

Abstract
Radioresistance is a major hurdle in the treatment of head and neck squamous cell carcinoma (HNSCC). Here, we report that concomitant treatment of HNSCCs with radiotherapy and mevalonate pathway inhibitors (statins) may overcome resistance. Proteomic profiling and comparison of radioresistant to radiosensitive HNSCCs revealed differential regulation of the mevalonate biosynthetic pathway. Consistent with this finding, inhibition of the mevalonate pathway by pitavastatin sensitized radioresistant SQ20B cells to ionizing radiation and reduced their clonogenic potential. Overall, this study reinforces the view that the mevalonate pathway is a promising therapeutic target in radioresistant HNSCCs.
Keywords: DNA damage response, HNSCC, mevalonate pathway, radiation sensitivity, senescence, statin
Abbreviations
- a.u.
arbitrary units
- DSB
double‐strand break
- FDPS
farnesyl diphosphate synthase
- FPP
farnesyl pyrophosphate
- GGPP
geranylgeranyl pyrophosphate
- GGPS
geranylgeranyl diphosphate synthase
- HMGCR
3‐hydroxy‐methylglutaryl HMG‐CoA reductase
- HMGCS1
3‐hydroxy‐3‐methylglutaryl‐CoA synthase 1
- HNSCC
head and neck squamous cell cancer
- IPP
isopentenyl diphosphate
- IR
ionizing radiation
- LC‐MS/MS
liquid chromatography tandem mass spectrometry
- LDL
low‐density lipoprotein
- LDLR
low‐density lipoprotein receptor
- LFQ
label‐free quantification
- LRP1
prolow‐density lipoprotein receptor‐related protein 1
- M5P
mevalonate‐5‐phosphate
- M5PP
mevalonate‐5‐diphosphate
- MFI
mean fluorescence intensity
- MVD
mevalonate decarboxylase
- MVK
mevalonate kinase
- NSDHL
NAD(P) dependent steroid dehydrogenase‐like
- PIT
pitavastatin
- PMVK
phosphomevalonate kinase
- SD
standard deviation
- STR
short tandem repeats
1. Introduction
Radiotherapy remains one of the most widely used treatments for cancer as nearly half of patients receive it alone or in combination with other types of therapies (Delaney et al., 2005). For some tumor types, evidence indicates that dose escalation can improve the efficacy of radiotherapy (Andrews et al., 2004) (Bartelink et al., 2007) (Kuban et al., 2008), but the consequence is an increase in normal tissue toxicity, which has been documented as a major limitation of chemoradiation therapy (Eisbruch et al., 2002).
Head and neck squamous cell carcinomas include a broad category of neoplasms that primarily develop in the oral cavity, pharynx, and larynx and are the sixth leading cause of cancer worldwide with approximately half a million new cases every year (Boyle and Levin, 2008) (Hunter et al., 2005). In the past decade, the survival rate for HNSCC has not significantly changed, with two‐thirds of HNSCC patients presenting with locally advanced disease and five‐year survival rates remaining below 50% (Carvalho et al., 2005). Radical radiotherapy in combination with platinum‐based chemotherapy remains the standard treatment for HNSCC with locoregionally advanced carcinoma (J. Pignon et al., 2000). Common acute toxicities include dysphagia, mucositis, dermatitis, and dysgeusia (altered taste sensation), with xerostomia (reduced salivary output) the most common and most adversely affecting quality of life in patients (Bjordal et al., 2001). Other cytotoxic cancer agents including targeted drugs can be combined with radiotherapy (Bonner et al., 2006) (J.‐P. Pignon et al., 2009), though toxicities may be dose‐limiting and resistance is common and associated with a poor prognosis.
An attractive alternative strategy is to identify agents that are nontoxic and likely to be ineffective on their own but can serve as radiosensitizers (Dickreuter et al., 2016) (Khan et al., 2010) (Leiker et al., 2015) (Su et al., 2013), potentially allowing treatment of patients with safer radiation doses. An ideal outcome would be increasing the efficacy of radiation on the tumor while at the same time decreasing its adverse effects on normal tissue. One promising class of drugs is the statins, a family of chemically related competitive inhibitors of 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase (HMGCR), the first committed enzyme of the mevalonate pathway (Hristov et al., 2009) (Stancu and Sima, 2001) (Alagona, 2010). As the mevalonate pathway serves a key role in cholesterol biosynthesis, statins are commonly used to treat dyslipidemia and to prevent cardiovascular disease. Statins lead to an overall decrease in circulating low‐density lipoprotein (LDL) cholesterol both by inhibiting cholesterol biosynthesis and increasing cholesterol uptake via compensatory upregulation of the LDL receptor. Their broad use and high overall safety have permitted large‐scale studies to identify additional impacts on patients, revealing potential benefits in cancer prevention and treatment. Retrospective studies to identify factors affecting success of radiotherapy for prostate cancer have found that incidental use of statins, as for treatment of dyslipidemia, confers improved responses (Hutchinson and Marignol, 2017). Along with extensive preclinical studies, the clinical data suggest that statins may display cancer selectivity, increasing radiosensitization in tumor cells while protecting normal tissue from late radiation effects.
In earlier work, we identified several statin drugs as candidate radiosensitizers in a screen for ionizing radiation‐induced foci (IRIF), a proxy for DNA double‐strand break (DSB) repair (Labay et al., 2011). Following up on this screen, we tested effects of several statins on irradiated mammary and melanoma tumor cells, finding that lipophilic statins delayed DNA repair and promoted therapy‐induced senescence in vitro and in vivo (Efimova et al., 2017). These results suggested examining the mevalonate pathway as a target to enhance radiation sensitivity in HNSCC, a malignancy where radiation resistance is a common clinical challenge. This stimulated a hypothesis‐driven proteomics analysis comparing two radioresistant HNSCC cell lines, JSQ3 and SQ20B (Weichselbaum et al., 1986, 1988), to a radiosensitive control, SCC61 (Weichselbaum et al., 1986), to explore contributions of the mevalonate pathway to the proteomic signature of radioresistance. Indeed, radioresistance was not only associated with dysregulated mevalonate pathway activity but also increased sensitivity to statins with respect to proliferation, viability, DNA damage response, and radiation sensitization. We found that cholesterol levels were increased in radioresistant cells, along with elevated LDL receptor abundance and uptake of extracellular LDL. Further, consistent with these in vitro results, in patients treated for HNSCC with radiotherapy, incidental use of statins was associated with improved local control of tumors. These studies suggest potential benefit to concomitantly prescribing lipophilic statin drugs along with radiation therapy in order to improve outcomes for head and neck cancers.
2. Materials and methods
2.1. Cell lines, cell culture, and agents
Head and neck squamous cell carcinoma cell lines SCC61 (radiosensitive) derived from a glossal tumor (Weichselbaum et al., 1986), JSQ3 (radioresistant) derived from a nasal vestibule tumor, and SQ20B (radioresistant) derived from a laryngeal tumor (Weichselbaum et al., 1988) were grown in DMEM/F‐12 media (Life Technologies, Grand Island, NY, USA) supplemented with 20% fetal bovine serum (Denville Scientific) and penicillin/streptomycin (100 units·mL−1, 100 mg·mL−1, Life Technologies). Cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2 until approximately 70% confluence was reached. Authenticity was confirmed by short tandem repeats (STR) profiling (Center for Genomic Research, University of Illinois at Chicago). All the cell lines tested negative for mycoplasma contamination.
Pitavastatin calcium (PIT) was obtained from Atomole. Simvastatin, pravastatin, atorvastatin, lovastatin, and rosuvastatin were from the NIH Clinical Collection (BioFocus, Mechelen, Belgium). FTI (FTI‐276) and GGTI (GGTI‐2147) inhibitors were from CalBiochem.
Cellular irradiation was delivered using a 60Co gamma irradiator (GammaCell, MDS Nordion) with a dose rate ranging from 9.4 to 7.6 cGy·s−1, depending on the date of the experiment.
2.2. LC‐MS/MS proteomics
2.2.1. Sample preparation
SCC61, JSQ3, and SQ20B were seeded at 106 cells in 100 mm Petri dishes and were grown under standard conditions for 48 h. Proteins were isolated with M‐PER (Thermo Scientific, Fitchburg, WI, USA) in the presence of protease and phosphatase inhibitors (Thermo Scientific). Thirty microgram of the extract was separated by SDS/PAGE electrophoresis on a 12% MOPS‐buffered gel (Thermo Scientific) run for 10 min at 200V resulting in a ~2‐cm ‘gel plug’. After staining with Imperial Protein Stain (Thermo Scientific), individual samples were excised by razor blade and chopped into ~1‐mm3 pieces. Each sample was washed in distilled H2O and destained using 100 mm NH4HCO3 pH 7.5 in 50% acetonitrile. A reduction step was performed by addition of 100 μL 50 mm NH4HCO3 pH 7.5 and 10 μL of 200 mm Tris (2‐carboxyethyl) phosphine HCl at 37 °C for 30 min. The proteins were alkylated by addition of 100 μL of 50 mm iodoacetamide prepared fresh in 50 mm NH4HCO3 pH 7.5 buffer and allowed to react in the dark at 20 °C for 30 min. The gel fragments were washed in water, then acetonitrile, and vacuum‐dried. In‐gel trypsin digestion was carried out overnight at 37 °C with 1 : 50–1 : 100 enzyme–protein ratio of sequencing grade‐modified trypsin (Promega, Fitchburg, WI, USA) in 50 mm NH4HCO3 pH 7.5, and 20 mm CaCl2. Peptides were extracted first with 5% formic acid, then with 75% ACN: 5% formic acid, combined, and vacuum‐dried.
2.2.2. High‐pressure liquid chromatography (HPLC)
All samples were re‐suspended in Burdick & Jackson HPLC‐grade water containing 0.2% formic acid (Fluka, Muskegon, Michigan, USA), 0.1% TFA (Pierce, Rockford, IL, USA), and 0.002% Zwittergent 3–16 (Calbiochem, San Diego, CA, USA), a sulfobetaine detergent that contributes the following distinct peaks at the end of chromatograms: MH+ at 392, and in‐source dimer [2 M + H+] at 783, and some minor impurities of Zwittergent 3–12 seen as MH+ at 336. The peptide samples were loaded onto a 0.25 μL C8 OptiPak trapping cartridge custom‐packed with Michrom Magic (Optimize Technologies, Oregon City, OR, USA) C8, washed, then switched in‐line with a 20 cm by 75 μm C18 packed spray tip nanocolumn packed with Michrom Magic C18AQ, for a 2‐step gradient. Mobile phase A was water/acetonitrile/formic acid (98/2/0.2), and mobile phase B was acetonitrile/isopropanol/water/formic acid (80/10/10/0.2). Using a flow rate of 350 nL·min−1, a 90 min, 2‐step LC gradient was run from 5% B to 50% B in 60 min, followed by 50–95% B over the next 10 min, 10 min at 95% B, then back to the starting conditions and re‐equilibrated.
2.2.3. LC‐MS/MS analysis
The samples were analyzed via electrospray tandem mass spectrometry (LC‐MS/MS) on a Thermo Q‐Exactive Orbitrap mass spectrometer, using a 70 000 RP survey scan in profile mode, m/z 360–2000 Da, with lockmasses, followed by 20 MS/MS HCD fragmentation scans at 17,500 resolution on doubly and triply charged precursors. Singly charged ions were excluded, and ions selected for MS/MS were placed on an exclusion list for 60 s.
Label‐free quantification (LFQ) sample sets were analyzed as described previously (Truman et al., 2012). All LC‐MS/MS *.raw data files were analyzed with maxquant version 1.5.2.8, searching against the SPROT Human database (Downloaded: 1/07/2016 with isoforms) using the following criteria: LFQ was selected for quantitation with a min of 1 high confidence peptide to assign LFQ intensities. Trypsin was selected as the protease with max miss cleavage set to 2. Carbamidomethyl(C) was selected as a fixed modification. Variable modifications were set to Oxidization (M), Formylation (n‐term), Acetyl (Protein n‐term), Deamidation (NQ), GlyGly (K), HNE (Budach et al., 2005), and Phosphorylation (STY). Orbitrap mass spectrometer was selected using an MS error of 20 ppm and a MS/MS error of 0.5 Da. 1% FDR cutoff was selected for peptide, protein, and site identifications.
Ratios were reported based on the LFQ intensities of protein peak areas determined by MaxQuant and reported in the proteinGroups.txt. Proteins were removed from this results file if they were flagged by MaxQuant as ‘Contaminants’, ‘Reverse’, or ‘Only identified by site’. Three complete biological replicates were performed. LFQ peak intensities were analyzed in each run to determine protein hits that fell into the category of single‐condition‐only hits and retained if they maintained this state across all runs. Proteins with two out of three observations were retained and quantitated based on LFQ intensity. Log2 transformation of ratios was performed, and a significance cutoff of 1.5‐fold change was implemented (±0.58 log2 ratio). Gene Ontology analysis was performed by submitting significant hits (≥log2 0.58 and ≤ log2 −0.58) and those only seen in either radioresistant (SQ20B or JSQ3) or radiosensitive (SCC61) lists to a statistical overrepresentation test using bioinformatics software (Panther 14.0) (The Gene Ontology Consortium, 2016) (Mi et al., 2013) (Muruganujan et al., 2016). Significant gene lists for (SQ20B/SCC61) and (JSQ3/SCC61) were compared to the Homo sapiens reference list using Fisher's exact test for determining P value and a Benjamini–Hochberg procedure to calculate false discovery rate (FDR). Results were sorted by fold‐enrichment for GO_BP (biological pathway), GO_MF (molecular function), GO_CC (cell component), and Reactome Pathway annotations results.
2.3. Western blotting
About 1 × 106 cells were seeded in 10‐cm‐diameter culture dishes, incubated overnight, harvested, and pelleted by centrifugation at 1000 g. Total cell lysates were prepared using ice‐cold RIPA buffer (Thermo Fisher) in the presence of protease inhibitors (Halt protein inhibitor cocktail, Thermo Fisher) including EDTA (Thermo Fisher). Protein concentration was determined by BCA assay (Thermo Fisher). About 20 μg of protein was loaded per well, separated using a 12% Bis‐Tris gel (Life Technologies), and electroblotted onto PVDF membrane (Bio‐Rad, Hercules, CA, USA). Equal loading was verified by actin detection (1:10 000 Sigma‐Aldrich, St. Louis, Missouri, USA, A‐5316). Immunoblotting was performed using a rabbit monoclonal primary antibody targeting HMGCR (1:1000, Abcam, Cambridge, UK, ab174830) followed by anti‐rabbit peroxidase‐conjugated secondary antibody (GE) followed by detection with chemiluminescent peroxidase substrate (ECL Femto, Thermo Fisher). Blot imaging was conducted using an iBright FL1000 Imaging System (Life Technologies).
For HMGCS1 and NSDHL 0.5 × 106 cells were plated in 10‐cm‐diameter culture dishes, and after the indicated treatments, total cell lysates were prepared using M‐PER cell lysis buffer (Thermo) in the presence of protease and phosphatase inhibitors (Thermo). About 20–30 μg of protein was loaded per well, separated using SDS/PAGE gels (Bio‐Rad), and electroblotted onto a PVDF membrane (Millipore, Burlington, MA, USA). Equal loading was verified by actin detection (Sigma‐Aldrich). Immunoblotting for targets was performed using primary rabbit, mouse, or goat anti‐human antibodies and species‐specific peroxidase‐conjugated secondary antibodies (Millipore, Jackson Laboratories) followed by detection with ECL Femto chemiluminescent peroxidase substrate (Thermo). Antibodies: HMGCS1 (1:200, Santa Cruz, sc‐32422), NSDHL (1:2000, Abcam, ab190353), Actin (1:10,000, Sigma‐Aldrich, A‐5316).
2.4. Cholesterol measurement
Total cholesterol content from 0.7 × 106 cells was measured per sample with a colorimetric assay kit (Cell Biolabs, Inc). Measurements were performed for three biological replicates per group. Cells were lysed in chloroform:isopropanol:NP‐40 (7:11:0.1) for 30 seconds using a mini bead beater (BioSpec). Homogenized cells were centrifuged at 15,000 × g for 10 min. The resulting organic phase layer was air‐dried at 50 °C, and the remaining organic solvent was removed by using a vacuum concentrator (Eppendorf) for 30 min. The lipid pellets were resuspended in the 1X assay diluent included in the kit. Following the kit protocol, relative cholesterol concentrations in the samples were determined by absorbance at 570 nm using a microplate reader (Synergy Neo, BioTek).
2.5. Colony formation assay
200 cells per well were plated in 6‐well plates the day before treatment. Pitavastatin was added at the indicated doses 1 h prior to irradiation. Two weeks later, cells were fixed and stained with 0.1% crystal violet (Sigma‐Aldrich) for 5 to 10 min. Wells were rinsed with water, dried, and imaged with a Nikon D70s camera. For quantitation plots, the number of cell colonies with greater than 50 cells was counted manually in each well.
2.6. Cell viability assays
For the ATP luminescence cell viability assay, 10,000 cells per well were plated in 96‐well white polystyrene plates (Thermo) in a final volume of 100 μL. 24 h after plating, cells were treated with different pitavastatin concentrations and some plates were exposed to different radiation doses. 48 h after treatment, an ATP‐based cell viability assay (Cell Titer‐Glo Kit, Promega) was used to measure the relative amount of ATP by luminescence following manufacturer's instructions. Luminescence was measured with a microplate reader (Synergy Neo, BioTek). Three biological replicates were measured.
For the flow cytometry‐based Calcein Violet viability assay, cells were plated overnight in 6‐well culture plates at 0.2 x 106 cells per well in complete culture medium as described above. The following day, cells were treated with pitavastatin (10 μm) for 1 h followed by irradiation (0 or 10 Gy). Cells were then returned to a 37 °C, 5% CO2 environment for 48 h prior to cell viability assay, conducted as follows. Cells were harvested by cell scraper into the medium contained in each culture well. The cell suspensions were transferred to sterile tubes, and cells were collected by centrifugation for 5 min at 1200 x g. Cell pellets were resuspended in Calcein Violet 450 AM viability stain (1 μm, eBiosciences) in 1% BSA‐DPBS for 20 min on ice in the dark. Flow cytometry analysis was then conducted using a Fortessa flow cytometer (BD) equipped with a 405‐nm excitation laser and a 450/50‐nm emission detector. Fluorescence data for 10,000 cells were acquired per sample. Cellular viability data were analyzed using FlowJo software (FlowJo).
2.7. LDL receptor immunostaining and LDL uptake assays
HNSCC cells were plated at 0.2 × 106 cells per well in 6‐well plates for 24 h in complete culture medium. For LDL receptor staining, cells were harvested by cell scraper to preserve cell surface receptors, counted, and resuspended in 0.5% BSA‐DPBS at 0.5 × 106 cells per sample for staining. 0.5% BSA‐DPBS served as the blocking agent for 10 min on ice. A fluorescently labeled antibody targeting LDLR (Abcam clone EP1553Y, Alexa Fluor 647 conjugate) was then added to cells at the titration recommended by manufacturer for 30 min on ice. Sytox Blue viability stain was added at 1 μm for 10 min on ice just prior to flow cytometry analysis. Two biological replicates were analyzed per cell line.
For LDL uptake assays, an LDL uptake assay kit was used according to manufacturer specifications (Cayman Chemical, Ann Arbor, MI, USA). Briefly, cells were plated at 0.1 × 106 cells per well in 6‐well plates for 48 h in complete culture medium. At 48 h, serum‐free medium was exchanged on all samples to provide a lipid‐free growth environment, to which fluorescent LDL‐DyLight 488 (Cayman Chemical) was added at manufacturer recommended dilution (1 : 200). Samples for microscopy were incubated for 6 h, washed 2X with kit assay buffer, and imaged using a fluorescent microscope with an appropriate fluorescence filter set (Zeiss). A set of biological replicates for flow cytometry were incubated for 6 h, harvested by cell scraper, pelleted by centrifugation for 5 min at 1,200 x g, and resuspended in kit assay buffer (Cayman Chemical). 7‐AAD viability stain was added for 10 min on ice just prior to flow cytometry analysis.
Flow cytometry was conducted using a Fortessa cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) equipped with excitation lasers and emission detectors appropriate for the fluorophores used in the experiment. Data for 10,000 cells per sample were acquired using FACSDiva software. Postacquisition analysis was conducted using FlowJo software (FlowJo, LLC).
2.8. Neutral comet assay
To analyze DSB, cells were plated at 0.2 x 106 cells per well in 6‐well plates the day before treatment. PIT was administered at the indicated doses 1 h prior to 10 Gy irradiation. After 20 h, cells were mixed with Comet LM agarose (Bio‐Rad) and single‐cell electrophoresis was performed on CometSlides (Trevigen). Slides were fixed, dried, stained with SYBR Green DNA stain (1/10 000×; Life Technologies), and imaged on an Axiovert 40 with a 20X Plan‐NeoFluar objective and an AxioCam monochromatic fluorescence camera. Images were analyzed using a Comet assay macro for ImageJ software (http://www.med.unc.edu/microscopy/resources/imagejplugins-and-macros/comet-assay).
2.9. Senescence‐associated β‐galactosidase (SA‐β‐Gal) assay
0.5 x 106 cells were plated in a 100‐mm dish the day before treatment. Pitavastatin (10 μm) was administered 1 h prior to 10 Gy irradiation. After 6 days, cells were washed with PBS, harvested by scraping, and resuspended in 1 ml of culture medium. Cells were incubated with Bafilomycin A1 (1 μm, Thermo) for 30 min at 37 °C with rotation to neutralize lysosomal pH. DDAO galactoside (10 μg·mL−1) was added, and cells were incubated for 1 h at 37 °C with rotation. After centrifugation at 4 °C for 5 min at 1200 x g, cells were resuspended in ice‐cold 1% BSA‐DPBS and centrifuged again. Supernatant was discarded, and cells were resuspended in serum‐free media with 1 μm Calcein Violet 450 AM (eBioscience) for 15 min on ice. Stained cells were with examined with a Fortessa flow cytometer and analyzed with FlowJo to measure DDAO to detect SA‐β‐Gal and calcein violet to identify viable cells.
2.10. Patient study population and analysis
We utilized a retrospective head and neck cancer database of 803 patients to identify patients with Stage III‐IVB disease treated for curative intent at the University of Illinois Medical Center at Chicago between 1990 and 2012. Patients treated with medications other than a statin or having Stage I‐II disease were excluded resulting in 518 patients for analysis. Analysis was performed under University of Illinois Medical Center IRB protocol 2011‐1075 in accordance with the ethical standards of the responsible committee on human experimentation and with the Helsinki Declaration of 1999, as revised in 2000. Data were collected from all available physical and electronic medical records. Time to local control (LC) or distant control (RC) was determined from last date of radiotherapy. Patterns of failure were determined as the first failure with any component of local, regional, or distant recurrence, respectively.
2.11. Statistical analysis
For cell‐based assays, statistical significance was determined using the unpaired t‐test (cholesterol assays), unpaired multiple t‐test (ATP viability assays), or Mann–Whitney U‐test (Comet assays and flow cytometry). Most experiments were performed in biological duplicate or triplicate. Flow cytometry experiments comprised n > 3000 viable cells per sample. P value ≤ 0.05 was considered statistically significant. Calculations were performed using Prism software (GraphPad).
For patient data analysis, discrete variables were compared with the Chi‐square test and differences in the medians were assessed using the Wilcoxon rank sum test. We used JMP version 10 (SAS Institute) to perform statistical analysis using two‐sided tests and defining significance as P < 0.05. For univariate and multivariate analyses, we used Cox proportional hazard models to compare differences in survival. Censoring is assumed to be noninformative. Variables with P value < 0.1 on univariate analysis were included on multivariate analysis. Survival curves were plotted using the Kaplan–Meier method, and significance was assessed using the Log Rank test.
2.12. Cholesterol uptake assay
For cholesterol uptake assays, an assay kit was used according to manufacturer's specifications (Cayman Chemical). Briefly, cells were plated at 0.2 x 106 cells per well in 6‐well plates for 24 h in complete culture medium. At 24 h, serum‐free medium was exchanged on all samples to provide a lipid‐free growth environment. NBD cholesterol was then added to the serum‐free media at the recommended dilution (20 μg·mL−1). At this point, inhibitors were added, including PIT (2.5 μm) and U‐18666A (1/1000X manufacturer stock solution, Cayman Chemical). Cells were incubated for 24 h, harvested by cell scraper, pelleted by centrifugation for 5 min at 1200 x g, and resuspended in kit assay buffer (Cayman Chemical) for flow cytometry analysis.
Flow cytometry was conducted using a Fortessa cytometer (Becton Dickinson) equipped with excitation lasers and emission detectors appropriate for the fluorophores used in the experiment. Data for 10,000 cells per sample were acquired using FACSDiva software. Postacquisition analysis was conducted using FlowJo software (FlowJo, LLC).
3. Results
3.1. Proteomic profiling of radioresistant HNSCCs
We set out to uncover molecular determinants of radiation resistance in HNSCCs by performing proteome profiling (Fig. 1A). Radiosensitive (SCC61) and radioresistant (JSQ3 and SQ20B) HNSCC human cancer cell lines were grown under standard culture conditions, and after protein isolation and digestion in biological triplicates, peptides were analyzed by LC‐MS/MS mass spectrometry. Using MaxQuant software and accepting only protein identifications with a minimum FDR of 1%, a total of 4700 different proteins were identified, of which 4392 were present in the SCC61 samples, 4471 in JSQ3, and 4463 in SQ20B (Fig. 1B).
Figure 1.
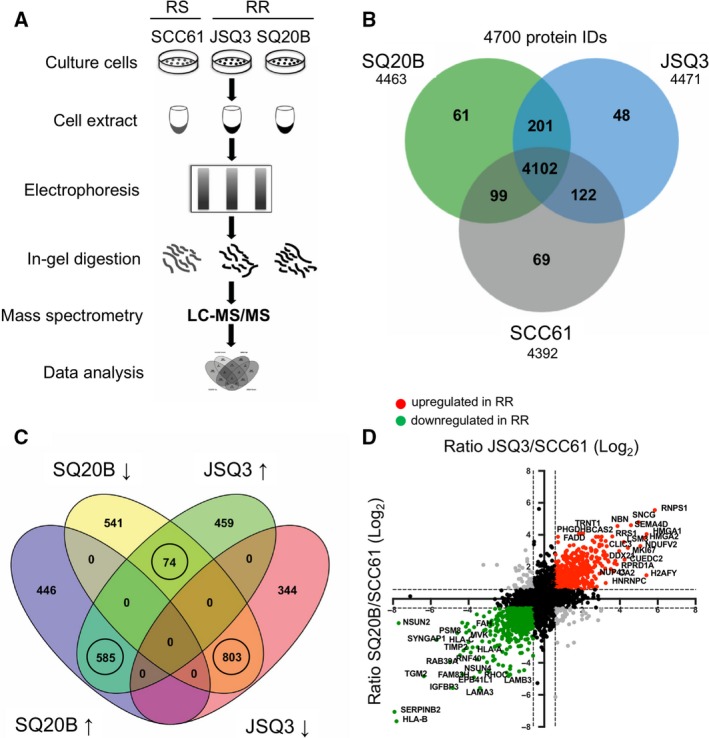
Proteomic analysis reveals a distinct radioresistant HNSCC cell proteome. (A) Protocol schematic for mass spectrometry assay. Whole‐cell protein lysates were prepared from SCC61 (radiosensitive), JSQ3 (radioresistant), and SQ20B (radioresistant) HNSCC cell lines. Proteins were separated by gel electrophoresis, digested with trypsin, and analyzed via LC‐MS/MS. (B) Venn diagram showing proteins uniquely identified in one or two cell lines, or proteins shared between all cell lines. 201 proteins were shared by the two radioresistant cell lines (SQ20B and JSQ3) and 69 proteins were unique to radiosensitive cells (SCC61), while 4102 proteins were shared by all three cell lines. (C) Venn diagram showing distribution of up‐ and downregulated proteins shared between the two radioresistant cell lines, SQ20B and JSQ3, compared to the radiosensitive cell line, SCC61. In general, most proteins found in both radioresistant cell lines were similarly regulated, with 585 shared proteins upregulated and 803 shared proteins downregulated. Only 74 proteins displayed variable regulation. Only circled groups were considered for further analysis. (D) Representation of quantitative mass spectrophotometric intensity ratios of SQ20B/SCC61 (x‐axis) and JSQ3/SCC61 (y‐axis) shown on log2 scale. A 1.5‐fold (0.58 log2) change was considered statistically significant. Upregulated proteins, red. Downregulated proteins, green. Variably regulated proteins, gray; nonsignificant proteins, black. The data show a linear trend, with proteins up‐ or down‐regulated in the SQ20B vs. SCC61 cell line tending to be similarly regulated in JSQ3 vs. SCC61 samples.
In order to analyze the proteomic signature of radioresistant cells, label‐free quantitation (LFQ) was conducted using the mass spectrometry intensities for all the protein hits identified in a minimum of 2 of 3 biological replicates. Radioresistant LFQ values for each protein ID were compared to those of the radiosensitive cell line. A 1.5‐fold (0.58 log2) change was considered statistically significant. We observed a total of 585 proteins significantly upregulated and 803 significantly downregulated in each of the radioresistant cell lines compared with the radiosensitive cell line (Fig. 1C). Individual proteins in these categories were then further evaluated (Fig. 1D). Proteins strongly upregulated in the radioresistant cell lines included RNPS1, SNCG, SEMA4D, HMGA1, HMGA2, and several others. Downregulated proteins in radioresistant cell lines included HLA‐B, SERPINB2, TGM2, IGFBP3, LAMA3, and others. The full proteomic data set was uploaded to the ProteomeXchange repository with identifier PXD007188. A supplementary data file including all significant protein hits and GO pathways was uploaded to the https://figshare.com/ under https://doi.org/10.6084/m9.figshare.8145971.
3.2. Proteomics analysis suggests a role for dysregulated mevalonate pathway activity in HNSCC radioresistance
To determine a proteomic ‘signature’ of radioresistant cells and evaluate potential links to mevalonate pathway activity, we first performed gene ontology (GO) analysis on the hit list of proteins that showed significant variability between radioresistant and radiosensitive cell lines. We then searched for GO terms of relevance to our hypothesis. Metabolism‐related GO terms of significance included glucose metabolic process (GO0006006, P = 2.06 × 10−2 and 5.12 × 10−3), protein metabolic process (GO0019538, P = 4.84 × 10−17 and 3.13 × 10−19), and lipid metabolic process (GO0006629, P = 1.73 × 10−2 and 9.31 × 10−5) (Fig. S1). We also evaluated the significance of GO terms specifically related to mevalonate biosynthesis and metabolism (Fig. 2A), including mevalonate pathway (GO0019287, P = 0.030 for SQ20B/SCC61 and 0.021 for JSQ3/SCC61), cholesterol biosynthetic process (GO0006695, P = 0.075 and 0.028), protein farnesylation (GO0018343, P = 0.043 and 0.033), and protein geranylgeranylation (GO0018344, P = 0.075, and 0.057).
Figure 2.
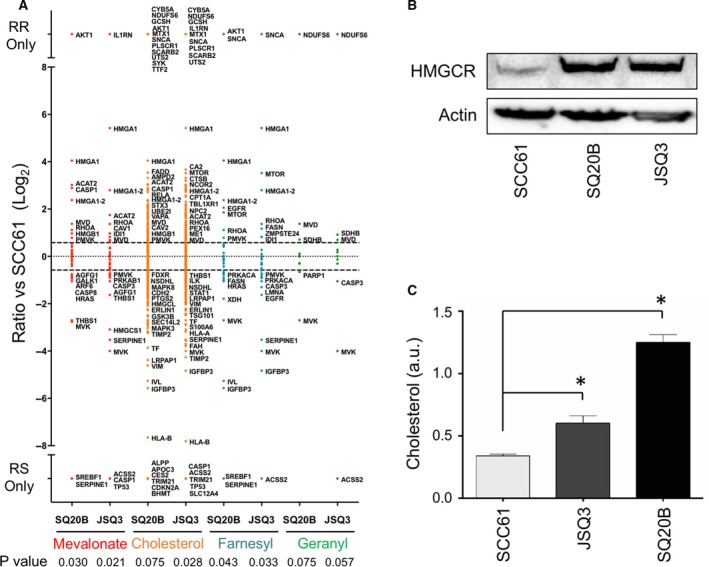
Gene ontology analysis of proteomic data of mevalonate pathway activity and substrate validation. (A) Proteins were separated into functional categories by gene ontology (GO) terms related to mevalonate pathway activity, including mevalonate biosynthetic pathway, cholesterol biosynthesis, protein farnesylation, and protein geranylgeranylation. Protein data are shown as the log2 fold‐change ratio for SQ20B/SCC61 (labeled SQ20B) and JSQ3/SCC61 (labeled JSQ3). (B) Amount of HMGCR protein is increased in radioresistant cells. Cell samples were collected during active growth, lysed, and a western blot was performed. (C) Mean cholesterol levels in SCC61, JSQ3, and SQ20B 48 h after cell passage (during active growth). 0.7 × 106 cells were assayed per sample. Statistical significance was determined using the parametric unpaired t‐test. Error bars: SD, n = 3 biological replicates. Significance is indicated by asterisks (*P < 0.05). Cholesterol levels were significantly increased in the radioresistant cell lines.
Expression profiles of individual protein hits identified to be differentially regulated in the mevalonate pathway (Fig. 2A) suggest dysregulation of this pathway in radioresistant HNSCC. According to LFQ intensities, HMGCS1, the initiator enzyme of the pathway, was significantly downregulated in the JSQ3 radioresistant cells. We confirmed the downregulation of HMGCS1 in both radioresistant cell lines by western blotting (Fig. S2). Following the conversion of acetoacetyl‐CoA to HMG‐CoA by HMGCS1, the enyzme HMGCR converts HMG‐CoA to mevalonate. HMGCR is a key rate‐limiting enzyme in the mevalonate pathway. HMGCR was not detected by LC‐MS/MS analysis, but western blotting revealed the enzyme to be upregulated in both radioresistant cell lines (Fig. 2B). Downstream from HMGCR are the enzymes MVK, PMVK, and MVD, which function to convert mevalonate to several intermediate metabolites (mevalonate‐5‐phosphate, M5P and mevalonate‐5‐diphosphate, M5PP) resulting in the formation of isopentenyl diphosphate (IPP). Of these enzymes, LC‐MS/MS analysis indicated that MVK was downregulated, PMVK was variably regulated, and MVD was upregulated in radioresistant cell lines. For enzymes required for conversion of IPP to geranyl pyrophosphate (GGPS), and then to farnesyl pyrophosphate (FDPS), differences across cell lines were insignificant. The enzyme NSDHL, which mediates the conversion of farnesyl pyrophosphate to cholesterol, was found to be downregulated in both radioresistant cell lines. NSDHL downregulation was confirmed by western blotting (Fig. S2). Taken together, these results implicate dysregulation of mevalonate pathway protein expression in radioresistant HNSCC.
An important metabolic outcome of mevalonate pathway activity is cholesterol biosynthesis. Suggesting a functional consequence of mevalonate pathway dysregulation on cell cholesterol content, analysis of cholesterol in untreated cell lysates (Fig. 2C) revealed significantly higher levels of cholesterol in the radioresistant cell lines (P < 0.05 by unpaired t‐test). These data raised the question whether cholesterol biosynthesis might offer a target to restore radiation sensitivity in radioresistant HNSCC.
3.3. Lipophilic statins inhibit cell proliferation in radioresistant HNSCC cell lines
To ascertain the role of the mevalonate pathway in survival of HNSCC cell lines, we tested a panel of FDA‐approved statin drugs, which block mevalonate generation. Statins included lipophilic pitavastatin (PIT), simvastatin, lovastatin, and atorvastatin, as well as the hydrophilic statins pravastatin and rosuvastatin. Using a colony formation assay (Fig. 3A), we evaluated whether any statin might suppress cellular proliferation in HNSCC cell lines. Indeed, growth of cells was suppressed by PIT at doses as low as 2.5 μm, as well as the other lipophilic statins simvastatin, lovastatin, and atorvastatin at higher doses (10 μm). These effects were particularly dramatic for the radioresistant cell lines. The hydrophilic statins pravastatin and rosuvastatin displayed only minimal effects. Given these results, PIT was selected as the most promising statin for further testing in the study.
Figure 3.
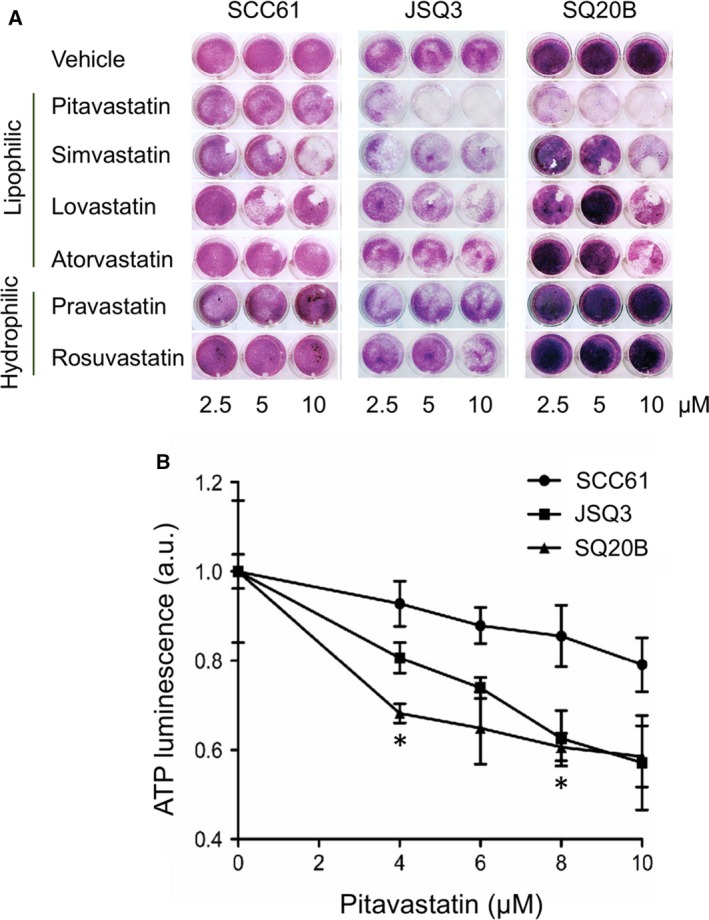
Inhibition of mevalonate pathway by pitavastatin inhibits cellular proliferation and cholesterol levels. (A) Colony formation assay of SCC61, SQ20B, and JSQ3 treated with statins. SCC61, SQ20B, and JSQ3 cells were treated with 2.5, 5, or 10 μm pitavastatin, simvastatin, lovastatin, atorvastatin (lipophilic), or pravastatin or rosuvastatin (hydrophilic) for 1 h prior to IR. After 4 days of culture, crystal violet staining was performed and plates were imaged. Lipophilic statins, notably pitavastatin, and reduced colony formation in cell lines. (B) ATP luminescence viability assay was performed on SCC61, JSQ3, and SQ20B cells to measure viability 48 h after exposing cells to 0, 4, 6, 8, or 10 μm PIT. Three biological replicates were measured; mean ATP luminescence and standard deviation are shown. Viability was reduced by PIT in all cell lines, but markedly so in the radioresistant cell lines, particularly SQ20B. Statistical significance was determined using the parametric unpaired multiple t‐test. Significant conditions are indicated by asterisks (*P < 0.05).
Results of an ATP‐based cell viability assay (Fig. 3B) further demonstrate that radioresistant cell lines have increased sensitivity to PIT (0 to 10 μm), as measured after 48 hours of treatment. While PIT treatment reduced viability in all cell lines, its effects were more dramatic on radioresistant cell lines. At 4 and 8 μm, PIT treatment significantly reduced viability in SQ20B cells compared with the same doses used on SCC61 cells (P < 0.05 by unpaired multiple t‐test, n = 3 biological replicates per sample). At 10 μm, PIT reduced viability in both radioresistant cell lines to approximately 50% of radiosensitive cells.
3.4. Cholesterol uptake via the LDL receptor is elevated in radioresistant HNSCC cell lines
Cellular cholesterol uptake is commonly mediated by endocytosis of low‐density lipoprotein (LDL) through the LDL receptor. Thus, we immunostained for cell surface LDL receptors (Fig. 4A) and examined uptake of fluorescently labeled LDL (Fig. 4B,C). Flow cytometry analysis revealed that levels of LDL receptors were upregulated (LDLRHI) on the surface of radioresistant (JSQ3, 30.6%; SQ20B, 68.8%) vs. (SCC61, 23.9%) HNSCC cells. In turn, the radioresistant cell lines demonstrated higher levels of LDL uptake (JSQ3, 98.4%; SQ20B, 99.0%; vs. SCC61, 48.3%). Differential uptake of fluorescently labeled LDL can also be seen in microscopy images taken with standardized exposure and equal cell density across cell lines (Fig. 4C). Statistical analysis of mean staining intensities for LDLR (Fig. 4D) and LDL‐Dy488 uptake (Fig. 4E) confirmed significant upregulation of these features in the radioresistant cell lines (P < 0.05 by Mann–Whitney U‐test, n > 3000 viable cells per sample).
Figure 4.
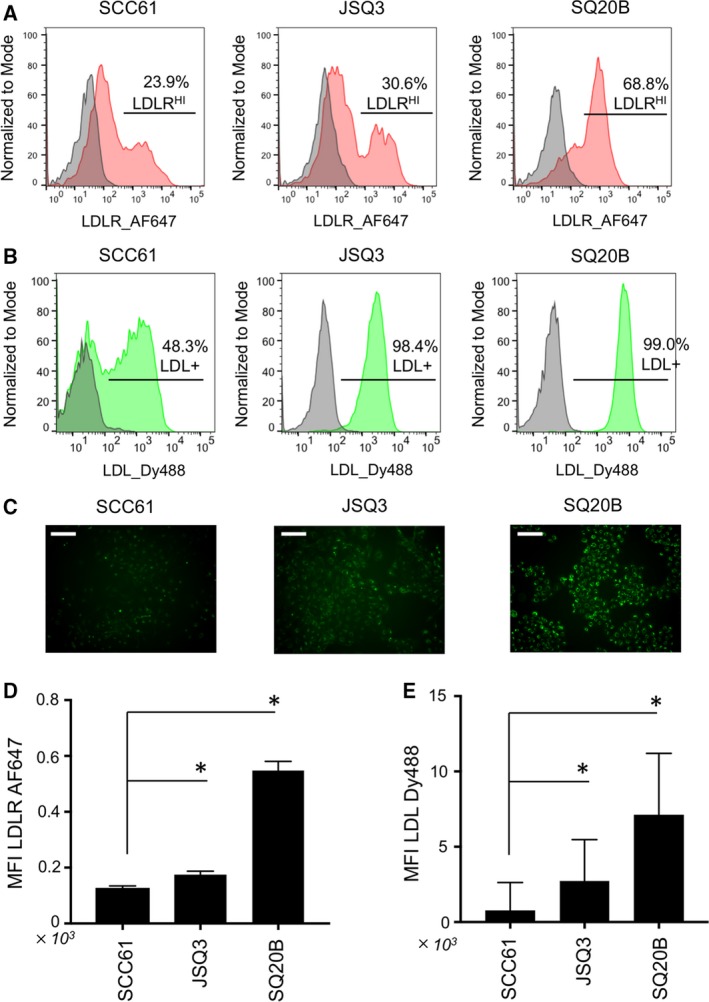
Elevation of LDL receptor levels and LDL uptake characterize radioresistant HNSCC cells. (A) Immunostaining of LDL receptor, LDLR, on living HNSCC cell lines analyzed by flow cytometry. (B) Uptake of fluorescently labeled LDL‐DyLight 488 by HNSCC cells analyzed by flow cytometry. (C) Microscopy images of biological replicates of cell populations treated as shown in (B); scale bars, 20 μm. Elevated levels of LDL receptors and LDL uptake are evident in the radioresistant cell lines JSQ3 and SQ20B. (D) Statistical analysis of LDLR staining intensity. (E) Statistical analysis of LDL‐Dy488 uptake intensity. Mean fluorescence intensities are shown; error bars = SEM. Significance is indicated by asterisks (*P < 0.05). Statistical significance of E and D was determined using Mann–Whitney U‐test.
3.5. Inhibition of mevalonate pathway sensitizes radioresistant HNSCC cell lines to radiation
To demonstrate the differences in radioresponse across cell lines, an ATP‐based cell viability assay was performed. After a single 10 Gy dose of IR, the radioresistant cell lines JSQ3 and SQ20B maintained viability according to cellular ATP assay for at least 50 h after irradiation (Fig. 5A), while the radiosensitive cell line SCC61 displayed a significant loss of viability over the same period (P < 0.05 by unpaired multiple t‐test, n = 3 biological replicates per sample). The ATP assay further demonstrated the radiosensitizing effects of PIT at concentrations from 1 to 10 μm (Fig. 5B), particularly in radioresistant cells. At 10 μm, PIT reduced ATP in irradiated cells by 15% (SCC61), 40% (SQ20B), or 50% (JSQ3). All dose conditions of PIT tested significantly reduced ATP levels in both radioresistant cell lines as compared to the same dose in the radiosensitive cell line (P < 0.05 by unpaired multiple t‐test, n = 3 biological replicates per sample).
Figure 5.
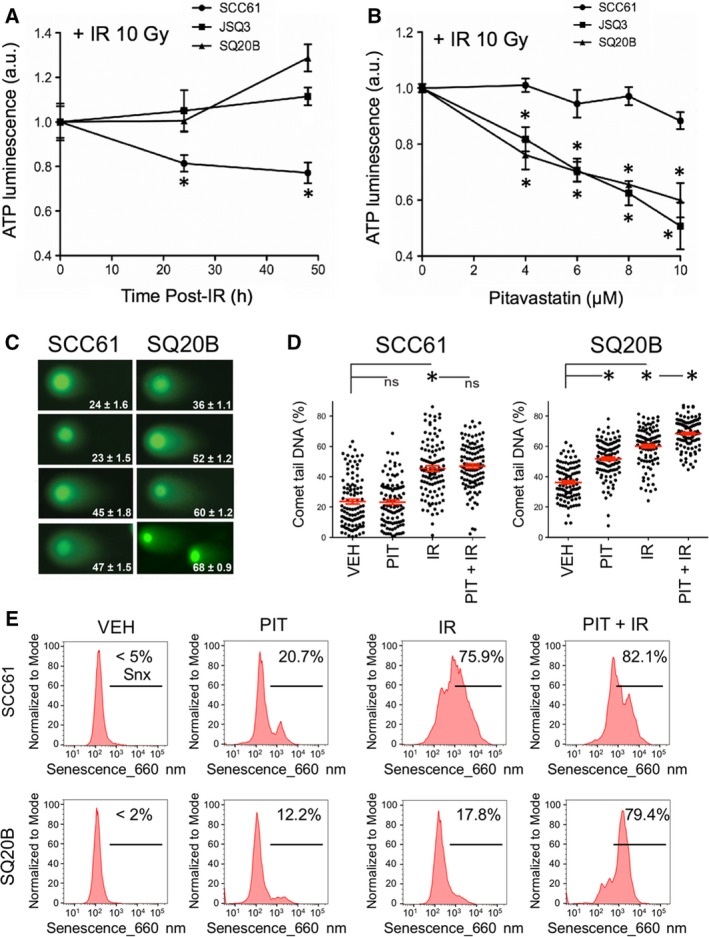
Inhibition of mevalonate pathway by pitavastatin reduces viability, increases DNA damage, and induces senescence upon ionizing radiation in radioresistant HNSCC. (A) ATP‐based cellular proliferation assay of cell lines treated with IR (10 Gy) to demonstrate cell line radiosensitivity. Radioresistant cell lines JSQ3 and SQ20B exhibited lower ATP levels at 48 hr than SCC61 (radiosensitive) cells, which continued proliferating normally (*P < 0.05 vs radiosensitive cells at same time point) Statistical significance was determined using the parametric unpaired multiple t‐test. (B) ATP‐based proliferation assay for cell lines treated with IR (10 Gy) ± PIT (0–10 μm). Radioresistant cell lines demonstrated significantly reduced ATP levels with PIT treatment (*P < 0.05 vs radiosensitive cells at same treatment condition). Statistical significance was determined using the parametric unpaired multiple t‐test. (C) Representative images of ‘Comet’ DNA double‐strand break (DSB) assay data with mean percent of DNA in comet tail ± SEM shown. (D) Plots representing mean percent of DNA in comet tail ± SEM for 100 cells per treatment condition. Significance is indicated by asterisks (*P < 0.05). Statistical significance was determined using Mann–Whitney U‐test. Results indicate that PIT enhances persistent DNA DSBs induced by IR in radioresistant SQ20B cells. (E) Flow cytometric senescence assay data of cells treated with 10 μm of PIT alone (PIT), IR only (10 Gy), or in combination with IR (PIT + IR). Vehicle‐treated cells (VEH) were stained as controls. Cells were analyzed by flow cytometry 6 days after treatment. The combination of PIT + IR dramatically elevated the percent of senescent cells above IR alone in the SQ20B radioresistant cells.
In prior studies of PIT as a radiosensitizer (Efimova et al., 2017), we observed that when MCF7 mammary carcinoma cells were treated with PIT and then irradiated, they displayed a marked delay in double‐strand break (DSB) repair. To examine whether PIT similarly affects DSB repair in HNSCC cells, we performed neutral comet assays after treatment with PIT and/or ionizing radiation (IR) (Fig. 5C,D). As expected, the combination of PIT + IR (68 ± 0.9% comet tail DNA) significantly increased the levels of persistent DSBs over that of IR alone (60 ± 1.2%) in SQ20B cells (P < 0.05 by Mann–Whitney U‐test). This effect was not seen in the sensitive cell line SCC61 (47 ± 1.5% for PIT + IR vs. 45 ± 1.8% for IR only). Further, even without radiation, PIT increased the basal level of DSBs in SQ20B cells. These effects were recapitulated in JSQ3 cells (Fig. S3A). These effects may underlie the loss of proliferation and viability induced by PIT in the radioresistant cell lines.
Along with increasing persistence of DNA damage, our prior work showed that combining PIT with radiation also promoted therapy‐induced senescence in melanoma and breast cancer cells (Efimova et al., 2017). Here, senescence of HNSCC cells treated with PIT with or without IR was measured by a conventional flow cytometry SA‐β‐Gal assay at six days after treatment (Fig. 5E). While treating radioresistant SQ20B cells with PIT or IR alone slightly increased senescence (12.2% for PIT alone, 17.8% for IR alone, compared to < 2% in vehicle only treated SQ20B cells), combining statin with radiation induced senescence in 79.4% of cells. A similar pattern was observed in radioresistant JSQ3 cells (Fig. S3B). The senescence response in radiosensitive SCC61 cells was comparable for combination‐treated cells above levels induced by IR treatment alone (75.9 vs 82.1%). Statistical analysis of mean senescence probe fluorescence (Fig. S3C) supports these conclusions (P < 0.05 for most conditions compared with controls, Mann–Whitney U‐test, n > 3000 viable cells per sample).
To examine effects of treatment with various statins on radioresponse, SCC61, JSQ3, and SQ20B cell lines were treated with statins before irradiation (IR) and assayed for colony formation (Fig. S4). PIT alone decreased colony formation in radioresistant cells. The lipophilic statins simvastatin, lovastatin, and atorvastatin potentiated the effect of radiation on SQ20B cells. The hydrophilic statins tested failed to confer toxicity or radiosensitization in any cell line.
3.6. Clinically approved lipophilic statin drugs are associated with better local tumor control in HNSCC patients
Radiotherapy is widely used in the treatment of head and neck cancers, and our previous results suggest that its combination with statins may have beneficial effects. Consequently, we analyzed 518 HNSCC patients on Stage III–IV treated with radiotherapy of which 53 patients were also taking with a statin drug.
Table 1 describes patient characteristics. Patients receiving statin were significantly older, had higher comorbidity scores but less alcohol use, and fewer T3–T4 tumors and N2‐3 nodal disease. Of the 53 patients incidentally taking a statin drug during their radiotherapy, 41 (77.4%) were taking a lipophilic statin and 3 (5.7%) were taking a hydrophilic statin. For 9 patients, the type of statin was not indicated. Table 2 shows univariate and multivariate analysis of survival. On univariate analysis, improved survival was associated with statin use (HR 0.47, 95% CI, 0.20–0.93; P = 0.03). On multivariate analysis, statin use remained associated with improved survival (HR 0.29, 95% CI, 0.09–0.71; P = 0.004).
Table 1.
Patient characteristics (n = 518).
| Statin (n = 53) | No statin (n = 465) | P value | |
|---|---|---|---|
| Median follow‐up (months) | 18.4 (11.4–38.7) | 18.7 (6.8–58.7) | 0.87 |
| Age | |||
| ≤57 | 14 (26.4%) | 258 (56.0%) | <0.0001 |
| >57 | 39 (73.6%) | 203 (44.0%) | |
| Gender | |||
| Male | 41 (77.4%) | 350 (75.3%) | 0.74 |
| Female | 12 (22.6%) | 115 (24.7%) | |
| Comorbidity | |||
| Medium | 16 (30.2%) | 366 (78.7%) | <0.0001 |
| High‐very high | 37 (69.8%) | 99 (21.2%) | |
| Performance status | |||
| <70 | 5 (9.4%) | 22 (4.7%) | 0.31 |
| ≥70 | 40 (75.5%) | 380 (81.7%) | |
| Not stated | 8 (15.1%) | 63 (13.6%) | |
| Alcohol use | |||
| ≥3 drinks/day | 26 (63.4%) | 276 (76.7%) | 0.06 |
| <3 drinks/day | 15 (36.6%) | 84 (23.3%) | |
| Smoking | |||
| ≥10 pack‐year | 39 (81.2%) | 350 (79.2%) | 0.74 |
| <10 pack‐year | 9 (18.8%) | 92 (20.8%) | |
| T‐stage | |||
| T1–2 | 21 (39.6%) | 119 (25.6%) | 0.04 |
| T3–4 | 32 (60.4%) | 346 (74.4%) | |
| N‐stage | |||
| N0–1 | 31 (58.5%) | 199 (42.8%) | 0.03 |
| N2–3 | 22 (41.5%) | 266 (57.2%) | |
| Primary site | |||
| Oral cavity | 15 (28.3%) | 116 (25.0%) | 0.36 |
| Oropharynx | 18 (34.0%) | 119 (25.6%) | |
| Larynx | 12 (22.6%) | 97 (20.9%) | |
| Hypopharynx | 3 (5.7%) | 48 (10.3%) | |
| Nasopharynx | 1 (1.9%) | 27 (5.8%) | |
| Other | 4 (7.6%) | 58 (12.5%) | |
| Induction chemotherapy | |||
| Yes | 9 (17.0%) | 161 (34.6%) | 0.006 |
| No | 44 (83.0%) | 304 (65.4%) | |
| Concurrent chemotherapy | |||
| Yes | 47 (88.7%) | 313 (67.3%) | 0.0005 |
| No | 6 (11.3%) | 152 (32.7%) | |
| Post‐operative radiotherapy | |||
| Yes | 18 (34.0%) | 180 (38.7%) | 0.50 |
| No | 35 (66.0%) | 285 (61.3%) | |
| Type of statin | |||
| Lipophilic | 41 (77.4%) | N.D. | |
| Hydrophilic | 3 (5.7%) | N.D. | |
| Not stated | 9 (17.0%) | N.D. | |
Included in propensity matching: age, gender, year of diagnosis, facility, path stage grouping, path T‐stage, path N‐stage, LN > 2, LVSI, LND and site. For patient data analysis (table 1) discrete variables were compared with the Chi‐square test and differences in the medians were assessed using the Wilcoxon Rank Sum test, significance was defined as P < 0.05. In patients under statin treatment, variables such as age, comorbidity, T and N‐stage and succes in induction and concurrent chemotherapy, were significantly changed compared to those who where not in treatment.
Table 2.
Univariate and multivariate analysis of survival (n = 518).
| Univariate analysis for survival | Multivariate analysis for survival | |||
|---|---|---|---|---|
| Hazard ratio | P value | Hazard ratio | P value | |
| Statin use | ||||
| No | Reference | Reference | ||
| Yes | 0.47 (0.20–0.93) | 0.03 | 0.29 (0.09–0.71) | 0.004 |
| Age | ||||
| ≤57 | Reference | Not done | ||
| >57 | 0.85 (0.60–1.19) | 0.34 | ||
| Comorbidity | ||||
| Medium | Reference | Reference | ||
| High‐very high | 1.62 (1.12–2.31) | 0.01 | 1.67 (1.07–2.54) | 0.02 |
| Performance status | ||||
| <70 | Reference | Not done | ||
| ≥70 | 0.62 (0.32–1.38) | 0.22 | ||
| Alcohol use | ||||
| <3 drinks/day | Reference | Reference | ||
| ≥3 drinks/day | 1.72 (1.06–2.95) | 0.003 | 1.60 (0.98–2.74) | 0.06 |
| Smoking | ||||
| <10 pack‐year | Reference | |||
| ≥10 pack‐year | 1.16 (0.76–1.83) | 0.50 | Not done | |
| T‐stage | ||||
| T1–2 | Reference | Reference | ||
| T3–4 | 1.75 (1.14–2.79) | 0.009 | 2.12 (1.23–4.00) | 0.006 |
| N‐stage | ||||
| N0–1 | Reference | Reference | ||
| N2–3 | 1.41 (1.00–2.00) | 0.05 | 1.26 (0.84–1.91) | 0.25 |
| Induction chemotherapy | ||||
| No | Reference | Not done | ||
| Yes | 1.23 (0.86–1.74) | 0.24 | ||
| Concurrent chemotherapy | ||||
| No | Reference | Not done | ||
| Yes | 1.21 (0.84–1.80) | 0.30 | ||
| Post‐operative radiotherapy | ||||
| No | Reference | Reference | ||
| Yes | 0.52 (0.35–0.75) | 0.0005 | 0.53 (0.32–0.84) | 0.006 |
For univariate and multivariate analyses (Table 2), Cox proportional hazard models was used to compare differences in survival. Variables with P value < 0.1 on univariate analysis were included on multivariate analysis. Significance was assessed using the Log Rank test. Use of statins, high ‐ very high comorbidity, the size of the tumor and the use of post‐operative radiotherapy have an impact on survival.
For univariate and multivariate analyses, Cox proportional hazard models was used to compare differences in survival. Variables with P value < 0.1 on univariate analysis were included on multivariate analysis. Significance was assessed using the Log Rank test. Use of statins, high ‐ very high comorbidity, the size of the tumor and the use of post‐operative radiotherapy have an impact on survival. The effect of statins on local and distant control by radiotherapy treatment of HNSCC tumors is shown in Fig. 6. A significant effect on local control of tumors was found for patients taking statins (P = 0.04), with ≥ 90% of patients on a statin drug achieving local control for up to 48 months. Rates of local control for patients not taking a statin fell to 70% by 12 months post‐treatment. Unfortunately, no significant effect was observed for distant control of HNSCC tumor metastasis (P = 0.28), implying that statin drugs are most effective in local rather than distant tumor control.
Figure 6.
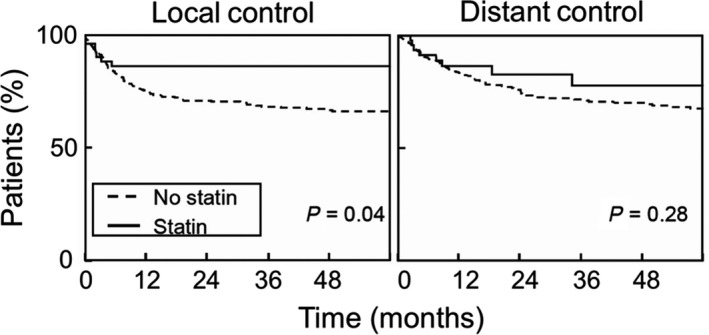
Statin use improves outcomes of radiotherapy in HNSCC patients. Analysis was conducted using data from 518 medical records of stage III‐IVB HNSCC patients, a fraction of whom were incidentally being treated with statins concomitantly with their radiotherapy. Time to local control or distant control was determined from last date of radiotherapy. Survival curves were plotted using the Kaplan–Meier method, and significance was assessed using the Log Rank test. Data indicate that incidental use of statins was statistically associated with better local, but not distant, control in HNSCC patients.
4. Discussion
Although many head and neck squamous cell cancers (HNSCC) initially regress upon treatment with conventional or targeted therapies and display a complete or partial response, a large fraction recur locally or at distant sites with metastatic disease. For recurrent HNSCC, the prognosis remains poor as these tumors typically display acquired therapy resistance. This pattern likely reflects the heterogeneity of epithelial tumors and treatment driving selection for cancer cells able to survive and proliferate even in the presence of therapy. When local recurrence occurs after radiotherapy, this can be particularly problematic, given the limited tolerance of normal tissue for reirradiation. Were it possible to prevent the selection of resistant clones and thus suppress the emergence of resistant disease, recurrence might be rarer and easier to treat. In the case of locoregional recurrence, sensitizers targeting resistance might permit reirradiation at tolerable doses.
This work identifies the mevalonate pathway as a promising target to overcome radiation resistance in HNSCC. Quantitative proteomic profiling to compare two radioresistant HNSCC cell lines, SQ20B and JSQ3, to the radiosensitive cell line SCC61 revealed altered expression of several mevalonate pathway enzymes. Further analysis revealed increased expression of HMG‐CoA reductase (HMGCR), the rate‐limiting enzyme of mevalonate synthesis, in the radioresistant cells. In turn, the radioresistant cells also displayed increased cholesterol levels. Treating SQ20B and JSQ3 with PIT only partly suppressed cholesterol levels, suggesting active uptake of cholesterol along with increased biosynthesis. Import of free cholesterol was similar between the three cell lines, but SQ20B and JSQ3 displayed higher expression of LDL receptors and greater LDL uptake compared with SCC61. Despite the moderate impact on cellular cholesterol, PIT and other lipophilic statins decreased cell proliferation and viability in the radioresistant HNSCC cells. Further, when combined with IR, PIT restored radiation sensitivity to SQ20B and JSQ3 cells, increasing cell death, DNA damage persistence, and senescence. These latter effects may be partly independent of cholesterol levels, potentially mediated by other metabolic pathways downstream of HMGCR and mevalonate biosynthesis such as protein prenylation as previously observed (Efimova et al., 2017). Even so, the active uptake of cholesterol by the radioresistant cell lines SQ20B and JSQ3 reinforces the impression that high cellular cholesterol may be critical for their survival and proliferation.
Prior studies have shown that the mevalonate pathway in cancer cells can impact cell division, tumor growth, and metastatic potential (Clendening et al., 2010) (Thurnher et al., 2012) and modulate immune response and mTOR signaling (Bekkering et al., 2018). Recently, novel links between the mevalonate pathway and apoptosis, cell cycle arrest, and DNA damage response have been observed (Horlbeck et al., 2018; Martin Sanchez et al., 2015; Stine et al., 2016). Our prior work demonstrated strong radiosensitization by statin drugs in breast cancer and melanoma cell lines both in vitro and in vivo (Efimova et al., 2017). While connections have been noted between the mevalonate pathway and tumor radioresistance in prostate and pancreatic cancer (Chen et al., 2018; Souchek et al., 2014), this study is the first to extend these links to HNSCC.
Given the strong influence of advanced age and smoking history on head and neck cancer, it is not surprising that a large fraction of patients also display intercurrent cardiovascular disease and/or hypercholesterolemia, leading to high rates of incidental statin use. In our analysis, among HNSCC patients treated with radiotherapy, taking statins appeared to improve locoregional control, suggesting radiosensitization in vivo. Additional studies would be useful to reduce potentially confounding variables such as HNSCC subtype, HPV status, and patient age. Expanding the analysis to include more patients taking statins would allow analysis of the potential impact of specific types of statin, if any. However, our data mining approach utilizing currently available patient data revealed promising findings regarding local control of tumors. We hope these findings will help to justify future clinical studies involving co‐treatment with lipophilic statins for HNSCC patients undergoing radiotherapy.
5. Conclusions
In radioresistant HNSCC cell lines, we observed dysregulation of proteins in the mevalonate pathway, as well as elevated cholesterol content, increased LDL receptor abundance, and high levels of LDL uptake. Radioresistant cells displayed loss of cell viability, decreased clonogenic potential, an increase in DNA damage, and induction of cell senescence when treated with the mevalonate pathway inhibitor pitavastatin along with radiation. As a clinical correlate, head and neck cancer patients taking statins displayed improved local tumor control after radiation therapy, suggesting value for administering lipophilic statins to HNSCC patients as a radiosensitization strategy.
Conflict of interest
S.J.K. is a founder of OncoSenescence. The other authors have no conflicts to declare.
Author contributions
NR, EVE, AWT, and SJK involved in the conception and design. NR, DW, AF, EVE, and SJK involved in the development of methodology. NR, DW, AF, EVE, AR, OKA, JB, and MTS involved in the acquisition of data. NR, DW, AF, EVE, AR, OKA, MTS, and SJK analyzed and interpreted the data. NR, AF, DW, EVE, AR, OKA, JB, AWT, MTS, and SJK wrote, reviewed, and/or revised the manuscript.
Supporting information
Fig. S1. Metabolism‐related Gene Ontology (GO) analysis of radioresistant HNSCC proteomes. Proteins were separated into functional categories and represented as the log2 ratio for JSQ3/SCC61 (left) and SQ20B/SCC61 (right). Significantly differentiated metabolism‐related GO categories found in both radioresistant cell lines include glucose metabolic process, protein metabolic process, and lipid metabolic process. P values for each GO category were determined using the Panther GO database and are shown in the figure.
Fig. S2. Downregulation of critical mevalonate pathway enzymes HMGCS1 and NSDHL. Cells were harvested in active growth phase, lysed, and subjected to Western blotting for targets as shown. Actin is shown as a protein loading control. HMGCS1 and NSDHL were both downregulated in radioresistant cell lines JSQ3 and SQ20B, consistent with proteomics data.
Fig. S3. Combination treatment of pitavastatin and irradiation induces persistent DNA damage and accelerated senescence in JSQ3 radioresistant cells. (A) Plots representing mean percent of DNA in comet tail ± SEM for 100 cells per treatment condition. Significance is indicated by *, P < 0.05 (Mann–Whitney U‐test). Results indicate that PIT alone enhances persistent DSBs in radioresistant JSQ3 cells. PIT + IR had a similar effect. (B) Flow cytometric senescence assay data of cells treated with 10 μm PIT (PIT), 10 Gy (IR), or PIT + IR. Vehicle‐treated cells (VEH) were stained as controls. PIT + IR significantly induced senescence in the radioresistant cells above IR or PIT only treatments. (C) Statistical significance was determined using Mann–Whitney U‐test. Mean fluorescent intensity (MFI) of the senescence probe is shown. Error bars, SEM. *, P < 0.05; ns, not significant. n > 3000 viable cells per sample.
Fig. S4. Colony formation assay of SCC61, JSQ3, and SQ20B treated with statin ± IR. Cells were treated with 2.5, 5 or 10 μm PIT, simvastatin, lovastatin, atorvastatin, pravastatin or rosuvastatin for 1 h prior to 10 Gy irradiation. After 4 d of culture, crystal violet staining was conducted and plates were imaged.
Acknowledgements
This work was supported by NIH R01s CA176843 to SJK and DE027445 to MTS. We thank Dr. Tanguy Siewert (Johns Hopkins) and Dr. Ralph Weichselbaum (University of Chicago) for providing cell lines. The University of Chicago Cytometry and Antibody Technology Core Facility, supported by P30 CA014599, provided flow cytometry instrumentation. The Office of Radiation Safety at University of Chicago assisted with irradiation of cell lines.
Amy Flor and Don Wolfgeher contributed equally to this work
Data Accessibility
Research data pertaining to this article is located at figshare.com: https://doi.org/10.6084/m9.figshare.8145971.
References
- Alagona P Jr (2010) Pitavastatin: evidence for its place in treatment of hypercholesterolemia. Core Evid 5, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews DW, Scott CB, Sperduto PW, Flanders AE, Gaspar LE, Schell MC and Bahary J‐P (2004) Whole brain radiation therapy with or without stereotactic radiosurgery boost for patients with one to three brain metastases: phase III results of the RTOG 9508 randomised trial. Lancet 363(9422), 1665–1672. [DOI] [PubMed] [Google Scholar]
- Bartelink H, Horiot J‐C, Poortmans PM, Struikmans H, Van den Bogaert W, Fourquet A and Wárlám‐Rodenhuis CC (2007) Impact of a higher radiation dose on local control and survival in breast‐conserving therapy of early breast cancer: 10‐year results of the randomized boost versus no boost EORTC 22881‐10882 trial. J Clin Oncol 25(22), 3259–3265. [DOI] [PubMed] [Google Scholar]
- Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, Popa CD, Ter Horst R, van Tuijl J, Netea‐Maier RT et al (2018) Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 172(1–2), 135–146.e139. 10.1016/j.cell.2017.11.025. [DOI] [PubMed] [Google Scholar]
- Bjordal K, Ahlner‐Elmqvist M, Hammerlid E, Boysen M, Evensen JF, Biörklund A, Jannert M, Westin T and Kaasa S (2001) A prospective study of quality of life in head and neck cancer patients. Part II: Longitudinal data. Laryngoscope 111(8), 1440–1452. [DOI] [PubMed] [Google Scholar]
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J et al (2006) Radiotherapy plus cetuximab for squamous‐cell carcinoma of the head and neck. N Engl J Med 354(6), 567–578. [DOI] [PubMed] [Google Scholar]
- Boyle P and Levin B (2008) World Cancer Report 2008: IARC. Press, International Agency for Research on Cancer. [Google Scholar]
- Budach V, Stuschke M, Budach W, Baumann M, Geismar D, Grabenbauer G, Lammert I, Jahnke K, Stueben G, Herrmann T et al (2005) Hyperfractionated accelerated chemoradiation with concurrent fluorouracil‐mitomycin is more effective than dose‐escalated hyperfractionated accelerated radiation therapy alone in locally advanced head and neck cancer: final results of the radiotherapy cooperative clinical trials group of the German Cancer Society 95‐06 Prospective Randomized Trial. J Clin Oncol 23(6), 1125–1135. [DOI] [PubMed] [Google Scholar]
- Carvalho AL, Nishimoto IN, Califano JA and Kowalski LP (2005) Trends in incidence and prognosis for head and neck cancer in the United States: a site‐specific analysis of the SEER database. Int J Cancer 114(5), 806–816. [DOI] [PubMed] [Google Scholar]
- Chen YA, Shih HW, Lin YC, Hsu HY, Wu TF, Tsai CH, Wu CL, Wu HY, Hsieh JT, Tang CH et al (2018) Simvastatin sensitizes radioresistant prostate cancer cells by compromising DNA double‐strand break repair. Front Pharmacol 9, 600 10.3389/fphar.2018.00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clendening JW, Pandyra A, Li Z, Boutros PC, Martirosyan A, Lehner R, Jurisica I, Trudel S and Penn LZ (2010) Exploiting the mevalonate pathway to distinguish statin‐sensitive multiple myeloma. Blood 115(23), 4787–4797. [DOI] [PubMed] [Google Scholar]
- Delaney G, Jacob S, Featherstone C and Barton M (2005) The role of radiotherapy in cancer treatment. Cancer 104(6), 1129–1137. [DOI] [PubMed] [Google Scholar]
- Dickreuter E, Eke I, Krause M, Borgmann K, van Vugt M and Cordes N (2016) Targeting of β1 integrins impairs DNA repair for radiosensitization of head and neck cancer cells. Oncogene 35(11), 1353–1362. [DOI] [PubMed] [Google Scholar]
- Efimova EV, Ricco N, Labay E, Mauceri HJ, Flor AC, Ramamurthy A, Sutton HG, Weichselbaum RR and Kron SJ (2017) HMG‐CoA reductase inhibition delays DNA repair and promotes senescence after tumor irradiation. Mol Cancer Therap Molcanther 0288, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisbruch A, Lyden T, Bradford CR, Dawson LA, Haxer MJ, Miller AE, Teknos TN, Chepeha DB, Hogikyan ND, Terrell JE et al (2002) Objective assessment of swallowing dysfunction and aspiration after radiation concurrent with chemotherapy for head‐and‐neck cancer. Int J Rad Oncol*Biol*Phys 53(1), 23–28. [DOI] [PubMed] [Google Scholar]
- Horlbeck MA, Xu A, Wang M, Bennett NK, Park CY, Bogdanoff D, Adamson B, Chow ED, Kampmann M, Peterson TR et al (2018) Mapping the genetic landscape of human cells. Cell 174(4), 953–967. e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristov AC, Cope L, Reyes MD, Singh M, Iacobuzio‐Donahue C, Maitra A and Resar L (2009) HMGA2 protein expression correlates with lymph node metastasis and increased tumor grade in pancreatic ductal adenocarcinoma. Mod Pathol 22(1), 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter KD, Parkinson EK and Harrison PR (2005) Profiling early head and neck cancer. Nat Rev Cancer 5(2), 127–135. [DOI] [PubMed] [Google Scholar]
- Hutchinson J and Marignol L (2017) Clinical potential of statins in prostate cancer radiation therapy. Anticancer Res 37(10), 5363–5372. [DOI] [PubMed] [Google Scholar]
- Khan K, Araki K, Wang D, Li G, Li X, Zhang J, Xu W, Hoover RK, Lauter S, O'Malley B Jr et al (2010) Head and neck cancer radiosensitization by the novel poly (ADP‐ribose) polymerase inhibitor GPI‐15427. Head Neck 32(3), 381–391. [DOI] [PubMed] [Google Scholar]
- Kuban DA, Tucker SL, Dong L, Starkschall G, Huang EH, Cheung MR, Lee AK and Pollack A (2008) Long‐term results of the MD Anderson randomized dose‐escalation trial for prostate cancer. Int J Rad Oncol*Biol*Phys 70(1), 67–74. [DOI] [PubMed] [Google Scholar]
- Labay E, Efimova EV, Quarshie BK, Golden DW, Weichselbaum RR and Kron SJ (2011) Ionizing radiation‐induced foci persistence screen to discover enhancers of accelerated senescence. Int J High Throughput Screen 2, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiker AJ, DeGraff W, Choudhuri R, Sowers AL, Thetford A, Cook JA, Van Waes C and Mitchell JB (2015) Radiation enhancement of head and neck squamous cell carcinoma by the dual PI3K/mTOR inhibitor PF‐05212384. Clin Cancer Res 21(12), 2792–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Sanchez C, Perez Martin JM, Jin JS, Davalos A, Zhang W, de la Pena G, Martínez‐Botas J, Rodríguez‐Acebes S, Suárez Y, Hazen MJ et al (2015) Disruption of the mevalonate pathway induces dNTP depletion and DNA damage. Biochim Biophys Acta 1851(9), 1240–1253. 10.1016/j.bbalip.2015.06.001. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT and Thomas PD (2013) Large‐scale gene function analysis with the PANTHER classification system. Nat Protoc 8, 1551 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muruganujan A, Mills C, Kang D, Tang H, Huang X, Mi H and Thomas PD (2016) PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 45(D1), D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignon J, Bourhis J, Domenge CO and Designe L (2000) Chemotherapy added to locoregional treatment for head and neck squamous‐cell carcinoma: three meta‐analyses of updated individual data. Lancet 355(9208), 949–955. [PubMed] [Google Scholar]
- Pignon J‐P, Le Maitre A, Maillard E and Bourhis J (2009) Meta‐analysis of chemotherapy in head and neck cancer (MACH‐NC): an update on 93 randomised trials and 17,346 patients. Radiother Oncol 92(1), 4–14. [DOI] [PubMed] [Google Scholar]
- Souchek JJ, Baine MJ, Lin C, Rachagani S, Gupta S, Kaur S, Lester K, Zheng D, Chen S, Smith L et al (2014) Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br J Cancer 111(6), 1139–1149. 10.1038/bjc.2014.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancu C and Sima A (2001) Statins: mechanism of action and effects. J Cell Mol Med 5(4), 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine JE, Guo H, Sheng X, Han X, Schointuch MN, Gilliam TP, Gehrig PA, Zhou C and Bae‐Jump VL (2016) The HMG‐CoA reductase inhibitor, simvastatin, exhibits anti‐metastatic and anti‐tumorigenic effects in ovarian cancer. Oncotarget 7(1), 946–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y‐C, Hung S‐K, Yu C‐C, Lee C‐C, Li S‐C and Lai H‐C (2013) The radiosensitization effect of mTOR inhibitior in human tongue squamous cell carcinoma. [Google Scholar]
- The Gene Ontology Consortium (2016) Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45(D1), D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurnher M, Nussbaumer O and Gruenbacher G (2012) Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clin Cancer Res 18(13), 3524–3531. [DOI] [PubMed] [Google Scholar]
- Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang H, Perrett S, Prodromou C, Jones GW and Kron SJ (2012) CDK‐dependent Hsp70 phosphorylation controls G1 cyclin abundance and cell‐cycle progression. Cell 151(6), 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichselbaum RR, Beckett MA, Schwartz JL and Dritschilo A (1988) Radioresistant tumor cells are present in head and neck carcinomas that recur after radiotherapy. Int J Rad Oncol*Biol*Phys 15(3), 575–579. [DOI] [PubMed] [Google Scholar]
- Weichselbaum RR, Dahlberg W, Beckett M, Karrison T, Miller D, Clark J and Ervin TJ (1986) Radiation‐resistant and repair‐proficient human tumor cells may be associated with radiotherapy failure in head‐and neck‐cancer patients. Proc Natl Acad Sci USA 83(8), 2684–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Metabolism‐related Gene Ontology (GO) analysis of radioresistant HNSCC proteomes. Proteins were separated into functional categories and represented as the log2 ratio for JSQ3/SCC61 (left) and SQ20B/SCC61 (right). Significantly differentiated metabolism‐related GO categories found in both radioresistant cell lines include glucose metabolic process, protein metabolic process, and lipid metabolic process. P values for each GO category were determined using the Panther GO database and are shown in the figure.
Fig. S2. Downregulation of critical mevalonate pathway enzymes HMGCS1 and NSDHL. Cells were harvested in active growth phase, lysed, and subjected to Western blotting for targets as shown. Actin is shown as a protein loading control. HMGCS1 and NSDHL were both downregulated in radioresistant cell lines JSQ3 and SQ20B, consistent with proteomics data.
Fig. S3. Combination treatment of pitavastatin and irradiation induces persistent DNA damage and accelerated senescence in JSQ3 radioresistant cells. (A) Plots representing mean percent of DNA in comet tail ± SEM for 100 cells per treatment condition. Significance is indicated by *, P < 0.05 (Mann–Whitney U‐test). Results indicate that PIT alone enhances persistent DSBs in radioresistant JSQ3 cells. PIT + IR had a similar effect. (B) Flow cytometric senescence assay data of cells treated with 10 μm PIT (PIT), 10 Gy (IR), or PIT + IR. Vehicle‐treated cells (VEH) were stained as controls. PIT + IR significantly induced senescence in the radioresistant cells above IR or PIT only treatments. (C) Statistical significance was determined using Mann–Whitney U‐test. Mean fluorescent intensity (MFI) of the senescence probe is shown. Error bars, SEM. *, P < 0.05; ns, not significant. n > 3000 viable cells per sample.
Fig. S4. Colony formation assay of SCC61, JSQ3, and SQ20B treated with statin ± IR. Cells were treated with 2.5, 5 or 10 μm PIT, simvastatin, lovastatin, atorvastatin, pravastatin or rosuvastatin for 1 h prior to 10 Gy irradiation. After 4 d of culture, crystal violet staining was conducted and plates were imaged.
Data Availability Statement
Research data pertaining to this article is located at figshare.com: https://doi.org/10.6084/m9.figshare.8145971.