

Abstract
The apurinic/apyrimidinic (AP) site is a common lesion of DNA damage. The levels of AP sites reported in the literature range widely, which is primarily due to the artefactual generation or loss of AP sites during processing of the DNA. Herein, we have developed a method to quantitate AP sites with a largely reduced level of artifacts by derivatizing AP sites before DNA isolation. A rapid digestion of nuclear protein was performed to minimize enzymatic DNA repair, followed by direct derivatization of AP sites in the nuclear lysate with O-(pyridin-3-yl-methyl)hydroxylamine (PMOA), yielding an oxime derivative that is stable through subsequent DNA processing steps. Quantitation was done using highly selective and sensitive liquid chromatography-tandem mass spectrometry, with a limit of quantitation at 2.2 lesions per 108 nucleotides (nts, 0.9 fmol on column). The method was applied in vivo to measure AP sites in rats undergoing oxidative stress [liver: 3.31 ± 0.47/107 nts (dosed) vs 0.91 ± 0.06/107 nts (control), kidney: 1.60 ± 0.07/107 nts (dosed) vs 1.13 ± 0.12/107 nts (control)]. The basal AP level was significantly lower than literature values. The method was also used to measure AP sites induced by the chemotherapeutic nitrogen mustard in vitro.
Graphical Abstract
Exogenously or endogenously induced DNA apurinic/apyrimidinic sites can be quantitated by direct derivatization of nuclei and liquid chromatography-tandem mass spectrometry.

INTRODUCTION
The apurinic/apyrimidinic (AP) site is a ubiquitous DNA lesion and the central intermediate in DNA base excision repair (BER) pathway.1 Various processes, such as alkylation and deamination of bases, oxidative stress, or radiation can lead to damaged DNA bases that are excised by DNA glycosylases and result in AP sites.2-4 Spontaneous cleavage of a glycosylic bond also generates AP sites, which happens slowly under physiological conditions but is greatly accelerated by heating or acidic pH.5 The non-coding abasic lesion can be cytotoxic and mutagenic if not efficiently repaired.6-9 In mammals, the major pathway for removing this lesion utilizes an AP endonuclease to incise the 5’ end of AP sites, resulting in a 5′-deoxyribose phosphate terminus that is subsequently repaired by DNA polymerase β.1,10 Isolated AP sites can be rapidly repaired by BER, while clustered lesions can persist in cells for over one day.11,12
AP sites exist in several equilibria forms.13,14 The aldehyde form can react with alkoxyamine agents and yield a relatively stable oxime. This chemical reaction is often employed as a primary strategy for AP site derivatization and quantitation. Many analytical methods have been developed for quantitating AP sites, including chemiluminescent or fluorescent probes,5,15-20 as well as mass spectrometry (MS) techniques.21-24 The former strategies, e.g., the commonly used aldehyde-reactive probe (ARP) assay, label all naturally occurring aldehydes and ketones present in DNA non-selectively. In comparison, MS is a more specific method for measuring AP sites where exact mass and product ions are used for an accurate quantitation.19
Literature values for the steady-state level of AP sites vary considerably, when assayed by different analytical methods. The ARP assay is a luminescent assay that reacts with aldehydes and ketones via a hydroxylamine group but does not provide further structural information. Oxidative damage to the sugar backbone of DNA leads to strand breaks that leave reactive aldehyde and ketone group on the remnants of the deoxyribose.25 The steady-state level of ARP reactive DNA lesions (including AP sites) has been estimated to range from <0.67 AP sites/106 nucleotides (nts) in human cell lines to ~1 AP sites/105 nts in mammalian tissues.12,15,26 A recently developed liquid chromatography-tandem mass spectrometry (LC-MS/MS) method estimated that the steady-state level of AP sites was ~1 AP site/106 nts in cell lines.24 The wide discrepancies in reported values of AP sites are likely due to the artifactual formation or the loss of AP sites during DNA isolation, digestion, or the derivatization step.
We surmise that the actual level of AP sites is considerably lower than reported. In this paper, we describe a method for quantitating AP sites in nuclear DNA isolated from mammalian tissue with reduced artifact formation. Our approach is to isolate the nuclei from tissue homogenate and immediately deactivate most BER enzymes with proteinase K digestion. The nuclear DNA is then derivatized with O-(pyridin-3-yl-methyl)hydroxylamine (PMOA) in the nuclear lysate matrix (Scheme 1). The alkoxyamine modified DNA is isolated, enzymatically digested, and measured by LC-MS/MS.
Scheme 1.
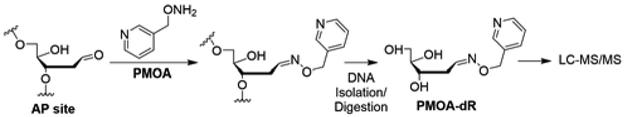
Quantitation of AP sites by PMOA derivatizing method.
The method was applied to measure oxidative stress and nitrogen mustard alkylation induced-AP site generation in vivo and in vitro. In a rat model, ferric nitrilotriacetate (Fe-NTA)-mediated oxidative stress leads to an over three-fold increase of AP sites. The induction of AP site formation in isolated DNA followed a linear relationship to the concentration of the chemotherapeutic nitrogen mustard.
MATERIAL AND METHODS
Caution:
Fe-NTA and N,N-bis(2-chloroethyl)ethylamine (NM) are carcinogens and must be handled in a well-ventilated fume hood with proper use of gloves and protective clothing. All reagents and instrumentation information are reported in supporting information.
Optimizing the derivatization efficiency of PMOA with AP sites in oligonucleotides
The experiments conducted with a single-stranded oligonucleotide are reported in supporting information. AP sites were prepared by uracil DNA glycosylase-catalyzed removal of uracil in double-stranded (5′-GCCGTUAGGTA-3’ • 3’-CGGCAATCCAT-5′) oligonucleotide.22 The oligonucleotide was reacted with 5 mM PMOA at 37 °C at pH 7.0 without a catalyst, or at pH 7.4 with 5 or 10 mM L-histidine.
To explore the catalytic effect of enzymes, the double-stranded 11-mer oligonucleotide (50 μL, 1.89 μg/μL), RNase A (15 μL, 150 μg), and RNase T1 (1 μL, Sigma R1003) were added to 500 μL of TE buffer [50 mM Tris-HCl buffer, 10 mM ethylenediaminetetraacetic acid (EDTA), pH 8.0]. PMOA (25 μL, 100 mM in water) was added and incubated for 30 min at 37 °C. Then, proteinase K (20 μL, 400 μg) was added and incubated for another 30 min. Finally, Puregene Protein Precipitation (PP) solution (250 μL, pH = 8, Qiagen) was added, and the mixture was incubated for an additional 30 min. At selected time points, aliquots were taken, and the unreacted oligonucleotide was quantitated with HPLC-UV at 260 nm.
Synthesis of (3S,4R)-3,4,5-trihydroxypentanal O-pyridin-3-yl-methyl oxime (PMOA-dR) and [13C5]-PMOA-dR.
PMOA (20 mg, 0.16 mmol) was added to 500 μL of 50 mM potassium phosphate buffer (pH 7.0) containing 2-deoxy-D-ribose (23 mg, 0.17 mmol) and incubated in a thermomixer at 37 °C for 1 h. The product was purified by HPLC (Agilent 1260 Infinity HPLC), with mobile phases (A) 5 mM ammonium bicarbonate (pH = 7.8) and (B) methanol. An XBridge Prep Phenyl column (10 × 250 mm, 5 μm particle size, Waters) was used to separate the compounds at a flow rate of 3 mL/min using a 50 min isocratic flow at 10% B. Fractions were collected and dried under high vacuum to yield the two geometric isomers as white solids.
Overall product yield: 92%. Yield of Z isomer: 16.8 mg. UV extinction coefficient at 260 nm (2 mM NH4HCO3): ε = 2.84 × 103 M−1·cm−1. Yield of E isomer: 18.5 mg, ε = 2.87 × 103 M−1·cm−1. The UV spectra and 1H-NMR are reported in supporting information. [13C5]-PMOA-dR was synthesized using [13C5]-2-deoxy-D-ribose and purified as an isomeric mixture (isotopic purity > 99.8%).
Derivatization of AP sites in isolated DNA.
Calf thymus (CT) DNA (30 μg) was incubated at 37 °C for 1.5 h in PBS buffer (pH 7.4) containing 5 mM histidine and 0.5 to 20 mM PMOA. The excess PMOA was consumed by adding butyraldehyde solution (1 M in 50% isopropanol/water) to a final concentration of 50 mM and incubated at 37 °C for 10 min.
The DNA was precipitated by adding 0.1 volume of 5 M NaCl and two volumes of chilled isopropanol, washed twice with 70% ethanol in 5 mM HEPES buffer (pH 8.0), and dried under vacuum centrifugation. The DNA pellet was reconstituted in 5 mM HEPES buffer (pH 8.0), and the concentration was determined by UV assuming that 50 μg/mL of double-stranded DNA is equal to an absorbance of 1.0 at 260 nm.
Enzymatic digestion of AP sites in DNA.
The [13C5]-PMOA-dR (stable isotopically labeled internal standard, 1.1 per 106 nt) was added to the DNA prior to enzymatic digestion. The DNA samples (28 μg) were incubated at 37 °C with DNase I (5 μg) and nuclease P1 (0.5 μg) for 3 h, followed by phosphodiesterase I (0.25 μg) and alkaline phosphatase (2 μg) for 15 h, and then incubated with adenosine deaminase (12 mU) at 25 °C for 1.5 h.
Solid phase extraction method (SP) method.
After DNA digestion, the derivatized AP sites were enriched by solid phase extraction (SPE) with a Strata-X polymeric cartridge (30 mg, 33 μm) preconditioned with CH3OH, followed by H2O. The cartridge was washed with 8% CH3OH in 2 mM NH4HCO3 (2.5 mL), and the PMOA-dR was eluted from the cartridge with 90% CH3OH in 2 mM NH4HCO3 (1 mL). The eluents were dried by vacuum centrifugation, reconstituted in 20 μL 2 mM NH4HCO3, and transferred to LC autosampler vials for LC-MS/MS analysis.
LC-MS/MS method for quantitation of PMOA-dR in DNA.
Quantitation of PMOA-dR was conducted on a Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an UltiMate™ 3000 RSLCnano System (Thermo Fisher Scientific, San Jose, CA) and an Advance CaptiveSpray source (Michrom bioresource Inc., Auburn, CA). A Magic C18AQ reversed-phase column (Michrom Bioresources, Inc., 0.3 × 150 mm, 3 μm particle size, 100 Å pore size) was employed for chromatographic separation. The solvents that constitute the mobile phases were (A) 2 mM NH4OAc and (B) 2 mM NH4OAc in 95% CH3CN. The sample (9 μL) was injected onto the analytical column with the following gradient elution profile: 0−2 min: 5 μL/min, 1% B; 2−2.5 min: 5−2 μL/min, 1% B; 2.5 to 20 min: 2 μL/min, 1 to 95 % B; 20−22 min: 2−5 μL/min, 95% B; 22−24 min: 5 μL/min, 95−1% B, 24−30 min: 5 μL/min, 1% B. Under this condition, the E and Z isomers of PMOA-dR coelute as a single peak. PMOA-dR was quantified in the positive ion mode. MS/MS parameters were as follows: collision-induced dissociation (CID): 30%, activation Q: 0.25, isolation width: 2.0. PMOA-dR (m/z 241.1 → 92.0, 108.0, 110.0, 125.0); [13C5]-PMOA-dR (m/z 246.1 → 92.0, 108.0, 110.0, 125.0).
A seven-point calibration curve was constructed with a CT DNA digestion matrix. The DNA was digested and cleaned up with SPE as described above, then the internal standard [13C5]-PMOA-dR was spiked at 100 fmol/30 μg DNA (1.1 per 106 nts), together with the synthesized PMOA-dR standard (1:1 isomeric mixture). The calibration curve ranged from 0, 0.22, up to 275 PMOA-dR per 107 and contained three replicates per calibrant level. The lower limit of quantitation (LLOQ) was 0.9 fmol of derivatized AP-site on column, or 2.2 PMOA-dR adducts per 108 nts in 13.5 μg of enzymatically digested CT DNA serving as the background matrix. The calibration curve was fitted with linear least-square regression analysis using 1/y weighting with GraphPad Prism software (V 6), R2 = 0.9989 (Figure S4).
Animal studies.
All protocols were reviewed and approved by the University of Minnesota Institutional Animal Care and Use Committee.
Fe-NTA was prepared according to reported method27,28 with modifications. Fe(NO3)3 (1 M, 5 mL) was mixed with HCl (1 N, 5 mL) and Na2-NTA (1 M, 10 mL). The pH was adjusted to 7.4 with Na2CO3 (1 M) and the volume was increased to 37.5 mL with Milli-Q water. Final concentration: 133 mM Fe(III), 7.5 mg Fe/mL. The negative control vehicle was prepared by adding Na2CO3 (1 M) to HCl (1 N, 5 mL) to adjust the pH to 7.4, and then the volume was increased to 37.5 mL with Milli-Q water. Male F344 rats aged three months, body weight range 240−266 g (Charles River Laboratories, Wilmington, MA), were housed in the animal facility under standard conditions on AIN-76A diet. After one week of acclimatization, the rats were dosed with vehicle or Fe-NTA at 15 mg Fe/kg body wt. via intraperitoneal injection (n = 4 in each group), and sacrificed 6 h after dosing.29 Kidneys and livers were immediately put on dry ice and frozen at −80 °C until sample workup.
For measuring spontaneous depurination rate, as well as AP site induced by DNA alkylation, healthy male F344 rats aged 70 days, body weight 204−218 g, were sacrificed one week after acclimatization.
Derivatization of AP sites in mammalian tissue.
Step 1. Fresh-frozen rat tissue from Fe-NTA treated and control rats were homogenized with a PRO 200 homogenizer (PRO Scientific, Oxford, CT) in HE lysis buffer [50 mM HEPES buffer, 10 mM EDTA, pH 8.0] on ice. The tissue homogenate (40 mg wet weight) was centrifuged at 3,000g at 4 °C, and the nuclear pellet was resuspended in HE buffer (500 μL). Proteinase K (20 μL, 20 mg/mL in water) and 10% SDS (60 μL) were added to the suspension and incubated in a thermomixer at 37 °C, 900 rpm for 1.5 h. Step 2. (A) Direct derivatization of the nuclear lysate: PMOA (32 μL of 100 mM stock solution was the optimized concentration and used for the rest of the experiments) was added to the nuclear lysate and incubated at 37 °C for 1.5 h. Butyraldehyde (30 μL, 1 M in 50% isopropanol/water) was then added to quench the remaining PMOA by further incubation for 10 min. The samples were kept on ice for 5 min, then 250 μL Puregene PP solution was added. The tubes were vortexed thoroughly and centrifuged at 2,000g for 10 min. The supernatant was transferred to a new 2 mL Eppendorf tubes, and the nucleic acid was precipitated with 0.1 volume of NaCl (5 M) and 1 mL isopropanol. The pellet was washed with 70% ethanol in 5 mM HEPES buffer (pH 8.0) and dried under vacuum centrifugation. (B) Derivatization of the isolated nucleic acid mixture: proteins were removed with Puregene PP solution, and the nucleic acid pellet was isolated by the method described above. The pellet was reconstituted in PBS buffer (pH 7.4) containing histidine (5 mM) and derivatized with PMOA (as described above) at 37 °C for 1.5 h, then quenched with butyraldehyde and isolated. Step 3. The derivatized pellets were reconstituted with 300 μL HE buffer, incubated at 37 °C with RNase A (150 μg) and RNase T1 (0.5 μg) for 1.5 h, and the enzymes were precipitated with Puregene PP solution. The DNA pellet was isolated, digested, and quantitated by LC-MS/MS as described above.
Determination of spontaneous depurination rate.
Liver tissue from healthy rats was homogenized, and the isolated nuclear pellet was resuspended in HE buffer (500 μL). Proteinase K (20 μL, 20 mg/mL in water) and 10% SDS (60 μL) were added to the suspension and incubated at 37 °C for 0, 1.5, 3, …, 47.5 h. After the incubation, PMOA (32 μL of 100 mM stock solution) was added to the nuclear lysate and incubated at 37 °C for 1.5 h, then the reaction was quenched with butyraldehyde, and the DNA was isolated and quantitated as described in method A.
Artifactual formation of AP sites during sample workup.
Liver tissue from healthy rats was homogenized, and the isolated nuclear pellet was resuspended in HE buffer (500 μL). The AP sites were derivatized under four conditions. Method A: Derivatization was performed in the nuclear lysate after proteinase K digestion, as described in method A. A1: Immediately after nuclei isolation, proteinase K (20 μL, 20 mg/mL in water), 10% SDS (60 μL), as well as PMOA (32 μL of 100 mM stock solution) were added to the suspension, incubated at 37 °C for 1.5 h, and quenched with butyraldehyde. The derivatized DNA was then isolated as described in method A. A2: The nuclear pellet suspension was treated with the same amount (as method A) of RNase A and RNase T1 for 1.5 h, followed by proteinase K and SDS for 2 h. Puregene PP solution was added to precipitate the proteins, and the DNA in the supernatant was precipitated with NaCl and chilled isopropanol, washed twice, and dried under vacuum. The isolated pellet was incubated in PBS buffer (200 μL, pH 7.4) containing histidine (5 mM) and PMOA (5 mM) at 37 °C for 1.5 h and the reaction was quenched with butyraldehyde. The derivatized DNA was precipitated, washed twice and dried under vacuum. A3: The nuclear lysate was treated with the same enzymes and reagents in method A2 except that the DNA pellet was precipitated under −20 °C overnight before derivatization. The AP sites were quantitated by LC-MS/MS as described above.
Quantitation of NM-induced AP sites in isolated DNA.
Nuclear DNA was isolated from fresh-frozen untreated rat liver tissue. Briefly, after tissue homogenization on ice, the nuclear pellet was immediately digested with proteinase K and SDS, and the nucleic acid was isolated with Puregene solution. RNase A/T1 digestion was performed, and the DNA pellet was isolated as described above. DNA (42 μg) was incubated in 500 μL PBS buffer (pH 7.4) containing freshly prepared N,N-Bis(2-chloroethyl)ethylamine (nitrogen mustard, NM) at 0, 3, 10, 30, or 100 μM at 37 °C for 1 h. The excess NM was removed with ethyl acetate extraction (500 μL × 3). The DNA was precipitated, washed twice with ethyl acetate, and dried. The DNA was reconstituted in 450 μL PBS buffer (pH 7.4) and incubated at 37 °C for 5 h to induce AP sites derived from N7-alkylated guanine adducts.30 The AP sites were then derivatized by adding histidine (5 mM) and PMOA (5 mM), and then quantitated by LC-MS/MS as described above.
RESULTS AND DISCUSSION
Optimization of PMOA derivatizing efficiency using oligonucleotides
We employed AP site-containing 11-mer oligonucleotides to optimize the derivatization reaction. Enzymatic removal of uracil in single- and double-stranded oligonucleotides quantitatively generated a single AP site per oligonucleotide.22 We chose the commercially available O-(pyridin-3-yl-methyl)hydroxylamine (PMOA) as the derivatization agent due to its product stability,31 moderate polarity, and the basic pyridine nitrogen enhances ionization efficiency.
The derivatization reaction with both the single- and double-stranded oligonucleotide was completed within 3 h at pH 7.0 and 37 °C, as measured by HPLC-UV (Figure S1/S2). The efficiency of the reaction largely depends on pH. Since oxime formation is an acid-catalyzed reaction,16,32 a lower pH gives a faster derivatization rate (Figure 1A, the reaction was completed within 3 h at pH 7.0 vs. 4 h at pH 7.4). Nevertheless, lower pH also promoted oxime hydrolysis (reverse reaction of oxime formation), as well as spontaneous depurination that generates artifactual AP sites.33,34 The PMOA-derivatized oligonucleotides hydrolyzed at pH 7.1 (15%) or pH 7.6 (5%) at 37 °C over 24 h, while the derivatized AP site was completely stable at pH 8.0 (Figure S3). Therefore, all buffers used after the derivatization reaction were kept at pH ≥ 8.0 to minimize hydrolytic loss.
Figure 1.
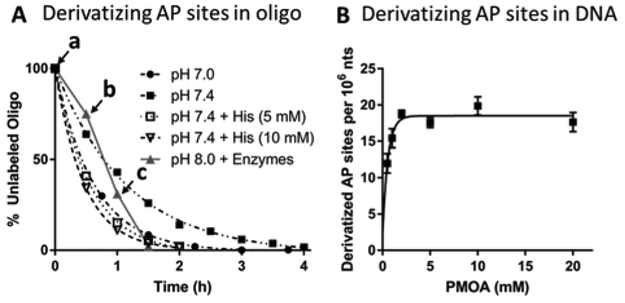
(A) Derivatization of AP sites in double-stranded oligonucleotide with 5 mM PMOA at 37 °C, with different buffers and catalysts. The percent unreacted oligonucleotide vs. time is reported. Points on the reaction rate curve at pH 8.0 containing sequential addition of enzymes employed during lysis of the nuclear pellet: a. RNase A & RNase T1 added. b. Proteinase K added, c. Puregene PP solution added, or reactions conducted at pH 7.0 or pH 7.4 without or with histidine. (B) Derivatization of AP sites in CT DNA with various concentrations of PMOA. Reaction condition: pH 7.4, 37 °C, 1.5 h. Each value represents the mean ± SD, n = 3.
We utilized histidine as a catalyst to optimize the PMOA derivatization reaction with an AP-site containing oligonucleotide duplex. The reaction was conducted at pH 7.4 and thus, minimized the hydrolysis of glycosidic bonds and formation of artefactual AP-site.33,34 The addition of 5 mM histidine35 reduced the derivatization time from 4 to 1.5 h. A further increase of the histidine concentration only slightly enhanced the efficiency of the reaction (Figure 1A). We then tested the PMOA derivatization reaction in the presence of the reagents required to process cellular DNA. We reacted PMOA with AP-containing duplex oligonucleotide in TE buffer at 37 °C in the presence of RNases for 30 min, then added proteinase K, followed by the Puregene PP solution (Figure 1A, triangles, and grey lines). Interestingly, the rate of derivatization increased after addition of proteinase K. Although we did not investigate the exact mechanism, we suspect the digestion of proteins generates sufficient quantities of amino acids to serve as acid/base catalysts that increase the reaction efficiency. This observation provides another choice of conditions for derivatization of the AP sites in nuclear lysate before DNA isolation, which we have investigated in the following experiments.
LC-MS/MS method to quantitate AP sites
An LC-MS/MS method was developed with the Velos Pro ion-trap mass spectrometer monitoring at the MS2 scan stage. The collision-induced dissociation (CID) of PMOA-dR and [13C5]-PMOA-dR produced product ions at m/z 92, 108, 110, 125, which were extracted for identification and quantitation of the AP site (Figure 2). The LLOQ value was 0.9 fmol of derivatized AP-site, or 2.2 PMOA-dR adducts per 108 nts in 13.5 μg of DNA assayed on column.
Figure 2.
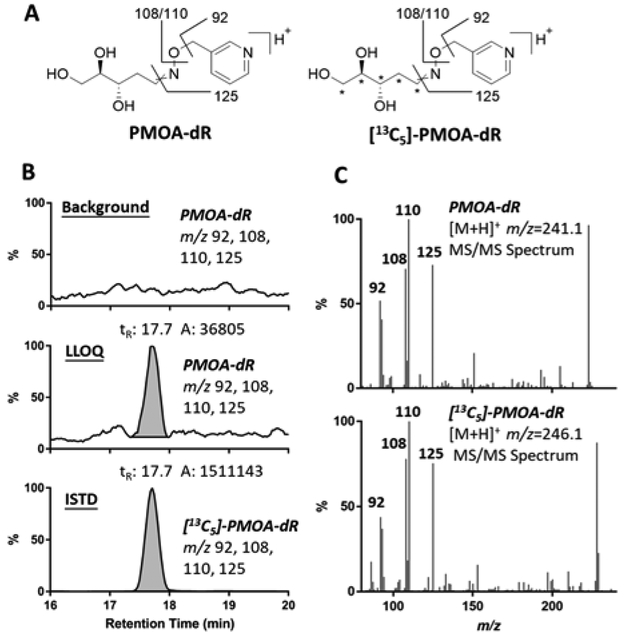
(A) Proposed MS/MS fragmentation of PMOA-dR ([M+H]+ m/z 241.1) and [13C5]-PMOA-dR ([M+H]+ m/z 246.1). (B) Extracted ion chromatogram (EIC) of PMOA-dR (at background level normalized against the base peak in LLOQ, and at LLOQ) and [13C5]-PMOA-dR (1.1 per 106 nts) in DNA digestion matrix and (C) PMOA-dR and [13C5]-PMOA-dR MS/MS spectra. The * represent the sites of the 13C atoms in the internal standard.
Stability and recovery of PMOA-dR DNA during enzymatic digestion and SPE processing
The stability of the derivatized AP-site during nuclease digestion of the DNA and SPE processing was determined by comparing the peak area ratio (PAR) of PMOA-dR/[13C5]-PMOA-dR in samples spiked with PMOA-dR after DNA digestion/SPE with the PAR of samples spiked with PMOA-dR prior to DNA digestion/SPE, where the internal standard was spiked after DNA digestion/SPE. The average recovery at three concentration levels (0.66, 5.5, 27.5 PMOA-dR per 107 nts) was 108 ± 7% (Table S1), indicating no significant loss of PMOA-dR occurred during the processing or enzymatic digestion of DNA. Moreover, we varied the quantity of the four nucleases by 4-fold (0.5x, 1x or 2x amounts of all enzymes) to digest PMOA-dR-modified DNA (Figure S5). The level of PMOA-dR recovered from DNA was constant as a function of enzyme concentration, demonstrating that the PMOA labeling did not compromise the efficacy of nuclease digestion of DNA.
Derivatization of AP sites in calf thymus DNA
We determined the derivatization efficiency of AP sites in CT DNA by reacting PMOA at 37 °C, pH 7.4, in the presence of 5 mM histidine catalyst. After incubation for 1.5 h, butyraldehyde32 was added to quench the reaction, followed by DNA precipitation, digestion, and SPE before LC-MS/MS analysis (Figure 1B). We found that 5 mM PMOA is sufficient to derivatize all AP sites within 1.5 h. Therefore, we used this concentration of PMOA to derivatize isolated nucleic acids as described below.
Stability of free and derivatized AP sites during DNA isolation
Artifactual formation or loss of AP sites during DNA isolation are likely factors for the large discrepancies of AP site levels reported in the literature. Potential sources of artifacts include BER enzyme activity, spontaneous depurination at high temperature and/or low pH, β-elimination, and reaction of the AP site with nucleophiles in the DNA isolation matrix. Commercial CT DNA contains a high level of free AP sites (~1 to 2 AP site/105 nts, vide supra), which was used as a model to test the AP site stability during DNA isolation. We observed a large variation and relative decrease of free AP site levels, when underivatized CT DNA was processed in the presence of DNA isolation buffers (containing Tris or HEPES, EDTA, ammonium sulfate, Puregene PP solution, isopropanol, etc.) and enzymes (RNases and proteinase K), (Figure S6). In contrast, the PMOA-derivatized CT DNA was highly stable when subjected to the entire DNA isolation process with an average recovery of 93 ± 3% derivatized AP sites compared to control group (no incubation performed) (Figure S7 & Table S2). The stability of AP sites is largely increased upon PMOA labeling because the derivatized oxime structure is less susceptible to potential reactions than the free AP sites with nucleophiles.. Therefore, to control artifacts in AP site quantitation in mammalian tissue samples, we performed the derivatization step before the isolation of DNA.
Derivatization of AP sites in mammalian tissue
The AP site quantitation method for mammalian tissue was optimized using fresh-frozen tissue of rats treated with Fe-NTA, an iron complex that catalytically generates reactive oxygen species (ROS).36 Increased ROS production leads to oxidative stress, which plays an essential role in Fe-NTA-mediated renal and hepatic tumor promotion after long-term administration.37-39 Oxidative stress induces DNA lesions that are primarily repaired by BER enzymes, leading to elevated levels of AP sites as the major BER intermediate.4,40,41 Therefore, the derivatization method was optimized using both the Fe-NTA-treated and control rats.
Ames and co-workers reported a method for in situ derivatization of AP sites designed to reduce artifact.12 They directly derivatized the aldehydic lesions with ARP in the nuclei of living cells, thoroughly washed the cells, and then isolated the DNA for quantitation. Although this strategy largely reduced artifacts from DNA isolation (they reported ≤0.67 AP sites per 106 nts as the steady-state level), the BER enzymes that catalyze the generation or excision of AP sites during derivatization were not completely inhibited. The authors observed more than a three-fold increase of aldehydic lesion when derivatizing the intact nuclei compared to lysed nuclei, where the nuclear glycosylases were released. Thus, herein, to minimize the BER enzyme activity, we immediately digested the nuclear proteins with proteinase K and SDS after isolating nuclei from rat tissue homogenate. Following proteinase K treatment for 1.5 h, we compared the efficiency of two derivatization methods: (A) directly adding PMOA to the nuclear lysate to derivatize the AP sites in the presence of proteinase K at pH 8.0; and (B) quickly precipitating proteins with Puregene PP solution, isolating the nucleic acids, and derivatizing with PMOA under pH 7.4. After quenching the reaction with excess butyraldehyde, for both method A and B, the derivatized nucleic acids were then treated with RNases to obtain pure DNA. As mentioned previously, the addition of proteinase K accelerated the rate of PMOA derivatization rate (Figure 1A). Indeed, the rate of derivatization of the nuclear lysate was similar to that of isolated nucleic acids (Figure 3A). The product yield of AP sites reached a plateau at 5 mM PMOA, signifying the reaction went to completion. The AP site level obtained by the two methods is the same, suggesting that the nuclear lysate matrix did not inhibit the efficiency of the reaction. Since the nucleic acid derivatization (method B) requires additional isolation steps, we chose the nuclear lysate derivatization (method A) with 5 mM PMOA for quantitation of AP sites in mammalian tissue.
Figure 3.
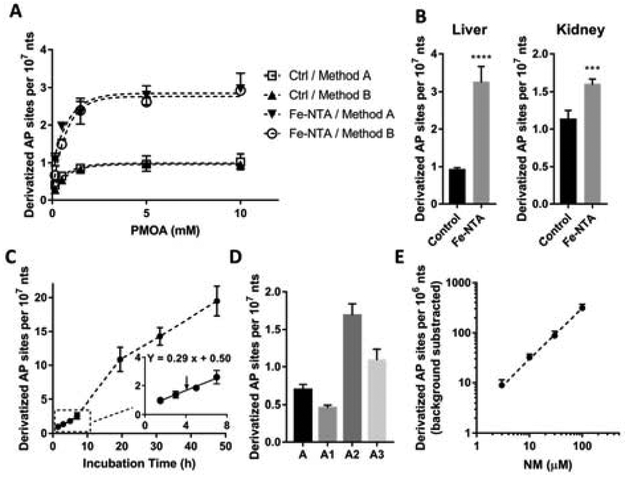
(A) Derivatization of AP sites in rat liver nuclei. Rats were sacrificed 6 h after intraperitoneal injection with Fe-NTA (15 mg Fe/kg) and vehicle (Ctrl group). AP sites were derivatized by the nuclear lysate-derivatization method (method A) or nucleic acid-derivatization method (method B). Each value represents the mean ± SD, n = 3. (B) AP site level in rat livers and kidneys after administration of Fe-NTA (grey bar) or vehicle (black bar). n = 4. Two-tailed t test p < 0.0001 (****), p < 0.001 (***). (C) Spontaneous depurination rate measured in untreated rat liver nuclei lysate (n=3). (D) AP site level in DNA isolated/derivatized by different methods. A. After nuclear isolation, conduct proteolysis for 1.5 h and then AP site derivatization for 1.5 h. A1: After nuclear isolation, conduct AP site derivatization and proteolysis together for 1.5 h. A2: Treat nuclei with RNases for 1.5 h, proteinase K for 2 h, and precipitate excess proteins. Precipitate DNA with salt/isopropanol and immediately derivatize the isolated DNA. A3: Unlike method A2, DNA pellet was precipitated under −20 °C overnight before derivatization. n = 4. (E) Isolated rat liver DNA was incubated with NM at 37 °C for 1 h, followed by neutral hydrolysis for 5 h (pH 7.4). n = 3. The background level of AP sites by neutral hydrolysis (6.5 ± 1.5/107 nts) was subtracted.
Precision of the method
The within-day and between-day precision of method A were determined using livers of four Fe-NTA treated rats and four control rats (treated with vehicle). Derivatization, isolation, and quantitation were performed with three independent analyses per animal per day on three different days. The average within-day/between-day coefficient of variation (CV) were 8.6%/9.8% for the Fe-NTA treated rats, and 8.9%/12.5% for the control rats (Table 1).
Table 1.
Within-day and Between-day Quantitation of AP Sites (per 107 nts) in Fe-NTA Treated Rats and Control Rats.
| Control Rats | Fe-NTA Treated Rats | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 |
Day 2 |
Day 3 |
CV% | CV% | Day 1 |
Day 2 |
Day 3 |
CV% | CV% | ||||
| Within- day |
Between- day |
Within- day |
Between- day |
||||||||||
| 1 | Mean | 0.90 | 1.01 | 0.81 | 8.9 | 11.3 | 1 | Mean | 2.86 | 2.77 | 3.04 | 11.0 | 4.8 |
| SD | 0.01 | 0.11 | 0.12 | SD | 0.55 | 0.13 | 0.28 | ||||||
| RSD% | 1.1 | 10.5 | 14.9 | RSD% | 19.1 | 4.8 | 9.2 | ||||||
| 2 | Mean | 0.78 | 0.88 | 0.81 | 10.8 | 6.4 | 2 | Mean | 2.62 | 3.08 | 3.00 | 4.7 | 8.5 |
| SD | 0.10 | 0.07 | 0.09 | SD | 0.12 | 0.08 | 0.21 | ||||||
| RSD% | 12.9 | 8.5 | 10.9 | RSD% | 4.6 | 2.7 | 6.9 | ||||||
| 3 | Mean | 0.78 | 1.05 | 1.00 | 8.1 | 15.1 | 3 | Mean | 3.23 | 4.07 | 3.76 | 5.6 | 11.6 |
| SD | 0.11 | 0.02 | 0.08 | SD | 0.19 | 0.28 | 0.15 | ||||||
| RSD% | 14.0 | 2.3 | 8.0 | RSD% | 6.0 | 6.8 | 3.9 | ||||||
| 4 | Mean | 0.79 | 1.12 | 0.99 | 7.8 | 17.4 | 4 | Mean | 3.16 | 3.86 | 4.22 | 13.2 | 14.3 |
| SD | 0.09 | 0.09 | 0.04 | SD | 0.53 | 0.40 | 0.52 | ||||||
| RSD% | 10.8 | 8.4 | 4.2 | RSD% | 16.9 | 10.4 | 12.3 | ||||||
AP-site formation in rats under oxidative stress conditions
The AP site levels in liver and kidney of Fe-NTA treated and control rats (n=4 for each group) were quantitated using method A and plotted in Figure 3B. As expected, the level of AP sites increased in both organs of Fe-NTA treated rats.. In rat liver, the AP sites of Fe-NTA-dosed group (3.31 ± 0.47 AP sites per 107 nts) was almost four-fold higher compared to control group (0.91 ± 0.06/107 nts). In rat kidney, dosed group also showed an elevated AP site level (1.60 ± 0.07/107 nts) compared to control group (1.13 ± 0.12/107 nts), although the difference was not as large as for liver. The lower level of AP-sites in kidney of Fe-NTA treated rats could be due to its faster AP site repair activity. The repair rate of 8-oxo-deoxyguanosine 6 h after treatment of Fe-NTA was reported to be much faster in rat kidney than in liver.42
Determination of spontaneous depurination rate of DNA
Using our optimized nuclear lysate derivatization method that minimized artifact formation, we observed a background level of AP sites (<1 AP site/107 nts in rat livers) that was significantly lower than literature values employing the ARP assay after DNA isolation (8−9 per 106 nts in rat livers).15 Our LC-MS/MS method is selective for the quantitation of AP sites, whereas the ARP assay is likely detecting a wider range of aldehydic sites in DNA. However, at this low level of AP site formation, we must consider the spontaneous rate of depurination as an artifact during nuclei-lysis and AP site derivatization. Although the rate of glycosidic bond hydrolysis is low at near-neutral pH,33,34 the rate of AP site formation in isolated DNA was reported to be 0.64/107 nts/h at 37 °C at pH 7.4, which is sufficiently high to introduce artifacts.5 Herein, we examined the spontaneous generation of regular AP sites in healthy (no treatment performed) rat liver DNA, which was incubated in the nuclear lysate containing proteinase K, SDS, and HE buffer (pH 8.0). As shown in Figure 3C, the AP sites increased linearly at 0.29/107 nts/h during the first 7 h. Then the rate was accelerated during the overnight incubation, which may be attributed to the diffusion of DNA to the lysate buffer following proteolysis of the histones. A linear regression curve-fitting of the first 7 h gave a Y-intercept of 0.50/107 nts (Figure 3C), providing an estimate of the background AP site level in healthy male rat liver. Therefore, considering this spontaneous depurination rate, the difference in the AP site level between Fe-NTA and vehicle-treated rats is likely greater than the actual values measured.
Artefactual formation of AP sites during sample workup
Although the spontaneous depurination is not completely inhibited, this optimized method A greatly reduces the artefactual AP site generation. We compared several different methods of DNA isolation and derivatization. If the nuclear pellet is simultaneously subjected to protein digestion and derivatization, the sample processing time can be shortened by 1.5 h, and the AP site level we obtained is slightly lower (Figure 3D, method A1). However, we cannot confirm whether the derivatization process is complete or if the incomplete digestion of histones restricted the accessibility of PMOA to react with some AP sites.
We also measured the AP site level in DNA isolated by conventional sample processing methods. The RNA in the nuclear pellet was first digested for 1.5 hours, followed by digestion of the protein for 2 hours, precipitation of protein, and salt/isopropanol precipitation of DNA. The DNA precipitation step can be done quickly (method A2) or incubated at −20 °C overnight (method A3). As shown in Figure 3D, conventional DNA isolation followed by derivatization resulted in a high level of artifacts. If the precipitated DNA is rapidly derivatized, the AP site level obtained is over two times of that obtained by method A. If the DNA is precipitated overnight before derivatization, the AP site level dropped due to the instability of underivatized AP site. In summary, method A minimizes the artifact formation for quantitation of the AP site in mammalian tissue.
Quantitation of NM-induced AP-sites in isolated DNA
We applied our method to measure AP site formation in vitro using rat liver DNA alkylated with nitrogen mustard(NM). The reactive intermediates of NM are N-alkylaziridinium ions, which react with the nucleophilic nitrogens on DNA bases, yielding mono-DNA adducts and crosslinks.43 The NM forms adducts at the endocyclic nitrogen atoms of nucleobases, such as N7 of guanine This cationic N7-guanine (NM-G) adduct readily undergoes depurination.43.43 We previously quantitated NM-DNA adducts for the reaction of CT DNA with 1 μM NM, yielding monomeric NM-G at 2 adducts/106 bases.30
We measured the generation of AP sites in isolated DNA treated with a range of NM concentrations. Since AP sites are abundant in CT DNA (vide supra), we used the DNA isolated from livers of healthy untreated rats. The alkylation reaction was performed with NM and then the AP sites were generated by neutral hydrolysis of N-alkylated DNA bases as described in Material and Methods.30 Without treatment with NM, the background level of AP sites was estimated at 6.5 ± 1.5/107 nts after neutral hydrolysis; this value was subtracted as the background level from NM-treated DNA. The NM-induced AP sites were plotted against the concentration of NM (Figure 3E), showing an excellent linear dose-response to the concentration of NM in the range from 3 to 100 μM. This experiment demonstrates the ability of the assay to quantitate AP sites over a wide range in isolated DNA.
Summary
We have developed a sensitive and robust mass spectrometry method for the quantitation of AP sites in DNA and mammalian tissue with minimal artifact formation. The LC-MS/MS method showed a limit of quantitation of 2.2 AP sites per 108 nts for 13.5 μg DNA assayed on column with a linearity over three orders of magnitude. By directly derivatizing AP sites in nuclear lysate, the background level of AP sites was significantly reduced compared to literature values. The method was successfully applied in vivo and in vitro to measure the AP site level in liver and kidney of rats undergoing oxidative stress and in isolated DNA damaged by alkylating agents.
Supplementary Material
ACKNOWLEDGMENT
This work is funded by the National Cancer Institute [P01 CA160032], and the National Center for Advancing Translational Sciences of the National Institutes of Health award number UL1TR000114. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support [CA 077598].
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
PDF: Materials and instrumentation, derivatization of AP sites in oligo, stability of PMOA-labeled AP sites in different buffers, calibration curve of PMOA-dR in CT DNA digest matrix, recovery of PMOA-dR in DNA digestion and SPE, recovery of PMOA-derivatized DNA as a function of different enzyme concentrations, stability of free and PMOA-derivatized AP sites during DNA isolation, UV-Vis and NMR spectra of PMOA-dR.
The authors declare no competing financial interest.
REFERENCES
- (1).Kim Y-J; Wilson DM Curr. Mol. Pharmacol 2012, 5, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lindahl T In Prog. Nucleic Acid Res. Mol. Biol, Cohn WE, Ed.; Academic Press, 1979, pp 135–192. [DOI] [PubMed] [Google Scholar]
- (3).Krokan HE; Standal R; Slupphaug G Biochem. J 1997, 325, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).De Bont R; van Larebeke N Mutagenesis 2004, 19, 169–185. [DOI] [PubMed] [Google Scholar]
- (5).Nakamura J; Walker VE; Upton PB; Chiang S-Y; Kow YW; Swenberg JA Cancer Res. 1998, 58, 222–225. [PubMed] [Google Scholar]
- (6).Kingma PS; Corbett AH; Burcham PC; Marnett LJ; Osheroff N J. Biol. Chem 1995, 270, 21441–21444. [DOI] [PubMed] [Google Scholar]
- (7).Boiteux S; Guillet M DNA Repair 2004, 3, 1–12. [DOI] [PubMed] [Google Scholar]
- (8).Kow YW; Bao G; Minesinger B; Jinks-Robertson S; Siede W; Jiang YL; Greenberg MM Nucleic Acids Res. 2005, 33, 6196–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wilson DM; Barsky D Mutat. Res.- DNA Repair 2001, 485, 283–307. [DOI] [PubMed] [Google Scholar]
- (10).Dianov GL; Sleeth KM; Dianova II; Allinson SL Mutat. Res-Fund. Mol. M 2003, 531, 157–163. [DOI] [PubMed] [Google Scholar]
- (11).Georgakilas AG; Bennett PV; Wilson IIIDM; Sutherland BM Nucleic Acids Res. 2004, 32, 5609–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Atamna H; Cheung I; Ames BN Proc. Natl. Acad. Sci., U.S.A 2000, 97, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lhomme J; Constant J-F; Demeunynck M Biopolymers 1999, 52, 65–83. [DOI] [PubMed] [Google Scholar]
- (14).Wilde JA; Bolton PH; Mazumder A; Manoharan M; Gerlt JA J. Am. Chem. Soc 1989, 111, 1894–1896. [Google Scholar]
- (15).Nakamura J; Swenberg JA Cancer Res. 1999, 59, 2522–2526. [PubMed] [Google Scholar]
- (16).Boturyn D; Constant J-F; Defrancq E; Lhomme J; Barbin A; Wild CP Chem. Res. Toxicol 1999, 12, 476–482. [DOI] [PubMed] [Google Scholar]
- (17).Fundador E; Rusling J Anal. Bioanal. Chem 2007, 387, 1883–1890. [DOI] [PubMed] [Google Scholar]
- (18).Kojima N; Takebayashi T; Mikami A; Ohtsuka E; Komatsu Y J. Am. Chem. Soc 2009, 131, 13208–13209. [DOI] [PubMed] [Google Scholar]
- (19).Liu C; Luo X; Chen Y; Wu F; Yang W; Wang Y; Zhang X; Zou G; Zhou X Anal. Chem 2018, 90, 14616–14621. [DOI] [PubMed] [Google Scholar]
- (20).Wei S; Shalhout S; Ahn Y-H; Bhagwat AS DNA Repair 2015, 27, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhou X; Liberman RG; Skipper PL; Margolin Y; Tannenbaum SR; Dedon PC Anal. Biochem 2005, 343, 84–92. [DOI] [PubMed] [Google Scholar]
- (22).Roberts KP; Sobrino JA; Payton J; Mason LB; Turesky RJ Chem. Res. Toxicol 2006, 19, 300–309. [DOI] [PubMed] [Google Scholar]
- (23).Li J; Leung EMK; Choi MMF; Chan W Anal. Bioanal. Chem 2013, 405, 4059–4066. [DOI] [PubMed] [Google Scholar]
- (24).Rahimoff R; Kosmatchev O; Kirchner A; Pfaffeneder T; Spada F; Brantl V; Müller M; Carell T J. Am. Chem. Soc 2017, 139, 10359–10364. [DOI] [PubMed] [Google Scholar]
- (25).Jaruga P Free Radic. Res 2012, 46, 382–419. [DOI] [PubMed] [Google Scholar]
- (26).Barbin A; Ohgaki H; Nakamura J; Kurrer M; Kleihues P; Swenberg JA Cancer Epidemiol. Biomarkers Prevent. 2003, 12, 1241–1247. [PubMed] [Google Scholar]
- (27).Hartwig A; Klyszcz-Nasko H; Schlepegrell R; Beyersmann D Carcinogenesis 1993, 14, 107–112. [DOI] [PubMed] [Google Scholar]
- (28).Toyokuni S; Uchida K; Okamoto K; Hattori-Nakakuki Y; Hiai H; Stadtman ER Proc. Natl. Acad. Sci. U.S.A 1994, 91, 2616–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gautier J-C; Holzhaeuser D; Markovic J; Gremaud E; Schilter B. t. ; Turesky RJ Free Radic. Biol. Med 2001, 30, 1089–1098. [DOI] [PubMed] [Google Scholar]
- (30).Gruppi F; Hejazi L; Christov PP; Krishnamachari S; Turesky RJ; Rizzo CJ Chem. Res. Toxicol 2015, 28, 1850–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kalia J; Raines RT Angew. Chem. Int. Ed 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kölmel DK; Kool ET Chem. Rev 2017, 117, 10358–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zoltewicz JA; Clark DF; Sharpless TW; Grahe G J. Am. Chem. Soc 1970, 92, 1741–1750. [DOI] [PubMed] [Google Scholar]
- (34).An R; Jia Y; Wan B; Zhang Y; Dong P; Li J; Liang X PLoS One 2015, 9, e115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Larsen D; Pittelkow M; Karmakar S; Kool ET Org. Lett. 2015, 17, 274–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).OKADA S Pathol. Int 1996, 46, 311–332. [DOI] [PubMed] [Google Scholar]
- (37).Toyokuni S Free Radic. Biol. Med 1996, 20, 553–566. [DOI] [PubMed] [Google Scholar]
- (38).Iqbal M; Giri U; Athar M Biochem. Biophys. Res. Commun 1995, 212, 557–563. [DOI] [PubMed] [Google Scholar]
- (39).Kawabata T; Ma Y; Yamador I; Okada S Carcinogenesis 1997, 18, 1389–1394. [DOI] [PubMed] [Google Scholar]
- (40).Nakamura J; La DK; Swenberg JA J. Biol. Chem 2000, 275, 5323–5328. [DOI] [PubMed] [Google Scholar]
- (41).Kryston TB; Georgiev AB; Pissis P; Georgakilas AG Mutat. Res-Fund. Mol. M 2011, 711, 193–201. [DOI] [PubMed] [Google Scholar]
- (42).Yamaguchi R; Hirano T; Asami S; Chung M-H; Sugita A; Kasai H Carcinogenesis 1996, 17, 2419–2422. [DOI] [PubMed] [Google Scholar]
- (43).Gates KS Chem. Res. Toxicol 2009, 22, 1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.