

Abstract
In the blood, mosaic somatic aneuploidy (mSA) of all chromosomes has been found to be associated with adverse health outcomes, including hematological cancer. Sex chromosome mSA in the blood has been found to occur at a higher rate than autosomal mSA. Mosaic loss of the Y chromosome is the most common copy number alteration in males, and has been found to be associated with Alzheimer’s disease (AD) in blood lymphocytes. mSA of the sex chromosomes has also been identified in the brain; however, little is known about its frequency across individuals. Using WGS data from 362 males and 719 females from the ROSMAP cohort, we quantified the relative rate of sex chromosome mSA in the dorsolateral prefrontal cortex (DLPFC), cerebellum and whole blood. To ascertain the functionality of observed sex chromosome mosaicism in the DLPFC, we examined its correlation with chromosome X and Y gene expression as well as neuropathological and clinical characteristics of AD and cognitive ageing. In males, we found that mSA of the Y chromosome occurs more frequently in blood than in the DLPFC or cerebellum. In the DLPFC, the presence of at least one APOE4 allele was associated with a reduction in read depth of the Y chromosome (p = 1.9e-02). In the female DLPFC, a reduction in chromosome X read depth was associated with reduced cognition at the last clinical visit and faster rate of cognitive decline (p = 7.8e-03; p = 1.9e-02 ). mSA of all sex chromosomes in the DLPFC were associated with aggregate measures of gene expression, implying functional impact. Our results provide insight into the relative rate of mSA between tissues and suggest that Y and female X chromosome read depth in the DLPFC is modestly associated with late AD risk factors and cognitive pathologies.
1 –. Introduction
Somatic mutations acquired throughout life represent a new frontier of genomic diversity. Long thought to only occur in the context of cancer, the genome of somatic cells has been shown to mutate at a rate twice that of the germline in humans (Milholland et al., 2017). This phenomenon--in which the genome in a portion of somatic cells differs from other cells of the same tissue--is termed somatic mosaicism. The functional impact of somatic mosaicism is highly tissue-specific. In the blood, aberrant clonal expansions of cells with post-zygotic chromosomal dose changes have been associated with leukemia (Forsberg et al., 2013; Jacobs et al., 2012; Laurie et al., 2012; MacHiela et al., 2016; Machiela and Chanock, 2017). In the skin, mitotic recombination of mutated keratin genes KRT10 and KRT1 in a portion of cells leads to Ichthyosis with confetti, a congenital skin disorder (Guerra et al., 2015).
Somatic mosaicism has been found to more frequently occur in the sex chromosomes than the autosomes (Machiela et al., 2016). Furthermore, independent genome-wide analyses of CNVs have found that mosaic loss of the Y chromosome (mLOY) is the most common copy number variation in lymphocytes, with a frequency of between 1.7 and 20% (Forsberg et al., 2014; Loftfield et al., 2018; Thompson et al., 2019). mLOY in lymphocytes has been associated with shorter survival, higher risk of cancer and Alzheimer’s disease and increased risk for Type II diabetes (Dumanski et al., 2016; Forsberg et al., 2014; Machiela et al., 2016; Bonnefond et al., 2013). To determine whether cancer causes mLOY or vice-versa, a recent study examined the association between 156 mLOY-associated autosomal genetic loci in lymphocytes and cancer outcomes in 100,000 females. Interestingly, these variants were also associated with increased risk of cancer in women, suggesting that mLOY may occur as a result of oncogenic processes (Thompson et al., 2019). Despite its relationship to various disease processes, only one study has investigated the impact of mLOY on gene expression, finding through single-cell RNA-seq that mLOY was associated with increased expression of the oncogene TCL1A (Thompson et al., 2019).
In the past decade, it has become increasingly apparent that somatic mosaicism occurs not just in tissues with high turnover, such as lymphocytes and sex-specific tissues, but also in the brain, including neurons in the frontal cortex and cerebellum (Cai et al., 2014; Iourov et al., 2009b, 2009a, 2006; Knouse et al., 2014; McConnell et al., 2013; Rehen, 2005; van den Bos et al., 2016; Yurov et al., 2007, 2005, 2014, 2008). In the brain, somatic mosaicism is proposed to contribute to neuronal diversity, allowing clonal populations of neurons to perform different functions (McConnell et al, 2017). Due to the small number of samples tested and differing sensitivity of molecular methods, estimates of aneuploidy in the cerebral cortex have varied widely (Rohrback et al., 2018). Several early characterization efforts used molecular techniques such as FISH, multicolor banding (MCB) and spectral karyotyping (SKY) to assay the prevalence of aneuploidy in chromosomes 1, 7, 8, 9, 10, 11, 14, 15–18, 21 and X/Y, estimating that aneuploidy occurs in 13–59% of cells (Iourov et al., 2009b, 2009a, 2006; Rehen, 2005;Yurov et al., 2014, 2008, 2007, 2005). More recently, single-cell whole genome sequencing (scWGS) methods have produced much lower estimates of the prevalence of aneuploidy, ranging from 0.7 to 4.9% across all chromosomes (Cai et al., 2014; Knouse et al., 2014; McConnell et al., 2013; van den Bos et al., 2016).
Although previous studies have investigated the prevalence of somatic mosaicism within tissues, little is known about the relative rate of aneuploidy between tissues (McConnell et al., 2017; Piotrowski et al., 2008). In a study of CNVs across fibroblasts and induced pluripotent stem cells (iPSC) derived neurons in the same individual, sub-chromosomal CNVs were found to occur at a lower rate in fibroblasts than in iPSC derived neurons, suggesting that neuronal cells exhibit greater genomic diversity (McConnell et al., 2017). Through subsequent single-cell sequencing of 110 neurons in the frontal cortex, they also found that the majority of these CNVs occurred in a minority of cells with highly aberrant genomes. These CNVs were unique to each cell, implying that CNVs in the frontal cortex primarily occur on a single cell level. A separate study of LOY in lymphocytes and bulk tissue from the DLPFC found that LOY occurs more commonly in the blood than in the brain. However, this finding was based on the expression of only two chromosome X/Y homologs in a small number of DLPFC samples (n = 26) (Kimura et al., 2018). Due to the small number of total aneuploid events measured in these experiments, and the conflicting evidence at the CNV and chromosomal level, it remains unclear whether aneuploidy occurs at a higher or lower rate in the brain. Understanding the relative rate of somatic mosaicism between the blood and brain in a larger sample would help clarify the role of somatic mosaicism in the disease context.
In this study, we investigate the relative prevalence of mosaic somatic aneuploidy (mSA) in the sex chromosomes in the blood and brain. Using WGS data from 1081 male and female individuals profiled through the Rush Religious Order Study and Rush Memory and Aging Project (ROSMAP), we compare the relative rate of mSA between whole blood, DLPFC and cerebellum. Further, we utilize RNA-seq and neuropathology data to assess the associations between mSA, gene expression and neuropathology in the DLPFC. Notably, this is the largest study comparing mSA in the brain and whole blood to date, with 718 of 1081 WGS samples generated from either the DLPFC or cerebellum. To quantify aneuploidy from WGS data in each tissue, we developed an approach that uses median read depth measurements in highly mappable locations along each chromosome to rank each sample by its relative chromosomal content. This method shows high correlation with other metrics currently used to assess mSA. To compare the relative rate of mosaicism between tissues, we developed a method that adjusts for sample-specific technical variation, facilitating comparison of chromosomal content between multiple tissue types. Through application of these methods to a large number of individuals, we are able to circumvent the lack of single-cell resolution in bulk sequencing to gain insight into the rate of mSA in the blood, DLPFC and cerebellum. Taken together, we find that mSA of the Y chromosome occurs more frequently in the blood than in the brain. In addition, we find that mSA of the Y and female X chromosome is associated with aggregate gene expression changes in the DLPFC as well as neuropathological and cognitive indicators of Alzheimer’s disease and cognitive ageing.
2 –. Methods
2.1. ROSMAP Cohort
The Rush Religious Order Study (ROS) and Rush Memory and Ageing Project (MAP) are longitudinal studies that aimed to characterize the clinical and pathological features underlying ageing, cognitive decline, Alzheimer’s and Parkinson’s disease. Although two separate studies, clinical follow-up procedures and specimen collection were standardized and jointly analyzed. At time of enrollment, both studies conducted a clinical evaluation of each individual and collected blood samples. Upon the individual’s death, autopsy of the brain was performed, allowing for the collection of an additional tissue sample from the brain, and for revaluation of neuropathology by board-certified neuropathologists. Clinical and neuropathological characterization of these cohorts has been reported elsewhere (Bennett et al., 2018). A summary of all information collected on the ROSMAP cohort is summarized in De Jager et al., 2018; and is available at www.radc.rush.edu.
2.2. Summary of all data analyzed in this study
Samples in the ROSMAP WGS dataset were sampled from a variety of brain tissues and whole blood samples. In order to maximum our power to detect mosaicism in each tissue, only tissues with greater than 50 individuals (i.e. the DLPFC, whole blood and cerebellum) were analyzed. The data used in this study includes WGS (males = 362, females = 719), SNP array genotyping (males = 306, females = 974) and RNA-sequencing (males = 175, females = 407) data from all male and female samples in the ROSMAP cohort. Of all WGS samples, 363 (males = 129, females = 234) were sampled from whole blood, 460 (males = 155, females = 305) were sampled from the DLPFC and 258 (males = 78, females = 180) were sampled from cerebellum (Figure 1). SNP array data was sampled from the blood. A summary of the origin, type and overlap between all data types used in this study is included in Figure 1.
Figure 1: Overview of ROSMAP study data.

Whole blood samples from males and females were analyzed through WGS (males = 129, females = 234) and Affymetrix Genotype Array 6.0 (males = 306, females = 974). Data from both WGS and SNP array was available for 40 males and 102 females. Samples from the dorsolateral prefrontal cortex (males = 155, females = 305) and cerebellum (males = 78, females = 180) were analyzed through WGS. RNA sequencing (RNA-seq) was performed on DLPFC samples from 175 males and 407 females. Joint WGS and RNA-seq profiling data from the DLPFC was available in 65 males and 110 females.
2.3. Quantifying aneuploidy from SNP array data
Genotype quantification
Processing of SNP genotype data was previously described (Ng et al, 2017). Briefly, 1261 samples (352 male) were genotyped using the Affymetrix GeneChip 6.0 platform (Santa Clara, CA) by the Broad Institute’s Center for Genotyping. The Affymetrix Power Tools bundle (v1.20.0) was used to genotype and call copy number variants from provided CEL files. For probe normalization and CN calling the apt-copynumber-workflow tool (v1.20.0) was used using default settings.
Genotypes were called using the birdseedv2 algorithm using the suggested APT workflow. Samples with contrast quality control (CQC) values < 0.40, median absolute pairwise difference (MAPD) values > 0.35, call rate < 0.95, or conflicting sex determination were removed, as per manufacturer instruction. Birdseed files which were converted to PLINK format (http://zzz.bwh.harvard.edu/plink/) for SNP QC. SNP probes with genotype call rates < 95%, minor allele frequency < 0.01, mishap test < 1e-9, and Hardy-Weinberg P < 0.001 were removed. After quality control, a total of 306 male and 974 female samples, 757,091 SNP probes and 908,043 CN probes were included in the study for further analysis. Normalized Log R Ratio values were produced using Affymetrix Power Tools.
Chromosome loss quantification
The median Log R Ratio in a given chromosome or region was used as a marker of chromosomal content. The Affymetrix Power Tools bundle outputs the Log R Ratio as log2(Robserved/Rreference), where the Rreference is the probe intensity in a reference population of HapMap individuals. All SNP and CN probes across each chromosome were used as markers of chromosomal content. In the Y chromosome, 8797 probes (Affymetrix Genotyping Array 6.0) located in the male-specific Y region (MSY), but not the pseudoautosomal regions 1 (Y:10,001–2,649,520; hg19) and 2 (Y:59,034,050–59,363,566; hg19) were used to quantify chromosomal content. In the X chromosome, 82,387 probes in all regions excluding the PAR 1 and 2 regions were used to quantify the presence of the X chromosome.
2.4. Quantifying aneuploidy from Whole Genome Sequencing (WGS) data
Libraries of whole blood and DLPFC samples were generated using the KAPA Library Preparation Kit. Final libraries were evaluated using fluorescent-based assays including qPCR with the Universal KAPA Library Quantification Kit and Fragment Analyzer (Advanced Analytics) or BioAnalyzer (Agilent 2100). Libraries were sequenced on an Illumina HiSeq X sequencer (v2.5 chemistry) using 2 × 150 bp cycles. Sequencing reads were aligned to GRCH37 using BWAmem (version 0.7.15). Aligned BAM files contain all reads (passing or failing vendor quality checks), whether or not they aligned. We marked duplicate reads using Picard’s MarkDuplicates module (version 2.4.1). For variant calling, local re-alignment was performed around indels to identify putative insertions or deletions in the region GATK’s (version 3.5) indel realignment tool. To compute read depth, we used the GATK DepthOfCoverage module and only included reads passing minimum mapping quality>20 and minimum normalized base quality greater than zero. For regions with excess coverage, we down sampled to include only 5000 reads in the region.
Read depth values were collected from only high mappability regions for individuals of European ancestry specified in the high mappability BED file from the Genome In a Bottle Consortium (sample NA12878, version NISTv3.3.2, build hg37). To improve granularity in the sex chromosomes, read depth was confined to 998bp length bins. All read depth bins were subject to additional filtering steps that selected for bins of high mappability (wgEncodeCrgMapabilityAlign50mer > 0.9), GC content percentage between 45 and 55. Additionally, bins within known CNVs observed in greater than 5% of European individuals (DGV.GS.March2016.50percent.GainLossSep.Final.hg19.gff3; retrieved from http://dgv.tcag.ca), and ENCODE excludable regions (wgEncodeDacMapabilityConsensusExcludable.bed) were removed.
Relative read depth (rRD) values for each chromosome and each individual was computed as the ratio of the per-chromosome median read depths in pre-defined but irregularly sized high mappability regions across the chromosome and the median read depth across the whole genome (note: no difference in results was noted when the mean whole genome read depth was used). Given the non-uniform distribution of bins in the autosomes, a weighted mean based on bin length and median read depth was used when calculating chromosomal and whole genome depth. In order to prevent auto-normalization, the chromosome undergoing normalization was excluded from the median whole genome read depth calculation.
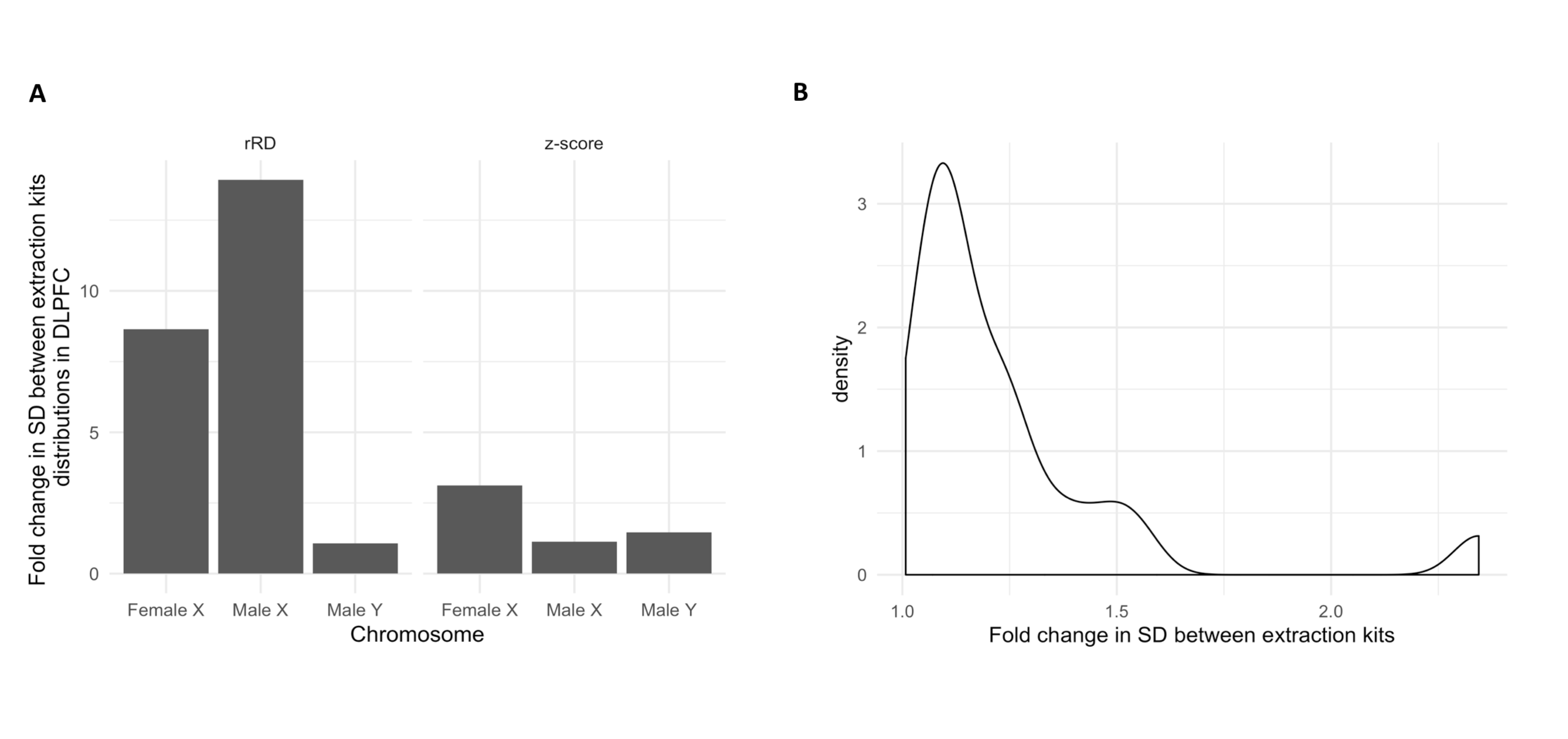
Because the standard deviation of relative read depth values were found to differ by extraction kit (Supplemental Figure 1), an additional per-sample metric accounting for the read depth distribution of each sample was developed to allow for comparison between kits and tissues (Figure 2). Briefly, 1–3kb bins in each chromosome were z-normalized to a sample-specific reference distribution composed of all 1–3kb bins in the autosomes.
Figure 2: Schematic depicting z-normalization procedure for sex chromosomes in a single sample.

Z-normalization was performed to adjust for technical artifacts due to the use of different DNA extraction kits in different tissues. 1) First, raw read counts in the autosomes were binned into irregularly sized, non-contiguous bins defined as high mappability regions by the Genome In a Bottle (GIAB) consortium. Sex chromosomes were binned into continuous 998bp bins along the non-PAR1 and PAR2 regions. 2) All bins with a mean ENCODE CRG mappability score less than 0.9 and GC content greater than 45 and less than 55 percent were removed. 3) All remaining 1–3kbp bins in the autosomes were isolated and 4) used to form a sample-specific reference distribution of expected count in 1–3kbp bins. 5) In each sex chromosome, per-bin read depth was z-normalized using the mean and standard deviation of the reference distribution. For haploid chromosomes, the mean and standard deviation were multiplied by 0.5. The median per-bin z-score in each chromosome was considered the z-score for that chromosome (referred to as “z-score”).
For bin i and sample j,
In the male haploid sex chromosomes, to account for differences in expected read depth and variation, z-normalization was performed using 0.5 times the mean and standard deviation of the reference distribution. For each sex chromosome in each sample, the median of the bin-level z-scores was considered the z-score for that particular chromosome/sample combination. As expected, the z-score distribution was approximately centered at 0 for all chromosomes (Figure 3B). When building the reference distribution, we excluded bins in chromosomes with an interquartile range larger than three standard deviations above the mean. This resulted in the exclusion of chromosome 19.
Figure 3: Estimation of chromosomal content in three tissues across 1081 individuals using WGS.

Each point in the boxplot is a single individual. A) Relative read depth (rRD) in each chromosome across different tissues. rRD was defined as the ratio of the median read depth along high mappability regions that passed filtering criteria (see Methods section 2.4) in each chromosome and the median whole genome read depth. Depicted data includes both male (n = 363) and female (n = 719) samples that originates from whole blood (males = 129, females = 234), DLPFC (males = 155, females = 305) and cerebellar (males = 78, females = 180) tissues. As expected, the normalized read depth for diploid chromosomes are approximately centered at 1, and haploid chromosomes are approximately centered at 0.5. B) Median bin-level z-score for each chromosome, as calculated by the pipeline depicted in Figure 2. The same individuals are profiled in A and B. Briefly, bins between 1–3kb in size along each chromosome were z-normalized to a reference distribution of all 1–3kb bins in the autosomes. In the male haploid sex chromosomes, to account for differences in expected read depth and variation, z-normalization was performed using 0.5 times the mean and standard deviation of the reference distribution. As expected, the z-score distribution is approximately centered at 0 for all chromosomes.
2.5. Concordance between WGS depth and SNP array data
To determine concordance between WGS and SNP array measures of aneuploidy, we characterized the similarity between all individuals with both SNP array and WGS data chromosomal content values. For all intersecting SNP array samples, we ranked mLRR values and rRD values per chromosome, and tested for ordinal association using the Kendall rank correlation coefficient.
2.6. Measuring skewness
The skewness of the distribution of normalized chromosomal read depths was calculated as the Pearson’s moment coefficient of skewness:
Where u3 is the third cumulant and u2 is the second cumulant.
2.7. RNA-Seq
RNA-seq data from 175 males and 407 females was analyzed. Tissue extraction, library preparation and sequencing have been described extensively elsewhere (De Jager, 2018). Gene-level expression estimates from RSEM were obtained from the ROSMAP AMPAD portal in units of Fragment per kilobase per million mapped reads (FPKM) (https://www.synapse.org/#!Synapse:syn3219045).
2.7.1. Expression ratio
Assuming that all chromosomes are transcribed at similar rates, a change in the copy number of a sex chromosome in a given sample will affect the sex chromosome to autosomal expression ratio in that sample, as a greater/smaller proportion of reads in the that sample’s library will map to the sex chromosome in question. In this study, we created such an expression ratio to determine whether rRD was correlated with overall chromosomal gene expression. The expression ratio of each sex chromosome was defined as the ratio of the median top 20 highly expressed genes on that chromosome to the median expression of a set of 440 highly expressed autosomal genes (20 from each autosome). The top 20 highly expressed genes in the X chromosome were defined as the genes with the highest median expression across the entire dataset (n = 582) (TMSB4Y, TMSB4X, PCSK1N, RPL10, RPL36A, GDI1, BEX3, TSPAN7, PLP1, SYP, RTL8C, PDZD4, GPM6B, RPS4X, TCEAL2, BEX2, BEX4, RPL9P7, TSPYL2, BCAP31, TCEAL4). None of these genes were identified as X inactivation escapees (Berletch et al., 2011). For the Y chromosome, only 13 Y chromosome genes located in the non-recombining region of the Y chromosome were quantified by RNA-seq, and therefore only these 13 were used in the expression ratio (genes: KDM5D, DDX3Y, ZFY, USP9Y, RPS4Y1, TXLNGY, TMSB4Y, NLG4N4Y, TTY14, UTY, EIF1AY, TTY15, AC010889.1).
2.8. Clinical and neuropathological indicators of Alzheimer’s disease
In this study, we quantified the relationship between rRD and APOE4 status, rate of change in global cognition, cognition at last clinical visit and amyloid content. A description of how this data was collected is summarized elsewhere (Bennett et al., 2018). APOE4 status is a binary variable that denotes the presence of the E4 allele of the APOE gene. Genotyping was performed in PBMCs or brain by Agencourt Bioscience Corporation utilizing high-throughput sequencing of codon 112 (position 3937) and codon 158 (position 4075) of exon 4 of the APOE gene on chromosome 19. The rate of change of global cognition is the estimated person-specific rate of change in the global cognition variable over time. It is determined through a linear mixed effects model with annual global cognitive function as the longitudinal outcome (De Jager et al., 2013). The model controls for age at baseline, sex, and years of education. Cognition at last visit is the z-score of the individual’s global cognitive function value at the time of last assessment before death. Amyloid-beta was identified by molecularly-specific immunohistochemistry and quantified by image analysis and is presented as the natural log of the average percent area occupied by amyloid-beta in eight regions of the brain. Because extraction kit was known to influence the rRD distribution, we performed two separate analyses: one analysis that included extraction kit as a covariate in a linear model (referred to as “combined” model), and another that treated each extraction kit as a separate dataset (“single” model). A criterion for considering the correlations identified in the combined analysis as internally valid was that in the single model, the direction of correlation was concordant between the two extraction kits and statistical significance was reached in at least one kit. In a series of linear models, each clinical/neuropathological variable was considered the dependent variable and chr Y/X rRD, age at death, APOE4 status (for analyses not included APOE4 as independent variable) and post mortem interval (PMI) and DNA extraction kit (if combined model) were included as independent variables.
2.9. HMMcopy
Chromosomes X and Y were analyzed for copy number variations using HMMcopy. Raw read depths in 998bp bins in the MSY of chromosome Y and the non-PAR regions of chromosome X were adjusted for GC content and mappability using QCDNAseq (Scheinin, 2014; v1.20.0) and provided as input to HMMcopy (Lai et al, 2019; v1.26.0). The output of HMMcopy was the number of segments identified. In the case of no CNVs, a single segment (the entire chromosome) would be the output.
2.10. Data availability
WGS z-scores (section 2.4) for each patient are available as supplementary data. Associated clinical and neuropathological data (section 2.8) are available through the ROSMAP portal (https://www.synapse.org/#!Synapse:syn3219045).
3 –. Results
In this study, we use WGS data from 363 whole blood samples (males = 129, females = 234), 460 DLPFC samples (males = 155, females = 305) and 258 cerebellum samples (males = 78, females = 180) to assess the relative rate of sex chromosome mSA in the blood, DLPFC and cerebellum (Figure 1). To examine the potential functionality of this mosaicism, we also assess the correlation between mSA and gene expression and clinical/neuropathological characteristics of AD. To be able to rank mSA across individuals within the same tissue, we developed a metric, termed relative read depth (rRD), that measures read depth across the chromosome in relation to whole genome read depth. To be able to compare the range of mSA observed between tissues, we developed a processing pipeline that adjusted for sample-specific technical variation by generating a z-score for each sample (Figure 2). This pipeline allows for tissue-level variation to be compared across individuals. We quantified rRD and z-score across all chromosomes. As expected, the average rRD for chromosomes X and Y was approximately half that of the autosomes and z-score distributions for each chromosome were centered at 0 (Figure 3A, B). Reassuringly, a copy number detection algorithm did not detect CNVs in any sex chromosomes, indicating that the differences in rRD and z-score observed in this study were not confounded by the presence of sub-chromosomal CNVs (Figure 4A–D). Manual inspection revealed a potential CNV in one sample, which was excluded from all further analyses.
Figure 4. Identification of copy number variants (CNVs) across the X and Y chromosome using HMMcopy.

To ensure that estimates of rRD and z-score for a specific individual were not confounded by the presence of sub-chromosomal CNVs, we used HMMcopy to identify CNVs in our sample. No CNVs were detected in any sample. The X and Y chromosome bin-level z-score and copy number (HMMcopy) profile are provided. The package QCDNAseq was used to normalize the expression data prior to input to HMMcopy. A) Z-score and QCDNAseq normalized copy numbers for the sample with the lowest median z-score in the blood in the Y chromosome. B) Z-score and QCDNAseq normalized copy numbers for the sample with the highest median z-score in the blood in the Y chromosome. C) Z-score and QCDNAseq normalized copy numbers for the sample with the highest median z-score in the blood in the X chromosome. D) Z-score and QCDNAseq normalized copy numbers (input to HMMcopy) for the sample with the lowest median z-score in the blood in the X chromosome. Each point represents the z-score/CN value of a bin. The red line represents the per-sample median of z-score/CN values across the entire chromosome. The black line demarcates a z-score/CN value of zero. The red line demarcates the median z-score/CN value in each sample.
3.1. Correlation between rRD and mLRR
To ensure validity of rRD as a metric for measuring sex chromosome mosaicism within each tissue, we investigated its relationship to artifacts of sequencing and mLRR, an array-based metric that has been previously used to assess population-level mosaicism in the Y chromosome (Forsberg et al, 2016; Dumanski et al, 2015). To ensure that differences in blood and brain rRD values were not influenced by total read depth per sample, we compared overall median read depths between samples from blood (n=363), DLPFC (n=460) and cerebellum (n = 258). No difference in whole genome read depth was observed between tissues (Kruskal-Wallis, p = 0.63). To further validate that the rRD metrics in chromosomes X and Y were not affected by technical bias, we utilized the fact that whole blood samples from 41 male and 102 female individuals had undergone both WGS and SNP array profiling. For these 143 individuals, we 1) determined the correlation between rRD values from WGS and median Log R Ratio (mLRR) values from SNP array and 2) compared the relative ranking of chromosomal content in each individual (see Methods, section 2.5). mLRR values correlated strongly with rRD values for the Y chromosome, and female X, but not for the male X (Chr Y: r = 0.86, p = 7.7e-13; Male Chr X: r = 0.32, p = 0.041; Female Chr X: 0.40, p = 3.6e-05) (Figure 5). Similarly, we found that the order of individuals was significantly similar in chromosome Y (τ=6.0e-01, p=2.22e-16) and female X (τ=2.2e-01, p= 1.1e-03) but not the male X (τ=1.5e-01, p=1.7e-01). Notably, restricting our analysis to Genome In A Bottle (GIAB) high confidence regions (regions that produce fewer false positive variant calls), highly mappable regions (Crg mappability > 0.9) and regions with GC content within 45–55% range was critical in improving the correlation between mLRR and rRD from 0.81 to 0.86 in the Y chromosome, and the correlation between mLRRX and chr X rRD in females from a baseline of 0.33 to 0.40. Applying these technical filters additionally decreased the average difference in mean absolute deviation (MAD) between samples across chromosomes from 9.5e-03 to 5.4e-03 (Supplemental Figure 2). Low correlation between mLRR and rRD values is also observed in the autosomes, suggesting that correlation between mLRR and rRD values is a function of the extent of the range of chromosomal content observed. Indeed, as noted by a recent report, and confirmed by our findings, the range of blood-based mosaicism observed in the male Y chromosome is larger than observed in either the male or female X, which is in turn greater than that observed in the autosomes (Machiela et al, 2016).
Figure 5: Correlation between median Log R ratios (mLRR) and relative read depth (rRD) measurements of the X and Y chromosome in males and females.

In order to assess the validity of the rRD metric, we assessed its correlation with HapMap-normalized mLRR values from the Affymetrix genotyping array 6.0 on the Y chromosome and X chromosome in 41 male and 102 female individuals. mLRR values for the male X chromosome are lower than those for the female because males and females were analyzed in the same Affymetrix workflow and the haploid males will have lower mLRR-X values than the diploid females.
3.2. Developing a metric for comparing mosaicism between samples
In order to compare samples across tissues, we needed to develop a robust pipeline to eliminate technical artifacts such as those arising from DNA extraction kit. To do this, we utilized the fact that two kits were used to extract DNA across DLPFC tissue samples, allowing us to ascertain the effect of technical differences in sample collection and processing on rRD across different chromosomes. We noticed that the variation observed in per-chromosome rRD distributions differed by extraction kit across the autosomes in the DLPFC. This observation was preserved across both male and female samples (AllPrepUniversal, N = 90, QIA Amp, N = 373; mean fold change (FC) = 4.52, max FC = 10.98, min FC = 1.37) (Supplemental Figure 1A). To avoid confounding tissue-level changes in mosaicism with technical artefacts of DNA extraction kit, we concluded that the standard deviation of rRD could not be used to compare mosaicism between tissues. Recent research has suggested that DNA extraction kit may induce changes in variance at the bin level (van Heesch et al., 2013). Therefore, to address this artefact, we devised a novel z-score metric that adjusts each sample based on variance observed across autosomal bins (see Methods, section 2.4). This metric is referred to as the z-score throughout our analysis. Fold change in SD of z-score distributions between extraction kits used in the same tissue were 3.67-fold lower than observed in rRD distributions, indicating that this metric effectively reduces extraction-kit specific variance (Supplemental Figure 1B). FC between extraction kits in the sex chromosomes also became more comparable (Supplemental Figure 3A). To account for the small fold change difference that could feasibly be due to technical variation, fold change in SD between extraction kit-specific z-score distributions were obtained for each chromosome, forming an expected technical error distribution (Supplemental Figure 3B). For all comparisons of SD and range between tissues, only FC differences greater than the maximum FC observed in the technical error distributions (2.34) were performed. For simplicity, this threshold was called the FC SD threshold. Because any measurable mosaicism may be confounded by this technical variation, the FC SD threshold was also used to create a confidence interval for our mosaicism measurements.
3.3. Estimating relative prevalence of mosaic somatic aneuploidy in the sex chromosomes
The relative prevalence of mosaic somatic aneuploidy (mSA) was determined in each sex chromosome by comparing the standard deviation of the z-score distributions across tissues (Figure 6A, B). For all comparisons involving the DLPFC, fold change in SD across both AllPrepUniversal and QIAamp extraction kits samples are reported and only comparisons for which the direction of fold change occurred in the same direction in both kits were considered internally valid. The observed FC in SD was greater than the FC SD threshold for two comparisons within the Y chromosome: whole blood vs DLPFC and whole blood vs cerebellum (Figure 6C). The SD in Y chromosomal content in whole blood is 3.40-fold (plus or minus 2.24) larger than in the DLPFC, and 3.9-fold (plus or minus 2.34) larger than in the cerebellum. In each tissue, z-score distributions were negatively skewed, implying that the Y chromosome most commonly undergoes hypoploidy rather than hyperploidy (Figure 6D). Together, these results suggest that the rate of mSA of the Y chromosome occurs at a higher rate in the blood than in the brain. In contrast, mSA in the male and female X did not differ at a rate higher than that attributable to technical variation between any tissues, implying that mosaicism is roughly equal between tissues, or that mosaicism occurs at a rate below the lower detection limit of mosaicism as measured through bulk sequencing (Forsberg et al, 2013; Forsberg et al., 2016; Jacobs et al., 2012).
Figure 6: Relative rate and direction (hypoploidy vs hyperploidy) of chromosome Y mSA in whole blood, DLPFC and cerebellar tissues.

A) z-score distribution for the male X, female X and male Y chromosomes as measured in whole blood, DLPFC and cerebellum. B) SD of z-score distributions in A. These values were used to compute the fold change differences in C. C) Fold change in SD between tissues for the Y chromosome (FC = larger SD/smaller SD). Green shading indicates that the FC in SD between tissues was larger than the maximum SD differences attributable to extraction kit alone (referred to as FC threshold, Supplemental Figure 4B). Standard deviation in Y chromosomal content is 3.40-fold (plus or minus 2.34) higher in whole blood than in DLPFC, and 3.9-fold (plus or minus 2.34) higher than in the cerebellum. D) Skew of Y chromosome z-score distribution in whole blood, DLPFC and cerebellum. In the Y chromosome, a greater negative skew is observed in the blood than in the brain. Because distributions varied by extraction kit, we present the skew of both the AllPrepUniversal and QIAamp samples separately.
3.4. Determining relationship between somatic mosaicism and gene expression in the DLPFC
In order to determine whether mSA was associated with gene expression in the DLPFC, we investigated the correlation between rRD and sex chromosome gene expression. Assuming equal transcription rates across all chromosomes, differences in chromosomal content in a sample would result in a change in the median expression of Y chromosome genes attributable to that particular sex chromosome. This ratio, termed expression ratio, was defined as the ratio of the median of highly expressed genes in a sex chromosome to the median of a set of highly expressed genes in the autosome (see Methods, section 2.7.1). If variation in rRD reflects variation in chromosomal content, a correlation between mosaicism and expression ratio would be expected. After correcting for RIN score, PMI and RNA sequencing batch, correlation between rRD and Y expression ratio and X expression ratio was observed in the male Y and female X sex chromosomes, but not the male X (Y Chr: r = 0.29, p = 2.42e-02, female X Chr: r = 0.36, p =1.2e-04; male X Chr: r = 0.23, p = 6.5e-02) (Figure 7A–C). This correlation between rRD and expression ratio suggests that mSA may exert a functional effect on gene expression in the DLPFC.
Figure 7: Relationship between sex chromosome rRD and proportion of gene expression attributable to that chromosome.

A, B, C) Here, rRD was used instead of z-score because rRD exhibits improved overall correlation with mLRR. Further, a single DNA extraction kit (QIAamp) was used to extract both the WGS and RNAseq data, therefore negating the need for kit-specific correction. Briefly, a measure of the proportion of gene expression attributable to the sex chromosomes was defined as the ratio of the median of the top 20 highly expressed genes on the sex chromosome in question (or top 13 on the Y) to the median of the top 440 highly expressed genes across all autosomes (20 from each autosome, see Methods, section 2.7.1). An increase in total amount of a sex chromosome in the sample would be expected to lead to an increase in the proportion of that chromosome’s gene expression relative to the autosomes. After correcting for RIN score, PMI and RNA sequencing batch, correlation between rRD and Y expression ratio and X expression ratio was observed in the male Y and female X chromosomes (Y chr: r = 0.29, p = 2.42e-02, female X chr: r = 0.36, p =1.2e-04; male X chr: r = 0.23, p = 6.5e-02). P-values from ANOVA are shown on graphs for ease of visualization. D) Distribution in skew across all WGS and RNAseq data. “all” signifies the skew in all available data for each data type, not just those individuals common to WGS and RNA-seq analyses. “Common” signifies the skew in the data common to both WGS and RNA-seq analyses. In the female and male X chromosomes, positive skew is observed at both the RNA and DNA level. In contrast, in the Y chromosome, conflicting skew is observed at the DNA and RNA level.
Since there are reports of both somatic mosaic hypo- and hyperploidy in the frontal cortex, we sought to determine if the direction of X and Y chromosome mosaicism at the DNA and mRNA level concurred. At the DNA level, rRD in the DLPFC is slightly negatively skewed, indicating a mild shift towards hypoploidy (Figure 7D). A corresponding negative skew is not observed at the mRNA level, suggesting that low Y chromosomal content is one of several factors dictating gene expression levels (Figure 7D). The significant correlation between age at death and decreasing Y chromosome rRD in the DLPFC indicates that Y chromosomal content decreases with age (r = 3.7e-01, p = 3.0e-05, Figure 8A). Notably, rRD in no other chromosome was associated with age across the DLPFC, meaning that this loss does not occur as a result of a general reduction in DNA content (Supplemental Figure 4A–B). In the cerebellum, reduced chromosome 9 rRD was weakly associated with age, suggesting that age-related loss is chromosome and brain-region specific (r = 5.4e-02, p = 2.0e-02) (Supplemental Figure 4C–D).
Figure 8. Associations between neuropathological and clinical markers of ageing and AD and chromosome Y/female chromosome X mSA.

A) Association between rRD in the Y chromosome with age and PMI (controlling for extraction kit). Y chromosome rRD was significantly negatively correlated with age (p = 2.92e-05), but not with PMI. B) A linear model was used to assess the relationship between rRD and APOE4 status, slope of global cognition, cognition at last visit and amyloid content, subsetting by extraction kit and controlling for age at death and PMI. Because extraction kit was known to influence the rRD distribution, we performed two separate analyses with clinical data: one analysis that included extraction kit as a covariate in a linear model (referred to as “combined” model), and another that treated each extraction kit as a separate dataset (“single” model). A criteria for considering the correlations identified in the combined analysis as internally valid was that in the single model, the direction of correlation was concordant between the two extraction kits and statistical significance was reached in at least one kit. Associations that meet both criteria are shaded red. Asterisks signify significance at p < 0.05.
In the male and female X chromosome, the direction of skew at the DNA and RNA level concurred, suggesting a mild shift towards hyperploidy. mSA of the X chromosome was not associated with age at sampling, indicating that mosaicism of the X chromosome in the DLPFC is not age-dependent (Supplemental Figure 4A–B). Although some evidence exists that mosaic loss of the female X chromosome preferentially affects the inactive X in blood lymphocytes, it is unclear whether a similar effect occurs in the DLPFC (Machiela et al, 2016). Because none of the X genes considered highly expressed have been found to escape X inactivation, the correlation observed here suggests that hyperploidy occurs as variation in chromosomal content on the active X.
3.5. Association between mosaicism and clinical and neuropathological indicators of Alzheimer’s disease
Given the evidence for functional mosaicism in the male Y and female X, we wanted to determine whether rRD of the Y and X chromosome correlated with neuropathological and clinical indicators of Alzheimer’s disease in the DLPFC (Figure 8B). We focused on the DLPFC specifically as neuropathological variables, such as amyloid-beta, were derived from this region. Because extraction kit was known to influence the rRD distribution, we performed two separate analyses: one analysis that included extraction kit as a covariate in a linear model (referred to as “combined” model), and another that treated each extraction kit as a separate dataset (“single” model). A criterion for considering the correlations identified in the combined analysis as internally valid was that in the single model, the direction of correlation was concordant between the two extraction kits and statistical significance was reached in at least one kit. After controlling for age, PMI and DNA extraction kit, chromosome Y rRD in the DLPFC was mildly negatively correlated with the presence of at least one E4 allele in the APOE gene (combined, p = 1.9e-02) and rate of change of cognitive functioning over time (combined, p = 4.6e-02).
Female X rRD was also positively correlated with rate of change of cognitive functioning over time in addition to global cognition at last visit (combined, p = 7.8e-03; p = 1.9e-02). Interestingly, female chromosome X rRD was not correlated with amyloid-beta levels (combined, p = 4.2e-03, but direction discordant in single analysis). Additionally, no associations between mosaic loss of the male X chromosome and cognitive or neuropathological indicators of Alzheimer’s were identified.
Because we noted a correlation between male chr Y and female chr X rRD and sex chromosome expression, we sought to determine if X and Y expression ratio was directly associated with APOE4, slope of random global cognition and cognition at last visit. Although we did not note a direct correlation between APOE4 and slope of global cognition in the Y chromosome, we noted a significant correlation between X chromosome and cognition at last visit in the female X after controlling for PMI, RIN, batch number and the presence/absence of AD (p = 2.7e-03).
4 –. Discussion
In this study, we developed a method that uses per-chromosome read depth values from WGS to quantify differences in mSA of the sex chromosomes across tissues. We applied this method to determine the relative rate of mSA of the sex chromosomes in the blood, DLPFC and cerebellum. On aggregate, we found that mosaic somatic aneuploidy of the Y chromosome occurs more frequently in whole blood than in the DLPFC or cerebellum. In the male and female X, fold change in mSA between tissues was within the range attributable to technical variation, therefore implying that the range of X chromosomal content is similar across blood, DLPFC and cerebellum, or that mosaicism of these chromosomes occurs at a rate below bulk sequencing’s limit of detection. Analysis of skew at the DNA level identified that both hypo- and hyperploidy occur in the DLPFC, echoing previous findings (McConnell et al., 2017; Rehen, 2005; Rehen et al., 2001; Westra et al., 2008; Yurov et al., 2007, 2005). In addition, we found that gene expression correlated with sex chromosome content, suggesting that mSA may have a functional impact at the mRNA level. Our study confirms in a much larger cohort (n = 1078 vs n = 26) the previously reported observation that Y chromosomal content varies at a higher rate in the blood than in the DLPFC. We additionally confirm the correlation between Y chromosome loss and age in the DLPFC (Kimura et al., 2018). Further, by using the entire MSY region rather than just two X/Y homologs to measure chromosomal content, our methodology provides a more robust measure of sex chromosomal content that correlates highly with array-based mosaicism metrics. We also ascertain the functional impact of this mosaicism in the DLPFC, and provide evidence that sex differences in the incidence of neuropathological diseases may be mediated by differences in sex chromosome content.
There are conflicting reports on the existence of aneuploidy in the Alzheimer’s disease (AD) brain, with a sizeable body of literature finding that aneuploidy is associated with AD pathogenesis (Arendt et al., 2010; Arendt et al., 2015; Boeras et al., 2008; Bushman et al., 2015; Potter, 1991; Geller and Potter, 1999; Chen et al., 2010; Iourov et al., 2009; Migliore et al., 1999; Mosch et al., 2007; Vincent et al., 1996; Yang et al., 2001; Yang et al., 2003b; Yurov et al., 2014), and a separate body of literature finding no connection between aneuploidy and AD (Westra et al, 2009; van den Bos et al, 2016). In this study, we found that, Y, but not female X chromosomal content decreases with age in the DLPFC, indicating that age-related neurodegeneration may affect the X and Y chromosomes differently. This correlation was not observed in the cerebellum, indicating that this mosaicism of the sex chromosomes is brain region-specific. We also found that after correcting age and PMI, APOE4 allele status is negatively correlated with male Y, but not X, chromosomal content, highlighting a possible link between the APOE4 allele and mosaic loss of the Y chromosome in the DLPFC. We also find that reduced Y chromosomal and female X chromosomal content is associated with a faster rate of cognitive decline and reduced cognition at last visit, a hallmark of neurodegeneration and AD. These results indicate that Y and X chromosomal loss is associated with clinical manifestations of Alzheimer’s disease and neurodegeneration in a manner independent of age. Recent studies have observed an increase in hyperploid cortical neurons during preclinical AD, which then decrease to euploid levels as hyperploid cells enter the cell cycle and undergo cell death (Arendt et al., 2010, Arendt et al., 2015). Further research is needed to determine whether the female X chromosome undergoes a similar process in the brain.
All studies that have previously investigated somatic aneuploidy using SNP genotyping arrays and next generation sequencing have grappled with identifying a threshold value at which to define an individual as mosaic for a particular chromosome (Dumanski et al., 2016; Forsberg et al., 2014; Thompson et al., 2019). This threshold value can dramatically alter the conclusions of an analysis, and can often depend on the data itself (Forsberg et al, 2019; Zhou et al, 2019). In this study, we developed a measure of chromosomal mosaicism that does not depend on a specific threshold. Our metric, referred to as z-score, corrects for sample-specific technical biases, allowing us to directly compare the variation of chromosomal content across chromosomes and across tissues without the use of a threshold. This method can be applied to any WGS data set. Skew of this metric within each tissue can be used to determine whether this mosaicism occurs as hyperploidy or hypoploidy.
The fraction of mosaicism detected in a bulk sequencing sample depends on the sequencing depth. At the depth to which samples were sequenced in this study (average of 31x), mosaicism would only be detected if occuring in greater than 10% of cells (Forsberg et al, 2013; Forsberg et al., 2016; Jacobs et al., 2012). To distinguish technical variation from biologically meaningful variation, we assessed the correlation between rRD and gene expression in each sex chromosome in the DLPFC, with the assumption that biologically meaningful mosaicism would be correlated with gene expression. The association between sex chromosome rRD and expression ratio indicates that the observed variation is in fact biologically meaningful. Given the limits of mosaicism detection by bulk sequencing, this suggests that mosaicism in the sex chromosomes on average occurs at a result equal to or higher than 10% in the DLPFC, a finding in line with previous estimates of aneuploidy through FISH and SKY-MCB methods, but not more recent single-cell analyses. However, it should be noted that recent single-cell analyses had limited coverage of the sex chromosomes (Cai et al., 2014; Knouse et al., 2014; McConnell et al., 2013; van den Bos et al., 2016).
In this study, we addressed several technical challenges associated with the comparison of WGS data between individuals in large cohort studies, namely the non-random association between rRD and DNA extraction kit. A literature search yielded only one paper reporting a similar finding, which described a wave-like disruption at the bin level caused by different DNA extraction kits (van Heesch et al., 2013). To address this technical artefact, we normalized per-bin rRD to a sample-specific reference distribution built in the autosomes. This allowed us to compare SD of the z-score distribution between tissues (Supplemental Figure 1). To determine the range of fold change values that could be attributable to technical differences, the fold change in SD and range of z-score distributions sampled using different extraction kits in a single tissue (DLPFC) were calculated for each autosome. Comparisons were only performed for tissues that surpassed the maximum fold change seen in this distribution (Supplemental Figure 3B,C). This normalization procedure adjusts for technical differences and allows the relative rate of somatic mosaic aneuploidy to be compared.
The origin and timescale of biological mechanisms capable of generating somatic mosaicism in the blood and brain may help explain observed differences in the rate of mosaic somatic aneuploidy. In the blood, lymphocytes—the most common cell type among the DNA-containing white blood cells—replicate on the order of days and are known to undergo clonal expansion in the context of normal ageing. Therefore, the greater magnitude of skew observed in the Y chromosome in the blood could be attributed to aberrant clonal expansions in our relatively elderly population of individuals.
In contrast, in the brain, somatic mitotic events are restricted to neurogenesis, a critical time in brain development in which mitotic division of neural progenitor cells and the subsequent generation of differentiated neurons occurs at a fast pace. During this time, like cells in the blood, mosaic aneuploidies can occur as a result of lagging chromosomes, micronuclei, supernumerary centrosomes and chromosomal nondisjunction (Rehen, 2005; Rehen et al., 2001; Westra et al., 2008; Yang et al., 2003; Yurov et al., 2005). Neural progenitor cells formed during neurogenesis give way to non-dividing differentiated cell types, approximately 70% of which are post-mitotic neurons, and 30% of which are monocytes, astrocytes, microglia, oligodendrocytes and pericytes. Subsequent pruning of neurons occurs over the life course. Given the timescale at which mitotic events can occur in the brain, the observed reduction in Y chromosome content over the lifespan could emerge as a result of two possible scenarios:
Mitotic aneuploidy events during neurogenesis, resulting in hyperploid Y cells. Over the lifespan, death of hyperploid cells due to caspase-mediated programmed cell death could result in an observed decrease in Y chromosomal content over time (Peterson et al., 2012). This decrease would bring the Y chromosome from the hyperploid state to the euploid state.
Preferential cell death of euploid Y cells, resulting in an increasing proportion of hypoploid cells over time.
X/Y chromosome dosage has been found to play a role in the functioning of specific regions of the cortex, striatum and cerebellum, highlighting its biological utility even in the post-neurogenesis brain (Raznahan et al., 2016). This implies that scenario 1) is most likely. However, further research into somatic mutations during neurogenesis and neural pruning over the life course would be needed to identify the most likely mechanism through which the observed copy number distributions were formed.
Resolving the rate of mSA between the blood and brain will require a combination of single cell and bulk sequencing methods. Bulk methods, such as the one used in this study, can allow for a greater understanding of the relative distribution of chromosome X and Y copy number in different tissues and across populations. On the other hand, single cell methods provide utility in establishing the distribution of aneuploid cells within a single individual. Used together, they would allow researchers to understand not only the relative range of sex chromosomal content in the brain across individuals, but also the type and number of individual cells affected within the same individual. Over the coming decade, the Brain Somaticism Network will utilize both bulk and single-cell approaches to quantify somatic mosaicism in the brain, helping shed light on the relative landscape of somatic mosaicism in blood and brain (McConnell et al., 2017).
Supplementary Material
Supplementary Figure 1: SD of A) rRD distribution and B) z-score distribution in DLPFC samples by two different extraction kits: All Prep Universal (A) and QIAamp (Q).
{kind=link}
Supplemental Figure 2. rRD distributions after each read depth filtering step. A) rRD in GIAB-defined irregularly sized regions of high confidence in each chromosome (MAD = 0.0095). B) Bins of A were filtered so that only bins with average mappability greater than 0.9 remained (MAD = 0.0086). C) Bins of B were further filtered so that only bins with GC content between 45–55% remained (MAD = 0.0054).
{kind=link}
Supplementary Figure 3. Z-score metric reduced standard deviation between extraction kits. A) Fold change (FC) between extraction kits used in the DLPFC (QIAamp and AllPrepUniversal) when using the RD and z-score based metric. Overall reduced FC in SD are observed when using the z-score metric. B) To determine the relationship between extraction kit and SD of z-score distribution, we calculated the FC in SD between extraction kits for all autosomes in the DLPFC (Supplemental Figure 1B). To reduce the likelihood of conflating tissue-specific differences in SD with extraction kit differences, tissue-specific differences in SD were only considered when larger than the max fold change seen in this distribution (2.34).
{kind=link}
Supplementary Figure 4: Correlation between age at death and rRD and z-score in each chromosome of the DLPFC and cerebellum. A) Correlation between age and rRD in DLPFC. B) Correlation between age and z-score in DLPFC. Age is only significantly associated with z-score and rRD in the Y chromosome (rRD, p = 9.0e-05; z-score, p = 1.0e-03). C) Correlation between age and rRD in cerebellum. D) Correlation between age and z-score in cerebellum. Age is significantly associated with rRD in chromosome 9 in the cerebellum (rRD, p = 2.0e-02; z-score, p = 4.8e-01).
{kind=link}
Highlights.
Somatic mosaic aneuploidy (mSA) of the sex chromosome occurs more frequently in the blood than in the brain.
mSA of the male Y and female X chromosomes is associated with aggregate changes in gene expression.
Reduced read depth of the Y chromosome (suggesting mosaic loss) is modestly associated with a genetic risk factor for Alzheimer’s disease: the presence of the APOE4 allele.
Reduced read depth of the Y and female X chromosome is associated with a faster rate of cognitive decline.
Reduced read depth in the female X chromosome is associated with cognitive characteristics of Alzheimer’s disease, including reduced cognition at the last clinical visit.
Acknowledgments
5 - Funding sources
This work was supported by NIH grants P30AG10161, R01AG15819, R01AG17917, U01AG46152, R01AG055909, and R21MH118576.
Footnotes
Declaration of Interest:
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6 – References
- Arendt T, Bruckner ML, Mosch B, Losche A, 2010. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J Pathol. 177 pp. 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Bruckner MK, Losche A, 2015. Regional mosaic genomic heterogeneity in the elderly and in Alzheimer’s disease as a correlate of neuronal vulnerability. Acta Neuropathol. 130 pp. 501–510 [DOI] [PubMed] [Google Scholar]
- Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA, 2018. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimer’s Dis. doi: 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berletch B, Yang F, Xu J, Carrel L, Disteche CM, 2011. Genes that escape from X inactivation. 130(2). pp 237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeras DI, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, Potter H, 2008. Alzheimer’s presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging. 29 pp. 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefond A, Skrobek B, Lobbens S, Eury E, Thuillier D, Cauchi S, Lantieri O, Balkau B, Riboli E, Marre M, Charpentier G, Yengo L, Froguel P, 2013. Nat. Gen. 45(9). pp. 1040–1043. [DOI] [PubMed] [Google Scholar]
- Bushman DM, Kaeser GE, Siddoway B, Westra JW, Rivera RR, Rehen SK, Yung YC, Chun J, 2015. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. Elife. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, Poduri A, Walsh CA, 2014. Single-Cell, Genome-wide Sequencing Identifies Clonal Somatic Copy-Number Variation in the Human Brain. Cell Rep. doi: 10.1016/j.celrep.2014.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J Cohen ML, Lerner AJ, Yang Y, Herrup K, 2010. DNA damage and cell cycle events implicate cerebellar dentate nucleus neurons as targets of Alzheimer’s disease. Mol Neurodegener. 5 p. 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D, Klein HU, White CC, Peters MA, Lodgson B, Nejad P, Tang A, Mangravite LM, Yu L, Gaiteri C, Mostafavi S, Schneider JA, Bennett DA, 2018. Data descriptor: A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci. Data 5, 1–13. doi: 10.1038/sdata.2018.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Shulman J, Chibnik L, Keenan B, Raj T, Wilson RS, Yu L, Luergans SE, Tran D, Aubin C, Anderson CD, Biffi A, Corneveaux JJ, Huentelman MJ, Alzheimer’s Disease Neuroimaging Initiative, Rosand J, Daly MJ, Myers AJ, Reiman EM, Bennett DA, Evans DA, 2013. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol. Aging. 33(5): 1017.e1–1017.e15. doi: 10.1016/j.neurobiolaging.2011.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanski JP, Lambert JC, Rasi C, Giedraitis V, Davies H, Grenier-Boley B, Lindgren CM, Campion D, Dufouil C, Pasquier F, Amouyel P, Lannfelt L, Ingelsson M, Kilander L, Lind L, Forsberg LA, 2016. Mosaic Loss of Chromosome y in Blood Is Associated with Alzheimer Disease. Am. J. Hum. Genet. 98, 1208–1219. doi: 10.1016/j.ajhg.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Absher D, Dumanski JP, 2013. Non-heritable genetics of human disease: Spotlight on post-zygotic genetic variation acquired during lifetime. Postgrad. Med. J. doi: 10.1136/postgradmedj-2012-101322rep [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Rasi C, Malmqvist N, Davies H, Pasupulati S, Pakalapati G, Sandgren J, De Ståhl TD, Zaghlool A, Giedraitis V, Lannfelt L, Score J, Cross NCP, Absher D, Janson ET, Lindgren CM, Morris AP, Ingelsson E, Lind L, Dumanski JP, 2014. Mosaic loss of chromosome y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat. Genet. 46, 624–628. doi: 10.1038/ng.2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Halvardosn J, Rychlicka-Buniowska E, Danielsson M, Moghadam B, Mattisson J, Rasi C, Davies H, Lind L, Giedraitis V, Lannfelt L, Kilander L, Ingelsson M, Dumanski J, 2019. Mosaic loss of chromosome Y in leukocytes matters. Nature Genetics, 51 (4–7). doi: 10.1038/s41588-018-0267-9 [DOI] [PubMed] [Google Scholar]
- Geller LN, Potter H, 1999. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer’s disease. Neurobiol Dis. 6 pp. 167–179 [DOI] [PubMed] [Google Scholar]
- Guerra L, Diociaiuti A, El Hachem M, Castiglia D, Zambruno G, 2015. Ichthyosis with confetti: Clinics, molecular genetics and management Rare skin diseases. Orphanet J. Rare Dis. doi: 10.1186/s13023-015-0336-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iourov IY, Liehr T, Vorsanova SG, Kolotii AD, Yurov YB, 2006. Visualization of interphase chromosomes in postmitotic cells of the human brain by multicolour banding (MCB). Chromosome. Res. doi: 10.1007/s10577-006-1037-6 [DOI] [PubMed] [Google Scholar]
- Iourov IY, Vorsanova SG, Liehr T, Kolotii AD, Yurov YB, 2009a. Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum. Mol. Genet. doi: 10.1093/hmg/ddp207 [DOI] [PubMed] [Google Scholar]
- Iourov IY, Vorsanova SG, Liehr T, Yurov YB, 2009b. Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: Differential expression and pathological meaning. Neurobiol. Dis. doi: 10.1016/j.nbd.2009.01.003 [DOI] [PubMed] [Google Scholar]
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner MJ, Cullen M, Epstein CG, Burdett L, Dean MC, Chatterjee N, Sampson J, Chung CC, Kovaks J, Gapstur SM, Stevens VL, Teras LT, Gaudet MM, Albanes D, Weinstein SJ, Virtamo J, Taylor PR, Freedman ND, Abnet CC, Goldstein AM, Hu N, Yu K, Yuan JM, Liao L, Ding T, Qiao YL, Gao YT, Koh WP, Xiang YB, Tang ZZ, Fan JH, Aldrich MC, Amos C, Blot WJ, Bock CH, Gillanders EM, Harris CC, Haiman CA, Henderson BE, Kolonel LN, Le Marchand L, McNeill LH, Rybicki BA, Schwartz AG, Signorello LB, Spitz MR, Wiencke JK, Wrensch M, Wu X, Zanetti KA, Ziegler RG, Figueroa JD, Garcia-Closas M, Malats N, Marenne G, Prokunina-Olsson L, Baris D, Schwenn M, Johnson A, Landi MT, Goldin L, Consonni D, Bertazzi PA, Rotunno M, Rajaraman P, Andersson U, Freeman LEB, Berg CD, Buring JE, Butler MA, Carreon T, Feychting M, Ahlbom A, Gaziano JM, Giles GG, Hallmans G, Hankinson SE, Hartge P, Henriksson R, Inskip PD, Johansen C, Landgren A, McKean-Cowdin R, Michaud DS, Melin BS, Peters U, Ruder AM, Sesso HD, Severi G, Shu XO, Visvanathan K, White E, Wolk A, Zeleniuch-Jacquotte A, Zheng W, Silverman DT, Kogevinas M, Gonzalez JR, Villa O, Li D, Duell EJ, Risch HA, Olson SH, Kooperberg C, Wolpin BM, Jiao L, Hassan M, Wheeler W, Arslan AA, Bueno-De-Mesquita HB, Fuchs CS, Gallinger S, Gross MD, Holly EA, Klein AP, Lacroix A, Mandelson MT, Petersen G, Boutron-Ruault MC, Bracci PM, Canzian F, Chang K, Cotterchio M, Giovannucci EL, Goggins M, Bolton JAH, Jenab M, Khaw KT, Krogh V, Kurtz RC, McWilliams RR, Mendelsohn JB, Rabe KG, Riboli E, Tjønneland A, Tobias GS, Trichopoulos D, Elena JW, Yu H, Amundadottir L, Stolzenberg-Solomon RZ, Kraft P, Schumacher F, Stram D, Savage SA, Mirabello L, Andrulis IL, Wunder JS, García AP, Sierrasesà maga L, Barkauskas DA, Gorlick RG, Purdue M, Chow WH, Moore LE, Schwartz KL, Davis FG, Hsing AW, Berndt SI, Black A, Wentzensen N, Brinton LA, Lissowska J, Peplonska B, McGlynn KA, Cook MB, Graubard BI, Kratz CP, Greene MH, Erickson RL, Hunter DJ, Thomas G, Hoover RN, Real FX, Fraumeni JF, Caporaso NE, Tucker M, Rothman N, Pérez-Jurado LA, Chanock SJ, 2012. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. doi: 10.1038/ng.2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Hishimoto A, Otsuka I, Okazaki S, Boku S, Horai T, Izumi T, Takahashi M, Ueno Y, Shirakawa O, Sora I, 2018. Loss of chromosome Y in blood, but not in brain, of suicide completers. PLoS One 13(1). doi: 10.1371/journal.pone.0190667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, Amon A, 2014. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. doi: 10.1073/pnas.1415287111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D, Ha G, Shah S. 2019. HMMcopy: Copy number prediction with correction for GC and mappability bias for HTS data. R package version 1.26.0 [Google Scholar]
- Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh EW, Amos C, Wei Q, Wang LE, Lee JE, Barnes KC, Hansel NN, Mathias R, Daley D, Beaty TH, Scott AF, Ruczinski I, Scharpf RB, Bierut LJ, Hartz SM, Landi MT, Freedman ND, Goldin LR, Ginsburg D, Li J, Desch KC, Strom SS, Blot WJ, Signorello LB, Ingles SA, Chanock SJ, Berndt SI, Le Marchand L, Henderson BE, Monroe KR, Heit JA, De Andrade M, Armasu SM, Regnier C, Lowe WL, Hayes MG, Marazita ML, Feingold E, Murray JC, Melbye M, Feenstra B, Kang JH, Wiggs JL, Jarvik GP, McDavid AN, Seshan VE, Mirel DB, Crenshaw A, Sharopova N, Wise A, Shen J, Crosslin DR, Levine DM, Zheng X, Udren JI, Bennett S, Nelson SC, Gogarten SM, Conomos MP, Heagerty P, Manolio T, Pasquale LR, Haiman CA, Caporaso N, Weir BS, 2012. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. doi: 10.1038/ng.2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftfield E, Zhou W, Graubard BI, Yeager M, Chanock SJ, Freedman ND, Machiela MJ, 2018. Predictors of mosaic chromosome Y loss and associations with mortality in the UK Biobank. Sci. Rep. doi: 10.1038/s41598-018-30759-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, Chanock SJ, 2017. The ageing genome, clonal mosaicism and chronic disease. Curr. Opin. Genet. Dev. doi: 10.1016/j.gde.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, Zhou W, Karlins E, Sampson JN, Freedman ND, Yang Q, Hicks B, Dagnall C, Hautman C, Jacobs KB, Abnet CC, Aldrich MC, Amos C, Amundadottir LT, Arslan AA, Beane-Freeman LE, Berndt SI, Black A, Blot WJ, Bock CH, Bracci PM, Brinton LA, Bueno-De-Mesquita HB, Burdett L, Buring JE, Butler MA, Canzian F, Carreón T, Chaffee KG, Chang IS, Chatterjee N, Chen C, Chen C, Chen K, Chung CC, Cook LS, Crous Bou M, Cullen M, Davis FG, De Vivo I, Ding T, Doherty J, Duell EJ, Epstein CG, Fan JH, Figueroa JD, Fraumeni JF, Friedenreich CM, Fuchs CS, Gallinger S, Gao YT, Gapstur SM, Garcia-Closas M, Gaudet MM, Gaziano JM, Giles GG, Gillanders EM, Giovannucci EL, Goldin L, Goldstein AM, Haiman CA, Hallmans G, Hankinson SE, Harris CC, Henriksson R, Holly EA, Hong YC, Hoover RN, Hsiung CA, Hu N, Hu W, Hunter DJ, Hutchinson A, Jenab M, Johansen C, Khaw KT, Kim HN, Kim YH, Kim YT, Klein AP, Klein R, Koh WP, Kolonel LN, Kooperberg C, Kraft P, Krogh V, Kurtz RC, Lacroix A, Lan Q, Landi MT, Marchand L Le Li, D., Liang X, Liao LM, Lin D, Liu J, Lissowska J, Lu L, Magliocco AM, Malats N, Matsuo K, McNeill LH, McWilliams RR, Melin BS, Mirabello L, Moore L, Olson SH, Orlow I, Park JY, Patinõ-Garcia A, Peplonska B, Peters U, Petersen GM, Pooler L, Prescott J, Prokunina-Olsson L, Purdue MP, Qiao YL., Rajaraman P, Real FX, Riboli E, Risch HA, Rodriguez-Santiago B, Ruder AM, Savage SA, Schumacher F, Schwartz AG, Schwartz KL, Seow A, Wendy Setiawan V, Severi G, Shen H, Sheng X, Shin MH, Shu XO, Silverman DT, Spitz MR, Stevens VL, Stolzenberg-Solomon R, Stram D, Tang ZZ, Taylor PR, Teras LR, Tobias GS, Van Den Berg D, Visvanathan K, Wacholder S, Wang JC, Wang Z, Wentzensen N, Wheeler W, White E, Wiencke JK, Wolpin BM, Wong MP, Wu C, Wu T, Wu X, Wu YL, Wunder JS, Xia L, Yang HP, Yang PC, Yu K, Zanetti KA, Zeleniuch-Jacquotte A, Zheng W, Zhou B, Ziegler RG, Perez-Jurado LA, Caporaso NE, Rothman N, Tucker M, Dean, Yeager M, Chanock SJ, 2016. Female chromosome X mosaicism is age-related and preferentially affects the inactivated X chromosome. Nat. Commun. doi: 10.1038/ncomms11843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, Gage FH, 2013. Mosaic copy number variation in human neurons. Science. doi: 10.1126/science.1243472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, Erwin JA, Fasching L, Flasch DA, Freed D, Ganz J, Jaffe AE, Kwan KY, Kwon M, Lodato MA, Mills RE, Paquola ACM, Rodin RE, Rosenbluh C, Sestan N, Sherman MA, Shin JH, Song S, Straub RE, Thorpe J, Weinberger DR, Urban AE, Zhou B, Gage FH, Lehner T, Senthil G, Walsh CA, Chess A, Courchesne E, Gleeson JG, Kidd JM, Park PJ, Pevsner J, Vaccarino FM, 2017. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science (80-. ). doi: 10.1126/science.aal1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore L, Botto N, Scarpato R, Petrozzi L, Cipriani GU Bonuccelli, U., 1999. Preferential occurrence of chromosome 21 malsegregation in peripheral blood lymphocytes of Alzheimer disease patients. Cytogenet Cell Genet. 87pp. 41–46 [DOI] [PubMed] [Google Scholar]
- Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J, 2017. Differences between germline and somatic mutation rates in humans and mice. Nat. Commun. doi: 10.1038/ncomms15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosch B, Morawski M, Mittag A Lenz D Tarnok A Arendt T Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. 2007. J Neurosci. 27. pp. 6859–6867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski A, Bruder CEG, Andersson R, De Ståhl TD, Menzel U, Sandgren J, Poplawski A, Von Tell D, Crasto C, Bogdan A, Bartoszewski R, Bebok Z, Krzyzanowski M, Jankowski Z, Partridge EC, Komorowski J, Dumanski JP, 2008. Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat. doi: 10.1002/humu.20815 [DOI] [PubMed] [Google Scholar]
- Potter H Review and hypothesis: Alzheimer disease and Down syndrome–chromosome 21 nondisjunction may underlie both disorders. 1991. Am J Hum Genet. 48. pp. 1192–1200 [PMC free article] [PubMed] [Google Scholar]
- Raznahan A, Lee NR, Greenstein D, Wallace GL, Blumenthal JD, Clasen LS, Giedd JN, 2016. Globally Divergent but Locally Convergent X- and Y-Chromosome Influences on Cortical Development. Cereb. Cortex. doi: 10.1093/cercor/bhu174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raznahan A, Parkshak N, Chandran V, Blumenthal J, Clasen L, Alexander-Blach A, Zinn A, Wangsa D, Wise J, Murphy D, Bolton P, Ried T, Ross J, Giedd J, Geschwind D, 2018. Sex chromosome dosage effects on gene expression in humans. PNAS. 10.1073/pnas.1802889115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehen SK, 2005. Constitutional Aneuploidy in the Normal Human Brain. J. Neurosci. doi: 10.1523/JNEUROSCI.4560-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J, 2001. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Natl. Acad. Sci. doi: 10.1073/pnas.231487398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrback S, Siddoway B, Liu CS, Chun J, 2018. Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. doi: 10.1002/dneu.22626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinin I, Sie D, Bengtsson H, van de Wiel MA, Olshen AB, van Thuijl HF, van Essen HF, Eijk PP, Rustenburg F, Meijer GA, Reijneveld JC, Wesseling P, Pinkel D, Albertson DG, Ylstra B., 2014. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Research. doi: 10.18129/B9.bioc.QDNAseq [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D, Genovese G, Halvardson J, Ulirsch J, Wright D, Terao C, Davidsson O, Day F, Sulem P, Jiang Y, Danielsson M, Davies H, Dennis J, Dunlop M, Easton D, Fisher V, Zink F, Houlston R, Ingelsson M, Kar S, Kerrison N, Kristjansson R, Li R, Loveday C, Mattisson J, McCarroll S, Murakami Y, Murray A, Olszewski P, Rychlicka-Buniowska E, Scott R, Thorsteinsdottir U, Tomlinson I, Torabi Moghadam B, Turnbull C, Wareham N, Gudbjartsson D, Kamatani Y, Finucane H, Hoffmann E, Jackson S, Stefansson K, Auton A, Ong K, Machiela M, Loh P-R, Dumanski J, Chanock S, Forsberg L, Perry J, 2019. Genetic predisposition to mosaic Y chromosome loss in blood is associated with genomic instability in other tissues and susceptibility to nonhaematological cancers. bioRxiv 514026. doi: 10.1101/514026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bos H, Spierings DCJ, Taudt AS, Bakker B, Porubský D, Falconer E, Novoa C, Halsema N, Kazemier HG, Hoekstra-Wakker K, Guryev V, den Dunnen WFA, Foijer F, Tatché MC, Boddeke HWGM, Lansdorp PM, 2016. Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. doi: 10.1016/j.jue.2005.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heesch S, Mokry M, Boskova V, Junker W, Mehon R, Toonen P, de Bruijn E, Shull JD, Aitman T, Cuppen E, Guryev V 2013. Systematic biases in DNA copy number originate from isolation procedures. Genome Biol. doi: 10.1186/gb-2013-14-4-r33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I Rosado M Davies P, 1996. Mitotic mechanisms in Alzheimer’s disease? J Cell Biol. 132 pp. 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra JW, Peterson SE, Yung YC, Mutoh T, Barral S, Chun J, 2008. Aneuploid mosaicism in the developing and adult cerebellar cortex. J. Comp. Neurol. doi: 10.1002/cne.21648 [DOI] [PubMed] [Google Scholar]
- Westra JW, Barral S, Chun J, 2009. A reevaluation of tetraploidy in the Alzheimer’s disease brain. Neurodegener Dis. 6. pp. 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K, 2001. DNA replication precedes neuronal cell death in Alzheimer’s disease.. J Neurosci. 21. pp. 2661–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Kaushal D, Rehen S, Kriedt K, Kingsbury M, McConnell M, Chun J, 2003. Chromosome segregation defects contribute to aneuploidy in normal neural progenitor cells. J Neurosci. doi:23/32/10454 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurov YB, Iourov IY, Monakhov VV, Soloviev IV, Vostrikov VM, Versanova SG, 2005. The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study, in: Journal of Histochemistry and Cytochemistry. doi: 10.1369/jhc.4A6430.2005 [DOI] [PubMed] [Google Scholar]
- Yurov YB, Iourov IY, Vorsanova SG, Demidova IA, Kravetz VS, Beresheva AK, Kolotii AD, Monakchov VV, Uranova NA, Vostrikov VM, Soloviev IV, Liehr T, 2008. The schizophrenia brain exhibits low-level aneuploidy involving chromosome 1. Schizophr. Res. doi: 10.1016/j.schres.2007.07.035 [DOI] [PubMed] [Google Scholar]
- Yurov YB, Iourov IY, Vorsanova SG, Liehr T, Kolotii AD, Kutsev SI, Pellestor F, Beresheva AK, Demidova IA, Kravets VS, Monakhov VV, Soloviev IV, 2007. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS One. doi: 10.1371/journal.pone.0000558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurov YB, Vorsanova SG, Liehr T, Kolotii AD, Iourov IY, 2014. X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. doi: 10.1186/1755-8166-7-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Machiela M, Freedman N, Rothman N, 2018. Reply to ‘Mosaic loss of chromosome Y in leukocytes matters’. Nature Genetics, 51(1). doi: 10.1038/s41588-018-0310-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: SD of A) rRD distribution and B) z-score distribution in DLPFC samples by two different extraction kits: All Prep Universal (A) and QIAamp (Q).
Supplemental Figure 2. rRD distributions after each read depth filtering step. A) rRD in GIAB-defined irregularly sized regions of high confidence in each chromosome (MAD = 0.0095). B) Bins of A were filtered so that only bins with average mappability greater than 0.9 remained (MAD = 0.0086). C) Bins of B were further filtered so that only bins with GC content between 45–55% remained (MAD = 0.0054).
Supplementary Figure 3. Z-score metric reduced standard deviation between extraction kits. A) Fold change (FC) between extraction kits used in the DLPFC (QIAamp and AllPrepUniversal) when using the RD and z-score based metric. Overall reduced FC in SD are observed when using the z-score metric. B) To determine the relationship between extraction kit and SD of z-score distribution, we calculated the FC in SD between extraction kits for all autosomes in the DLPFC (Supplemental Figure 1B). To reduce the likelihood of conflating tissue-specific differences in SD with extraction kit differences, tissue-specific differences in SD were only considered when larger than the max fold change seen in this distribution (2.34).
Supplementary Figure 4: Correlation between age at death and rRD and z-score in each chromosome of the DLPFC and cerebellum. A) Correlation between age and rRD in DLPFC. B) Correlation between age and z-score in DLPFC. Age is only significantly associated with z-score and rRD in the Y chromosome (rRD, p = 9.0e-05; z-score, p = 1.0e-03). C) Correlation between age and rRD in cerebellum. D) Correlation between age and z-score in cerebellum. Age is significantly associated with rRD in chromosome 9 in the cerebellum (rRD, p = 2.0e-02; z-score, p = 4.8e-01).
Data Availability Statement
WGS z-scores (section 2.4) for each patient are available as supplementary data. Associated clinical and neuropathological data (section 2.8) are available through the ROSMAP portal (https://www.synapse.org/#!Synapse:syn3219045).