

Abstract
Maternal embryonic leucine-zipper kinase (MELK) overexpression impacts survival and proliferation of multiple cancer types, most notably glioblastomas and breast cancer. This makes MELK an attractive molecular target for cancer therapy. Yet, the molecular mechanisms underlying the involvement of MELK in tumorigenic processes are unknown. MELK participates in numerous protein-protein interactions that affect cell cycle, proliferation, apoptosis, and embryonic development. Here we used both in vitro and in-cell assays to identify a direct interaction between MELK and arrestin-3. A part of this interaction involves the MELK kinase domain, and we further show that the interaction between the MELK kinase domain and arrestin-3 decreases the number of cells in S-phase, as compared to cells expressing the MELK kinase domain alone. Thus, we describe a new mechanism of regulation of MELK function, which may contribute to the control of cell fate.
Keywords: Maternal embryonic leucine-zipper kinase (MELK), arrestin, cell fate signaling, protein-protein interactions
Graphical Abstract
INTRODUCTION
Maternal embryonic leucine-zipper kinase (MELK) is a serine/threonine kinase of the AMPK/Snf1 family [1]. Unlike other family members, MELK does not require an upstream kinase activator and can self-activate via auto-phosphorylation in vitro [2]. MELK is highly conserved across species and is thought to play a role in cell cycle regulation, proliferation, and mitosis [1].
While the functions and regulatory mechanisms of MELK are just starting to be elucidated, MELK has been linked to several cell fate-related processes. For example, it has been demonstrated that in mice MELK induces phosphorylation and activation of apoptosis signal-regulating kinase 1 (ASK1), which activates cell death pathways [3]. In contrast, overexpression of MELK in breast cancer cell lines results in aggressive cell proliferation and suggests that the kinase may function as a driver oncogene [4, 5]. These contradictory findings suggest that the role of MELK in pro- and anti-apoptotic pathways may be context-dependent.
Many signaling proteins have been identified as potential regulators of MELK activity. MELK has been proposed to increase transcription of oncogenes through interactions with transcription factors (FOXM1 [6, 7], EIF4B [8], ZPR9 [9], and NIPP1 [9]), aid in DNA damage repair via regulation of p53 and ATM [7, 10], and act as a cell cycle regulator [11]. MELK may also activate proteins that induce cell death, such as ASK1 [3] and Smads [12]. While these interacting partners and overexpression of MELK in various cancer types suggest the physiological importance of MELK, the molecular mechanisms underlying these interactions and their biological consequences remain unclear. More studies are needed to determine the exact role of MELK in disease progression and whether other regulators of its activity exist.
Here, we identify a novel interacting partner for MELK, arrestin-33. While the arrestin proteins are best known for their role in the desensitization and internalization of G protein-coupled receptors (GPCRs) [13–16], they also interact with, and affect the activity of, other proteins, including Src family tyrosine kinases [17, 18], mitogen-activated protein kinases and their upstream activators [19–23], and E3 ubiquitin ligases [24, 25]. A key regulatory role of the arrestin proteins is their ability to act as scaffolds or adaptors for kinases and phosphatases [26]. Mechanistically, the arrestins likely both bring kinases or phosphatases and their substrates into proximity and orient these optimally to facilitate signaling [23, 27, 28]. Many of these kinases and phosphatases that interact with arrestin are implicated in cell-fate signaling [29], which suggests that the MELK/arrestin-3 interaction as a target for therapeutic development.
We used a combination of biophysical techniques and in-cell assays to show that arrestin-3 interacts with MELK and that the interaction with the MELK kinase domain is biologically relevant. Conceivably, this interaction influences the subcellular localization of MELK, which may explain how it affects cell fate signaling pathways.
RESULTS
Pull-down assay of the MELK kinase domain with arrestin-31–393.
Because arrestin scaffolding of mitogen activated protein kinases (MAPKs) depends upon interactions with the MAPK kinase domain [27, 30, 31], we reasoned that MELK could similarly interact via its kinase domain and used the isolated kinase domain of MELK (residues 1–326 out of 651 residues) for initial studies. We combined this with the T167E mutant (denoted MELK1−326,T167E). MELK1−326,T167E is a well-characterized mimic of the constitutively phosphorylated state [32] that is expected to predominate in the cell as a result of MELK autophosphorylation [9]. This mutation likely results in MELK favoring the activated conformation that is biologically the most probable interactor with scaffolds; the phosphorylation of Thr167 and Ser171 is required for full activation [9]. This MELK1−326,T167E was used in two separate in vitro pull-down assays with truncated arrestin-3 (residues 1–393, denoted below as arrestin-31−393), which is an established model for both GPCR-dependent and GPCR-independent arrestin activation [19, 22, 23, 27, 33–35]. This C-terminal truncation of arrestin-3 shifts the equilibrium from a “basal” to an “active” conformation while preserving known kinase binding sites.
To test whether MELK directly interacts with arrestin-3, we used an in vitro pull-down assay with purified proteins. This assay is useful in the detection of high-affinity interactions [36]. We evaluated both human and mouse MELK1−326,T167E binding to arrestin-31−393 (Fig. 1A). Within the kinase domain of MELK, these homologs share 94% sequence identity. The MELK kinase domain of both species exhibited equally strong binding to arrestin-31−393 (Fig. 1A). Therefore, for subsequent experiments we used the human isoform of MELK1−326,T167E.
Figure 1. Pull-down of purified MELK1−326,T167E with purified arrestin-31–393.

(A) Analysis of mouse MELK1−326,T167E (10 μg; mMELK) and human MELK1−326,T167E (10 μg; hMELK) binding to bovine arrestin-31−393 (10 μg). Top: Representative Western blot of the eluates using a polyclonal anti-arrestin antibody (1:10,000, F431 [58]). Middle: Representative Coomassie staining of the eluates. Bottom: Quantification of arrestin-3 binding using densitometry. Intensity was compared to the negative control (beads alone) and binding is shown as a percentage of total arrestin-3 applied (n=3). Statistical analysis was performed using Student’s t-test (**, p<0.01). (B) Analysis of human MELK1−326,T167E (10 μg) binding to bovine arrestin-31−393 (10 μg) in the absence or presence of 2 mM ATP and 4 mM MgCl2. Top: Representative Western blot of the eluates using an anti-arrestin-3 antibody (1:10,000, F431). Middle: Representative Coomassie staining of the eluates. Bottom: Quantification of arrestin-3 binding using densitometry. Intensity was compared to the negative control (beads alone) and binding is displayed as a percentage of total arrestin-3 applied (n=7). Statistical analysis was performed using Kruskal-Wallis test with multiple comparisons (**, p<0.01; ***, p<0.001).
Prior work that evaluated the interaction between arrestin and MAPKs revealed that the presence of ATP significantly increased the affinity and suggested that the arrestin-binding elements of kinases included regions that changed conformation upon ATP binding [27]. Therefore, we tested whether the presence of physiological concentrations of ATP and MgCl2 affected the binding of human MELK1−326,T167E to arrestin-31–393. In the presence of ATP and MgCl2 the MELK1−326,T167E interaction with arrestin-31−393 was unchanged (Fig. 1B). Thus, the effect of ATP on the conformation of the kinase domain does not affect its interaction with arrestin-31–393. Because the ATP-binding pocket is located between the N- and C-lobes of the MELK kinase domain, and ATP likely affects the conformation of this interdomain region, we conclude that arrestin does not bind between the two kinase lobes [32].
Affinity of MELK1−326,T167E for arrestin-31–393.
MicroScale Thermophoresis (MST) quantifies biomolecular interactions using the directed movement of molecules along a temperature gradient [37]. To measure the affinity of the kinase domain of human MELK1−326,T167E for arrestin-31−393, we used increasing concentrations of arrestin-31−393 (0–40 μM) in the presence of fluorescently-tagged MELK1−326,T167E (50 nM) and physiological concentrations of ATP and MgCl2. In this assay, MELK1−326,T167E exhibited high affinity for arrestin-31−393 with a Kd of 130 ± 35 nM (Fig. 2). This interaction is likely biologically relevant, as MELK1−326,T167E binding to arrestin-3 is much tighter than observed in a highly-validated set of interactions between arrestin and members of the JNK3 cascade (~1–20 μM) [27].
Figure 2. The affinity of MELK1−326,T167E for arrestin-31−393 in the presence of ATP.

A binding curve for bovine arrestin-31−393 with human MELK1−326,T167E in the presence of 1 mM ATP and 2 mM MgCl2 is shown with the average Kd value (n=3). Microscale thermophoresis was performed at a constant concentration of Tris-NTA-labeled His-MELK1−326,T167E (50 nM) with a serial dilution of arrestin-31−393 (0–40 μM). The binding isotherm was calculated using preset T-jump in the software PALMIST [59, 60], and the graph was created in the program GUSSI.
Mapping the arrestin-3 binding site for MELK1−326,T167E.
Defining the interaction interfaces between proteins is crucial both for understanding the mechanism of binding and for targeted manipulation of signaling pathways. Protein-protein interactions are studied by a wide variety of techniques and can be probed using full-length proteins, separated protein domains, or peptides. Two distinct advantages of using peptides to study protein-protein interactions are the ease of synthesis and the ability to identify specific interaction sites [38]. Peptide arrays allow the identification of specific binding residue(s) within a complex interaction surface [39].
To identify the regions of arrestin-3 that are important for the interaction with MELK1−326,T167E, we performed a 15-mer peptide scan of full-length arrestin-3 using 1 amino acid shifts (Fig. 3A, Supplementary Table 1). We classified peptides by the strength of their binding to MELK and mapped interaction regions onto the crystal structure of arrestin-31−393 (PDB entry 3P2D) [40] (Fig. 3B). Most of the interacting peptides were localized on the non-receptor-binding surface in folded arrestin-31−393, suggesting that GPCR-bound arrestin-3 can interact with MELK. Several peptides appear to be critical for the interaction. The first set of peptides encompasses residues A87-L109, previously termed T2A [35]. This region includes the α-helix of arrestin involved in the three-element interaction, which is part of the phosphate sensor [40]. The second group of peptides corresponds to arrestin residues T187-S203, Y209-T226 (previously T4 [35]), and V272-D291; these peptides encompass several β-strands on the non-receptor-binding surface of the C-domain (Fig. 3C,D). Third, a group of peptides corresponding to residues V165-Q173 also displayed strong binding. However, surface inaccessibility in the context of folded arrestin [33, 38, 40] suggests that these may be false positives. Indeed, this region is fairly hydrophobic, which could result in non-specific binding of these peptides to MELK. Collectively, these results suggest that MELK1−326,T167E binds elements in both arrestin domains, so that the observed high affinity interaction likely involves multiple sites (Fig. 2). This is similar to our previous finding that each kinase in the ASK1-MKK4/7-JNK3 and cRaf-MEK1-ERK1/2 cascades [41], as well as E3 ubiquitin ligase parkin [24] engage both arrestin domains.
Figure 3. MELK1−326,T167E binding sites on arrestin-3.

(A) Representative peptide blot (15-mer) of bovine arrestin-3-derived peptides (residues 1–393, 1 amino acid shifts). (B) Map of interaction sites for MELK1−326,T167E on arrestin-3 (PDB 3P2D [40]). Peptides calculated to be in the top 1% for binding to MELK1−326,T167E are highlighted in red and top 10% in salmon. The peptide V165-Q173 (shown in orange-yellow) demonstrated binding within the top 10% but is likely a false positive due to its surface inaccessibility in both basal [40] and active [33] arrestin-3 structures. (C) Quantified binding of MELK1−326,T167E to arrestin-3-derived peptides. Binding was measured as a percentage of total intensity on the membrane. A 15-mer glycine peptide was used to determine non-specific interactions. Inset: Peptide segments corresponding to the non-receptor surface of arrestin-3. The corresponding regions are circled in blue on the quantification. (D) Heat map using a single gradient to display ranges of binding was calculated using GraphPad Prism 8.0.2. Peptides corresponding to the non-receptor-binding surface of arrestin-3 are shown above.
Mapping the MELK site for arrestin-31−393 binding.
To characterize the arrestin-3 binding site on MELK, we used a peptide array corresponding to full-length MELK (residues 1–651) using 3 amino acid shifts (Fig. 4A, Supplementary Table 2). This strategy revealed several regions of MELK that could be responsible for the interaction in the context of the folded protein (Fig. 4B). The highest binding was detected for residues N562-L582 in the KA-1 domain. It has been suggested that this domain must be disrupted to prevent autoinhibition of the kinase and maintain its activity [42, 43]. The KA-1 domain was also implicated in membrane localization of MELK [43]. In addition, a second region of strong binding involved residues Y88–E108 of the kinase domain. Binding was also detected across several residues in the kinase (Fig. 4C, D). Of note, no interaction with arrestin-31−393 was detected in the activation segment of MELK (residues D150-E178), which is a region of the kinase that is important for activity [44]. Because an interaction with the activation segment might be anticipated to block kinase activity, this suggests that the kinase can retain biological function in complex with arrestin.
Figure 4. Arrestin-31−393 binding sites on MELK1–651.

(A) Representative peptide blot using 15-mers derived from human MELK (residues 1–651, 3 amino acid shifts). (B) Map of interaction sites for arrestin-31−393 on MELK (PDB 4BL1; to be published [45]). Peptides were analyzed using conditional formatting and those that exhibited binding to arrestin-31−393 within the top 10% are highlighted in salmon, while those binding within the top 5% are shown in red. The crystal structure of the MELK kinase domain used a construct that is truncated after residue 347 (peptide 107 in the array). Therefore, the high binding peptides from the C-terminal KA1 domain (188–190; sequence NVTTTRLVNPDQLLNEIMSIL), cannot be mapped onto the crystal structure. (C) Quantified binding of arrestin-31−393 to MELK-derived peptides. Arrestin-31−393 binding was measured as a percentage of total intensity of all spots. (D) Heat map using a single gradient to display ranges of binding was calculated using GraphPad Prism 8.0.2. Corresponding MELK domains are shown above.
Mapping of these peptides is somewhat limited because only the isolated kinase domain of MELK has been crystallized (PDB 4IXP [42, 45–48]), so it is not known whether residues N562-L582 of the KA-1 domain and Y88-E108 of the kinase domain form a contiguous surface in full-length MELK. Mapping Y88-E108 and other peptides with more moderate signal onto the crystal structure of the MELK kinase domain (PDB 4IXP, [42]) identifies that these peptides localize to the C-lobe (highlighted in red in Fig. 4B).
Mutational analysis of arrestin-3- and MELK-derived peptides pinpoints key interacting residues.
To test whether the high-interaction peptides identified by array analysis contribute to the binding between arrestin-3 and MELK, we performed additional peptide array experiments using alanine scanning, charge reversal, and alanine substitution (Supplementary Figs. 1 and 2). First, we measured MELK1−326, T167E binding to the wild type (WT) and mutant T2A peptides (Supplementary Fig. 1). We found that most mutations weakened the MELK interaction compared to the control T2A peptide; however, a single charge reversal from lysine to glutamate at the twenty-second amino acid greatly enhanced the interaction (Supplementary Fig. 3A). To test MELK-derived peptides for binding with arrestin-3, we chose peptides from the kinase domain (YCPGGELFDYIISQDRLS) and the KA-1 domain (NVTTTRLVNPDQLLNEIMSIL), which demonstrated high binding in the initial screen (Supplementary Fig. 2). Again, most mutations weakened the arrestin-31−393 interaction compared to the control peptides (Supplementary Figs. 3B, C).
Arrestin-31−393 coimmunoprecipitates with full-length MELK and MELK1−340 in cells.
To validate our in vitro findings in a more biological context we assessed the MELK/arrestin-31−393 interaction in living cells using overexpression. This overexpression in HEK293 cells resulted in a 4.6-fold increase in arrestin-3 as compared to the endogenous levels of arrestin-3 in the same cell line (Supplementary Fig. 4). It should be noted that arrestin has uneven subcellular distribution and the effect of overexpression on this distribution is unknown. The MELK constructs used in these experiments did not carry the T167E mutation and could therefore be in an inactive or active state, depending on whether or not they were phosphorylated. We first used coimmunoprecipitation to test the ability of full-length MELK and arrestin-31−393 to interact (Fig. 5). We observed a robust binding of arrestin-31−393 with full-length MELK, suggesting that the interaction is likely biologically relevant.
Figure 5. Co-immunoprecipitation of wild-type MELK1−651 with arrestin-31–393.

(A) Western analysis of coimmunoprecipitation. HEK293 arrestin-2/3 knockout cells were transfected with empty vector (20 μg), HA-arrestin-31−393 (10 μg), FLAG-MELK1−651 (10 μg), or both plasmids encoding arrestin-3 and MELK at a 1:1 DNA ratio for 48 h prior to immunoprecipitation with FLAG primary antibody. Western analysis was performed using anti-arrestin (1:10,000; F431) and anti-FLAG (1: 1,000; F3165 Sigma) antibodies. (B) Arrestin-31−393 co-immunoprecipitation with MELK1−651 was quantified using densitometric analysis of arrestin-3 blots. Empty vector and non-specific arrestin-3 binding (no bait control) are also shown. Statistical analysis was performed using One-way ANOVA followed by Dunnett’s post hoc test with correction for multiple comparisons (****, p<0.0001).
To assess whether the separated kinase domain binds arrestin-31−393 in cells, we repeated our assay using several DNA ratios with a truncated MELK construct, MELK1−340 (Fig. 6). We detected a modest binding of arrestin-31−393 and MELK1−340 at a 1:1 DNA transfection ratio; however, it was not as strong as that observed for the full-length protein. This suggests that regions of MELK outside of the kinase domain also interact with arrestin, a conclusion consistent with the in vitro peptide array data (Fig. 4).
Figure 6. Co-immunoprecipitation of wild-type MELK1−340 with arrestin-31–393.

(A) Western analysis of coimmunoprecipitation. HEK293 arrestin-2/3 knockout cells were transfected with HA-arrestin-31−393 (1 μg), FLAG-MELK1−340 (1 μg), or both plasmids encoding arrestin-3 and MELK at indicated DNA ratios for 48 h prior to immunoprecipitation with FLAG primary antibody. Western analysis was performed using anti-arrestin (1:10,000; F431) and anti-FLAG (1: 1,000; F3165 Sigma) antibodies. (B) Arrestin-31−393 co-immunoprecipitation with MELK1−340 was quantified using densitometric analysis of arrestin-3 blots. Non-specific arrestin-3 binding (no bait control) is also shown. Statistical analysis was performed using One-way ANOVA followed by Tukey’s post hoc test with correction for multiple comparisons (*, p<0.05).
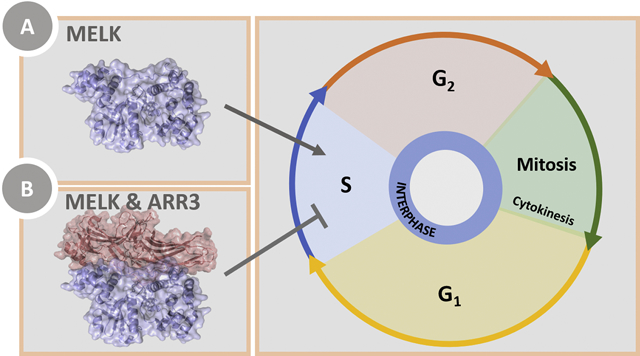
The number of S-phase cells is decreased by co-expression of arrestin-31−393 with MELK1–340.
Over-expression of MELK has been demonstrated in several cancer types and is often correlated with poor prognosis [4]. As our in vitro and in-cell studies support an interaction between the MELK kinase domain and arrestin-3, we chose to measure the effect of arrestin-31−393, a MELK fragment containing the kinase and UBA domain (MELK1−340; residues 1–340), and arrestin-31−393/MELK1−340 on the cellular transition to S-phase. To this effect, we directly measured DNA synthesis using 5-ethynyl-2’-deoxyuridine (EdU) incorporation as a readout. EdU is a nucleoside analog of thymidine that is readily incorporated into replicating DNA and can be detected using fluorescent azides [49]. HEK293 arrestin-2/3 knockout cells [50] were incubated in media with or without EdU following transfection. Cells were then fixed and incubated with Alexa Flour 488 picolyl azide, whereupon flow cytometry was used to determine the percentage of S-phase cells in the population (Fig. 7). Untransfected cells with EdU treatment were used as a negative control for gating purposes (gray trace in Fig. 7A, gating schematic shown in Fig. 7B) and yielded 22 ± 10% S-phase cells. Three replicates yielded the following percentages of S-phase cells: 17 ± 5% for arrestin-31−393 transfection, 37 ± 6% for MELK1−340 transfection, and 15 ± 6% for cells expressing both proteins. Thus arrestin-31−393 significantly reduced the observed increase in S-phase cells associated with MELK1−340 expression. Conceivably, arrestin-3, which directly binds ASK1 [19, 22, 41, 51], facilitates MELK-dependent ASK1 activation, which would suppress proliferation.
Figure 7. The effect of wild-type MELK1−340 and arrestin-31−393 expression on the fraction of cells in S-phase.

HEK293 arrestin-2/3 knockout cells were transfected with HA-arrestin-31−393 (1 μg) (red), FLAG-MELK1−340 (1 μg) (blue), or a fixed DNA ratio of plasmids encoding the two proteins (1 μg:1 μg) (orange), and the number of cells in S-phase was measured using the Click-iT™ EdU Alexa Fluor 488 Imaging Kit (ThermoFisher Scientific), followed by flow cytometry. (A) A representative histogram shows data from cells labeled with Alexa Fluor 488 picolyl azide analyzed using a 3-laser LSRII (BD Biosciences). The inset shows the relative heights of the second distribution (Alexa Fluor 488 positive cells) with the percentages of cells in S-phase for combined experiments (n=3). Analysis was completed on FlowJo 10.1.5 (FlowJo LLC). Statistical analysis was performed using One-way ANOVA followed by Tukey’s post-hoc test (*, p<0.05 to 1:1 Arr3:MELK). The first distribution represents cells that are not proliferating, while the second distribution represents cells in S-phase. (B) The gating strategy used for histogram generation. Cells were initially sorted by scatter (SSC-A by FSC-A) (P1) to determine the population of living cells, then by single cells (SSC-W by SSC-H) (P2), then singlets (FSC-W by FSC-H) (P3), and finally by cells positive for Alexa Fluor 488 (SSC-A by Alexa Fluor 488) (P4).
DISCUSSION
Previous proteomic analysis of the non-visual arrestins activated by the angiotensin 1 receptor showed >100 effectors that bind arrestins either directly or indirectly [26]. This list, however, did not include MELK as a potential binding partner. In contrast, our data indicate a direct (Fig. 1), high affinity (Fig. 2) interaction between the MELK kinase domain (MELK1−326,T167E) and arrestin-31–393. This observed difference could be due to the limitations of the methods, or the differences in protein constructs used. For example, the proteomics study used the angiotensin II type 1a receptor to measure proteins bound to arrestin after agonist stimulation; the detected molecules are expected to be biased toward the effectors activated by this receptor.
Consideration of how this protein-protein interaction might affect MELK activity draws from known roles of arrestin. Arrestins can act as adaptors or scaffold proteins with multiple biological roles. For example, arrestin scaffolds are commonly thought to regulate signaling of cytoplasmic proteins, a process that includes both kinase activation and signal amplification [26]. In considering this role within the context of the data presented here, multiple studies have demonstrated that MELK undergoes autophosphorylation [9, 45], and our data show that the phosphomimetic variant of MELK binds robustly to arrestin-3 (Fig. 2). Moreover, the interaction with arrestin-3 appears to influence the biological function of the active MELK kinase domain, by pushing toward anti-proliferative, rather than proliferation pathways (Fig. 7). Together, this suggests that the arrestin-3-MELK interaction is not involved in the initial phosphorylation and activation of MELK.
A second proposed role for arrestins is to control the subcellular localization of bound effectors, as is demonstrated for MAPK cascades [22, 23, 30, 41, 52]; [53], and ubiquitin ligase Mdm2 [53, 54]. One possibility is that arrestin localizes MELK to a specific region in the cell in order to direct MELK activity to particular substrates. MELK exhibits cell-cycle dependent localization and has been shown to associate with the scaffold protein anillin along with other proteins involved in cytokinesis [55]. The KA-1 domain of MELK has also been shown to regulate MELK association with cellular membranes [9, 43, 56]. This could explain the prior controversial findings that the cellular effects of MELK activity are context dependent [3–5]. Given the role of MELK activity in controlling the cell cycle, understanding how the arrestin-3/MELK interaction influences MELK function could provide a new avenue for modulating MELK activity.
It should be emphasized that we observe in vitro binding between arrestin-31−393 and MELK1−326,T167E (Fig. 1), which corresponds to the kinase domain of MELK. Although our peptide array data (Fig. 4) and pull downs from cells (compare Fig. 5 and Fig. 6) suggest that the interaction between arrestin and MELK involves both the kinase domain and other regions outside of this domain, most notably the TP dipeptide-rich domain and the KA-1 domain, the interaction between arrestin-3 and the MELK kinase domain appears to be sufficient to alter cell fate (Fig. 7). In addition to regulating membrane association, the KA-1 domain has been implicated in autoinhibition of MELK through an interaction with the UBA region [9, 43, 56]. While the hypotheses regarding possible binding mechanisms are limited by the absence of a structure of full-length MELK, it is tempting to speculate that arrestin-31−393 binding to the KA-1 domain prevents MELK autoinhibition and promotes kinase activity. If this activity is directed towards ASK1, an established arrestin-3 partner, this would explain arrestin-3-dependent reduction in S-phase cells (Fig. 7). Further experimentation is necessary to test this hypothesis.
EXPERIMENTAL PROCEDURES
Purified proteins.
For this study, we used a fragment of MELK including its kinase domain and the flanking ubiquitin-associated domain (residues 1–326). This fragment retains full catalytic activity [9] and is easier to purify than full-length MELK. The MELK construct used also contained a T167E mutation (denoted MELK1−326,T167E), a well-characterized mimic of the constitutively phosphorylated state [32] mimicking its activation (phosphorylation at Thr167 and Ser171 is required for full activation). The phosphorylated kinase is expected to predominate in the cell as a result of MELK autophosphorylation [9].
We used a truncated arrestin-3 (residues 1–393, denoted below as arrestin-31−393) construct for all analyses. This truncated version of arrestin-3 is an established model for both GPCR-dependent and GPCR-independent arrestin activation [19, 22, 23, 27, 33–35] as the C-terminal deletions in arrestin shift the equilibrium from a “basal” to an “active” conformation. This construct eliminates the need for arrestin activation via GPCRs and allows arrestin-3 to sample multiple active conformations.
Arrestin-31−393 was purified as previously described [57, 58]. For MELK expression, a BL21 DE3 pLysS glycerol stock of MELK T167E (residues 1–326) in pET21b was used to inoculate a 120 mL starter culture overnight at 30°C, 205 rpm. Each 1 L was inoculated using 10 mL of overnight culture. Expression was induced with 500 μM IPTG after cells reached an OD600 0.6, and the temperature was reduced to 17 °C for ~18 h. The cells were pelleted by centrifugation at 6,000 × g, 10 min. The pellets were subjected to one cycle of freeze-thaw before adding the lysis buffer (20 mM Tris-HCl pH 7.5, 300 mM NaCl, 50 mM imidazole, 25 μL DNAse, 500 μL Pepstatin A, 100 μL leupeptin, 2 mM TCEP). The crude lysate was sonicated for 4 min (5 secs on, 10 secs off, 75% amplitude (FB505 Sonic Dismembrator, Fisher Scientific) before pelleting debris at 18,000 × g for 30 min.
The clarified lysate was loaded onto a HisTrap column (GE) at 2.5 mL/min in wash buffer (20 mM Tris-HCl pH 7.5, 300 mM NaCl, 50 mM imidazole). Bound protein was eluted with a 0–200 mM imidazole gradient in elution buffer (20 mM Tris-HCl pH 7.5, 300 mM NaCl). The fractions were analyzed by SDS-PAGE. Pooled fractions containing MELK1−326 were concentrated and subjected to size exclusion chromatography on a Superdex S200 Increase 10/300 GL column equilibrated with 25 mM HEPES pH 7.3, 300 mM NaCl, and 2 mM TCEP.
In vitro pull-down assay.
Ni-NTA agarose resin (Qiagen) was rinsed three times with wash buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 10 mM imidazole pH 7.5, 0.1 mg/ml BSA) and 25 μL of the 50% slurry was aliquoted into each tube. His-MELK1−326,T167E (10 μg in 25 μL wash buffer) was immobilized on agarose resin for 1 h at 4°C with gentle rotation. Arrestin-31−393 (10 μg in 50 μL wash buffer) was added to the suspension and incubated with slow rotation for 2 h at 4°C in the presence or absence of 2 mM ATP and 4 mM MgCl2. Samples were transferred to centrifuge filters (Durapore® PVDF-0.65 μm) and washed quickly with 900 μL 50 mM HEPES pH 7.3 and 150 mM NaCl. Proteins were eluted with 100 μL 50 mM HEPES pH 7.3, 150 mM NaCl, and 100 mM imidazole. The eluates were immediately precipitated with methanol (90% final concentration), air dried, and dissolved in SDS sample buffer (40 μL Laemmli 2×). The samples were analyzed using SDS-PAGE and Western blotting, as described [35].
Microscale thermophoresis.
MST experiments were performed using the Monolith NT.115 instrument (NanoTemper Technologies GmbH). The Monolith™ His-Tag Labeling Kit RED-tris-NTA was used to label human His-MELK1−326,T167E following the manufacturer’s protocol (100 nM final concentration of His-MELK1−326,T167E in PBS-T buffer). Purified arrestin-31−393 was diluted to a starting concentration of 80 μM in MST buffer (20 mM MOPS pH 7.5, 150 mM NaCl, 2 mM TCEP, 0.05% Tween-20). A 16-step dilution series was prepared by aliquoting 10 μL of MST buffer to 15 tubes. A 20 μL aliquot of the prepared arrestin-31−393 sample was placed in the first tube, and 10 μL was transferred to the second tube and mixed well by pipetting (1:1 dilution series). The final tube did not contain any arrestin-31−393 and served as a blank. To all tubes, 10 μL of labeled His-MELK1−326,T167E in the presence of 1 mM ATP and 2 mM MgCl2 was added. After 15–30 min incubation on ice, the samples were transferred to Standard Monolith NT™ Capillaries. Each capillary was scanned using the MST instrument (40% LED, 40% MST power) and data were analyzed using PALMIST [59, 60]. Measured fluorescence was normalized and averaged, and the data were fit using a 1:1 binding model. The preset T-jump was used to calculate binding isotherms. All data were displayed using GUSSI.
Peptide Array Synthesis.
The ResPep SL peptide synthesizer was used with standardized SPOT synthesis protocols using 15-residue peptides, as previously described [61]. A 15-mer glycine peptide was used as a negative control in all experiments. Peptide assembly was conducted on solid support by the ResPep SL via a peptide amide linker using Fmoc-chemistry. The sequences were derived from either the full-length bovine arrestin-3 or full-length human MELK. After peptide synthesis, the amino protecting group was removed using piperidine (20% (v/v) solution in dimethylformamide) and extensive washing. The membranes were immersed for 1 h in a solution of 95% trifluoroacetic acid (TFA) and 3% triisopropylsilane with slight agitation. After the TFA solution was removed, the membranes were washed in dichloromethane four times 10 min each, after which they were washed in dimethylformamide four times 10 min each, and finally washed in ethanol two times 2 min each. The membranes were dried under the hood and stored at 4°C.
Peptide Membrane Blots.
Dried membranes were activated by soaking in 100% ethanol for 5 min at room temperature and rehydrated by rinsing two times 5 min in water. The membranes were then blocked in a 20 mL solution of 5% non-fat dry milk in Tris-buffered saline with 0.1% Tween-20 (TBS-T) for 1 h. Then, the membranes were washed three times 5 min each in TBS-T before overnight incubation with prey protein (0.2 μM MELK1−326,T167E or 0.3 μM arrestin-31−393) in binding buffer (MELK: 20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 5 mM DTT; arrestin-3: 20 mM MOPS pH 7.5, 150 mM NaCl, 2 mM TCEP). The following day the membranes were washed three times for 5 min each in TBS-T at room temperature and then incubated with primary antibody (1:1,000 Aviva anti-MELK or 1:5,000 anti-arrestin rabbit polyclonal antibody F431 [58]) for 1 h at room temperature. The membranes were then washed three times, 5 min each, in TBS-T, incubated for 1 h with the corresponding HRP-conjugated secondary antibody (1: 4,000 dilution; Abcam) in TBS-T, and washed three times 5 min each in TBS-T. The membranes were developed using a peroxide solution (SuperSignal West Pico) and visualized using a Bio-Rad Gel Doc Imager. Individual dots were analyzed by densitometry using a protein array analyzer (ImageJ). Individual dot density was expressed as a percentage of total binding across the entire membrane. Peptides were mapped onto either the arrestin-3 structure (PDB 3P2D [40]) or MELK structure (PDB 4BL1; to be published [45]) using conditional formatting in Excel (color scales) to identify the highest and lowest intensity values in relation to one another.
Co-immunoprecipitation of arrestin-31−393 with full-length MELK or MELK1–340.
HEK293 arrestin-2/3 knockout cells [50, 62] were transfected with HA-arrestin-3 (10/1 μg), FLAG-MELK/FLAG-MELK-340-WT (10/1 μg), or a fixed ratio of the two plasmids for 48 h. Cells were washed in phosphate-buffered saline (PBS) before incubation with RIPA buffer (Sigma) for 1 h. Lysed cells were centrifuged for 10 min at maximum speed (10,000×g) at 4°C and the supernatant was precleared by 50 μL of suspension of Protein G Sepharose (supplied as aqueous ethanol suspension; Sigma; washed with PBS) for 1 h. Samples were then centrifuged at low speed (3200×g) to remove the beads, and the supernatant was incubated overnight with primary antibody (Anti-FLAG; 1:1,000; F3165 Sigma). The next morning, the samples were incubated with Protein G Sepharose for 2 h to capture the FLAG-MELK/FLAG-MELK1–340. The beads were then washed with lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate). Finally, the samples were eluted with 100 μL SDS sample buffer. Both cell lysates and co-immunoprecipitated samples were subjected to Western blotting using antibodies against arrestin (1:10,000; polyclonal rabbit F431 [58]) and MELK (1:1,000; F3165 Sigma).
Cellular Proliferation Assay.
HEK293 arrestin-2/3 knockout cells were either left untransfected or transfected with HA-arrestin-3 (1 μg), FLAG-MELK1−340 (1 μg), or a fixed ratio of the two plasmids for 46 h prior to treatment with 10 mM EdU (5-ethynyl-2’-deoxyuridine) for 2 h. Cells were then detached from support using 0.025% trypsin, and the protease was quenched with DMEM medium (10% FBS, 1% Pen/Strep). Cells were immediately washed with 3 mL of 1% bovine serum albumin (BSA) in PBS and centrifuged at 700×g. The pellet was then fixed for 15 min at room temperature with 4% paraformaldehyde in PBS and the wash step was repeated. A 100 μL saponin-based permeabilization and wash reagent was used to prepare the cells for the Click-iT™ reaction (ThermoFisher Scientific).
A Click-iT™ reaction mixture was prepared using PBS, copper protectant, reaction buffer additive, and picolyl azide fluorescent dye following manufacturer’s instructions (ThermoFisher Scientific). Cells were incubated with 500 μL of the mixture for 30 min prior to a final wash with saponin-based permeabilization and wash reagent. Cells were resuspended in 500 μL buffer and analyzed using flow cytometry. Flow analysis was performed using a 3-laser LSRII (BD Biosciences). Analysis of flow cytometry data was performed using FlowJo 10.1.5 (FlowJo LLC, Ashland, OR).
Statistical analysis.
Statistical methods used are indicated in figure legends.
Supplementary Material
HIGHLIGHTS.
We identified a novel interacting partner for arrestin-3: maternal embryonic leucine-zipper kinase (MELK).
Arrestin-3 interaction with MELK decreases S-phase cells compared to MELK expression alone.
Arrestin-3 interaction with MELK could be targeted to regulate cell fate signaling and may be an attractive cancer therapeutic.
Acknowledgements
The authors are grateful to Dr. Asuka Inoue for the generous gift of the arrestin-2/3 knockout HEK293 cells and Dr. Chad Brautigam for discussions of MST data analysis. The authors also thank Brittany Matlock for technical assistance with flow cytometry experiments, which were performed in the VMC Flow Cytometry Shared Resource.
Funding
This work was supported by American Heart Association 16PRE30180007 (NAP) and 18PRE34030017 (NAP), NIH grants R35 GM122491 (VVG), GM120569 (TMI), GM123252 (KND), DA043680 (TMI/VVG), Vanderbilt Discovery Award (TMI/VVG), Cancer Research and Prevention Institute of Texas (CPRIT) (RP160657) (KND), and the Welch Foundation (F-1390) (KND). The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
We use the systematic names of arrestins, which are based on the order of cloning: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- [1].Ganguly R, Mohyeldin A, Thiel J, Kornblum HI, Beullens M, Nakano I. MELK—a conserved kinase: functions, signaling, cancer, and controversy. Clinical and Translational Medicine. 2015;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jung H, Seong H-A, Ha H. Murine Protein Serine/Threonine Kinase 38 Activates Apoptosis Signal-regulating Kinase 1 via Thr838 Phosphorylation. Journal of Biological Chemistry. 2008;283:34541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Giuliano CJ, Lin A, Smith JC, Palladino AC, Sheltzer JM. MELK expression correlates with tumor mitotic activity but is not required for cancer growth. eLife. 2018;7:e32838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Speers C, Zhao SG, Kothari V, Santola A, Liu M, Wilder-Romans K, et al. Maternal Embryonic Leucine Zipper Kinase (MELK) as a Novel Mediator and Biomarker of Radioresistance in Human Breast Cancer. Clinical Cancer Research. 2016;22:5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells. 2013;31:1051–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Beke L, Kig C, Linders JTM, Boens S, Boeckx A, Van Heerde E, et al. MELK-T1, a small-molecule inhibitor of protein kinase MELK, decreases DNA-damage tolerance in proliferating cancer cells. Bioscience Reports. 2015:BSR20150194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang Y, Begley M, Li Q, Huang H-T, Lako A, Eck MJ, et al. Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proceedings of the National Academy of Sciences. 2016;113:9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beullens M, Vancauwenbergh S, Morrice N, Derua R, Ceulemans H, Waelkens E, et al. Substrate Specificity and Activity Regulation of Protein Kinase MELK. Journal of Biological Chemistry. 2005;280:40003–11. [DOI] [PubMed] [Google Scholar]
- [10].Gu C, Banasavadi-Siddegowda YK, Joshi K, Nakamura Y, Kurt H, Gupta S, et al. Tumor-specific activation of the C-JUN/MELK pathway regulates glioma stem cell growth in a p53-dependent manner. Stem Cells. 2013;31:870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jiang P, Zhang D. Maternal embryonic leucine zipper kinase (MELK): a novel regulator in cell cycle control, embryonic development, and cancer. Int J Mol Sci. 2013;14:21551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Seong H-A, Manoharan R, Ha H. Smad proteins differentially regulate obesity-induced glucose and lipid abnormalities and inflammation via class-specific control of AMPK-related kinase MPK38/MELK activity. Cell Death & Disease. 2018;9:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ferguson SSG, Zhang J, Barak LS, Circ MG. Role of β-Arrestins in the IntracelIular Trafficking of G-Protein-Coupled Receptors In: Goldstein DS, Eisenhofer G, McCarty R, editors. Advances in Pharmacology: Academic Press; 1997. p. 420–4. [Google Scholar]
- [14].Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- [15].Goodman OB, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–50. [DOI] [PubMed] [Google Scholar]
- [16].Goodman OB, Krupnick JG, Gurevich VV, Benovic JL, Keen JH. Arrestin/clathrin interaction. Localization of the arrestin binding locus to the clathrin terminal domain. J Biol Chem. 1997;272:15017–22. [DOI] [PubMed] [Google Scholar]
- [17].Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–61. [DOI] [PubMed] [Google Scholar]
- [18].Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, et al. Regulation of tyrosine kinase activation and granule release through β-arrestin by CXCR1. Nature Immunology. 2000;1:227. [DOI] [PubMed] [Google Scholar]
- [19].Breitman M, Kook S, Gimenez LE, Lizama BN, Palazzo MC, Gurevich EV, et al. Silent Scaffolds: INHIBITION OF c-Jun N-TERMINAL KINASE 3 ACTIVITY IN CELL BY DOMINANT-NEGATIVE ARRESTIN-3 MUTANT. Journal of Biological Chemistry. 2012;287:19653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cellular Signalling. 2009;21:462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kook S, Zhan X, Kaoud TS, Dalby KN, Gurevich VV, Gurevich EV. Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J Biol Chem. 2013;288:37332–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, et al. Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–7. [DOI] [PubMed] [Google Scholar]
- [23].Zhan X, Kaoud TS, Kook S, Dalby KN, Gurevich VV. JNK3 Enzyme Binding to Arrestin-3 Differentially Affects the Recruitment of Upstream Mitogen-activated Protein (MAP) Kinase Kinases. Journal of Biological Chemistry. 2013;288:28535–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ahmed MR, Zhan X, Song X, Kook S, Gurevich VV, Gurevich EV. Ubiquitin Ligase Parkin Promotes Mdm2–Arrestin Interaction but Inhibits Arrestin Ubiquitination. Biochemistry. 2011;50:3749–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Girnita L, Shenoy SK, Sehat B, Vasilcanu R, Girnita A, Lefkowitz RJ, et al. β-Arrestin Is Crucial for Ubiquitination and Down-regulation of the Insulin-like Growth Factor-1 Receptor by Acting as Adaptor for the MDM2 E3 Ligase. Journal of Biological Chemistry. 2005;280:24412–9. [DOI] [PubMed] [Google Scholar]
- [26].Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, et al. Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proceedings of the National Academy of Sciences. 2007;104:12011–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perry NA, Kaoud TS, Ortega OO, Kaya AI, Marcus DJ, Pleinis JM, et al. Arrestin-3 scaffolding of the JNK3 cascade suggests a mechanism for signal amplification. Proceedings of the National Academy of Sciences. 2018:201819230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Coffa S, Breitman M, Hanson SM, Callaway K, Kook S, Dalby KN, et al. The Effect of Arrestin Conformation on the Recruitment of c-Raf1, MEK1, and ERK1/2 Activation. PLoS ONE. 2011;6:e28723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Luttrell LM, Miller WE. Chapter Five - Arrestins as Regulators of Kinases and Phosphatases. In: Luttrell LM, editor. Progress in Molecular Biology and Translational Science: Academic Press; 2013. p. 115–47. [DOI] [PubMed] [Google Scholar]
- [30].Guo C, Whitmarsh AJ. The beta-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J Biol Chem. 2008;283:15903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li X, MacLeod R, Dunlop AJ, Edwards HV, Advant N, Gibson LCD, et al. A scanning peptide array approach uncovers association sites within the JNK/βarrestin signalling complex. FEBS Letters. 2009;583:3310–6. [DOI] [PubMed] [Google Scholar]
- [32].Cho Y-S, Yoo J, Park S, Cho H-S. The structures of the kinase domain and UBA domain of MPK38 suggest the activation mechanism for kinase activity. Acta Crystallographica Section D: Biological Crystallography. 2014;70:514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen Q, Perry NA, Vishnivetskiy SA, Berndt S, Gilbert NC, Zhuo Y, et al. Structural basis of arrestin-3 activation and signaling. Nature Communications. 2017;8:1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Miller WE, Lefkowitz RJ. Expanding roles for β-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Current Opinion in Cell Biology. 2001;13:139–45. [DOI] [PubMed] [Google Scholar]
- [35].Zhan X, Perez A, Gimenez LE, Vishnivetskiy SA, Gurevich VV. Arrestin-3 binds the MAP kinase JNK3a2 via multiple sites on both domains. Cellular signalling. 2014;26:766–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Perry NA, Zhan X, Gurevich EV, Iverson TM, Gurevich VV. Using In Vitro Pull-Down and In-Cell Overexpression Assays to Study Protein Interactions with Arrestin In: Scott MGH, Laporte SA, editors. Beta-Arrestins: Methods and Protocols. New York, NY: Springer New York; 2019. p. 107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. Molecular Interaction Studies Using Microscale Thermophoresis. Assay and Drug Development Technologies. 2011;9:342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Amartely H, Iosub-Amir A, Friedler A. Identifying Protein-protein Interaction Sites Using Peptide Arrays. Journal of Visualized Experiments : JoVE. 2014:52097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Katz C, Levy-Beladev L, Rotem-Bamberger S, Rito T, Rüdiger SGD, Friedler A. Studying protein–protein interactions using peptide arrays. Chemical Society Reviews. 2011;40:2131–45. [DOI] [PubMed] [Google Scholar]
- [40].Zhan X, Gimenez LE, Gurevich VV, Spiller BW. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol. 2011;406:467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Song X, Coffa S, Fu H, Gurevich VV. How Does Arrestin Assemble MAPKs into a Signaling Complex? Journal of Biological Chemistry. 2009;284:685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cao LS, Wang J, Chen Y, Deng H, Wang ZX, Wu JW. Structural basis for the regulation of maternal embryonic leucine zipper kinase. PLoS One. 2013;8:e70031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Moravcevic K, Mendrola JM, Schmitz KR, Wang Y-H, Slochower D, Janmey PA, et al. Kinase Associated-1 Domains Drive MARK/PAR1 Kinases to Membrane Targets by Binding Acidic Phospholipids. Cell. 2010;143:966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nolen B, Taylor S, Ghosh G. Regulation of Protein Kinases: Controlling Activity through Activation Segment Conformation. Molecular Cell. 2004;15:661–75. [DOI] [PubMed] [Google Scholar]
- [45].Canevari G, Re Depaolini S, Cucchi U, Bertrand JA, Casale E, Perrera C, et al. Structural Insight into Maternal Embryonic Leucine Zipper Kinase (MELK) Conformation and Inhibition toward Structure-Based Drug Design. Biochemistry. 2013;52:6380–7. [DOI] [PubMed] [Google Scholar]
- [46].Johnson CN, Berdini V, Beke L, Bonnet P, Brehmer D, Coyle JE, et al. Fragment-Based Discovery of Type I Inhibitors of Maternal Embryonic Leucine Zipper Kinase. ACS Medicinal Chemistry Letters. 2015;6:25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Touré BB, Giraldes J, Smith T, Sprague ER, Wang Y, Mathieu S, et al. Toward the Validation of Maternal Embryonic Leucine Zipper Kinase: Discovery, Optimization of Highly Potent and Selective Inhibitors, and Preliminary Biology Insight. Journal of Medicinal Chemistry. 2016;59:4711–23. [DOI] [PubMed] [Google Scholar]
- [48].Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig P-A, et al. The target landscape of clinical kinase drugs. Science. 2017;358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proceedings of the National Academy of Sciences. 2008;105:2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun. 2018;9:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Seo J, Tsakem EL, Breitman M, Gurevich VV. Identification of arrestin-3-specific residues necessary for JNK3 activation. J Biol Chem. 2011;286:27894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhan X, Stoy H, Kaoud TS, Perry NA, Chen Q, Perez A, et al. Peptide mini-scaffold facilitates JNK3 activation in cells. Scientific Reports. 2016;6:21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song X, et al. Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol. 2007:in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–13. [DOI] [PubMed] [Google Scholar]
- [55].Chartrain I, Le Page Y, Hatte G, Körner R, Kubiak JZ, Tassan J-P. Cell-cycle dependent localization of MELK and its new partner RACK1 in epithelial versus mesenchyme-like cells in Xenopus embryo. Biology open. 2013;2:1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tochio N, Koshiba S, Kobayashi N, Inoue M, Yabuki T, Aoki M, et al. Solution structure of the kinase-associated domain 1 of mouse microtubule-associated protein/microtubule affinity-regulating kinase 3. Protein Science. 2006;15:2534–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhan X, Kaoud TS, Dalby KN, Gurevich VV. Non-visual arrestins function as simple scaffolds assembling the MKK4–JNK3a2 signaling complex. Biochemistry. 2011;50:10520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Vishnivetskiy SA, Zhan X, Chen Q, Iverson TM, Gurevich VV. Arrestin expression in E. coli and purification. Curr Protoc Pharmacol. 2014;67:Unit 2.111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tso S-C, Chen Q, Vishnivetskiy SA, Gurevich VV, Iverson TM, Brautigam CA. Using two-site binding models to analyze microscale thermophoresis data. Analytical Biochemistry. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Scheuermann TH, Padrick SB, Gardner KH, Brautigam CA. On the acquisition and analysis of microscale thermophoresis data. Analytical Biochemistry. 2016;496:79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yim YY, Betke K, Hamm H. Using Peptide Arrays Created by the SPOT Method for Defining Protein-Protein Interactions In: Meyerkord CL, Fu H, editors. Protein-Protein Interactions: Methods and Applications. New York, NY: Springer New York; 2015. p. 307–20. [DOI] [PubMed] [Google Scholar]
- [62].Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, et al. Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J Biol Chem. 2016;291:27147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.