

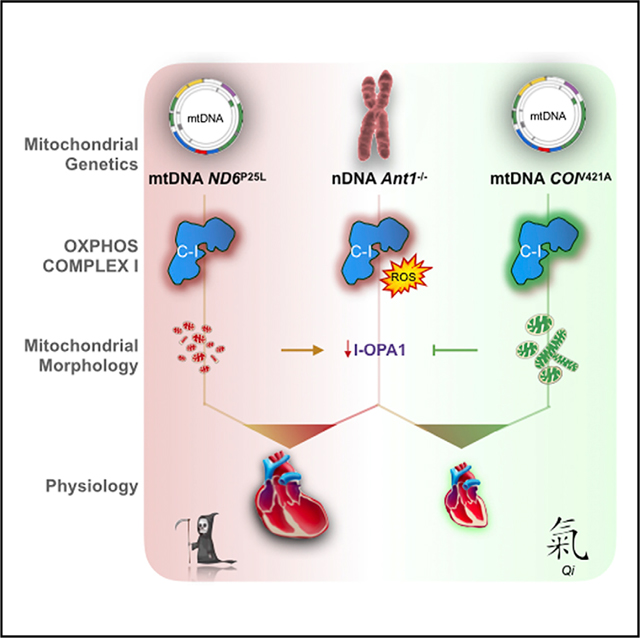
SUMMARY
Nuclear-encoded mutations causing metabolic and degenerative diseases have highly variable expressivity. Patients sharing the homozygous mutation (c.523delC) in the adenine nucleotide translocator 1 gene (SLC25A4, ANT1) develop cardiomyopathy that varies from slowly progressive to fulminant. This variability correlates with the mitochondrial DNA (mtDNA) lineage. To confirm that mtDNA variants can modulate the expressivity of nuclear DNA (nDNA)-encoded diseases, we combined in mice the nDNA Slc25a4 −/− null mutation with a homoplasmic mtDNA ND6P25L or COlV421A variant. The ND6P25L variant significantly increased the severity of cardiomyopathy while the COlV421A variant was phenotypically neutral. The adverse Slc25a4−/− and ND6P25L combination was associated with impaired mitochondrial complex I activity, increased oxidative damage, decreased l-Opa1, altered mitochondrial morphology, sensitization of the mitochondrial permeability transition pore, augmented somatic mtDNA mutation levels, and shortened lifespan. The strikingly different phenotypic effects of these mild mtDNA variants demonstrate that mtDNA can be an important modulator of autosomal disease.
In Brief
McManus et al. investigate mitochondrial-nuclear interactions in human disease by developing a murine model combining the Slc25a4 cardiomyopathy null gene mutation with mild mtDNA variants. Differences in mtDNA genotype can strongly influence autosomal gene mutations; conversely, accumulation of somatic mtDNA mutations can augment the deleterious consequences of mitochondrial-nuclear interactions.
Graphical Abstract
INTRODUCTION
Mitochondrial dysfunction may play a critical role in the pathophysiology of complex metabolic and degenerative diseases (Wallace, 2013b). One such disease is cardiomyopathy, in which mitochondrial dysfunction has been demonstrated in both pediatric and adult cases (Murphy et al., 2016; Porter et al., 2011; Zaragoza et al., 2011).
The mitochondrial proteome is encoded by 1,000 to 2,000 nDNA genes and by hundreds to thousands of copies of the maternally inherited mtDNA. The mtDNA encodes for 13 essential polypeptides required for energy production via oxidative phosphorylation (OXPHOS). The energetically favorable transfer of electrons through OXPHOS complexes I-IV is used to generate an electrochemical gradient across the mitochondrial inner membrane that is utilized by complex V (H+-pumping F1,F0-ATP synthase) to produce ATP. Mitochondrial matrix ATP is then exchanged for cytosolic ADP by the inner membrane adenine nucleotide translocators (ANTs). In addition to ATP/ADP translocation, the ANTs regulate the mitochondrial permeability transition pore (mtPTP) in distinct ways depending upon the isoform (Bauer et al., 1999; Chevrollier et al., 2011; Jang et al., 2008; Kokoszka et al., 2004; Zamora et al., 2004a, 2004b). Humans have four ANT isoforms, while mice have three, but both species express the heart-muscle-brain isoform, ANT1 (SLC25A4), and the systemic isoform, ANT2 (SLC25A5), in the heart (Kokoszka et al., 2016). Hence, inactivation of the SLC25A4 gene will result in a partial cardiac ANT defect.
Inactivating mutations in ANT1 cause autosomal recessive myopathy and cardiomyopathy (Echaniz-Laguna et al., 2012; Palmieri et al., 2005). However, the severity of ANT1-deficient cardiomyopathy can be variable, and this variability has been correlated with the mtDNA lineage (Strauss et al., 2013).
There are three types of clinically relevant mtDNA variations: functional polymorphisms, which are associated with ancient mtDNA lineages called haplogroups, recent deleterious mutations that can result in maternally inherited disease, and somatic mutations that accumulate in tissues over time. The phenotypic consequences of these different types of mtDNA variation are interdependent (Ji et al., 2012) and may be modulated by interactions with nDNA variants, as well as the environment (Wallace, 2013a). Due to these complex interactions, the etiological significance of mtDNA variation in common, age-related diseases has been difficult to delineate (Zaragoza et al., 2011). Therefore, the significance of mtDNA contribution to cardiomyopathy in which patients harbor mtDNA variants along with nDNA contractile protein mutations (Arbustini et al., 1998a, 1998b) or Anti mutations (Strauss et al., 2013) remains to be clarified.
To address this knowledge gap, we have developed a mouse model of mitochondrial cardiomyopathy by combining the Slc25a4 (Anti) null gene mutation (Graham et al., 1997; Narula et al., 2011) with two different mild mtDNA variants. These mtDNA variants are the NADH dehydrogenase subunit 6 (ND6) gene nucleotide 13997G>A missense mutation (ND6P25L) causing a partial complex I defect (Lin et al., 2012) and the cytochrome c oxidase subunit 1 gene (COI) nucleotide 6589T>C missense mutation (COIV421A) resulting in a partial complex IV defect (Acin-Perez et al., 2003; Fan et al., 2008).
Here, we report the physiological effects of six nDNA-mtDNA combinations of wild-type (WT) and mutant nDNA Anti and mtDNA COIV421A and ND6P25L alleles in C57BL/6JEiJ mice (Navarro et al., 2012). This investigation has established that mild differences in the mtDNA genotype can strongly influence the expressivity of autosomal gene mutations. Furthermore, the accumulation of somatic mtDNA mutations can augment the consequences of unfavorable mitochondrial-nuclear interactions.
RESULTS
Transcriptional Profile of the Ant1-Deficient Heart
To clarify the molecular basis of the Slc25a4 mutation-associated cardiomyopathy, we performed RNA sequencing on left ventricular myocardium of WT and Ant1−/− mice. Changes in functionally annotated gene families were assessed by gene set enrichment analysis. Significantly downregulated pathways included extracellular matrix, intracellular signaling, chemotaxis, and mitochondrial antioxidant and fatty acid metabolism genes (Figures 1A and S1). Upregulated pathways included mitochondrial OXPHOS enzymes, canonical skeletal muscle proteins, and membrane-associated voltage-gated ion channels (Figures 1B and S1).
Figure 1. ANT1-Deficiency Induces Transcriptional Changes Associated with Pathological Remodeling of the Heart.
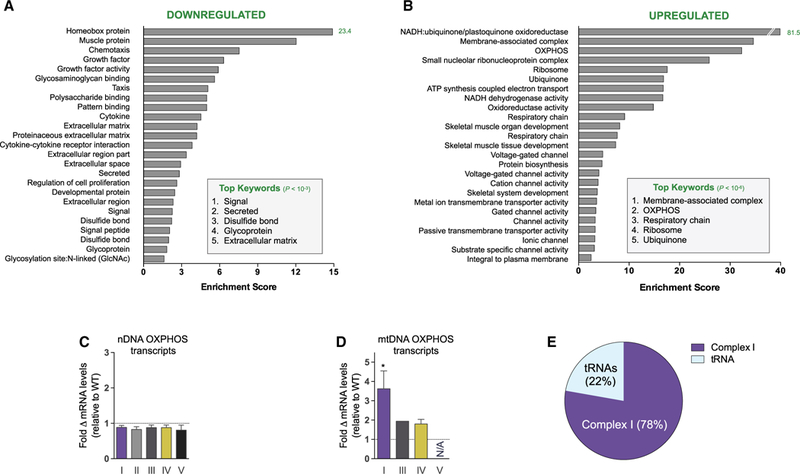
(A and B) Twenty-five most significantly downregulated (>2-fold, total = 459; A) and upregulated (<0.4-fold, total = 363; B) functionally annotated gene categories in Ant1-null myocardium compared with WT. Searched categories include Gene Ontology, Protein Information Resource, Sequence (Seq) Features, Kyoto Encyclopedia of Genes and Genomes, InterPro protein sequence, and analysis classification. Analysis performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID, v6.7).
(C and D) (C) Fold change in mRNA transcripts from nDNA and (D) mtDNA OXPHOS genes by complex relative to WT (*p < 0.05).
(E) Portion of differentially expressed RNA transcripts from mtDNA. n = 4.
The loss of structural extracellular components and the ectopic induction of skeletal muscle proteins in the myocardium are consistent with other forms of cardiomyopathy, as well as aging (Aronow et al., 2001; Houtkooper et al., 2011). The fibrotic alterations and cardiac remodeling from hypertrophic to dilated cardiomyopathy are also consistent with the observed pathology of the Ant1−/− mouse (Burke et al., 2016; Graham et al., 1997; Narula et al., 2011).
The induction of OXPHOS components is particularly note-worthy, with complex I (NADH-ubiquinone/plastoquinone oxidoreductase) genes being among the most strongly upregulated gene families in the Ant1-null heart (Figure 1B and Table S1). While the average change of all nDNA-coded OXPHOS polypeptide genes was slightly downregulated (Figure 1C), the mtDNA-coded OXPHOS complex I subunits were induced (Figures 1D and 1E). The preferential upregulation of mtDNA complex I genes may represent a compensatory response to a partial impairment of complex I by the Ant1 mutation.
mtDNA Variants Alter Ant1−/− Murine Lifespan and the Response to Environmental Stress
Given the alterations in complex I expression induced by the Ant1 mutation, we investigated the effects of combining the Ant1 mutation with mtDNA mutations that resulted in partial defects in complex I, ND6P25L, versus complex IV, COIV421A. Ant1 WT (Ant1+/+) and Ant1 null (Anti−/−) mice were crossed with mice harboring normal, ND6P25L, or COIV421A mtDNAs. This resulted in six nDNA-mtDNA combinations: WT nDNA and WT mtDNA (WT); WT nDNA and ND6P25L mtDNA (ND6); WT nDNA and COIV421A mtDNA (COI); Ant1−/− and WT mtDNA (ANT1); Ant1−/− and ND6P25L mtDNA (ANTI/ND6); and Ant1−/− and COIV421A mtDNA (ANTI/COI). In all cases, the nDNA background was derived from C57BL/6J into which the WT Nnt allele was reintroduced (C57BL6/JEiJ) (Navarro et al., 2012).
In isolation, the Ant1−/−, ND6P25L, and COIV421A mutations had a modest effect on mouse longevity relative to WT: the COIV42iA variant reduced lifespan by 9%, the ND6P25L variant by 16%, and the Ant1−/− by 22%. Combining the Ant1−/− with the COIV421A variant had minimal effect on the Ant1−/− life-span, but combining the Ant1−/− with ND6P25L reduced the lifespan by 49%, more than twice that of the Ant1−/− alone (Figure 2A). Therefore, combining Ant1−/− with the ND6P25L variant was significantly more deleterious than combining Ant1−/− with COIV421A.
Figure 2. Effects of Six nDNA-mtDNA Anti and COIV421A and ND6P25L Combination Strains on Longevity, Activity, and Thermal Tolerance.
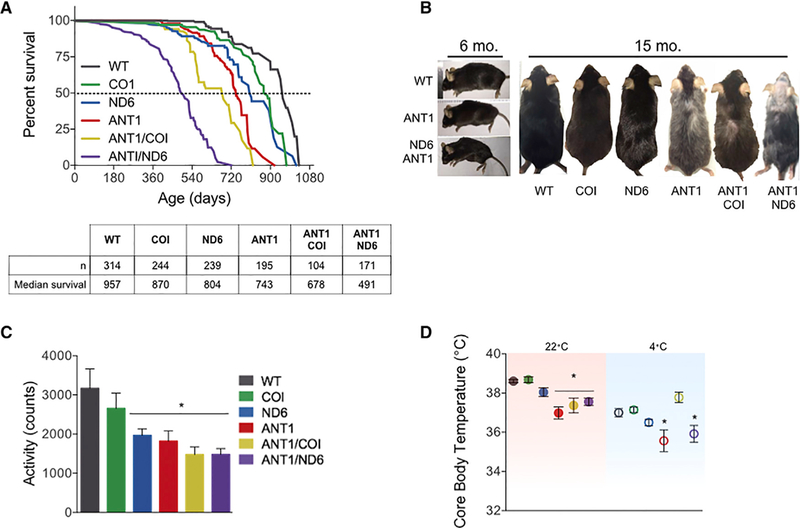
(A) Kaplan-Meier analysis of six nDNA-mtDNA combinations. Median lifespan and n are depicted in the chart below.
(B) Progeroid morphology evident in ANT1 and ANTI/ND6 mice as early kyphosis at 6 months, which progresses to gray hair, alopecia, and advanced kyphosis at 15 months compared with WT and ANTI/COI.
(C) Indirect calorimetry recordings of activity counts during the dark cycle of all strains at 6 months.
(D) Core body temperature for 6-month-old mice measured at 22°C (filled symbols; red box) showing reduction of all Ant1-null strains. Core body temperature after 4° Ccold stress (open symbols; blue box) showing that the ANT1 and ANTI/ND6 strains were unable to maintain normal body temperature while the ANTI/COI mice were unaffected by cold stress.
*p < 0.02 versus WT; n = 6. See also Figure S2.
The ANT1 and ANTI/ND6 mice exhibited morphological features of premature aging starting with kyphosis at 6 months (Figure 2B), but COI, ND6, and ANTI/COI were indistinguishable from WT (not shown). By 15 months, ANT1 and ANTI/ND6 mice had pronounced kyphosis, gray hair, and alopecia, while ANTI/COI mice appeared less affected (Figure 2B). Basal motor activity levels of the ND6, ANT1, ANTI/COI, and ANTI/ND6 mice were significantly reduced in young mice, but COI mice were again indistinguishable from WT (Figure 2C).
To assess the effects of nDNA-mtDNA interaction on thermoregulation, core body temperature was first determined in mice housed at 22°C, which is ~10°C lower than their thermoneutral zone. This mild stress demands an ~60% increase in metabolic rate to maintain thermoregulation relative to thermoneutral temperature (Cannon and Nedergaard, 2011). WT, COI, and ND6 mice maintained their core body temperature under these standard housing conditions. However, the core body temperatures of ANT1, ANTI/COI, and ANTI/ND6 mice were reduced by an average of ~1.3°C relative to WT (Figure 2D). Of note, the body weights of these mice were indistinguishable (Figure S2). Metabolic stress was then increased by lowering the temperature to 4°C for 6 hr. Core body temperature of WT, COI, and ND6 mice decreased 1°C-2°C, while the ANT1 and ANTI/ND6 mice lost ~3°C (Figure 2D). Surprisingly, the ANTI/COI strain maintained the same core body temperature in these extreme conditions as at 22°C (Δ0.31°C ± 0.07°C). Hence, the Ant1 mutation has the greatest impact on thermoregulation in mild conditions, which is sustained by the mtDNA ND6P25L, but ameliorated by the mtDNA COIV421A variant under more extreme environmental stress.
mtDNA Variation Alters the Expressivity of Age-Dependent Cardiomyopathy
Since ANT1-deficient humans (Strauss et al., 2013) and mice (Narula et al., 2011) manifest age-related, progressive cardiomyopathy, we next investigated the relative cardiac weight and morphology of the six nDNA-mtDNA mouse strains over their lifespans (Figure 3A). The relative heart weight of WT mice increased 0.06 ± 0.008 mg g−1•month−1 (r2 = 0.32; p < 0.0001). The mtDNA COIV421A variant did not perturb this trend (p = 0.32), while the ND6P25L variant reduced the WT rate by ~21% (p = 0.0003; Figure 3A).
Figure 3. Progression of Cardiomyopathy and Left Ventricular Mechanics in the nDNA-mtDNA Combination Strains.
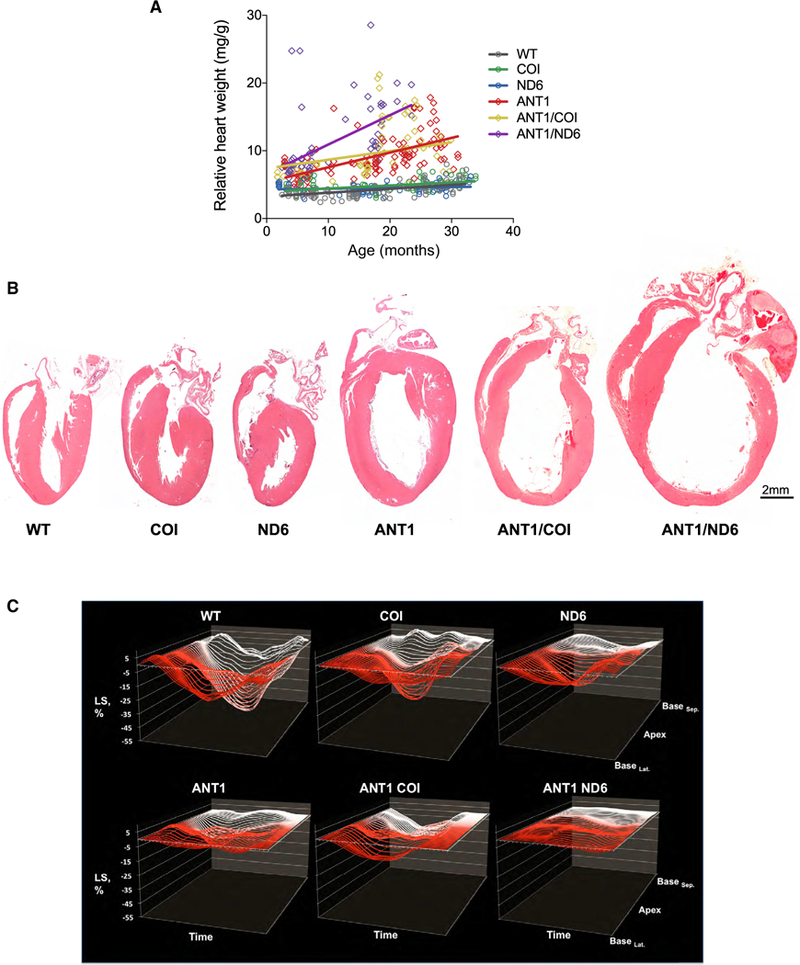
(A) Correlation between the relative rate of cardiac enlargement (heart weight/body weight) over the lifespan for each strain (p < 0.0001; n = 65–111).
(B) Gross morphology of hearts by H&E stain at 12 months of age.
(C) Cardiac contractility measured by two-dimensional speckle-tracking echocardiography (2D-STE). Representative strain curves obtained from the B-mode long-axis view of the left ventricle over the cardiac cycle (x axis) showing longitudinal strain (% deformation; y axis) and region of the left ventricle (z axis). Each panel shows 49 regional strain curves topographically extending from infero-lateral base (BaseLat.) toward LV apex and back toward antero-septal base (BaseSep ). Note the progressive variations in magnitude and timing of the strain curves between different nDNA-mtDNA combinations. n = 10–34.
See also Table S2.
Deletion of Anti increased the rate and extent of the age-related cardiac hypertrophy (Figure 3A). Again, the CO1V421A variant did not affect this trend (p = 0.18 for ANTI/COI versus ANT1). However, combination of ND6P25L with Ant1−/− accelerated the rate of cardiac enlargement ~200% over that of the Ant1−/− hearts (p = 0.003). By 15 months, the relative heart weight of the ANTI/ND6 mice had increased 6.5-fold due to hypertrophic dilation (Figure 3B).
To further delineate the structural and functional consequences in the heart of each nDNA-mtDNA combination, we employed echocardiography (ECG) with M mode and speckle tracking in B mode for strain analysis. Strain reflects the total deformation of the ventricular myocardium during a cardiac cycle as a percentage of its initial length. Speckle-tracking-based strain analysis detects intrinsic contractile function, allowing assessment of myocardial pathophysiology prior to overt cardiac dysfunction (Biering-Sorensen et al., 2017; Geyer et al., 2010). Notably, there were no significant differences in mice harboring either the mtDNA COIV421A or ND6P25L variant related to WT across all ECG parameters examined (Table S2). The mtDNA variants only affected cardiac structure or function when the nDNA Anti defect was also present. Addition of COIV421A to Ant1−/− decreased left ventricular posterior wall thickness at systole (PWTs), but had no functional consequences on the Ant1−/− heart. Conversely, addition of the ND6P25L (ANTI/ND6) adversely affected every ECG measure, leading to dramatic ventricular dilation and an 80% reduction in cardiac contractility (Figure 3C; Table S2). Thus, the mtDNA ND6P25L variant markedly increased the severity of the Ant1−/− cardiomyopathy, while the mtDNA COIV421A variant had no effect.
Opposing Effects of mtDNA ND6P25L and COIV421A on Mitochondrial Morphology
Abnormal mitochondrial morphology is commonly found in primary mitochondrial diseases, as well as in idiopathic cardiomyopathies. To determine how these genetic alterations could influence mitochondrial morphology within the heart, we performed ultrastructural analysis of the left ventricle. The results revealed highly disordered myofibrils in all but the WT and COI strains (Figures 4A–4F), altered mitochondrial morphology (Figures 4A’–4F’), and increased lipofuscin aggregates (Figure 4G) in strains with the shortest lifespan. While the mtDNA variants alone did not significantly alter mitochondrial content, Ant1−/− led to a 60% increase in mitochondrial number (Figure 4H), and ANTI/COI and ANTI/ND6 hearts had over twice the amount of mitochondria as WT (Figure 4H). ND6, ANT1, and ANTI/ND6 ventricles had increased mitochondrial fragmentation, with the ANTI/ND6 mitochondria reduced to half the size of all other strains (Figures 4I, 4J, and S3). Notably, the COIV421A variant had the reverse effect, leading to significant mitochondrial enlargement, which counterbalanced mitochondrial fragmentation in the Ant1−/− heart (p = 0.64 for ANTI/COI versus WT; Figures 4I and S3).
Figure 4. Opposing Effects of mtDNA Variation in Complexes I and IV on Mitochondrial Morphology and Cristae Architecture.
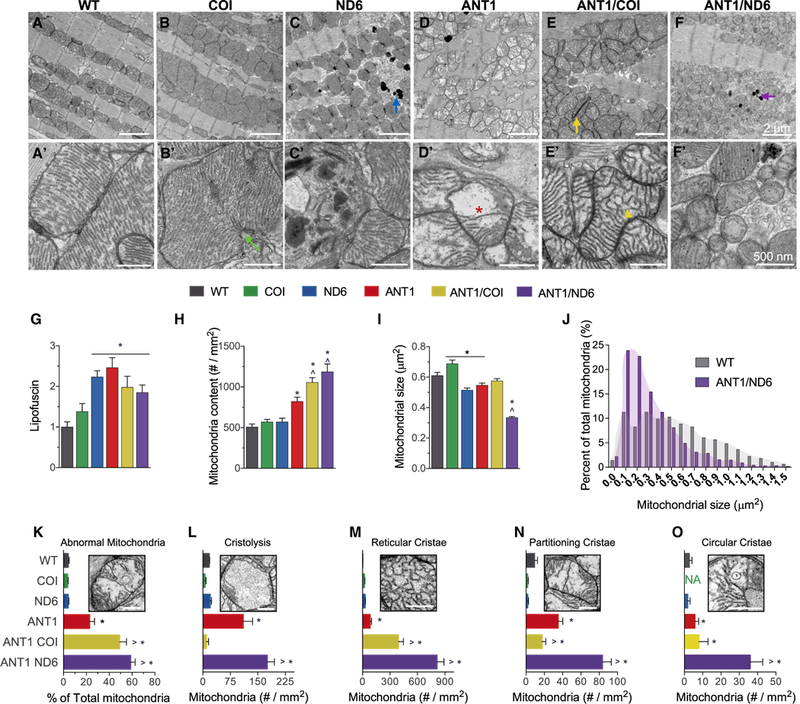
(A-F) Representative electron micrographs of (A-F) ventricular cardiomyocytes and (A’-F’) mitochondria from each nDNA-mtDNA combination (n = 3). Scale bars, 2 μm (A-F); 500 nm (A’-F’). Sarcomeric and mitochondrial alignment in (A) WT and (B) COI versus structural disarray in all other nDNA-mtDNA combinations (C-F). (B and B’) Mitochondrial enlargement in COI myocardium, suggestive of hyperfusion or impaired fission (green arrow; B’). Mitochondrial fragmentation, autophagic vesicles, and lipofuscin accumulation (blue and purple arrows) in (C and C’) ND6 and (F and F’) ANTI/ND6 myocardium. (D-F) Mitochondrial proliferation and (D’-F’) cristae abnormalities, paracrystalline inclusions (yellow arrow; E, ANTI/COI), cristolysis (red asterisk; D’, ANT1), and reticular morphology (yellow arrowhead; E’, ANTI/COI), present in all Ant1-null strains.
(G-I) (G) Quantification of age-related lipofuscin deposits, normalized to WT. Ultrastructural quantification of (H) mitochondrial content and (I) average size per strain (n = 496–1,541).
(J) Percentage distribution of mitochondrial size showing the shift in mitochondrial morphology by the ANTI/ND6 compared with WT.
(K) Percent of abnormal mitochondria counted in each strain.
(L-O) Average number of mitochondria per mm2 with the following most common defects in cristae: (L) cristolysis, and (M) reticular, (N) partitioning, and (O) circular morphologies.
*p < 0.05 versus WT; ^p < 0.001 versus ANT1. Data are represented as means ± SE. See also Figures S3 and S4.
While the WT, COI, and ND6 strains had similar intramitochondrial ultrastructure, all of the Ant1−/− strains were highly abnormal. The most common intramitochondrial abnormalities were electron dense inclusions, hypodense compartments, and cristae malformations (Figures 4K–4O and S4). Cristae morphology regulates mitochondrial functions that are crucial for cardiac homeostasis, such as respiratory efficiency, Ca2+ buffering, reactive oxygen species (ROS) release, and apoptotic signaling (Cogliati et al., 2016). Accordingly, the strains with the most severe dilated cardiomyopathy (ANT, ANT/COI, and ANT/ ND6) had the highest percentage of mitochondria with irregular cristae morphology (Figures 4L–4O and S4). The incidence of abnormal cristae was consistently higher in the ANTI/ND6 mitochondria than the ANTI/COI mitochondria.
Functional OXPHOS Consequences of Ant1 and mtDNA Mutations
Modulation of cristae morphology allows mitochondrial OXPHOS machinery to adapt to cellular substrate supply and metabolic demand by shifting form to meet function. The master regulator of cristae dynamics is the dynamin-related GTPase OPA1 (Glytsou et al., 2016). OPA1 localizes to the IMM, where its multiple isoforms coordinate mitochondrial fusion, cristae integrity, respiratory complex assembly and function, and mtDNA maintenance (Chen et al., 2012; Cogliati et al., 2013). When OPA1 is lost, or the isoforms are imbalanced, mitochondria fragment, undergo cristolysis, and OXPHOS assembly fails. Moreover, OPA1 overexpression proves protective for mitochondrial disease by correcting cristae structure and restoring OXPHOS function (Civiletto et al., 2015). To determine if altered OPA1 was associated with ultrastructural variation between the different nDNA-mtDNA combinations, OPA1 levels were quantified in cardiac mitochondria from each strain (Figure 5A). The presence of ND6P25L or Ant1−/− was associated with decreased OPA1 (Figure 5B), attributable to the specific loss of long OPA1 isoforms (l-OPAs; Figure 5C). Conversely, COIV421A mitochondria maintained OPA1 levels equivalent to WT, and blocked the loss of l-OPA1 associated with Ant1-/−.
Figure 5. Consequences of Mitochondrial-Nuclear Interaction on OPA1 Processing and Mitochondrial OXPHOS Complexes.
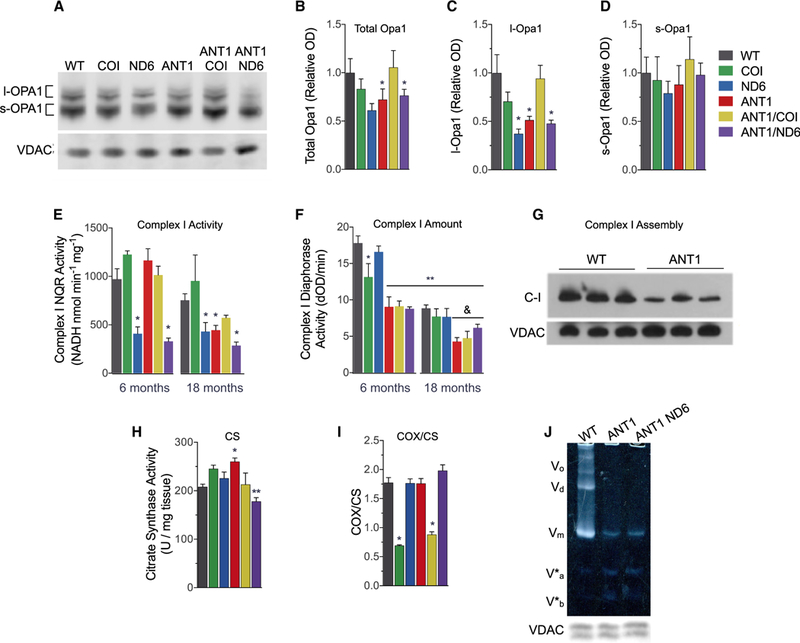
(A-D)(A) Representative western blot of OPA1 and VDAC from isolated heart mitochondria. Densitometric analysis of (B) total OPA1, (C) long Opal (I-OPA1), and (D) short OPA1 (s-OPA1) isoforms, normalized to VDAC and shown as fold change from WT.
(E and F) (E) Complex I (C-1) NADH:quinone oxidoreductase (NQR) activity determined by rotenone-sensitive NADH oxidation in the presence of coenzyme Q1 using isolated heart mitochondria (25 μg). (F) Age-dependent decline in C-I diaphorase activity (dOD/min) determined by C-I immunocaptured from 10 μg of isolated heart mitochondria (**p < 0.0001 versus WT at 6 months; &p < 0.05 versus WT at 18 months).
(G) C-I assembly measured by blue native electrophoresis of heart mitochondria (20 μg) and immunodetection with anti-NDUFA1.
(H) Citrate synthase (CS) activity per mg myocardial tissue as a marker of mitochondrial content at 6 months (**p < 0.01 versus ANT1).
(I) Cytochrome c oxidase (COX) activity from the same samples normalize to CS activity. Data are represented as means ± SE.
(J) Resolution of oligomeric states of complex V (C-V) F1F0-ATPase by clear native PAGE. Oligomers were undetectable in Ant1−/− mitochondria solubilized in digitonin (3% w/v). V0, oligomers; Vd, dimers; Vm, monomers; V*a and V*b, partial C-V components. Each well was loaded with 30 μg of mitochondrial protein, evident by the anti-VDAC loading control from a duplicate gel.
*p ≤ 0.05, **p < 0.001 versus age-matched WT, except as noted in (F) and (H); n = 3–7. See also Figure S5.
Loss of OPA1 and cristae integrity are associated with an age-dependent decrease in the specific activities and assembly of OXPHOS complexes I, IV, and V (Chen et al., 2012; Cogliati et al., 2013; Patten et al., 2014). To evaluate this relationship in the six nDNA-mtDNA combination strains, we analyzed the activity, amount, and structural assembly of these heart OXPHOS complexes. The ND6P25L variant decreased complex I (C-I), rotenone-sensitive, NADH:quinone oxidoreductase (NQR) specific activity by 58% in 6-month-old mice (Figure 5E), which was reduced an additional 10% when combined with Ant1-/−. At 18 months of age, C-I NQR activity in the ANT1 heart was decreased to the same low level as the ND6, and their combination (ANT1/ND6) decreased C-I NQR activity an additional ~50% (Figure 5E). In contrast, C-I NQR activity in the COI mice remained normal at both 6 and 18 months, and C-I activity in the ANT1/COI heart was higher than that of the ND6, ANT1, or ANT1/ND6 hearts at 18 months (Figure 5E). COIV421A also increased C-I subunits in Ant1−/− hearts at 6 months of age and prevented the age-associated loss of C-I subunits that occurred in all other nDNA-mtDNA mutant strains (Figure S5). Thus, the COIV421A mtDNA variant partially compensates for the Ant1−/− complex I defect.
To determine whether the decline in NQR activity could be attributed to a decreased amount of C-I, we measured the diaphorase activity of holoenzyme C-I (Figure 5F). While ND6P25L decreased C-I NQR specific activity by half, this variant had no effect on the amount of holoenzyme (Figure 5F). Surprisingly, Ant1−/− reduced holoenzyme levels by ~51% in 6-month-old hearts without affecting NQR specific activity. Both age and genotype significantly influenced C-I content, accounting for 42% and 34% of the total variance, respectively, but age was the dominant factor (p < 0.0001; two-way ANOVA for age × genotype). The decreased amount of assembled C-I in ANT1 hearts was confirmed by blue native gel electrophoresis (Figure 5G).
Complex IV (COX) activity was measured by cytochrome c oxidation in myocardial homogenates, and the specific activity normalized to citrate synthase (CS) activity (Figures 5H and 5I). CS activity was increased by 25% in Ant1-deficient myocardium but decreased by the addition of ND6P25L (Figure 5H). COX/CS activity was decreased to 39% of control by the mtDNA CO/V421A mutation, but the additional loss of Anti (ANT1/COI) had no appreciable effect. Ant1−/−, ND6P25L, or their combination, had no effect on the COX/CS activity up to 18 months of age (Figure 5I).
Since all Ant1−/− hearts have an abnormal cristae structure, which is only partially alleviated by the retention of l-Opa1 in ANT1/COI mitochondria, and complex V(F1F0ATPase) dimerization is obligatory for proper cristae formation (Davies et al., 2012; Paumard et al., 2002; Strauss et al., 2008), we hypothesized that F1F0-ATPase structural assembly might be affected by loss of Ant1. The dimer equilibrium of the F1F0-ATPase was determined by clear native gel electrophoresis under mild solubilizing conditions (Wittig et al., 2010). F1F0-ATPase dimers and oligomers were drastically decreased in ANT1 and ANTI/ND6 heart mitochondria (Figure 5J). Analysis using more stringent detergent revealed that the ATP synthase monomers (V) are also labile and give rise to subcomplexes V*a and V*b in all Ant1−/− hearts (Figure S5), which resemble those reported in cells harboring mtDNA ATP6 and ATP8 mutations (Carrozzo et al., 2006; Mourier et al., 2014; van der Westhuizen et al., 2010).
Mitochondrial Respiration, ROS Production, and mtPTP Activation
To determine how these genetic defects alter mitochondrial physiology, we analyzed isolated heart mitochondria from 6- to 18-month-old animals. Ant1−/− increased the mitochondrial membrane potential ~25%−30% in young ANT1, ANT1/COI, and ANT1/ND6 hearts relative to WT, COI, and ND6 hearts (Figure 6A), possibly due to the subassembled F1F0-ATPase (Mourier et al., 2014) or its diminished activity due to limited availability of matrix ADP (Graham et al., 1997).
Figure 6. Mitochondrial Bioenergetics, ROS Production, and mtPTP Stability in Isolated Cardiac Mitochondria from Six nDNA-mtDNA Genetic Combinations.
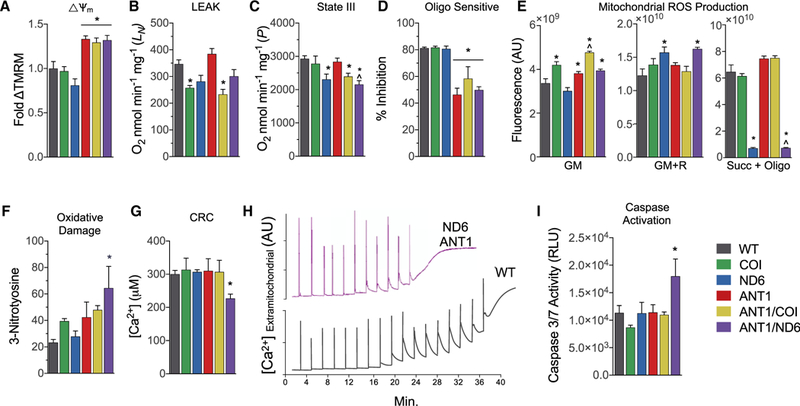
(A) Mitochondrial membrane potential determined bytetramethylrhodamine methyl ester fluorescence respiring on glutamate and malate (GM), graphed relative to WT.
(B) Mitochondrial oxygen consumption rate metabolizing glutamate and malate (GM) in the absence of ADP or uncoupler (state II or LEAK rate).
(C) Mitochondrial oxygen consumption rate metabolizing GM in the presence of ADP (state III or P rate).
(D) Relative inhibition of respiration by oligomycin in mitochondria during state III.
(E) Hydrogen peroxide (H2O2) production detected by Amplex red in isolated mitochondria incubated with GM, rotenone (R), or succinate (Succ) and oligomycin (Oligo).
(F) Nitroxidative damage determined by 3-nitrotyrosine (3NT) protein adducts in heart tissue from 12-month-old mice.
(G) Ca2+ levels required to activate the mtPTP and collapse the mitochondrial membrane potential.
(H) Representative traces of extramitochondrial Ca2+ following 20 μM Ca2+ pulses delivered every 2 min until the spontaneous release of mitochondrial Ca2+, marking the onset of mtPTP opening in WT and ANTI/ND6 mice.
(I) Activation of intrinsic apoptosis determined by effector caspase-3 and −7 activities.
Data are represented as means ± SE. *p ≤ 0.01 versus WT; ^p ≤ 0.01 versus ANT1; n = 3–5. See also Figure S6.
Oxygen consumption was then measured in heart mitochondria respiring on the C-I, NADH-linked substrates glutamate and malate (GM). The mtDNA mutations affected different aspects of mitochondrial respiration: COIV421A impaired the LEAK (L) state (in the absence of ADP or uncoupler; Figure 6B), while ND6P25L impaired the ADP-stimulated (P) state (Figure 6C). Interestingly, Anti−/− did not impair ADP-stimulated respiration in young animals, possibly due to compensation by ANT2 (Brand et al., 2005).
To determine the importance of the F1F0-ATPase on state III (ADP-stimulated; P) respiration, we treated mitochondria respiring on GM and ADP with the F1F0-ATPase inhibitor oligomycin. Oligomycin decreased the state III respiration rate of WT, COI, and ND6 mitochondria 80%, consistent with mitochondrial respiration being predominantly coupled to the F1F0- ATPase through use of the proton gradient to synthesize ATP (Figure 6D). Conversely, oligomycin only decreased Anti-null mitochondrial respiration by 50%−60% (Figure 6D). The diminished effect of oligomycin may be due to the preexisting inhibition of respiration due to limited mitochondrial ADP import caused by the loss of ANT1 and impaired F-i F0-ATPase function and assembly (Mourier et al., 2014).
To determine how the nDNA-mtDNA genetic combinations influence mitochondrial ROS production in the heart, we analyzed H2O2 emission using the Amplex red assay (Figure 6E). When mitochondria were respiring on GM, H2O2 emission was increased 18%−48% in mitochondria harboring the COIV421A variant, as well as all Ant1-null mitochondria (Figure 6E, GM). Treatment with rotenone (R), which binds to the C-I CoQ binding site downstream of the site of NADH reduction and enhances ROS production via forward electron transport (FET) (Murphy, 2009), decreased CO/V421A ROS production but increased ND6P25L ROS production. When the complex II substrate, succinate, is added as the electron donor and the membrane potential maximized by blocking the F1F0-ATPase with oligomycin, the electrons are driven backward from CoQ into C-I, resulting in ROS production by reverse electron transport (RET) (Brand, 2010; Chouchani et al., 2014; Murphy, 2009). In this case, ROS production from ND6 and ANTI/ND6 heart mitochondria was abolished (Figure 6E, Succ + Oligo). By contrast, WT, ANT1, and COI mitochondria showed robust ROS production by RET. To determine if these different modes of ROS production were associated with oxidative stress in vivo, we measured 3-nitrotyrosine protein adducts and found the highest level of oxidative damage in ANT1/ND6 hearts, which only produce ROS by FET (Figure 6F).
Excessive mitochondrial ROS production via FET induces ryanodine receptor-dependent Ca2+ release and sensitizes the mtPTP, which impairs cardiac contractility and increases mitochondrial permeability transition (Zorov et al., 2000). Since ANTI/ND6 hearts have the most diminished contractility and the highest FET-associated ROS and oxidized proteins, we hypothesized that their mitochondria would also be most susceptible to Ca2+-mediated permeability transition. Indeed, the addition of ND6P25L to Ant1−/− increased Ca2+ sensitivity of the mtPTP, rendering ANTI/ND6 hearts more prone to permeability transition (Figures 6G and 6H) and the release of apoptogenic proteins into the cytosol, culminating in effector caspase activation (Figure 6I) (McManus et al., 2014).
mtDNA Somatic Mutation Load Correlates with the Progression of Ant1−/− Cardiomyopathy
Since the ANT1 and COI and ND6 alterations are present at birth, the progression of the cardiomyopathy of the ANTI/ND6 mutant heart implies the presence of an additional age-related mitochondrial factor. We hypothesized that this additional factor may be the accumulation of somatic mtDNA mutations due to persistent oxidative stress and/or impaired mitochondrial turnover (Dai et al., 2010). We analyzed the relative levels of deleted mtDNAs in ventricular tissue from the six nDNA-mtDNA combinations using long extension PCR (LX-PCR) (Figures 7A and 7B) and random mutation capture quantitative PCR (Figure 7C) (Vermulst et al., 2007, 2008a, 2008b). The frequency of mtDNA deletions per mitochondrial genome increased linearly over time in all strains (Figure 7C). The somatic mtDNA mutation rate was 2-fold higher in ANT1 hearts and almost 6-fold higher in ANTI/ND6 hearts (versus WT, p < 0.01), which correlated with increased COX-deficient myocardial fibers (Figure S7), and severity of cardiomyopathy and mortality induced by the different mtDNA-nDNA combinations (Figures 7D and 7E).
Figure 7. Effect of Mitochondrial-Nuclear Interaction on Somatic mtDNA Mutation Accumulation.
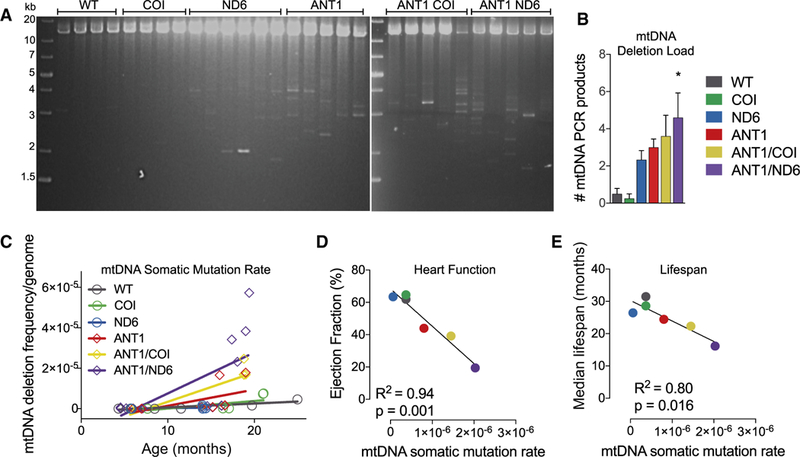
(A and B) Long-range amplification of mtDNA(12.7 kb) from each nDNA-mtDNAcombination at 12–15monthsindicating multiple, large-scaledeletions (*p = 0.01; n = 4–6).
(C) Linear regression analysis of the frequency of mtDNA deletions per genome detected by qPCR of the most common deletion hotspot flanking two 15-bp repeats in mtDNA of mice from each strain over 4–25 months of age (p < 0.001; n = 8–14).
(D and E) Correlation between (D) median lifespan or (E) cardiac function and the mtDNA somatic mutation rate, as determined by random mutation capture assay.
See also Figure S7.
DISCUSSION
The goal of this study was to determine whether mtDNA is a crucial modulator of autosomal gene mutations with variable expressivity and to provide insight into how mitochondrial-nuclear interactions contribute to cardiovascular disease and heart failure (Arbustini et al., 1998b; Strauss et al., 2013). We first confirmed that mtDNA variants can modulate the expressivity of nDNA gene mutations by analyzing cardiac function and mitochondrial physiology of mice sharing the nuclear Anti null mutation (Graham et al., 1997; Narula et al., 2011), but with different mtDNAs (WT, ND6P25L, or COIV421A). Importantly, neither the mtDNA ND6P25L nor COIV421A variant alone has an appreciable effect on cardiac physiology in C57BL/6JEiJ mice. The COIV421A variant also has minimal effect on the Ant1−/− cardiac pathophysiology. By contrast, the mtDNA ND6P25L variant accelerates the Ant1−/− phenotype, resulting in a rapidly progressive, and ultimately fatal, dilated cardiomyopathy. The markedly different effects of the COIV421A and ND6P25L variants on Ant1−/−-induced cardiomyopathy is proof of principle that mtDNA can exert modifier effects on the expressivity of autosomal gene mutations.
Further evidence that nDNA-mtDNA interactions can affect the severity of cardiomyopathy is found in our concurrent discovery that the nDNA Nnt genotype exerts a marked modifier effect on the cardiac phenotype of the mtDNA COIV421A variant. C57BL/6J mice are homozygous for a truncated Nnt allele (Nnt−/−), which itself causes no overt cardiac defects in mice (Freeman et al., 2006; Huang et al., 2006; Toye et al., 2005). As we reported previously, the mtDNA COIV421A variant on the C57BL/6J Nnt−/− background causes hypertrophic cardiomyopathy (Fan et al., 2008). However, when the mtDNA COIV421A was transferred onto the C57BL/6JEiJ Nnt+/+ nuclear background, the COIV421A variant had little effect on the cardiac function (Figure 3C). Hence, the COIV421A variant acts synergistically with the Nnt−/− null mutation, while the ND6P25L mutation acts synergistically with the Ant1−/− null mutation.
Having established that mitochondrial-nuclear interactions do modulate both mtDNA and nDNA expressivity, we investigated the pathophysiological basis of this relationship. Our analysis revealed four major mechanisms by which mtDNA variation modulates Ant1 expressivity: OXPHOS complex I function, differential ROS production, mPTP sensitivity, and the somatic mtDNA mutation rate. First, Ant1−/− and ND6P25L both perturb complex I, with Ant1−/− reducing the amount of structurally assembled complex I, and the ND6P25L variant reducing complex I specific activity. Second, the ND6P25L variant generates comparable total ROS production as Ant1−/− and COIV421A, but the ND6P25L variant produces ROS only by FET, as opposed to both FET and RET for the Ant1−/− and COIV421A variants. ROS produced by RET induces mitochondrial hormesis, which is protective of age-related disease and extends lifespan (Scialo et al., 2016). Since ND6P25L blocks RET, this cytoprotective effect would be lost for the mtDNA ND6P25L but not for the COIV421A. Third, the Ant1−/−/ND6P25L combination sensitizes cardiac mitochondria to Ca2+-induced mtPTP permeability transition while the Ant1−/−/COIV421A combination does not, which is consistent with increased oxidative damage and defective cardiac contractility in the compound Ant1−/−/ND6P2SL genotype (Zorov et al., 2000). Finally, the Ant1−/−/ND6P2SL combination results in a 2.5-fold higher mtDNA somatic mutation rate than the Ant1−/−/co/v421a combination, which may amplify the inherited mitochondrial defects over time and accelerate the progression of cardiomyopathy (Vermulst et al., 2007, 2008b).
The distinctive alterations in mitochondrial ultrastructure conferred by the mtDNA variants are consistent with their respective biochemical consequences on Ant1 expressivity, which appear to converge at the mitochondrial dynamics regulator, OPA1. The effect of mtDNA COIV421Aand ND6P25Lon mitochondrial morphology is diametrically opposed: COIV421A increases mitochondrial size, while ND6P25L causes severe mitochondrial fragmentation. Interestingly, ND6P25L and Ant1−/− each lead to loss of the long OPA1 isoforms (l-OPA1) responsible for mitochondrial fusion (Figure 5C), supporting a key role for complex I inhibition and ROS production in aberrant OPA1 processing and downstream mitochondrial fragmentation (Gray et al., 2013; Norton et al., 2014; Ramonet et al., 2013). Mitochondrial fragmentation not only induces more ROS and mtDNA damage (Yu et al., 2008), it also impairs the mixing of multiple mtDNAs within the same mitochondrion, thereby blocking the benefits of inter-mtDNA complementation (Chen et al., 2010). Accordingly, increased de novo somatic mtDNA mutations due to Ant1−/− (Figures 7A–7C) would be more deleterious when combined with ND6P25Lthan with COIV421A. Indeed, the COIV421A variant prevents Anti−/−-induced complex I impairment and loss of l-OPA1 and blocks mitochondrial fragmentation, thus preventing the decline in Ant1−/− mitochondrial function with age (Figure S6).
In conclusion, we have shown that the severity of the cardiomyopathy caused by a nuclear gene mutation can be directly modulated by the presence of otherwise sub-pathogenic mtDNA variants. This was true both for the differential effects of the ND6P25L and COIV421A mtDNA variants on the Anti−/− back-ground, as well as for the COIV421A versus WT mtDNA on the Nnt−/− background. These results demonstrate that nDNA-mtDNA interactions can play a critical role in modulating the phenotypes of nDNA gene mutations.
Limitations of Study
While we feel that mtDNA-nDNA interactions will be important in modulating the manifestations of various nDNA gene mutations, the specific phenotypic manifestations described here undoubtedly reflect the specific mouse mtDNA sequences and the Anti−/− mutant used in this study.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Douglas C. Wallace (wallaced1@email.chop.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Genetics
All mice were maintained on the C57Bl/6JEiJ background, which was derived from the Jackson Laboratory C56Bl/6J line into which the wild-type (WT) nicotinamide nucleotide transhydrogenase (Nnt) gene was reintroduced (Navarro et al., 2012), herein designated as WT. These mice were compared to mice harboring the mtDNA NADH dehydrogenase subunit 6 gene (ND6) nt 13997G>A P25Lor cytochrome c oxidase subunit I gene (COI) nt 6598T>C V421 mutations generated via the female embryonic stem (ES) cell fusion method (Fan et al., 2008; Lin et al., 2012). The phenotypes of these mtDNA variants, as well as the adenine nucleotide translocator isoform 1 (Ant1)-null gene (Graham et al., 1997; Narula et al., 2011), were previously reported on the C56Bl/6J Nnt1−/− strain. We transferred the Ant1−/−, ND6P25L, and COIV421A mutations in to the C57Bl/6JEiJ strain by repeated backcrosses to remove the confounding influence of the nuclear modifier, Nnt (Huang etal., 2006; Kim etal., 2010). The C57Bl/6JEiJ Ant1−/− males were then crossed with female C57Bl/6JEiJ ND6P25Lor COIV421A to attain C57Bl/6JEiJ Ant1−/− ND6P25L and C57Bl/6JEiJ nDNA-Ant1−/− COIV41A mice. The WT and Ant1−/− mice were maintained by brother-sister matings. The mtDNA ND6P25L and COIV421A mutant mice were maintained by crossing female mtDNA mutant mice with WT males. The Ant1−/− ND6P25L and Ant1−/− COIV421A mutant mice were maintained by crossing the Ant1−/− ND6P25L and Ant1−/− COIV421A females with Anti−1- males.
Mouse Maintenance and Longevity
All experimental procedures involving mice were conducted in accordance to approved Institutional Animal Care and Use Committee (IACUC) protocols at the Children’s Hospital of Philadelphia. To eliminate confounding effects of estrogen on mitochondrial physiology, only male mice were considered in our analyses (Duckles et al., 2006; Eichner and Giguere, 2011). Male mice were pooled at weaning to attain 3–5, genotype-, sex- and age-matched mice per cage with ad libitum access to food and water, on a 13:11 light:dark cycle. A minimum of 3 subjects per strain were used for all experiments, except assessment of respiratory complex I assembly by blue-native electrophoresis (n = 2). Additional subjects were included, as noted, based on the availability of age- and strain-matched mice and nature of the assay (Lin et al., 2012; Narula et al., 2011). During sample preparation, subjects were randomly numbered to blind the experimenter for subsequent tests and analysis. The effect of specific mitochondrial genetic variation on longevity was determined by Kaplan-Meier survival curves constructed from the known date of birth and death of at least one hundred mice per genotype. Apart from cage changes and daily health checks, these mice were left undisturbed. When mice were determined moribund by the veterinary staff, they were humanely euthanized and the date of death recorded. Differences between groups were evaluated using the log-rank (Mantel-Cox) test (GraphPad, La Jolla, CA, USA).
METHOD DETAILS
RNA Sequencing and Bioinformatics
Total RNA was extracted in Trizol with a motorized pestle from 30–60 mg of left myocardium (n = 4) and processed with the PureLink RNA-Easy kit (Life technologies #12183018A), and affinity-depleted of ribosomal RNA (rRNA) with magnetic bead RiboMinus Eukaryote System v2 (Life technologies # A15026). PolyA enrichment was avoided to maximize recovery of mtDNA-encoded transcripts. Purified RNA was quantified and quality checked with RNA 6000 Pico chip (Agilent #5067–1513) on Agilent Bioanalizer2100, and spiked with the ERCC RNA Spike-In Mix (4456740). Library preparation was performed from 800 ng of rRNA-depleted RNA and processed with the Ion Total RNA-Seq Kit v2 (Life technologies #4475936) and multiplexed-sequenced with Ion PI Sequencing 200 Kit v2 chemistry (Life Techologies #4485149) on the Ion Proton platform. Two samples were pooled per chip, yielding on average >25 million reads per sample, and an average read length of ~80 bp. After trimming, transcript counts were generated by a STAR alignment to the reference mouse genome, mapping to a total of 14,642 annotated genes, and normalized using ERCC spike-ins counts. Differential expression, fold change, and statistical significance for each gene was established using the DESeq Bioconductor package in R. DESeq. Data were subsequently analyzed for individual genes of interest, or using DAVID (Data-base for Annotation, Visualization and Integrated Discovery v6.7) to derive functional significance of up- and down-regulated pathways of interest.
Cardiac Size and Histopathology
Hearts were excised, weighed, and fixed in 4% paraformaldehyde for at least 48 hr. Relative heart weight was determined by the ratio of heart to body weight over the lifespan of each strain (n = 65 −111). Longitudinal sections of paraffin embedded tissues were sliced and stained with hematoxylin and eosin stain (H&E; n = 3).
Calorimetry
Energy expenditure was assessed using indirect calorimetry (Oxymax; Columbus Instruments). Mice were singly housed with water and food ad libitum (n = 6). After a 3-day acclimation period, oxygen consumption and carbon dioxide production were measured over 30 hr using an air flow of 600 mL/min at 22°C. For the cold stress challenge, mice were placed in 4°C for 6 hr. Core body temperature was measured using a rectal probe (Harvard Apparatus).
Speckle Tracking Echocardiography of Left Ventricular Mechanics
Images were obtained using the Vevo2100 equipped with a MS550D transducer (Visual Sonics, Toronto, ON, Canada). The mice were lightly anaesthetized using 1.5% isofluorane mixed with 100% O2 during the time of imaging. Electrocardiography leads were applied to monitor heart rate and trigger echo image acquisitions. The images were obtained from the B-mode long-axis view and the M-mode of the parasternal short-axis view. Speckle tracking echocardiography (STE) was performed as a sensitive indicator of myocardial contractility using Vevostrain software (Visual Sonics, Toronto, ON, Canada) incorporated into the Vevo2100 from the movies acquired from the B-mode long-axis view. The tracking quality was visually inspected, and the tracing was confirmed as acceptable when the traced line moved along with the moving heart image for at least three cardiac cycles. These cardiac cycles were used for the analysis.. Strain analysis was performed using speckle tracking algorithms applied on high-frequency ultrasound images. Parasternal long-axis view provided longitudinal strain, whereas, parasternal short axis view was used for assessing circumferential strain. M-mode images at mid-LV were used to determine left ventricular internal dimension at end-diastole (LVIDd) and LV dimension internal dimension at end-systole (LVIDs). The LV ejection fraction (EF) was calculated. Interventricular septum (IVS) wall thickness and left ventricular posterior wall (LVPW) thickness were obtained (n = 10 – 34).
Mitochondrial Morphology
Thin slices of left ventricular myocardium were excised from 6 month old animals of each genotype and immediately immersed in fixation buffer containing 2% glutaraldehyde and 0.1 M cacodylate (pH 7.4) (Picard et al., 2015). Samples were post-fixed in 2.0% osmium tetroxide for 1 h at room temperature and rinsed in distilled H2O before in-bloc staining with 2% uranyl acetate. After dehydration through a graded ethanol series, each sample was embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Cardiomyocyte orientation and quality were first checked in 1 mm thick sections stained with 1% toluidine blue. Thin sections (90 mm) were then mounted on filmed copper grids and stained with uranyl acetate and lead citrate and examined on a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT Advantage image capture software. Myocardial mitochondria were manually traced from at least 8 calibrated images per subject at ×12,000 indirect magnification using Image J (National Institutes of Health, Bethesda, MD). To produce frequency distributions of morphological parameters, the area of each mitochondrion was assigned to one of twenty bins of equal size. Lipofuscin granules were counted per image and normalized to the mean of WT. Abnormal mitochondria were counted and expressed as percent of total mitochondria per image. The most common types of cristae defects found across all strains (i.e., reticular, partitioning, circular, concentric, or paracrystalline structures and cristolysis) were manually quantified. Statistical significance was evaluated based on 95% confidence interval (C.I.) of the mean.
Mitochondrial Isolation
Mitochondria were isolated from murine hearts (Palmer et al., 1977). The entire procedure was performed on ice or in a cold room (4°C). Ventricular tissue was excised, blotted, weighed, rinsed, and diced in a small beaker containing 2 mL of ice-cold organ preservation solution (BIOPS) comprised of 2.77 mM CaK2 EGTA buffer, 7.23 mM K2 EGTA buffer, 0.1 μM free calcium, 20 mM imidazole, 20 mM taurine, 50 mM 2-(N-morpholino) ethanesulfonic acid hydrate (MES), 0.5 mM dithiothreitol, 6.56 mM MgCl2 • 6H2O, 5.77 mM ATP, and 15 mM phosphocreatine (pH 7.1). The buffer was decanted, and tissue digested in 0.015% trypsin for 10 min. The protease was neutralized by 5x dilution in isolation buffer (50 mM MOPS, 100 mM KCl, 1 mM EGTA, 5 mM MgSO4, 1mM ATP) containing 10 mg/ml fatty acid-free BSA. The tissue was electrically homogenized using a Eurostar Power B (1100 rpm × 7–8 strokes) and mitochondria isolated by differential centrifugation. The final pellet was resuspended in the appropriate experimental buffer.
Complex I Activity
Complex I NADH-ubiquinone reductase (NQR) activity (Ji et al., 2014) was assayed using 25 mg/ml of isolated mitochondria using the complex I assay buffer (250 mM sucrose, 10 mM Hepes, 0.2 mM EDTA, 2.5 mM MgCl2, pH 7.2) containing 40 μg/ mLalamethicin to permeabilize the mitochondria inner membrane. After 1 min, the reaction was started by addition of 100 μM coenzyme Q1, and 200 μM NADH. NADH oxidation was monitored at 340 nm using a Cary300 dual-beam spectrophotometer at 30°C, and rotenone sensitive activity calculated. NQR activity was determined by subtracting the basal (no substrate) and the rotenone (4 μM) insensitive rates from the NADH:CoQ1 rate. The results are shown as nmol NADH mg−1 min−1 using the extinction coefficient of NADH at 340 nm (6.22 mM−1 cm−1). The assay was performed in 3 independent trials (n = 3 – 7).
The amount of assembled complex I was estimated by assaying diaphorase-type activity from complex I immunocaptured from isolated heart mitochondria (10 μg/ml) (Abcam; ab109721). The assay was performed in 2 independent trials (n = 3 – 6).
Native Gel Electrophoresis and Immunodetection
Mitochondrial respiratory complexes were separated by native electrophoresis for analysis of complex I assembly (Yadava et al., 2002). Fresh mitochondrial pellets equivalent to 400 μg of protein were solubilized with 800 μg of dodecyl-β-D-maltoside (DDM; Sigma) in 5 mM 6-aminohexanoic acid, 50 mM imidazole-HCl (pH 7.0) and 10% glycerol. Coomassie Brilliant Blue G-250 (Serva) was added to the solubilized samples at a dye/detergent ratio of 1:5 (w/w). The samples were loaded on a 4%−13% acrylamide gradient gel and electrophoresed overnight. Proteins were transferred to PVDF membranes from the native gels and western blotting performed using standard transfer conditions for 3 hr. The blots were de-stained by washing with 100% methanol and then probed with NDUFA1 antisera to determine complex I assembly (n = 5; antibody provided by Scheffler IE; validated in (Yadava et al., 2002)).
The assembly and oligomerization state of F1F0 ATPase were determined using clear native electrophoresis (CNE) (Wittig et al., 2007). To quantify the holo-complexV(Vm) stability, mitochondrial protein was solubilized on ice for 15 min with 2.5% DDM. The samples were then centrifuged for 30 min at 25000 g, 4°C, and equal mitochondrial protein loaded on CNE gels. The complexes were electrophoresed overnight at 100 – 120 mV; the gel incubated in 35 mM Tris (0.42%), 270 mM Glycine (2.02%), pH 8.3 for 2h; and washed in the same buffer supplemented with 14 mM MgSO4, 0.2% Pb(NO3)2, and 8 mM ATP until the white lead phosphate precipitated. The reaction was stopped with 50% methanol, and the gel imaged. Western blots of duplicate gels were probed with an anti-mouse VDAC monoclonal antibody as a loading control (Abcam; ab14734). The assay was performed in 4 independent trials (n = 4).
Cytochrome-c Oxidase and Citrate Synthase Activity
Frozen samples from the left ventricular myocardium were thawed on ice and approximately 10 mg of each was diced and homogenized with a Teflon pestle at 1:30 (w/v) in 50 mM triethanolamine with 1 mM EDTA (pH 7.4). The samples were solubilized in potassium phosphate buffer and 0.1% (w/v) n-dodecylmaltoside (pH 7.5). The reaction was started by the addition of 0.1 mM reduced ferrocytochrome c (0.1 mM) and the initial rate of oxidation determined by following the decrease in absorbance at 550 nm in a 96-well plate at 30°C (Capaldi et al., 1995). To control for differences in myocardial mitochondrial content, citrate synthase (CS) activity was determined from the same tissue homogenates (Mofarrahi et al., 2013). The samples were diluted 1:26 in CS buffer (100 mM Tris, 0.2 mM acetylCoA, 0.2 mM 5,5-dithio-bis-2-nitrobenzoic acid (DTNB), 70 μM oxaloacetate) and the change in absorbance of DTNB was measured at 412 nm. Parallel samples in buffer lacking oxaloacetate were included as negative controls to ensure specificity of the reaction. The molar extinction coefficients used were 29.5 L mol−1 cm−1 for ferrocytochrome c and 13.6 L mol−1 cm−1 for DTNB. Each assay was performed twice, n = 4 – 6.
Western Blotting and ELISA
Isolated mitochondria were resuspended in 2% SDS (50 mM Tris pH 7.4), rotated at 4C for 30 min, centrifuged at 16,000×g for 10min, and the protein concentration of the supernatant determined by the BCA assay. Aliquots, 30 μg each, were run through a 4%−12% SDS-PAGE and electroblotted onto a nitrocellulose membrane. The membrane was then probed with anti-OPA1 (Cell Signaling), Total OXPHOS Rodent WB Antibody Cocktail (Abcam; ab110413), or anti-VDAC (Cell Signaling; D73D12) primary antibodies, followed by the appropriate secondary antibody (Fluorescent TrueBlot: Anti-Mouse or Rabbit Ig DyLight 800 (Rockland)), imaged on an Odyssey System, and quantified using Image Studio. Each sample was first normalized to VDAC, and then to the mean of the age-matched WT control, n = 3 – 6, in four independent experiments.
Nitrotyrosine concentration was measured by enzyme-linked immunosorbent assay (ELISA) using heart tissue homogenate from 12 mo. old animals according to the OxiSelect Nitrotyrosine EL/SA Kit protocol (STA-305, Cell Biolabs, San Diego, CA, USA), n = 3–4.
Respiration
High-resolution respirometry of isolated mitochondria was performed in ice-cold mitochondrial respiration medium 5 (MiR05:0.5 mM EGTA, 3 mM MgCl2, 60 mM potassium lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, and 1 g/L BSA essentially fatty acid free, adjusted to pH 7.1) at 25°C using the Oxygraph-2k (Oroboros, Innsbruck, Austria). Sequential oxygen consumption rates were determined in mitochondria respiring first on malate (2 mM) and glutamate (10 mM) (state II; L = LEAK), followed by addition of 2.5 mM ADP (state III; P state), then the addition of oligomycin (2 μg/mL) (state 4), and lastly addition of rotenone and antimycin A (residual oxygen consumption; ROX). The quality of the mitochondrial preparations was confirmed by the absence of a cytochrome c effect on oxygen consumption in the P state (data not shown). Respirometry experiments were conducted in pairs of over 18 independent experiments, n = 3.
Mitochondrial Reactive Oxygen Species (mtROS) and Membrane Potential (MMP)
The rate of mtROS production was determined by monitoring the oxidation of the fluorogenic indicator Amplex Red by H2O2 in the presence of horseradish peroxidase as previously described (Lin et al., 2012). Mitochondria (0.05 mg/ml) were incubated in assay medium [125 mM KCl, 20 mM Hepes, 2 mM K2HPO4, 1 mM MgCl2, 0.1 mM EGTA, 0.025% BSA (pH 7.2) at 37°C]. Glutamate and malate (5 mM each) were used to induce forward electron transport. For reverse electron transport experiments, 5 mM succinate and 1 μg/ml oligomycin were included. Amplex Red (1 μM) and 5 U mL-1 horseradish peroxidase were added to initiate the reaction. Fluorescence was recorded at excitation 560 nm and emission 590 nm. Mitochondrial membrane potential (MMP) was assessed on 0.025 mg mitochondria using the potentiometric fluorescent dye TMRM (100 nM) (535 nm excitation, 600 nm emission) while metabolizing the complex I substrates malate and glutamate. The results were normalized by comparison with a parallel experiment but with the FCCP (carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone) uncoupler added. Each sample was run in triplicate, n = 3, in two independent experiments, and results are shown as fold change from WT per condition.
Calcium Retention Capacity
The mitochondrial calcium retention capacity (CRC) of mitochondrial preparations (Tiepolo et al., 2009) was assessed by monitoring Ca2+ uptake and release using the Ca2+ indicator, Calcium Green-5N (1 μM; excitation, 505 nm; emission, 535 nm), monitored using a Fluoromax spectrofluorometer (Horiba; Kyoto Japan) equipped with magnetic stirring rod. The incubation medium contained 0.2 M sucrose, 10 mM Tris-MOPS, 5 mM glutamate-Tris, 2.5 mM malate-Tris, 1 mM. Final volume was 2 mL, pH 7.4, 25°C. All the experiments were started with the addition of 0.5 mg/ml mitochondria followed 1 min later by the indicated pulses of Ca2+. Each sample was run in duplicate. Traces are representative for the group (n = 3 – 4).
mtDNA Deletion Analysis
Whole genomic DNA was isolated from the ventricular myocardium using phenol-chloroform-isoamyl alcohol extraction and diluted to 2.5 ng/μl. mtDNA deletions were detected by amplification of a 12.8 kB mtDNA fragment using Accuprime Hi Fi (Invitrogen 12346068) and the following primer pairs: ND2 Forward: 5’-CTGGAATTCAGCCTACTAGCAATTATCC-3’; 12s Reverse: 5’-TTTAGGTTTATGGCTAAGCATAGTGGGG- 3’. The long template PCR products were equally loaded and run in 2 TBE (0.8%) gels and the number of bands per sample (n = 4 – 6) quantified by ImageJ.
To quantify the deletion burden at a particular site, the random mutation capture (RMC) assay was adapted to detect large deletions in the major arc between two 15-bp direct repeats previously shown to be hotspots for mtDNA deletions (Vermulst et al., 2007, 2008a, 2008b). Briefly, mtDNA was first digested by Taq1 to remove WT mtDNA. The number of mtDNA molecules with a deletion were then determined using the following qPCR primers, spaced several kb apart, flanking multiple TaqI restriction sites: Deletion Forward: 5’-AGGCCACCACACTCCTATTG-3’, position 8810–8829; Deletion Reverse: 5’-AATGCTAGGCGTTTGATTGG-3’, position 13098–13117. The total number of mtDNA molecules per sample was determined using the following control primers that do not include a Taq1 site: Control Forward: 5’-TCGGCGTAAAACGTGTCAAC-3’, position 350–369; Control Reverse: 5’-CCGCCAAGTCCTTTGAGTTT-3’ position 579–598. The ratio of deleted mtDNA molecules to total mtDNA molecules was used to determine the deletion frequencyin the myocardium of mice 4–25 mo. of age. The assay was performed in two independent experiments, n = 8 −14.
Caspase-3/7 Activity
To detect caspase-3/7 activity, mouse hearts were homogenized in hypotonic extraction buffer (25 mM HEPES [pH 7.5], 5 mM MgCl2,1 mM EGTA) on ice. The homogenates were cleared by centrifugation at 13,000 rpm for 15 min at 4°C. Protein concentrations were adjusted to 1 mg/ml, and an equal volume of 10 μg/ml added to the Caspase-Glo Reagent (Promega G8091). The assays were incubated for 1 hr at room temperature before reading on a luminometer (SpectraMax Paradigm; Eugene, OR). n = 4, performed in two independent experiments.
COX-SDH Histochemistry
To identify mitochondrial respiratory chain-deficient cardiomyocytes, we performed sequential cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) reactions on frozen sections from the myocardium of mice at 6 and 18 mo. of age. Tissue samples were frozen in isopentane/liquid nitrogen. Cryostat sections were then stained for simultaneous COX and SDH activity and imaged on a Zeiss Axio Imager.M2, using the 20x objective.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-VDAC | Abcam | Cat#ab14734; RRID: AB_443084 |
| Rabbit monoclonal anti-VDAC | Cell Signaling Technology | Cat#D73D12; RRID: AB_10557420 |
| Mouse Monoclonal total OXPHOS rodent antibody cocktail | Abcam | Cat#ab110413; RRID: AB_2629281 |
| Mouse monoclonal anti-NDUFAl | Yadava et al., 2002 | N/A |
| Mouse monoclonal anti-OPA1 | BD Biosciences | Cat#612606; RRID: AB_399888 |
| Fluorescent TrueBlot: Anti-Mouse Ig DyLight 800 | Rockland | Cat#18–4517-32; RRID: AB_2610842 |
| Rabbit IgG (H&L) Antibody DyLight 800 Conjugated Pre-Adsorbed | Rockland | Cat#611–145-122; RRID: AB_1057618 |
| Critical Commercial Assays | ||
| Ion Total RNA-Seq Kit v.2 | Life Technologies | Cat#4475936 |
| Ion PI Sequencing 200 Kit v.2 chemistry | Life Technologies | Cat#4485149 |
| PureLink RNA-Easy kit | Life Technologies | Cat#12183018A |
| RiboMinus Eukaryote System v.2 | Life Technologies | Cat #A15026 |
| OxiSelect Nitrotyrosine ELISA Kit | Cell Biolabs | Cat#STA-305 |
| CaspaseGlo 3/7 | Promega | Cat#G8090 |
| Deposited Data | ||
| Raw sequence files and associated metadata | This paper | SRA: SUB2425516 |
| Reproducible code for generating the values presented in this paper | This paper | https://github.com/chop-dbhi/mcmanus_ant1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: WT: C57Bl/6JEiJ | Navarro et al., 2012 | N/A |
| Mouse: COI: COIV421A C57Bl/6JEiJ | This paper | N/A |
| Mouse: ND6: ND6P25L C57Bl/6JEiJ | This paper | N/A |
| Mouse: ANT1: Ant1−/− C57Bl/6JEiJ | Morrow et al., 2017 | N/A |
| Mouse: ANT1/COI: Ant1−/−, COIV421A C57Bl/6JEiJ | This paper | N/A |
| Mouse: ANT1/ND6: Ant1−/−, ND6P25L C57Bl/6JEiJ | This paper | N/A |
| Oligonucleotides | ||
| Primers for mtDNA LT-PCR ND2 Forward: 5’CTGGAA TTCAGCCTACTAGCAATTATCC-3’; 12s Reverse: 5’-TTTAGGTTTATGGCTAAGCATAGTGGGG- 3’ |
This paper | N/A |
| Primers for mtDNA deletions flanking multiple TaqI restriction sites: Deletion Forward: 5’-AGGCCAC CACACTCCTATTG-3’, position 8810–8829; Deletion Reverse: 5’-AATGCTAGGCGTTTGATTGG-3’, position 13098–13117 |
Vermulst et al., 2008b | N/A |
| Primers for mtDNA Taq1 controls: Control Forward: 5’-TCGGCGTAAAACGTGTCAAC-3’, position 350–369; Control Reverse: 5’-CCGCCAAGTCCTTTGAGTTT-3’ position 579–598 | Vermulst et al., 2008b | N/A |
| Software and Algorithms | ||
| Speckle tracking echocardiography: Vevostrain software | Visual Sonics | https://www.visualsonics.com/product/software/vevo-lab |
| Electron micrograph quantification: Image J | National Institutes of Health | https://imagej.nih.gov/ij/download.html |
| Western Analysis: Image Studio | LI-COR | https://www.licor.com/bio/products/software/image_studio_lite/ |
Highlights.
Mitochondrial-nuclear interactions modulate autosomal mutation expressivity
mtDNA complex I and IV variants have opposing effects on Ant1−/− pathophysiology
Ant1−/− decreases OXPHOS complex I amount and complex V structural assembly
Somatic mtDNA mutations augment the effects of unfavorable mtDNA-nDNA interactions
ACKNOWLEDGMENTS
This work was supported by NIH grants5R01-NS021328–30,1R01MH108592– 01A1, 1R01MN110185–01A1, RO1NS41850, and R01OD010944–05 and DOD grant PR150585.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.cmet.2018.08.002.
DATA AND SOFTWARE AVAILABILITY
Raw sequence files and associated metadata have been deposited in NCBI SRA: SUB2425516. Reproducible code for generating the values presented in this paper is located at https://github.com/chop-dbhi/mcmanus_ant1.
QUANTIFICATION AND STATISTICAL ANALYSIS
The data were quantified in Prism 6.0 (GraphPad, La Jolla, CA, USA), and the appropriate statistical analysis performed using one-way or two-way ANOVAs, corrected for multiple comparisons by the Sidak post hoc test, unless noted otherwise. The Brown-For-sythe test was used to determine differences in standard deviations among groups, and the Geisser-Greenhouse correction applied for sphericity. Further statistical details of experiments can be found in the figure legend and results sections.
REFERENCES
- Acin-Perez R, Bayona-Bafaluy MP, Bueno M, Machicado C, Fernandez-Silva P, Perez-Martos A, Montoya J, Lopez-Perez MJ, Sancho J, and Enriquez JA (2003). An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum. Mol. Genet. 12, 329–339. [DOI] [PubMed] [Google Scholar]
- Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, Banchieri N, Bellini O, Dal Bello B, Pilotto A, Magrini G, et al. (1998a). Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am. J. Pathol. 153, 1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbustini E, Fasani R, Morbini P, Diegoli M, Grasso M, Dal Bello B, Marangoni E, Banfi P, Banchieri N, Bellini O, et al. (1998b). Coexistence of mitochondrial DNA and beta myosin heavy chain mutations in hypertrophic cardiomyopathy with late congestive heart failure, [published erratum appears in Heart 1999 Mar;81(3):330]. Heart 80, 548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronow BJ, Toyokawa T, Canning A, Haghighi K, Delling U, Kranias E, Molkentin JD, and Dorn GW 2nd (2001). Divergent transcriptional responses to independent genetic causes of cardiac hypertrophy. Physiol. Genomics 6, 19–28. [DOI] [PubMed] [Google Scholar]
- Bauer MK, Schubert A, Rocks O, and Grimm S (1999). Adenine nucleo-tidetranslocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J. Cell Biol. 147, 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biering-Sorensen T, Biering-Sorensen SR, Olsen FJ, Sengelov M, Jorgensen PG, Mogelvang R, Shah AM, and Jensen JS (2017). Global longitudinal strain by echocardiography predicts long-term risk of cardiovascular morbidity and mortality in a low-risk general population: the Copenhagen City Heart Study. Circ. Cardiovasc. Imaging 10, e005521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD (2010). The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, and Cornwall EJ (2005). The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 392, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke MA, Chang S, Wakimoto H, Gorham JM, Conner DA, Christodoulou DC, Parfenov MG, DePalma SR, Eminaga S, Konno T, et al. (2016). Molecular profiling ofdilated cardiomyopathythat progresses to heart failure. JCI Insight 1, e86898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2011). Nonshivering thermogenesis and its adequate measurement in metabolic studies. J. Exp. Biol. 214, 242–253. [DOI] [PubMed] [Google Scholar]
- Capaldi RA, Marusich MF, and Taanman JW (1995). Mammalian cytochrome-c oxidase: characterization of enzyme and immunological detection of subunits in tissue extracts and whole cells. Methods Enzymol. 260, 117–132. [DOI] [PubMed] [Google Scholar]
- Carrozzo R, Wittig I, Santorelli FM, Bertini E, Hofmann S, Brandt U, and Schagger H (2006). Subcomplexes of human ATP synthase mark mitochondrial biosynthesis disorders. Ann. Neurol. 59, 265–275. [DOI] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, and Chan DC (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V, Cortopassi G, Chiamvimonvat N, Bers DM, Votruba M, et al. (2012). OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 1, e003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrollier A, Loiseau D, Reynier P, and Stepien G (2011). Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim. Biophys. Acta 1807, 562–567. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, Lamperti C, Viscomi C, Scorrano L, and Zeviani M (2015). Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 21, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Enriquez JA, and Scorrano L (2016). Mitochondrial cristae: where beauty meets functionality. Trends Biochem. Sci. 41, 261–273. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, and Rabinovitch PS (2010). Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 9, 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KM, Anselmi C, Wittig I, Faraldo-Gomez JD, and Kuhlbrandt W (2012). Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. USA 109, 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckles SP, Krause DN, Stirone C, and Procaccio V (2006). Estrogen and mitochondria: a new paradigm for vascular protection? Mol. Interv. 6, 26–35. [DOI] [PubMed] [Google Scholar]
- Echaniz-Laguna A, Chassagne M, Ceresuela J, Rouvet I, Padet S, Acquaviva C, Nataf S, Vinzio S, Bozon D, and Mousson de Camaret B (2012). Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J. Med. Genet. 49, 146–150. [DOI] [PubMed] [Google Scholar]
- Eichner LJ, and Giguere V (2011). Estrogen related receptors (ERRs): a new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion 11, 544–552. [DOI] [PubMed] [Google Scholar]
- Fan W, Waymire K, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, MacGregor GR, and Wallace DC (2008). A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman HC, Hugill A, Dear NT, Ashcroft FM, and Cox RD (2006). Deletion of nicotinamide nucleotide transhydrogenase: a new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 55, 2153–2156. [DOI] [PubMed] [Google Scholar]
- Geyer H, Caracciolo G, Abe H, Wilansky S, Carerj S, Gentile F, Nesser HJ, Khandheria B, Narula J, and Sengupta PP (2010). Assessment of myocardial mechanics using speckle tracking echocardiography: fundamentals and clinical applications. J. Am. Soc. Echocardiogr. 23, 351–369, quiz 453–355. [DOI] [PubMed] [Google Scholar]
- Glytsou C, Calvo E, Cogliati S, Mehrotra A, Anastasia I, Rigoni G, Raimondi A, Shintani N, Loureiro M, Vazquez J, et al. (2016). Optic atrophy 1 is epistatic to the core MICOS component MIC60 in mitochondrial cristae shape control. Cell Rep. 17, 3024–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, and Wallace DC (1997). A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/skeletal muscle isoform of the adenine nucleotide translocator. Nat. Genet. 16, 226–234. [DOI] [PubMed] [Google Scholar]
- Gray JJ, Zommer AE, Bouchard RJ, Duval N, Blackstone C, and Linseman DA (2013). N-terminal cleavage of the mitochondrial fusion GTPase OPA1 occurs via a caspase-independent mechanism in cerebellar granule neurons exposed to oxidative or nitrosative stress. Brain Res. 1494,28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, and Auwerx J (2011). The metabolic footprint of aging in mice. Sci. Rep. 1, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ, and Epstein CJ (2006). Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum. Mol. Genet. 15, 1187–1194. [DOI] [PubMed] [Google Scholar]
- Jang JY, Choi Y, Jeon YK, Aung KC, and Kim CW (2008). Over-expression of adenine nucleotide translocase 1 (ANT1) induces apoptosis and tumor regression in vivo. BMC Cancer 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji F, Sharpley MS, Derbeneva O, Alves LS, Qian P, Wang Y, Chalkia D, Lvova M, Xu J, Yao W, et al. (2012). Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc. Natl. Acad. Sci. USA 109, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Liang M, Zhang J, Zhang M, Zhu J, Meng X, Zhang S, Gao M, Zhao F, Wei QP, et al. (2014). Mitochondrial haplotypes may modulate the phenotypic manifestation of the LHON-associated ND1 G3460A mutation in Chinese families. J. Hum. Genet. 59, 134–140. [DOI] [PubMed] [Google Scholar]
- Kim A, Chen CH, Ursell P, and Huang TT (2010). Genetic modifier of mitochondrial superoxide dismutase-deficient mice delays heart failure and prolongs survival. Mamm. Genome 21, 534–542. [DOI] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Flierl A, Sweeney KM, Angelin A, MacGregor GR, and Wallace DC (2016). Deficiency in the mouse mitochondrial adenine nucleotide translocator isoform 2 gene is associated with cardiac noncompaction. Biochim. Biophys. Acta 1857, 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, and Wallace DC (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun A, Carelli V, Ross-Cisneros FN, Baciu P, Sung E, McManus MJ, et al. (2012). A mouse mtDNA mutant model of Leber’s hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA109, 20065–20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MJ, Murphy MP, and Franklin JL (2014). Mitochondria-derived reactive oxygen species mediate caspase-dependent and -independent neuronal deaths. Mol. Cell. Neurosci. 63, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mofarrahi M, Guo Y, Haspel JA, Choi AM, Davis EC, Gouspillou G, Hepple RT, Godin R, Burelle Y, and Hussain SN (2013). Autophagic flux and oxidative capacity of skeletal muscles during acute starvation. Autophagy 9, 1604–1620. [DOI] [PubMed] [Google Scholar]
- Morrow RM, Picard M, Derbeneva O, Leipzig J, McManus MJ, Gouspillou G, Barbat-Artigas S, Dos Santos C, Hepple RT, Murdock DG, et al. (2017). Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc. Natl. Acad. Sci. USA114, 2705–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier A, Ruzzenente B, Brandt T, Kuhlbrandt W, and Larsson NG (2014). Loss of LRPPRC causes ATP synthase deficiency. Hum. Mol. Genet. 23, 2580–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW 2nd, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, et al. (2016). Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ. Res. 118, 1960–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula N, Zaragoza MV, Sengupta PP, Li P, Haider N, Verjans J, Waymire K, Vannan M, and Wallace DC (2011). Adenine nucleotide trans-locase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC Cardiovasc. Imaging 4, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro SJ, Trinh T, Lucas CA, Ross AJ, Waymire KG, and Macgregor GR (2012). The C57BL6J mouse strain background modifies the effect of a mutation in Bcl2l2. G3 (Bethesda) 2, 99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton M, Ng AC, Baird S, Dumoulin A, Shutt T, Mah N, Andrade-Navarro MA, McBride HM, and Screaton RA (2014). ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci. Signal. 7, ra10. [DOI] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, and Hoppel CL (1977). Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 252, 8731–8739. [PubMed] [Google Scholar]
- Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, Santoro A, Scarcia P, Fontanesi F, Lamantea E, et al. (2005). Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum. Mol. Genet. 14, 3079–3088. [DOI] [PubMed] [Google Scholar]
- Patten DA, Wong J, Khacho M, Soubannier V, Mailloux RJ, Pilon-Larose K, MacLaurin JG, Park DS, McBride HM, Trinkle-Mulcahy L, et al. (2014). OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 33, 2676–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brethes D, di Rago JP, and Velours J (2002). The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 21, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, McManus MJ, Csordas G, Varnai P, Dorn Ii GW, Williams D, Hajnoczky G, and Wallace DC (2015). Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat. Commun. 6, 6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter GA Jr., Hom J, Hoffman D, Quintanilla R, de Mesy Bentley K, and Sheu SS (2011). Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatr. Cardiol. 31, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramonet D, Perier C, Recasens A, Dehay B, Bove J, Costa V, Scorrano L, and Vila M (2013). Optic atrophy 1 mediates mitochondria remodeling and dopaminergic neurodegeneration linked to complex I deficiency. Cell Death Differ. 20, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialo F, Sriram A, Fernandez-Ayala D, Gubina N, Lohmus M, Nelson G, Logan A, Cooper HM, Navas P, Enriquez JA, et al. (2016). Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KA, Dubiner L, Simon M, Zaragoza M, Sengupta PP, Li P, Narula N, Dreike S, Platt J, Procaccio V, et al. (2013). Severity of cardio-myopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc. Natl. Acad. Sci. USA 110, 3253–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss M, Hofhaus G, Schroder RR, and Kuhlbrandt W (2008). Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiepolo T, Angelin A, Palma E, Sabatelli P, Merlini L, Nicolosi L, Finetti F, Braghetta P, Vuagniaux G, Dumont JM, et al. (2009). Thecyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1−/− myopathic mice. Br. J. Pharmacol. 157, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, et al. (2005). A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48, 675–686. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen FH, Smet J, Levanets O, Meissner-Roloff M, Louw R, Van Coster R, and Smuts I (2010). Aberrant synthesis of ATP synthase resulting from a novel deletion in mitochondrial DNA in an African patient with progressive external ophthalmoplegia. J. Inherit. Metab. Dis. 33 (Suppl 3 ), S55–S562. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, and Loeb LA (2007). Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 39, 540–543. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, and Loeb LA (2008a). Quantification of random mutations in the mitochondrial genome. Methods 46, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, and Loeb LA (2008b). DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 40, 392–394. [DOI] [PubMed] [Google Scholar]
- Wallace DC (2013a). Bioenergetics in human evolution and disease: implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20120267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2013b). Mitochondrial bioenergetic etiology of disease. J. Clin. Invest. 123, 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I, Karas M, and Schagger H (2007). High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol. Cell. Proteomics 6, 1215–1225. [DOI] [PubMed] [Google Scholar]
- Wittig I, Meyer B, Heide H, Steger M, Bleier L, Wumaier Z, Karas M, and Schagger H (2010). Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim. Biophys. Acta 1797, 1004–1011. [DOI] [PubMed] [Google Scholar]
- Yadava N, Potluri P, Smith EN, Bisevac A, and Scheffler IE (2002). Species-specific and mutant MWFE proteins. their effect on the assembly of a functional mammalian mitochondrial complex I. J. Biol. Chem. 277, 21221–21230. [DOI] [PubMed] [Google Scholar]
- Yu T, Sheu SS, Robotham JL, and Yoon Y (2008). Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 79, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora M, Granell M, Mampel T, and Vinas O (2004a). Adenine nucleotide translocase 3 (ANT3) overexpression induces apoptosis in cultured cells. FEBS Lett. 563, 155–160. [DOI] [PubMed] [Google Scholar]
- Zamora M, Merono C, Vinas O, and Mampel T (2004b). Recruitment of NF-kappaB into mitochondria is involved in adenine nucleotide translocase 1 (ANT1)-induced apoptosis. J. Biol. Chem. 279, 38415–38423. [DOI] [PubMed] [Google Scholar]
- Zaragoza MV, Brandon MC, Diegoli M, Arbustini E, and Wallace DC (2011). Mitochondrial cardiomyopathies: howto identify candidate pathogenic mutations by mitochondrial DNA sequencing, MITOMASTER and phylogeny. Eur. J. Hum. Genet. 19, 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, and Sollott SJ (2000). Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.