

Abstract
INTRODUCTION:
Immunotherapy targeting the programmed cell death protein–1 (PD-1) axis elicits durable antitumor responses in multiple cancer types. However, clinical responses vary, and biomarkers predictive of response may help to identify patients who will derive the greatest therapeutic benefit. Clinically validated biomarkers predictive of response to the anti–PD-1 monoclonal antibody pembrolizumab include PD-1 ligand 1 (PD-L1) expression in specific cancers and high microsatellite instability (MSI-H) regardless of tumor type. Tumor mutational burden (TMB) and T cell–inflamed gene expression profile (GEP) are emerging predictive biomarkers for pembrolizumab. Both PD-L1 and GEP are inflammatory biomarkers indicative of a T cell–inflamed tumor microenvironment (TME), whereas TMB and MSI-H are indirect measures of tumor antigenicity generated by somatic tumor mutations. However, the relationship between these two categories of biomarkers is not well characterized.
RATIONALE:
This study assessed the potential for TMB and a T cell–inflamed GEP to jointly predict clinical response to pembrolizumab in >300 patient samples with advanced solid tumors and melanoma across 22 tumor types from four KEYNOTE clinical trials. To assess the individual and joint clinical utility of TMB and GEP, patients were stratified in four biomarker–defined clinical response groups [GEP low and TMB low (GEPlo TMBlo), GEP low and TMB high (GEPlo TMBhi), GEPhi TMBlo, and GEPhi TMBhi] based on predefined cutoffs for TMB and GEP. These patient–defined biomarker groups were further used to guide transcriptome and exome analyses of tumors in a large molecular database [The Cancer Genome Atlas (TCGA)] (n = 6384 tumors) to identify targetable patterns of biology that may modulate response and resistance.
RESULTS:
TMB and GEP exhibited only modest correlation and were independently predictive of response across the KEYNOTE clinical datasets. We found that objective response rates were strongest in patients with GEPhi TMBhi (37 to 57%), moderate in those with GEPhi TMBlo (12 to 35%) and GEPlo TMBhi (11 to 42%), and reduced or absent in those with GEPlo TMBlo (0 to 9%) (see the figure). Additionally, longer progression–free survival times were seen in patients with higher levels of both TMB and GEP. Findings were comparable when TMB and PD-L1 expression were jointly assessed. Within TCGA database, GEP and TMB again had a low correlation, demonstrating the potential to jointly stratify transcriptomic and genomic features across cancer types. Specific gene expression patterns reflective of TME biology showed significant associations with TMB, GEP, or both. In particular, gene set enrichment analysis identified proliferative and stromal, myeloid, and vascular biology corresponding to specific TMB-defined subgroups within GEPhi tumors. In TMBhi tumors, indication-dependent somatic DNA alterations in key cancer driver genes showed a strong negative association with GEP.
CONCLUSION:
This analysis shows that TMB and inflammatory biomarkers (T cell–inflamed GEP and PD-L1 expression) can jointly stratify human cancers into groups with different clinical responses to pembrolizumab monotherapy and identify patterns of underlying, targetable biology related to these groups. TMB and inflammatory biomarkers independently predict response and may capture distinct features of neoantigenicity and T cell activation, respectively. This approach may provide a precision medicine framework for rationally constructing and evaluating anti–PD-1– and/or –PD-L1–based combination therapy regimens.
Programmed cell death protein–1 (PD-1) and programmed cell death ligand–1 (PD-L1) checkpoint blockade immunotherapy elicits durable antitumor effects in multiple cancers, yet not all patients respond.We report the evaluation of >300 patient samples across 22 tumor types from four KEYNOTE clinical trials.Tumor mutational burden (TMB) and a Tcell–inflamed gene expression profile (GEP) exhibited joint predictive utility in identifying responders and nonresponders to the PD-1 antibody pembrolizumab.TMB and GEP were independently predictive of response and demonstrated low correlation, suggesting that they capture distinct features of neoantigenicity and Tcell activation. Analysis of The Cancer Genome Atlas database showed TMB and GEP to have a low correlation, and analysis by joint stratification revealed biomarker-defined patterns of targetable-resistance biology.These biomarkers may have utility in clinical trial design by guiding rational selection of anti–PD-1 monotherapy and combination immunotherapy regimens.
Graphical Abstract
Biomarker-defined responses to pembrolizumab monotherapy identify targetable resistance biology.(A) Tumors have low TMB and low neoantigenicity and lack a T cell–inflamed TME. (B) Tumors can evade the immune response despite high TMB and high neoantigenicity. (C) Although T cells are present, stromal and/or endothelial factors in the TME, low TMB, and low neoantigenicity impede their activity. (D) Tumors have high TMB, high neoantigenicity, and a T cell–inflamed TME, typified by activated T cells and other immune cells with cytolytic roles.
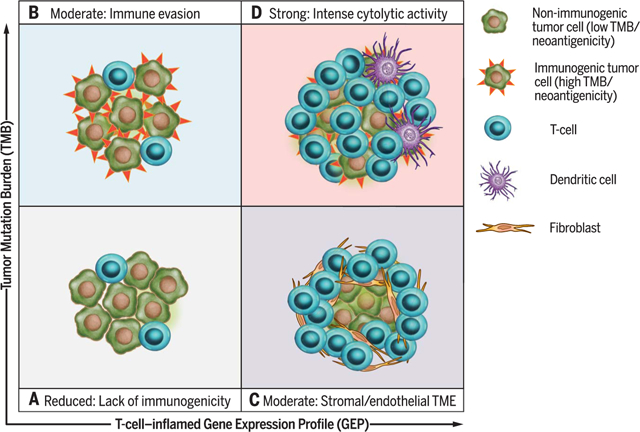
Emerging immune-relevant biomarkers for checkpoint blockade immunotherapy response can be placed broadly into two categories: those related to tumor neoepitope burden, such as microsatellite instability (MSI) or high tumor mutational burden (TMB), and those indicative of a T cell–inflamed tumor microenvironment (TME). The latter include programmed cell death ligand–1 (PD-L1) protein expression on tumor and immune cells, which in many cases is up-regulated in response to local T cell–derived interferon-γ (IFN-γ), and gene signatures of activated T cells (1–3). TMB is correlated with clinical response to cytotoxic T lymphocyte–associated antigen–4 blockade in advanced melanoma (4–6) and with anti–programmed cell death protein–1 (PD-1) and/or PD-L1 blockade in melanoma (7), non–small cell lung cancer (NSCLC) (8, 9), colorectal and gastric cancers (10,11), and urothelial cancer (12). Similarly, tumors with MSI that have high levels of both single-nucleotide and frameshift mutations [high MSI (MSI-H)] are responsive to anti–PD-1 therapy in colorectal cancer and other malignancies (10,11). Expression of genes related to immune cytolytic activity have also been shown to be associated with clinical response to checkpoint blockade in certain tumors (13,14). Recently, a T cell–inflamed gene expression profile (GEP) was shown to predict response to anti–PD-1–directed therapy (15). However, the interplay between these two distinct categories of biomarkers has not been well characterized across cancer types with respect to their ability either to independently or jointly predict response to immunotherapy or to reveal underlying genomic and/or transcriptomic features of tumor antigenicity and TME.
We evaluated the relationship between somatic TMB and clinical response to anti–PD-1 immunotherapy with pembrolizumab. Twenty-two cancer types were included in the discovery and validation cohorts and were analyzed for the independent and joint predictive values of TMB and T cell–inflamed GEP. Additionally, by using large molecular databases [e.g., The Cancer Genome Atlas (TCGA) (16)], we explored transcriptomic and genetic features associated with the presence or absence of either of these two markers.
Study cohorts and tumor and mutation types
The predictive values of TMB and the T cell–inflamed GEP were first assessed separately by rigorous stepwise testing in four cohorts of patients across the pembrolizumab clinical development program (one discovery, one pan-tumor validation, and two single-indication summary cohorts). TMB was evaluated by whole-exome sequencing (WES) of germline and tumor DNA, and the T cell–inflamed GEP was analyzed by targeted gene expression profiling of tumor RNA (with the NanoString platform) from formalin-fixed, paraffin-embedded (FFPE) pretreatment samples. The initial discovery cohort for TMB comprised patients with PD-L1–positive head and neck squamous cell carcinoma (HNSCC) from a phase 1b clinical trial (KEYNOTE-012 B1 cohort; n = 34 patients), and the pan-tumor validation cohort consisted of patients with PD-L1–positive advanced solid tumors (n = 119 patients) from two multicohort phase 1b trials across 20 cancer types [KEYNOTE-028 (17 cohorts; n = 80 patients) and KEYNOTE-012 (A, C, and D cohorts; n = 39 patients)]. The HNSCC single-indication cohort (n = 107 patients) included patients in the phase 1b KEYNOTE-012 B1 cohort and additional patients with PD-L1-unselected HNSCC (n = 73 patients) from the KEYNOTE-012 B2 cohort. The melanoma single-indication cohort included patients with advanced melanoma from the phase 1b (KEYNOTE-001; n = 30 patients) and the phase 3 (KEYNOTE-006 pembrolizumab arm; n = 59 patients) trials. The clinical characteristics of each cohort are listed in table S1, and the characteristics of all patients included in this study are listed in table S2.
The distribution of tumor mutational signatures across the study cohorts largely reflected recognized cancer subtype–dependent determinants of mutagenesis (17) (table S3 and fig. S1). The dominant mutational signatures varied across tumor types in the pan-cancer cohort, with higher TMB associated with tissue-specific signatures, such as smoking in small cell lung cancer; apolipoprotein B mRNA editing enzyme, catalytic polypeptide–like (APOBEC) in genitourinary tumors; and mismatch repair (MMR) in gastrointestinal cancer. Within the pan-cancer validation cohort, the DNA polymerase epsilon catalytic subunit (PolE) signature and the Val411 mutation in POLE were observed in an endometrial carcinoma tumor that had the highest TMB (5464). Dominant signatures in the single-indication cohorts were more homogenous, with an APOBEC signature in the HNSCC cohort (61% of tumors) and an ultraviolet (UV) light exposure signature in melanoma (in 78% of the tumors, >30% of mutations were UV light induced).
Association of TMB and T cell–inflamed GEP with clinical response
Clinical response associations were assessed on the basis of best overall response (BOR) and progression-free survival (PFS) by RECIST 1.1. BOR and PFS associations with TMB and the T cell–inflamed GEP were assessed in all patients who had WES and transcriptomic data available.
We first assessed the predictive value of each individual genomic biomarker separately across the different cohorts. In the HNSCC B1 discovery cohort, higher TMB predicted a greater frequency of clinical response (BOR) (P = 0.0123). This was validated by using the pan-tumor cohort, in which TMB was again associated with BOR (P < 0.001) (Fig. 1A). Higher T cell–inflamed GEP scores were also positively associated with BOR in the pantumor cohort (P < 0.01) (Fig. 1B), showing that a T cell–activated tumor environment also affects response in addition to TMB. Similarly, both TMB and T cell–inflamed GEP scores were positively associated with BOR in the single-indication cohorts of HNSCC (P < 0.05 and P < 0.001, respectively) and melanoma (P < 0.05 for both) patients (Fig. 1, A and B). In this study, we did not evaluate the effect of human papillomavirus (HPV) antigens on the association of TMB with response in the HNSCC cohort; however, we have previously described the association of TMB with clinical outcome in a larger, overlapping group of HNSCC patients (KEYN0TE-012 B1 and B2 cohorts) stratified by HPV status (18). Although we found that TMB was more strongly associated with BOR in HPV-negative patients than in HPV-positive patients, those exploratory findings await validation in larger, independent studies.
Fig. 1. Individual association of TMB or T cell–inflamed GEP with anti–PD-1 response across multiple patient cohorts.
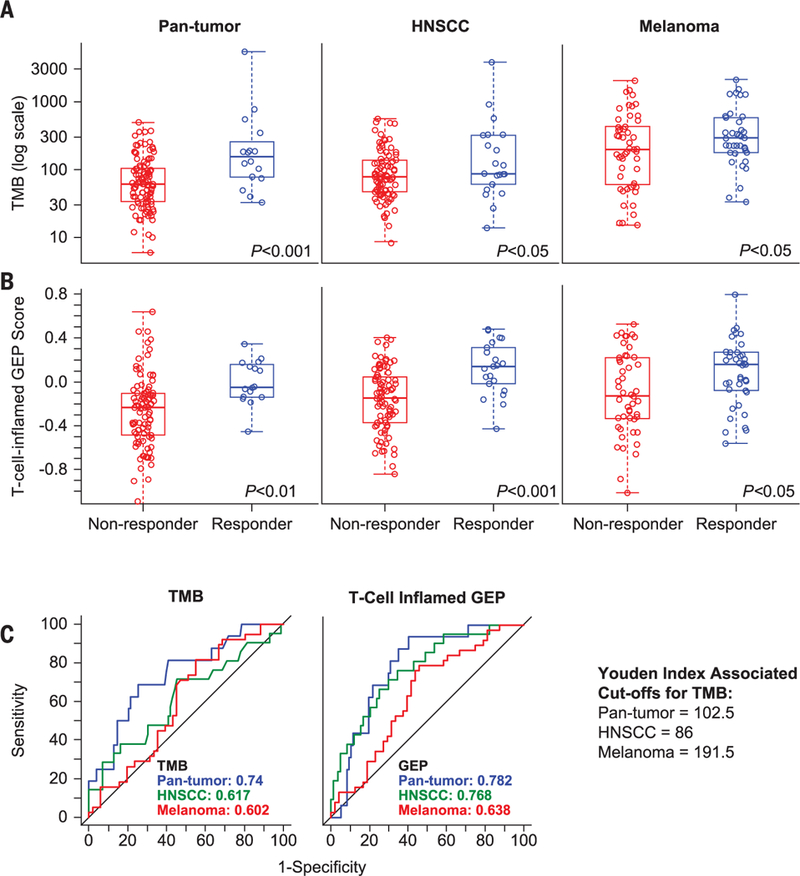
(A and B) The association of (A) TMB, defined as the sum of somatic nonsynonymous mutations, and (B) T cell–inflamed GEP with BOR was assessed in pan-tumor, HNSCC, and melanoma cohorts by central radiology review for all-patients-as-treated populations in all cohorts. A responder is defined as having a partial response (PR) or a complete response (CR); a nonresponder is defined as having no PR or CR. Nonresponders and responders for TMB, respectively, were n = 103 and n = 16 for pan-tumor, n = 86 and n = 21 for HNSCC, and n = 51 and n = 38 for melanoma cohorts. For GEP score analysis, nonresponders and responders were n = 97 and n = 16 for pan-tumor, n = 84 and n = 21 for HNSCC, and n = 48 and n = 38 for melanoma cohorts. For both (A) and (B), raw data are displayed in standard box plots with medians and interquartile ranges. (C) AUROCs for TMB and T cell–inflamed GEP in the three patient cohorts. Youden Index–associated cutoffs for TMB in each cohort are shown.
The clinical utility of TMB in predicting BOR was generally high, and degrees of utility were similar across cancer types, with areas under the receiver operating characteristic curves (AUROCs) of 0.740, 0.617, and 0.602 in the pan-tumor, HNSCC, and melanoma cohorts, respectively. Similar results were observed for the T cell–inflamed GEP across the cohorts (AUROCs = 0.782,0.768, and 0.638, respectively) (Fig. 1C). The potential performance of a targeted sequencing–based TMB assay was simulated by using the genes in the Foundation Medicine targeted sequencing platform (19). The corresponding AUROC across the cohorts was comparable to that observed by using WES (0.721), suggesting potential translatability to a targeted panel diagnostic. Taken together, these data imply that both TMB and the T cell–inflamed GEP have comparable performance characteristics and potential diagnostic utility.
We next evaluated the joint utility of the two genomic biomarkers in predicting response. The correlation between TMB and GEP was low in the pan-tumor and melanoma cohorts (Spearman correlation coefficient r = 0.221, P < 0.05, and r = 0.252, P < 0.05, respectively), and there was no correlation in the HNSCC cohort (r = −0.020, P = 0.841) (Fig. 2A). This lack of correlation, combined with the observed individual predictive values, suggested that TMB and the T cell–inflamed GEP are independent predictive measures of response to pembrolizumab. When tested in a multivariate model adjusted for each measure, both TMB and T cell–inflamed GEP retained significant predictive value in the pan-tumor (P = 0.0028 and 0.0051, respectively) and HNSCC (P = 0.0013 and 0.0004) cohorts, whereas only GEP remained significant in the melanoma cohort (P = 0.1644 and 0.026). Although a portion of the patients in this study were PD-L1 selected, these relationships were observed even in those cohorts of patients that were not PD-L1 selected.
Fig. 2. Joint relationship of TMB or T cell–inflamed GEP with anti–PD-1 response across multiple patient cohorts.
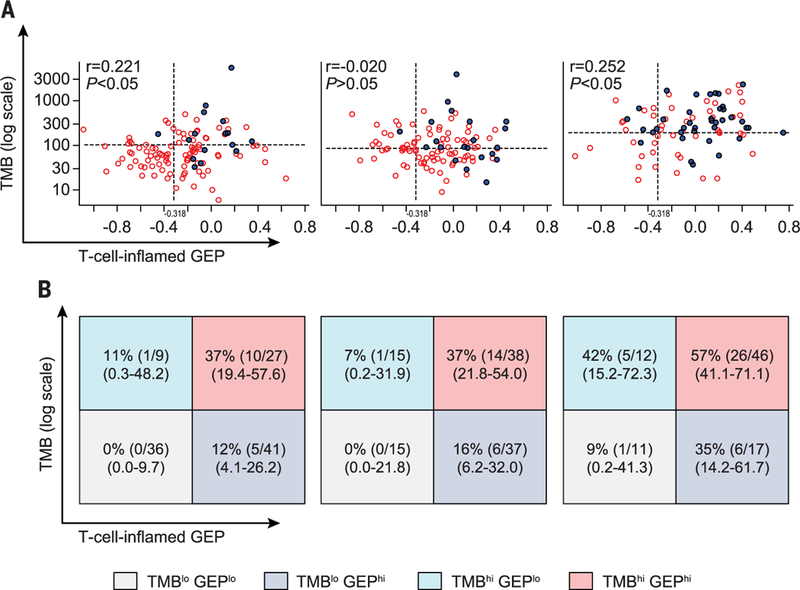
(A) Relationships of both TMB and T cell–inflamed GEP signatures with BOR. A responder is defined as having a PR or CR (filled circles); a nonresponder has no PR or CR (open circles). Dashed horizontal lines represent the Youden Index–associated cutoffs for TMB in each cohort as derived from AUROCs in Fig. 1C. Dashed vertical lines represent a discovery cutoff for the T cell–inflamed GEP selected via analysis of pan-cancer data. (B) Response (PR or CR) rates [expressed as a percentage calculated as the number of responders divided by the number in the cutoff-defined group, with 95% confidence intervals (CI)] per TMB cutoff status and Tcell–inflamed GEP cutoff status as designated in (A). TMBhi and TMBlo response groups are defined by values greater than or equal to and less than Youden Index–associated cut points (102.5, 86, and 191.5 for pan-cancer, HNSCC, and mela-noma cohorts, respectively); GEPhi and GEPlo groups are defined by cutoffs greater than or equal to and less than −0.318, respectively.
We evaluated the association of the genomic biomarkers with PD-L1 immunohistochemistry (IHC) scores (fig. S2). TMB was significantly but moderately correlated with PD-L1 in the pantumor cohort [combined positive score (CPS), r = 0.330; P = 0.0038] and showed no association with PD-L1 in the HNSCC cohort (CPS, r = 0.020; P = 0.8084) or in the melanoma cohort [mela-noma (MEL) score, r = 0.049; P = 0.6473]. In contrast, GEP was more significantly correlated with PD-L1 in the pan-tumor, HNSCC, and melanoma cohorts (r = 0.49, 0.51, and 0.53, respectively; all P values < 0.001), consistent with the known regulation of PD-L1 gene expression by T cell–derived IFN-γ (1–3). This correlation suggests that a PD-L1 IHC–based assay is relevant in assessing a T cell–inflamed TME. As seen with high TMB (TMBhi) and high GEP scores (GEPhi), responses in patients who had both TMBhi and greater PD-L1 expression (PD-L1+; CPS ≥ 1) were greater than those in patients who had low levels of both TMB and PD-L1 expression.
We next studied the potential joint utility of TMB and GEP for patient stratification and treatment outcome prediction. Clinical response was evaluated on the basis of cut points associated with the Youden Index (derived from the AUROCs for TMB in each cohort) and a discovery cutoff of −0.318 for the T cell–inflamed GEP score (selected via analysis of pan-cancer data) (15). Rates of response to pembrolizumab were greater in patients with TMBhi (greater than or equal to Youden Index cut points) than in those with low TMB (TMBlo) (less than Youden Index cut points) and were similarly greater for those with higher T cell–inflamed GEP scores (greater than or equal to the cutoff of −0.318) than for those with lower scores (less than the −0.318 cutoff) (Fig. 2B). The highest objective response rate was observed for patients within each cohort who had both TMBhi and GEPhi. Additionally, among patients with both TMBlo and low T cell–inflamed GEP scores (GEPlo) no responses were observed in the pan-tumor and HNSCC cohorts and only one response was observed in the melanoma cohort, suggesting greater sensitivity for the combination of bio-markers. Patients who had high scores for only one of the biomarkers (TMBlo GEPhi and TMBhi GEPlo) had moderate responses (Fig. 2B). These data suggest the potential for greater positive and negative predictive value when these bio-markers are used together in the setting of PD-1–directed monotherapy.
Patient stratification by TMB and GEP was also differentially associated with PFS. In all three cohorts, hazard ratios associated with PFS were <1.0 (implying PFS benefit) among patients with high versus low TMB and high versus low T cell–inflamed GEP scores. The most pronounced PFS-associated hazard ratios were observed for TMBhi GEPhi tumors in the pan-tumor (Fig. 3A), HNSCC (Fig. 3B), and melanoma cohorts (Fig. 3C). The greatest differential was observed in each cohort for patients with TMBhi GEPhi versus patients with TMBlo GEPlo. Patients who had greater levels of either TMB or GEP (TMBhi or GEPhi) versus low levels of these biomarkers (TMBlo or GEPlo) also had longer PFS.
Fig. 3. Relationship between TMB and T cell–inflamed GEP signatures and PFS after anti-PD-1 treatment across multiple patient cohorts.
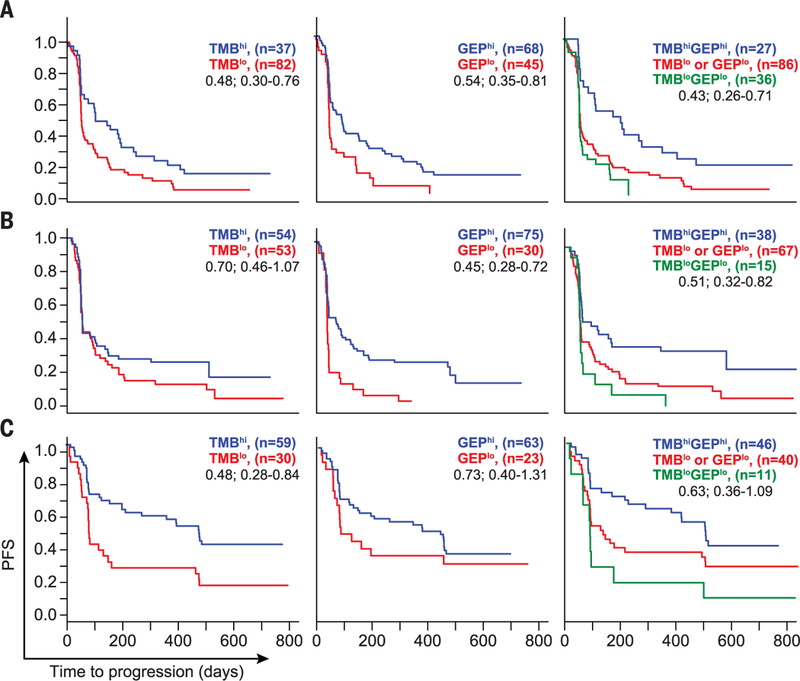
Relationships of TMB and T cell–inflamed GEP with PFS in all patients as treated per TMB cutoff and GEP cutoff as described in the legend to Fig. 2. Median PFS times in days for (A) pan-tumor, (B) HNSCC, and (C) melanoma cohorts for TMBhi versus TMBlo were 115 versus 59 (hazard ratio, 0.48; 95% CI, 0.30 to 0.76), 64 versus 64 (0.70; 0.46 to 1.07), and 502 versus 85 (0.48; 0.28 to 0.84); those for GEPhi versus GEPlo were 96 versus 57 (0.54; 0.35 to 0.81), 103 versus 57 (0.45; 0.28 to 0.72), and 418 versus 90 (0.73; 0.40 to 1.31); those for TMBhi GEPhi versus TMBlo GEPlo or TMBlo GEPlo were 189 versus 59 (0.43; 0.26 to 0.71), 110 versus 62 (0.51; 0.32 to 0.82), and 504 versus 123 (0.63; 0.36 to 1.09). Kaplan-Meier plots are shown, and median survival was estimated on the basis of Kaplan-Meier estimates. Hazard ratios with 95% CI were derived from a Cox proportional model fit, with adjustment for baseline ECOG score and protocol where relevant.
We also explored the feasibility and potential clinical value of identifying a pan-cancer threshold for TMB across our cohorts that maximizes its joint predictive utility with GEP by using a method similar to that of Panda et al. (20). A TMB cutoff of ≥123 mutations per exome maximized the effect size of the difference in GEP distributions between tumors having TMB less than and greater than the cutoff. The response rates to pembrolizumab in the TMB-GEP–defined groups of each clinical cohort were comparable to those observed by using the cohort-specific cut points for TMB reported above (fig. S3). The hazard ratios observed for PFS were also generally similar with the use of the TMB cutoff of ≥123 mutations per exome (fig. S4). A pan-tumor threshold may be further optimized with the availability of additional data beyond those in our study. For example, a pan-tumor TMB threshold of ≥175 mutations per exome was recently reported for response to pembrolizumab (21).
Association of other DNA-based measures with response
The predictive value of other DNA-based measures of mutation status in relation to response was also evaluated in these cohorts, including predicted neoantigen signature, smoking status, APOBEC-driven mutations, UV light exposure, DNA transversions, homologous recombination deficiency, and MSI. Aside from MSI, none of these specific measures of genetic alteration provided additional meaningful improvement in predictive value over TMB assessment alone. The predicted neoantigen load was highly correlated with TMB in the pan-tumor, HNSCC, and melanoma cohorts (r = 0.87, 0.83, and 0.90, respectively), as expected (fig. S5). In the pan-tumor cohort, most measures of mutagenic processes were significantly associated with BOR (e.g., predicted neoantigen load and smoking; both P values = 0.001), with similar relevant trends toward significant association with PFS (table S4). By using a WES-based method to infer MSI (22), two patients with MSI-H tumors (gastric and biliary tract carcinomas) were identified, and both were responders; the MSI status of these patients was confirmed with standard MSI polymerase chain reaction (PCR) methods. In the melanoma cohort, the percentage of UV light–induced mutations correlated with TMB (r = 0.77; P < 1 × 10−10) (fig. S1) and was significantly associated with response (P = 0.02). These data suggest that nonsynonymous mutations arising from a wide variety of mutagenic processes are capable of enhancing the antigenicity of tumors, with comparable effects on the response to PD-1 checkpoint blockade.
Somatic mutation clonality and copy number variation (CNV) have previously been reported to positively and negatively associate, respectively, with response to PD-1 checkpoint blockade (23,24). In an analysis of clonal versus nonclonal tumors (clonality of 1 versus <1, respectively), the treatment response rates were numerically higher in clonal tumors in the pan-tumor cohort (18% versus 10%) but not different in the HNSCC (21% versus 23%) or melanoma (44% versus 41%) cohort. A low and nonsignificant overall correlation was observed between clonality and TMB (r = 0.05; P > 0.05) in the pooled dataset, suggesting a potential utility of including clonality assessment in the application of a TMB-based biomarker. Higher levels of CNV trended toward negative associations with response but approached statistical significance only in the HNSCC and melanoma cohorts (AUROCs = 0.48, 0.35, and 0.42; P = not significant, 0.1, and 0.1 for the pan-tumor, HSNCC, and melanoma cohorts, respectively). Correlations between TMB and CNV load were low in the pan-tumor (r = −0.03), HNSCC (r = 0.16), and melanoma (r = −0.12) cohorts (P > 0.05 for all), suggesting a potential complementary role of CNV in biomarker-based prediction of responders versus nonresponders.
TMB and Tcell–inflamed GEP relationships can be applied to a wide range of tumor types across genomic databases
To explore the generalizability of our findings and the utility of our stratification schema across tumor types, the relationship among TMB, T cell–inflamed GEP, and related genomic features was further explored in TCGA (n = 9963 patients with transcriptomic data, 6384 of which also had WES data) (16). Patients were stratified by TMB (WES score ≤ 100 mutations per exome) and T cell–inflamed GEP score (below the top tertile of data) by using cutoffs equivalent in terms of prevalence to those that were used to define the clinical response groups in the pantumor cohort (Fig. 4A). Consistent with our clinical data, TMB and the T cell–inflamed GEP were found to have low but significant correlations (r = 0.30; P < 1 × 10−4), as did TMB and PD-L1 gene expression (r = 0.16; P < 1 × 10−4) and TMB and PD-L2 gene expression (r = 0.22; P < 1 × 10−4). By contrast, both PD-L1 expression and PD-L2 expression, which are induced by IFN-γ from activated Th1 and cytotoxic T cells (1–3), were highly correlated with the T cell–inflamed GEP (r = 0.61 and 0.72; P < 1 × 10−10). MSI-H tumors made up a subset of tumors with TMBhi in both T cell–inflamed and noninflamed tumors. Even in these tumors, which exhibit very high mutational burdens, the modest correlation between GEP and TMB was preserved. The frequency of the TMBhi GEPhi subgroup, which was identified as the most clinically responsive population in our datasets, varied across cancer types (Fig. 4B), with enrichment among patients with tumors that are generally more responsive to pembrolizumab, such as melanoma and NSCLC (25, 26), and underrepresentation among patients with tumors such as prostate cancer and glioblastoma that are typically more resistant to immunotherapy (27,28).
Fig. 4. Relationships of TMB, GEP, and other key biomarkers with gene expression across tumor types in TCGA.
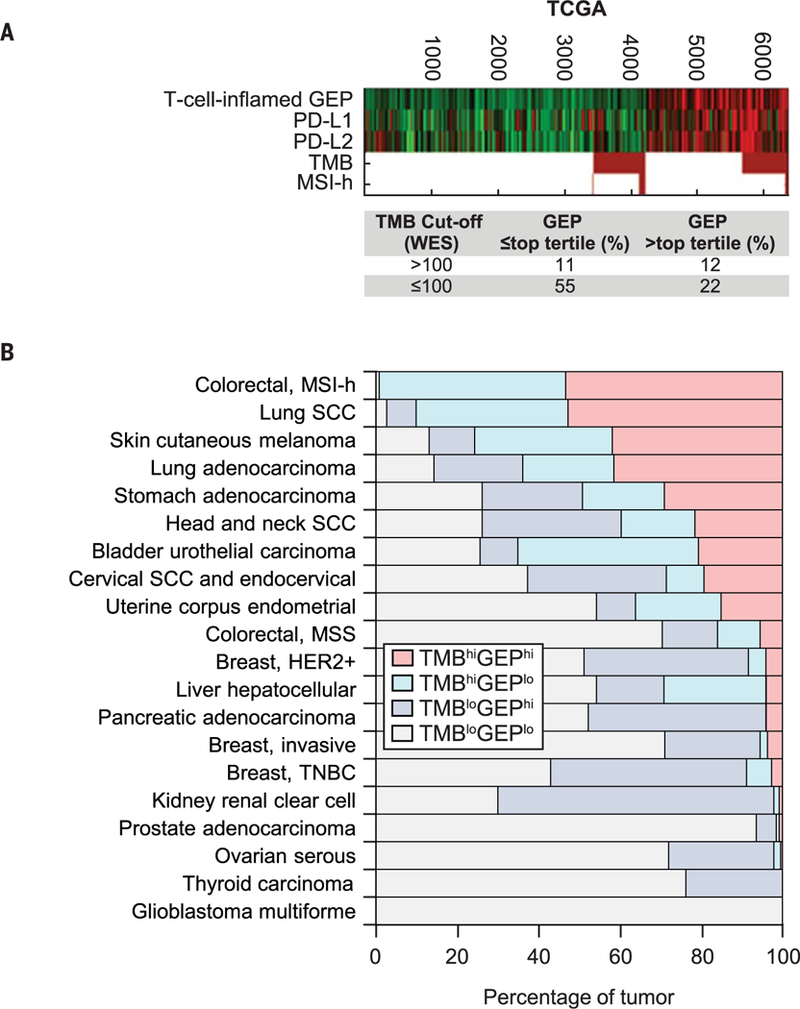
(A) Data are stratified by TMB and GEP cutoffs, which are equivalent in terms of prevalence to those that define the clinical response groups in the pan-tumor cohort of patients treated with pembrolizumab from the KEYNOTE studies. The WES cutoff of >100 mutations per exome for TMB was chosen to match the Youden Index–associated TMB cutoff defined for the pan-tumor cohort. The GEP cutoff was chosen as the top pan-cancer tertile value. Columns represent individual tumors, and rows represent genomic features. Red and green represent elevated and decreased expression, respectively (versus the median, in black), for continuous variables, and red and white represent true and false for Boolean (binary) variables. In the absence of MSI evaluation across cancer types, MSI-H status was determined by loss of MLH1 gene expression by using cutoffs determined by the bimodality in the distribution of expression. (B) Percentages of tumors in each cancer type in biomarker-defined response groups as defined in (A) in TCGA database. SCC, squamous cell carcinoma; MSS, microsatellite stable; TNBC, triple-negative breast cancer.
Rooted in the well-studied field of T cell inflammation and cytolytic process (13, 29–31), the T cell–inflamed GEP signature was derived by a stepwise process of discovery, validation, and refinement of candidate gene sets associated with patient response to pembrolizumab across multiple solid tumors with the use of a NanoString platform enriched in immune genes (15) and thus represents a universal signature. Notably, in TCGA dataset, we observed a strong correlation (r > 0.9) between the GEP and several other previously published transcriptional signatures reflective of a T cell–inflamed TME associated with cytolytic processes (Fig. 5A).
Fig. 5. Transcriptomic and genomic features defined by the GEP and TMB biomarker–based stratification in TCGA database.
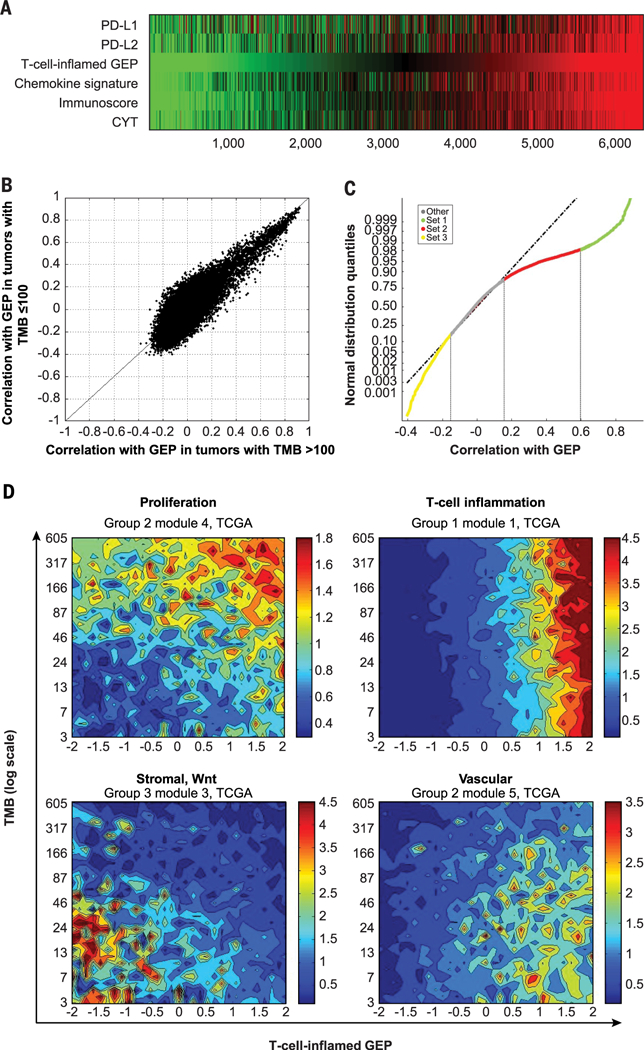
(A) Association of T cell–inflamed GEP (15) with other key markers and expression signatures representative of T cell inflammation and a cytolytic environment, including chemokine signature (29), Immunoscore (30), and cytolytic activity (CYT) (13). (B) Association between T cell–inflamed GEP and expression of each gene in TCGA for tumors with a TMB of >100 mutations per exome (x axis) and in tumors with a TMB of ≤100 mutations per exome (y axis). (C) Each gene in the transcriptome is assigned to one of four clusters determined by cutoffs obtained from the distribution of correlation with the T cell-inflamed GEP. The cutoffs used were the inflection point where the distribution deviates from normal on the positive side (0.15; 83rd quantile), the cut point that selects T cell–inflamed GEP genes (0.6; 98% quantile), and the inflection point where the distribution deviates from normal on the negative side (−0.15; 15th quantile). Vertical lines represent cutoffs for gene sets 1, 2, and 3 (r > 0.6, r = 0.15 to 0.6, and r < −0.15, respectively); gene sets are color coded on the regression line. (D) Gene set annotation in each cluster suggested enrichment for biological patterns with distinct relevance for the individual biomarker-based groups. Contour plots illustrate the association with TMB and GEP of selected patterns of TME and cellular biology represented by gene expression modules formed by genes coexpressed in TCGA database. Blue and red represent under- and overexpression, respectively.
Stratification of additional genomic features by TMB and T cell–inflamed GEP
The patient groups defined by TMB and GEP status show notable differences in clinical response to pembrolizumab. In particular, the two groups with only one positive biomarker indicative of potential for pembrolizumab response (TMBhi GEPlo or TMBlo GEPhi) have markedly lower response rates than the TMBhi GEPhi group, suggesting that mechanisms of resistance to pembrolizumab may exist that are specific to each respective group. In order to identify potential mechanisms of resistance, we assessed molecular differences among tumors that belong to different TMB- and T cell–inflamed GEP–defined groups through analyses in TCGA molecular database.
First, we compared the correlation of genes in the transcriptome with GEP in TMBhi and in TMBlo tumors separately. Both distributions of correlations diverged from a normal distribution because of a pattern of significant skewing toward positive correlations with the T cell–inflamed GEP, consistent with robust coregulation of gene expression markers of cell types present in a cytolytic TME. However, there were no major differences in the correlations of individual genes with the T cell–inflamed GEP between TMBhi (TMB > 100 mutations per exome) and TMBlo (TMB ≤ 100 mutations per exome) tumors (r = 0.76; P < 1 × 10−20) (Fig. 5B), suggesting a lack of qualitative difference in T cell inflammation markers as a function of tumor neoantigenicity. Notably, much smaller deviations from a normal distribution were observed in the negative range of correlations with GEP in both TMBhi and TMBlo tumors, suggesting the absence of major pan-cancer transcriptional signatures strongly associated with T cell exclusion.
To understand the origin of the skewness toward positive correlations with the T cell–inflamed GEP, genes positively correlated with the T cell–inflamed GEP (r > 0.15) were classified into two sets by using cutoffs defined by deviations from a normal distribution of the correlation with the T cell–inflamed GEP at 83% and 98% quantiles, respectively (Fig. 5C). Set 1 comprised genes that had a Spearman correlation r > 0.6 with the T cell–inflamed GEP (the lower bound for the correlation of individual genes in the signature with the signature as a whole), whereas set 2 genes had correlations with GEP that ranged between 0.15 and 0.6. Additionally, genes negatively correlated with the T cell–inflamed GEP and divergent from a normal distribution (r < −0.15 at 14% quantile) were grouped in set 3.
As expected, a strong enrichment of genes related to T cell–inflamed cytolytic processes was observed in set 1 (table S5). By contrast, set 2 showed enrichment in genes specific to other cell types in the TME, including vascular endothelium and myeloid infiltrate, but did not show enrichment of genes for T cell–inflamed cytolytic processes or tumor cell–intrinsic pathways. Genes in set 1 and set 2 were further grouped as modules of gene coexpression by K-means clustering (K = 10 for set 2, and K = 4 for set 1). Modules in set 1 did not show a strong association with TMB, consistent with the weak associations between TMB and the T cell–inflamed GEP described above. However, several modules in set 2 (table S6) displayed distinct patterns of correlation or anticorrelation with TMB. Annotation of the genes in the modules that were most strongly correlated and anticorrelated with TMB (modules 4 and 5, respectively), revealed enrichment in biology related to cell proliferation (module 4) and vasculature (module 5). These data suggest that distinct patterns of underlying biology can be identified by using TMB and the T cell–inflamed GEP to categorize tumors (Fig. 5D). The association of the average expression of these gene modules (modules 4 and 5) with TMB and T cell–inflamed GEP is represented in Fig. 5D in the upper left and lower right panels, respectively, by using the cytolytic module 1 from set 1 in the upper right panel as a reference.
The group of genes in set 3 that were anti-correlated with the T cell–inflamed GEP (r < −0.15) was also investigated; however, the biological annotation of the resulting coexpression modules was less informative than that for genes positively correlated with the T cell–inflamed GEP. However, some modules in this group were anticorrelated with TMB as well as with T cell–inflamed GEP. In particular, a module enriched in stromal and Wnt signaling elements was identified in tumors with both TMBlo and T cell–inflamed GEPlo (Fig. 5D, lower left panel).
An additional analysis was performed by interrogating the entire transcriptome for genes associated with TMB in T cell–inflamed tumors, independently of the GEP-based clustering approach described above. Similar to the analysis of modules, this analysis showed that genes that positively correlated with TMB were enriched for proliferation whereas those that were anticorrelated with TMB were related to vascular and stromal biology (table S7). Consistent with these analyses, the distribution of previously identified signatures of stromal biology, proliferation, cytolytic activity, and Wnt signaling (13, 32–34) also showed similar patterns of association with TMB and the T cell–inflamed GEP (fig. S6). However, in this analysis, we were not able to identify a gene expression signature of TMBhi that was as predictive as TMB itself for response to pembrolizumab.
A complementary approach was used to identify genomic determinants of low cytolytic transcriptomic activity (absence of a T cell–inflamed GEP) in tumors with TMBhi as potential drivers of immune evasion in a mutagen-rich context. As described above, the transcriptomic correlation of the T cell–inflamed GEP in TMBhi tumors (Fig. 5B) showed a distribution that skewed toward positive correlation with GEP, suggesting the absence of a robust transcriptome signal in tumors with TMBhi and GEPlo. Therefore, DNA alterations in TCGA were explored to reveal potential negative associations of somatic mutations with GEP by using a previously reported approach (13) but focusing specifically on tumors with TMBhi. Among known cancer drivers, serine-threonine kinase 11 (STK11) [also known as liver kinase B1 (LKB1)] mutation in lung adenocarcinoma, Kelch-like ECH-associated protein 1 (KEAP1) mutation in lung adenocarcinoma and lung squamous cell carcinoma, and adenomatous polyposis coli (APC) mutation in colorectal cancer showed highly significant negative associations with the T cell–inflamed GEP (Fig. 6). Notably, none of these associations passed the nominal significance level (P < 0.01) in the pan-cancer analysis, suggesting a potential cancer type–specific role for these somatic alterations. Other genes demonstrating negative associations with the T cell–inflamed GEP were either of low frequency or were not known cancer drivers (Fig. 6B).
Fig. 6. Cancer driver genes associated with immune evasion in selected tumor types.
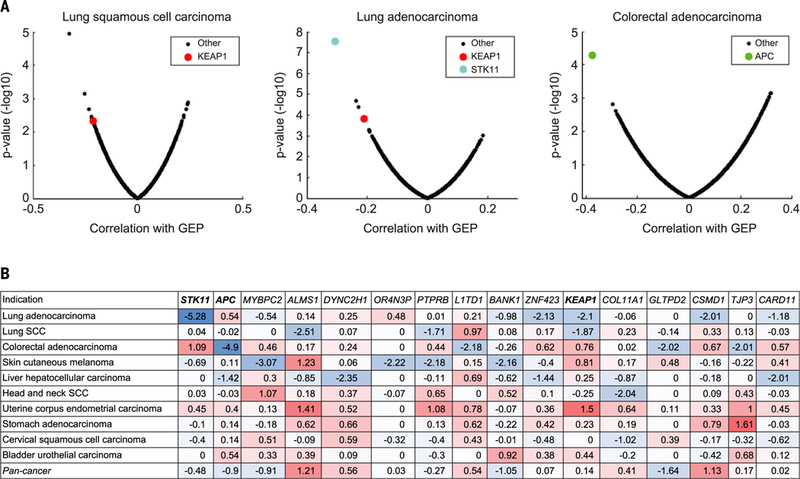
(A) Volcano plots of AUROC and rank sum P values illustrating the association of somatic SNV mutations with GEP in lung squamous cell carcinoma, lung adenocarcinoma, and colorectal adenocarcinoma in TCGA database. Analysis was restricted to cancer types having >20% of tumors with TMBhi (>100 mutations per exome). For each cancer type, the negative logi0-transformed rank sum P value between GEP and mutations was calculated for each gene. (B) Rank sum P values of association between GEP and mutations in selected genes. The selection was made on the basis of a nominal P value of <0.01 for negative association with GEP in any cancer type and an alteration frequency of ≥10% in that cancer type. Negative and positive associations are represented in blue and red, respectively. Negative associations for known cancer driver genes are shown in boxes.
Discussion
Several studies have shown that either TMBhi or cytolytic elements of the TME are associated with clinical response to checkpoint blockade immunotherapy in some tumor types (4–9,11–13,15). However, the relationship between these two central aspects of tumor immunobiology and their combined association with clinical response to checkpoint blockade immunotherapy has not been well-studied across multiple cancer types. Here, we show that TMB and a T cell–inflamed GEP are tissue-agnostic measures of distinct aspects of tumor immunobiology and independently predict response to anti–PD-1 therapy in multiple tumors. In particular, limited clinical responses to pembrolizumab occurred in patients with low levels of both TMB and T cell–inflamed GEP, whereas the greatest response rates were seen in patients with high levels of both biomarkers. Similarly, improved responses were seen in patients who had high levels of both PD-L1IHC expression and TMB, reflective of the relationship of PD-L1 and GEP to a T cell–inflamed TME. These observations suggest that using inflammatory biomarkers such as the T cell–inflamed GEP or PD-L1 jointly with TMB may help to identify patients who are responsive to anti–PD-1 therapies. Additional IHC assays have been developed that measure protein markers of a cytolytic T cell environment, and evaluating their performance characteristics in conjunction with TMB in future studies may be useful (14,35). More broadly, our study demonstrates the orthogonal relationship between universal measures of tumor antigenicity and tumor infiltration that can occur by activated T cells (14, 36–38). Although these are upstream and downstream components, respectively, of a robust antitumor T cell response, there is sufficient intervening biology such that biomarkers for each process can provide complementary information.
As an increasing number of PD-1– and PD-L1– based combination regimens show clinical benefit, it will become challenging to determine the relative utility of each regimen for an individual patient. A refined set of biomarker tools that can stratify underlying patterns of tumor immunobiology may enable rational and biology-driven personalization of these various treatment regimens, such as selection of patients with tumors typically less responsive to immunotherapy. Our data demonstrate that TMB and a T cell–inflamed GEP can be used to categorize tumors into discrete subgroups that exhibit distinct patterns of potentially targetable biology to enhance clinical response. These patterns include tumor type–agnostic signatures of proliferative, vascular, myeloid, and stromal biology, as well as tumor type–specific dysregulation of tumor cell–intrinsic signaling pathways. Although the utility of TMB, T cell–inflamed GEP, and PD-L1, as well as other emerging tumor-agnostic biomarkers, will need to be prospectively validated for use in predicting response to various immunotherapy regimens, including combination therapies, the findings reported here suggest a rationale for further exploring the utility of these biomarkers as guides for precision cancer immunotherapy.
Materials and methods
Clinical tumor samples
Associations of TMB and the T cell–inflamed GEP with BOR and PFS were evaluated by using tumor samples from subgroups of patients treated with pembrolizumab in clinical trials who had WES data available. These included a discovery cohort of patients with HNSCC (KEYNOTE-012 B1), a pan-tumor validation cohort (KEYNOTE-012/028), and single-indication cohorts of patients with HNSCC (KEYNOTE-012 B1+B2) and melanoma (KN001 and 006). The discovery cohort included 34 of 297 total enrolled patients with PD-L1–selected (≥1%, modified proportion score or interface pattern, QualTek IHC) (39) HNSCC (B1 cohort). The pan-tumor cohort comprised patients with PD-L1-positive (≥1%, modified proportion score or interface pattern, QualTek IHC) (39) advanced solid tumors pooled from two multi-cohort trials, including 39 of 297 total enrolled patients in KEYNOTE-012 (cohorts A, C, and D: triple-negative breast cancer, urothelial cancer, and gastric cancer, respectively) and 80 of 450 total enrolled patients in KEYNOTE-028 (17 of 20 cohorts with anal, biliary, carcinoid, cervical, colorectal, endometrial, esophageal, estrogen receptor–positive human epidermal growth factor receptor-2–negative breast, pancreatic, salivary gland, prostate, small cell lung, thyroid, and vulvar cancers and neuroendocrine tumors, mesothelioma, and leiomyosarcoma). Single-indication cohorts included 107 HNSCC patients from the KEYNOTE-012 PD-L1–positive (≥1%, modified proportion score or interface pattern, QualTek IHC) (39) B1 (n = 34) and PD-L1–unselected B2 (n = 73) cohorts (40,41) and patients with advanced melanoma from the pembrolizumab arms of the KEYNOTE-001 (n = 30 of 668 total enrolled patients) and KEYNOTE-0006 (n = 59 of 834 total enrolled patients) studies (26, 42). Tissue specimens were obtained with the approval of the institutional review boards, and patients provided informed consent [clinical trial registration: KEYN0TE-012 (NCT01848834); KEYN0TE-028 (NCT02054806); KEYN0TE-001 (NCT01295827); KEYN0TE-006 (NCT01866319)].
Clinical end points
BOR was assessed in the discovery HNSCC, pantumor, and HNSCC cohorts by central radiology review and in the melanoma cohort by integrated radiology and oncologist assessment. For BOR, a responder was defined as a patient with a partial response (PR) or complete response (CR), and PFS was defined as the time from the start of treatment to documented evidence of progressive disease or death. BOR and PFS were both assessed in the all-patients-as-treated populations, defined as those who had received ≥1 dose of study drug, in each cohort.
Processing of tissue samples
DNA sequencing (WES) and RNA analysis (gene expression profiling) were performed by using FFPE sections of pretreatment tumor samples from the above-listed studies. WES was performed on both germline and tumor samples, and gene expression profiling was performed on tumor samples. With a fresh scalpel, the tissue was either macrodissected from the marked tumor area (tissue containing <20% tumor) or scraped from the entire section and transferred to a 1.5-ml tube containing 200 μl of 100% ethanol.
Gene expression (RNA) profiling: NanoString methodology
The previously described T cell–inflamed GEP was derived by using a stepwise derivation process of discovery, validation, and refinement of candidate gene sets across a wide variety of solid tumors (15). The GEP was composed of 18 inflammatory genes related to antigen presentation, chemokine expression, cytolytic activity, and adaptive immune resistance, including CCL5, CD27, CD274 (PD-L1), CD276 (B7-H3), CD8A, CMKLR1, CXCL9, CXCR6, HLA-DQA1, HLA-DRB1, HLA-E, IDO1, LAG3, NKG7, PDCD1LG2 (PDL2), PSMB10, STAT1, and TIGTT. For GEP analysis, total RNA was isolated from 5-μm-thick FFPE sections of tumor tissue fixed on positively charged slides (Ambion RecoverAll total nucleic acid isolation kit for FFPE; catalog no. AM1975) at ALMAC, United Kingdom. Total RNA concentrations were measured using the NanoDrop ND1000 (Thermo Fisher Scientific) in 1.5 μl of test sample.
Gene expression analysis was conducted on the NanoString nCounter gene expression platform (NanoString Technologies, Seattle, WA) as described previously (15). Per sample, 50 ng of total RNA was mixed in a final volume of 5 to 7 μl with a 3′-biotinylated capture probe and 5′-reporter probe tagged with a fluorescent barcode, from the desired custom gene expression codeset (HUIMR680_V2_C2406+PLS_SPI-KE80_C2765 for Batch 1 and HUIMR800_C3176 for Batch 2), containing probes designed to function as positive and negative hybridization controls. Probes and target transcripts were hybridized overnight at 65°C for 14 to 18 hours as per manufacturers’ recommendations. Hybridized samples were run on the NanoString nCounter preparation station by using a high-sensitivity protocol where excess capture and reporter probes were removed and transcript-specific ternary complexes were immobilized on a streptavidin-coated cartridge. The cartridge samples were scanned at maximum resolution by using the nCounter digital analyzer. GEP scores were calculated as a weighted sum of normalized expression values for the 18 genes. Quality control of the gene expression data followed an approach similar to that of the NanoString clinical-grade assay, with the use of joint criteria that assessed the relationships between housekeeping genes and the negative control probes plus a weighted score evaluating the GEP gene counts versus background-subtracted counts. For housekeeping normalization, raw counts for the individual genes were log10 transformed and then normalized by subtracting the arithmetric mean of the log10 counts for a set of 11 housekeeping genes.
WES pipeline
Somatic single-nucleotide variant (SNV) calling
Whole-exome sequence reads were aligned to reference human genome GRCh37 by using bwa mem (43) followed by preprocessing steps including duplicate marking, indel realignment, and base recalibration with Picard (v1.114) and GATK (Genome Analysis Toolkit, v2) (44) to generate analysis-ready BAM files. MuTect was used to generate somatic SNV calls using default parameters by comparing BAM files from tumor and matched normal samples (45). MuTect-called SNVs present in the Single Nucleotide Polymorphism Database (dbSNP, v141) (46) but not in the Catalogue of Somatic Mutations in Cancer (COSMIC, v68) (47) were filtered out. The SNVs with mutant reads of <4 in tumor samples were also eliminated. TMB for a subject was defined as the sum of somatic nonsynonymous SNVs that passed all the filters described.
HLA class I typing
HLA-I major loci, A, B and C, were typed at four-digit resolution by using OptiType (v1.0) (48).
For output typed alleles not found in the NetMHC (v3.4) (49) input list, the corresponding supertype was identified for each allele (50, 51) and the supertype-representative allele was used for NetMHC.
SNV annotation and neoantigen detection
Somatic mutations were annotated with VEP (Variant Effect Predictor) (52), and nonsynonymous mutations in protein coding regions were counted for TMB. All possible 9-mer peptide sequences with mutated amino acid inside for each nonsynonymous mutation locus were extracted, and binding affinities for patient HLA-A and HLA-B alleles were computed by using NetMHC (v3.4). The 9-mer peptide with the highest binding affinity with the HLA alleles from a nonsynonymous mutation locus was selected as the representative antigen for the mutation. Representative antigens with HLA-A or -B binding affinity of <50 nM were considered neoantigens.
Microsatellite instability (MSI) calling
MSI phenotype was detected by applying mSINGS on WES data from tumor samples (22). The stability of each mononucleotide microsatellite locus was evaluated, and the proportion of unstable microsatellite loci was determined as the MSI score. Samples with an MSI score of more than 20% were classified as MSI-high (MSI-H) positive. MSI was confirmed by PCR by using the Promega MSI analysis system, version 1.2.
Mutation signature analysis
Mutational signature analysis was performed by using the deconstructSigs package (v1.6.0) in R that selects the combination of known mutational signatures that can account for the observed mutational profile in each sample (53). Exome regions were defined by Agilent Sureselect V5 target region. Only somatic mutations in exome regions were considered, and trinucleotide counts were normalized by the number of times each trinucleotide context was observed in the exome region. Mutational signatures as defined by Alexandrov et al. (54) and named as signatures. nature 2013 were the target signature set to be screened. The relationships of these various mutational signatures, including specific nucleotide changes, DNA repair, smoking, neoantigen, TP53, and APOBEC, with BOR and PFS were evaluated in patient samples in the pan-tumor cohort.
Allele-specific copy number and purity estimation
VarScan2 (55) output copy number ratio and SNP were input to Sequenza (56) to provide a maximum a posteriori estimation for cellularity and segmented allele-specific copy number for each sample.
Clonality
For each sample, MuTect-called somatic SNVs with variant allele frequency information, combined with Sequenza output allele-specific copy number and cellularity estimation, were input to PyClone to estimate cellular prevalence for all somatic SNVs. Mutational clonality was also inferred through the clustering process of PyClone (57).
PD-L1 expression
PD-L1 expression levels were evaluated in pretreatment samples by IHC staining by using the PD-L1 IHC 22C3 pharmDx kit (Agilent Technologies) in the pan-tumor and HNSCC cohorts (39); expression levels were reported as the CPS, defined as the number of PD-L1–positive cells (tumor cells, lymphocytes, macrophages) divided by the total number of tumor cells × 100. CPS was previously reported as a percentage and is now reported as an equivalent unitless measure. This assay differs from the one used to determine PD-L1 positivity (≥1%, modified proportion score or interface pattern, QualTek IHC) for enrollment eligibility as described above for the pan-tumor and HNSCC clinical cohorts (58). For the mela-noma cohort, PD-L1 levels were assessed by IHC by using the MEL score, and positivity was defined as a score of ≥2 membranous PD-L1 staining in at least 1% of tumor and tumor immune cells (59).
TCGA molecular data
Gene expression data for 9963 tumors and somatic alterations data for 6384 tumors were obtained through TCGA portal (16) as of September 2015.
Statistical methods
The retrospective, statistical analysis of clinical samples in this study was prespecified and performed in a blinded fashion, with genomic end points generated without access to clinical out-comes. Associations with BOR were tested by using logistic regression, and associations with PFS were examined by using Cox proportional hazards models. All models (logistic regression and Cox models) were adjusted for baseline Eastern Cooperative Oncology Group (ECOG) score performance. One-sided nominal P values were reported. Associations between continuous variables were assessed by using Spearman correlation, and associations between continuous variables and binary variables (e.g., BOR) were further assessed by using AUROC and rank sum P values. Statistical analyses and visualizations were performed with Matlab R2010 or with R3.4.1. TMB cutoffs for the pan-tumor and single-indication clinical cohorts were the Youden Index values derived in AUROC analysis. An additional, exploratory, pan-tumor TMB threshold was derived by using TMB and GEP data across each cohort, similar to a previously described method (20).
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge D. Li, E. Rubin, and J.Yuan for critical review of the manuscript and valuable input and S. Erespe for editorial assistance, all of Merck, Kenilworth, NJ.
Funding: This work was supported by Merck, Kenilworth, NJ.
Footnotes
Competing interests: M.A., A.L., J.K.L., T.K.M., E.M., and M.N. are inventors on patent W0/2016/094377A1, submitted by Merck Sharp and Dohme, which covers “System and methods for deriving gene signature biomarkers of response to PD-1 antagonists.” E.R.P. has served as a consultant or scientific advisor for AstraZeneca, Bristol-Myers Squibb, Clovis, Eli Lilly, Exelexis, Genentech, Horizon Pharma, Inovio, Novartis, Pfizer, and Roche and has received research funding from Agensys, AstraZeneca, Bristol-Myers Squibb, Merck, Peloton, Pfizer, and Genentech. P.A.O. has served as a consultant for Alexion Pharmaceuticals, Amgen, Bristol-Myers Squibb, Celldex, CytomX Therapeutics, Genentech, and Neon Therapeutics and has received research funding from ARMO BioSciences, AstraZeneca/Medlmmune, Bristol-Myers Squibb, Celldex, and Merck. T.Y.S. has received honoraria from Amgen, AstraZeneca, Bayer/Onyx, Bristol-Myers Squibb, Merck, and Merck Serono and also research funding from Boehringer Ingelheim and Genentech/Roche. A.R. has served as a consultant-advisor for Merck, and his institution received research funding from Merck. R.C., R.M., M.A., A.A., E.M., J.Y., X.S., X.Q.L., H.L., M.N., C.Z., J.K.L., A.J., J.C., A.L.W., N.I., T.K.M., J.E.T., A.L., and D.K. are employees or former employees of Merck Sharp and Dohme, a subsidiary of Merck, Kenilworth, NJ, and may hold stock options in the company.
Data and materials availability: Anonymized WES genomic data from tumor and normal specimens from patients in the KEYNOTE trials included in this study are available through the NCBI Database of Genotypes and Phenotyes (dbGaP) under accession number phs001572.v1.p1. Requests for access to patient-level clinical data from the KEYNOTE trials in this study can be submitted through the EngageZone site (http://engagezone.msd.com/ds_documentation.php) or via email (dataaccess@merck.com) per Merck’s data sharing policy; those requests pertaining to the validation of work in the study will be reviewed in an expedited manner.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Lee SJ et al. , Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 580, 755–762 (2006). doi: 10.1016/j.febslet.2005.12.093; pmid: [DOI] [PubMed] [Google Scholar]
- 2.Lu C, Redd PS, Lee JR, Savage N, Liu K, The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 5, e1247135 (2016). doi: 10.1080/2162402X.2016.1247135; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taube JM et al. , Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 20, 5064–5074 (2014). doi: 10.1158/1078-0432.CCR-13-3271; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder A et al. , Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014). doi: 10.1056/NEJMoa1406498; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Allen EM et al. , Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015). doi: 10.1126/science.aad0095; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hugo W et al. , Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016). doi: 10.1016/j.cell.2016.02.065; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson DB, Pollack MH, Sosman JA, Emerging targeted therapies for melanoma. Expert Opin. Emerg. Drugs 21, 195–207 (2016). doi: 10.1080/14728214.2016.1184644; pmid: [DOI] [PubMed] [Google Scholar]
- 8.Rizvi NA et al. , Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). doi: 10.1126/science.aaa1348; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kowanetz M et al. , Tumor mutation load assessed by FoundationOne (FM1) is associated with improved efficacy of atezolizumab (atezo) in patients with advanced NSCLC. Ann. Oncol. 27, 77P (2016). doi: 10.1093/annonc/mdw363.25 [DOI] [Google Scholar]
- 10.Le DT et al. , Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). doi: 10.1126/science.aan6733; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le DT et al. , PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015). doi: 10.1056/NEJMoa1500596; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenberg JE et al. , Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 387,1909–1920 (2016). doi: 10.1016/S0140-6736(16)00561-4; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N, Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015). doi: 10.1016/j.cell.2014.12.033; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tumeh PC et al. , PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). doi: 10.1038/nature13954; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayers M et al. , IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 127, 2930–2940 (2017). doi: 10.1172/JCI91190; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Cancer Genome Atlas, TCGA Data Portal; https://tcga-data.nci.nih.gov/docs/publications/tcga/.
- 17.Alexandrov LB, Stratton MR, Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 24, 52–60 (2014). doi: 10.1016/j.gde.2013.11.014; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haddad R et al. , Genomic determinants of response to pembrolizumab in head and neck squamous cell carcinoma. J. Clin. Oncol. 35 (Suppl.), 6009 (2017). doi: 10.1200/JC0.2017.35.15_suppl.6009 [DOI] [Google Scholar]
- 19.Johnson DB et al. , Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 4, 959–967 (2016). doi: 10.1158/2326-6066.CIR-16-0143; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Panda A et al. , Identifying a clinically applicable mutational burden threshold as a potential biomarker of response to immune checkpoint therapy in solid tumors. JCO Precis. Oncol. 2017, 10.1200/P0.17.00146 (2017). pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seiwert TY et al. , “Biomarkers predictive of response to pembrolizumab in head and neck cancer (HNSCC),” in Proceedings of the American Association for Cancer Research Annual Meeting 2018, Chicago, IL, 14 to 18 April 2018 (American Association for Cancer Research, 2018), abstract no. LB-339. [Google Scholar]
- 22.Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC, Microsatellite instability detection by next generation sequencing. Clin. Chem. 60, 1192–1199 (2014). doi: 10.1373/clinchem.2014.223677; pmid: [DOI] [PubMed] [Google Scholar]
- 23.Roh W et al. , Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 9, eaah3560 (2017). doi: 10.1126/scitranslmed.aah3560; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGranahan N et al. , Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016). doi: 10.1126/science.aaf1490; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garon EB et al. , Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015). doi: 10.1056/NEJMoa1501824; pmid: [DOI] [PubMed] [Google Scholar]
- 26.Robert C et al. , Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015). doi: 10.1056/NEJMoa1503093; pmid: [DOI] [PubMed] [Google Scholar]
- 27.Neagu MR, Reardon DA, An update on the role of immunotherapy and vaccine strategies for primary brain tumors. Curr. Treat. Options Oncol. 16, 54 (2015). doi: 10.1007/s11864-015-0371-3; pmid: [DOI] [PubMed] [Google Scholar]
- 28.Vaishampayan U, Therapeutic options and multifaceted treatment paradigms in metastatic castrate-resistant prostate cancer. Curr. Opin. Oncol. 26, 265–273 (2014). doi: 10.1097/CC0.0000000000000066; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coppola D et al. , Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am. J. Pathol. 179, 37–45 (2011). doi: 10.1016/j.ajpath.2011.03.007; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galon J, Angell HK, Bedognetti D, Marincola FM, The continuum of cancer immunosurveillance: Prognostic, predictive, and mechanistic signatures. Immunity 39, 11–26 (2013). doi: 10.1016/j.immuni.2013.07.008; pmid: [DOI] [PubMed] [Google Scholar]
- 31.Łuksza M et al. , A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551, 517–520 (2017). pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai H et al. , A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 65, 4059–4066 (2005). doi: 10.1158/0008-5472.CAN-04-3953; pmid: [DOI] [PubMed] [Google Scholar]
- 33.Spranger S, Bao R, Gajewski TF, Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). doi: 10.1038/nature14404; pmid: [DOI] [PubMed] [Google Scholar]
- 34.Yoshihara K et al. , Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013). doi: 10.1038/ncomms3612; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang AC et al. , T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). doi: 10.1038/nature22079; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robbins PF et al. , Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor- reactive T cells. Nat. Med. 19, 747–752 (2013). doi: 10.1038/nm.3161; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segal NH et al. , Epitope landscape in breast and colorectal cancer. Cancer Res. 68, 889–892 (2008). doi: 10.1158/0008-5472.CAN-07-3095; pmid: [DOI] [PubMed] [Google Scholar]
- 38.Matsushita H et al. , Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404 (2012). doi: 10.1038/nature10755; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dolled-Filhart M et al. , Development of a companion diagnostic for pembrolizumab in non–small cell lung cancer using immunohistochemistry for programmed death ligand-1. Arch. Pathol. Lab. Med. 140, 1243–1249 (2016). doi: 10.5858/arpa.2015-0542-OA; pmid: [DOI] [PubMed] [Google Scholar]
- 40.Chow LQ et al. , A phase Ib study of pembrolizumab (pembro; MK-3475) in patients (pts) with human papillomavirus virus (HPV)-positive and negative head and neck cancer (HNC). Ann. Oncol. 25 (Suppl. 4), 14 (2014). doi: 10.1093/annonc/mdu438.32 [DOI] [Google Scholar]
- 41.Seiwert TY et al. , Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE- 012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 17, 956–965 (2016). doi: 10.1016/S1470-2045(16)30066-3; pmid: [DOI] [PubMed] [Google Scholar]
- 42.Hamid O et al. , Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 369, 134–144 (2013). doi: 10.1056/NEJMoa1305133; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). doi: 10.1093/bioinformatics/btp324; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna A et al. , The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). doi: 10.1101/gr.107524.110; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cibulskis K et al. , Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013). doi: 10.1038/nbt.2514; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sherry ST, Ward M, Sirotkin K, dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 9, 677–679 (1999). pmid: [PubMed] [Google Scholar]
- 47.Forbes SA et al. , COSMIC (the Catalogue of Somatic Mutations in Cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 38, D652–D657 (2010). doi: 10.1093/nar/gkp995; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szolek A et al. , OptiType: Precision HLA typing from next- generation sequencing data. Bioinformatics 30, 3310–3316 (2014). doi: 10.1093/bioinformatics/btu548; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nielsen M et al. , Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017 (2003). doi: 10.1110/ps.0239403; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lund O et al. , Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics 55, 797–810 (2004). doi: 10.1007/s00251-004-0647-4; pmid: [DOI] [PubMed] [Google Scholar]
- 51.Sidney J, Peters B, Frahm N, Brander C, Sette A, HLA class I supertypes: A revised and updated classification. BMC Immunol. 9, 1 (2008). doi: 10.1186/1471-2172-9-1; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLaren W et al. , The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016). doi: 10.1186/s13059-016-0974-4; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C, DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016). doi: 10.1186/s13059-016-0893-4; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alexandrov LB et al. , Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). doi: 10.1038/nature12477; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koboldt DC et al. , VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). doi: 10.1101/gr.129684.111; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Favero F et al. , Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 26, 64–70 (2015). doi: 10.1093/annonc/mdu479; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roth A et al. , PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 11, 396–398 (2014). doi: 10.1038/nmeth.2883; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dolled-Filhart M et al. , Development of a prototype immunohistochemistry assay to measure programmed death ligand-1 expression in tumor tissue. Arch. Pathol. Lab. Med. 140, 1259–1266 (2016). doi: 10.5858/arpa.2015-0544-OA; pmid: [DOI] [PubMed] [Google Scholar]
- 59.Daud AI et al. , Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J. Clin. Oncol. 34, 4102–4109 (2016). doi: 10.1200/JC0.2016.67.2477; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.