

Abstract
Vitamin D is an essential nutrient that is metabolized in the body to generate an active metabolite (1,25(OH)2D) with hormone-like activity and highly diverse roles in cellular function. Vitamin D deficiency (VDD) is a prevalent but easily preventable nutritional disturbance. Emerging evidence demonstrates the importance of sufficient vitamin D concentrations during fetal life with deficiencies leading to long-term effects into adulthood. Here, we provide a detailed review and perspective of evidence for the role of maternal VDD in offspring long term health, particularly as it relates to Developmental Origins of Health and Disease (DOHaD). We focus on roles in neurobehavioral and cardiometabolic disorders in humans and highlight recent findings from zebrafish and rodent models that probe potential mechanisms linking early life VDD to later life health outcomes. Moreover, we explore evidence implicating epigenetic mechanisms as a mediator of this link. Gaps in our current understanding of how maternal VDD might result in deleterious offspring outcomes later in life are also addressed.
Keywords: Vitamin D deficiency, DOHAD, metabolic disorders, autism, neurobehavioral disorders, animal models
INTRODUCTION
Vitamin D is obtained through various plant and animal dietary sources and can also be synthesized in the skin through sunlight exposure (Figure 1). All forms of vitamin D are metabolized in the liver via 25-hydroxylases to generate 25 hydroxyvitamin D, 25(OH)D, the predominant but inactive circulating form of vitamin D. (Figure 1). As shown, the kidney is the primary site of conversion of 25(OH)D to the active form 1,25(OH)2D via the 1-alpha-hydroxylase CYP27B1. However, extrarenal expression of Cyp27b1 also generates 1,25(OH)2D in numerous target tissues including the placenta and brain (Adams & Hewison 2012) (Townsend et al. 2005). Vitamin D deficiency (VDD) is commonly defined as 25(OH)D concentration <50 nmol/L (Holick et al. 2011) and represents one of the most widespread but treatable nutrient deficiencies (Holick et al. 2011). VDD has been attributed to dietary depletion, insufficient sunlight exposure, adiposity, and genetic variants (Ross 2011). Pregnant women are especially at high risk (Fiscaletti et al. 2017), and global rates range from 7% in Southern Africa up to 100% in some parts of Northern Europe (Hossein-nezhad & Holick 2013). In the United States (US), up to 80% of pregnant women, including those prescribed prenatal vitamins, are estimated to have low vitamin D status (McAree et al. 2013).
Figure 1.

Synthesis and functions of active hormonal vitamin D metabolite 1,25(OH)2D. As shown, the liver is essential in converting vitamin D to 25(OH)D. Within the kidney, it is then further metabolized to 1,25(OH)2D, although it is increasingly being recognized that other organs can convert 25(OH)D to 1,25(OH)2D. Once formed, 1,25(OH)2D can induce both genomic and non-genomic effects mediated by VDR, some of which are illustrated in this diagram. (Modified from (Vuolo et al. 2012; Lim & Kim 2014)).
VDD during pregnancy is associated with increased risk of gestational diabetes and preeclampsia, which can indirectly affect offspring health (Bodnar et al. 2007; Brannon 2012; Senti et al. 2012). It is also associated with direct effects on offspring health such as low birth weight, poor skeletal health, impaired brain development, autoimmune disease, obesity and insulin resistance (Krishnaveni et al. 2011; Crozier et al. 2012; Reichetzeder et al. 2014; Hawes et al. 2015; Gur et al. 2016; Maia-Ceciliano et al. 2016). Recent randomized controlled trials involving vitamin D supplementation in high-risk pregnancies demonstrated decreased cesarean section rate and maternal hospitalization, decreased macrosomia, and hospitalization in newborns of women with gestational diabetes (Karamali et al. 2016). Favorable effects on insulin metabolism parameters, serum HDL-cholesterol, and total cholesterol concentrations in women with pre-eclampsia risk factors were also reported (Karamali et al. 2015).
Maternal VDD likely impacts offspring health by altering fetal and newborn vitamin D availability. Although 1,25-dihydroxyvitamin D [1,25(OH)2D], the primary bioactive form, does not readily cross the placenta, umbilical cord concentrations of its precursor, 25(OH)D, are similar to maternal concentrations (Hillman & Haddad 1974; Fleischman & Cole 1980; Gray et al. 1981; Marshall et al. 2013). Findings from a recent human study provide evidence that the placenta, working as the interface between mother and fetus, modulates circulating vitamin D metabolites in pregnant women and that it favors the uptake of DBP-bound 25(OH)D3 through a specific receptor system (LRP2-CUBN) (Park et al. 2017). In the placenta, CYP27B1 activity is detectable in both maternal decidua and fetal trophoblast, which then allows for local production of 1,25(OH)2D from maternal 25(OH)D via 1-alpha-hydroxylase (Ganguly et al. 2018). Likewise, both decidua and trophoblast also express VDR (Liu & Hewison 2012). Placental production of 1,25(OH)2D is proposed to be essential for immunosuppressive effects required for immune tolerance of implantation. However, ex vivo and in vitro studies provide evidence for a more extensive role of vitamin D in placental function, including trophoblastic differentiation and extravillous trophoblast invasion of the decidua and myometrium (Ganguly et al. 2018).
Studies are ongoing to better understand potential genomic and non-genomic mechanisms responsible for effects of maternal VDD on offspring health (Figure 1). Vitamin D has both calciotrophic and noncalciotrophic functions (Maalouf 2008). It plays a major role in calcium ion homeostasis by regulating calcium transport and bone mineralization. Noncalciotropic functions of vitamin D occur via vitamin D receptor (VDR) activity in many tissues across the body. 1,25(OH)2D acts as a nuclear receptor ligand primarily binding to the vitamin D receptor (VDR) – retinoic X receptor (RXR) complex to activate or repress gene transcription at vitamin D response elements across the genome (Figure 1). The calciotropic function of vitamin D does not sufficiently explain all of the adverse effects of VDD on offspring health since calcium absorption during pregnancy is increased in an estrogen-dependent and vitamin D-independent manner in vitamin D receptor (VDR) knockout mice (Heaney & Skillman 1971; Van Cromphaut et al. 2003; Fudge & Kovacs 2010). Additionally, maternal-fetal calcium transfer in both VDD rats and VDR knockout mice is unaffected (Glazier & Sibley 1995; Kovacs et al. 2005). Thus, VDD-related offspring health outcomes are likely in part due to non-calciotropic functions of vitamin D. However, whether these are direct vs. indirect effects and mediated by VDR genomic activity remains unclear.
Recently, the focus on the relationship between maternal VDD and long term offspring health has shifted to roles of vitamin D as a regulatory agent in “fetal programming” of disease in later life, also termed developmental origins of health and disease (DOHaD) (Brannon 2012; Reichetzeder et al. 2014; Chango & Pogribny 2015). Animal models have been designed to elucidate the DOHaD effects of this secosteroid hormone-nutrient. The purpose of this review is to discuss the findings, strengths, and limitations of DOHaD focused VDD studies in current and emerging rodent and zebrafish models, primarily as they relate to human metabolic and neurobehavioral health. We also discuss emerging evidence of epigenetic activity of vitamin D and implications of a mechanistic role in DOHaD.
NEUROBEHAVIORAL HEALTH
Recent findings link low vitamin D status to adverse neurological outcomes such as autism spectrum disorder (ASD), schizophrenia, depression, multiple sclerosis, and dementia (Eyles et al. 2011; 2013; Yates et al. 2018). Accumulating evidence suggests that vitamin D is metabolized in various cells within the central nervous system, such as glial cells, neurons, and astrocytes (Cui et al. 2017). 1,25(OH)2D genomic activity is responsible for activating transcription of genes critical for early brain development (Eyles et al. 2011). VDR’s regulation of the dopaminergic system implicates a direct association with neurodegenerative diseases (Stednitz et al. 2015). Low vitamin D status, at early developmental stages and throughout adulthood, results in impaired dopamine (DA) turnover (Kesby et al. 2009; Pertile et al. 2016). Impaired DA turnover can cause oxidative stress, neuron degradation, and hyperactivity (Beninger 1983; Nowicki et al. 2015; Takeshima et al. 2016). Given the essential role of vitamin D in promoting healthy brain development, VDD has been proposed as a contributing factor to neurobehavioral health outcomes such as ASD and attention deficit hyperactivity disorder (ADHD) (Berridge 2018; El-Ansary et al. 2018).
Human studies
Autism and ADHD
In an extensive epidemiological study, Childhood Autism Risks from Genetics and the Environment (CHARGE), showed an association between autism spectrum disorders (ASD) and several functional polymorphisms in genes within the vitamin D pathway, including Bsm1, Taq1, Cdx2, and FokI in the vitamin D receptor (VDR) gene (Schmidt et al. 2015). Importantly, this study pinpointed that the genotype AA/A-allele of GC rs4588, encoding the vitamin D binding protein, was strongly associated with ASD in children (Schmidt et al. 2015). Additionally, supplementation with high doses of vitamin D (ranging from 300 IU/kg/day to 5000 IU/day) in two open-label trials has been found to improve clinical core symptoms of autism in ~75% of affected children (Cannell 2017).
Assessment of the Generation R study, a population-based cohort of mother-child dyads (n = 4229) from Rotterdam, The Netherlands, revealed that gestational VDD (defined by the study as maternal 25(OH)D < 25 nmol/L) was associated with an increase in autism-related traits in 6-year-old offspring (Vinkhuyzen et al. 2018). Analogous findings were reported with a birth cohort that included 4,334 individuals where mid-gestational VDD was associated with increased risk of ASD (Vinkhuyzen et al. 2018). A prospective study with 1,650 mother-children pairs included as part of the INMA Project (Spain 1997–2008) assessed maternal 25(OH)D concentrations in relation to child ADHD-like symptoms (attention deficit and hyperactivity-impulsivity) evaluated using the ADHD Criteria of Diagnostic and Statistical Manual of Mental Disorders fourth edition form. It was found that increasing maternal 25(OH)D concentrations were linked with a lower risk of ADHD-like symptoms in resulting children (Morales et al. 2015).
Other Neurological Disorders
Assessment of later receptive language ability in children age 5 and 10 born to Caucasian women in Perth, Western Australia discovered that maternal VDD (≤ 46 nmol/L of 25(OH)D) during pregnancy was associated with higher incidence of later language difficulties in children (Whitehouse et al. 2012). It has also been hypothesized that maternal VDD during pregnancy may increase offspring risk for later adult-onset schizophrenia (McGrath 1999). Two studies, Finnish Maternity Cohort and the Nurses’ Health Study II, collectively sampled ~ 36,000 mothers and identified a positive link between maternal VDD and risk of adult offspring developing multiple sclerosis (MS) (Mirzaei et al. 2011; Munger et al.).
Polymorphisms in vitamin D pathway genes such as VDR have been shown to affect binding to calciferol and contribute to VDD (Zumaraga et al. 2017). Therefore, several studies have also assessed the link between MS and genetic variants in vitamin D pathway genes. However, not all reports agree. A study of Canadian MS patients and unaffected relatives did not detect any significant associations between vitamin D pathway genetic variants that were previously linked to serum 25(OH)D concentrations, and disease status (Orton et al. 2011). A meta-analysis with 303 Caucasian Spanish MS patients and 310 healthy Caucasian Spanish controls also did not find any association with VDR variants rs2228570 (Fok1) or rs731236 (Taq1) and risk of MS (García-Martín et al. 2013). In contrast, another study that examined 726 MS patients and 604 controls from the United Kingdom (UK) found that the VDR Taq1 polymorphism rs731236 was associated with MS such that there was overrepresentation of the minor allele (C) in MS patients (Cox et al. 2012). This latter study also suggested a trend of increasing risk for MS specifically in individuals homozygous for the HLA-DRB1*501 allele in association with the Fok1 allele known to generate a more active form of VDR. Genetic variants in CYP27B1, which catalyzes the conversion of 25(OH)D to 1,25(OH)2D, have also been linked with MS susceptibility. A genome-wide association study performed with 1,618 MS patients of European ancestry from Australia and New Zealand and 3,412 controls of European ancestry from the UK and US found alleles at the CYP27B1 variant rs703842 were associated with increased risk of MS (Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene) 2009). Alleles at rs703842 were associated with circulating 25(OH)D levels in a study comparing twins concordant or discordant for MS (Orton et al. 2008).
Rodent Models
Rodent models of VDD during pregnancy reproduce the human condition by depleting the maternal diet of vitamin D and establishing an in utero environment deficient in vitamin D. Mouse dams were fed diets lacking vitamin D (0 IU/g of vitamin D) or gradient levels of reduced vitamin D (described below) to induce VDD during critical windows of pre- and perinatal development. VDD effects were assessed by comparing mice on VDD diet to mice on vitamin D sufficient diets. The exact amount of vitamin D in sufficient diets varies by diet. Various rodent strains have been tested depending on the phenotype of interest to be examined. Outbred (not genetically identical) strains such as Sprague-Dawley rats, Wistar rats, or Swiss Webster mice were presumably used to maximize phenotypic robustness, while genetically identical (inbred) mouse strains such as C57BL/6, BALB/c, or BTBR were likely used to ensure biological reproducibility. Strains from recombinant inbred mouse lines such as the Collaborative Cross (CC) both maximize phenotypic robustness and ensure reproducibility(Churchill et al. 2004). Lines carrying targeted/naturally occurring genetic mutations in genes of interest have also been used to elucidate effects of specific genetic changes in response to VDD.
Learning and Memory
Progeny of VDD C57BL/6J dams exhibited later in life impairments in learning and memory, as assessed by an olfactory-based test, and smaller lateral ventricles (Fernandes de Abreu et al. 2010). Pre- and post-natal VDD increased learning and memory deficits and resulted in elevated depressive-like and anxiety-like behaviors in male mice (Fu et al. 2017). Similar learning and memory deficits were reported in male rats subjected to VDD during the perinatal period (Yates et al. 2018). Deviations in pup ultrasonic vocalizations reduced social behaviors, and increase stereotypical behaviors were also detected in this group, which might be attributed to reduced maternal care provided by VDD dams. One report, however, suggests inbred mice differ in prenatal VDD effects on later neurobehavioral disruptions (Langguth et al. 2018). Assessment of the effect of prenatal VDD on autism-related behaviors (ultrasonic vocalizations, social interaction, hyperlocomotion, grooming, rearing, and spatial learning and memory) in several inbred mouse models demonstrate significant differences between strains for the behaviors examined (Langguth et al. 2018). For example, the C57BL/6J strain and (C57BL/6J x BTBR)F1 hybrid strain were seemingly susceptible to prenatal VDD, while the BALB/c and BTBR mice were less so. C57BL/6J offspring exhibited decreased preference for novel social interactions and the F1 offspring exhibiting hypolocomotive behavior when tested in an open field test (Langguth et al. 2018). Such behavioral deficits though were absent in the other two strains. The collective data suggest offspring sex and genotype can interact with prenatal VDD to influence later neurobehavioral outcomes, including cognitive and social impairments.
Neuro-affective disorders
Juvenile progeny of VDD Sprague Dawley rat dams treated from mating until weaning showed heightened anxiety-like behaviors, as measured by significantly increased grooming frequency compared to other groups when tested in the elevated plus maze (EPM, measures anxiety-like and exploratory behaviors) (Pan et al. 2014). Interestingly, juvenile offspring of dams provided an excessive vitamin D diet (10.0 IU/g) also showed significantly increased grooming frequency during social behavior and learning tests (Pan et al. 2014). Effects of maternal VDD on anxiety-related behavior appear to be independent of the hypothalamic-pituitary-axis (HPA) with one study showing normal function in rats born to VDD dams (Eyles et al. 2006). Instead, the fetal mesencephalon and dopaminergic neurons might be affected by Nurr1 and p57Kip2 expression reduced in this brain region of this group (Cui et al. 2010).
Emerging Zebrafish Models
Initial evidence for a canonical role for VDR and the vitamin D endocrine system in calcium ion homeostasis and skeletal maintenance has been demonstrated for some teleost species (Eyles et al. 2005). It is likely that aquatic vertebrates also exhibit non-calcemic roles of vitamin D, including induction of xenobiotic metabolism, regulation of cell proliferation and differentiation, regulation of neurodevelopment and immune system function, (Nagpal et al. 2005) (Kollitz et al. 2016). Most teleost species, express two distinct VDR paralogs (VDRα and VDRβ) (Howarth et al. 2008). These receptors exhibit similar functionality to mammalian VDRs, having high-affinity ligand binding and potent receptor transactivation efficacies with 1,25(OH)2D and lithotomic acid as ligands (Kollitz et al. 2016).
Emerging zebrafish models of VDD during development pose as valuable tools to study the effects of VDD on neurobehavioral health. Small fish models offer unique advantages over traditional rodent models, including low maintenance and husbandry costs, high fecundity, and ease of developing genetic mutants with the added benefit of reduced animal welfare concerns (Planchart et al. 2016). In this regard, the zebrafish model has become a highly utilized animal model for investigating molecular mechanisms of neurodevelopment, cardiovascular disease, and obesity.
With regards to neurodevelopment, zebrafish models can be used to assess important behavioral phenotypes due to their well-defined behavioral responses at larval and adult stages, skin transparency during embryonic and early larval developmental stages, and a large number of genetic mutants available for testing (Norton 2013). More importantly, since zebrafish have external fertilization and development, a DOHaD zebrafish model of VDD will specifically allow for dissecting important effects of embryonic & early developmental VDD that are independent of maternal metabolism.
METABOLIC HEALTH
Human Studies of Later Life Effects of VDD
In 2012, the WHO estimated that 2.8 million deaths were attributable to obesity and its metabolic sequelae every year (WHO 2016). Although the etiology of obesity is often simplified as an issue of excess caloric intake, there is mounting evidence that in utero environment can determine offspring susceptibility to obesity and related metabolic complications in later life (Saffery & Novakovic 2014).
There is no universally-accepted definition of the broad term “metabolic health”, however, a combination of assessment criteria including measurements of body weight/composition, blood pressure, serum lipid profile, insulin resistance (IR), and systemic inflammation are frequently used for clinical and research purposes (Phillips & Perry 2013). When using these criteria as terms in searching the existing literature, there exists only a paucity of data on the influence of maternal VDD on offspring metabolic health beyond the neonatal/first year of life. The findings are mixed but provide some evidence for a relationship between poor maternal vitamin D status and abnormal metabolic health outcomes such as excess adiposity and insulin resistance.
In the Southampton Women’s Survey of 977 pregnant women and their offspring, lower pregnancy 25(OH)D concentrations were significantly-linked to higher fat mass assessed at six years of age (Crozier et al. 2012). Similarly, findings from the Amsterdam Born Children and their Development (ABCD) study of 1882 mother-child pairs observed an inverse relationship between maternal 25(OH)D and offspring percent body fat at five years of age (Hrudey et al. 2015). This same study also showed inverse associations between maternal 25(OH)D and markers of IR in children whose mothers were overweight during pregnancy (Hrudey et al. 2015). Maternal VDD in another large birth cohort involving Indian children likewise predicted higher IR at 9.5 years old (Krishnaveni et al. 2011).
Not all cohort data support a connection between pregnancy vitamin D status and metabolic health. In a longitudinal prospective UK study, a weak inverse association was detected between circulating 25(OH)D concentrations during pregnancy and offspring C reactive protein measured in childhood and adolescence (Williams et al. 2013). However, no consistent associations were detected between pregnancy serum 25(OH)D concentrations and serum lipids, fasting glucose or fasting insulin at either time point (Williams et al. 2013). Neither the Danish D-Tect Case-Cohort Study or the 1988–89 Danish Cohort Study found associations between 25(OH)D concentrations at birth and risk of being overweight at seven years of age (Jensen et al. 2017); or associations between maternal 25(OH)D and offspring cardio-metabolic risk factors (body weight, serum lipids, inflammatory markers, and IR) in a 20 year follow-up (Rytter et al. 2016).
Metabolomic evidence from a randomized controlled trial
Recently, metabolomic data were used to identify downstream metabolites that are influenced by or interact with 25(OH)D (Blighe 2017). They used metabolomic data from 3-year-olds enrolled in the Vitamin D Antenatal Asthma Reduction Trial (VDAART) (Litonjua et al. 2014). They identified three distinct subsets/clusters of biologically meaningful metabolic profiles that were distinguished by concentrations of inflammatory mediators (predominantly fatty acids and amines), and maternal vitamin D status postsupplementation. Individuals either had (1) high fatty acids, high amines, and low maternal vitamin D status; (2) high amines, intermediate fatty acids, and normal maternal vitamin D status; or (3) low fatty acids and amines and normal maternal vitamin D status (Blighe 2017). Importantly, maternal vitamin D status had a significant influence on cluster membership. This finding suggests in utero vitamin D exposure may be an important predictor of susceptibility to inflammation and other undesirable metabolic disorders later in life (Blighe 2017).
Important considerations
Maternal weight and fetal growth must be considered in the interpretation of offspring metabolic data collected later in life. Maternal obesity is an independent risk factor for the development of obesity and other metabolic complications in offspring later life (Gaillard 2015; Maffeis & Morandi 2017). Furthermore, pregnant women with excess adiposity are more likely to exhibit VDD as there is a well-established inverse association between body fat mass and serum 25(OH)D concentrations (Lagunova et al. 2009; Delle Monache et al. 2018). Thus, characterizing metabolic health effects of maternal obesity vs. those of maternal VDD is confounded and appears to depend on the outcomes examined. The ABCD study found that only the offspring of overweight women exhibited an association between low maternal 25(OH)D (<29.9nmol/L) and increased insulin resistance; and there were no interactive effects of pre-pregnancy BMI in the association between low maternal 25(OH)D and increased % body fat in offspring (Hrudey et al. 2015).
One theme dominating the literature on maternal VDD is its influence on fetal growth. Although, the specific effects of maternal VDD on fetal growth are dependent on the severity and timing of the deficiency, the preponderance of data show VDD is associated with decreased birth weight and higher risk for ‘small for gestational age’ (SGA) (Leffelaar et al. 2010; Burris et al. 2012; Schneuer et al. 2014; Eckhardt et al. 2015; Miliku et al. 2016; Bi et al. 2018). This influence complicates the interpretation of results examining the prenatal programming effects of maternal VDD since fetal growth restriction, regardless of its origin, and its subsequent “catch-up” weight gain are also well known to be related to metabolic health complications (Hales & Ozanne 2003). Thus the application of animal models can be quite useful in elucidating these complex relationships (Hales & Ozanne 2003).
Rodent models
Support for the role of maternal VDD in fetal programming of later metabolic disorders has been documented in several studies using rodent models subjected to in utero VDD (Reichetzeder et al. 2014). The majority of these investigations show that offspring of mothers with low serum 25(OH)D during pregnancy present with abnormal metabolic phenotypes later in life most commonly increased fat mass, insulin resistance, hypertension, and/or altered fat metabolism.
In one of the earliest transgenerational mouse vitamin D fetal programming studies, compared with controls, 6-month-old F1 generation offspring of VDD dams displayed greater body and epididymal fat mass (Nascimento et al. 2013). These mice also had increased insulin secretion when given an oral glucose challenge, and showed marked hepatic steatosis. An in utero VDD Sprague-Dawley rat model expanded these findings showing a positive correlation between serum inflammatory markers and HOMA-IR in adult (16 week old) offspring, suggesting that IR might be a result of persistent inflammation (Zhang et al. 2014). Furthermore, the finding that progeny of VDD C57BL/6 dams exhibited impaired pancreatic development (smaller) and islet structure (smaller with diminished beta cell mass although overall morphology remained intact) and reduces insulin secretion, suggests that impaired insulin sensitivity in VDD offspring is due to alterations in structure and metabolism of the pancreas (Maia-Ceciliano et al. 2016). In support of this conclusion, VDD offspring exhibited a decrease in protein expression of PDK-1, an essential mediator of insulin-signaling in beta-cells (Maia-Ceciliano et al. 2016).
1,25 (OH)2 vitamin D is well-known to inhibit the renin-angiotensin system through suppression of renin secretion in the kidney (Li et al. 2002) and in pancreatic islets (Cheng et al. 2011). However, there are limited data available on the effects of maternal VDD on kidney function. A few rodent studies show elevation in blood pressure in adult offspring of VDD dams (Nascimento et al. 2012) (Meems et al. 2016). This increased blood pressure in progeny of VDD Swiss Webster dams was shown to be related to significant changes in kidney development (poor maturation of glomeruli) and decreased expression of the podocin protein, a key component of the renin-angiotensin system (Nascimento et al. 2012). Remarkably, both the F1 generation and F2 generation were negatively affected by maternal VDD (F0 generation). Alternatively, a Sprague-Dawley rat model demonstrated that increased susceptibility for hypertension in offspring of VDD parents is linked to impaired endothelial relaxation in the large vessels and that reduced Panx1 gene expression may contribute to the observed loss of endothelial relaxation (Meems et al. 2016).
Another condition related to maternal VDD and metabolic disturbances in offspring is nonalcoholic fatty liver disease (NAFLD) (Hyppönen & Boucher 2018). Since both vitamin D status and NAFLD are associated with excess adiposity and IR, interest in examining their potential links has grown. This interest has extended into potential DOHaD effects. A recent study used a VDD Wistar rat model to explore this area. F1 generation adult (34-weeks-old) female offspring exhibited severe hepatic steatosis, abnormal lipid metabolism, increased markers of inflammation and oxidative stress, and down-regulation of hepatic Ppara and Ucp2 expression (Sharma et al. 2017). Interestingly, male F1 offspring were affected to a lesser extent with only mild hepatic steatosis but showed similar downregulation of Ucp2 and the positive regulator of oxidative stress superoxide dismutase 1 (Sod1), demonstrating sex differences.
Adipogenesis and DOHaD effects
There is evidence that in utero adipogenic programming may be associated with obesity later in life (Desai & Ross 2011). In most mammals, including humans, adipose tissue forms throughout the lifespan (Rosen et al. 2000). Adipogenesis during fetal development is a complex, yet tightly-regulated process involving the proliferation and differentiation of progenitor cells into preadipocytes (vs. other cell types such myocytes), and eventually into mature adipocytes when stimulated (Desai et al. 2013). Perturbations to the intrauterine environment caused by suboptimal maternal nutrition (e.g., restricted energy and protein intakes) have been shown to lead to dramatic DOHaD effects on this process (Li et al. 2010). The specific consequences of maternal VDD, however, are largely unclear and contradictory, although the importance of VDR signaling in adipogenesis is undeniable as VDR knockout mice exhibit drastic changes in adipose tissue development including reduced adiposity and resistance to diet-induced obesity (Narvaez et al. 2009). Moreover, 1,25(OH)2 D has been shown to influence several key adipogenic genes and transcription factors (Ding et al. 2012). Our lab recently completed a mouse study on the molecular mechanisms by which maternal VDD might influence adipose tissue in offspring (Belenchia et al. 2017). In that analysis, relative to controls, 19-week old male mice born to VDD dams experienced rapid weight gain in the early weeks post-weaning and acquired a higher proportion of body fat as depots associated with metabolic complications (visceral fat). Additionally, offspring of VDD dams also had higher expression of perigonadal adipose Pparg, a transcript with integral roles in adipogenesis and lipid storage.
Interestingly, a recent VDR knockout study report did not support a fundamental role for vitamin D in adipogenesis (Schutkowski et al. 2018). While VDR knockouts represent a valuable genetic model of completely obliterated genomic vitamin D activity, these models may not recapitulate the consequences of diet-induced VDD, which better mimic human VDD with loss of both genomic and non-genomic effects, allowing also for potential physiological adaptations to low vitamin D provision (Narvaez et al. 2009; Kong et al. 2013; Narvaez et al. 2013). Therefore, caution should be exercised in comparing dietary and genetic models of VDD including systemic depletion, KO, or disruption of any gene involved in vitamin D metabolism, transport or function.
Emerging Zebrafish models
A putative relationship between vitamin D and obesity in zebrafish has recently been investigated using a cyp2r1−/− mutant line (Peng et al. 2017). Cyp2r1 is a liver microsomal hydroxylase essential for the conversion of vitamin D into 25(OH)D. Human genetic variants in CYP2R1 have been correlated with serum 25(OH)D concentrations in recent genome-wide association studies (Ahn et al. 2010; Wang et al. 2010)(Ahn et al., 2010; Wang et al., 2010). Deficits in this metabolic conversion would be expected to limit serum levels of 1,25(OH)2D since 25(OH)D is the direct precursor. However, despite a >50% reduction in circulating 25(OH)D concentrations in mouse cyp2r1−/− mutants, 1,25(OH)2D levels in cyp2r1−/− mutants were seemingly unaffected and mice were relatively healthy (Zhu et al. 2013). This finding strongly implicates additional contributors to this metabolic step in vitamin D activation. Additionally, there are likely localized and tissue-specific differences in 1-alpha-hydroxylase activities that may impart specified paracrine or autocrine functionalities like those observed in human brain pericytes and the epidermis which exhibit functionally active CYP2R1 enzyme activities (El-Atifi et al. 2015; Christakos et al. 2016).
In contrast to the mouse cyp2r1−/− mutants, zebrafish cyp2r1−/− mutants exhibited an almost 70% reduction in circulating 1,25(OH)2D that was completely rescued by 25(OH)D treatment (Peng et al. 2017). Zebrafish cyp2r1−/− mutants also exhibited an obesity-like phenotype with increased central adiposity (e.g., visceral fat). However, no significant differences in liver triglyceride concentrations, bone density or bone volume were observed (Peng et al. 2017), likely due to the incomplete depletion of 1,25(OH)2D. While these studies do suggest that vitamin D insufficiency may precede obesity, a causal mechanism has not been established. The ability of vitamin D to impact both preadipocytes and mesenchymal stem cells differentiation towards adipocytes may be one putative mechanism however there are conflicting results on the pro or anti-adipogenic effects of vitamin D depending upon the model utilized (Abbas 2017). Differing effects of VDD on obesity between the mouse and zebrafish cyp2r1−/− mutant models highlights the value of assessing and comparing multiple animal models of VDD for the purpose of understanding human health.
EVIDENCE FOR THE ROLE OF EPIGENETIC MECHANISMS
Epigenetic regulation of the genome through changes in DNA and histone modifications and noncoding RNA is an essential component of fetal development, and both the non-genomic and genomic actions of vitamin D can affect such responses. During development, dynamic changes in epigenetic markers such as DNA methylation, DNA hydroxymethylation, posttranslational covalent modifications (e.g., addition of methyl or acetyl groups to histone residues), and numerous types of short and long RNAs regulate the transcriptional activity of genes as cells acquire specific functions. Although select epigenetic marks are malleable throughout development, a subset of epigenetic states is programmed early on and stably maintained into adulthood. These epigenetic programs regulate essential cellular functions including cell metabolism, survival, proliferation, and activity (Hemberger et al. 2009). Environmental dysregulation leading to the inability to establish or maintain these essential stable epigenetic programs has significant implications for long term health outcomes (Walker & Ho 2012; Martinez et al. 2015; Chen et al. 2016; Faa et al. 2016).
The role of diet in modulating epigenetic state is well documented. Individual nutrients can act as donors of epigenetic markers, cofactors of epigenetic regulatory enzymes, and signaling molecules of epigenetic regulation of transcription (Ideraabdullah & Zeisel 2018). Epigenetic dysregulation as a result of maternal restriction of individual macro- and micro-nutrients has been demonstrated (Ideraabdullah & Zeisel 2018). In some cases, this affects multiple generations (Wolff et al. 1998; Radford et al. 2014; Nowacka-Woszuk et al. 2018). Developing research shows that maternal vitamin D status during pregnancy may determine offspring epigenetic state although whether there are specific windows of susceptibility has not been examined. As illustrated in Figure 2, VDD-induced epigenetic dysregulation during development represents a potential mechanistic link between maternal VDD and long-term offspring health.
Figure 2.
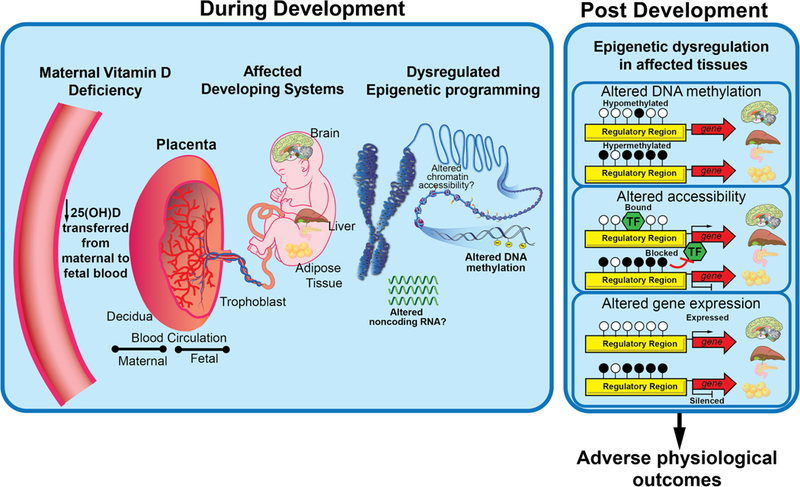
Model of in utero VDD-induced epigenetic effects on long-term health outcomes in offspring. Potential pathway of events connecting maternal vitamin D deficiency to offspring health outcomes via epigenetically regulated organ/tissue dysfunction. Decreased availability of maternal 25(OH)D leads to decreased 25(OH)D entering the placenta and thus decreased availability of the 1,25(OH)2D required for genomic (VDR) and non-genomic signaling responses in the fetus. Consequently, fetal VDD potentially disrupts epigenetic programming during development of target organs/tissues such as brain, liver, and adipose tissue. Dysregulated epigenetic programming leads to long term altered epigenetic states, which as shown, can disrupt genomic accessibility of regulatory machinery and downstream gene expression resulting in adverse long-term health outcomes in offspring, including neurobehavioral and metabolic disorders.
Human studies
Human studies measuring the relationship between maternal vitamin D status and offspring epigenetic states have had differing results. A study performed in cohorts from Norway and the United Kingdom failed to find significant associations between maternal vitamin D status during the second trimester and cord blood DNA methylation at birth (Suderman et al. 2016). However, a more recent case/control study of a Midwestern US cohort, reported that maternal supplementation with vitamin D (3,800 IU/day vs. 400 IU/day) during pre- and post-natal fetal development (starting in the second trimester) resulted in numerous DNA methylation changes in infant leukocytes (Anderson et al. 2018). These changes localized to genes involved collagen metabolism, apoptosis, lung and bone development, and immune function including loss of DNA methylation at HLA-A, an antigen of the major histocompatibility complex (Anderson et al. 2018). The differences in outcome between these two studies are possibly due to study design (supplemented vs. basal serum vitamin D concentrations, geographical population, cell type assessed, and cohort size) or differences in VDD prevalence within the cohort. While the Suderman et al. (2016) study reported mean serum 25(OH)D concentrations well within the currently recommended levels of sufficiency (73.5±23.42 ng/ml and 68.3±32.3 ng/ml), Anderson et al. (2018) reported much lower mean serum 25(OH)D concentration that decreased for control mothers (32.4ng/ml to 21.9ng/ml postpartum) and subtly increased for supplemented mothers (31.4ng/ml to 36ng/ml postpartum).
Association between maternal VDD and offspring epigenetic status at imprinted genes was also assessed recently in humans. Imprinted genes play vital roles in pre- and post-natal development and health (including growth, adiposity and brain development). Assessment of mother-infant dyads in the Newborn Epigenetics Study (NEST) study cohort found that maternal 25(OH)D status measured in the first or second trimester, was inversely associated with birthweight z-scores, and positively associated with 1yr old weight-for-length and three-year-old BMI z-score (Neelon et al. 2018). However, no significant association was detected between maternal 25(OH)D concentrations and cord blood DNA methylation levels at imprinted genes H19, IGF2, MEG3, MEG3- IG, MEST, NNAT, PEG3, PLAGL1 and SGCE/PEG10 (Neelon et al. 2018).
These studies were limited to easily accessible biospecimens like maternal, infant or cord blood. Interpreting biological relevance of blood epigenetic changes is challenging due to wide variation in cell composition and often hampered by the lack of specific relevance to physiologically affected tissues. Animal models represent a valuable research tool, allowing for a controlled environment and precise dietary manipulation including assessment of discrete exposure windows and collection of tissues/cells from progressive developmental stages.
Rodent Models
One of the earliest studies in rodents reported that progeny of VDD Sprague Dawley dams exhibited a significant increase in global DNA methylation in adult liver (Zhang et al. 2014). This corresponded with changes in expression of DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b) and glycemic dysregulation including lower glucose tolerance, lower insulin sensitivity and higher levels of hepatic markers of inflammation (Zhang et al. 2014). Further examination implicated the role of epigenetically regulated decreases in mRNA and protein levels of nuclear factor κB inhibitor α (IκBα, a key regulator of NFκB-mediated inflammation) (Zhang et al. 2014).
A later epigenome-wide DNA methylation study in Sprague Dawley rats found that offspring of VDD dams exhibited differential methylation in adipose tissue at >800 loci linked to 305 genes (Wen et al. 2018). Differential expression was detected at 141 of the differentially methylated genes, including two validated lipid metabolism genes, Vldlr and Hif1α. These epigenetic and transcriptional changes were linked to obesity- and metabolic dysfunction- related phenotypes in the offspring (Wen et al. 2018). A separate epigenome-wide DNA methylation study also in Sprague Dawley rats reported that progeny of VDD dams exhibited differential methylation at ten genes in the kidney including gain of methylation at Pannexin-1, Panx1, which is abundantly expressed in heart, skeletal muscle, and vasculature (Meems et al. 2016). No corresponding change in Panx1 expression was detected in kidney, however, expression was altered in aorta but without any corresponding change in DNA methylation.
Our work recently showed that VDD in the mouse during gestation and lactation, followed by vitamin D sufficient diet at weaning, resulted in DNA methylation changes at imprinted loci across two generations (Xue et al. 2016). Furthermore, by comparing offspring from reciprocal crosses of inbred collaborative cross strains (CC001/Unc and CC011/Unc), we also showed that this effect is likely dependent on the maternal strain (Xue et al. 2016). For reciprocal crosses, Cross 1 mice were generated by mating CC001 dams to CC011 sires, while Cross 2 mice were generated by mating CC011 dams to CC001 sires. Thus, the maternal genomes and associated uterine environmental conditions differ between crosses, but the offspring are genetically identical (except at mitochondrial DNA and Y chromosome). First generation offspring from Cross 1 exhibited loss of methylation at H19/Igf2ICR and Grb10DMR in liver and the latter was correlated with increased bodyweight. However, the genetically identical reciprocal cross (Cross 2) offspring showed no change in bodyweight or methylation at H19/Igf2ICR and Grb10DMR but instead showed loss of methylation at IG-DMR in sperm (Xue et al. 2016). It will be important to determine whether the parent of origin dependent outcomes are due to an imprinted effect or a maternal effect and whether bodyweight and adiposity changes are linked to subsequent metabolic dysfunction.
Ex vivo models
The impact of vitamin D status on epigenetic mechanisms beyond DNA methylation has been explored mainly in cell culture. 1,25(OH)2D is proposed to interact with and modulate activity of epigenetic regulatory proteins including chromatin remodeling complexes and histone modifying enzymes (Fetahu et al. 2014). For example, when not bound to its preferred ligand, 1,25(OH)2D, VDR has been reported to associate with repressive chromatin remodelers including NCoR1, SMRT/NCoR2, and HDACs (Malinen et al. 2008). This would suggest that VDD might create an environment that is conducive to repressive epigenetic effects. On the other hand, supplementation in colon cancer cell lines led to increased expression of JMJD3/KDM6B, which demethylates repressive H3K27me2/3 marks (Pereira et al. 2011). However, since global H3K27me3 levels were unchanged (Pereira et al. 2012), functional impact on histone modifications is unclear. Vitamin D has also been proposed to mediate CTCF-dependent chromatin activity since supplementation in a human leukemic monocyte line (THP-1) caused transient genome-wide changes in chromatin accessibility at 8,979 loci, 14% of which overlapped CTCF binding sites (Seuter et al. 2016).
CONCLUSIONS
The current animal and human data provide robust evidence that VDD during development exerts important effects on later offspring neurobehavioral and metabolic health. Furthermore, these studies demonstrate the effectiveness of rodent models and potential for emerging zebrafish models as valuable tools poised to answer key mechanistic questions about developmental time points and in target tissues inaccessible in most human studies. To pinpoint how VDD affects individual organs such as the brain, development of conditional genetic models (e.g., VDR-Cre) in rodents and zebrafish will be useful. Epigenetic mechanisms present a promising link between VDD and offspring metabolic and neurobehavioral health outcomes (Figure 2). Characterizing the impact of VDD-induced epigenetic changes in the genome is important for understanding and developing potential preventative/treatment measures to address VDD-induced effects. The mechanism by which maternal VDD alters offspring epigenetic status and physiological impact of such epigenetic modifications remains uncertain. Nonetheless, human and rodent studies strongly support the role of vitamin D as an effector of DNA methylation with potential dose-dependent effects (Zhu et al. 2016) and the ability to rescue methylation-dependent developmental phenotypes (Liu et al. 2017). Future studies are needed to elucidate windows of susceptibility, effective timing for intervention by supplementation, and genetic population-specific differences in susceptibility. Furthermore, although we have alluded to the impact of VDD on offspring as a direct effect, the indirect role of maternal/intrauterine effects and adverse pregnancy outcomes are also important considerations.
In conclusion, VDD critically affects developmental programming of short and long term offspring metabolic and neurobehavioral health, potentially via epigenetic mechanisms. Whether these effects are due to canonical non-genomic or genomic actions of vitamin D remains unclear. Understanding causal mechanisms leading to adverse health in sons and daughters born to VDD mothers is essential for early diagnoses and improving treatment during pregnancy to prevent later adverse DOHaD effects in at-risk offspring.
ACKNOWLEDGEMENTS
We thank Donald Conner, University of Missouri, for illustrating Figure 1 and 2.
FUNDING AND SUPPORT
This work was supported by the National Institutes of Environmental Health Science (1R01ES025547 to C.S.R., P30ES025128 to S.W.K., and KES023849A to F.Y.I); the UNC Nutrition Obesity Research Center (P30DK056350 to F.Y.I) and the US Environmental Protection Agency (STAR 835541 to S.W.K).
Footnotes
DECLARATION OF INTERESTS
Disclosures: The authors have no conflicts of interest to disclose
REFERENCES
- Abbas MA 2017. Physiological functions of Vitamin D in adipose tissue. The Journal of Steroid Biochemistry and Molecular Biology 165 369–381. (doi: 10.1016/j.jsbmb.2016.08.004) [DOI] [PubMed] [Google Scholar]
- Adams JS & Hewison M 2012. Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase. Archives of Biochemistry and Biophysics 523 95–102. (doi: 10.1016/j.abb.2012.02.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Yu K, Stolzenberg-Solomon R, Simon KC, McCullough ML, Gallicchio L, Jacobs EJ, Ascherio A, Helzlsouer K, Jacobs KB et al. 2010. Genome-wide association study of circulating vitamin D levels. Human Molecular Genetics 19 2739–2745. (doi: 10.1093/hmg/ddq155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Gillespie SL, Thiele DK, Ralph JL & Ohm JE 2018. Effects of Maternal Vitamin D Supplementation on the Maternal and Infant Epigenome. Breastfeeding Medicine : the Official Journal of the Academy of Breastfeeding Medicine 13 371–380. (doi: 10.1089/bfm.2017.0231) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene) 2009. Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nature Genetics 41 824–828. (doi: 10.1038/ng.396) [DOI] [PubMed] [Google Scholar]
- Belenchia AM, Johnson SA, Ellersieck MR, Rosenfeld CS & Peterson CA 2017. In utero vitamin D deficiency predisposes offspring to long-term adverse adipose tissue effects. The Journal of Endocrinology 234 301–313. (doi: 10.1530/JOE-17-0015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beninger RJ 1983. The role of dopamine in locomotor activity and learning. Brain Research Reviews 6 173–196. (doi: 10.1016/0165-0173(83)90038-3) [DOI] [PubMed] [Google Scholar]
- Berridge MJ 2018. Vitamin D deficiency: infertility and neurodevelopmental diseases (attention deficit hyperactivity disorder, autism, and schizophrenia). Am J Physiol Cell Physiol 314 C135–C151. (doi: 10.1152/ajpcell.00188.2017) [DOI] [PubMed] [Google Scholar]
- Bi WG, Nuyt AM, Weiler H, Leduc L, Santamaria C & Wei SQ 2018. Association between vitamin D supplementation during pregnancy and offspring growth, morbidity, and mortality: A systematic review and meta-analysis. JAMA Pediatrics 172 635–645. (doi: 10.1001/jamapediatrics.2018.0302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blighe K 2017. Vitamin D prenatal programming of childhood metabolomics profiles at age 3 y. The American Journal of Clinical Nutrition 106 1092–1099. (doi: 10.3945/ajcn.117.158220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar LM, Catov JM, Simhan HN, Holick MF, Powers RW & Roberts JM 2007. Maternal vitamin D deficiency increases the risk of preeclampsia. The Journal of Clinical Endocrinology and Metabolism 92 3517–3522. (doi: 10.1210/jc.2007-0718) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannon PM 2012. Vitamin D and adverse pregnancy outcomes: beyond bone health and growth. The Proceedings of the Nutrition Society 71 205–212. (doi: 10.1017/S0029665111003399) [DOI] [PubMed] [Google Scholar]
- Burris HH, Rifas-Shiman SL, Camargo CAJ, Litonjua AA, Huh SY, Rich-Edwards JW & Gillman MW 2012. Plasma 25-hydroxyvitamin D during pregnancy and small-for-gestational age in black and white infants. Ann Epidemiol 22 581–586. (doi: 10.1016/j.annepidem.2012.04.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell JJ 2017. Vitamin D and autism, what’s new? Rev Endocr Metab Disord 18 183–193. (doi: 10.1007/s11154-017-9409-0) [DOI] [PubMed] [Google Scholar]
- Chango A & Pogribny IP 2015. Considering maternal dietary modulators for epigenetic regulation and programming of the fetal epigenome. Nutrients 7 2748–2770. (doi: 10.3390/nu7042748) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Liu H-X, Yan H- Y, Wu D- M & Ping J 2016. Developmental origins of inflammatory and immune diseases. Molecular Human Reproduction 22 858–865. (doi: 10.1093/molehr/gaw036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Li YC, Boucher BJ & Leung PS 2011. A novel role for vitamin D: modulation of expression and function of the local renin-angiotensin system in mouse pancreatic islets. Diabetologia 54 2077–2081. (doi: 10.1007/s00125-011-2100-1) [DOI] [PubMed] [Google Scholar]
- Christakos S, Dhawan P, Verstuyf A, Verlinden L & Carmeliet G 2016. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiological Reviews 96 365–408. (doi: 10.1152/physrev.00014.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W et al. 2004. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nature Genetics 36 1133–1137. (doi: 10.1038/ng1104-1133) [DOI] [PubMed] [Google Scholar]
- Cox MB, Ban M, Bowden NA, Baker A, Scott RJ & Lechner-Scott J 2012. Potential association of vitamin D receptor polymorphism Taq1 with multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 18 16–22. (doi: 10.1177/1352458511415562) [DOI] [PubMed] [Google Scholar]
- Crozier SR, Harvey NC, Inskip HM, Godfrey KM, Cooper C, Robinson SMSWS Study Group 2012. Maternal vitamin D status in pregnancy is associated with adiposity in the offspring: findings from the Southampton Women’s Survey. The American Journal of Clinical Nutrition 96 57–63. (doi: 10.3945/ajcn.112.037473) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Pelekanos M, Burne TH, McGrath JJ & Eyles DW 2010. Maternal vitamin D deficiency alters the expression of genes involved in dopamine specification in the developing rat mesencephalon. Neurosci Lett 486 220–223. (doi: 10.1016/j.neulet.2010.09.057) [DOI] [PubMed] [Google Scholar]
- Cui X, Gooch H, Petty A, McGrath JJ & Eyles D 2017. Vitamin D and the brain: Genomic and non-genomic actions. Molecular and Cellular Endocrinology 453 131–143. (doi: 10.1016/j.mce.2017.05.035) [DOI] [PubMed] [Google Scholar]
- Delle Monache S, Di Fulvio P, Iannetti E, Valerii L, Capone L, Nespoli MG, Bologna M & Angelucci A 2018. Body mass index represents a good predictor of vitamin D status in women independently from age. Clinical Nutrition. (doi: 10.1016/j.clnu.2018.02.024) [DOI] [PubMed] [Google Scholar]
- Desai M & Ross MG 2011. Fetal programming of adipose tissue: effects of intrauterine growth restriction and maternal obesity/high-fat diet. Seminars in Reproductive Medicine 29 237–245. (doi: 10.1055/s-0031-1275517) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai M, Beall M & Ross MG 2013. Developmental origins of obesity: programmed adipogenesis. Current Diabetes Reports 13 27–33. (doi: 10.1007/s11892-012-0344-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C, Gao D, Wilding J, Trayhurn P & Bing C 2012. Vitamin D signalling in adipose tissue. British Journal of Nutrition 108 1915–1923. (doi: 10.1017/S0007114512003285) [DOI] [PubMed] [Google Scholar]
- Eckhardt CL, Gernand AD, Roth DE & Bodnar LM 2015. Maternal vitamin D status and infant anthropometry in a US multi-centre cohort study. Ann Hum Biol 42 215–222. (doi: 10.3109/03014460.2014.954616) [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Ansary A, Cannell JJ, Bjorklund G, Bhat RS, Dbass Al AM, Alfawaz HA, Chirumbolo S & Al-Ayadhi L 2018. In the search for reliable biomarkers for the early diagnosis of autism spectrum disorder: the role of vitamin D. Metab Brain Dis 33 917–931. (doi: 10.1007/s11011-018-0199-1) [DOI] [PubMed] [Google Scholar]
- El-Atifi M, Dreyfus M, Berger F & Wion D 2015. Expression of CYP2R1 and VDR in human brain pericytes: the neurovascular vitamin D autocrine/paracrine model. Neuroreport 26 245–248. (doi: 10.1097/WNR.0000000000000328) [DOI] [PubMed] [Google Scholar]
- Eyles D, Burne T & McGrath J 2011. Vitamin D in fetal brain development. Seminars in Cell & Developmental Biology 22 629–636. (doi: 10.1016/j.semcdb.2011.05.004) [DOI] [PubMed] [Google Scholar]
- Eyles DW, Burne TH & McGrath JJ 2013. Vitamin D, effects on brain development, adult brain function and the links between low levels of vitamin D and neuropsychiatric disease. Front Neuroendocrinol 34 47–64. (doi: 10.1016/j.yfrne.2012.07.001) [DOI] [PubMed] [Google Scholar]
- Eyles DW, Smith S, Kinobe R, Hewison M & McGrath JJ 2005. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat 29 21–30. (doi: 10.1016/j.jchemneu.2004.08.006) [DOI] [PubMed] [Google Scholar]
- Eyles DW, Rogers F, Buller K, McGrath JJ, Ko P, French K & Burne THJ 2006. Developmental vitamin D (DVD) deficiency in the rat alters adult behaviour independently of HPA function. Psychoneuroendocrinology 31 958–964. (doi: 10.1016/j.psyneuen.2006.05.006) [DOI] [PubMed] [Google Scholar]
- Faa G, Manchia M, Pintus R, Gerosa C, Marcialis MA & Fanos V 2016. Fetal programming of neuropsychiatric disorders. Birth Defects Research Part C: Embryo Today: Reviews 108 207–223. (doi: 10.1002/bdrc.21139) [DOI] [PubMed] [Google Scholar]
- Fernandes de Abreu DA, Nivet E, Baril N, Khrestchatisky M, Roman F & Feron F 2010. Developmental vitamin D deficiency alters learning in C57Bl/6J mice. Behav Brain Res 208 603–608. (doi: 10.1016/j.bbr.2010.01.005) [DOI] [PubMed] [Google Scholar]
- Fetahu IS, Höbaus J & Kállay E 2014. Vitamin D and the epigenome. Frontiers in Physiology 5 164. (doi: 10.3389/fphys.2014.00164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiscaletti M, Stewart P & Munns CF 2017. The importance of vitamin D in maternal and child health: a global perspective. Public Health Rev 38 19. (doi: 10.1186/s40985-017-0066-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischman AR & Cole J 1980. Maternal and fetal serum 1,25-dihydroxyvitamin D levels at term. J Pediatr 97 640–642. [DOI] [PubMed] [Google Scholar]
- Fu L, Chen YH, Chen X, Xu S, Yu Z & Xu DX 2017. Vitamin D deficiency impairs neurobehavioral development in male mice. Physiol Behav 179 333–339. (doi: 10.1016/j.physbeh.2017.07.017) [DOI] [PubMed] [Google Scholar]
- Fudge NJ & Kovacs CS 2010. Pregnancy up-regulates intestinal calcium absorption and skeletal mineralization independently of the vitamin D receptor. Endocrinology 151 886–895. (doi: 10.1210/en.2009-1010) [DOI] [PubMed] [Google Scholar]
- Gaillard R 2015. Maternal obesity during pregnancy and cardiovascular development and disease in the offspring. Eur J Epidemiol 30 1141–1152. (doi: 10.1007/s10654-015-0085-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A, Tamblyn JA, Finn-Sell S, Chan SY, Westwood M, Gupta J, Kilby MD, Gross SR & Hewison M 2018. Vitamin D, the placenta and early pregnancy: effects on trophoblast function. The Journal of Endocrinology 236 R93–R103. (doi: 10.1530/JOE-17-0491) [DOI] [PubMed] [Google Scholar]
- García-Martín E, Agúndez JAG, Martínez C, Benito-León J, Millán-Pascual J, Calleja P, Díaz-Sánchez M, Pisa D, Turpín-Fenoll L, Alonso-Navarro H et al. 2013. Vitamin D3 receptor ( VDR ) gene rs2228570 (Fok1) and rs731236 (Taq1) variants are not associated with the risk for multiple sclerosis: results of a new study and a meta-analysis. PLoS ONE 8 e65487. (doi: 10.1371/journal.pone.0065487) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazier JD & Sibley CP 1995. Calbindin-D9K gene expression in rat chorioallantoic placenta is not regulated by 1,25-dihydroxyvitamin D3. Pediatric Research 37 720–725. (doi: 10.1203/00006450-199506000-00008) [DOI] [PubMed] [Google Scholar]
- Gray TK, Lowe W & Lester GE 1981. Vitamin D and pregnancy: the maternal-fetal metabolism of vitamin D. Endocrine Reviews 2 264–274. (doi: 10.1210/edrv-2-3-264) [DOI] [PubMed] [Google Scholar]
- Gur EB, Gur MS, Ince O, Kasap E, Genc M, Tatar S, Bugday S, Turan GA & Guclu S 2016. Vitamin D deficiency in pregnancy may affect fetal thymus development. Ginekol Pol 87 378–383. (doi: 10.5603/GP.2016.0008) [DOI] [PubMed] [Google Scholar]
- Hales CN & Ozanne SE 2003. The dangerous road of catch-up growth. The Journal of Physiology 547 5–10. (doi: 10.1113/jphysiol.2002.024406) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes JE, Tesic D, Whitehouse AJ, Zosky GR, Smith JT & Wyrwoll CS 2015. Maternal vitamin D deficiency alters fetal brain development in the BALB/c mouse. Behav Brain Res 286 192–200. (doi: 10.1016/j.bbr.2015.03.008) [DOI] [PubMed] [Google Scholar]
- Heaney RP & Skillman TG 1971. Calcium metabolism in normal human pregnancy. The Journal of Clinical Endocrinology and Metabolism 33 661–670. (doi: 10.1210/jcem-33-4-661) [DOI] [PubMed] [Google Scholar]
- Hemberger M, Dean W & Reik W 2009. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nature Reviews Molecular Cell Biology 10 526–537. (doi: 10.1038/nrm2727) [DOI] [PubMed] [Google Scholar]
- Hillman LS & Haddad JG 1974. Human perinatal vitamin D metabolism. I. 25-Hydroxyvitamin D in maternal and cord blood. J Pediatr 84 742–749. [DOI] [PubMed] [Google Scholar]
- Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, Murad MH, Weaver CM & Endocrine S 2011. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology and Metabolism 96 1911–1930. (doi: 10.1210/jc.2011-0385) [DOI] [PubMed] [Google Scholar]
- Hossein-nezhad A & Holick MF 2013. Vitamin D for health: a global perspective. 88 720–755. (doi: 10.1016/j.mayocp.2013.05.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth DL, Law SHW, Barnes B, Hall JM, Hinton DE, Moore L, Maglich JM, Moore JT & Kullman SW 2008. Paralogous vitamin D receptors in teleosts: transition of nuclear receptor function. Endocrinology 149 2411–2422. (doi: 10.1210/en.2007-1256) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrudey EJ, Reynolds RM, Oostvogels AJJM, Brouwer IA & Vrijkotte TGM 2015. The association between maternal 25-hydroxyvitamin D concentration during gestation and early childhood cardio-metabolic outcomes: Is there interaction with pre-pregnancy BMI? PLoS ONE 10 e0133313. (doi: 10.1371/journal.pone.0133313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyppönen E & Boucher BJ 2018. Adiposity, vitamin D requirements, and clinical implications for obesity-related metabolic abnormalities. Nutrition Reviews 76 678–692. (doi: 10.1093/nutrit/nuy034) [DOI] [PubMed] [Google Scholar]
- Ideraabdullah FY & Zeisel SH 2018. Dietary Modulation of the Epigenome. Physiological Reviews 98 667–695. (doi: 10.1152/physrev.00010.2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CB, Lundqvist M, Sorensen TIA & Heitmann BL 2017. Neonatal Vitamin D Levels in Relation to Risk of Overweight at 7 Years in the Danish D-Tect Case-Cohort Study. Obes Facts 10 273–283. (doi: 10.1159/000471881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamali M, Beihaghi E, Mohammadi AA & Asemi Z 2015. Effects of high-dose vitamin D supplementation on metabolic status and pregnancy outcomes in pregnant women at risk for pre-eclampsia. Hormone and Metabolic Research = Hormon- Und Stoffwechselforschung = Hormones Et Métabolisme 47 867–872. (doi: 10.1055/s-0035-1548835) [DOI] [PubMed] [Google Scholar]
- Karamali M, Asemi Z, Ahmadi-Dastjerdi M & Esmaillzadeh A 2016. Calcium plus vitamin D supplementation affects pregnancy outcomes in gestational diabetes: randomized, double-blind, placebo-controlled trial. Public Health Nutr 19 156–163. (doi: 10.1017/S1368980015000609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesby JP, Cui X, Ko P, McGrath JJ, Burne THJ & Eyles DW 2009. Developmental vitamin D deficiency alters dopamine turnover in neonatal rat forebrain. Neurosci Lett 461 155–158. (doi: 10.1016/j.neulet.2009.05.070) [DOI] [PubMed] [Google Scholar]
- Kollitz EM, Zhang G, Hawkins MB, Whitfield GK, Reif DM & Kullman SW 2016. Evolutionary and functional diversification of the vitamin D receptor-lithocholic acid partnership. PLoS ONE 11 e0168278. (doi: 10.1371/journal.pone.0168278) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Chen Y, Zhu G, Zhao Q & Li YC 2013. 1,25-Dihydroxyvitamin D3 upregulates leptin expression in mouse adipose tissue. The Journal of Endocrinology 216 265–271. (doi: 10.1530/JOE-12-0344) [DOI] [PubMed] [Google Scholar]
- Kovacs CS, Woodland ML, Fudge NJ & Friel JK 2005. The vitamin D receptor is not required for fetal mineral homeostasis or for the regulation of placental calcium transfer in mice. AJP: Endocrinology and Metabolism 289 E133–E144. (doi: 10.1152/ajpendo.00354.2004) [DOI] [PubMed] [Google Scholar]
- Krishnaveni GV, Veena SR, Winder NR, Hill JC, Noonan K, Boucher BJ, Karat SC & Fall CHD 2011. Maternal vitamin D status during pregnancy and body composition and cardiovascular risk markers in Indian children: the Mysore Parthenon Study. The American Journal of Clinical Nutrition 93 628–635. (doi: 10.3945/ajcn.110.003921) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagunova Z, Porojnicu AC, Lindberg F, Hexeberg S & Moan J 2009. The dependency of vitamin D status on body mass index, gender, age and season. Anticancer Research 29 3713–3720. [PubMed] [Google Scholar]
- Langguth M, Fassin M, Alexander S, Turner KM & Burne THJ 2018. No effect of prenatal vitamin D deficiency on autism-relevant behaviours in multiple inbred strains of mice. Behav Brain Res 348 42–52. (doi: 10.1016/j.bbr.2018.04.004) [DOI] [PubMed] [Google Scholar]
- Leffelaar ER, Vrijkotte TG & van Eijsden M 2010. Maternal early pregnancy vitamin D status in relation to fetal and neonatal growth: results of the multi-ethnic Amsterdam Born Children and their Development cohort. 104 108–117. (doi: 10.1017/S000711451000022X) [DOI] [PubMed] [Google Scholar]
- Li CCY, Maloney CA, Cropley JE & Suter CM 2010. Epigenetic programming by maternal nutrition: shaping future generations. Epigenomics 2 539–549. (doi: 10.2217/epi.10.33) [DOI] [PubMed] [Google Scholar]
- Li YC, Kong J, Wei M, Chen Z-F, Liu SQ & Cao L-P 2002. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. Journal of Clinical Investigation 110 229–238. (doi: 10.1172/JCI15219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S-J & Kim SH 2014. Vitamin D in cancer: effects of pharmaceutical drugs on the vitamin D pharmacokinetics. Journal of Pharmaceutical Investigation 44 317–328. (doi: 10.1007/s40005-014-0147-y) [DOI] [Google Scholar]
- Litonjua AA, Lange NE, Carey VJ, Brown S, Laranjo N, Harshfield BJ, O’Connor GT, Sandel M, Strunk RC, Bacharier LB et al. 2014. The Vitamin D Antenatal Asthma Reduction Trial (VDAART): rationale, design, and methods of a randomized, controlled trial of vitamin D supplementation in pregnancy for the primary prevention of asthma and allergies in children. 38 37–50. (doi: 10.1016/j.cct.2014.02.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chen Z, Li W, Huang L & Zhang Y 2017. Vitamin D Enhances Alveolar Development in Antenatal Lipopolysaccharide-Treated Rats through the Suppression of Interferon-γ Production. 8 1923. (doi: 10.3389/fimmu.2017.01923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu NQ & Hewison M 2012. Vitamin D, the placenta and pregnancy. Archives of Biochemistry and Biophysics 523 37–47. (doi: 10.1016/j.abb.2011.11.018) [DOI] [PubMed] [Google Scholar]
- Maffeis C & Morandi A 2017. Effect of Maternal Obesity on Foetal Growth and Metabolic Health of the Offspring. Obes Facts 10 112–117. (doi: 10.1159/000456668) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia-Ceciliano TC, Barreto-Vianna AR, Barbosa-da-Silva S, Aguila MB, Faria TS & Mandarim-de-Lacerda CA 2016. Maternal vitamin D-restricted diet has consequences in the formation of pancreatic islet/insulin-signaling in the adult offspring of mice. Endocrine 54 60–69. (doi: 10.1007/s12020-016-0973-y) [DOI] [PubMed] [Google Scholar]
- Malinen M, Saramäki A, Ropponen A, Degenhardt T, Vaïsänen S & Carlberg C 2008. Distinct HDACs regulate the transcriptional response of human cyclin-dependent kinase inhibitor genes to Trichostatin A and 1alpha,25-dihydroxyvitamin D3. Nucleic Acids Research 36 121–132. (doi: 10.1093/nar/gkm913) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall I, Mehta R & Petrova A 2013. Vitamin D in the maternal-fetal-neonatal interface: clinical implications and requirements for supplementation. Journal of Maternal-Fetal and Neonatal Medicine 26 633–638. (doi: 10.3109/14767058.2012.746306) [DOI] [PubMed] [Google Scholar]
- Martinez SR, Gay MS & Zhang L 2015. Epigenetic mechanisms in heart development and disease. Drug Discovery Today 20 799–811. (doi: 10.1016/j.drudis.2014.12.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAree T, Jacobs B, Manickavasagar T, Sivalokanathan S, Brennan L, Bassett P, Rainbow S & Blair M 2013. Vitamin D deficiency in pregnancy - still a public health issue. Maternal & Child Nutrition 9 23–30. (doi: 10.1111/mcn.12014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J 1999. Hypothesis: is low prenatal vitamin D a risk-modifying factor for schizophrenia? Schizophrenia Research 40 173–177. [DOI] [PubMed] [Google Scholar]
- Meems LM, Mahmud H, Buikema H, Tost J, Michel S, Takens J, Verkaik-Schakel RN, Vreeswijk-Baudoin I, Mateo-Leach IV, van der Harst P et al. 2016. Parental vitamin D deficiency during pregnancy is associated with increased blood pressure in offspring via Panx1 hypermethylation. Am J Physiol Heart Circ Physiol 311 H1459–H1469. (doi: 10.1152/ajpheart.00141.2016) [DOI] [PubMed] [Google Scholar]
- Miliku K, Vinkhuyzen A, Blanken LM, McGrath JJ, Eyles DW, Burne TH, Hofman A, Tiemeier H, Steegers EA, Gaillard R et al. 2016. Maternal vitamin D concentrations during pregnancy, fetal growth patterns, and risks of adverse birth outcomes. The American Journal of Clinical Nutrition 103 1514–1522. (doi: 10.3945/ajcn.115.123752) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaei F, Michels KB, Munger K, O’Reilly E, Chitnis T, Forman MR, Giovannucci E, Rosner B & Ascherio A 2011. Gestational vitamin D and the risk of multiple sclerosis in offspring. Annals of Neurology 70 30–40. (doi: 10.1002/ana.22456) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales E, Julvez J, Torrent M, Ballester F, Rodríguez-Bernal CL, Andiarena A, Vegas O, Castilla AM, Rodriguez-Dehli C, Tardón A et al. 2015. Vitamin D in Pregnancy and Attention Deficit Hyperactivity Disorder-like Symptoms in Childhood. Epidemiology 26 458–465. (doi: 10.1097/EDE.0000000000000292) [DOI] [PubMed] [Google Scholar]
- Munger KL, Aivo J, JAMA KH2016 Vitamin D status during pregnancy and risk of multiple sclerosis in offspring of women in the Finnish maternity cohort. Jamanetwork.com [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal S, Na S & Rathnachalam R 2005. Noncalcemic actions of vitamin D receptor ligands. Endocrine Reviews 26 662–687. (doi: 10.1210/er.2004-0002) [DOI] [PubMed] [Google Scholar]
- Narvaez CJ, Brunton J, Salinero A, Chittur SV & Welsh JE 2013. Induction of STEAP4 correlates with 1,25-dihydroxyvitamin D3 stimulation of adipogenesis in mesenchymal progenitor cells derived from human adipose tissue. Journal of Cellular Physiology 228 2024–2036. (doi: 10.1002/jcp.24371) [DOI] [PubMed] [Google Scholar]
- Narvaez CJ, Matthews D, Broun E, Chan M & Welsh J 2009. Lean phenotype and resistance to diet-induced obesity in vitamin D receptor knockout mice correlates with induction of uncoupling protein-1 in white adipose tissue. Endocrinology 150 651–661. (doi: 10.1210/en.2008-1118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento FAM, Ceciliano TC, Aguila MB & Mandarim-de-Lacerda CA 2012. Maternal vitamin D deficiency delays glomerular maturity in F1 and F2 offspring. PLoS ONE 7 e41740. (doi: 10.1371/journal.pone.0041740) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento FAM, Ceciliano TC, Aguila MB & Mandarim-de-Lacerda CA 2013. Transgenerational effects on the liver and pancreas resulting from maternal vitamin D restriction in mice. Journal of Nutritional Science and Vitaminology 59 367–374. [DOI] [PubMed] [Google Scholar]
- Neelon SEB, White AJ, Vidal AC, Schildkraut JM, Murtha AP, Murphy SK, Kullman SW & Hoyo C 2018. Maternal vitamin D, DNA methylation at imprint regulatory regions and offspring weight at birth, 1 year and 3 years. International Journal of Obesity (2005) 42 587–593. (doi: 10.1038/ijo.2017.160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton WHJ 2013. Toward developmental models of psychiatric disorders in zebrafish. Frontiers in Neural Circuits 7 79. (doi: 10.3389/fncir.2013.00079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowacka-Woszuk J, Szczerbal I, Malinowska AM & Chmurzynska A 2018. Transgenerational effects of prenatal restricted diet on gene expression and histone modifications in the rat. PLoS ONE 13 e0193464. (doi: 10.1371/journal.pone.0193464) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicki M, Tran S, Chatterjee D & Gerlai R 2015. Inhibition of phosphorylated tyrosine hydroxylase attenuates ethanol-induced hyperactivity in adult zebrafish (Danio rerio). Pharmacology, Biochemistry, and Behavior 138 32–39. (doi: 10.1016/j.pbb.2015.09.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orton S-M, Morris AP, Herrera BM, Ramagopalan SV, Lincoln MR, Chao MJ, Vieth R, Sadovnick AD & Ebers GC 2008. Evidence for genetic regulation of vitamin D status in twins with multiple sclerosis. The American Journal of Clinical Nutrition 88 441–447. (doi: 10.1093/ajcn/88.2.441) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orton S-M, Ramagopalan SV, Para AE, Lincoln MR, Handunnetthi L, Chao MJ, Morahan J, Morrison KM, Sadovnick AD & Ebers GC 2011. Vitamin D metabolic pathway genes and risk of multiple sclerosis in Canadians. J Neurol Sci 305 116–120. (doi: 10.1016/j.jns.2011.02.032) [DOI] [PubMed] [Google Scholar]
- Pan P, Jin DHS, Chatterjee-Chakraborty M, Halievski K, Lawson D, Remedios D, Smetka C, Pinto V, Parra E & Fleming AS 2014. The effects of vitamin D₃ during pregnancy and lactation on offspring physiology and behavior in sprague-dawley rats. Developmental Psychobiology 56 12–22. (doi: 10.1002/dev.21086) [DOI] [PubMed] [Google Scholar]
- Park H, Wood MR, Malysheva OV, Jones S, Mehta S, Brannon PM & Caudill MA 2017. Placental vitamin D metabolism and its associations with circulating vitamin D metabolites in pregnant women. The American Journal of Clinical Nutrition 106 1439–1448. (doi: 10.3945/ajcn.117.153429) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Shang G, Wang W, Chen X, Lou Q, Zhai G, Li D, Du Z, Ye Y, Jin X et al. 2017. Fatty Acid Oxidation in Zebrafish Adipose Tissue Is Promoted by 1α,25(OH)2D3. Cell Reports 19 1444–1455. (doi: 10.1016/j.celrep.2017.04.066) [DOI] [PubMed] [Google Scholar]
- Pereira F, Barbáchano A, Silva J, Bonilla F, Campbell MJ, Munoz A & Larriba MJ 2011. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Human Molecular Genetics 20 4655–4665. (doi: 10.1093/hmg/ddr399) [DOI] [PubMed] [Google Scholar]
- Pereira F, Barbáchano A, Singh PK, Campbell MJ, Munoz A & Larriba MJ 2012. Vitamin D has wide regulatory effects on histone demethylase genes. Cell Cycle (Georgetown, Tex) 11 1081–1089. (doi: 10.4161/cc.11.6.19508) [DOI] [PubMed] [Google Scholar]
- Pertile RAN, Cui X & Eyles DW 2016. Vitamin D signaling and the differentiation of developing dopamine systems. Neuroscience 333 193–203. (doi: 10.1016/j.neuroscience.2016.07.020) [DOI] [PubMed] [Google Scholar]
- Phillips CM & Perry IJ 2013. Does inflammation determine metabolic health status in obese and nonobese adults? The Journal of Clinical Endocrinology and Metabolism 98 E1610–E1619. (doi: 10.1210/jc.2013-2038) [DOI] [PubMed] [Google Scholar]
- Planchart A, Mattingly CJ, Allen D, Ceger P, Casey W, Hinton D, Kanungo J, Kullman SW, Tal T, Bondesson M et al. 2016. Advancing toxicology research using in vivo high throughput toxicology with small fish models. 33 435–452. (doi: 10.14573/altex.1601281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford EJ, Ito M, Shi H, Corish JA, Yamazawa K, Isganaitis E, Seisenberger S, Hore TA, Reik W, Erkek S et al. 2014. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 345 1255903–1255903. (doi: 10.1126/science.1255903) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichetzeder C, Chen H, Foller M, Slowinski T, Li J, Chen YP, Lang F & Hocher B 2014. Maternal vitamin D deficiency and fetal programming--lessons learned from humans and mice. Kidney & Blood Pressure Research 39 315–329. (doi: 10.1159/000355809) [DOI] [PubMed] [Google Scholar]
- Rosen ED, Walkey CJ, Puigserver P & Spiegelman BM 2000. Transcriptional regulation of adipogenesis. Genes & Development 14 1293–1307. [PubMed] [Google Scholar]
- Ross AC 2011. The 2011 report on dietary reference intakes for calcium and vitamin D. Public Health Nutr 14 938–939. (doi: 10.1017/S1368980011000565) [DOI] [PubMed] [Google Scholar]
- Rytter D, Bech BH, Halldorsson TI, Henriksen TB, Grandström C, Cohen A & Olsen SF 2016. Maternal Vitamin D Status at Week 30 of Gestation and Offspring Cardio-Metabolic Health at 20 Years: A Prospective Cohort Study over Two Decades. PLoS ONE 11 e0164758. (doi: 10.1371/journal.pone.0164758) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffery R & Novakovic B 2014. Epigenetics as the mediator of fetal programming of adult onset disease: what is the evidence? Acta Obstetricia Et Gynecologica Scandinavica 93 1090–1098. (doi: 10.1111/aogs.12431) [DOI] [PubMed] [Google Scholar]
- Schmidt RJ, Hansen RL, Hartiala J, Allayee H, Sconberg JL, Schmidt LC, Volk HE & Tassone F 2015. Selected vitamin D metabolic gene variants and risk for autism spectrum disorder in the CHARGE Study. Early Human Development 91 483–489. (doi: 10.1016/j.earlhumdev.2015.05.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneuer FJ, Roberts CL, Guilbert C, Simpson JM, Algert CS, Khambalia AZ, Tasevski V, Ashton AW, Morris JM & Nassar N 2014. Effects of maternal serum 25-hydroxyvitamin D concentrations in the first trimester on subsequent pregnancy outcomes in an Australian population. The American Journal of Clinical Nutrition 99 287–295. (doi: 10.3945/ajcn.113.065672) [DOI] [PubMed] [Google Scholar]
- Senti J, Thiele DK & Anderson CM 2012. Maternal vitamin D status as a critical determinant in gestational diabetes. J Obstet Gynecol Neonatal Nurs 41 328–338. (doi: 10.1111/j.1552-6909.2012.01366.x) [DOI] [PubMed] [Google Scholar]
- Seuter S, Neme A & Carlberg C 2016. Epigenome-wide effects of vitamin D and their impact on the transcriptome of human monocytes involve CTCF. Nucleic Acids Research 44 4090–4104. (doi: 10.1093/nar/gkv1519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SS, Jangale NM, Harsulkar AM, Gokhale MK & Joshi BN 2017. Chronic maternal calcium and 25-hydroxyvitamin D deficiency in Wistar rats programs abnormal hepatic gene expression leading to hepatic steatosis in female offspring. The Journal of Nutritional Biochemistry 43 36–46. (doi: 10.1016/j.jnutbio.2017.01.008) [DOI] [PubMed] [Google Scholar]
- Stednitz SJ, Freshner B, Shelton S, Shen T, Black D & Gahtan E 2015. Selective toxicity of L-DOPA to dopamine transporter-expressing neurons and locomotor behavior in zebrafish larvae. Neurotoxicology and Teratology 52 51–56. (doi: 10.1016/j.ntt.2015.11.001) [DOI] [PubMed] [Google Scholar]
- Suderman M, Stene LC, Bohlin J, Magnus MC, Joubert BR & Wu MC 2016. 25-Hydroxyvitamin D in pregnancy and genome wide cord blood DNA methylation in two pregnancy cohorts (MoBa and ALSPAC). The Journal of Steroid Biochemistry and Molecular Biology 159 102–109. (doi: 10.1016/j.jsbmb.2016.03.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima M, Miyazaki I, Murakami S, Kita T & Asanuma M 2016. l-Theanine protects against excess dopamine-induced neurotoxicity in the presence of astrocytes. 59 93–99. (doi: 10.3164/jcbn.16-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend K, Evans KN, Campbell MJ, Colston KW, Adams JS & Hewison M 2005. Biological actions of extra-renal 25-hydroxyvitamin D-1alpha-hydroxylase and implications for chemoprevention and treatment. The Journal of Steroid Biochemistry and Molecular Biology 97 103–109. (doi: 10.1016/j.jsbmb.2005.06.004) [DOI] [PubMed] [Google Scholar]
- Van Cromphaut SJ, Rummens K, Stockmans I, Van Herck E, Ederveen AGH, Carmeliet P, Verhaeghe J & Bouillon R 2003. Intestinal calcium transporter genes are upregulated by estrogens and the reproductive cycle through vitamin D receptor-independent mechanisms. Journal of Bone and Mineral Research : the Official Journal of the American Society for Bone and Mineral Research 18 1725–1736. (doi: 10.1359/jbmr.2003.18.10.1725) [DOI] [PubMed] [Google Scholar]
- Vinkhuyzen AAE, Eyles DW, Blanken LME, Kruithof CJ, Verhulst F, Tiemeier H & McGrath JJ 2018. Gestational vitamin D deficiency and autism-related traits: the Generation R Study. Molecular Psychiatry 23 240–246. (doi: 10.1038/mp.2016.213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuolo L, Di Somma C, Faggiano A & Colao A 2012. Vitamin D and cancer. Frontiers in Endocrinology 3 58. (doi: 10.3389/fendo.2012.00058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CL & Ho S-M 2012. Developmental reprogramming of cancer susceptibility. Nature Reviews Cancer 12 479–486. (doi: 10.1038/nrc3220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Zhang F, Richards JB, Kestenbaum B, van Meurs JB, Berry D, Kiel DP, Streeten EA, Ohlsson C, Koller DL et al. 2010. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. The Lancet 376 180–188. (doi: 10.1016/S0140-6736(10)60588-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Hong Q, Wang X, Zhu L, Wu T, Xu P, Fu Z, You L, Wang X, Ji C et al. 2018. The effect of maternal vitamin D deficiency during pregnancy on body fat and adipogenesis in rat offspring. Scientific Reports 8 365. (doi: 10.1038/s41598-017-18770-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse AJ, Holt BJ, Serralha M, Holt PG, Kusel MM & Hart PH 2012. Maternal serum vitamin D levels during pregnancy and offspring neurocognitive development. Pediatrics 129 485–493. (doi: 10.1542/peds.2011-2644) [DOI] [PubMed] [Google Scholar]
- Williams DM, Fraser A, Fraser WD, Hyppönen E, Davey Smith G, Deanfield J, Hingorani A, Sattar N & Lawlor DA 2013. Associations of maternal 25-hydroxyvitamin D in pregnancy with offspring cardiovascular risk factors in childhood and adolescence: findings from the Avon Longitudinal Study of Parents and Children. Heart (British Cardiac Society) 99 1849–1856. (doi: 10.1136/heartjnl-2013-303678) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff GL, Kodell RL, Moore SR & Cooney CA 1998. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB Journal 12 949–957. [PubMed] [Google Scholar]
- World Health Organization. Obesity. Situation and trends: Global Health Observatory (GHO) data. 2016. http://www.who.int/gho/ncd/risk_factors/obesity_text/en/ (accessed 15 Sept 2018).
- Xue J, Schoenrock SA, Valdar W, Tarantino LM & Ideraabdullah FY 2016. Maternal vitamin D depletion alters DNA methylation at imprinted loci in multiple generations. Clinical Epigenetics 8 107. (doi: 10.1186/s13148-016-0276-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates NJ, Tesic D, Feindel KW, Smith JT, Clarke MW, Wale C, Crew RC, Wharfe MD, Whitehouse AJO & Wyrwoll CS 2018. Vitamin D is crucial for maternal care and offspring social behaviour in rats. The Journal of Endocrinology 237 73–85. (doi: 10.1530/JOE-18-0008) [DOI] [PubMed] [Google Scholar]
- Zhang H, Chu X, Huang Y, Li G, Wang Y, Li Y & Sun C 2014. Maternal vitamin D deficiency during pregnancy results in insulin resistance in rat offspring, which is associated with inflammation and Ikappabalpha methylation. Diabetologia 57 2165–2172. (doi: 10.1007/s00125-014-3316-7) [DOI] [PubMed] [Google Scholar]
- Zhu H, Bhagatwala J, Huang Y, Pollock NK, Parikh S, Raed A, Gutin B, Harshfield GA & Dong Y 2016. Race/Ethnicity-Specific Association of Vitamin D and Global DNA Methylation: Cross-Sectional and Interventional Findings. PLoS ONE 11 e0152849. (doi: 10.1371/journal.pone.0152849) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JG, Ochalek JT, Kaufmann M, Jones G & DeLuca HF 2013. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proceedings of the National Academy of Sciences 110 15650–15655. (doi: 10.1073/pnas.1315006110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumaraga MP, Medina PJ, Recto JM, Abrahan L, Azurin E, Tanchoco CC, Jimeno CA & Palmes-Saloma C 2017. Targeted next generation sequencing of the entire vitamin D receptor gene reveals polymorphisms correlated with vitamin D deficiency among older Filipino women with and without fragility fracture. The Journal of Nutritional Biochemistry 41 98–108. (doi: 10.1016/j.jnutbio.2016.12.003) [DOI] [PubMed] [Google Scholar]