

Abstract
The PIWI‐interacting RNA (piRNA) pathway preserves genomic integrity by repressing transposable elements (TEs) in animal germ cells. Among PIWI‐clade proteins in Drosophila, Piwi transcriptionally silences its targets through interactions with cofactors, including Panoramix (Panx) and forms heterochromatin characterized by H3K9me3 and H1. Here, we identified Nxf2, a nuclear RNA export factor (NXF) variant, as a protein that forms complexes with Piwi, Panx, and p15. Panx–Nxf2–P15 complex formation is necessary in the silencing by stabilizing protein levels of Nxf2 and Panx. Notably, ectopic targeting of Nxf2 initiates co‐transcriptional repression of the target reporter in a manner independent of H3K9me3 marks or H1. However, continuous silencing requires HP1a and H1. In addition, Nxf2 directly interacts with target TE transcripts in a Piwi‐dependent manner. These findings suggest a model in which the Panx–Nxf2–P15 complex enforces the association of Piwi with target transcripts to trigger co‐transcriptional repression, prior to heterochromatin formation in the nuclear piRNA pathway. Our results provide an unexpected connection between an NXF variant and small RNA‐mediated co‐transcriptional silencing.
Keywords: heterochromatin formation, nuclear RNA export factor, RNA silencing, transcriptional regulation, transposable element
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; RNA Biology
Introduction
Transposable elements (TEs) act as endogenous mobile mutagens to alter the sequence and structure of the genome, thereby changing the transcriptome and chromatin structure. TEs are often deleterious to the host by, for example, disrupting a gene, but may also be adaptive and drive genome evolution (Han & Boeke, 2005; Chuong et al, 2017). TEs comprise nearly half of the human genome and approximately 30% of the genome of Drosophila melanogaster; thus, the successively arising families of TEs are the main drivers of genome expansion. In metazoans, TEs are silenced by the piRNA pathway, in which Piwi proteins are guided by Piwi‐interacting RNAs (piRNAs) to their targets (Iwasaki et al, 2015; Ernst et al, 2017). The piRNA pathway is also essential for germline development and fertility in animals (Lin & Spradling, 1997; Cox et al, 1998; Carmell et al, 2007; Houwing et al, 2007).
In the Drosophila ovary, two cytoplasmic PIWI paralogs, AGO3 and Aubergine (Aub), engage an amplification loop termed the “ping‐pong cycle” to cleave both TE transcripts and long piRNA precursor transcripts arising from piRNA clusters, which comprise a large number as well as various types of fragmented TEs, leading to the post‐transcriptional silencing of TEs and production of piRNAs (Brennecke et al, 2007; Gunawardane et al, 2007). These piRNAs can in turn trigger the production of phased piRNAs from piRNA precursors, which generates a dazzling variety of piRNAs and is coupled to the activity of a third Piwi protein (Han et al, 2015; Homolka et al, 2015; Mohn et al, 2015). Phased piRNAs are also produced in ovarian somatic cells by a ping‐pong cycle‐independent mechanism, which are in turn also loaded onto Piwi (Han et al, 2015; Mohn et al, 2015). This loading initiates the transport of Piwi into the nucleus where it drives the transcriptional silencing of target TEs, by inducing specific histone modification and/or facilitating the folding of chromatin into a higher‐order structure (Iwasaki et al, 2016).
Lack of the nuclear Piwi activity results in de‐repression of TEs, which is concomitant with decreases in both H3K9me3 repressive epigenetic marks and RNA polymerase II (Pol II) occupancy on target TE loci (Sienski et al, 2012; Donertas et al, 2013; Huang et al, 2013; Le Thomas et al, 2013; Ohtani et al, 2013; Rozhkov et al, 2013). This suggests a scenario of how the nuclear piRNA pathway works: Piwi and its bound piRNAs scan for target TEs by complementary base‐pairing with their nascent transcripts. Upon targeting, Piwi recruits chromatin factors including H3K9me3 methyltransferases such as Eggless (also called dSetDB1) to initiate heterochromatin formation. The H3K9me3 modification is then bound by HP1a to maintain and propagate epigenetic silencing. However, thus far, direct association of the Piwi–piRNA complex with target transcripts and/or H3K9me3 methyltransferases has not been demonstrated.
Furthermore, depletion of Maelstrom (Mael), a Piwi cofactor in the nuclear piRNA pathway, does not decrease H3K9me3 levels at Piwi‐targeted TE loci, suggesting that the repressive histone mark per se is not the final silencing mark for transcriptional gene silencing mediated by Piwi–piRNA complexes (Sienski et al, 2012). Recently, we identified linker histone H1 as a component of a nuclear Piwi complex and found that depletion of H1 de‐represses Piwi‐targeted TEs and their surrounding genes without affecting the density of H3K9me3 marks and HP1a at target TE loci (Iwasaki et al, 2016). Instead, we demonstrated that the chromatin accessibility at Piwi‐targeted TE loci is modulated by H1. These findings suggested a model in which Piwi recruits H1 and forces it to stay on target TE loci to induce chromatin compaction, thereby repressing target TE transcription, and that the nuclear piRNA pathway adopts collaborated actions of H1 and H3K9me3 mark to maintain silencing of the TE state by modulating the chromatin state.
In addition to Mael and H1, biochemical and genetic analyses have also identified a number of putative Piwi cofactors including DmGTSF1/Asterix (Arx) and Panoramix (Panx)/Silencio (Brower‐Toland et al, 2007; Wang & Elgin, 2011; Sienski et al, 2012, 2015; Czech et al, 2013; Donertas et al, 2013; Le Thomas et al, 2013; Muerdter et al, 2013; Ohtani et al, 2013; Yu et al, 2015). These findings together suggest that multiple pathways leading to Piwi‐mediated TE silencing may exist, and raise the question of whether these pathways are independently initiated or have a common initial event.
To investigate this issue, we reanalyzed Panx, which interacts with Piwi and promotes the deposition of H3K9me3 marks on target TE chromatin by H3K9me3 histone methyltransferase, Eggless (Sienski et al, 2015; Yu et al, 2015). Here, we biochemically isolated Nxf2, nuclear RNA export factor (NXF) variant, as a component of Panx‐associated complexes. Nxf2 further associates with p15 (also called Nxt1), a co‐adaptor for nuclear RNA export (Fribourg et al, 2001; Herold et al, 2001; Kerkow et al, 2012). The NXF family comprises four members in Drosophila, among which Nxf1 is an essential mRNA nucleocytoplasmic export factor (Izaurralde, 2002; Stutz & Izaurralde, 2003; Bjork & Wieslander, 2014; Wickramasinghe & Laskey, 2015). However, other members (Nxf2‐4) are gonad‐specific and their functions are not known, although they share common domain structures with Nxf1 (Herold et al, 2000, 2001, 2003). Detailed analysis of Nxf2 function in the nuclear piRNA pathway revealed that the interactions between Panx, Nxf2, and p15 are necessary to maintain the protein stability of Nxf2 and Panx. Moreover, ectopic targeting of Nxf2 initiates co‐transcriptional repression of the target reporter gene prior to heterochromatin formation, and H3K9me3 marks and H1 are required at later time points. Notably, the RNA binding domain of Nxf2 is essential for recruitment of the complex to target TEs. CLIP experiments demonstrated that both Piwi and Nxf2 directly interact with Piwi‐targeted TE transcripts and that the association of Nxf2 with target TE transcripts is Piwi‐dependent. These results suggest that Nxf2 enforces the association of Piwi–Panx–Nxf2–p15 (PPNP) complexes with the nascent transcripts of target TEs and triggers co‐transcriptional repression in the nuclear silencing pathway.
Results
Nxf2 forms a complex with Piwi and Panx, and plays an essential role in the piRNA pathway
To reveal the precise mechanism by which Piwi–piRNA complexes transcriptionally silence TEs, we raised a monoclonal antibody that specifically recognizes Panx (Fig EV1A). Using this antibody, Panx‐associated complexes were immunopurified from OSCs, a cultured ovarian somatic cell line, in which piRNA‐guided silencing operates (Saito et al, 2009). Mass spectrometry analysis of the purified complexes (Fig 1A, Appendix Table S1) revealed that, in addition to Piwi, Nxf2 is present in the associated complexes. Nxf2 is a variant of the nuclear RNA export factor (NXF) family, which harbors similar domain structures (Herold et al, 2000, 2001, 2003; see also Fig EV1H). Among NXF variants, Nxf1 is highly conserved, ubiquitously expressed, and well known to be involved in the nuclear export of various mRNAs (Izaurralde, 2002; Stutz & Izaurralde, 2003; Bjork & Wieslander, 2014; Wickramasinghe & Laskey, 2015). However, Nxf2 and Nxf3 are almost exclusively expressed in the ovary, while Nxf4 is specifically expressed in the testis (Gramates et al, 2017). NXF variants other than Nxf1 are not involved in general mRNA export (Herold et al, 2001, 2003), and their functions remain to be revealed. By using a specific monoclonal antibody generated against Nxf2 (Fig EV1B), we confirmed the interaction among Panx, Piwi, and Nxf2 in both OSCs and Drosophila ovary (Fig EV1C and D). Immunoprecipitation followed by nuclear extraction also detected Piwi‐Panx‐Nxf2 complex (Figs 1B and EV1E and F). In addition, Panx and Nxf2 co‐localized at the nucleus (Fig EV1G), suggesting that the complex is formed within the nucleus. The interaction of Panx with NXF variants is restricted to Nxf2 (Fig EV1H and I). Thus, we focused on the function of Nxf2 in the Piwi–piRNA pathway.
Figure EV1. Nxf2 associates with Panx and Piwi to regulate piRNA target TEs.
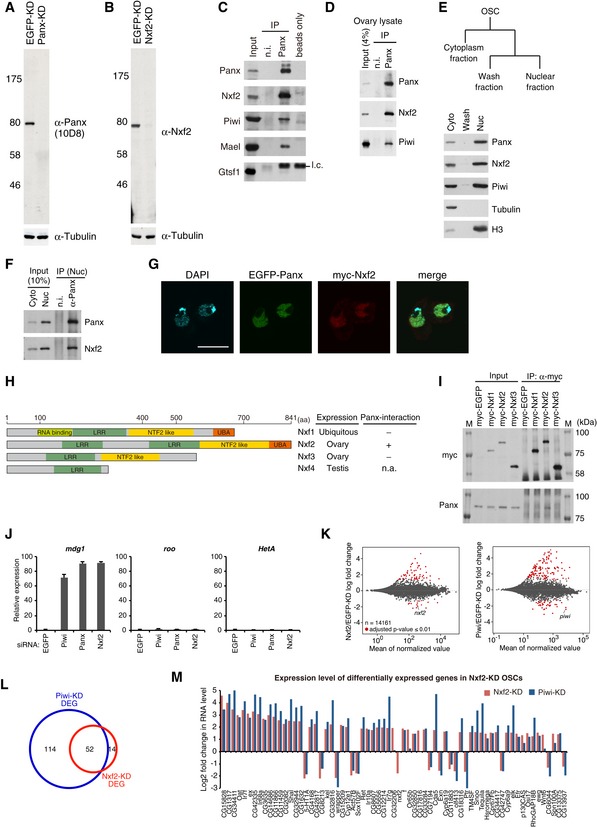
- Western blotting (WB) shows the specificity of anti‐Panx monoclonal antibodies raised in this study. Tubulin was used as a loading control.
- WB shows the specificity of anti‐Nxf2 monoclonal antibodies raised in this study.
- Immunoprecipitation (IP) from OSC lysate using anti‐Panx antibody, followed by WB of Panx, Nxf2, Piwi, Mael, and Gtsf1. IP‐WB was performed under the same conditions as the silver staining described in Fig 1A. Mouse immunoglobulin G (IgG) (n.i.) was used for control IP. l.c.: light chain from the antibody.
- IP from fly ovary lysate using anti‐Panx antibody, followed by WB of Panx, Nxf2, and Piwi.
- Scheme of preparation of cytoplasmic and nuclear fractions of OSCs. WB using Panx, Nxf2, Piwi, Tubulin, and histone H3 antibodies shows the level of each protein in the separate fractions. Tubulin was detected in the cytoplasmic fraction, where histone H3 was enriched in the nuclear fraction.
- IP from nuclear OSC lysate using anti‐Panx antibody, followed by WB.
- Immunofluorescence of OSCs co‐transfected with EGFP‐Panx‐ and myc‐Nxf2‐expressing vectors, using myc antibody (red). EGFP is detected as Panx signal, and DAPI staining (blue) shows the location of nuclei. EGFP‐Panx and myc‐Nxf2 co‐localize in the nucleus. Scale bar: 10 μm.
- Schematic of NXF variants. LRR: leucine‐rich repeat, NTF2‐like: nuclear transport factor 2‐like domain, UBA: ubiquitin‐associated domain (left panel). Nxf1 is ubiquitously expressed, while Nxf2 and Nxf3 are almost exclusively expressed in ovary, and Nxf4 is specifically expressed in testis. The interaction between Nxf4 and Panx was not analyzed, since Nxf4 expression is limited to testis, indicated as n.a. (not available) (right panel).
- IP from lysate of OSCs expressing myc‐tagged NXF variants, followed by WB using anti‐myc and anti‐Panx antibodies. M indicates protein markers. The results are summarized in the right panel in (H). Among NXF variants, only Nxf2 can interact with Panx.
- RNA levels of mdg1, roo, and HetA were quantified by qRT–PCR upon depletion of EGFP (control), Piwi, Panx, or Nxf2. Expression levels are normalized by the expression of RP49. Error bars represent SD (n = 3). Piwi–piRNA‐targeted TE was specifically de‐silenced by Piwi‐, Panx‐, and Nxf2‐KD, confirming the results of RNA‐seq.
- MA plot of RPKM values (log10 scale) for mRNAs in the indicated KD samples, based on RNA‐seq. Differentially expressed genes (DEGs) are in red.
- Venn diagram displaying the number of DEGs upon depletion of Nxf2 (red) or Piwi (blue).
- Log2 fold changes in mRNA levels of 67 DEGs in Nxf2‐KD OSCs. mRNA levels calculated from RNA‐seq data are used for Nxf2‐ and Piwi‐KD OSCs. Most mRNAs whose expression was altered upon Nxf2‐KD were also affected by Piwi‐KD, suggesting that Nxf2 does not globally affect mRNA levels.
Figure 1. Nxf2 is required for Piwi–piRNA‐mediated TE silencing in OSCs.
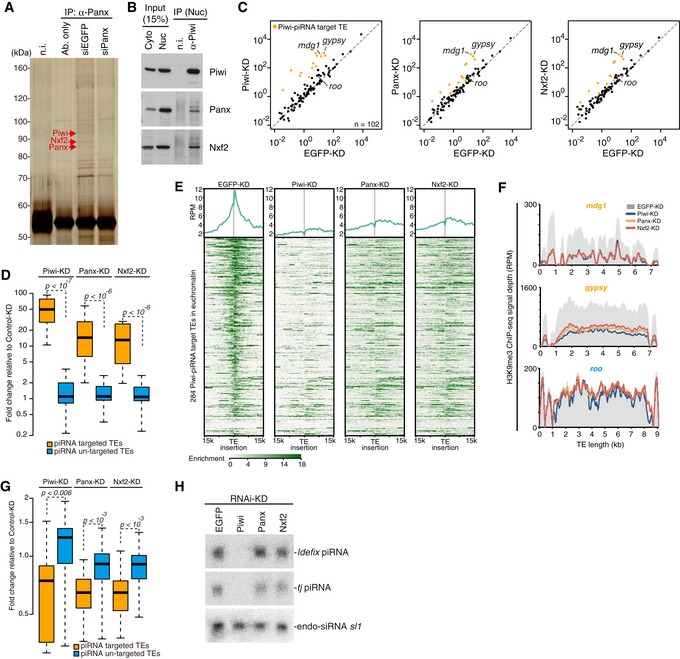
- Immunoprecipitation (IP) from siRNA‐transfected OSC lysate using anti‐Panx antibody, followed by silver staining. Protein bands indicated by red arrows were identified by mass spectrometry analyses (see also Appendix Table S1). Mouse immunoglobulin G (IgG) (n.i.) was used for control IP.
- IP from OSC lysate using anti‐Piwi antibody, followed by Western blotting. Cyto: cytoplasmic fraction of OSCs. Nuc: nuclear fraction of OSCs.
- Scatterplot of RPKM values for TEs in EGFP‐ (control), Panx‐, or Nxf2‐KD samples, based on RNA‐seq. TEs for which the expression level differed from that of the control by more than tenfold in Piwi‐KD (Piwi–piRNA‐targeted TEs) are plotted in orange. Both x‐axis and y‐axis are a log10 scale.
- Boxplots showing fold changes in the expression of Piwi–piRNA‐targeted and un‐targeted TEs based on RNA‐seq upon the indicated KD. Piwi–piRNA‐targeted TEs are as defined previously (Iwasaki et al, 2016). Boxplot central bands, boxes, and whiskers show median, third quartile, first quartile, maxima, and minima, respectively. P‐values were calculated by Wilcoxon rank‐sum test, and the y‐axis is a log2 scale.
- Meta‐plot and heatmap indicating H3K9me3 levels within 15 kb of the Piwi–piRNA‐targeted TE insertion are shown for OSCs with the indicated KD. Heatmap is sorted by the decrease of H3K9me3 levels in Piwi‐KD against EGFP‐KD.
- Density plots for normalized H3K9me3 ChIP‐seq signals over the consensus sequence from mdg1, gypsy (targeted by Piwi–piRNA, orange), and roo (not targeted by Piwi–piRNA, blue) TEs in EGFP‐, Piwi‐, Panx‐, and Nxf2‐KD OSCs.
- Boxplots as in (D) showing fold changes in the H3K9me3 levels of Piwi–piRNA‐targeted and un‐targeted TEs upon the indicated KD.
- Northern blotting for Idefix‐piRNA, traffic jam (tj)‐piRNA, and esiRNA sl‐1 (control) on total RNA isolated from OSCs with the indicated KD.
Source data are available online for this figure.
To identify transcripts regulated by Nxf2, we performed mRNA‐seq analysis of OSCs under Nxf2 knockdown (KD) and compared the results with those obtained for Piwi‐ and Panx‐KD OSCs (Figs 1C and D, and EV1J). TEs that were de‐silenced upon Piwi depletion (Iwasaki et al, 2016) were also specifically de‐silenced upon depletion of Panx or Nxf2. For example, mdg1 and gypsy were among the most highly de‐silenced TEs in Nxf2‐, Panx‐, and Piwi‐KD experiments. Panx‐ and Nxf2‐KD resulted in nearly identical patterns of TE de‐silencing, albeit to a lesser extent than Piwi‐KD, possibly because Piwi functions not only in TE silencing but also in piRNA biogenesis (see also Fig 1H). These results suggest that Nxf2 regulates TEs in the Piwi–piRNA pathway, in a manner resembling Panx, and are consistent with previous reports describing that Nxf2 was included in the list of candidate cofactors for the piRNA pathway identified by RNAi screening (Czech et al, 2013; Handler et al, 2013; Muerdter et al, 2013).
Depletion of Piwi has been shown to result in the reduction of H3K9me3 marks and H1 in the regions neighboring TE insertions, resulting in de‐repression of coding genes located near TEs (Sienski et al, 2012; Iwasaki et al, 2016). Coding genes that were de‐repressed upon Nxf2‐KD were highly similar to those observed in Piwi‐KD, suggesting that Nxf2 also regulates genes located near TE insertions. The population of differentially expressed genes (DEGs) and their fold change were smaller in Nxf2‐KD (Fig EV1K–M), in line with the lower extent of TE de‐silencing in Nxf2‐KD (Fig 1C and D). These results indicate that Nxf2 functions in the Piwi–piRNA pathway and does not affect the expression pattern of bulk mRNA, consistent with the previous finding that Nxf1 is the only member of the Drosophila NXF family that mediates the export of mRNAs (Herold et al, 2001, 2003).
We further analyzed the H3K9me3 levels in these corresponding regions. ChIP‐seq analysis of H3K9me3 marks at the euchromatic insertions of piRNA target TEs revealed that the depletion of Nxf2 decreased the H3K9me3 levels at these loci, similar to the case in Piwi or Panx depletion (Fig 1E–G). Notably, a severe decrease of H3K9me3 marks was observed not only at the TE insertions but also at the regions surrounding TE insertions (Fig 1E). The effect of Nxf2 or Panx depletion was weaker than that of Piwi depletion, and the results of Nxf2 and Panx depletion highly resembled each other, consistent with the effects on the expression levels of TEs observed by RNA‐seq (Fig 1C and D). The analysis focusing on the TE consensus sequence confirmed that the effect on the H3K9me3 level was limited to the TEs targeted by Piwi–piRNA complexes (Fig 1G). Furthermore, the depletion of Nxf2 in OSCs did not affect the expression levels of piRNAs (Fig 1H), indicating that piRNA biogenesis including piRNA precursor export to the cytoplasm occurs in the absence of Nxf2. Together, these results indicate that Nxf2 is a key factor required for transcriptional silencing in the Piwi–piRNA pathway.
Formation of Panx–Nxf2–P15 complex is essential for stabilization of Panx and Nxf2 protein levels
To further characterize the Piwi‐Panx‐Nxf2 complex, we immunopurified flag‐tagged Nxf2 expressed in OSCs (Fig EV2A), which was then subjected to shotgun proteome analysis (Fig EV2B, Appendix Table S2). This analysis not only confirmed the presence of Panx and Piwi, but further detected p15, also known as Nxt1, in Nxf2‐associated complexes (see also Fig EV6C). p15 is a co‐adaptor for nuclear RNA export and has been shown to form a heterodimeric complex with NXF proteins (Fribourg et al, 2001; Herold et al, 2001; Kerkow et al, 2012). We confirmed the interaction of Nxf2 with p15 in OSCs (Fig 2A), and p15 was also co‐immunoprecipitated with Panx (Fig EV2C). These results suggest the formation of a Piwi‐Panx–Nxf2–p15 complex, which we hereafter call the PPNP complex. Both Nxf1 and p15 are highly conserved from yeast to mammals, and the Nxf1–p15 complexes bind to mRNAs without strong sequence preference and have the ability to export mRNAs through the nuclear pore complex (NPC; Herold et al, 2001; Levesque et al, 2001; Wilkie et al, 2001; Wiegand et al, 2002). Although a previous study showed that recombinant p15 can associate with Nxf1, Nxf2, and Nxf3 proteins, only the Nxf1–p15 complexes function in mRNA export, and the role of p15's association with Nxf2 and Nxf3 remains unknown (Herold et al, 2001). We therefore analyzed the involvement of p15 in Nxf2‐mediated transcriptional silencing.
Figure EV2. p15 interacts with Nxf2 at its NTF2‐like domain.
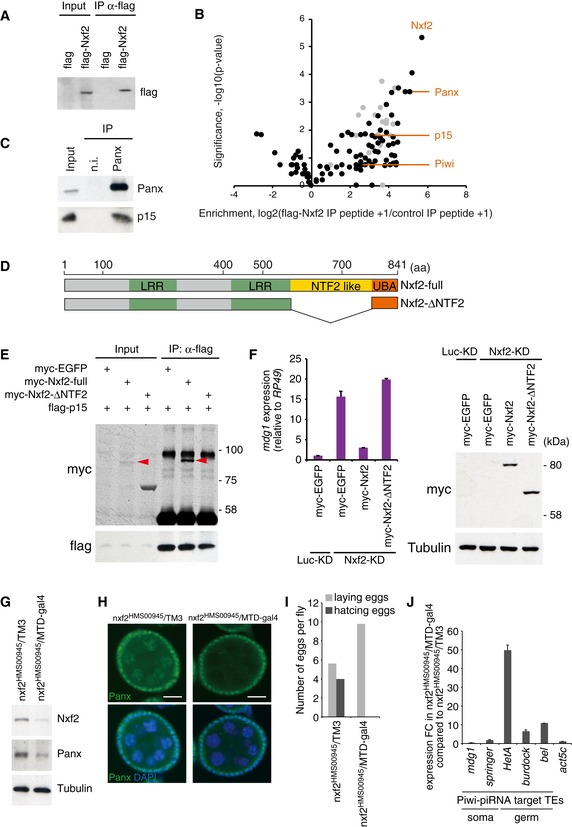
- Western blot (WB) showing flag–Nxf2 immunoprecipitant from OSCs expressing either flag only (control) or flag–Nxf2, using flag antibody. The immunoprecipitant was subjected to proteome analysis.
- Summary of the proteome analysis of peptide‐eluted flag–Nxf2 component. The plot shows the enrichment of peptide obtained by flag‐Nxf2 immunoprecipitation (IP) over control IP (log2 value of peptide count + 1) on the x‐axis, and significance calculated by replicate experiments as the Student's t‐test P‐value (−log10) on the y‐axis. Enrichment and significance of each protein are listed in Appendix Table S2. Ribosomal proteins are plotted in gray.
- IP from OSC lysate using anti‐Panx antibody, followed by WB of Panx and p15. IP was performed under the same conditions as silver staining described in Fig 1A.
- Schematic of full‐length Nxf2 protein and deletion mutant of NTF2‐like domain (Nxf2‐ΔNTF2).
- Flag‐IP performed using lysate of flag‐p15‐ and myc‐EGFP‐ or myc‐Nxf2‐ΔNTF2‐expressing OSCs, followed by WB using myc or flag antibody. myc‐Nxf2‐full is indicated by a red arrowhead. p15 cannot interact with the Nxf2 construct lacking NTF2‐like domain.
- myc‐EGFP, myc‐Nxf2, and myc‐Nxf2‐ΔNTF2 were expressed in Luc‐ (control) or Nxf2‐depleted OSCs. mdg1 expression levels were monitored by qRT–PCR. Expression values are normalized by the expression of RP49. Error bars indicate SD (n = 3) (left panel). WB using an antibody against myc and tubulin using lysates from transfected OSCs (right). The ΔNTF2 deletion mutant, which cannot interact with p15, could not rescue the silencing of TE.
- WB of ovaries from nxf2HMS00945/MTD‐gal4 flies using Nxf2, Panx, and tubulin (control) antibody. Note that the expression of shRNA is limited to germline cells by MTD‐gal4. Depletion of Nxf2 in the germline cells also results in a decrease of Panx protein level.
- Immunofluorescence of ovaries from nxf2HMS00945 flies using Panx antibody (green). DAPI staining (blue) shows the location of nuclei. Decrease of Panx in Nxf2‐depleted ovaries was observed specifically in the germ cells. Scale bars: 20 μm.
- Numbers of eggs laid and hatched per fly are indicated. Nxf2 depletion in Drosophila ovaries results in sterility phenotype.
- mRNA levels of Piwi–piRNA‐targeted TEs and actin 5c (control) were quantified by qRT–PCR. Expression values are normalized by the expression of RP49. Error bars represent SD (n = 3). Piwi–piRNA‐targeted TEs in the germ cells were specifically de‐silenced upon germline‐specific Nxf2 depletion.
Figure 2. Panx–Nxf2–P15 complex formation is essential for the protein stability of Panx and Nxf2.
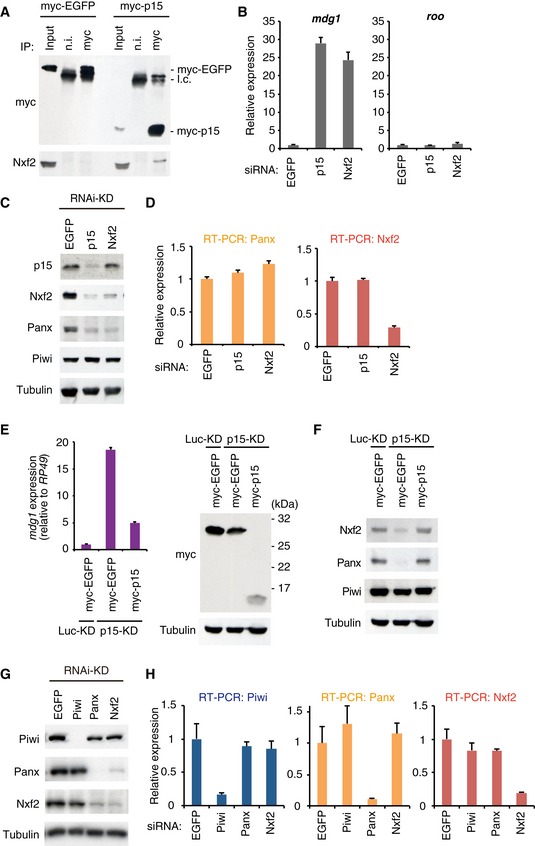
- Western blot (WB) showing Nxf2 protein levels in the complex immunoprecipitated from OSCs expressing either myc‐EGFP or myc–p15, using myc antibody. Mouse immunoglobulin G (IgG) (n.i.) was used for control immunoprecipitation.
- RNA levels of mdg1 and roo were quantified by qRT–PCR upon depletion of EGFP (control), p15, or Nxf2. Error bars indicate SD (n = 3). Note that KD was performed for 2 days with a single siRNA transfection for this experiment.
- WB showing p15, Nxf2, Panx, Piwi, and Tubulin (loading control) protein levels upon EGFP (control)‐, p15‐, or Nxf2‐KD.
- RNA levels of Panx and Nxf2 were quantified by qRT–PCR upon depletion of EGFP (control), p15, or Nxf2. Error bars indicate SD (n = 3).
- myc‐EGFP or myc–p15 was expressed in Luc‐ (control) or p15‐depleted OSCs, and mdg1 expression levels were monitored by qRT–PCR (left panel). Expression values are normalized by the expression of RP49. Error bars indicate SD (n = 3). Using the same samples, WB was performed using antibodies against myc and Tubulin (right panel).
- myc‐EGFP or myc–p15 was expressed in Luc‐ (control) or p15‐depleted OSCs, and WB was performed using antibodies against Nxf2, Panx, Piwi, and Tubulin.
- WB showing Piwi, Panx, Nxf2, and Tubulin (loading control) protein levels upon EGFP (control)‐, Piwi‐, Panx‐, or Nxf2‐KD.
- RNA levels of Piwi, Panx, and Nxf2 were quantified by qRT–PCR upon depletion of EGFP (control), Piwi, Panx, or Nxf2. Error bars represent SD (n = 3).
Source data are available online for this figure.
Depletion of p15 resulted in specific de‐repression of Piwi–piRNA‐targeted TEs (Fig 2B). It was previously shown that p15 heterodimerizes with human and C. elegans Nxf1 at its NTF2 (nuclear transport factor 2)‐like domains (Fribourg et al, 2001; Klenov et al, 2014). We therefore analyzed the interaction between p15 and Nxf2 mutant without NTF2‐like domain (Fig EV2D and E). This mutant could not associate with p15, and the rescue experiment showed that the NTF2‐like domain of Nxf2 is indispensable for the silencing of mdg1 TE (Fig EV2F). These results suggest that the NTF2‐like domain of Nxf2 associates with p15, as in the case of Nxf1, and this association is necessary for TE silencing.
Interestingly, depletion of p15 resulted in decreased levels of both Nxf2 and Panx proteins (Fig 2C). Since the RNA levels of Nxf2 and Panx remained unchanged in the p15‐depleted samples (Fig 2D), p15 likely affects the stability of Nxf2 and Panx proteins. The protein level of p15 was unaffected by Nxf2‐KD, possibly because p15 can bind to the other NXF proteins once Nxf2 is depleted (Fig 2C). De‐silencing of the TEs and protein levels of Nxf2 and Panx could be rescued by expressing myc–p15 protein in p15‐depleted OSCs (Fig 2E and F). Thus, p15 is an essential partner protein for Nxf2 to transcriptionally regulate Piwi–piRNA‐targeted TEs, by stabilizing protein levels of Panx and Nxf2.
Notably, the knockdown of Nxf2 resulted in a severe decrease in the level of Panx protein (Fig 2C and G). A similar finding was observed in Panx‐KD, where the level of Nxf2 protein was greatly decreased (Fig 2G). In contrast, neither Nxf2‐ nor Panx‐KD affected the level of Piwi protein. RNA levels of Piwi, Panx, and Nxf2 in KD samples were determined by qRT–PCR, which showed that the levels of Panx and Nxf2 transcripts remained unchanged upon the knockdown of Nxf2 or Panx (Fig 2H). Destabilization of Panx upon Nxf2‐KD was also observed in the germ cells of the ovary (Fig EV2G and H). Although the morphology of the ovary was not affected by the silencing of Nxf2 in the germline cells (Fig EV2H), severe fertility defects were observed: Flies with Nxf2 knockdown could lay eggs like the control flies, but none of them hatched (Fig EV2I). This phenotype was similar to that observed in flies expressing Piwi only in ovarian somatic cells but not germ cells (Jin et al, 2013). Additionally, TEs targeted by piRNAs in the germline were specifically de‐silenced in Nxf2‐depleted flies (Fig EV2J). These results together show that the Panx–Nxf2–P15 complex formation stabilizes the protein levels of Nxf2 and Panx (see also Figs 5 and EV5), and this is essential for TE silencing.
Tethering of Nxf2 to nascent mRNA leads to co‐transcriptional silencing
We made use of reporter constructs to analyze the precise steps that the PPNP complex takes to mediate transcriptional silencing. First, we constructed reporter constructs whose transcripts are targeted by piRNAs derived from the flam locus, a prototype piRNA cluster (Brennecke et al, 2007; Post et al, 2014) (Fig EV3A). The luc reporter gene containing flam fragments in sense orientation or in antisense orientation relative to the luc transcripts was transfected into OSCs; it was shown that only the antisense reporters were strongly silenced (Fig EV3B–D). The antisense reporters were de‐silenced in OSCs upon Piwi‐KD (Fig EV3E) or depletion of Piwi cofactors, including Panx and Nxf2 (Fig EV3F), indicating that these factors were required for the co‐transcriptional silencing in the Piwi–piRNA‐targeted reporter system.
Figure EV3. The PPNP complex is required for the co‐transcriptional silencing in the Piwi‐piRNA‐targeted reporter system.
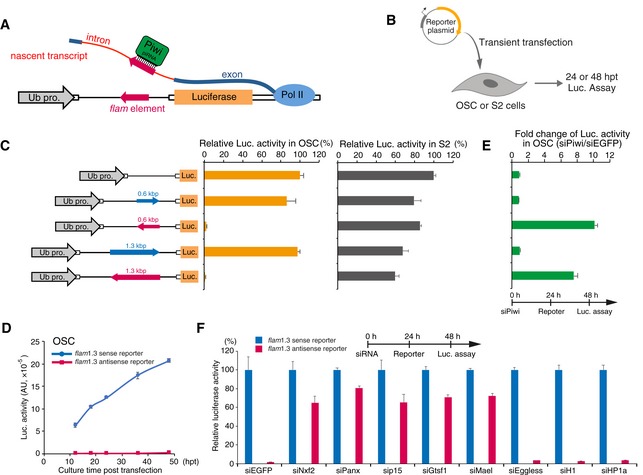
- Schematic of piRNA reporter gene assay. Reporter plasmid carries flam element in its intron. Luc mRNA harboring flam element in the antisense direction is targeted by Piwi carrying piRNAs derived from the flam locus.
- Experimental scheme. OSCs or S2 cells were transfected with reporter plasmids transiently and harvested at 24 or 48 hpt.
- Left panel shows reporter gene constructs harboring flam elements (0.6 kbp or 1.3 kbp) inserted in the intron. Blue and red arrows indicate sense and antisense directions, respectively. Middle graph shows luciferase activity relative to that of reporter gene without flam elements in OSC. Right graph shows luciferase activity relative to that of reporter gene without flam elements in S2 cells. Error bar indicates SD (n = 4). The sense reporters were expressed similar to control reporter gene lacking the flam element, whereas the antisense (piRNA‐targeted) reporters were completely silenced, even though we introduced the reporter gene as a plasmid into OSCs, suggesting that the co‐transcriptional silencing occurs independent of the chromatin context.
- Time course of luciferase activity in OSCs. Blue and red arrows indicate reporter gene bearing a flam element (1.3 kbp) in the sense or antisense direction, respectively. Error bar indicates SD (n = 3).
- Silencing of reporter gene harboring flam element (1.3 kbp, antisense direction) occurs in a Piwi‐dependent manner. Graph shows fold change of luciferase activity in OSCs treated with siPiwi or siEGFP (control). Error bar indicates SD (n = 3).
- Effects of knockdown of the indicated genes on piRNA reporter activity (1.3 kbp, flam). Red bar shows the luciferase activities of the antisense reporter relative to that of the sense reporter (blue). Error bars indicate SD (n = 3). Transfection schedule of siRNA and reporter plasmids is shown at the top of the figure. Depletion of Nxf2, Panx, p15, and known cofactors Gtsf1 and Mael, de‐silenced the piRNA‐targeted reporter in OSCs. In contrast, Eggless‐KD, H1‐KD, and HP1‐KD had no impact on the silencing.
Previously, it was shown that the artificial recruitment of Panx can induce transcriptional silencing in Drosophila ovary (Sienski et al, 2015; Yu et al, 2015). For further functional analysis of Nxf2 in the Piwi–piRNA silencing pathway, we took advantage of the λN‐boxB tethering system, in which λN fusion proteins are delivered to a reporter RNA via protein–RNA interaction (Baron‐Benhamou et al, 2004). We established a tethering system using OSCs carrying a genome‐integrated luciferase (luc) reporter driven by the ubiquitin promoter, which harbors 14 boxB sites in its intron (Fig 3A). Expression of λN‐HA‐tagged Nxf2 or Panx led to reporter silencing in a manner dependent on the λN peptide fused to Nxf2 or Panx (Figs 3B and EV4A). Silencing induced by tethered Panx or Nxf2 results in strongly reduced luc RNA levels (Fig 3C). Knockdown of Panx and p15 weakened the repression by the enforced tethering of Nxf2, showing that Nxf2 needs Panx and p15 for efficient repression (Fig 3D). Consistent with previous reports (Sienski et al, 2015; Yu et al, 2015), the tethering of Piwi to the reporter failed to induce silencing (Fig 3B and C). λN‐HA‐myc‐tagged Piwi localized at the nucleus (Fig EV4B), indicating that λN‐HA‐myc‐tagged Piwi harbored piRNA in OSCs (Saito et al, 2010; Yashiro et al, 2018). Because Piwi, which is guided to the target reporter via loaded piRNA, represses its targets (Fig EV3), it is likely that only piRNA‐guided Piwi is able to recruit silencing machinery to the target (see also Appendix Fig S2I).
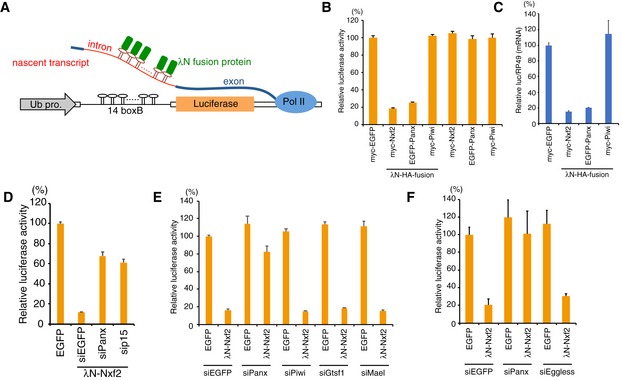
Figure 3. Nxf2 tethered to nascent mRNA causes co‐transcriptional silencing.
-
ASchematic of boxB‐λN tethering system in OSCs. A genome‐integrated luciferase reporter with 14 copies of boxB sites located within the intronic region of ubiquitin (Ub). λN fusion proteins are recruited to luciferase reporter RNA via 14× boxB.
-
BEffects on luciferase activity of the proteins indicated below. OSCs carrying the reporter gene were transfected with plasmids expressing the indicated proteins and were harvested at 48 h post‐transfection (hpt). Luciferase activities were normalized by the total protein amount. Bar graph shows luciferase activity relative to that of myc‐EGFP (control). Error bars indicate SD (n = 4).
-
CqRT–PCR showing effects of the indicated λN fusion proteins on Luc mRNA level at 48 hpt. Levels of Luc mRNA were normalized by RP49 mRNA. Bar graph shows the level of Luc mRNA relative to myc‐EGFP (control). Error bars indicate SD (n = 6).
-
DSilencing by the enforced tethering of Nxf2 shows Panx‐ and p15‐dependent manner. Bar graph shows luciferase activity relative to that of the sample transfected with EGFP expression vector (control). Error bars indicate SD (n = 3).
-
E, FEffects of knockdown of the indicated genes on luciferase activity upon λN‐Nxf2 expression at 48 hpt. Bar graph shows luciferase activity relative to that of the sample co‐transfected with myc‐EGFP and siEGFP (control). Error bars indicate SD (n = 3).
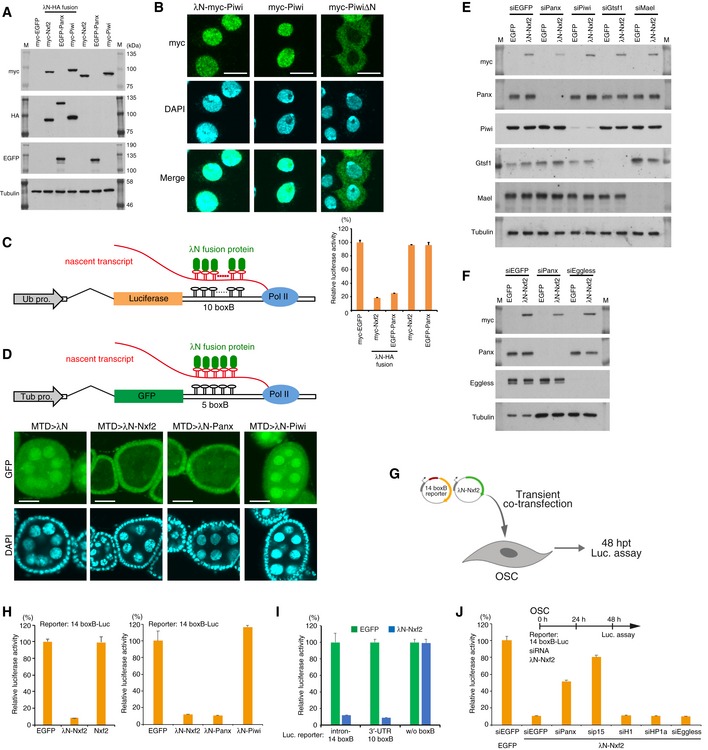
Figure EV4. Enforced tethering of Nxf2 to nascent mRNA causes co‐transcriptional silencing.
-
AWestern blotting (WB) associated with Fig 3B shows the exogenous expression of λN‐fused proteins. Nxf2 and Piwi were tagged with λN‐HA and myc, whereas Panx was tagged with λN‐HA and EGFP, since the expression level of EGFP–Panx was higher than the level of myc‐tagged Panx, which is quite unstable. M indicates protein markers.
-
BImmunofluorescence of OSCs transfected with λN‐myc‐Piwi‐, myc‐Piwi‐, or myc‐PiwiΔN‐expressing vectors, using myc antibody (Green). DAPI staining (blue) shows the location of nuclei. myc‐PiwiΔN lacking nuclear localization signal (NLS) was distributed in cytoplasm (Saito et al, 2010; Yashiro et al, 2018). λN‐myc‐Piwi localized in the nucleus similarly to myc‐Piwi, indicating that λN‐myc‐Piwi harbored piRNA in OSCs, even though it failed to induce silencing of the boxB reporter gene (Fig 3B, see also Fig EV4D and H). This may be due to only Piwi, which is guided to the target via its loaded piRNAs, potentially being able to recruit silencing machinery to the target (see also Fig EV3C and Appendix Fig S2I). Scale bars: 10 μm.
-
CSchematic of boxB‐λN tethering system in OSCs shows a genome‐integrated luciferase reporter with 10 copies of boxB sites located within the 3′ UTR of luc mRNA (left). Bar graph shows relative luciferase activity normalized by total protein amount at 48 h post‐transcription (right). Error bars indicate SD (n = 4). Tethering of Nxf2 and Panx to the 3′ UTR of luc mRNA leads to silencing in OSCs.
-
DSchematic of boxB‐λN tethering system in fly ovary shows a genome‐integrated GFP reporter with five copies of boxB sites located within the 3′ UTR of GFP mRNA. λN‐fused proteins are driven by MTD‐Gal4. Tub pro: α‐Tubulin gene promoter (top panel). Confocal images depict GFP fluorescence and DAPI signals in egg chambers expressing the indicated λN fusion proteins in the germline. Expression of λN‐Nxf2 and Panx leads to reporter silencing in germ cells, but that of λN‐Piwi does not. This is consistent with our data in Fig 3B and previous reports (Sienski et al, 2015; Yu et al, 2015). Scale bars: 20 μm.
- E, F
-
GExperimental design. According to Fig EV3B, the reporter genes do not have to be integrated into the genome, suggesting that the co‐transcriptional silencing occurs independent of the chromatin context. To examine this issue, we carried out co‐transfection of reporter plasmid (14 boxB‐Luc) and expression plasmids for λN fusion protein to OSCs, transiently. Cells were harvested at 48 hpt.
-
HEffect on luciferase activity of the proteins indicated below. Error bars indicate SD (n = 4). Even if a plasmid with 14 boxB reporter sites was transiently introduced into OSCs, λN‐Nxf2 repressed luciferase activity.
-
IλN‐Nxf2 represses luciferase activity of reporter plasmid harboring 10 boxB sites in its 3′ UTR, but not that of reporter plasmid without boxB. Error bars indicate SD (n = 4).
-
JEffects of knockdown of the indicated genes on boxB reporter activity upon λN‐Nxf2 expression. Bar graph shows luciferase activity relative to that of the sample co‐transfected with myc‐EGFP and siEGFP (control). Error bars indicate SD (n = 4). Transfection schedule of siRNA and reporter plasmids is shown at the top of the figure. Although knockdown of Panx and p15 weakened the repression by the forced tethering of Nxf2, the effects of H1‐ and HP1a‐KD on the λN‐Nxf2‐mediated silencing were negligible, which is consistent with the results from Figs 4D and EV3F.
To rule out the possibility that λN‐Nxf2 interferes with the splicing of the reporter mRNA, we constructed a luc reporter with 10 boxB sites in the 3′ untranslated region (Fig EV4C). Tethering of Nxf2 to the reporter mRNA also led to silencing in a λN peptide‐dependent manner. To corroborate the results, we used an in vivo RNA‐tethering system in the Drosophila ovary (Sienski et al, 2015). The expression of λN‐Nxf2 with the MTD‐Gal4 driver (Fig EV4D) led to potent reporter silencing in germline cells but not in somatic cells of the ovary. Panx and Nxf2 proteins need to associate with each other to ensure their stability (Figs 2G and EV2G); therefore, it is likely that tethering of Panx results in the recruitment of Nxf2, and vice versa. These results indicate that the Nxf2–Panx complex is sufficient to induce co‐transcriptional silencing, when targeted to nascent RNA.
To dissect the mechanism of the observed co‐transcriptional gene silencing in the tethering system in OSCs, we depleted the expression of genes known to be associated with Panx (Fig EV1C) and involved in piRNA‐directed transcriptional gene silencing. Depletion of Piwi or known Piwi cofactors (Gtsf1 (Sienski et al, 2012; Donertas et al, 2013; Ohtani et al, 2013) and Mael (Sienski et al, 2012)) instead had no impact on the Nxf2‐mediated repression (Figs 3E and EV4E). Thus, Nxf2 acts with Panx most likely downstream from Piwi and Gtsf1. Deficiency of Mael, however, does not result in decrease of H3K9me3 marks (Sienski et al, 2012), and thus, Mael may act in parallel to the silencing mediated by the Piwi‐Panx‐Nxf2 complex; knockdown of each component of the complex all results in depletion of H3K9me3 marks.
Furthermore, we examined the roles of chromatin‐related factors in co‐transcriptional silencing mediated by Piwi–piRNA complexes. Depletion of Eggless had no effect on the silencing by λN‐Nxf2 in the tethering system using OSCs (Figs 3F and EV4F). In addition to Eggless‐KD, H1‐KD and HP1‐KD also had no impact on the silencing by Piwi–piRNA and λN‐Nxf2 in the reporter system based on plasmids (Figs EV3F and EV4G–J). While this result is consistent with a report of a study using a plasmid‐based reporter assay to detect Piwi‐directed silencing in Drosophila ovary somatic sheet cells (Clark et al, 2017), it is at odds with previous reports demonstrating that the methyltransferase Eggless is required for Panx‐mediated silencing using the tethering system in the Drosophila ovary (Sienski et al, 2015; Yu et al, 2015). These conflicting results are probably due to the different expression systems of λN fusion proteins. λN fusion proteins are driven by MTD‐Gal4 constitutively in the tethering system in the ovary, whereas they are expressed transiently by DNA transfection in the tethering system using OSCs. The tethering system in the ovary probably enables us to observe late phases of a suppressed state of the reporter gene. In contrast, transient expression of λN fusion proteins makes it possible to analyze the mechanism of transition from initiation to the fully suppressed state of the reporter gene in our tethering system using OSCs. The results of Eggless‐KD upon tethering of λN‐Nxf2 suggest that the Nxf2–Panx pair elicits co‐transcriptional repression prior to heterochromatin formation on TEs in the Piwi–piRNA pathway.
Nxf2 triggers co‐transcriptional repression prior to heterochromatin formation
To elucidate the precise mechanism by which λN‐Nxf2 represses the reporter gene integrated in the genome, we examined the time course of silencing by λN‐Nxf2, which was expressed in OSCs by transient transfection of expression vector. The expression level of λN‐Nxf2 gradually decreased with a peak at 36 h post‐transfection (hpt), and it had dropped below the detection limit at 96 hpt (Fig 4A). The luciferase activity reached its bottom at 48 hpt and remained suppressed at 96 hpt despite the absence of λN‐Nxf2. Consistent with these observations, the level of Pol II association with the reporter gene decreased at 48 and 96 hpt (Fig 4B). Notably, no significant difference was observed in the occupancies of H1 and H3K9me3 at 48 hpt, despite λN‐Nxf2 repressed the reporter gene (Fig 4A and C). This can be interpreted to mean that Nxf2‐triggered co‐transcriptional repression is independent of chromatin factors. By contrast, H1 and H3K9me3 accumulated on the reporter gene at 96 hpt (Fig 4C), suggesting that heterochromatin is formed at this time point on the reporter gene in the absence of λN‐Nxf2, leading to continued silencing. To verify the role of heterochromatin formation in the gene silencing induced by λN‐Nxf2, we knocked down H1 or HP1a after expression of λN‐Nxf2. The effects of H1‐ and HP1a‐KD on the λN‐Nxf2‐mediated silencing were negligible at 48 hpt (Fig 4D). To examine the roles of H1 and HP1a in the λN‐Nxf2‐mediated silencing at the latter time point, we determined luciferase activity at 96 hpt of the λN‐Nxf2 expression vector followed by H1‐ and HP1a‐KD. The depletion of H1 or HP1a partially restored luciferase activities upon tethering of λN‐Nxf2 at 96 hpt (Fig 4E). These findings together suggest that λN‐Nxf2 may initiate co‐transcriptional repression prior to heterochromatin formation mediated by H1 and HP1a, which is required for maintaining the suppressed state of the reporter gene in the latter stage of silencing when little or no λN‐Nxf2 is present. In other words, the silencing mode mediated by tethered Nxf2 may switch from Nxf2–Panx‐dependent to heterochromatin‐dependent.
Figure 4. Nxf2 initiates co‐transcriptional silencing before heterochromatin formation.
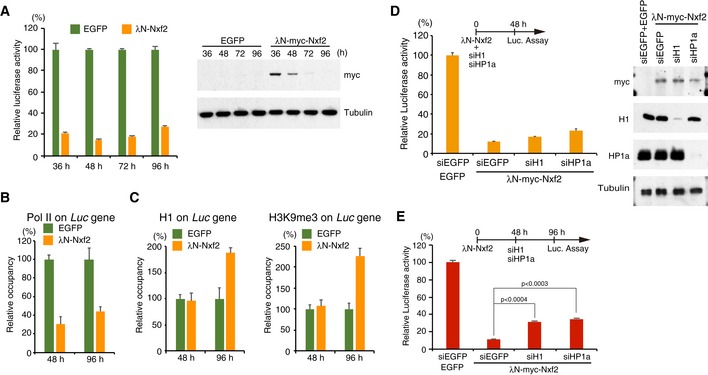
- Time course of luciferase activity upon λN‐Nxf2 tethering. Bar graph shows luciferase activity relative to that of EGFP at the indicated incubation time. Error bars indicate SD (n = 3) (left panel). Expression levels of myc‐tagged λN‐Nxf2 were monitored using anti‐myc antibody. Tubulin was used as a loading control (right panel).
- ChIP‐qPCR analysis of RNA polymerase II occupancy on the Luc gene upon λN‐Nxf2 tethering. Bar graph shows the occupancy relative to that of the sample transfected with a plasmid expressing EGFP. Error bars indicate SD (n = 3).
- ChIP‐qPCR analysis of histone H1 and H3K9me3 occupancy on the Luc gene. Bar graph shows the occupancy relative to that of the sample transfected with a plasmid expressing EGFP. Error bars indicate SD (n = 3).
- Effects of H1 and HP1a on luciferase activity upon λN‐Nxf2 tethering at 48 hpt. OSCs were co‐transfected with protein expression vectors and the indicated siRNAs and cultured for 48 h in accordance with the experimental scheme at the top of the figure. Bar graph shows luciferase activity relative to that of the sample expressing EGFP (control). Error bars indicate SD (n = 3) (left panel). Western blots showing the protein levels of λN‐myc‐Nxf2, H1, HP1a, and Tubulin (loading control) in the same samples (right panel).
- Effects of H1 and HP1a on luciferase activity upon λN‐Nxf2 at 96 hpt. Transfection schedule of plasmids and siRNAs is shown at the top of the figure. siRNA transfection was carried out at 48 h, since the depletion effect reaches maximum after 48 hpt. Bar graph shows luciferase activity relative to that of the sample expressing EGFP (control). Error bars indicate SD (n = 3). P‐values were calculated by the Student's t‐test.
The first LRR region of Nxf2 harbors RNA binding activity and is necessary for piRNA‐mediated transcriptional silencing
To reveal the role of Nxf2 in the PPNP complex‐mediated co‐transcriptional silencing, we characterized the regions of Panx and Nxf2 proteins necessary for their function. This revealed that the middle region (300–399 aa) of Panx is necessary for its interaction with Nxf2 (Fig 5A and B), whereas its N‐terminal region (1–169 aa) interacts with Piwi (Fig 5A and B). Rescue experiments and ectopic tethering experiments with the deletion mutants confirmed that both regions of Panx are indispensable for the transcriptional regulation (Fig EV5A and B). We also produced a series of Nxf2 deletion mutants (Fig 5C) and found that the C‐terminal UBA (ubiquitin‐associated) domain of Nxf2 is necessary for interaction with Panx (Fig 5D). Although Nxf1 also harbors a UBA domain, we did not detect interaction between Nxf1 and Panx, indicating that Panx specifically recognizes the UBA domain of Nxf2 (Fig EV1I). These results suggest that the UBA domain of Nxf2 and middle region of Panx interact with each other, and N‐terminal region of Panx is linked to Piwi.
Figure 5. Nxf2 associates with Piwi–piRNA silencing complex to target RNA via its first LRR region.
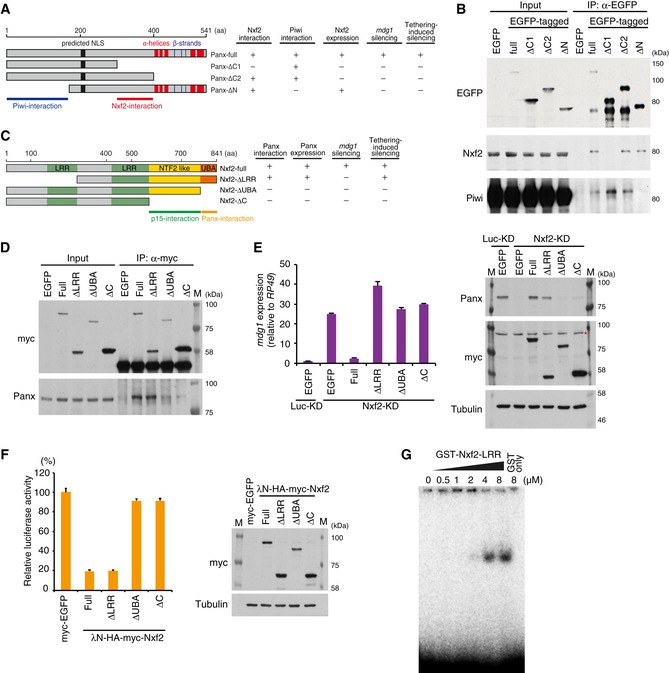
- Schematic of full‐length Panx protein and deletion mutants. Predicted NLS and α‐helices and β‐strands are indicated (left panel). The results of rescue experiments are summarized (right panel).
- Western blot (WB) of EGFP‐Immunoprecipitation (IP) product from lysate of EGFP‐tagged Panx deletion construct‐expressing OSCs, using EGFP, Nxf2, and Piwi antibody.
- Schematic of full‐length Nxf2 and deletion mutant proteins. LRR: leucine‐rich repeat domain, NTF2‐like: nuclear transport factor 2‐like domain, UBA: ubiquitin‐associated domain (left panel). The results of rescue experiments and tethering assay are summarized (right panel).
- IP using anti‐myc antibody from lysate of OSCs expressing myc‐tagged Nxf2 proteins, followed by WB using anti‐myc or anti‐Panx antibody.
- mdg1 expression levels were monitored by qRT–PCR in OSCs expressing exogenous Nxf2 proteins treated with either siLuc (control) or siNxf2 (left panel). Error bars indicate SD (n = 3). Exogenous Nxf2 mRNAs are resistant to siRNA for Nxf2. Expression levels of Panx‐ and myc‐tagged Nxf2 proteins were confirmed by WB using anti‐myc and anti‐Panx antibodies. Tubulin was used as a loading control. Asterisk indicates the background signal (right panel).
- Effects of λN fusion Nxf2 proteins on luciferase activity at 48 hpt. Error bars indicate SD (n = 3) (left panel). Expression levels of exogenous λN‐HA–myc‐Nxf2 proteins (right panel).
- Electrophoretic mobility shift assay (EMSA) showing the binding of the first LRR region of Nxf2 (1–285 aa) to single‐stranded RNA. The indicated amount of recombinant protein was mixed with 1 nM single‐stranded RNA (16 nt).
Figure EV5. Both N‐terminal and C‐terminal regions of Panx are essential for TE silencing.
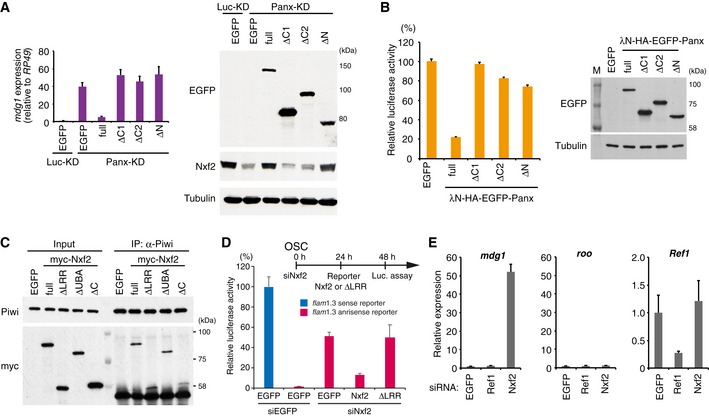
- EGFP‐tagged Panx deletion constructs were expressed in Luc‐ (control) or Panx‐depleted OSCs. mdg1 expression levels were monitored by qRT–PCR. Expression values are normalized by the expression of RP49. Error bars indicate SD (n = 3) (left panel). Western blotting (WB) with an antibody against EGFP, Nxf2, and Tubulin using lysates from transfected OSCs (right panel). The deletion mutants that cannot interact with Nxf2 or Panx could not induce silencing of TE. In addition, the C‐terminal region of Panx (400–541 aa) is required for silencing of TE and stable expression of Nxf2.
- Luciferase fluorescence level in OSCs expressing boxB reporter luciferase and the indicated λN fusions (Panx deletion mutants). Error bars indicate SD (n = 4) (left panel). WB of lysates from OSCs expressing boxB reporter luciferase and the indicated λN fusions using EGFP and tubulin antibody (right panel). The deletion mutants that cannot interact with Nxf2 or Panx could not induce silencing upon recruitment to reporter RNA.
- Immunoprecipitation (IP) from lysate of OSCs expressing myc‐tagged Nxf2 proteins using anti‐Piwi antibody, followed by WB using anti‐myc and ‐Piwi antibodies. All mutant proteins of Nxf2 can interact with Piwi in OSCs.
- Silencing of reporter gene harboring flam element (1.3 kbp, antisense) occurs in an Nxf2‐dependent manner. Transfection schedule of siRNA and plasmids is shown at the top of the figure. Exogenous expression of Nxf2 protein repressed reporter gene activity in OSCs, but that of Nxf2‐ΔLRR protein did not. Error bar indicates SD (n = 3).
- RNA levels of mdg1, roo, and Ref1 were quantified by qRT–PCR upon depletion of EGFP (control), Ref1, or Nxf2. Expression levels are normalized by the expression of RP49. Error bars represent SD (n = 3). Although Ref1 knockdown significantly decreased the expression level of Ref1, mdg1 TE was unaffected.
Using the deletion mutants of Nxf2 (Fig 5C), we further performed rescue experiments by expressing each deletion mutant in Nxf2‐depleted OSCs (Fig 5E). The expression of Panx protein could be stabilized not only by full‐length Nxf2 but also by Nxf2‐ΔLRR (leucine‐rich repeat), showing that mutants that interact with Panx can rescue its protein level. However, Nxf2‐ΔLRR, which associates with Panx and also Piwi (Figs 5D and EV5C), could not recover the silencing of mdg1 TE, suggesting that the LRR region of Nxf2 has an essential function other than formation of the PPNP complex. Consistent with this, piRNA‐targeted reporters that were de‐silenced upon depletion of Nxf2 (Fig EV3F) could be rescued by full‐length Nxf2, but not by Nxf2‐ΔLRR (Fig EV5D). We next investigated which of the Nxf2 mutants could induce silencing, when tethered to the reporter gene (Fig 5F). Interestingly, in addition to full‐length Nxf2, Nxf2‐ΔLRR mutant could induce silencing, indicating that the LRR region is dispensable once Nxf2 is tethered to the target gene. These results suggest that the LRR domain of Nxf2 is required to maintain recruitment of the PPNP complex to its targets.
It has been shown that Nxf1, the other NXF member in Drosophila, associates with bulk mRNA at its N‐terminal RNA binding domain to induce its export (Viphakone et al, 2012). Additionally, a human homolog of Nxf2 associates with RNA through its N‐terminal region (1–377 aa; Herold et al, 2000). Meanwhile, both Nxf1 and Nxf2 in human use the N‐terminal region to bind Alyref, a key factor for recruitment of Nxf1 to mRNA (Herold et al, 2000). To investigate the possible function of N‐terminal region of Drosophila Nxf2 in piRNA pathway, we first preformed knockdown of Alyref homolog in Drosophila. There are two homologs of Alyref in Drosophila, and Ref2 is not expressed in ovary nor OSCs (Gramates et al, 2017). We therefore performed knockdown of Ref1, which did not result in de‐silencing of mdg1 TE (Fig EV5E). We further performed an electrophoretic mobility shift assay (EMSA) to analyze the RNA binding activity of the N‐terminal LRR domain of Nxf2 and observed an association of single‐stranded RNA with the purified GST‐tagged first LRR domain of Nxf2 (1–285 aa) (Fig 5G). These results suggest that Nxf2 requires the LRR domain, which harbors RNA binding activity, in addition to the other domains necessary for formation of the PPNP complex, to tether the silencing complex to its target transcripts.
Nxf2 directly interacts with piRNA‐targeted TE transcripts in a Piwi‐dependent manner
The RNA binding activity of Nxf2 prompted us to elucidate the mechanistic details of how Nxf2 is recruited to TE transcripts. We investigated cellular transcripts associated with Nxf2 and/or Piwi via UV cross‐linking and immunoprecipitation (CLIP) assay (Flynn et al, 2015, Van Nostrand et al, 2016). We first raised a stable cell line of OSCs that express siRNA‐resistant myc‐Nxf2, since our Nxf2 antibody is not suitable for immunoprecipitation. The expression level of myc‐Nxf2 is slightly higher than that of endogenous Nxf2, and Nxf2‐siRNA specifically depletes endogenous Nxf2 (Fig EV6A). myc‐Nxf2 forms a complex identified using antibody against endogenous Panx and is capable of silencing mdg1 TEs (Fig EV6B and C). We therefore used this stable line to analyze the RNA population targeted by Nxf2.
Figure EV6. Stably expressed myc‐Nxf2 associates with transcripts of Piwi‐piRNA target TEs.
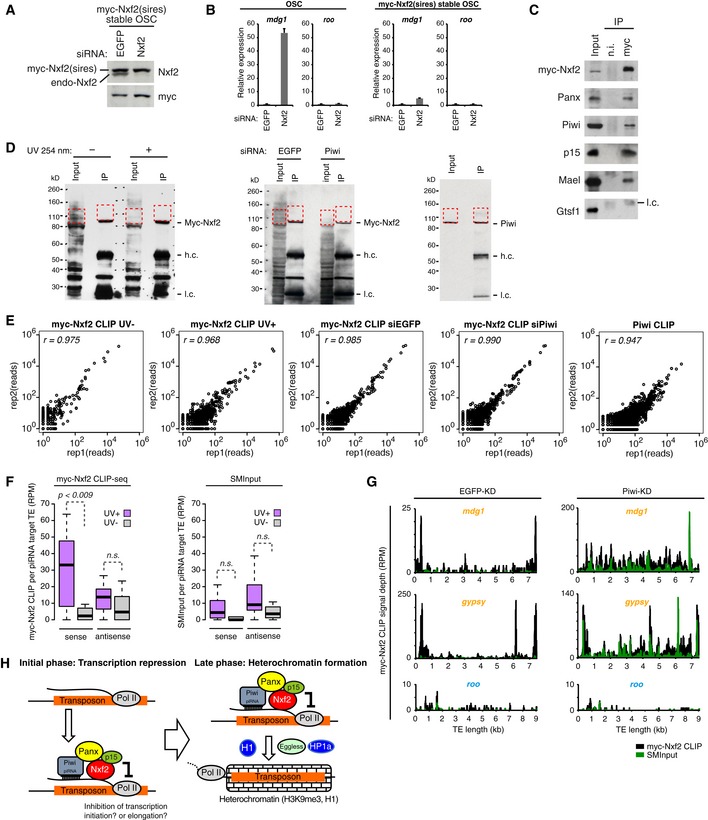
- Western blotting (WB) of myc‐Nxf2 stable OSC lysate, using Nxf2 and myc antibody upon knockdown of EGFP (control) or Nxf2. Note that expressed myc‐Nxf2 is resistant to siRNA.
- RNA levels of mdg1 and roo were quantified by qRT–PCR upon depletion of EGFP (control) or Nxf2. Expression levels are normalized by the expression of RP49. Error bars represent SD (n = 3). In OSCs without myc‐Nxf2 expression, mdg1 is de‐silenced upon Nxf2‐KD, whereas in the myc‐Nxf2‐expressing stable line, mdg1 remains silenced.
- Immunoprecipitation (IP) from myc‐Nxf2‐expressing OSC lysate using anti‐myc antibody, followed by WB of myc‐Nxf2 (by myc antibody), Panx, Piwi, p15, Mael, and Gtsf1. l.c.: light chain from the antibody.
- WB of myc‐Nxf2 IP during CLIP from non‐irradiated or UV‐irradiated cells (left panel). WB of myc‐Nxf2 IP during CLIP from EGFP or Piwi knockdown cells (middle panel). WB of Piwi IP during CLIP (right panel). Red dotted line indicates the region excised for CLIP library preparation.
- Scatter plots of obtained CLIP reads in two biological replicates of indicated CLIP experiments. Each dot represents the read count of the peak called using the Piranha peak‐calling algorithm (Uren et al, 2012).
- Boxplots showing piRNA‐targeted TE‐mapped read counts (RPM) obtained from myc‐Nxf2 CLIP or SMInput, based on CLIP‐seq performed on UV‐irradiated and non‐irradiated OSCs. Reads mapped in sense and antisense directions were calculated separately. Boxplot central bands, boxes, and whiskers show median, third quartile, first quartile, maxima, and minima, respectively. P‐values were calculated by Wilcoxon rank‐sum test.
- Density plots for myc‐Nxf2 CLIP signal depth over the consensus sequence from mdg1, gypsy (targeted by Piwi–piRNA, in orange letters), and roo (not targeted by Piwi–piRNA, in blue letters) TEs in EGFP‐, Piwi‐KD OSCs. Reads obtained in myc‐Nxf2 CLIP samples are indicated in black, where SMInput is indicated in green.
- Schematic model showing the two‐phase regulation of TEs by Piwi–piRISC. Nxf2 forms a complex with Panx, Piwi, and p15 and associates with nascent RNA of target transposable elements. This complex regulates transcription of the TE by the inhibition of Pol II (initial phase). The co‐transcriptionally regulated TE shifts to heterochromatin formation, mediated by H3K9me3 marks and H1 (late phase).
We generated CLIP libraries using RNA isolated from myc‐Nxf2 or Piwi immunoprecipitants. Immunoprecipitants were run on standard protein gels and transferred to nitrocellulose membranes, and a region 50 kDa above the protein size was excised for RNA isolation (Fig EV6D), referring to the eCLIP method (Van Nostrand et al, 2016). The same region was excised for the input sample in order to generate a size‐matched input (SMInput) library, which enables efficient background normalization and then leads to a more accurate measure of the enrichment of CLIP signals (Van Nostrand et al, 2016). Each CLIP experiment was performed in two biological replicates, and the reads were merged after checking the correlation of read counts of annotated peaks (Fig EV6E). We first performed CLIP assay of myc‐Nxf2 protein in UV‐crosslinked and non‐UV‐crosslinked samples, to examine whether RNA associated with myc‐Nxf2 could be captured by this assay. We observed a significant decrease in the amount of CLIP tags obtained for piRNA‐targeted TE transcripts when CLIP was performed on non‐UV‐crosslinked samples (Fig EV6F), indicating that we can specifically capture target RNA by the myc‐Nxf2 CLIP assay.
Based on these results, we further performed myc‐Nxf2 CLIP assay upon control (EGFP) or Piwi knockdown. Endogenous Nxf2 was also depleted, in order to increase the ratio of functioning myc–Nxf2. We first compared the enrichment of obtained CLIP tags to that of SMInput control, for the reads mapped in the sense or antisense direction of TEs (Fig 6A). This revealed that there was some fraction of TEs with enrichment of CLIP tags mapped in the sense direction, whereas this enrichment was not observed for the reads mapped in the antisense direction. Additionally, we plotted the TEs targeted by Piwi–piRNA complexes, which showed that TEs with enriched CLIP tags were the targets of piRNAs (Fig 6A). Notably, the specific enrichment of myc‐Nxf2 CLIP tags on piRNA‐targeted TEs was lost upon the knockdown of Piwi (Figs 6B and EV6G, see also Fig 6D), indicating that Piwi is required for Nxf2 to specifically associate with piRNA‐targeted transcripts.
Figure 6. Nxf2 associates with nascent transcript of Piwi–piRNA‐targeted TEs.
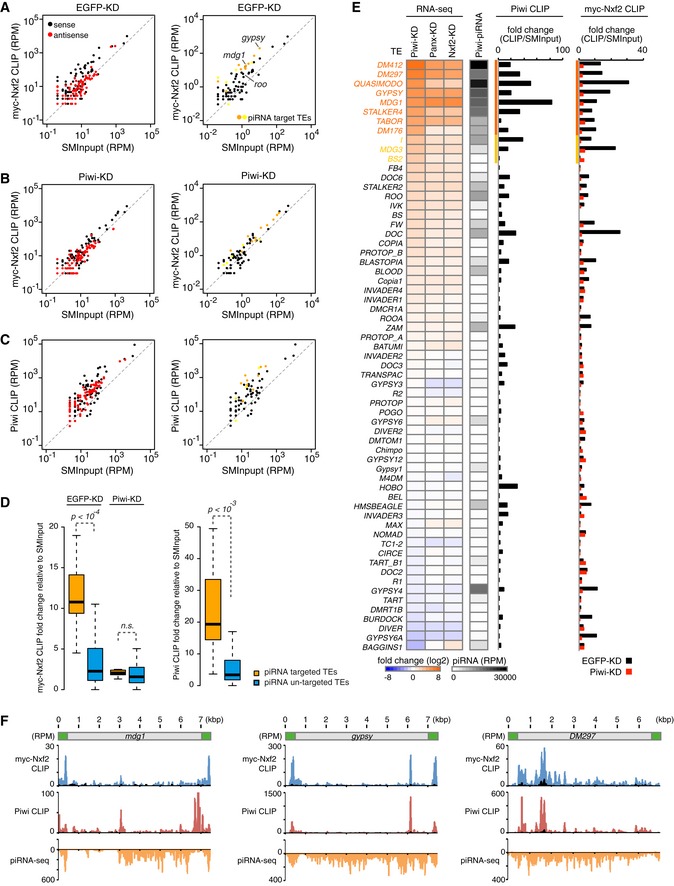
- Scatterplot of RPM values for myc‐Nxf2 CLIP tags (y‐axis) and SMInput reads (x‐axis) mapped to TEs in EGFP (control)‐KD samples. Both x‐axis and y‐axis are log10 scales. Sense reads are plotted in black and antisense reads in red (left panel). TEs for which the expression level differed from that of the control by more than tenfold or fivefold in Piwi‐KD (Piwi–piRNA‐targeted TEs) are plotted in orange or yellow, respectively (right panel).
- Scatterplot as in (A). The plot was created using myc‐Nxf2 CLIP data from Piwi‐KD samples.
- Scatterplot as in (A). The plot was created using Piwi CLIP data. Note that RNAs used for creating Piwi CLIP libraries are in the size range of 43–73 nt, and therefore, piRNAs are excluded.
- Boxplots showing fold changes of the indicated CLIP tags to SMInput, for Piwi–piRNA‐targeted and un‐targeted TEs. Piwi–piRNA‐targeted TEs are defined based on RNA‐seq analysis (Fig 1). Reads mapped to sense direction of indicated TEs were used for calculation of CLIP tag fold change, and TEs with SMInput signal under 0.5 RPM were eliminated. Boxplot central bands, boxes, and whiskers show median, third quartile, first quartile, maxima, and minima, respectively. P‐values were calculated by the Wilcoxon rank‐sum test. n.s., not significant, P > 0.01.
- Heatmaps displaying fold changes of TE expression with indicated siRNA knockdown (normalized to EGFP‐KD), and Piwi‐associated piRNA levels obtained from small RNA‐seq (RPM). The CLIP‐seq diagram indicates fold changes of Piwi CLIP tags, and myc‐Nxf2 CLIP tags relative to SMInput, obtained upon EGFP‐ or Piwi‐KD. Reads mapped to sense direction of indicated TEs were used for calculation of CLIP tag fold change, and TEs with SMInput signal under 0.5 RPM were eliminated. Piwi‐bound piRNA levels were determined by using reads mapped to antisense of the indicated TEs. TEs for which the expression level differed from that of the control by more than tenfold or fivefold in Piwi‐KD (Piwi–piRNA‐targeted TEs) are indicated in orange or yellow, respectively.
- Density plots for myc‐Nxf2 CLIP tags (blue plots), Piwi CLIP tags (red plots), and Piwi–piRNA reads (orange plots) over the consensus sequence from mdg1, gypsy, and DM297 (Piwi–piRNA target TEs). The LTR region of each TE is illustrated in green. SMInput is overlaid in black at each plot; y‐axis has been adjusted to 100 for the plot of Piwi CLIP tags mapped to mdg1.
To further investigate the relationship between RNA targeting of Nxf2 and Piwi, we performed Piwi CLIP assay. As in the case of myc‐Nxf2 CLIP, we observed the specific enrichment of CLIP tags against SMInput, specifically for those mapped in the sense direction of piRNA‐targeted TEs (Fig 6C and D). We compared the expression level of TEs under Piwi‐, Panx‐, and Nxf2‐KD, and also piRNA reads from Piwi‐immunoprecipitant (Fig 6E). This revealed that TEs with enriched myc‐Nxf2 and Piwi CLIP tags were de‐silenced upon the depletion of Piwi, Panx, or Nxf2, and they were also targeted by piRNAs. A density plot of myc‐Nxf2 and Piwi CLIP tags, along with piRNA reads, showed that myc‐Nxf2 and Piwi CLIP tags had similar distributions, while piRNAs that can target wider regions of TEs were produced (Fig 6F). This indicates the possibility that specific piRNAs can silence certain transposons more efficiently than others. Since CLIP peaks tend to be detected on LTR regions or 5′ regions of transposons, piRNAs may silence their targets as soon as they are transcribed, and therefore, downstream RNAs may not be produced. These results, together with the finding that PPNP association is essential in order to stabilize the protein level of Nxf2 (Fig 2), suggest that the PPNP complex is recruited to nascent transcripts by base‐pairing of Piwi‐associated piRNAs with targets, and further enforced through the RNA binding activity of Nxf2.
Discussion
Here, we propose a model in which the engagement of the PPNP complex with nascent TE transcripts initiates co‐transcriptional silencing prior to heterochromatin formation (Fig EV6H). Although the heterochromatin factors are not required for the initial step of the silencing (Fig 4), they play a significant role in maintenance of the repressed status in the later stages of TE silencing. Because the depletion of Piwi activity rapidly results in the de‐repression of TEs (Fig 1), this also suggests that silencing initiation and heterochromatin formation undergo continuous cycling to enforce a silenced state of TEs.
Four groups including ours have obtained the similar findings regarding the essential role of Nxf2 in the Piwi–piRNA pathway rather than in nuclear RNA export (preprint: Batki et al, 2019; Fabry et al, 2019, preprint: Zhao et al, 2019). We all have identified a new complex of Panx–Nxf2–P15, which associates with Piwi. Although Batki et al and Fabry et al did not observe significant amount of Piwi peptides in their shotgun MS analysis of Panx immunoprecipitants, they showed the interaction between Panx‐Piwi in immunoprecipitation followed by Western blot analysis and their former studies (preprint: Batki et al, 2019; Fabry et al, 2019; Sienski et al, 2015; Yu et al, 2015). The inconsistency could be due to the immunoprecipitation conditions and/or antibodies used. Additionally, we all have shown that the UBA domain of Nxf2 associates with Panx, and the NTF2 domain associates with p15. Our group and Batki et al have shown that the first LRR domain with RNA binding activity is needed for the regulation of TEs (preprint: Batki et al, 2019), and we have further shown direct association of TE transcripts with Nxf2 (Fig 6). Among these four groups, we were the only one which was able to observe the initial steps of the silencing. This is because we performed our analysis using cultured cell line OSCs, and took different timepoints after expression of Nxf2 (Fig 4). The present study is the only one to consider time points as early as 48 h. The regulation observed at 96 h after transfection, representing later stages of TE silencing, was consistent with the observation by Batki et al (preprint: Batki et al, 2019). An important issue to be clarified is the mechanism by which the PPNP complex initiates the silencing of TEs.
In C. elegans, NRDE‐2 (nuclear RNAi defective‐2) associates with the Argonaute protein NRDE‐3 in the nucleus and is recruited by the NRDE‐3/siRNA complex to nascent transcripts. This complex inhibits Pol II during the elongation and directs the accumulation of Pol II at genomic loci targeted by RNAi (Guang et al, 2010). We investigated whether λN‐Nxf2 directs the accumulation of Pol II at boxB sites in the co‐transcriptional silencing (Appendix Fig S1A–C). Although the time course of Pol II occupancy upon λN‐Nxf2 was examined by ChIP‐qPCR at the boxB sites, we could not observe any accumulation of Pol II along the reporter gene.
Recently, it was reported that Pol II‐associated proteins, PAF1 and RTF1, antagonize Piwi‐directed silencing (Clark et al, 2017), which are factors involved in the modulation of promoter release, elongation, and termination of Pol II (Chen et al, 2015; Van Oss et al, 2017; Vos et al, 2018). The Panx–Nxf2–P15 complex might interfere with Pol II via the PAF1 complex, considering that fission yeast PAF1 represses AGO1/siRNA‐directed silencing (Kowalik et al, 2015). Although we examined the effect of PAF1 and RTF1 on the co‐transcriptional silencing, PAF1 and RTF1 do not appear to be involved in the co‐transcriptional silencing mediated by λN‐Nxf2 (Appendix Fig S1D–I). These results suggest that Pol II regulation by the PPNP complex differs from that of the small RNA‐mediated Pol II regulation model proposed previously. It may be necessary to identify key factors involved in this regulation, possibly by performing proteome analysis of the unknown factors tethered to our reporter recruitment system.
We found a decrease in the active histone H3K4me3 marks on the reporter gene even at an earlier time point (48 hpt) at which the level of H3K9me3 marks remained low (Appendix Fig S2A and B, Fig 4C). Two reports about H3K4 methylation in Piwi‐mediated silencing have been published. The level of H3K4 methylation on TEs is increased upon Piwi‐KD (Klenov et al, 2014). The expression of artificial piRNAs that target a reporter locus induced transcriptional silencing associated with a decrease in the active H3K4‐methylation marks (Le Thomas et al, 2013). In addition, the depletion of LSD1, which removes H3K4‐methylation marks from promoters, had significant effects on the ability of Panx to repress the reporter gene (Yu et al, 2015). Therefore, we performed LSD1‐KD and examined the effect of LSD1 on co‐transcriptional silencing by the enforced tethering of Nxf2. However, the depletion of LSD1 had a limited impact on the silencing, whereas the level of mdg1 was increased 27‐fold upon LSD1‐KD (Appendix Fig S2C–G). These findings together suggest that the decrease observed in H3K4me3 levels at 48 hpt in the tethering assay may not have been due to LSD1.
Nxf2 is the core factor of the PPNP complex, and our results suggest that the nuclear export factor variant has been co‐opted to repress TEs in the Piwi–piRNA pathway. We speculate that the co‐option of Nxf2 into TE silencing is not coincidental but may reflect previous TE adaptation to exploit cellular mRNA transport pathways promoting the export of TE transcripts and replication/transposition (Zhang et al, 2012; Checkley et al, 2013). Why is it necessary for Piwi to use the RNA binding activity of Nxf2 to silence their targets? Piwi, as a member of the Argonaute family, is thought to search for targets through random interactions between Piwi–piRNA and transcripts in the ocean of the transcriptome, many of which likely show partial complementarity with Piwi‐loaded piRNAs (Klein et al, 2017). To distinguish “scanning Piwi” and “target‐engaged Piwi”, the silencing pathway may need an engaged‐in system to repress only targets and avoid unnecessary silencing of other cellular transcripts. It is possible that Nxf2 plays a role in supporting the association of Piwi that has found its bona fide targets (Appendix Fig S2H). Panx preferentially associates with Piwi, which loads piRNAs targeting TEs (Sienski et al, 2015), suggesting that there may be a system for Panx–Nxf2 to form the PPNP complex, specifically with Piwi that is engaged with its target TEs. Our CLIP analysis shows that Nxf2 cannot associate with target TEs without the activity of Piwi (Fig 6), indicating that the Panx–Nxf2–P15 complex itself does not recognize target transcripts, but rather recognizes target‐engaged Piwi. In line with this, while piRNA‐targeted Piwi can repress the reporter gene (Fig EV3), λN‐tethered Piwi cannot (Fig 3). This may be due to the Panx–Nxf2–p15 complex recognizing only piRNA‐directed target‐engaged Piwi, but not scanning or λN‐tethered Piwi (Appendix Fig S2H and I). Further studies will be necessary to confirm this speculation and elucidate the precise mechanism by which the Panx–Nxf2–P15 complex selectively recognizes only the “target‐engaged Piwi” and forms a PPNP complex to induce silencing of its targets.
Materials and Methods
Cell culture and transfection
Ovarian somatic cells (OSCs) were established in the previous studies (Niki et al, 2006; Saito et al, 2009). Ovarian somatic cells were cultured in Shield and Sang M3 Insect Medium (Sigma) supplemented with 10% fly extract, 10% fetal bovine serum, 0.6 mg/ml glutathione, and 10 μg/ml insulin as described previously. For the transfection of small interfering (si)RNA into OSCs, 40–300 pmol siRNA duplex was nucleofected into 3.0 × 106 cells using the Cell Line 96‐well Nucleofector Kit SF (Lonza) and program DG150 of the 96‐well Shuttle Device (Lonza). The siRNAs were transfected twice for 4‐day KD, and once for 2‐day KD. The siRNA sequences are listed in Appendix Table S3. Co‐transfection of siRNA and plasmid DNA was performed using the Cell Line Nucleofector Kit V (Lonza) and program T‐029 of the Nucleofector II Device (Lonza). For immunoprecipitation, expression vectors were transfected using Xfect Transfection Reagent (TaKaRa Clontech), in accordance with the manufacturer's instructions.
Drosophila strains
Oregon R (OR) was employed as a wild‐type strain. Lines harboring shRNA for Nxf2 (y[1] sc[*] v[1]; P{y[+t7.7] v[+t1.8] = TRiP.HMS00945}attP2/TM3, Sb[1]) and MTD‐Gal4 lines (P{w[+mC] = otu‐GAL4::VP16.R}1, w[*]; P{w[+mC] = GAL4‐nos.NGT}40; P{w[+mC] = GAL4::VP16‐nos.UTR}CG6325[MVD1]) were obtained from Bloomington Drosophila Stock Center (Stock# 33985 and 31777, respectively). For the tethering assay in fly ovary, GFP reporter line (If/CyO; tub>EGFP_5xBoxB_SV40 [attP2]/TM3, Ser), λN line as a control (pUASp>lambdaN‐HA [attP40]/CyO; tub>EGFP_5xBoxB_SV40 [attP2]/TM3, Ser), λN‐Piwi line (pUASp>lambdaN‐HA‐Piwi [attP40]/CyO; tub>EGFP_5xBoxB_SV40 [attP2]/TM3, Ser), and λN‐Panx line (pUASp>lambdaN‐HA‐CG9754 [attP40]/CyO; tub>EGFP_5xBoxB_SV40 [attP2]/TM3, Ser) were obtained from Vienna Drosophila Resource Center (VDRC) (Stock# 313408, 313390, 313392, and 313393, respectively). All stocks were maintained at 25°C. For calculation of the egg hatching rate, 20 females were mated with Oregon R males in yeasted apple agar plates. The females were left in the plate to continue laying eggs after mating. Laid eggs were counted after 24 h, and hatched eggs were counted after another 24 h.
Construction of expression plasmids
To produce expression vectors, full‐length cDNA was amplified using primers described in Appendix Table S3. First‐strand cDNA was synthesized from total RNA isolated from the OR ovaries or OSCs. PCR products were then inserted into the pGEX‐5X expression vector (GE Healthcare Bioscience), pET28 expression vector (Novagen), pAcM vector (Saito et al, 2006), pAcF vector, or pAcEGFP vector (EGFP cloned into the myc tag region of the pAcM vector), using restriction enzyme cloning or In‐Fusion HD Cloning Kit (Clontech) (described in Appendix Table S3). The pGEX‐5X or pET28 expression vector was used to express GST‐ or HIS‐fused proteins, and the pAcM, pAcF, or pAcEGFP vector was used to express myc‐, Flag‐, or EGFP‐tagged proteins in OSCs. To yield siRNA‐resistant mutants and mutants for domain mapping experiments, inverse PCR was performed using the described oligonucleotides (Appendix Table S3). The inserted fragment for the pAcF‐Nxf2 vector was obtained by HindIII/EcoRI restriction enzyme digestion of the pAcM‐Nxf2 vector and ligated into the pAcF vector. Note that plasmid construction for the tethering assay is described in the “Tethering assay using OSCs” and “Tethering assay in fly ovary” sections.
Production of antibodies (anti‐Panx, anti‐Nxf2, anti‐Eggless, anti‐p15)
The middle fragment of the Panx protein (181–361 aa), N‐terminal region of the Eggless protein (1–405 aa), or full‐length p15 protein (1–134 aa) fused with glutathione S‐transferase (GST), and full‐length Nxf2 (1–841 aa) fused with histidine (HIS) were used as antigens to immunize mice. The anti‐Panx, ‐Nxf2, and ‐Eggless monoclonal antibodies were produced essentially as described previously (Ishizuka et al, 2002; Saito et al, 2006). For the anti‐p15 antibody, serum from immunized mice was used for a polyclonal antibody.
Immunoprecipitation
To obtain whole‐cell lysate, OSCs were washed in PBS and lysed in IP buffer (50 mM HEPES‐KOH pH 7.4, 200 mM KCl, 1 mM EDTA, 1% Triton X‐100, 0.1% Na‐deoxycholate or 30 mM HEPES‐KOH pH 7.4, 150 mM KOAc, 5 mM MgOAc, 5 mM DTT, 0.1% NP‐40) followed by probe‐sonication (BRANSON, SFX150, or Bioruptor). After centrifugation, the supernatant was used for immunoprecipitation. To prepare nuclear lysate, OSCs were washed in PBS and suspension buffer (10 mM HEPES‐KOH pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT) and then homogenized in suspension buffer by passing through a syringe with a 25G needle. After centrifugation, supernatant was recovered as cytoplasmic lysate. The nuclear pellet was washed once and then resuspended in IP buffer, followed by probe‐sonication. The nuclear lysate was centrifuged and the supernatant was kept for immunoprecipitation. Antibodies were immunized on Dynabeads Protein G (Thermo Fisher) and incubated with lysates for 1–2 h, at 4°C. Then, beads were washed three to five times in IP buffer. Control immunoprecipitation using mouse nonimmune IgG (IBL) was conducted in parallel.
Western blotting
Western blotting was performed as described previously (Saito et al, 2006). Besides the antibodies generated in this study, anti‐Piwi (Saito et al, 2006; hybridoma supernatant), anti‐DmGTSF1/Arx (Ohtani et al, 2013) (hybridoma supernatant), anti‐Mael (Sato et al, 2011) (hybridoma supernatant), anti‐H1 (Iwasaki et al, 2016) (hybridoma supernatant), anti‐Tubulin (E7, hybridoma supernatant), anti‐myc (9E10, hybridoma supernatant), anti‐flag (M2, sigma) (1:1,000), anti‐H3 (ab1791, Abcam) (1:2,000), anti‐HP1a (C1A9, DSHB) (1:1,000), anti‐HA (HA‐7, Sigma) (1:1,000), and anti‐GFP (11814460001, Roche) (1:1,000) antibodies were used.
Silver staining and mass spectrometry analysis
Silver staining was performed using the SilverQuest Silver Staining Kit (Invitrogen), in accordance with the manufacturer's instructions. The bands shown in Fig 1A were excised, de‐stained, and sent for liquid chromatography–tandem mass spectrometry (MS) analysis to identify the peptides (Support Center for Advanced Medical Sciences, The University of Tokushima, Japan). The results were analyzed using Mascot (Matrix Science), and Scaffold4 (Proteome Software Inc.) was used to validate MS/MS‐based peptides.
Immunofluorescence
Immunofluorescence of OSCs and Drosophila ovaries was performed using anti‐Panx IgG1 (hybridoma supernatant) or anti‐myc (9E10, hybridoma supernatant) antibody as described previously (Ishizuka et al, 2002). Alexa Fluor 546‐ or 488‐conjugated anti‐mouse IgG1 (Molecular Probes) (1:1,000 dilution) was used as secondary antibody. Slides were mounted using VECTASHIELD Mounting Medium with DAPI (Vector Laboratories).
Quantitative reverse‐transcription PCR and northern blotting
qRT–PCR analysis and northern blotting were performed as previously described (Iwasaki et al, 2016). Oligonucleotides used for qRT–PCR primers and northern blotting probes are described in Appendix Table S3. Previously described primers (Sienski et al, 2015) were used for qRT–PCR analysis of TE expression levels in ovary.
Shotgun proteome analysis
Immunoprecipitated proteins were eluted with 100 mM Tris–HCl (pH 8) and 2% sodium dodecyl sulfate (SDS). To remove the SDS from the eluted samples, the methanol–chloroform protein precipitation method was used. Briefly, four volumes of methanol, one volume of chloroform, and three volumes of water were added to the eluted sample and mixed thoroughly. Samples were centrifuged at 20,000 g for 10 min, the water phase was carefully removed, and then, four volumes of methanol were added to the samples, followed by centrifugation at 20,400 g for 10 min. The supernatant was removed, and the pellet was washed once with 100% ice‐cold acetone. Precipitated proteins were re‐dissolved in guanidine hydrochloride and reduced with Tris(2‐carboxyethyl)phosphine hydrochloride, alkylated with iodoacetamide, and then digested with lysyl endopeptidase and trypsin. Digested peptides were analyzed using a direct nanoflow liquid chromatography system coupled to a time‐of‐flight mass spectrometer (QSTAR Elite, Sciex). Mass spectrometry and tandem mass spectrometry spectra were obtained in the information‐dependent acquisition mode and were queried against the NCBI non‐redundant database with an in‐house Mascot server (Natsume et al, 2002) (version 2.2.1; Matrix Science).
Shotgun proteome data analysis
Shotgun proteome was performed in two biological replicates, and two technical replicates per each sample (in total of four measurements per sample). The obtained peptide numbers for four measurements were used to calculate Enrichment and Specificity of obtained protein candidate. Enrichment was calculated by dividing the total number of peptides obtained in Flag‐Nxf2‐IP samples by control samples, and taking log2. Specificity was calculated as P‐value obtained from t‐test of peptide counts obtained for Flag‐Nxf2‐IP samples against control IP samples.
EMSA
For EMSA, 16‐mer single‐stranded RNA (5′‐AGCACCGUAAAGACGC‐3′) described previously (Vourekas et al, 2015) was 5′‐end‐radiolabelled by p32 and 1 nM was incubated with the indicated amount of GST protein for 15 min at room temperature in 10 μl of RNA binding buffer [50 mM Tris–HCl (pH 7.5), 50 mM KOAc, 2 mM MgCl2, 1 mM DTT, and 1 U/μl RNasin]. After incubation, 1 μl of 50 mg/ml heparin was added and incubated for another 10 min, and the reactions were analyzed using 5% native PAGE (Tris‐glycine gel) at 4°C. The results were visualized by phospho‐imaging.
Tethering assay using OSCs
We established a tethering assay system in OSCs based on the system in fly ovary, which was reported by Hannon's group (Yu et al, 2015). To obtain firefly luciferase reporter constructs, a 5× boxB fragment was amplified from fly genomic DNA (VDRC #313408) and cloned into an EcoRI and XhoI sites of pAc5.1B, followed by insertion of luciferase fragment amplified from pCMV‐luc at a KpnI and EcoRI site, resulting in pAc‐Fluc‐5boxB. To obtain pAc‐Fluc‐10boxB, PCR‐amplified 5× boxB carrying a SalI and XhoI site was inserted into a XhoI site of pAc‐Fluc‐5boxB. To produce pUb‐Fluc‐10boxB, we replaced the actin 5c promoter of pAc‐Fluc‐10boxB with a ubiquitin promoter at BglII and KpnI sites. The ubiquitin promoter was amplified from fly genomic DNA. For the intronic boxB reporter construct, we first removed the XhoI site in pAc‐Fluc and replaced its actin 5c promoter with the ubiquitin promoter. Subsequently, 5× boxB fragments were inserted twice into the XhoI site within the intron of the ubiquitin promoter. In this process, a reporter construct with 14× boxB in the intron, pUb‐14boxB‐Fluc, was incidentally obtained, and its sequence was confirmed. For λN‐HA fusion protein expression constructs, a λN‐HA fragment was amplified from a plasmid, pUASp‐λN‐Empty (kindly provided by Dr. Brennecke), and inserted into an NheI site of pAcM‐based and pAcEGFP‐based expression plasmids. The oligonucleotides used for plasmid construction are described in Appendix Table S3.
Isolation of the OSC line carrying reporter gene was performed essentially as described previously (Sumiyoshi et al, 2016). OSCs were co‐transfected with a reporter construct and a plasmid carrying a blasticidin‐resistance gene, using ScreenFect A (Wako), followed by selection with 25 μg/ml blasticidin for 24 h. Subsequently, 1 × 105 cells were passaged in a 6‐cm dish and allowed to form colonies in medium supplemented with or without a lower concentration of blasticidin (4–8 μg/ml). Some colonies were isolated and selected using a luciferase assay.
For the tethering assay, 3 × 106 OSCs were transfected with 5 μg of plasmid DNA using the Cell Line Nucleofector Kit V (Lonza) and Program T‐029 of the Nucleofector II Device (Lonza) and passaged in four wells of a 24‐well tissue culture plate. Cells were lysed in 150 μl of 1× Glo Lysis Buffer (Promega). Aliquots of cell lysates were used to measure firefly luciferase activity using ONE‐Glo Ex Luciferase Assay system (Promega) and Cytation 5 (BioTek). Total protein concentration of cell lysates was measured by a Bradford assay (Bio‐Rad).
Tethering assay in fly ovary
Based on previous reports, we carried out a tethering assay in fly ovary (Sienski et al, 2015; Yu et al, 2015). For the RNA‐tethering system in fly ovary, a DNA fragment of myc‐Nxf2 was inserted at NheI and EcoRI sites in pUASp‐λN‐MCS, resulting in pUASp‐λN‐HA‐myc‐Nxf2. To obtain pUASp‐λN‐MCS, a λN fragment including multiple cloning sites was inserted between KpnI and NotI sites in pUASp‐λN‐Empty (kindly provided by Dr. Brennecke). Oligonucleotides used for plasmid construction are described in Appendix Table S3. A transgenic line was established by phiC31‐mediated integration of pUASp‐λN‐HA‐myc‐Nxf2 into the attP40 landing site (Bischof et al, 2007; Markstein et al, 2008), resulting in a transgenic line (y'w1118;attp40{lambdaN‐HA‐myc‐Nxf2}/CyO). Transgenic and GFP reporter flies were crossed with a double balancer (Sp/CyO; Pr/TM3, Ser, sb), and then, the progeny were crossed together, resulting in a transgenic line carrying GFP reporter gene and λN‐HA‐myc‐Nxf2 gene (y'w1118; attp40{lambdaN‐HA‐myc‐Nxf2}/CyO; tub>EGFP_5xBoxB_SV40 [attP2]/TM3). Fly ovaries were fixed in 4% formaldehyde and stained with DAPI, followed by microscopic observation to detect GFP signal in the egg chamber.
piRNA reporter gene assay
We established a piRNA reporter assay system in OSCs, based on the reports by Lau's group (Post et al, 2014). We selected 0.6 kbp and 1.3 kbp flam elements, from which a number of piRNAs are derived, were cloned into the intron of pUb‐Fluc in sense orientation or in antisense orientation relative to the luciferase transcripts. These flam elements were amplified using primers described in Appendix Table S3. DNA and siRNA transfection, and luciferase assays in OSCs and S2 cells were performed as described in the “Tethering assay using OSCs” section.
ChIP‐qPCR analysis
For ChIP using anti‐RNA polymerase II antibody (8WG16; Santa Cruz), OSCs were fixed in 0.3% formaldehyde (methanol‐free; Thermo Fisher) and lysed in sonication buffer (50 mM Tris–HCl pH 7.9, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.1% Na‐deoxycholate), followed by probe‐sonication (BRANSON, SFX150). After centrifugation, the supernatant was passed through a 0.22‐μm syringe filter (Membrane Solutions) and used for immunoprecipitation. For ChIP using anti‐H1 (Iwasaki et al, 2016) (hybridoma supernatant), anti‐H3K9me3 (0319, Active motif), and anti‐H3K4me3 (ab8580, Abcam), OSCs were fixed in 1% formaldehyde and lysed in ChIP buffer (50 mM Tris–HCl pH 7.9, 5 mM EDTA, 0.1% SDS), followed by probe‐sonication. After centrifugation, the supernatant was mixed with dilution buffer (50 mM Tris–HCl pH 7.9, 250 mM NaCl, 1.6% Triton X‐100, 0.16% Na‐deoxycholate), passed through a 0.22‐μm syringe filter, and used for immunoprecipitation. Lysates were incubated with antibodies on Dynabeads Protein G for 3 h, at 4°C. Beads were washed in a series of wash buffers: low‐salt wash buffer (20 mM Tris–HCl pH 7.9, 150 mM NaCl, 2 mM EDTA, 1% Triton X‐100, 0.1% Na‐deoxycholate), high‐salt wash buffer (20 mM Tris–HCl pH 7.9, 500 mM NaCl, 2 mM EDTA, 1% Triton X‐100, 0.1% Na‐deoxycholate), LiCl wash buffer (10 mM Tris–HCl pH 7.9, 250 mM LiCl, 1 mM EDTA, 1% NP‐40, 0.5% Na‐deoxycholate), and TE (10 mM Tris–HCl pH 7.9, 1 mM EDTA). Protein–DNA complexes were eluted from beads in elute buffer (50 mM Tris–HCl pH 7.9, 10 mM EDTA, 1% SDS) at 65°C for 10 min, followed by reversing cross‐linking at 65°C for over 12 h in the presence of 200 mM NaCl. DNA was recovered and quantified with the Dice Real Time System III (TaKaRa) using TB Green Fast qPCR Mix (TaKaRa) and primer sets for the reporter gene (described in Appendix Table S3).
mRNA‐seq analysis
Preparation and sequencing of Poly(A)+ RNA libraries and computational analyses were performed as described previously (Iwasaki et al, 2016). Libraries were prepared from total RNA obtained from OSCs with 4‐day knockdown of the indicated genes and analyzed by Illumina HiSeq. This yielded 10–17 million reads per sample. For computational analysis of the obtained reads, Bowtie 2.2.9 (Langmead & Salzberg, 2012) was used with the default parameters to map sequences to the Drosophila genome (dm3). Reads mapped to the genome were then mapped to the Drosophila melanogaster TE consensus sequence from Repbase (Jurka, 1998), using Bowtie 1.0.1, and those that were uniquely mapped were extracted. Expression levels (RPKM) of TEs were calculated using reads that were mapped to the TE consensus sequence. Expression levels (RPKM) of coding genes were calculated using TopHat 2.0.14 (Trapnell et al, 2009) and DESeq2 (Love et al, 2014). Drosophila melanogaster (dm3) GTF files obtained from Illumina iGenomes were used to define known gene structures. Analysis of coding genes was performed using duplicate samples. Duplicate data were obtained for EGFP‐, Nxf2‐, and Panx‐KD samples, and previously published data (Iwasaki et al, 2016) were used as duplicate data for Piwi‐KD samples.
ChIP‐seq analysis
ChIP was performed as described previously (Iwasaki et al, 2016). Briefly, OSCs (3 × 107) were fixed and lysed, and their nuclei were isolated using truChIP Chromatin Shearing Kits (Covaris), in accordance with the manufacturer's instructions. Sodium deoxycholate was added to a final concentration of 0.4% prior to shearing. DNA was sonicated to ~300 bases using Bioruptor (Cosmobio) and then diluted with ChIP buffer [1 × 50 mM HEPES‐KOH pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, Halt Protease Inhibitor Cocktail (Thermo Scientific)]. DNA–protein complexes were incubated with 3 μg of anti‐H3K9me3 antibody (61013; Active Motif) on Dynabeads Protein G for 4 h, at 4°C. Beads were washed with ChIP buffer, high‐salt lysis buffer (50 mM HEPES‐KOH pH 7.4, 450 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.1% SDS), and wash buffer (50 mM Tris–HCl pH 8.0, 1 mM EDTA, 250 mM lithium chloride, 0.5% NP‐40, 0.5% SDS) followed by TE (10 mM Tris–HCl pH 8.0, 1 mM EDTA). After reversing cross‐linking for 12–16 h at 65°C, samples were treated with RNase for 30 min at 37°C and Proteinase K for 60 min at 55°C. DNA was then recovered. To prepare ChIP‐seq libraries, DNA fragments from the ChIP experiment were sheared to ~200 bases using Covaris S220 (Covaris). These were used for library preparation with the NEBNext Ultra DNA Library Prep Kit for Illumina II (NEB), following the manufacturer's protocol. ChIP‐seq libraries were sequenced with Illumina MiSeq using MiSeq Reagent Kit v3 for 150 cycles (Illumina). For ChIP‐seq analysis, adapters were removed from the reads using Cutadapt, and subsequent reads shorter than 50 nt were discarded. ChIP‐seq reads were mapped to the Drosophila genome (dm3) using Bowtie 1.0.1 (Langmead et al, 2009), using ‐v 2 –best parameters. Reads mapped to the genome were extracted and mapped to Drosophila melanogaster TE consensus sequences from Repbase (Jurka, 1998), permitting only those that mapped uniquely to the TE consensus sequence. Metaplots of ChIP signals were calculated using ngs.plot (Shen et al, 2014). Coordinates of TE insertions and the euchromatin/heterochromatin distribution within OSCs were obtained from a previous publication (Sienski et al, 2012). For peak calling of H3K9me3 signals, MACS‐1.4.2 (Zhang et al, 2008) was used with the default parameters. ChIP‐seq analysis was performed in duplicates for Panx and Nxf2 4‐day knockdown samples, and EGFP (control) and Piwi 4‐day knockdown samples were obtained from a previous study (Iwasaki et al, 2016).
CLIP‐seq
For the preparation of preadenylated 3′ adapter, DNA oligonucleotide (5′‐/5phos/TGGAATTCTCGGGTGCCAAGG/3Bio/‐3′;/5′ phos/ = 5′ phosphorylation,/3′ Bio/ = 3′ biotin) was custom‐synthesized by IDT and then preadenylated using the 5′ DNA Adenylation Kit (NEB) following the manufacturer's protocol. The reaction was ethanol‐precipitated and dissolved in water to a final concentration of 20 μM. cDNA synthesis primer (5′‐/5phos/NNAACCNNNGATCGTCGGACTGTAGAACTCTGAA/iSp18/CACTCA/iSp18/ATCTCCTTGGCACCCGAGAATTCCA‐3′;/iSp18/ = Carbon‐18 spacer) and Illumina sequencing primers [RP1: AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA, RPIx: CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA, 6‐bases index sequence in bold (see Illumina customer service letter for other index sequences)] were custom‐synthesized by IDT.
The iCLIP libraries were prepared as previously described (Flynn et al, 2015; Van Nostrand et al, 2016) with the following modifications. For iCLIP from stable cell line of myc‐Nxf2, three confluent 15‐cm plates (Nunc) per replicate were used. Cells were UV‐crosslinked with irradiance of 200 mJ/cm2 at 254 nm using UV Stratalinker 1800 (Stratagene). Cells were harvested and lysed with 2 ml of lysis buffer [20 mM HEPES‐KOH (pH7.3), 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.5% NP‐40, 2 μg/ml pepstatin, 2 μg/ml leupeptin, 0.5% aprotinin]. Cell extracts were treated with 1 U/μl RNase T1 (Roche) for 15 min at room temperature. The lysate was cleared by centrifugation at 16,500 g for 15 min at 4°C. The resulting supernatant was then collected and cooled on ice. 10 μl was reserved for size‐matched input (SMInput; Van Nostrand et al, 2016). RNA–protein complexes were immunoprecipitated using 40 μg of mouse monoclonal antibodies (anti‐myc 9E10, DSHB) and 200 μl of Protein G Dynabeads (Invitrogen) for 2 h, at 4°C. Beads were then washed three times with IP wash buffer [20 mM HEPES‐KOH (pH7.3), 300 mM NaCl, 1 mM DTT, 0.05% NP‐40, 2 μg/ml pepstatin, 2 μg/ml leupeptin, 0.5% aprotinin] and three times with high‐salt wash buffer [20 mM HEPES‐KOH (pH7.3), 500 mM NaCl, 1 mM DTT, 0.05% NP‐40, 2 μg/ml pepstatin, 2 μg/ml leupeptin, 0.5% aprotinin]. Washed beads were resuspended in 30 μl of 1×NuPAGE LDS sample buffer (Thermo Fisher) with 50 mM DTT and incubated at 70°C for 10 min. Released RNA–protein complexes were separated on the 4–12% NuPAGE Bis‐Tris gel (Thermo Fisher) with MOPS running buffer (Thermo Fisher) at 200 V for 1 h. After the run, RNA–protein complexes from the gel were transferred to a nitrocellulose membrane using the Novex wet transfer apparatus following the manufacturer's protocol (Thermo Fisher). The transfer was performed for 1 h at 30 V using NuPAGE transfer buffer (Thermo Fisher). Western blotting was performed using SNAP i.d. 2.0 System (Merck) following the manufacturer's instruction to visualize bands and determine regions for isolating RNA–protein complexes. Briefly, the membrane was blocked with 0.5% skim milk in PBS‐0.1% Tween‐20, incubated with 1:1,000 anti‐myc antibody, washed with PBS‐0.1% Tween‐20, and incubated with 1:5,000 HRP‐conjugated secondary antibody (Cappel). Membranes were developed with ECL substrate (GE) and exposed using film. A region 50 kDa above the protein size was cut into 1‐ to 2‐mm‐narrow strips and then transferred to a 1.5‐mL tube. To each tube, 200 μl of Proteinase K buffer [100 mM Tris–HCl (pH 7.5), 150 mM NaCl, 12.5 mM EDTA, 2% (w/v) SDS] and 10 μl of Proteinase K (Roche) were added and incubated for 30 min at 55°C. The RNA was recovered by acidic phenol/chloroform extraction followed by a chloroform extraction and an ethanol precipitation with Pellet Paint NF Co‐Precipitant (Merck Millipore). Finally, the pellet was dissolved in 16 μl of nuclease‐free water. The recovered RNA was mixed with dephosphorylation mix [2 μl of 10 × PNK buffer pH 6.0 (500 mM imidazole–HCl pH 6.0, 100 mM MgCl2, 50 mM DTT), 1 μl of RNasin (Promega), 1 μl of T4 PNK (NEB)] and incubated for 30 min at 37°C. The sample was incubated with 10 μl of SPRIselect (Beckman Coulter) and 30 μl of isopropanol for 5 min and then separated on the magnetic stand for 5 min. The supernatant was discarded, and the beads were washed twice with 500 μl of 85% ethanol. The beads were then air dried for 5 min and eluted with 10.2 μl of nuclease‐free water. The recovered RNA was mixed with 3′ adapter ligation mix (1 μl of 20 μM preadenylated 3′ adapter, 2 μl of 10 × T4 RNA ligase buffer, 4.8 μl of 50% PEG8000, 1 μl of RNasin, 1 μl of T4 RNA ligase2, truncated KQ (NEB)) and incubated overnight at 16°C. Ligation products were purified using 10 μl of SPRIselect and 10 μl of isopropanol in the same procedure as above, and eluted with 16 μl of nuclease‐free water. The sample was mixed with 5′ deadenylation mix (2 μl of 10 × NEB2 buffer, 1 μl of RNasin, 1 μl of 5′ Deadenylase (NEB)) and incubated 30 min at 30°C. To digest unligated adapters, 5 μl of RecJf exonuclease (NEB) was added and incubated 30 min at 37°C. Ligation products were purified using 12.5 μl of SPRIselect and 12.5 μl of isopropanol in the same procedure as above, and eluted with 12.2 μl of nuclease‐free water. 1 μl of 10 μM cDNA synthesis primer was added to the whole isolated RNA and annealed to the adapter by incubation at 70°C for 5 min and at 25°C for 1 min. RT mix [4 μl of 5 × Superscript III RT buffer, 0.8 μl of dNTP mix (10 mM each), 1 μl of 0.1 M DTT, 1 μl of Superscript III RT (Thermo Fisher)] was added, and a reverse transcription reaction was performed using the following program: 42°C for 40 min, 50°C for 15 min, and 4°C for hold. After reverse transcription reaction, 0.5 μl of RNase (Roche) and 0.5 μl of RNaseH (NEB) were added to each sample and incubated at 37°C for 15 min. During this time, 5 μl of Dynabeads M‐270 Streptavidin (Invitrogen) was washed twice with 200 μl of StrepBead wash buffer (100 mM Tris–HCl, pH 7, 1 M NaCl, 10 mM EDTA, 0.1% Tween‐20), for each sample. Each 5 μl volume of beads was finally resuspended in 40 μl of StrepBead wash buffer. After RNase digestion, 40 μl of pre‐washed beads was added to each sample and incubated at 25°C for 30 min with rotation. After incubating, the samples were applied to a magnetic stand and the buffer was discarded. Each sample was washed four times with 100 μl of wash buffer (100 mM Tris–HCl pH 7, 4 M NaCl, 10 mM EDTA, 0.2% Tween‐20) and twice with 100 μl of NT2 buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP‐40). The purified cDNA was then circularized by adding 20 μl of CircLigase Reaction Mix (2 μl of 10 × CircLigaseII Reaction Buffer, 1 μl of 50 mM MnCl2, 1 μl of CircLigaseII ssDNA Ligase (Epicentre), 16 μl of nuclease‐free water) to each sample, and incubating for 1 h at 60°C, 10 min at 80°C, and hold at 4°C. Samples were then applied to a magnetic stand, the reaction buffer was collected (be sure not to discard the reaction buffer), and another 20 μl of elution buffer (10 mM Tris pH 7.5 and 1 μM PRIx primer) was added to the beads and heated at 95°C for 2 min. Samples were then applied to a magnetic stand and the elution buffer was removed and combined with reaction buffer, collected earlier (20 μl). The elution was repeated for two times in total obtaining a final volume of 60 μl. cDNA was purified using 30 μl of SPRIselect and 60 μl of isopropanol in the same procedure as above, and eluted with 29.5 μl of nuclease‐free water. PCR mix [2.5 μl of 10 μM RP1 primer, 2.5 μl of 10 μM RPIx primer, 10 μl of 5 × Q5 Reaction Buffer, 5 μl of dNTP mix (2 mM each), 0.5 μl of Q5 DNA Polymerase (NEB)] was added to the cDNA, and PCR was performed using the following program: 98°C for 30 s; cycles of 98°C for 10 s, 60°C for 30 s, and 72°C for 15 s, followed by 4°C hold). DNA was amplified with 10 and 12 cycles of PCR for SMInput samples and immunoprecipitated samples, respectively. Amplified DNA was captured with 1.8 volumes of AMPure XP beads (Beckman Coulter) following the manufacturer's protocol and size‐selected on 6% PAGE gels. DNA was visualized with SybrGold (Thermo Fisher), and those with sizes between 170 and 200 bp were isolated. The PAGE gel was crushed, and the DNA was eluted in 400 μl of 0.4 M NaCl at 4°C on rotation overnight. The eluted DNA was ethanol‐precipitated with Pellet Paint NF Co‐Precipitant and then re‐amplified with the same PCR program and for 8 cycles each. PCR products were captured with 1.8 volumes of AMPure XP beads, size‐selected on 6% PAGE gels, and purified as described above. 1 μl of libraries was quantitated by HS‐DNA Bioanalyzer. Libraries were sequenced on the Illumina Miseq platform.
For Piwi iCLIP from OSCs, one confluent 10‐cm plate per replicate was used. Cells were lysed in 1 ml of lysis buffer, and immunoprecipitation was performed using 10 μg of anti‐Piwi antibody and 50 μl of Protein G Dynabeads. The rest of the protocol was identical to that of myc‐Nxf2.
Isolation of myc‐Nxf2 stable line OSCs
Myc‐Nxf2 OSC line was isolated using the methodology described in “Tethering assay using OSCs” section, for isolation of OSC line carrying reporter gene. Briefly, OSCs were co‐transfected with pAcM‐Nxf2‐sires plasmid and the plasmid carrying a blasticidin‐resistant gene, and selection was performed by blasticidin.
Small RNA‐seq analysis
Small RNA cloning was performed as previously described (Iwasaki et al, 2017), with modifications. In summary, co‐immunoprecipitated piRNA was extracted from Piwi immunoprecipitates using phenol and chloroform, and purified piRNAs were cloned using the NEXTflex Small RNA Sequencing kit v3 (NEXTflex). The libraries were analyzed on a Miseq platform (Illumina).
For small RNA‐seq data analysis, 4 nt random nucleotides and adapter sequences were removed from the obtained reads using Cutadapt, and reads out of 20–35 nt size range were removed for further analysis. Obtained reads were mapped to the Release 3 assembly of the Drosophila genome using Bowtie (Langmead et al, 2009), allowing up to single mismatch. Genome‐mapped reads were further mapped to TE consensus sequences from Repbase (Jurka, 1998) using Bowtie (Langmead et al, 2009), allowing no mismatch and unique mapping. The reads were normalized to RPM by the number of Drosophila genome‐mapped reads. Reads mapped to antisense direction of TE consensus sequences were used to generate heatmap in Fig 6E, and reads mapped to both directions were used to generate density plot in Fig 6F. Sense reads were plotted in positive direction, where antisense reads were plotted in negative direction.
CLIP‐seq data analysis
First, we trimmed 3′ adapter sequences using Cutadapt and discarded reads shorter than 27 bp. Next, we removed PCR duplicates by collapsing all identical reads containing the same random barcode at the 5′ end with fastx‐collapser from the FASTX‐Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html). Finally, we trimmed the 5′‐end random barcode located at positions 1–9 of the reads. Mapping was then performed against the Release 3 assembly of the Drosophila melanogaster genome with STAR (Dobin et al, 2013). Alignment output BAM files were sorted and indexed by SAMtools (Li et al, 2009). Peaks were called using Piranha peak‐calling algorithm (Uren et al, 2012), and reads within the peaks were counted for two replicate samples. Of two replicate samples, peak position of the sample that had higher number of annotated peaks was used for counting reads. Dot plots were generated based on the read counts, and correlation coefficient (r) was calculated to check for the reproducibility (shown in Fig EV6E). After checking for reproducibility, samples were merged for downstream analysis using SAMtools (Li et al, 2009). Genome‐mapped reads were further mapped to TE consensus sequences from Repbase (Jurka, 1998) using Bowtie allowing only unique mapping reads. Mapped reads were counted for each consensus TE and normalized to RPM by the number of Drosophila genome‐mapped reads. Reads mapped to sense and antisense direction of TE consensus sequences were counted separately, when calculating RPM. Based on the results shown in Figs 6A–C and EV6F, sense‐mapped read counts were used for analysis shown in Fig 6D–F. Note that piRNAs were not included in the reads mapped to antisense direction of TEs, since the libraries were generated using RNA in the size range of 43–73 nt (~50 kDa above the protein size). In case of taking ratio of CLIP tags and SMInput, TEs that had RPM lower than 0.5 in SMInput were eliminated. Maximum x‐axis for the density plot of Piwi CLIP tags in mdg1 (Fig 6F) was adjusted to 100, since there was single high peak at position 6,870 bp (~600 RPM), which masks the other peaks. Dot plot, boxplot, and density plot were generated using the methodology described in “RNA‐seq” and “ChIP‐seq” section. Heatmaps were depicted using Java Treeview (Saldanha, 2004). Transcriptome‐wide CLIP data are available at NCBI‐GEO, accession number GSE131950.
Author Contributions
KM and YWI designed and performed most of the experiments with HI, AM, and AS, SK and KS generated mutant flies for tethering assays, SA and TN performed LC‐MS/MS analysis, and SS and MCS generated Eggless antibody. YWI and HS conceived the study, and KM, YWI, and HS wrote the paper with input from the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Acknowledgements
We thank Wataru Ito for experimental support and critical discussions. We also thank Julius Brennecke for sharing plasmids; Kazumichi Nishida, Ryo Ohnishi, and Tatsuki Kinoshita for sharing protocols; and Soichiro Yamanaka for critical reading of the paper. This work was supported by funding from JSPS KAKENHI Grant Number 17K08644 and Kato Memorial Bioscience Foundation to K.M.; from JSPS KAKENHI Grant Numbers 15H05583, 17H05603, 18H02421, and 19H05268, Senri Life Science Foundation, NOVARTIS Foundation (Japan), and the Naito Foundation to Y.W.I.; from JSPS KAKENHI Grant Number 17H05610 to S.A.; from Takeda Science Foundation to T.N.; and from JSPS KAKENHI Grant Number 25221003 to H.S.
The EMBO Journal (2019) 38: e102870
Contributor Information
Yuka W Iwasaki, Email: iwasaki@keio.jp.
Haruhiko Siomi, Email: awa403@keio.jp.
Data availability
The datasets produced in this study are available in the following database: RNA‐seq, ChIP‐seq, CLIP‐seq, and small RNA‐seq data: Gene Expression Omnibus GSE131950 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131950).
References
- Baron‐Benhamou J, Gehring NH, Kulozik AE, Hentze MW (2004) Using the lambdaN peptide to tether proteins to RNAs. Methods Mol Biol 257: 135–154 [DOI] [PubMed] [Google Scholar]
- Batki J, Schnabl J, Wang J, Handler D, Andreev VI, Stieger CE, Novatchkova M, Lampersberger L, Kauneckaite K, Xie W et al (2019) The nascent RNA binding complex SFiNX licenses piRNA‐guided heterochromatin formation. Nat Struct Mol Biol 26: 720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ‐line‐specific phiC31 integrases. Proc Natl Acad Sci USA 104: 3312–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork P, Wieslander L (2014) Mechanisms of mRNA export. Semin Cell Dev Biol 32: 47–54 [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ (2007) Discrete small RNA‐generating loci as master regulators of transposon activity in Drosophila . Cell 128: 1089–1103 [DOI] [PubMed] [Google Scholar]
- Brower‐Toland B, Findley SD, Jiang L, Liu L, Yin H, Dus M, Zhou P, Elgin SCR, Lin HF (2007) Drosophila PIWI associates with chromatin and interacts directly with HP1a. Genes Dev 21: 2300–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmell MA, Girard A, van de Kant HJ, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ (2007) MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12: 503–514 [DOI] [PubMed] [Google Scholar]
- Checkley MA, Mitchell JA, Eizenstat LD, Lockett SJ, Garfinkel DJ (2013) Ty1 gag enhances the stability and nuclear export of Ty1 mRNA. Traffic 14: 57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, Smith ER, Shiekhattar R, Shilatifard A (2015) PAF1, a molecular regulator of promoter‐proximal pausing by RNA polymerase II. Cell 162: 1003–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C (2017) Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet 18: 71–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JP, Rahman R, Yang N, Yang LH, Lau NC (2017) Drosophila PAF1 modulates PIWI/piRNA silencing capacity. Curr Biol 27: 2718–2726.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DN, Chao A, Baker J, Chang L, Qiao D, Lin H (1998) A novel class of evolutionarily conserved genes defined by piwi are essential for stem cell self‐renewal. Genes Dev 12: 3715–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Preall JB, McGinn J, Hannon GJ (2013) A transcriptome‐wide RNAi screen in the Drosophila ovary reveals factors of the germline piRNA pathway. Mol Cell 50: 749–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donertas D, Sienski G, Brennecke J (2013) Drosophila Gtsf1 is an essential component of the Piwi‐mediated transcriptional silencing complex. Genes Dev 27: 1693–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C, Odom DT, Kutter C (2017) The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat Commun 8: 1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabry MH, Ciabrelli F, Munafò M, Eastwood EL, Kneuss E, Falciatori I, Falconio FA, Hannon GJ (2019) piRNA‐guided co‐transcriptional silencing coopts nuclear export factors. eLife 8: e47999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Martin L, Spitale RC, Do BT, Sagan SM, Zarnegar B, Qu K, Khavari PA, Quake SR, Sarnow P et al (2015) Dissecting noncoding and pathogen RNA‐protein interactomes. RNA 21: 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribourg S, Braun IC, Izaurralde E, Conti E (2001) Structural basis for the recognition of a nucleoporin FG repeat by the NTF2‐like domain of the TAP/p15 mRNA nuclear export factor. Mol Cell 8: 645–656 [DOI] [PubMed] [Google Scholar]
- Gramates LS, Marygold SJ, Santos GD, Urbano JM, Antonazzo G, Matthews BB, Rey AJ, Tabone CJ, Crosby MA, Emmert DB et al (2017) FlyBase at 25: looking to the future. Nucleic Acids Res 45: D663–D671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S (2010) Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature 465: 1097–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC (2007) A slicer‐mediated mechanism for repeat‐associated siRNA 5’ end formation in Drosophila . Science 315: 1587–1590 [DOI] [PubMed] [Google Scholar]
- Han JS, Boeke JD (2005) LINE‐1 retrotransposons: modulators of quantity and quality of mammalian gene expression? BioEssays 27: 775–784 [DOI] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, Zamore PD (2015) Noncoding RNA. piRNA‐guided transposon cleavage initiates Zucchini‐dependent, phased piRNA production. Science 348: 817–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler D, Meixner K, Pizka M, Lauss K, Schmied C, Gruber FS, Brennecke J (2013) The genetic makeup of the Drosophila piRNA pathway. Mol Cell 50: 762–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold A, Suyama M, Rodrigues JP, Braun IC, Kutay U, Carmo‐Fonseca M, Bork P, Izaurralde E (2000) TAP (NXF1) belongs to a multigene family of putative RNA export factors with a conserved modular architecture. Mol Cell Biol 20: 8996–9008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold A, Klymenko T, Izaurralde E (2001) NXF1/p15 heterodimers are essential for mRNA nuclear export in Drosophila . RNA 7: 1768–1780 [PMC free article] [PubMed] [Google Scholar]
- Herold A, Teixeira L, Izaurralde E (2003) Genome‐wide analysis of nuclear mRNA export pathways in Drosophila . EMBO J 22: 2472–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolka D, Pandey RR, Goriaux C, Brasset E, Vaury C, Sachidanandam R, Fauvarque MO, Pillai RS (2015) PIWI slicing and RNA elements in precursors instruct directional primary piRNA biogenesis. Cell Rep 12: 418–428 [DOI] [PubMed] [Google Scholar]
- Houwing S, Kamminga LM, Berezikov E, Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz E, Moens CB et al (2007) A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 129: 69–82 [DOI] [PubMed] [Google Scholar]
- Huang XA, Yin H, Sweeney S, Raha D, Snyder M, Lin H (2013) A major epigenetic programming mechanism guided by piRNAs. Dev Cell 24: 502–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, Siomi H (2002) A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev 16: 2497–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki YW, Siomi MC, Siomi H (2015) PIWI‐interacting RNA: its biogenesis and functions. Annu Rev Biochem 84: 405–433 [DOI] [PubMed] [Google Scholar]
- Iwasaki YW, Murano K, Ishizu H, Shibuya A, Iyoda Y, Siomi MC, Siomi H, Saito K (2016) Piwi modulates chromatin accessibility by regulating multiple factors including histone H1 to repress transposons. Mol Cell 63: 408–419 [DOI] [PubMed] [Google Scholar]
- Iwasaki YW, Ishino K, Siomi H (2017) Deep sequencing and high‐throughput analysis of PIWI‐associated small RNAs. Methods 126: 66–75 [DOI] [PubMed] [Google Scholar]
- Izaurralde E (2002) A novel family of nuclear transport receptors mediates the export of messenger RNA to the cytoplasm. Eur J Cell Biol 81: 577–584 [DOI] [PubMed] [Google Scholar]
- Jin Z, Flynt AS, Lai EC (2013) Drosophila piwi mutants exhibit germline stem cell tumors that are sustained by elevated Dpp signaling. Curr Biol 23: 1442–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J (1998) Repeats in genomic DNA: mining and meaning. Curr Opin Struct Biol 8: 333–337 [DOI] [PubMed] [Google Scholar]
- Kerkow DE, Carmel AB, Menichelli E, Ambrus G, Hills RD Jr, Gerace L, Williamson JR (2012) The structure of the NXF2/NXT1 heterodimeric complex reveals the combined specificity and versatility of the NTF2‐like fold. J Mol Biol 415: 649–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M, Chandradoss SD, Depken M, Joo C (2017) Why argonaute is needed to make microRNA target search fast and reliable. Semin Cell Dev Biol 65: 20–28 [DOI] [PubMed] [Google Scholar]
- Klenov MS, Lavrov SA, Korbut AP, Stolyarenko AD, Yakushev EY, Reuter M, Pillai RS, Gvozdev VA (2014) Impact of nuclear Piwi elimination on chromatin state in Drosophila melanogaster ovaries. Nucleic Acids Res 42: 6208–6218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik KM, Shimada Y, Flury V, Stadler MB, Batki J, Buhler M (2015) The Paf1 complex represses small‐RNA‐mediated epigenetic gene silencing. Nature 520: 248–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Thomas A, Rogers AK, Webster A, Marinov GK, Liao SE, Perkins EM, Hur JK, Aravin AA, Toth KF (2013) Piwi induces piRNA‐guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev 27: 390–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque L, Guzik B, Guan T, Coyle J, Black BE, Rekosh D, Hammarskjold ML, Paschal BM (2001) RNA export mediated by tap involves NXT1‐dependent interactions with the nuclear pore complex. J Biol Chem 276: 44953–44962 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Spradling AC (1997) A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development 124: 2463–2476 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M, Pitsouli C, Villalta C, Celniker SE, Perrimon N (2008) Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat Genet 40: 476–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Handler D, Brennecke J (2015) Noncoding RNA. piRNA‐guided slicing specifies transcripts for Zucchini‐dependent, phased piRNA biogenesis. Science 348: 812–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muerdter F, Guzzardo PM, Gillis J, Luo Y, Yu Y, Chen C, Fekete R, Hannon GJ (2013) A genome‐wide RNAi screen draws a genetic framework for transposon control and primary piRNA biogenesis in Drosophila . Mol Cell 50: 736–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Yamauchi Y, Nakayama H, Shinkawa T, Yanagida M, Takahashi N, Isobe T (2002) A direct nanoflow liquid chromatography‐tandem mass spectrometry system for interaction proteomics. Anal Chem 74: 4725–4733 [DOI] [PubMed] [Google Scholar]
- Niki Y, Yamaguchi T, Mahowald AP (2006) Establishment of stable cell lines of Drosophila germ‐line stem cells. Proc Natl Acad Sci USA 103: 16325–16330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani H, Iwasaki YW, Shibuya A, Siomi H, Siomi MC, Saito K (2013) DmGTSF1 is necessary for Piwi‐piRISC‐mediated transcriptional transposon silencing in the Drosophila ovary. Genes Dev 27: 1656–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post C, Clark JP, Sytnikova YA, Chirn GW, Lau NC (2014) The capacity of target silencing by Drosophila PIWI and piRNAs. RNA 20: 1977–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov NV, Hammell M, Hannon GJ (2013) Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev 27: 400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Nishida KM, Mori T, Kawamura Y, Miyoshi K, Nagami T, Siomi H, Siomi MC (2006) Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev 20: 2214–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Inagaki S, Mituyama T, Kawamura Y, Ono Y, Sakota E, Kotani H, Asai K, Siomi H, Siomi MC (2009) A regulatory circuit for piwi by the large Maf gene traffic jam in Drosophila . Nature 461: 1296–1299 [DOI] [PubMed] [Google Scholar]
- Saito K, Ishizu H, Komai M, Kotani H, Kawamura Y, Nishida KM, Siomi H, Siomi MC (2010) Roles for the Yb body components Armitage and Yb in primary piRNA biogenesis in Drosophila . Genes Dev 24: 2493–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004) Java Treeview–extensible visualization of microarray data. Bioinformatics 20: 3246–3248 [DOI] [PubMed] [Google Scholar]
- Sato K, Nishida KM, Shibuya A, Siomi MC, Siomi H (2011) Maelstrom coordinates microtubule organization during Drosophila oogenesis through interaction with components of the MTOC. Genes Dev 25: 2361–2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, Nestler E (2014) ngs.plot: Quick mining and visualization of next‐generation sequencing data by integrating genomic databases. BMC Genom 15: 284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Donertas D, Brennecke J (2012) Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151: 964–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Batki J, Senti KA, Donertas D, Tirian L, Meixner K, Brennecke J (2015) Silencio/CG9754 connects the Piwi‐piRNA complex to the cellular heterochromatin machinery. Genes Dev 29: 2258–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz F, Izaurralde E (2003) The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol 13: 319–327 [DOI] [PubMed] [Google Scholar]
- Sumiyoshi T, Sato K, Yamamoto H, Iwasaki YW, Siomi H, Siomi MC (2016) Loss of l(3)mbt leads to acquisition of the ping‐pong cycle in Drosophila ovarian somatic cells. Genes Dev 30: 1617–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren PJ, Bahrami‐Samani E, Burns SC, Qiao M, Karginov FV, Hodges E, Hannon GJ, Sanford JR, Penalva LO, Smith AD (2012) Site identification in high‐throughput RNA‐protein interaction data. Bioinformatics 28: 3013–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin‐Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K et al (2016) Robust transcriptome‐wide discovery of RNA‐binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13: 508–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oss SB, Cucinotta CE, Arndt KM (2017) Emerging insights into the roles of the Paf1 complex in gene regulation. Trends Biochem Sci 42: 788–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viphakone N, Hautbergue GM, Walsh M, Chang CT, Holland A, Folco EG, Reed R, Wilson SA (2012) TREX exposes the RNA‐binding domain of Nxf1 to enable mRNA export. Nat Commun 3: 1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, Cramer P (2018) Structure of activated transcription complex Pol II‐DSIF‐PAF‐SPT6. Nature 560: 607–612 [DOI] [PubMed] [Google Scholar]
- Vourekas A, Zheng K, Fu Q, Maragkakis M, Alexiou P, Ma J, Pillai RS, Mourelatos Z, Wang PJ (2015) The RNA helicase MOV10L1 binds piRNA precursors to initiate piRNA processing. Genes Dev 29: 617–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Elgin SC (2011) Drosophila Piwi functions downstream of piRNA production mediating a chromatin‐based transposon silencing mechanism in female germ line. Proc Natl Acad Sci USA 108: 21164–21169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe VO, Laskey RA (2015) Control of mammalian gene expression by selective mRNA export. Nat Rev Mol Cell Biol 16: 431–442 [DOI] [PubMed] [Google Scholar]
- Wiegand HL, Coburn GA, Zeng Y, Kang Y, Bogerd HP, Cullen BR (2002) Formation of Tap/NXT1 heterodimers activates Tap‐dependent nuclear mRNA export by enhancing recruitment to nuclear pore complexes. Mol Cell Biol 22: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie GS, Zimyanin V, Kirby R, Korey C, Francis‐Lang H, Van Vactor D, Davis I (2001) Small bristles, the Drosophila ortholog of NXF‐1, is essential for mRNA export throughout development. RNA 7: 1781–1792 [PMC free article] [PubMed] [Google Scholar]
- Yashiro R, Murota Y, Nishida KM, Yamashiro H, Fujii K, Ogai A, Yamanaka S, Negishi L, Siomi H, Siomi MC (2018) Piwi nuclear localization and its regulatory mechanism in Drosophila ovarian somatic cells. Cell Rep 23: 3647–3657 [DOI] [PubMed] [Google Scholar]
- Yu Y, Gu J, Jin Y, Luo Y, Preall JB, Ma J, Czech B, Hannon GJ (2015) Panoramix enforces piRNA‐dependent cotranscriptional silencing. Science 350: 339–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang J, Xu J, Zhang Z, Koppetsch BS, Schultz N, Vreven T, Meignin C, Davis I, Zamore PD et al (2012) UAP56 couples piRNA clusters to the perinuclear transposon silencing machinery. Cell 151: 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Cheng S, Miao N, Xu P, Lu X, Zhang Y, Wang M, Ouyang X, Yuan X, Liu W et al (2019) A Pandas complex adapted for piRNA‐guided transposon silencing. bioRxiv 10.1101/608273 [PREPRINT] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Data Availability Statement
The datasets produced in this study are available in the following database: RNA‐seq, ChIP‐seq, CLIP‐seq, and small RNA‐seq data: Gene Expression Omnibus GSE131950 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131950).