

Abstract
Delafloxacin, a fluoroquinolone, has activity against Gram‐positive organisms including methicillin‐resistant S aureus and fluoroquinolone‐susceptible and ‐resistant Gram‐negative organisms. The intravenous formulation of delafloxacin contains the excipient sulfobutylether‐β‐cyclodextrin (SBECD), which is eliminated by renal filtration. This study examined the pharmacokinetics and safety of SBECD after single intravenous (IV) infusions in subjects with renal impairment. The study was an open‐label, parallel‐group, crossover study in subjects with normal renal function or mild, moderate, or severe renal impairment, and those with end‐stage renal disease undergoing hemodialysis. Subjects received 300 mg delafloxacin IV or placebo IV, containing 2400 mg SBECD, in 2 periods separated by ≥14‐day washouts. SBECD total clearance decreased with decreasing renal function, with a corresponding increase in area under the concentration‐time curve (AUC0‐∞). After IV delafloxacin 300 mg administration, SBECD mean total clearance was 6.28 and 1.24 L/h, mean AUC0‐∞ was 387 and 2130 h·μg/mL, and mean renal clearance was 5.36 and 1.14 L/h in normal and severe renal subjects, respectively. Similar values were obtained after IV placebo administration. In subjects with end‐stage renal disease, delafloxacin 300 mg IV produced mean SBECD AUC0‐48 values of 2715 and 7861 h·μg/mL when dosed before and after hemodialysis, respectively. Total SBECD clearance exhibited linear relationships to estimated glomerular filtration rate and creatinine clearance. Single doses of IV delafloxacin 300 mg and IV placebo were well tolerated in all groups. In conclusion, decreasing renal function causes reduced SBECD clearance and increased exposures, but SBECD continues to exhibit a good safety and tolerability profile in IV formulations.
Keywords: Delafloxacin, Sulfobutylether‐β‐cyclodextrin, Pharmacokinetics, Renal Dysfunction, Hemodialysis
Delafloxacin, a broad‐spectrum anionic fluoroquinolone, recently received FDA approval for the treatment of acute bacterial skin and skin structure infections due to Gram‐positive infections, including MRSA, as well as Gram‐negative pathogens.1 Subjects with acute bacterial skin and skin structure infections were successfully treated with delafloxacin in 2 phase 3 studies.2, 3 Delafloxacin is currently being studied for the treatment of hospitalized community‐acquired bacterial pneumonia.4
The delafloxacin intravenous formulation contains the excipient sulfobutylether‐β‐cyclodextrin (SBECD) as an aid to solubility.5 The intravenous dose of delafloxacin is 300 mg twice a day, which comprises 2400 mg of SBECD (4800 mg SBECD daily). Because SBECD is not metabolized and is cleared virtually exclusively by renal filtration,6 there was concern about its clearance and possible accumulation in patients with compromised renal function. This study was designed to characterize the pharmacokinetics and tolerance of SBECD in subjects with renal impairment, including subjects with end‐stage renal disease (ESRD) undergoing hemodialysis. Delafloxacin pharmacokinetics, as well as overall safety from this study, has been previously reported.7 This analysis specifically assesses SBECD pharmacokinetics and safety in patients with varying degrees of renal impairment.
Methods
This study was conducted at DaVita Clinical Research, Minneapolis, MN, and approval was obtained from the principal investigator's institutional review board (Western Institutional Review Boards, Olympia, Washington) before the beginning of the study. All subjects voluntarily signed informed consent prior to admission into the study. The study was conducted according to the International Conference on Harmonisation guideline Good Clinical Practice: Consolidated Guideline; and the United States Code of Federal Regulations.
Study Design
This was a phase 1, open‐label, parallel‐group, single‐dose, crossover, single‐site study assessing the pharmacokinetics and tolerance of delafloxacin and SBECD in subjects with renal impairment, including ESRD, undergoing hemodialysis. Eight subjects were planned for each renal impairment category, defined as follows:
Group A: Healthy subjects (estimated glomerular filtration rate [eGFR] >80 mL/min/1.73 m2)
Group B: Subjects with mild renal impairment (eGFR >50 to 80 mL/min/1.73 m2)
Group C: Subjects with moderate renal impairment (eGFR >30 to 50 mL/min/1.73 m2)
Group D: Subjects with severe renal impairment (eGFR ≤30 mL/min/1.73 m2)
Group E: Subjects with ESRD on hemodialysis
At screening, subjects were assigned to a study group based on their eGFR, calculated using the isotope dilution mass spectrometry‐traceable Modification of Diet in Renal Disease8 formula as follows:
Subject creatinine clearance (CLCR) was also calculated using the Cockcroft‐Gault formula9 as follows:
For inclusion in the study, each subject was required to meet all of the following criteria: male or female between 18 and 80 years of age; a body mass index between 18.5 and 40 kg/m2; baseline laboratory values within reference ranges or deemed not clinically significant by the investigator; subjects with renal impairment had laboratory values consistent with their disease; not child‐bearing, rendered not child‐bearing (eg, vasectomy, hysterectomy), or use of an adequate method of contraception from screening through 12 weeks after the last dose of study drug. Renally impaired subjects were accepted if they had been taking medications that did not affect study drug pharmacokinetics.
Subjects were excluded if they had a clinically or laboratory significant abnormality in their past medical history or at screening (excluding renal insufficiency) that in the investigator's opinion might have placed the subject at risk; any surgical or medical condition or medication history (active or chronic) that might have interfered with the absorption, distribution, metabolism, or excretion of delafloxacin or production and/or excretion of SCr; a functioning renal transplant; abnormal vital signs (systolic and diastolic blood pressure <90 or >200 mm Hg and <45 or >110 mm Hg, respectively, heart rate <50 or >120 bpm); a hemoglobin value <10 g/dL, blood/plasma donation within 30 days before dosing, or had lost >1200 mL of blood within 4 months before the first dose of study drug; a history of or current hepatitis, or subject was a carrier of hepatitis B surface antigen and/or hepatitis C antibodies (a subject with hepatitis C who had normal liver function test results was allowable with investigator approval); a history or evidence of drug abuse within 6 months of screening, positive serum drug screen or alcohol breathalyzer result at screening or admission to study center; HIV/AIDS; a history of an investigational drug within 30 days or 5 half‐lives, whichever was longer; or a baseline QTc interval ≥480 or ≥500 milliseconds for males and females, respectively.
Study Medication
Delafloxacin for injection, 300 mg/vial, is formulated as a sterile, nonpyrogenic, lyophilized powder. Each vial contains the following ingredients: 433 mg delafloxacin meglumine equivalent of 300 mg free acid, 58.56 mg meglumine, 2.4 g sulfobutylether‐β‐cyclodextrin (SBECD; Captisol®), and 3.4 mg ethylene diamine tetra‐acetate disodium (equivalent to 2.6 mg EDTA). Placebo for intravenous (IV) delafloxacin was supplied as a frozen solution in 20‐mL vials, which contained the same amounts of excipients per vial as the active vials. Study medications were further diluted with 250 mL D5W.
Study Drug Administration
The subjects in groups A through D participated in 3 treatment periods separated by a washout period of ≥14 days:
Period 1: Delafloxacin 300 mg IV infusion containing 2400 mg SBECD over 1 hour
Period 2: Placebo containing 2400 mg SBECD IV infusion over 1 hour
Period 3: Oral delafloxacin 400 mg (no SBECD). (This report focuses on periods 1 and 2, which included SBECD administration. Period 3 is not discussed further.)
ESRD subjects in group E participated in 2 treatment periods separated by a washout period of ≥14 days There was no placebo/SBECD administration in these subjects.
Period 1: Delafloxacin 300 mg IV infusion containing 2400 mg SBECD over 1 hour starting immediately before a hemodialysis session
Period 2: Delafloxacin 300 mg IV infusion containing 2400 mg SBECD over 1 hour followed immediately by a hemodialysis session
Subjects fasted for 4 hours before and after dosing (or after the end of the infusion for the IV treatments).
Pharmacokinetic Samples
For the IV treatments (periods 1 and 2), blood for plasma samples was collected before dosing and at 0.33, 0.66, 1 (end of infusion), 1.083, 1.167, 1.33, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 hours after the start of the infusion. Urine samples were collected in intervals from ‐2 to 0 hours and after dosing from 0 to 12, 12 to 24, 24 to 36, and 36 to 48 hours. For ESRD subjects, the entire volume of dialysis fluid was collected during hemodialysis sessions conducted after delafloxacin dosing (period 1).
Plasma, urine, and dialysate samples were analyzed for SBECD with validated methods for each matrix utilizing liquid chromatography coupled with detection by tandem mass spectrometry. (Sample analyses were performed by ABC Laboratories, Columbia, Missouri). The samples (plasma, urine, dialysate) were extracted using protein precipitation and/or dilution with methanol and analyzed by HPLC separation on a Waters Atlantis HILIC Silica column, and tandem mass spectrometry detection (Sciex API 4000; Framingham, Massachusetts). For the plasma and dialysis methods, the lower limit of quantitation was 2.00 μg/mL, and the standard curve range was 2.00 to 200 μg/mL. For the urine method, the lower limit of quantitation was 5.00 μg/mL, and the standard curve range was 5.00 to 200 μg/mL. The methods demonstrated acceptable linearity, accuracy, and precision. Adequate SBECD stability was demonstrated in standard freeze/thaw, room temperature, and frozen storage (–70°C or lower) stability tests.
Pharmacokinetic Analyses
SBECD pharmacokinetic parameters were calculated using Phoenix® WinNonlin® Versions 6.2.1 or 7.0 (Certara, Princeton, New Jersey).
The following pharmacokinetic parameters were calculated: area under the plasma concentration‐time curve (AUC) by the trapezoidal rule, including AUC0‐∞ (time 0 extrapolated to infinity) and AUC0‐t (time 0 to time of last quantifiable concentration); maximum observed plasma concentration (Cmax) and time to reach Cmax (Tmax); apparent terminal elimination rate constant and terminal half‐life (t½); total body clearance (CLtot) from the ratio of dose to AUC0‐∞; area under the IV plasma concentration‐time moment curve (AUMC); IV mean residence time from the ratio of AUMC to AUC0‐∞, corrected for the infusion time (MRT); and IV volume of distribution at steady‐state (Vss) calculated by the product of CLtot and mean residence time.
The following pharmacokinetic parameters were calculated from urine concentrations of SBECD: amount of SBECD excreted in urine over various time periods from time 0 to 48 hours after dosing (Ae); fraction of the dose excreted in urine from time 0 to 48 hours (Fe); and renal clearance (CLr) from the ratio of Ae over 48 hours to AUC0‐t.
Data from 2 subjects exhibited implausible SBECD concentration‐time profiles. High SBECD concentrations were observed from the IV infusion period (0.33, 0.66, and 1 hour) for Subject 1404 (severe renal impairment group) after dosing with IV delafloxacin. High SBECD concentrations were observed from the IV infusion period (0.33, 0.66, and 1 hour) for Subject 1507 (ESRD group) after dosing with IV delafloxacin both before and after hemodialysis. The high concentrations were determined to be outliers using a statistical outlier test (Extreme Studentized Deviate) performed using R (FindOutliersESDtest in the package Climtrends; R Foundation, Vienna, Austria). The entire profiles in question were excluded from the final plasma pharmacokinetic analyses and plots.
Safety Assessments
Safety assessments included evaluation of treatment‐emergent adverse events (TEAEs), clinical laboratory results (hematology [including coagulation], serum chemistry [including liver function parameters], and urinalysis), vital sign measurements (blood pressure, heart rate, and body temperature), 12‐lead electrocardiographic measurements, and physical examination findings. Safety from period 3 (oral delafloxacin, no SBECD) has previously been discussed7 and is not reviewed in this article.
Statistical Analysis
Summary statistics were prepared with WinNonlin, and SAS® software Version 9.1.3 (SAS Institute, Cary, North Carolina). Linear regressions were conducted using GraphPad Prism version 5.01 (GraphPad Software, La Jolla, California). Linear regressions of the IV pharmacokinetic parameters were performed without the implausible data from the severe impairment subject indicated above. There were 2 analysis populations: the pharmacokinetic population, which included all subjects who had sufficient concentration data to calculate reliable estimates of pharmacokinetic parameters; and the safety population, which included all subjects who received at least 1 dose of study drug (delafloxacin or placebo).
Results
Study Population Demographics
Forty‐four subjects were enrolled. Two subjects were discontinued early because of TEAEs. The 2 subjects were replaced and were not included in the pharmacokinetic population; all 44 subjects were included in the safety population. Forty subjects who received SBECD were considered evaluable and included in the pharmacokinetics population. Subject demographic and baseline characteristics are summarized in Table 1. With the exception of renal status, overall mean demographic characteristics were similar across groups.
Table 1.
Subject Demographics and Baseline Characteristics
| Normal | Mild Impairment | Moderate Impairment | Severe Impairment | ESRD | |
|---|---|---|---|---|---|
| N (completed) | 8 | 8 | 8 | 8 | 10 |
| N (discontinued) | 1 | 0 | 0 | 1 | 0 |
| Safety population | 9 | 8 | 8 | 9 | 10 |
| Pharmacokinetic population | 8 | 8 | 8 | 8 | 8 |
| Age (y) | 52 ± 4 | 56 ± 10 | 57 ± 9 | 54 ± 9 | 51 ± 17 |
| Weight (kg) | 84 ± 13 | 94 ± 15 | 98 ± 25 | 92 ± 18 | 83 ± 24 |
| Height (cm) | 174 ± 13 | 174 ± 8 | 175 ± 9 | 170 ± 9 | 171 ± 11 |
| BMI (kg/m2) | 28 ± 2 | 31 ± 3 | 32 ± 7 | 32 ± 5 | 28 ± 6 |
| eGFR (mL/min/1.73 m2) | 92 ± 11 | 63 ± 8 | 39 ± 5 | 22 ± 6 | 6 ± 2 |
| CLCR (mL/min) | 121 ± 19 | 87 ± 16 | 58 ± 15 | 35 ± 10 | 10 ± 4 |
Data shown as mean ± SD. BMI indicates body mass index; CLCR, clearance of creatinine; eGFR, estimated glomerular filtration rate; ESRD, end‐stage renal disease.
SBECD Pharmacokinetics in Subjects With Normal Renal Function to Severe Renal Impairment
Following IV infusion to patients with normal to severely impaired renal function, the plasma concentration profiles of SBECD were biphasic, exhibiting a short distribution phase followed by a monophasic decline (Figure 1). SBECD total exposure increased as the degree of renal impairment worsened (Table 2). Compared with the normal renal function group, mean AUC0‐∞ values for the severe renal impairment group were 5.5‐fold and 5.4‐fold greater, respectively, after IV delafloxacin and IV placebo (2130 vs 387, and 1989 vs 371 h·μg/mL). In contrast, Cmax did not vary significantly across the renal impairment groups. Mean apparent terminal t½ increased with worsening renal function (for IV delafloxacin and IV placebo, respectively, 1.8 and 1.7 hours in normal renal function and 10.8 and 9.7 hours in severe renal impairment). Mean Vss values increased somewhat with increasing degree of renal impairment. The fraction of the dose of SBECD excreted in urine during the 48 hours after dosing was similar across the renal function groups. Most urinary excretion of SBECD occurred in the first 12 hours after dosing (Figure 2).
Figure 1.
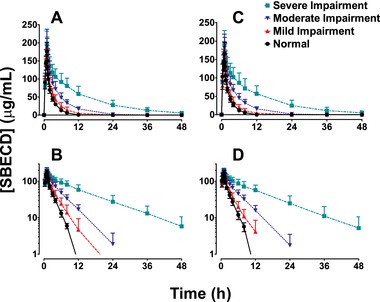
Mean (SD) plasma concentration‐time profiles in subjects with varying degrees of renal function after IV infusion of 2400 mg SBECD from either 300 mg IV delafloxacin (panels A and B) or IV placebo (panels C and D). Upper panels (A and C), linear scale. Lower panels (B and D), semi‐log scale. IV indicates intravenous; SBECD, sulfobutylether‐β‐cyclodextrin.
Table 2.
Mean (%CV) SBECD Pharmacokinetic Parameters Following IV Administration of 2400 mg SBECD Alone or With 300 mg Delafloxacin
| Plasma Pharmacokinetics | ||||||||
|---|---|---|---|---|---|---|---|---|
| Normal | Mild Impairment | Moderate Impairment | Severe Impairment | |||||
| Dose | Delafloxacin 300 mg IV | Placebo IV | Delafloxacin 300 mg IV | Placebo IV | Delafloxacin 300 mg IV | Placebo IV | Delafloxacin 300 mg IV* | Placebo IV |
| Cmax (μg/mL) | 177 (14.0) | 174 (14.9) | 167 (10.1) | 160 (10.5) | 191 (26.9) | 181 (20.9) | 197 (35.9) | 191 (19.8) |
| t½ (h) | 1.8 (8.5) | 1.7 (9.6) | 2.5 (33.0) | 2.4 (26.6) | 4.0 (19.3) | 4.0 (18.8) | 10.8 (25.9) | 9.7 (30.6) |
| AUC0‐t (μg·hr/mL) | 375 (11.9) | 359 (13.6) | 493 (31.8) | 427 (20.1) | 816 (26.5) | 760 (27.3) | 2010 (32.2) | 1878 (38.9) |
| AUC0‐∞ (μg·hr/mL) | 387 (11.6) | 371 (13.2) | 508 (31.5) | 446 (23.1) | 852 (25.4) | 802 (25.8) | 2130 (32.7) | 1989 (40.4) |
| CL (L/h) | 6.28 (12.5) | 6.57 (13.1) | 5.08 (26.0) | 5.60 (20.0) | 3.00 (27.3) | 3.15 (23.2) | 1.24 (32.7) | 1.38 (36.2) |
| Vss (L) | 12.8 (14.3) | 12.8 (14.8) | 14.4 (12.5) | 15.3 (15.2) | 15.0 (30.1) | 15.7 (26.5) | 16.4 (24.2) | 16.3 (18.2) |
| Urine pharmacokinetics | ||||||||
| Ae0‐48 (mg) | 1970 (8.7) | 1932 (19.4) | 2152 (24.5) | 2983 (100) | 2250 (21.3) | 2026 (23.4) | 1994 (23.8) | 1857 (19.6) |
| Fe0‐48 (%) | 82.1 (8.7) | 80.5 (19.4) | 89.7 (24.5) | 124 (100) | 93.8 (21.3) | 84.4 (23.4) | 83.1 (23.8) | 77.4 (19.6) |
| CLr (L/h) | 5.36 (18.9) | 5.43 (20.4) | 4.65 (36.4) | 6.54 (78.5) | 2.91 (29.7) | 2.87 (36.2) | 1.14 (47.7) | 1.13 (44.7) |
Ae indicates amount excreted; AUC, area under concentration‐time curve; CL, clearance; Cmax, peak concentration; %CV, coefficient of variation; Fe, fraction excreted; IV, intravenous; SBECD, sulfobutylether‐β‐cyclodextrin; t½, half‐time of elimination; Vss, volume of distribution at steady state.
Figure 2.
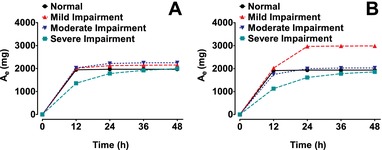
Cumulative mean urinary excretion (Ae) of SBECD in subjects with varying degrees of renal function after IV dosing of 2400 mg SBECD from either 300 mg IV delafloxacin (A) or IV placebo (B). IV indicates intravenous; SBECD, sulfobutylether‐β‐cyclodextrin.
The presence of delafloxacin did not have a significant effect on the pharmacokinetics of SBECD (Table 3). The 90%CI for the IV‐delafloxacin:IV‐placebo ratios of AUC0‐t, AUC0‐∞, and Cmax were within the 0.80 to 1.25 CI boundary often recognized as an equivalence interval in clinical studies.
Table 3.
Statistical Comparison of SBECD PK Parameters From IV Delafloxacin 300 mg vs IV Placebo
| Parameter (Unit) | Treatment | N | Geometric LS Means | Ratio of Geometric LS Means (300 mg IV/Placebo IV) | 90%CI of the Ratio |
|---|---|---|---|---|---|
| AUC0‐t (μg·h/mL) | 300 mg IV | 32 | 708.8 | 1.068 | (1.028, 1.110) |
| Placebo IV | 32 | 663.8 | |||
| AUC0‐inf (μg·h/mL) | 300‐mg IV | 32 | 738.6 | 1.063 | (1.026, 1.100) |
| Placebo IV | 32 | 695.2 | |||
| Cmax (μg/mL) | 300‐mg IV | 32 | 179.4 | 1.031 | (0.996, 1.068) |
| Placebo IV | 32 | 174.0 |
AUC0‐inf indicates area under concentration‐time curve from 0 to infinity; AUC0‐t, area under concentration‐time curve from 0 to last measurement; Cmax, peak concentration; IV, intravenous; LS, least‐squares; PK, pharmacokinetic; SBECD, sulfobutylether‐β‐cyclodextrin.
Relationship Between Renal Function and Pharmacokinetic Parameters for Subjects With Normal Renal Function to Severe Renal Impairment
The relationships between renal function (eGFR) and plasma pharmacokinetic parameters of SBECD were assessed by linear regression (Table 4). Plots of AUC0‐∞ and CLtot vs eGFR for IV delafloxacin and IV placebo are shown in Figure 3 and Figure 4, respectively.
Table 4.
Linear Regression of SBECD Pharmacokinetic Parameter Estimates vs eGFR
| PK Parameter | Drug | N | Slope Estimate | 95%CI of Slope Estimate | r2 | P Value |
|---|---|---|---|---|---|---|
| CLtot (L/h) | Delafloxacin 300 mg IV | 31 | 0.0700 | 0.0580 to 0.0819 | 0.832 | <.0001 |
| Placebo IV | 32 | 0.0738 | 0.0624 to 0.0852 | 0.854 | <.0001 | |
| Cmax (μg/mL) | Delafloxacin 300 mg IV | 31 | −0.320 | −0.785 to 0.144 | 0.064 | .169 |
| Placebo IV | 32 | −0.280 | −0.688 to 0.128 | 0.061 | .172 | |
| Vss/Weight (L/kg) | Delafloxacin 300 mg IV | 31 | −0.000422 | −0.000711 to –0.000133 | 0.235 | .0057 |
| Placebo IV | 32 | −0.000378 | −0.000631 to –0.000125 | 0.237 | .0047 |
CLtot indicates total clearance; Cmax, peak concentration; eGFR, estimated glomerular filtration rate; IV, intravenous; PK, pharmacokinetic; Vss, volume of distribution at steady state.
Figure 3.
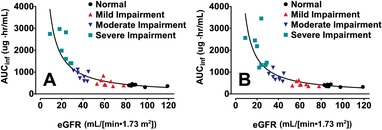
AUCinf vs eGFR after IV dosing of 2400 mg SBECD from either 300 mg IV delafloxacin (A) or IV placebo (B). Solid lines are hyperbolic line regressions. AUCinf indicates area under the concentration‐time curve to infinity; eGFR, estimated glomerular filtration rate; IV, intravenous; SBECD, sulfobutylether‐β‐cyclodextrin.
Figure 4.
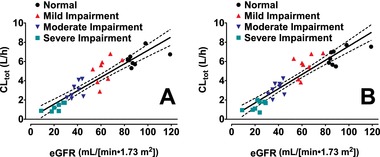
Linear regression of total clearance vs eGFR after IV dosing of 2400 mg SBECD from either 300 mg IV delafloxacin (A) or IV placebo (B). Solid lines are linear regressions of total clearance vs eGFR; dashed lines are the 95%CIs of the linear regressions. CLtot indicates total clearance; eGFR, estimated glomerular filtration rate; IV, intravenous; SBECD, sulfobutylether‐β‐cyclodextrin.
For IV delafloxacin or IV placebo, statistically significant slopes were obtained for the SBECD CLtot and Vss/body weight parameters with P values for the slope estimates <0.01. For the linear relationships between CLtot and eGFR, r2 were >0.83, and for the relationships between Vss/body weight and eGFR, r2 was about 0.24. The relationships between Cmax values and eGFR were not significant, with P values for the slope estimates >0.16, and r2 values <0.07.
SBECD Pharmacokinetics in ESRD Subjects
When IV delafloxacin was given after hemodialysis, the plasma SBECD profiles exhibited a short distribution phase followed by slow, monophasic decline (Figure 5). When IV delafloxacin was administered immediately before hemodialysis, a steep decline in plasma SBECD concentrations was observed during hemodialysis, after which a rebound in SBECD concentrations was observed, followed by slow, monophasic declines (Figure 5). SBECD exposures were markedly reduced by hemodialysis (Table 5). For dosing after and before hemodialysis, mean (coefficient of variation [CV%]) Cmax was 304 (28.3) and 238 (24.6) μg/mL, respectively, and mean (CV%) AUC0‐48 was 7861 (31.6) and 2715 (58.3) h·μg/mL, respectively. Hemodialysis immediately after dosing reduced mean AUC0‐48 by 65%. The majority of the ESRD subjects did not have urinary output, so urinary pharmacokinetic parameters are not reported.
Figure 5.
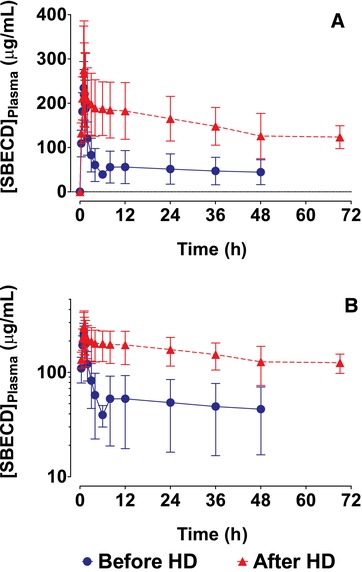
Mean (SD) plasma concentration‐time profiles in ESRD subjects after IV infusion of 2400 mg SBECD from 300 mg IV delafloxacin either before or after hemodialysis. A, Linear scale. B, Semi‐log scale. ESRD indicates end‐stage renal disease; IV, intravenous; SBECD, sulfobutylether‐β‐cyclodextrin.
Table 5.
Mean (%CV) SBECD Pharmacokinetic Parameters Following IV Administration of Delafloxacin 300 mg (Containing 2400 mg SBECD) to ESRD Patients
| Plasma Pharmacokinetics | Dosing Before Hemodialysis | Dosing After Hemodialysis |
|---|---|---|
| Cmax (μg·hr/mL) | 238 (24.6) | 304 (28.3) |
| AUC0‐48 | 2715 (58.3) | 7861 (31.6) |
AUC indicates area under the concentration‐time curve; Cmax, peak concentration; %CV, coefficient of variation; ESRD, end‐stage renal disease; IV, intravenous; SBECD, sulfobutylether‐β‐cyclodextrin.
The mean (CV%) amount of SBECD removed during a nominal 4‐hour hemodialysis session was 1347 (44.0) mg, which was a mean (CV%) 56.1% (44.0%) of the 2400‐mg dose of SBECD. Mean (CV%) dialysis clearance (CLd) was 4.74 (53.8) L/h.
Safety
After single 300‐mg delafloxacin/SBECD IV infusions, TEAEs were reported in 44.1% (15/34) of patients across groups A‐D (patients with normal renal function and with mild, moderate, or severe renal impairment). TEAEs were reported by 2 of 9 subjects (22.2%) in the healthy group, 5 of 8 subjects (62.5%) in the mild renal impairment group, 3 of 8 subjects (37.5%) in the moderate renal impairment group, and 5 of 9 subjects (55.6%) in the severe renal impairment group. The only events reported in 2 or more patients after delafloxacin/SBECD infusion across groups A‐D were headache (5 reports), diarrhea (3 reports), and dizziness, nausea, and hypoglycemia (2 reports each). Of these, headache and diarrhea were reported to be potentially treatment related in 3 patients each, and nausea and hypoglycemia were potentially treatment related in 2 patients each. The hypoglycemic events were reported more than 30 hours after the IV dose. The majority of events were mild or moderate in severity.
After single IV infusions of placebo/SBECD in groups A‐D, TEAEs were reported in 24.2% (8/33) of patients. TEAEs were reported by 1 of 9 subjects (11.1%) in the healthy group, 4 of 8 subjects (50.0%) in the mild renal impairment group, 3 of 8 subjects (37.5%) in the moderate renal impairment group, and no subjects in the severe renal impairment group. In placebo/SBECD treatment, there was no TEAE reported in more than 1 patient, and only 1 event, pruritus, was thought to be potentially related to treatment.
For the ESRD group, TEAEs were reported by 8 of 10 subjects (80.0%) when the 300‐mg delafloxacin/SBECD IV infusion was administered 1 hour before hemodialysis and by 7 of 10 subjects (70.0%) when it was administered 1 hour after hemodialysis. There was no placebo/SBECD comparison group. Diarrhea, nausea, vomiting, and headache were reported in 2 patients each across the 2 ESRD treatment options.
Two subjects were discontinued because of TEAEs. After receiving 300 mg IV delafloxacin/SBECD in period 1 and IV placebo/SBECD in period 2, 1 female subject in the healthy group (group A) was discontinued on day 4 of period 2 due to a severe adverse event of C difficile considered unrelated to the study drug by the investigator. This 52‐year‐old subject did not meet all inclusion/exclusion criteria as she had received amoxicillin within the 30‐day restriction period. Another subject, a 52‐year‐old man in the severe renal impairment group (group D), received a single dose of IV delafloxacin/SBECD in period 1 and discontinued from the study due to bursitis in the left shoulder, which was considered unrelated to the study drug by the investigator. No deaths or serious TEAEs occurred during the study.
Discussion
SBECD is a modified cyclodextrin excipient used to improve the solubility, stability, bioavailability, and dosing of drugs.5 SBECD is an enabling excipient in 10 FDA‐approved parenteral drug formulations, including intravenous delafloxacin. Studies indicate SBECD distribution is virtually limited to extracellular fluid, and it is cleared systemically at a rate similar to the glomerular filtration rate in all species studied.6 Results from the current report are consistent with these reported characteristics.
In normal subjects who received IV delafloxacin, SBECD systemic clearance was 6.6 L/h, whereas renal clearance was 5.4 L/h, with a Vss of 12.8 L and a terminal half‐life of 1.7 hours. Similar results were obtained after dosing with IV placebo. In comparison, in 2 published studies of SBECD pharmacokinetics in human subjects with normal renal function, total clearance was about 7.6 to 8.5 L/h, renal clearance was about 6.8 to 7.1 L/h, Vss was about 13 to 15 L, and terminal half‐life was 1.4 to 1.8 hours.6, 10
After dosing with IV delafloxacin, mean SBECD AUC0‐∞ increased 1.3‐, 2.2‐, and 5.5‐fold in subjects with mild, moderate, and severe renal impairment, respectively. Similar ratios were observed after dosing with IV placebo. In contrast, Cmax did not vary significantly with decreasing renal function, as would be expected for a parameter dependent more on volume of distribution and the time course of administration and distribution than on systemic clearance. In a published report on SBECD pharmacokinetics in subjects with compromised renal function using a marketed intravenous formulation of voriconazole (Vfend®), 4.8‐fold and 1.5‐fold increases in SBECD AUC and Cmax values, respectively, were observed in subjects with moderate renal impairment, together with a commensurate 4.7‐fold decrease in systemic clearance and increase in terminal half‐life from 1.8 to 8.9 hours.10 Volumes of distribution were essentially unchanged. These results indicated a more pronounced effect of renal impairment than seen in our study. The definition of moderate renal impairment (eGFR >30 to 50 mL/[min·1.73 m2]) was the same in both studies. In our study the distribution of renal function for subjects included lower, middle, and higher eGFR values within the defined range, whereas the uniformity of the distribution of subjects in the Abel study10 is unknown, and this could have contributed to their results.
In ESRD subjects, hemodialysis immediately following administration of IV delafloxacin reduced AUC0‐48 on average by 65% compared to dosing immediately after hemodialysis, and an average of 56% of the administered dose of SBECD was recovered in effluent fluid when hemodialysis was performed immediately after dosing. These observations are consistent with previous reports on dialysis of SBECD. From a model of SBECD pharmacokinetics in ESRD subjects receiving IV voriconazole every 12 hours, it was predicted that hemodialysis (2‐ to 6‐hour sessions) would remove on average 46% of the total systemic load of SBECD.11 No adverse events in this study were attributed to SBECD. Another model of SBECD pharmacokinetics estimated 67% of the systemic load of SBECD would be removed by a 6‐hour hemodialysis session after a single dose of IV voriconazole.12 The authors stated that infusion of voriconazole and SBECD was well tolerated in all patients without serious adverse events. In another report, 85% of the total dose of SBECD was recovered in the effluent fluid from subjects on continuous venovenous hemofiltration receiving IV voriconazole.13 The authors stated that no evidence of hepatic or renal disease attributable to SBECD was observed. In contrast, intermittent hemodialysis did not remove SBECD from patients receiving IV voriconazole in another study, but this was possibly related to the conditions of hemodialysis.14 Even though accumulation of SBECD occurred, no toxic effects were observed.
In our study, after administration of IV placebo, only 1 subject had a TEAE that was considered possibly related to the infusion of placebo. This suggests that SBECD did not cause a significant rate of TEAEs after a single IV placebo infusion. Taken together, the clinical experience with SBECD in subjects with renal impairment continues to demonstrate that it is well tolerated.
This study is limited in assessment of SBECD safety in patients with ESRD because there was no comparison using placebo/SBECD. In addition, this single‐dose study could not directly assess SBECD accumulation and safety with multiple dosing. However, pharmacokinetic modeling and simulation based on the data from this study suggest minor accumulation (<1.5‐fold) of SBECD would be expected in severe renal impairment with every‐12‐hour IV dosing for 14 days (on file at Melinta Therapeutics, New Haven, Connecticut).
In 2 phase 3 registration trials on the treatment of skin and skin structure infections, intravenous delafloxacin was administered twice daily for up to 14 days to 754 patients.3 Enrollment in the first of these trials was limited to patients with CLCR >30 mL/min, and in the second, to patients with CLCR >15 mL/min. Patients on hemodialysis were excluded. In the second trial, patients with severe renal impairment (CLCR >15 to 30 mL/min) were included, but the dose of IV delafloxacin was reduced from delafloxacin 300 mg (3200 mg SBECD) every 12 hours to delafloxacin 200 mg (2400 mg SBECD) every 12 hours in order that the exposure to delafloxacin not exceed the efficacious range. Delafloxacin with the SBECD‐containing formulation was effective and well tolerated in the 122 delafloxacin‐treated patients with renal impairment. The efficacy and safety outcomes in patients with renal impairment were comparable to those in the overall population. This assessment of TEAEs cannot distinguish between effects attributable to delafloxacin and effects attributable to SBECD, but it can be inferred that greater SBECD exposures from multiple IV dosing in renally impaired patients did not cause a markedly different rate of treatment‐related TEAEs.
Delafloxacin IV has received US FDA approval with a recommended dose reduction to 200 mg every 12 hours in patients with severe renal impairment, compared to 300 mg IV every 12 hours in patients with normal and mildly or moderately impaired renal function.1 There are insufficient data to make dosing recommendations in patients with ESRD.
Conclusion
The pharmacokinetics of the cyclodextrin excipient SBECD were investigated in subjects with renal impairment in a single‐dose crossover study using an IV formulation of delafloxacin compared with IV placebo. SBECD systemic and renal clearances decreased in parallel with decreased renal function, with resultant increases in exposure (as measured by AUC) and terminal half‐life. SBECD was extensively dialyzed in ESRD patients on hemodialysis. Increased SBECD exposures did not result in any noticeable increase in drug‐related TEAEs in this study. IV and oral delafloxacin have received FDA approval, with a recommended IV dose adjustment in patients with severe renal impairment (CLCR 15 to <30 mL/min) from 300 mg every 12 hours to 200 mg every 12 hours. No dosing recommendations were made in patients with ESRD, primarily due to insufficient multiple‐dose data in these patients.
Four authors (L.L., M.Q., D.R.L., and S.K.C.) were employed by Melinta Therapeutics, Inc, during this study, and all research was funded by Melinta Therapeutics, Inc.
The copyright line for this article was changed on 01 August 2019 after original online publication.
References
- 1. Delafloxacin Prescribing Information . https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208610s000,208611s000lbl.pdf. Accessed August 18, 2017.
- 2. O'Riordan W, McManus A, Teras J, et al. A global phase 3 study of delafloxacin (DLX) compared to vancomycin/aztreonam (VAN/AZ) in patients with acute bactyerial skin and skin structure infections (ABSSSI). Melinta Therapeutics Web Site. http://melinta.com/wp-content/uploads/2016/10/IDWEEK-1347-Baxdela-vs-VAN-AZ-302-Ph3-Results.pdf. Accessed August 18, 2017.
- 3. Beasley R, Oguchi G, Liang S, Lawrence L, Cammarata S. Delafloxacin (DLX) is effective and well‐tolerated in treatment of patients with renal impairment with acute bacterial skin and skin structure infections (ABSSSI) versus vancomycin/aztreonam (VAN/AZ). www.Melinta.com. http://melinta.com/wp-content/uploads/2017/04/ECCMID-2017-Renal.pdf. Accessed August 18, 2017.
- 4. Study to compare delafloxacin to moxifloxacin for the treatment of adults with community‐acquired bacterial pneumonia. www.clinicaltrials.gov. https://clinicaltrials.gov/ct2/show/NCT02679573?term=delafloxacin&rank=3. Accessed August 18, 2017.
- 5. Captisol® dosing in clinical and commercial products. Ligand Technology. www.Captisol.com. http://www.captisol.com/. Accessed August 18, 2017.
- 6. Luke D, Tomaszewski K, Damle B, Schlamm H. Review of the basic and clinical pharmacology of sulfobutylether‐β‐cyclodextrin (SBECD). J Pharm Sci. 2010;99:3291–3301. [DOI] [PubMed] [Google Scholar]
- 7. Hoover RK, Alcorn H, Lawrence L, Paulson S, Quintas M, Cammarata S. Delafloxacin pharmacokinetics in subjects with varying degrees of renal function [published online ahead of print December 18, 2017]. J Clin Pharmacol. 10.1002/jcph.1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levey A, Bosch J, Lewis J, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease study group. Ann Intern Med. 1999;130:461–470. [DOI] [PubMed] [Google Scholar]
- 9. Cockcroft D, Gault M. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. [DOI] [PubMed] [Google Scholar]
- 10. Abel S, Allan R, Gandelman K, Tomaszewski K, Webb D, Wood N. Pharmacokinetics, safety and tolerance of voriconazole in renally impaired subjects. Two prospective, multicentre, open‐label, parallel‐group volunteer studies. Clin Drug Invest. 2008;28:409–420. [DOI] [PubMed] [Google Scholar]
- 11. Luke D, Wood N, Tomaszewski K, Damle B. Pharmacokinetics of sulfobutylether‐β‐cyclodextrin (SBECD) in subjects on hemodialysis. Nephrol Dial Transpl. 2012;27:1207–1212. [DOI] [PubMed] [Google Scholar]
- 12. Hafner V, Czock D, Burhenne J, et al. Pharmacokinetics of sulfobutylether‐β‐cyclodextrin and voriconazole in patients with end‐stage renal failure during treatment with two hemodialysis systems and hemodiafiltration. Antimicrob Agents Chemother. 2010;54:2596–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kiser T, Fish D, Aquilante C, et al. Evaluation of sulfobutylether‐β‐cyclodextrin (SBECD) accumulation and voriconazole pharmacokinetics in critically ill patients undergoing continuous renal replacement therapy. Crit Care. 2015;19:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. von Mach M, Burhenne J, Weilemann L. Accumulation of the solvent vehicle sulphobutylether beta cyclodextrin sodium in critically ill patients treated with intravenous voriconazole under renal replacement therapy. BMC Clin Pharmacol. 2006;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]