicles in the tumour microenvironment. Philos. Trans. R. Soc. Lond. B 373:20170066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Podbielska M, Szulc ZM, Kurowska E, Hogan EL, Bielawski J, et al. 2016. Cytokine-induced release of ceramide-enriched exosomes as a mediator of cell death signaling in an oligodendroglioma cell line. J. Lipid Res 57:2028–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Puhka M, Takatalo M, Nordberg ME, Valkonen S, Nandania J, et al. 2017. Metabolomic profiling of extracellular vesicles and alternative normalization methods reveal enriched metabolites and strategies to study prostate cancer-related changes. Theranostics 7:3824–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Qazi MA, Vora P, Venugopal C, Sidhu SS, Moffat J, et al. 2017. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol 28:1448–56 [DOI] [PubMed] [Google Scholar]
- 112.Ragusa M, Barbagallo C, Cirnigliaro M, Battaglia R, Brex D. 2017. Asymmetric RNA distribution among cells and their secreted exosomes: biomedical meaning and considerations on diagnostic applications. Front. Mol. Biosci 4:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, et al. 2006. Salvage radioimmunotherapy with murine iodine-131-labeled antitenascin monoclonal antibody 81C6 for patients with recurrent primary and metastatic malignant brain tumors: phase II study results. J. Clin. Oncol 24:115–22 [DOI] [PubMed] [Google Scholar]
- 114.Redzic JS, Ung TH, Graner MW. 2014. Glioblastoma extracellular vesicles: reservoirs of potential biomarkers. Pharmacogenom. Pers. Med 7:65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reynes G, Vila V, Fleitas T, Reganon E, Font de Mora J, et al. 2013. Circulating endothelial cells and procoagulant microparticles in patients with glioblastoma: prognostic value. PLOS ONE 8:e69034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rich JN, Guo C, McLendon RE, Bigner DD, Wang XF, Counter CM. 2001. A genetically tractable model of human glioma formation. Cancer Res 61:3556–60 [PubMed] [Google Scholar]
- 117.Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, et al. 2018. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv 4: eaar 2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ricklefs FL, Mineo M, Rooj AK, Nakano I, Charest A, et al. 2016. Extracellular vesicles from highgrade glioma exchange diverse pro-oncogenic signals that maintain intratumoral heterogeneity. Cancer Res 76:2876–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rooj AK, Ricklefs F, Mineo M, Nakano I, Chiocca EA, et al. 2017. MicroRNA-mediated dynamic bidirectional shift between the subclasses of glioblastoma stem-like cells. Cell Rep 19:2026–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Santos PM, Butterfield LH. 2018. Dendritic cell-based cancer vaccines. J. Immunol 200:443–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sartori MT, Della Puppa A, Ballin A, Campello E, Radu CM, et al. 2013. Circulating microparticles of glial origin and tissue factor bearing in high-grade glioma: a potential prothrombotic role. Thromb. Haemost 110:378–85 [DOI] [PubMed] [Google Scholar]
- 122.Sartori MT, Della Puppa A, Ballin A, Saggiorato G, Bernardi D, et al. 2011. Prothrombotic state in glioblastoma multiforme: an evaluation of the procoagulant activity of circulating microparticles. J. Neurooncol 104:225–31 [DOI] [PubMed] [Google Scholar]
- 123.Sharif S, Ghahremani MH, Soleimani M. 2018. Delivery of exogenous miR-124 to glioblastoma multiform cells by Wharton’s jelly mesenchymal stem cells decreases cell proliferation and migration, and confers chemosensitivity. Stem Cell Rev 14:236–46 [DOI] [PubMed] [Google Scholar]
- 124.Simon T, Pinioti S, Schellenberger P, Rajeeve V, Wendler F, et al. 2018. Shedding of bevacizumab in tumour cells-derived extracellular vesicles as a new therapeutic escape mechanism in glioblastoma. Mol. Cancer 17:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Simpson RJ, Mathivanan S. 2012. Extracellular microvesicles: the need for internationally recognised nomenclature and stringent purification criteria. J. Proteom. Bioinform 5: ii [Google Scholar]
- 126.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, et al. 2008. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol 10:1470–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Skotland T, Sandvig K, Llorente A. 2017. Lipids in exosomes: current knowledge and the way forward. Prog. Lipid Res 66:30–41 [DOI] [PubMed] [Google Scholar]
- 128.Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. 2015. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 199:145–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Spinelli C, Montermini L, Meehan B, Brisson AR, Tan S, et al. 2018. Molecular subtypes and differentiation programmes of glioma stem cells as determinants of extracellular vesicle profiles and endothelial cell-stimulating activities. J. Extracell. Vesicles 7:1490144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stegmayr B, Ronquist G. 1982. Promotive effect on human sperm progressive motility by prostasomes. Urol. Res 10:253–57 [DOI] [PubMed] [Google Scholar]
- 131.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med 352:987–96 [DOI] [PubMed] [Google Scholar]
- 132.Sullivan BA, Kronenberg M. 2005. Activation or anergy: NKT cells are stunned by αgalactosylceramide. J. Clin. Investig 115:2328–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sun X, Ma X, Wang J, Zhao Y, Wang Y, et al. 2017. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget 8:36137–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sundarraj N, Schachner M, Pfeiffer SE. 1975. Biochemically differentiated mouse glial lines carrying a nervous system specific cell surface antigen (NS-1). PNAS 72:1927–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Svensson KJ, Kucharzewska P, Christianson HC, Skold S, Lofstedt T, et al. 2011. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. PNAS 108:13147–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Taheri B, Soleimani M, Aval SF, Memari F, Zarghami N. 2018. C6 glioma-derived microvesicles stimulate the proliferative and metastatic gene expression of normal astrocytes. Neurosci. Lett 685:173–78 [DOI] [PubMed] [Google Scholar]
- 137.Thaler J, Ay C, Mackman N, Bertina RM, Kaider A, et al. 2012. Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J. Thromb. Haemost 10:1363–70 [DOI] [PubMed] [Google Scholar]
- 138.Thompson EM, Frenkel EP, Neuwelt EA. 2011. The paradoxical effect of bevacizumab in the therapy of malignant gliomas. Neurology 76:87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Todorova D, Simoncini S, Lacroix R, Sabatier F, Dignat-George F. 2017. Extracellular vesicles in angiogenesis. Circ. Res 120:1658–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trams EG, Lauter CJ, Salem N Jr., Heine U. 1981. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta 645:63–70 [DOI] [PubMed] [Google Scholar]
- 141.Treps L, Edmond S, Harford-Wright E, Galan-Moya EM, Schmitt A, et al. 2016. Extracellular vesicletransported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 35:2615–23 [DOI] [PubMed] [Google Scholar]
- 142.Treps L, Perret R, Edmond S, Ricard D, Gavard J. 2017. Glioblastoma stem-like cells secrete the proangiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 6:1359479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 9:654–59 [DOI] [PubMed] [Google Scholar]
- 144.van der Pol E, Boing AN, Gool EL, Nieuwland R. 2016. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost 14:48–56 [DOI] [PubMed] [Google Scholar]
- 145.van der Vos KE, Abels ER, Zhang X, Lai C, Carrizosa E, et al. 2016. Directly visualized glioblastomaderived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro-Oncology 18:58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.van Es N, Bleker S, Sturk A, Nieuwland R. 2015. Clinical significance of tissue factor-exposing microparticles in arterial and venous thrombosis. Semin. Thromb. Hemost 41:718–27 [DOI] [PubMed] [Google Scholar]
- 147.Vega EA, Graner MW, Sampson JH. 2008. Combating immunosuppression in glioma. Future Oncol 4:433–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Waldowska M, Bojarska-Junak A, Rolinski J. 2017. A brief review of clinical trials involving manipulation of invariant NKT cells as a promising approach in future cancer therapies. Cent. Eur. J. Immunol 42:181–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang K, Ye L, Lu H, Chen H, Zhang Y, et al. 2017. TNF-α promotes extracellular vesicle release in mouse astrocytes through glutaminase. J. Neuroinflamm 14:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wei JW, Cai JQ, Fang C, Tan YL, Huang K, et al. 2017. Signal peptide peptidase, encoded by HM13, contributes to tumor progression by affecting EGFRvIII secretion profiles in glioblastoma. CNS Neurosci. Ther 23:257–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Weller M, Roth P, Preusser M, Wick W, Reardon DA, et al. 2017. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat. Rev. Neurol 13:363–74 [DOI] [PubMed] [Google Scholar]
- 153.Willms E, Johansson HJ, Mager I, Lee Y, Blomberg KE, et al. 2016. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep 6:22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Witwer KW, Soekmadji C, Hill AF, Wauben MH, Buzas EI, et al. 2017. Updating the MISEV minimal requirements for extracellular vesicle studies: building bridges to reproducibility. J. Extracell. Vesicles 6:1396823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wolf P 1967. The nature and significance of platelet products in human plasma. Br. J. Haematol 13:269–88 [DOI] [PubMed] [Google Scholar]
- 156.Wong ML, Prawira A, Kaye AH, Hovens CM. 2009. Tumour angiogenesis: its mechanism and therapeutic implications in malignant gliomas. J. Clin. Neurosci 16:1119–30 [DOI] [PubMed] [Google Scholar]
- 157.Wurdinger T, Deumelandt K, van der Vliet HJ, Wesseling P, de Gruijl TD. 2014. Mechanisms of intimate and long-distance cross-talk between glioma and myeloid cells: how to break a vicious cycle. Biochim. Biophys. Acta 1846:560–75 [DOI] [PubMed] [Google Scholar]
- 158.Xu R, Greening DW, Rai A, Ji H, Simpson RJ. 2015. Highly-purified exosomes and shed microvesicles isolated from the human colon cancer cell line LIM1863 by sequential centrifugal ultrafiltration are biochemically and functionally distinct. Methods 87:11–25 [DOI] [PubMed] [Google Scholar]
- 159.Yan K, Yang K, Rich JN. 2013. The evolving landscape of glioblastoma stem cells. Curr. Opin. Neurol 26:701–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, et al. 2015. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 4:27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yang Y, Boza-Serrano A, Dunning CJR, Clausen BH, Lambertsen KL, Deierborg T. 2018. Inflammation leads to distinct populations of extracellular vesicles from microglia. J. Neuroinflamm 15:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zabeo D, Cvjetkovic A, Lasser C, Schorb M, Lotvall J, Hoog JL. 2017. Exosomes purified from a single cell type have diverse morphology. J. Extracell. Vesicles 6:1329476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang G, Zhang Y, Cheng S, Wu Z, Liu F, Zhang J. 2017. CD133 positive U87 glioblastoma cellsderived exosomal microRNAs in hypoxia-versus normoxia-microenviroment. J. Neurooncol 135:37–46 [DOI] [PubMed] [Google Scholar]
- 164.Zhang H, Freitas D, Kim HS, Fabijanic K, Li Z, et al. 2018. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol 20:332–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhang J, Williams BM, Lawman S, Atkinson D, Zhang Z, et al. 2017. Non-destructive analysis of flake properties in automotive paints with full-field optical coherence tomography and 3D segmentation. Opt. Express 25:18614–28 [DOI] [PubMed] [Google Scholar]
- 166.Zhao C, Wang H, Xiong C, Liu Y. 2018. Hypoxic glioblastoma release exosomal VEGF-A induce the permeability of blood-brain barrier. Biochem. Biophys. Res. Commun 502:324–31 [DOI] [PubMed] [Google Scholar]