

Abstract
Background
Crohn’s disease (CD) is a chronic inflammatory disease of the gastrointestinal tract, and immune response modulation is the main treatment strategy to induce remission in active CD. Certolizumab pegol (CZP) is a tumor necrosis factor‐alfa (TNF‐α) inhibitor which regulates impaired immune response.
Objectives
The primary objectives were to evaluate the efficacy and safety of CZP for the induction of remission in CD.
Search methods
We searched MEDLINE, Embase, CENTRAL, the Cochrane IBD group specialized register, trials registers and other sources from inception to 28 January 2019. Moreover, we contacted the pharmaceutical company that manufactures CZP.
Selection criteria
We included randomized controlled trials comparing CZP with placebo or no treatment in active CD patients.
Data collection and analysis
We used standard Cochrane methodological procedures. The main outcomes selected for GRADE analysis were clinical remission at week 8 (Crohn’s Disease Activity Index [CDAI] ≤150), clinical response at week 8 (CDAI reduction ≥ 100 or clinical remission), and serious adverse events. The Mantel‐Haenszel random‐effects method was applied for the statistical analyses. For dichotomous outcomes, we calculated the risk ratio (RR) and corresponding 95% confidence interval (95% CI).
Main results
Four studies involving 1485 participants with moderate to severe CD met the inclusion criteria and were used in the meta‐analyses. All studies included active CD patients with CDAI ranging from 220 to 450. Most patients were adults over 18 years of age. One study was identified as high risk of bias due to a non‐identical placebo while the other studies were judged to be at low risk of bias.
CZP (100 mg to 400 mg every 2 to 4 weeks) was shown to be superior to placebo for achieving clinical remission at week 8 (RR 1.36, 95% CI 1.11 to 1.66; moderate certainty evidence). The raw numbers of participants achieving clinical remission at week 8 were 26.9% (225/835) and 19.8% (129/650) in the CZP and the placebo groups, respectively.
CZP was shown to be superior to placebo for achieving clinical response at week 8 (RR 1.29, 95% CI 1.09 to 1.53; moderate certainty evidence). In raw numbers, clinical response at week 8 was achieved in 40.2% (336/835) and 30.9% (201/650) of participants in the CZP and the placebo groups, respectively.
In raw numbers, serious adverse events were observed in 8.7% (73/835) and 6.2% (40/650) of participants in the CZP and the placebo groups, respectively (RR 1.35, 95% CI 0.93 to 1.97; moderate certainty evidence). Serious adverse events included worsening Crohn's disease, infections, and malignancy.
Authors' conclusions
Moderate certainty evidence suggests that CZP is effective for induction of clinical remission and clinical response in participants with active CD patients. It is uncertain whether the risk of serious adverse events differs between CZP and placebo as the 95% CI includes the possibility of a small decrease or doubling of events. Future studies are needed to evaluate the long‐term efficacy and safety of CZP in CD patients.
Plain language summary
Certolizumab pegol for the treatment of active Crohn’s disease
Review question
We reviewed the evidence about the benefits and harms of using certolizumab pegol in people with active Crohn’s disease.
Background
Crohn’s disease is a chronic inflammatory disease that mainly affects the gastrointestinal tract such as the small and large intestine. Common symptoms of Crohn’s disease are chronic diarrhea, abdominal pain, and weight loss. When patients with Crohn’s disease have symptoms, the condition is considered to be 'active'. When in 'remission', patients do not have symptoms.
Certolizumab pegol is a biologic medication used to modify the excessive immune response that causes chronic inflammation in Crohn’s disease. Certolizumab pegol is usually injected under the skin every 2 to 4 weeks.
Study characteristics
The literature was searched up to 28 January 2019. Four studies involving 1485 patients compared certolizumab pegol with placebo (a dummy drug). All studies included patients with active Crohn’s disease. Most patients were adults over 18 years of age, except for six patients aged 16 or 17 years old. All studies were funded by the drug manufacturer.
Key results
In a combined analysis of the four studies, patients with active Crohn’s disease who received certolizumab pegol at a dose ranging from 100 mg to 400 mg every 2 to 4 weeks, responded to the treatment and achieved remission at 8 weeks more often than patients taking placebo. No remarkable difference in the rate of serious side effects was observed between certolizumab pegol and placebo. Serious side effects included worsening Crohn's disease, infections, and malignancy (i.e. cancer).
Quality of the evidence
Moderate certainty evidence suggests that certolizumab pegol is beneficial in terms of achieving remission in people with moderate to severe Crohn's disease. Because of a low number of serious side effects, the certainty of evidence about harms of certolizumab pegol was moderate.
Conclusions
Moderate certainty evidence suggests that certolizumab pegol is effective for induction of clinical remission and clinical response in people with active Crohn's disease. It is uncertain whether the risk of serious side effects differs between certolizumab pegol and placebo. Future studies are needed to evaluate the long‐term benefits and harms of certolizumab pegol in people with Crohn's disease.
Summary of findings
Summary of findings for the main comparison. Certolizumab pegol compared to placebo for induction of remission in Crohn's disease.
| Certolizumab pegol compared to placebo for induction of remission in Crohn's disease | ||||||
|
Patient or population: Patients with active Crohn's disease Settings: Outpatient Intervention: Certolizumab pegol Comparison: Placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Certolizummab pegol | |||||
|
Clinical remission Follow‐up: 8 weeks |
198 per 1000 |
270 per 1000 (220 to 329) |
RR 1.36 (1.11 to 1.66) |
1485 (4 studies) | ⊕⊕⊕⊝ MODERATE1 | Certolizumab pegol was shown to be superior to placebo regarding clinical remission at week 8 Clinical remission was defined as a CDAI < 150 |
|
Clinical response Follow‐up: 8 weeks |
309 per 1000 |
399 per 1000 (337 to 473) |
RR 1.29 (1.09 to 1.53) |
1485 (4 studies) |
⊕⊕⊕⊝ MODERATE1 | Clinical response was defined as CDAI reduction ≥ 100 from baseline |
|
Serious adverse events Follow‐up: 8 weeks |
62 per 1000 |
83 per 1000 (57 to 121) |
RR 1.35 (0.93 to 1.97) |
1485 (4 studies) |
⊕⊕⊕⊝ MODERATE2 | Reported serious adverse events included worsening Crohn's disease, infections, and malignancy |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio; CDAI: Crohn's disease activity index | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded one level due to high risk of bias in one study in the pooled analysis
2 Downgraded one level due to imprecision (113 events)
Background
Description of the condition
Crohn’s disease (CD) is a chronic inflammatory disease that mainly affects the gastrointestinal tract. CD is more common in North America and Europe than in other regions. Nonetheless, both the incidence and prevalence of CD are increasing worldwide. The highest annual incidence of CD was reported to be 5.0, 12.7, and 20.2 per 100,000 person‐years in Asia and the Middle East, Europe and North America, respectively. Moreover, the highest reported prevalence of CD was 67.9, 322, and 318.5 per 100,000 people in Asia and the Middle East, Europe and North America, respectively (Molodecky 2012).
Common symptoms of CD include chronic diarrhea, abdominal pain, and weight loss (Torres 2016), and patients are typically diagnosed with CD in their 20s to 30s (Cosnes 2011). Up to one third of patients with CD had complications such as stricturing and penetrating disease at diagnosis, and half of these patients needed surgery within 10 years of diagnosis (Peyrin‐Biroulet 2010). After the initial surgery, one‐quarter of the patients required a second surgery within five years (Frolkis 2014). Moreover, the age‐adjusted risk of mortality in patients with CD was 50% higher than that of the general population (Canavan 2007).
The etiology of CD is unknown, but abnormal mucosal immune response and impaired barrier function are considered to play an important role in the pathogenesis of CD. Altered intestinal microflora and environmental factors, such as food and smoking, have been postulated to cause immune system dysfunction in genetically susceptible individuals (Torres 2016). Regulating impaired immune response is the key to CD treatment.
Description of the intervention
The current treatment strategy for inducing remission in active CD is based on immune response modulation. Pharmacologic treatments for induction of remission in CD include corticosteroids, tumor necrosis factor‐alpha (TNF‐α) inhibitors, antibodies to α4β7 integrin, and antibodies to interleukin‐12/23p40. TNF‐α is a pro‐inflammatory cytokine and plays a central role in the inflammatory cascade of CD. Regulating impaired immune response with TNF‐α inhibitors may be key for treatment of CD (Baumgart 2012; Nielsen 2013; Olesen 2016; Torres 2016).
Certolizumab pegol (CZP) is a TNF‐α inhibitor. Unlike other TNF‐α inhibitors such as infliximab (IFX) and adalimumab (ADA), CZP is a polyethylene glycolated Fab fragment of a humanized anti‐TNF‐α monoclonal antibody with high affinity for TNF‐α. CZP has no Fc portion and has a different mechanistic profile than other TNF‐ α inhibitors because of its unique structure. The drug lacks the ability to induce regulatory macrophage formation, antibody‐dependent cellular cytotoxicity, complement‐dependent cytotoxicity, and apoptosis via reverse signaling. However, CZP can inhibit inflammatory mediators and increase regulatory T cell activity as effectively as IFX and ADA (Gomollon 2017; Nesbitt 2007; Olesen 2016; Shao 2009; Torres 2016).
TNF‐α inhibitors including CZP are recommended for moderately‐to‐severely active CD (Gomollon 2017; Talley 2011; Terdiman 2013; Torres 2016). CZP is approved for the treatment of CD in the United States and Switzerland (Olesen 2016; Torres 2016). CZP is a subcutaneously delivered drug and can be self‐administered. Potential serious adverse events of CZP are anaphylactic reaction, lymphoproliferative disorder, tuberculosis reactivation, and opportunistic infection (Gomollon 2017).
How the intervention might work
CZP inhibits TNF‐α receptor activation by neutralizing both the transmembrane form of TNF‐α (tmTNF) and soluble form of TNF‐α (sTNF). Currently, tmTNF signaling is considered to have a central role in the pathogenesis of CD, and CZP can bind to both sTNF and tmTNF. CZP regulates impaired immune response through the following possible mechanisms of action: increased regulatory T cell frequency and activity, inflammatory mediator suppression in immune cells, decreased inflammatory mediators by reverse signaling in tmTNF‐expressing cells, and nonapoptotic cytotoxicity and apoptosis by blocking tmTNF‐mediated TNF‐α receptor activation (Olesen 2016).
Why it is important to do this review
Recent meta‐analyses have shown inconsistent results, which may be due to differences in methodology and in the selected time points for the assessment of clinical remission (Ford 2011; Kawalec 2013). In one review (Ford 2011), there was no difference between CZP and placebo in the proportion of participants who failed to achieve remission at weeks 6 to 12 (risk ratio [RR] 0.95, 95% confidence interval [CI] 0.90 to 1.01). Another review (Kawalec 2013) found a benefit for induction of remission for CZP over placebo at week four (RR 1.63, 95% CI 1.32 to 2.13). We are also aware of at least one unpublished trial (NCT00291668). The current review will summarize and properly integrate all of the available evidence, including unpublished randomized controlled trials (RCTs), to provide the best available evidence to assess the efficacy and safety of CZP for induction of remission in CD.
Objectives
The primary objectives were to evaluate the efficacy and safety of CZP for induction of remission in CD.
Methods
Criteria for considering studies for this review
Types of studies
This review included RCTs irrespective of publication status. No language status restrictions were applied.
Types of participants
Participants included patients (≥ 16 years old) with active CD that was diagnosed by standard clinical, endoscopic, radiographic, and histopathological assessment. Active CD was defined as having a Crohn’s Disease Activity Index (CDAI) score greater than 150 or a Harvey‐Bradshaw Index (HBI) score greater than 4 (Best 1976; Harvey 1980).
Types of interventions
Subcutaneous administration of any dose of CZP every two to four weeks compared to placebo or no treatment. Active comparators such as conventional treatment (including 5‐aminosalicylic acid, immunomodulators, or corticosteroids) were not included in this review.
Types of outcome measures
Primary outcomes
The primary outcome was the proportion of CD patients achieving remission at week eight after CZP administration. We selected week eight because this week is the recommended time for switching to maintenance dosing according to the approved dosing regimen (Schreiber 2011). If the outcome was not assessed at week 8, we selected the nearest week between weeks 4 and 12 as the outcome assessment point. If only dates from two time points equally distant from week 8, such as weeks 6 and 10, were available despite inquiring with the original investigators, the earlier point (week 6) was planned to be selected. Remission was defined as CDAI ≤ 150 or HBI ≤ 4. If both CDAI and HBI were reported in the primary studies, the CDAI was planned to be used for outcome assessment. The proportion of participants in remission was calculated in accordance with the intention‐to‐treat (ITT) principle; the denominator was the number of the randomized patients in each arm. Participants with missing data for the primary outcome were assumed to be treatment failures.
Secondary outcomes
Secondary outcomes included the proportion of participants with:
clinical response at week eight;
C‐reactive protein (CRP) improvement at week eight;
health‐related quality of life at week eight;
endoscopic improvement at week twelve;
fistula closure at week eight;
adverse events;
serious adverse events; and
withdrawals due to adverse events.
Clinical response was defined as a CDAI reduction from baseline of greater than or equal to 100 or remission (CDAI ≤ 150) or an HBI reduction from baseline of greater than or equal to 3 or remission (HBI ≤ 4) (Vermeire 2010). For outcomes that were not available for week 8, we selected the nearest week between weeks 4 and 12. If only two assessment points equally distant from week 8 were available, such as weeks 6 and 10, despite inquiring with the original investigators, the earlier point (week 6) was planned to be selected. We followed this procedure for all outcomes that we intended to assess at eight weeks. The assessment for C‐reactive protein (CRP) improvement at week eight was based on log scales of geometric mean CRP ratios between baseline and week eight. The assessment for health‐related quality of life at week eight was based on the mean change in quality of life scores from baseline as measured by a validated instrument including the Inflammatory Bowel Disease Questionnaire (IBDQ) (Guyatt 1989) or the Medical Outcomes Study Short‐Form 36 (SF‐36) questionnaire (Ware 1992). If endoscopic outcomes were not reported for week 12, we planned to select the nearest week between weeks 8 and 26. We also planned to assess endoscopic improvement by calculating the mean change from baseline in the Crohn’s Disease Endoscopic Index of Severity (CDEIS), the Simplified Endoscopic Activity Score for Crohn’s Disease (SES‐CD), or the Rutgeerts score (Daperno 2004; Mary 1989; Rutgeerts 1990). If only two assessment points equally distant from week 12 were available, such as weeks 10 and 14, despite inquiring with the original investigators, the earlier point (week 10) was planned to be selected. Adverse and serious adverse events were based on what was reported in the primary studies.
Search methods for identification of studies
Electronic searches
We conducted a comprehensive literature search without language restrictions. We searched the following databases to identify relevant RCTs:
MEDLINE (inception to date);
Embase (inception to date);
CENTRAL;
The Cochrane IBD Group Specialized Register (inception to date);
http://ClinicalTrials.gov (trial registry);
https://www.clinicaltrialsregister.eu/ (EU Clinical Trials Register);
http://apps.who.int/trialsearch/ (International Clinical Trials Registry Platform); and
http://www.ucb.com/our‐science/Our‐clinical‐studies/cimzia‐certolizumab‐pegol (web site of a pharmaceutical company producing CZP).
The search strategies are reported in Appendix 1.
Searching other resources
To identify additional studies, we searched the following resources manually or through personal contacts:
Abstracts of Digestive Disease Week, United European Gastroenterology Week, European Crohn’s and Colitis Organization Congress, and Advances in Inflammatory Bowel Diseases (2000 to date);
References from published articles; and
Pharmaceutical companies and experts involved in the development of CZP.
Data collection and analysis
Selection of studies
Two authors (HY and RS) independently screened titles and abstracts, and selected potential eligible studies based on the above criteria. In addition, these authors independently read the full‐text articles of the potential eligible studies and decided which studies should be included in the review. In cases of insufficient information, we contacted the authors of the primary studies to evaluate eligibility for inclusion. In the event of a disagreement regarding study selection, HY and RS discussed the matter together to reach a consensus. NW acted as the arbitrator when consensus was not reached.
Data extraction and management
Two authors (HY and RS) independently extracted data using data extraction forms to record data from the selected studies. Any disagreements were resolved through discussion. NW was the arbitrator when consensus was not reached.
We extracted the following data:
Characteristics of the primary studies: publication year, country, study recruitment period, study completion date, study type, and risk of bias items;
Participant characteristics: country, total number of participants, number of participants randomized, number of participants analyzed in each group, age, sex, ethnicity, body mass index, disease duration, disease site, smoking status, CDAI score, HBI score, CDEIS, SES‐CD, Rutgeerts score, IBDQ score, SF‐36 score, CRP, fistula, concurrent CD treatment, previous CD treatment, inclusion criteria, and exclusion criteria;
Intervention characteristics: dose, delivery route, and administration schedule of CZP;
Comparator characteristics: placebo or no treatment control;
Outcomes: proportion of participants who achieved clinical remission at week 8, proportion of participants with clinical response at week 8, CDAI score at week 8, HBI score at week 8, CDEIS at week 12, SES‐CD at week 12, Rutgeerts score at week 12, IBDQ score at week 8, SF‐36 score at week 8, CRP at week 8, fistula closure at week 8, any adverse events, adverse events leading to withdrawal, serious adverse events, time of outcome assessment in primary studies, length of follow‐up, number of participants lost to follow‐up, reasons for loss to follow‐up, number of participants who did not complete treatment, reasons for incomplete treatment, and criteria for evaluating outcomes in primary studies.
We contacted the authors of the primary studies and the pharmaceutical company that manufactures CZP if information in the published reports was insufficient.
Assessment of risk of bias in included studies
Two authors (HY and RS) independently assessed the quality of included studies using the Cochrane risk of bias tool (Higgins 2011). Any disagreements were resolved by consensus with a third author (NW). Primary studies were rated as high, low, or unclear risk of bias. We assessed the following risk of bias items: random sequence generation (selection bias), allocation concealment (selection bias), blinding of participants and personnel (performance bias), blinding of outcome assessment (detection bias), incomplete outcome data (attrition bias), selective reporting (reporting bias), and other potential sources of bias.
We rated random sequence generation as low risk of bias if the method for random sequence generation was described as a random number table, computer‐generated, coin tossing, shuffling cards or envelopes, throwing dice, drawing of lots or minimization. We rated random sequence generation as high risk of bias if the method of generation was not random. Examples included a systematic approach, such as date or record number, or a non‐systematic approach, such as preference and availability. We rated random sequence generation as unclear risk of bias if insufficient information was reported to allow for a judgement.
We rated allocation concealment as low risk of bias if allocation could not be foreseen by participants and investigators. Adequate methods of allocation concealment included centralized allocation such as telephone, web‐based, or pharmacy‐controlled randomization, sequentially numbered drug containers of the same appearance, or sequentially numbered, opaque, sealed envelopes. We rated allocation concealment as high risk of bias if the allocation sequence was likely to be foreseen. Examples included an open random allocation schedule or envelopes without safeguards. We rated allocation concealment as unclear risk of bias if insufficient information was reported to allow for a judgement.
We rated blinding of participants and personnel and blinding of outcome assessors as low risk of bias if proper methods were employed to prevent knowledge of treatment assignment (e.g. double‐blinding with an identical placebo) or if no blinding or incomplete blinding of participants or personnel was unlikely to affect assessment of the outcome (e.g. a serious adverse event resulting in death). We rated blinding of participants and personnel and blinding of outcomes assessors as high risk of bias if blinding was likely to be broken and this could affect outcome assessment, or if there was no blinding or incomplete blinding of participants or personnel or outcome assessors which could affect outcome assessment. We rated blinding of participants and personnel and blinding of outcome assessors as unclear risk of bias if insufficient information was reported to allow for a judgement.
We rated incomplete outcome data as low risk of bias when there were no missing outcome data; when missing outcome data were unlikely to be related to the true outcome; when the number of dropouts and reasons for withdrawal were balanced between treatment groups; when compared to the observed event, the proportion of missing outcomes did not have a clinically relevant impact on the effect estimate for dichotomous outcomes; when the expected effect size among missing outcomes did not have clinically relevant impact on the observed effect size for continuous outcome data; or when missing data were imputed using proper methods. We rated incomplete outcome data as high risk of bias when missing outcome data were likely to be related to the true outcome; when numbers or reasons for missing data were imbalanced across treatment groups; when compared with the observed event, the proportion of missing outcomes had a clinically relevant impact on the effect estimate for dichotomous outcomes; when the expected effect size among missing outcomes had a clinically relevant impact on the observed effect size for continuous outcomes; when an 'as‐treated analysis' was substantially performed; and when missing data were imputed using improper methods (e.g. simple imputation). We rated incomplete outcome data as unclear risk of bias when insufficient information was reported to allow for a judgement.
We rated selective reporting as low risk of bias when the protocols of primary studies were available, and all of the primary and secondary study outcomes related to this systematic review, were reported in a pre‐defined way; and when the study protocols were unavailable, but all of the expected outcomes, related to this systematic review, were reported. We rated selective outcome reporting as high risk of bias when pre‐defined primary outcomes related to this systematic review were not thoroughly reported; when those primary outcomes were measured or analyzed in a way that was different to the protocol; when reported primary outcomes related to this systematic review were different from those in the protocol; when the outcomes were not completely reported; and when key outcomes were not included in the primary studies. We rated selective outcome reporting as unclear risk of bias when insufficient information was reported to allow for a judgement.
We rated studies as low risk of bias for other sources of bias when the study appeared to be free of other potential sources of bias. We rated studies as high risk of bias for other sources of bias when other sources of bias could have an impact on the study outcomes. For example, fraudulent studies or baseline imbalances in demographic factors. We rated studies as unclear risk of bias for other sources of bias when the study reported insufficient details to allow for a judgement. We contacted study authors for additional information to clarify the risk of bias when the study reports did not provide enough detail to allow for a clear judgement.
Measures of treatment effect
For dichotomous outcomes, we calculated the risk ratio (RR) and corresponding 95% confidence interval (CI). ITT analyses were conducted for dichotomous outcomes, whereby all dropouts were assumed to be treatment failures. We calculated the mean difference (MD) and 95% CI for continuous outcomes. When different scales were used to measure the same construct, we planned to calculate the standardized mean difference (SMD) and 95% CI.
Unit of analysis issues
We collected outcomes per randomized participant. For cross‐over trials, we planned to use data from the first phase before the cross‐over. Cluster RCTs were not included in this review. If events occurred more than once (e.g. adverse events), we reported on the proportion of participants who experienced at least one event. To avoid double counting of the comparator for multi‐arm studies (multiple dose groups), the number of patients in the comparator group (i.e. placebo or no treatment control) were divided across the number of eligible CZP arms. To deal with multiple observations for the same outcome in primary studies, we precisely defined the outcome assessment points for both primary and secondary outcomes.
Dealing with missing data
We contacted authors of the primary studies and the pharmaceutical company that manufactures CZP to obtain missing data and the reason for missing data. If it was not possible to obtain the missing data, we reported as such in the results. For dichotomous outcomes, all missing data were treated as treatment failures in the ITT analyses. We conducted sensitivity analyses using available case data to assess the impact on the effect estimate. For continuous outcomes, we did not use any imputation methods, and used the available data only.
Assessment of heterogeneity
Clinical heterogeneity was first assessed with regard to patient characteristics, such as previous treatment and concurrent medication. If the studies were clinically homogenous, statistical heterogeneity was evaluated using the Chi² test and I² statistic. A P value of less than or equal to 0.10 for the Chi² test was considered to show statistically significant heterogeneity. The I² statistic estimates the degree of statistical heterogeneity. We considered an I² value of 25% to indicate low heterogeneity, a value of 50% to indicate moderate heterogeneity and a value of 75% to indicate high heterogeneity. If statistical heterogeneity existed, we planned to perform a visual inspection of the forest plots to identify potential outliers causing the heterogeneity. Moreover, sensitivity and subgroup analyses were planned to explore potential sources of heterogeneity when significant or moderate‐high heterogeneity was identified (Higgins 2003; Higgins 2011).
Assessment of reporting biases
We searched for both registered and published trials, and reported on the proportion of registered trials that were unpublished. We contacted the investigators of the unpublished trials and the pharmaceutical company that manufactures CZP to provide data related to outcomes in this systematic review. If we could not obtain these data, we reported as such in this review. When there were 10 or more eligible trials in a pooled analysis, we planned to generate funnel plots to evaluate potential publication bias. Moreover, when we found unclear or high risk of bias for selective reporting, we contacted the study authors and the pharmaceutical company that manufactures CZP to provide unpublished outcome data. If we could not obtain these data, we reported so in the results.
Data synthesis
When included studies were sufficiently similar from the clinical and statistical viewpoints, we conducted meta‐analyses. Data were analyzed using Review Manager 5.3, and a random‐effects model was used for the meta‐analyses. A P value of less than 0.05 was considered to be statistically significant.
On the basis of the characteristics of participants, interventions, and outcomes in primary studies, clinical similarity was determined by consensus between HY and KM. In cases of disagreement between HY and KM, the authors consulted with TK to resolve the disagreement. In cases of high heterogeneity (I² statistic ≥ 75%), meta‐analyses were not planned, and each study was planned to be described in detail.
Subgroup analysis and investigation of heterogeneity
If sufficient data were available, the following subgroup analyses were planned for the primary outcomes:
Disease severity at baseline (150 < CDAI < 220, 220 ≤ CDAI ≤ 450, CDAI > 450);
CRP levels at baseline (CRP levels < 10 mg/L, CRP levels ≥ 10 mg/L);
Doses of CZP (CZP < 200 mg, 200 mg ≤ CZP < 400 mg, 400 mg ≤ CZP < 600 mg, and CZP ≥ 600 mg); and
Previous treatment with other TNF‐α inhibitors (yes, no).
Sensitivity analysis
We planned to perform the following sensitivity analyses for primary outcomes:
Excluding studies judged to be at high risk of bias for any domain of the risk of bias tool;
Excluding studies judged to be at high or unclear risk of bias for any domain of the risk of bias tool;
Using available case data instead of ITT analysis for missing dichotomous outcome data;
Selecting later outcome assessment points if only dates from two time points equally distant from the defined outcome assessment points were available despite inquiring with the original investigators. For example, if only dates from two time points equally distant from week 8, such as weeks 6 and 10, were available, week 10 was planned to be selected in the sensitivity analysis.
Limiting the included studies that administered CZP strictly in accordance with the approved regimen which is subcutaneous administration of 400 mg CZP at weeks 0, 2, and 4, and then every 4 weeks.
Summary of findings tables
We produced 'Summary of findings' tables using the GRADEpro Guideline Development Tool for the following outcomes: clinical remission, clinical response, and serious adverse events.
The Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach was used to evaluate the certainty of the evidence supporting each outcome. Evidence from RCTs starts as high quality, but can be downgraded due to risk of bias, inconsistency across studies, indirectness of evidence, imprecision of effect estimate, and publication bias (Schünemann 2011). If serious limitations were present, we downgraded the evidence level by one. Moreover, very serious limitations can lead to downgrading of the evidence by two levels (Schünemann 2011). HY and RS independently assessed the certainty of evidence for each outcome and the overall quality of the evidence was rated as:
High: We are very confident that the true effect lies close to that of the effect estimate;
Moderate: We are moderately confident in the effect estimate: the true effect is likely to be close to the effect estimate, but it could be substantially different;
Low: Our confidence in the effect estimate is limited: the true effect may be substantially different from the effect estimate; or
Very low: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the effect estimate.
In cases of disagreement between HY and RS, we consulted with NW.
Results
Description of studies
Results of the search
The literature search was conducted on 28 January 2019 and 623 studies were identified. After removing duplicates, 423 reports remained for the title and abstract screening. Two authors independently screened the 423 reports, and 26 full‐text reports were assessed for further eligibility. Twelve reports of four studies (1485 participants) were finally included in this systematic review and a total of 14 reports of 12 studies were excluded (see Figure 1).
1.
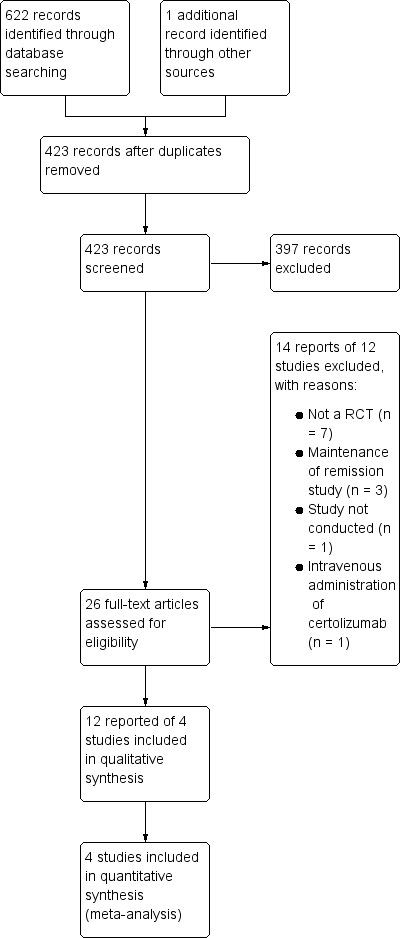
Study flow diagram.
Included studies
The four studies included in this systematic review are summarized in the Characteristics of included studies table. All four studies were randomized, double‐blind, placebo‐controlled multicenter trials sponsored by UCB Inc, the manufacturer of CZP. All the study participants were active CD patients whose CDAI was between 220 and 450. Follow‐up periods ranged between 6 weeks and 28 weeks.
Schreiber 2005 compared three different doses (100 mg, 200 mg, or 400 mg) of CZP with placebo in adult patients (18 to 75 years, N = 292). CZP was administered subcutaneously at weeks zero, four and eight. The primary outcome was clinical response (> 100 points CDAI decrease) or remission (CDAI ≤ 150) at week 12.
Sandborn 2007 compared 400 mg of CZP with placebo in adult patients (≥ 18 years, N = 660). CZP was administered subcutaneously at weeks zero, two and four, and then every four weeks. The primary outcome was clinical response (> 100 points CDAI decrease) at week 6 and at both weeks 6 and 26 in patients with a baseline serum CRP ≥ 10 mg/L. Clinical remission (CDAI ≤ 150) at week 6 was a secondary outcome.
Ogata 2009 compared two different doses (200 mg or 400 mg) of CZP with placebo in patients with CRP ≥ 10 mg/L (16 to 65 years, N = 94). Only two participants were aged 16 or 17 years old in each group. CZP was administered subcutaneously at weeks zero, two and four. The primary outcome was clinical response (> 100 points CDAI decrease) or remission (CDAI ≤ 150) at week 6.
Sandborn 2011 compared 400 mg of CZP with placebo in adult patients without previous treatment with TNF‐α inhibitors (18 to 75 years, N = 439). CZP was administered subcutaneously at weeks zero, two and four. The primary outcome was clinical remission (CDAI ≤ 150) at week 6.
We contacted UCB Inc and obtained the following information: a protocol of Schreiber 2005; a presentation poster from the United European Gastroenterology 2009, detailed information from Ogata 2009, and CRP and IBDQ data from Schreiber 2005, Sandborn 2007, Ogata 2009, and Sandborn 2011. We confirmed via the UCB Inc website (https://www.ucb.com/our‐science/Our‐clinical‐studies/cimzia‐certolizumab‐pegol) that all RCTs conducted by UCB Inc were included in this review.
Excluded studies
The characteristics of excluded studies by full‐text assessment are shown in the Characteristics of excluded studies table. We found no ongoing studies.
Risk of bias in included studies
Figure 2 and Figure 3 report a summary of risk of bias in the included studies.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
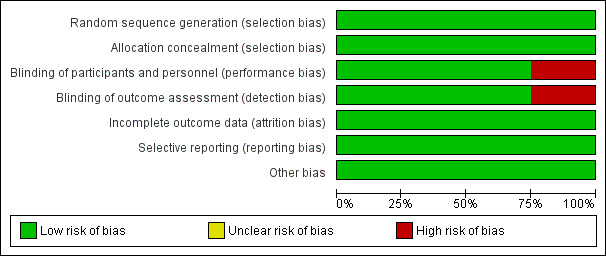
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Centralized randomization schemes with randomization code were used in all studies. Therefore, we considered all studies to be at low risk of selection bias.
Blinding
Schreiber 2005 used a placebo that did not have the same color or viscosity of CZP, and patients may have been able to distinguish between CZP and the placebo. Moreover, outcomes were assessed based on patients' daily diaries. Therefore, we considered this study to have high risk of performance and detection bias. The other three studies used an identical placebo and were considered as low risk of bias regarding both performance and detection bias (Ogata 2009;Sandborn 2007; Sandborn 2011).
Incomplete outcome data
The proportion of loss to follow‐up was less than 2% and was balanced between CZP and placebo in all studies. Therefore, we rated all studies as low risk of attrition bias.
Selective reporting
All of the studies reported our prespecified outcomes properly. Therefore, all studies were rated as having low risk of reporting bias.
Other potential sources of bias
We did not find any other potential sources of bias in all studies. Therefore, we rated all studies as having low risk of other potential sources of bias
Effects of interventions
See: Table 1
Main comparison
See Summary of findings table 1: Certolizumab pegol compared to placebo for induction of remission in Crohn’s disease.
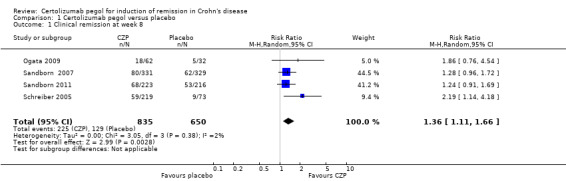
Clinical remission at week 8, the only primary outcome in this review, was assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In fact, we could not obtain week eight clinical remission data for two studies involving 533 CD patients (Ogata 2009; Sandborn 2011). As defined in our protocol, we used week six clinical remission data from Ogata 2009 and Sandborn 2011, and these data were combined with week eight data from the other two studies involving 952 CD patients (Sandborn 2007; Schreiber 2005). Heterogeneity was low ( I² = 2%). Clinical remission was achieved in 26.9% (225/835) and 19.8% (129/650) in the CZP and placebo groups, respectively. In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP (100 mg to 400 mg every 2 to 4 weeks) were significantly more likely to achieve clinical remission at week 8 than those treated with placebo (RR 1.36, 95% CI 1.11 to 1.66; see Figure 4). The certainty of evidence was moderate according to the GRADE system.
4.
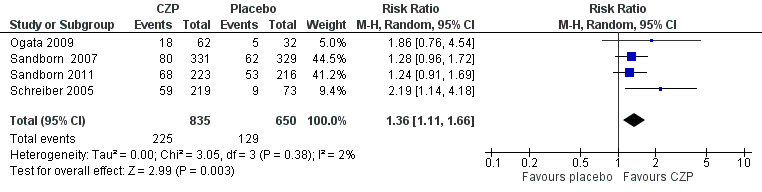
Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.1 Clinical remission at week 8.
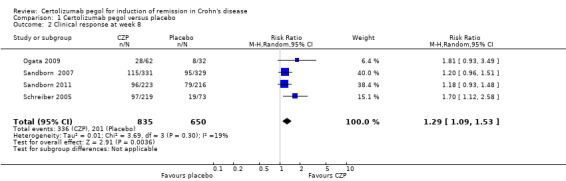
Clinical response at week 8, a secondary outcome in this review, was assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). Clinical response was achieved in 40.2% (336/835) and 30.9% (201/650) in the CZP and placebo groups respectively. In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP were significantly more likely to achieve clinical response at week 8 than those with treated with placebo (RR 1.29, 95% CI 1.09 to 1.53; see Figure 5). The certainty of evidence was moderate according to the GRADE system.
5.
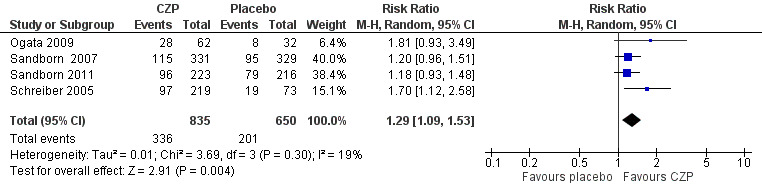
Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.2 Clinical response at week 8.
Serious adverse events, a secondary outcome in this review, were assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). Serious adverse events such as worsening CD, various infections, and malignancy occurred in 8.7% (73/835) and 6.2% (40/650) in CZP and placebo, respectively. In the meta‐analysis using the Mantel‐Haenszel random‐effects method, it is uncertain whether the risk of serious adverse events differs between CZP and placebo (RR 1.35, 95% CI: 0.93 to 1.97; see Figure 6). According to GRADE system, the certainty of evidence was moderate because the number of events was low, and showed imprecision in the results.
6.
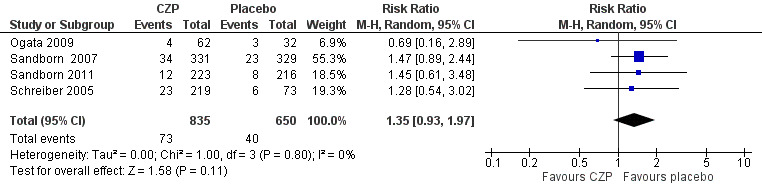
Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.6 Serious adverse events.
Efficacy outcomes
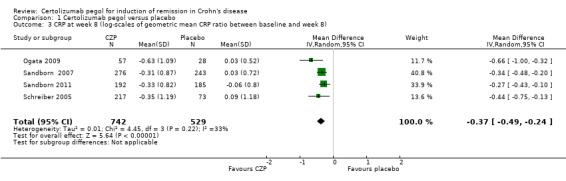
The log scales of the geometric mean CRP ratio between baseline and week 8 were assessed in four studies involving 1271 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the inverse variance random‐effects method, patients treated with CZP were significantly more likely to have improvement in CRP at week 8 than those treated with placebo (MD ‐0.37, 95% CI ‐0.49 to ‐0.24).
The mean change in IBDQ score from baseline was assessed in four studies involving 1315 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the inverse variance random‐effects method, CZP did not appear to make a clear difference in IBDQ at week 8 (MD 2.12, 95% CI ‐1.27 to 5.50).
We did not find any eligible studies assessing endoscopic improvement or fistula closure.
Safety outcomes
Adverse events were assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, CZP did not appear to make any clear difference in the risk of having adverse events (RR 1.03, 95% CI 0.97 to 1.10).
Withdrawals due to adverse events were assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, there was no clear difference between CZP and placebo in the risk of withdrawals due to adverse events (RR 1.01, 95% CI: 0.57 to 1.78).
Subgroup analyses
We conducted three prespecified subgroup analyses to evaluate clinical remission at week 8 based on CZP doses, previous treatment with TNF‐α inhibitors, and CRP levels at baseline. We did not conduct a prespecified subgroup analysis based on disease severity because disease severity was similar among all of the included studies.
Clinical remission at week 8 with CZP 100 mg was assessed in a study involving 99 CD patients (Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, it was uncertain whether clinical remission at week 8 differs between CZP 100 mg and placebo (RR 2.48, 95% CI: 0.81 to 7.58). Similarly, it was unclear whether clinical remission at week 8 differs between CZP 200 mg and placebo in 2 studies involving 142 CD patients (RR 1.84, 95% CI: 0.75 to 4.50) (Ogata 2009; Schreiber 2005). However, patients treated with CZP 400 mg were significantly more likely than placebo participants to achieve clinical remission at week 8 (RR 1.30, 95% CI: 1.06 to 1.60, 4 studies; 1244 CD patients) (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). We did not conduct a subgroup analysis of patients with CZP ≥ 600 mg because there were no studies using this dose.
Clinical remission at week 8 among patients with no previous treatment with TNF‐α inhibitors was assessed in a study involving 439 CD patients (Sandborn 2011). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, it was uncertain whether clinical remission at week 8 differs between CZP and placebo patients (RR 1.24, 95% CI: 0.91 to 1.69). We did not conduct a subgroup analysis of CD patients who had previous treatment with TNF‐α inhibitors due to lack of data.
Clinical remission at week 8 among patients with baseline CRP ≥ 10 mg/L was assessed in 4 studies involving 702 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method among patients with baseline CRP ≥ 10 mg/L, patients treated with CZP were significantly more likely to achieve clinical remission at week 8 than those treated with placebo: RR 1.59 (95% CI: 1.17 to 2.16). However, it was uncertain whether clinical remission at week 8 differs between CZP and placebo among patients with baseline CRP < 10 mg/L in three studies involving 762 CD patients (RR 1.15, 95% CI 0.88 to 1.50) (Sandborn 2007; Sandborn 2011; Schreiber 2005).
Sensitivity analyses
We conducted three prespecified sensitivity analyses to evaluate clinical remission at week eight based on excluded studies with high risk of bias, studies using available case data instead of ITT analysis, and studies with the approved regimen of CZP. We did not conduct a sensitivity analysis of selecting later outcome assessment points because we obtained outcomes at prespecified assessment points.
We excluded a study with high risk of bias (Schreiber 2005), and clinical remission at week 8 was assessed in 3 studies involving 1193 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP were significantly more likely to achieve clinical remission at week 8 than those treated with placebo (RR 1.29, 95% CI: 1.05 to 1.59). We found no studies with unclear risk of bias in any domain of the risk of bias evaluation.
For the available case data analysis, clinical remission at week 8 was assessed in 4 studies involving 1463 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP were significantly more likely to achieve clinical remission at week 8 than those treated with placebo (RR 1.36, 95% CI: 1.11 to 1.67).
Clinical remission at week 8 with the approved regimen of CZP was assessed in 3 studies involving 1147 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP were significantly more likely to achieve clinical remission at week 8 than those treated with placebo (RR 1.28, 95% CI 1.03 to 1.57).
Funnel plots
We did not generate funnel plots to evaluate potential publication bias because there were less than 10 eligible trials for each pooled analysis.
Discussion
Summary of main results
We included 4 studies with 1485 patients in our meta‐analyses of the main outcomes. Patients treated with CZP were significantly more likely to achieve clinical remission (CDAI ≤150) at week 8 than those treated with placebo. The efficacy of CZP was robust in a sensitivity analysis limited to studies with a low risk of bias. Similarly, patients treated with CZP were significantly more likely to achieve clinical response (CDAI reduction ≥ 100 or clinical remission) at week 8 than those treated with placebo. These results suggest a clear benefit of CZP for active CD patients. Higher CRP improvement in patients with CZP than those with placebo is in accordance with this evidence. With respect to safety, there may be no clear difference in adverse events and withdrawal due to adverse events. Moreover, it is uncertain whether the risk of serious adverse events differs between CZP and placebo as the 95% CI includes the possibility of a small decrease or doubling of events.
Overall completeness and applicability of evidence
We included both published and unpublished data in this review. All studies were funded by the pharmaceutical company, and we contacted the company to acquire the data not reported in the published articles. To our knowledge based on our systematic literature search, this review included all the existing data to evaluate the efficacy and safety of CZP for induction of remission in CD.
All study participants had moderate to severe CD (CDAI 220 to 450), and the evidence cannot be applied to people with mild CD (CDAI < 220) and extremely severe CD (CDAI > 450). Moreover, most of the patients were adults (≥ 18 years), and this review does not provide any evidence regarding pediatric CD patients. Finally, long‐term efficacy of more than 8 weeks and safety of more than 28 weeks were not evaluated in this review. A future review to evaluate maintenance of remission is warranted.
Quality of the evidence
According to the GRADE system, the overall certainty of evidence was moderate for both clinical remission and clinical response at week 8. Three out of four included studies were rated as low risk of bias for all domains (Ogata 2009; Sandborn 2007; Sandborn 2011), and only Schreiber 2005 was judged to be high risk of bias regarding both performance and detection bias. We confirmed the robust beneficial effect of CZP in the sensitivity analysis, which excluded Schreiber 2005. Due to the sparse data, the overall certainty of evidence was moderate for serious adverse events. We chose not to downgrade serious adverse events for high risk of bias (i.e. due to inclusion of Schreiber 2005 study in pooled analysis). Due to the objective nature of this outcome, we did not think that the potential for unblinding of treatment assignment would have an impact on participants experiencing a life threatening adverse event. We found no other reasons to downgrade the certainty of evidence regarding clinical remission, clinical response, and serious adverse events.
Potential biases in the review process
We systematically searched all the available resources and have contacted the pharmaceutical company that manufactures CZP. Based on this systematic search, two authors independently evaluated studies for inclusion, extracted data from the included study, assessed risk of bias for each study, and conducted GRADE evaluation for main outcomes. Because data were extracted from line graphs in Schreiber 2005 due to a lack of detailed data, there might be slight differences between these data and the actually observed data. With respect to safety, we included data between week 0 and week 12 in Schreiber 2005 because we could not obtain the total number of adverse events and serious adverse events for the overall follow‐up period of 20 weeks.
Agreements and disagreements with other studies or reviews
The primary result of this review, the significant effect of CZP on induction of clinical remission at week 8, was not consistent with Ford 2011 but was consistent with Kawalec 2013. Although both systematic reviews (Ford 2011; Kawalec 2013), included the same four studies (Sandborn 2007; Sandborn 2011; Schreiber 2005; Winter 2004), the time points used to evaluate clinical remission were different. CZP was shown to be effective in Kawalec 2013 (at week 4), but week 4 is still in the induction dosing period (frequent administration period). It is important to be in remission when patients start the maintenance dose at week 8 (Schreiber 2011). Ford 2011 selected more appropriate time points to assess clinical remission (at weeks 6 to 12), but the efficacy of CZP was not shown. In our review, we selected week 8 because that week is the recommended time to switch to maintenance dosing according to the approved regimen (Schreiber 2011). Moreover, we did not include Winter 2004 based on our protocol, because CZP in that study was administered intravenously rather than subcutaneously. More importantly, we found an unpublished study (Ogata 2009) through contact with UCB Inc and included this in our meta‐analysis (Ogata 2009). Therefore, the results of this review can be considered to be the most comprehensive review with proper assessment time points for this topic.
Authors' conclusions
Implications for practice.
Moderate certainty of evidence suggests that CZP is effective for induction of clinical remission and clinical response in people with moderate to severe CD. It is uncertain whether the risk of serious adverse events differs between CZP and placebo as the 95% CI includes the possibility of a small decrease or doubling of events.
Implications for research.
The primary outcome of this review was based on the CDAI, which is a largely subjective measure. This review included CRP improvement as an objective outcome, and we found higher CRP improvement in patients with CZP than those with placebo. We could not analyze endoscopic improvement because there was no relevant study reporting this outcome. Further studies investigating objective outcomes such as endoscopic improvement are needed.
This review focuses on induction of remission with CZP in CD as defined in the protocol, and we did not investigate maintenance therapy with CZP for CD. Future systematic reviews are required to evaluate long‐term efficacy (maintenance of remission) and safety of CZP in CD patients.
Acknowledgements
The authors thank John K MacDonald and Tran M Nguyen for their assistance.
The authors thank Emma Barber and the Cochrane Japan Centre for assisting with English editing.
This project was supported by the Japanese Society for Inflammatory Bowel Disease.
Funding for the Cochrane IBD Group (May 1, 2017 ‐ April 30, 2022) has been provided by Crohn's and Colitis Canada (CCC).
Appendices
Appendix 1. Search strategies
MEDLINE (inception to date)
1. random$.tw.
2. factorial$.tw.
3. (crossover$ or cross over$ or cross‐over$).tw.
4. placebo$.tw.
5. single blind.mp.
6. double blind.mp.
7. triple blind.mp.
8. (singl$ adj blind$).tw.
9. (double$ adj blind$).tw.
10. (tripl$ adj blind$).tw.
11 assign$.tw.
12. allocat$.tw.
13. randomized controlled trial/
14. or/1‐13
15. exp Crohn disease/ or Crohn*.mp.
16. (inflammatory bowel disease* or IBD).mp.
17. 15 or 16
18. exp certolizumab pegol/
19. (CDP870 OR 'CDP 870' OR CDP‐870 OR 'certolizumab pegol' OR certolizumab OR cimzia).mp.
20. 18 or 19
21. 14 and 17 and 20
Embase (inception to date)
1. random$.tw.
2. factorial$.tw.
3. (crossover$ or cross over$ or cross‐over$).tw.
4. placebo$.tw.
5. single blind.mp.
6. double blind.mp.
7. triple blind.mp.
8. (singl$ adj blind$).tw.
9. (double$ adj blind$).tw.
10. (tripl$ adj blind$).tw.
11 assign$.tw.
12. allocat$.tw.
13. crossover procedure/
14. double blind procedure/
15. single blind procedure/
16. triple blind procedure/
17. randomized controlled trial/
18. or/1‐17
19. exp Crohn disease/ or Crohn*.mp.
20. (inflammatory bowel disease* or IBD).mp.
21. 19 or 20
22. exp certolizumab pegol/
23. (CDP870 OR 'CDP 870' OR CDP‐870 OR 'certolizumab pegol' OR certolizumab OR cimzia).mp.
24. 22 or 23
25. 18 and 21 and 24
CENTRAL (inception to date)
#1 MeSH descriptor: [Inflammatory bowel diseases] explode all trees
#2 MeSH descriptor: [Crohn Disease] explode all trees
#3 Crohn
#4 #1 or #2 or #3
#5 MeSH descriptor: [Certolizumab pegol] explode all trees
#6 CDP870 OR 'CDP 870' OR CDP‐870 OR 'certolizumab pegol' OR certolizumab OR cimzia
#7 #5 or #6
#8 #4 and #7
Cochrane IBD Group Specialized Register (inception to date)
#1 (CDP870 OR 'CDP 870' OR CDP‐870 OR 'certolizumab pegol' OR certolizumab OR cimzia).ti.
#2 Crohn.ti.
#3 1 and 2
Data and analyses
Comparison 1. Certolizumab pegol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical remission at week 8 | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.11, 1.66] |
| 2 Clinical response at week 8 | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [1.09, 1.53] |
| 3 CRP at week 8 (log‐scales of geometric mean CRP ratio between baseline and week 8) | 4 | 1271 | Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.49, ‐0.24] |
| 4 IBDQ total score at week 8 (mean change from baseline) | 4 | 1315 | Mean Difference (IV, Random, 95% CI) | 2.12 [‐1.27, 5.50] |
| 5 Adverse events | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.97, 1.10] |
| 6 Serious adverse events | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.93, 1.97] |
| 7 Withdrawals due to adverse events | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.57, 1.78] |
| 8 Clinical remission at week 8 (Subgroup analysis based on CZP doses) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Certolizumab pegol 100mg | 1 | 99 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [0.81, 7.58] |
| 8.2 Certolizumab pegol 200mg | 2 | 142 | Risk Ratio (M‐H, Random, 95% CI) | 1.84 [0.75, 4.50] |
| 8.3 Certolizumab pegol 400mg | 4 | 1244 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [1.06, 1.60] |
| 9 Clinical remission at week 8 (Subgroup analysis of no previous treatment with TNF‐α inhibitors) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Clinical remission at week 8 (Subgroup analysis of CRP levels at baseline) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 CRP ≥ 10 mg/L | 4 | 702 | Risk Ratio (M‐H, Random, 95% CI) | 1.59 [1.17, 2.16] |
| 10.2 CRP < 10 mg/L | 3 | 762 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.88, 1.50] |
| 11 Clinical remission at week 8 (Sensitivity analysis of excluding studies with high risk of bias) | 3 | 1193 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [1.05, 1.59] |
| 12 Clinical remission at week 8 (sensitivity analysis of using available case data) | 4 | 1463 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.11, 1.67] |
| 13 Clinical remission at week 8 (Sensitivity analysis of studies with the approval dosing regimen) | 3 | 1147 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [1.03, 1.57] |
1.1. Analysis.
Comparison 1 Certolizumab pegol versus placebo, Outcome 1 Clinical remission at week 8.
1.2. Analysis.
Comparison 1 Certolizumab pegol versus placebo, Outcome 2 Clinical response at week 8.
1.3. Analysis.
Comparison 1 Certolizumab pegol versus placebo, Outcome 3 CRP at week 8 (log‐scales of geometric mean CRP ratio between baseline and week 8).
1.4. Analysis.
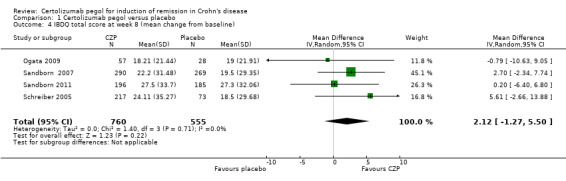
Comparison 1 Certolizumab pegol versus placebo, Outcome 4 IBDQ total score at week 8 (mean change from baseline).
1.5. Analysis.
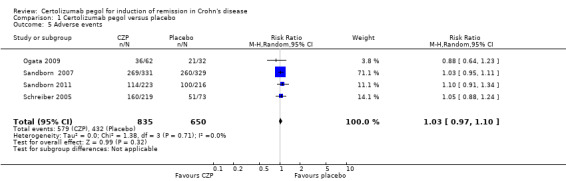
Comparison 1 Certolizumab pegol versus placebo, Outcome 5 Adverse events.
1.6. Analysis.
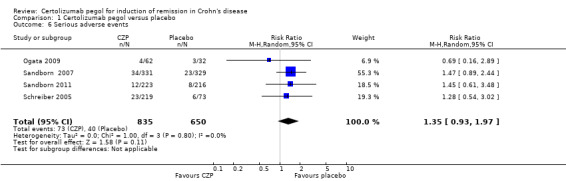
Comparison 1 Certolizumab pegol versus placebo, Outcome 6 Serious adverse events.
1.7. Analysis.
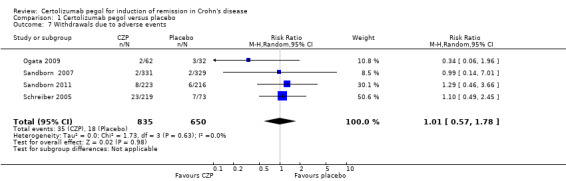
Comparison 1 Certolizumab pegol versus placebo, Outcome 7 Withdrawals due to adverse events.
1.8. Analysis.
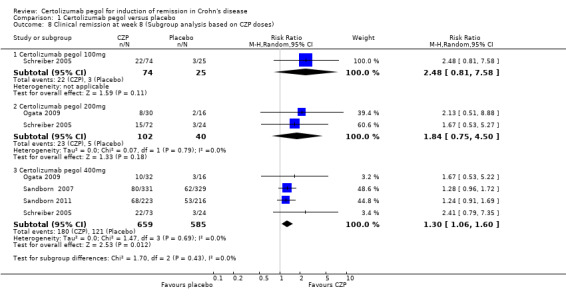
Comparison 1 Certolizumab pegol versus placebo, Outcome 8 Clinical remission at week 8 (Subgroup analysis based on CZP doses).
1.9. Analysis.
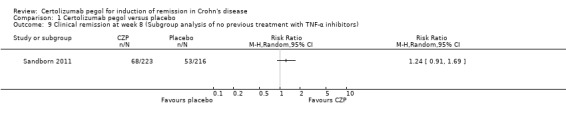
Comparison 1 Certolizumab pegol versus placebo, Outcome 9 Clinical remission at week 8 (Subgroup analysis of no previous treatment with TNF‐α inhibitors).
1.10. Analysis.
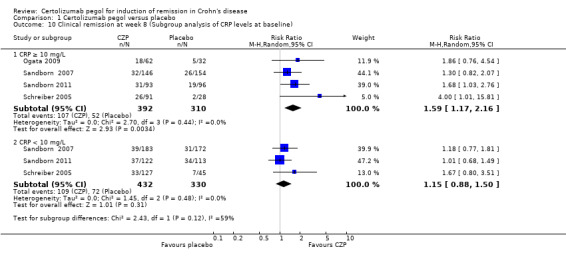
Comparison 1 Certolizumab pegol versus placebo, Outcome 10 Clinical remission at week 8 (Subgroup analysis of CRP levels at baseline).
1.11. Analysis.
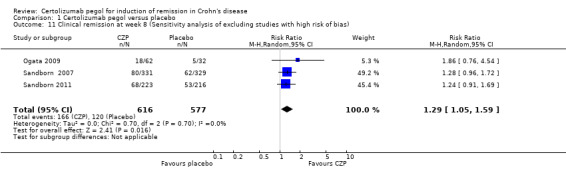
Comparison 1 Certolizumab pegol versus placebo, Outcome 11 Clinical remission at week 8 (Sensitivity analysis of excluding studies with high risk of bias).
1.12. Analysis.
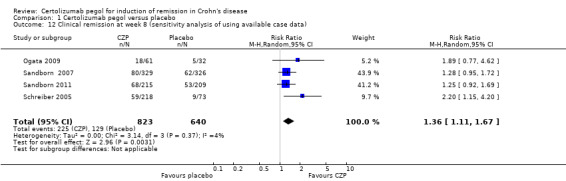
Comparison 1 Certolizumab pegol versus placebo, Outcome 12 Clinical remission at week 8 (sensitivity analysis of using available case data).
1.13. Analysis.
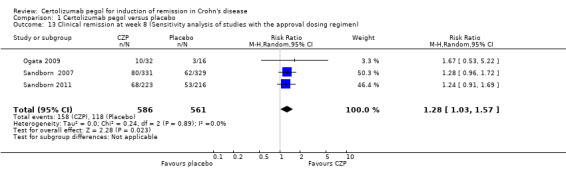
Comparison 1 Certolizumab pegol versus placebo, Outcome 13 Clinical remission at week 8 (Sensitivity analysis of studies with the approval dosing regimen).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ogata 2009.
| Methods | Randomized, double‐blind, placebo‐controlled, multicenter trial | |
| Participants | Patients (16‐65 years) with active Crohn's disease (CDAI: 220‐450) and C‐reactive protein value ≥ 10 mg/L (N = 94) | |
| Interventions | Subcutaneous administration at week 0, 2, and 4 Group 1: Placebo (n = 32) Group 2: 200 mg of Certolizumab pegol (n = 30) Group 3: 400 mg of Certolizumab pegol (n = 32) |
|
| Outcomes | Primary outcome: Clinical response (> 100 points CDAI decrease) or remission (CDAI ≤ 150) at week 6 Secondary outcomes: 1. CDAI at weeks 2, 4, 6 2. > 70 points CDAI decrease at weeks 2, 4, 6 3. Remission at weeks 2, 4, 6 4. Clinical response at weeks 2, 4, 6 5. IBDQ at weeks 2, 4, 6 6. CRP at weeks 2, 4, 6 | |
| Notes | This study was conducted between March 2006 and November 2007 The follow‐up period was 6 weeks. Adverse events were followed for 28 weeks Funding source was UCB Inc, which manufactures Certolizumab pegol All authors were from Japan Conflict of interest was reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralized randomization schemes with randomization code were used |
| Allocation concealment (selection bias) | Low risk | Centralized randomization schemes with randomization code were used |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Masking: Triple (Participant, Care Provider, Investigator) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Masking: Triple (Participant, Care Provider, Investigator) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No loss to follow‐up |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Sandborn 2007.
| Methods | Randomized, double‐blind, placebo‐controlled, multicenter trial | |
| Participants | Adult patients (≥ 18 years) with active Crohn's disease (CDAI: 220‐450) (N = 660) | |
| Interventions | Subcutaneous administration at week 0, 2, 4 and then every 4 weeks Group 1: Placebo (n = 329) Group 2: 400 mg of Certolizumab pegol (n = 331) |
|
| Outcomes | Primary outcome: Clinical response (> 100 points CDAI decrease) at week 6 and at both weeks 6 and 26 in patients with a baseline serum CRP ≥10 mg/L Secondary outcomes: In the patients with a baseline serum CRP ≥10 mg/L 1. Clinical remission (CDAI ≤ 150) at week 6 2. Clinical remission at both week 6 and week 26 3. IBDQ response (≥ 16 points total score increase) at week 6 4. IBDQ response at both week 6 and 26 In all of the patients 1. Clinical response at week 6 2. Clinical remission at week 6 3. Clinical response at both week 6 and 26 4. Clinical remission at both week 6 and 26 This study reported both clinical remission and response at week 2, 4, 6, 8, 12, 16, 20, 24, and 26 |
|
| Notes | This study was conducted between December 2003 and May 2005 The follow‐up period was 26 weeks Funding source was UCB Inc which manufactures certolizumab pegol. Other funding sources were the National Center for Research Resources, a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research; and by a grant for infrastructure from the German Federal Ministry of Education and Research’s competence network for chronic inflammatory bowel disease Authors were from 7 countries: USA, Canada, Bulgaria, South Africa, Belgium, UK, and Germany Conflict of interest was reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralized randomization schemes with randomization code were used |
| Allocation concealment (selection bias) | Low risk | Centralized randomization schemes with randomization code were used |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Data were collected from diaries kept by patients who were blinded to treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The number of patients lost to follow‐up was only two in the certolizumab pegol group |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Sandborn 2011.
| Methods | Randomized, double‐blind, placebo‐controlled, multicenter trial | |
| Participants | Adult patients (18‐75 years) with active Crohn's disease (CDAI: 220‐450) (N = 439) No previous treatment with TNF‐α inhibitors |
|
| Interventions | Subcutaneous administration at week 0, 2, and 4 Group 1: Placebo (n = 216) Group 2: 400 mg of Certolizumab pegol (n = 223) |
|
| Outcomes | Primary outcome: Clinical remission (CDAI ≤ 150) at week 6
Secondary outcomes: In all of the patients 1. Clinical response (> 100 points CDAI decrease) at weeks 2, 4, and 6 2. IBDQ remission (total score ≥ 170) at weeks 2, 4, and 6 3. Change in total CDAI score from week 0 to weeks 2, 4, and 6 4. Change in HBI score from week 0 to week 6 5. Clinical remission at weeks 2 and 4 In the patients with a baseline serum CRP ≥10 mg/L 1. Clinical remission at week 6 2. Clinical response at week 6 In the patients with a baseline serum CRP <10 mg/L 1. Clinical remission at week 6 2. Clinical response at week 6 |
|
| Notes | This study was conducted between March 2008 and June 2009 The follow‐up period was 6 weeks Funding source was UCB Inc, which manufactures certolizumab pegol Authors were from 6 countries: USA, Germany, Canada, UK, Belgium, and Netherlands Conflict of interest was reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralized randomization schemes with randomization code were used |
| Allocation concealment (selection bias) | Low risk | Centralized randomization schemes with randomization code were used |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Masking: Quadruple (Participant, Care Provider, Investigator, Outcomes Assessor) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Masking: Quadruple (Participant, Care Provider, Investigator, Outcomes Assessor) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The number of patients lost to follow‐up was only one in the certolizumab pegol group |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Schreiber 2005.
| Methods | Randomized, double‐blind, placebo‐controlled, multicenter trial | |
| Participants | Adult patients (18‐75 years) with active Crohn's disease (CDAI: 220‐450) (N = 292) | |
| Interventions | Subcutaneous administration at week 0, 4, and 8 Group 1: Placebo (n = 73) Group 2: 100 mg of certolizumab pegol (n = 74) Group 3: 200 mg of certolizumab pegol (n = 72) Group 4: 400 mg of certolizumab pegol (n = 73) |
|
| Outcomes | Primary outcome: Clinical response (> 100 points CDAI decrease) or remission (CDAI ≤ 150) at week 12 Secondary outcomes: 1. Clinical response or remission at weeks 2, 4, 6, 8, and 10 2. Remission at weeks 2, 4, 6, 8, 10, and 12 |
|
| Notes | This study was conducted between February 2001 and March 2002 The follow‐up period was 20 weeks Funding source was Celltech R&D, Ltd (now UCB Inc). Additional support was provided by the German Federal Ministry for Education and Research Competence Network “Inflammatory Bowel Disease" Authors were from 5 countries: Germany, Belgium, Canada, UK, and Denmark Conflict of interest was reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralized randomization schemes with randomization code were used |
| Allocation concealment (selection bias) | Low risk | Centralized randomization schemes with randomization code were used |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The color or viscosity was different between Certolizumab pegol and placebo although patients received the treatment from independent healthcare workers |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Full blinding to the patients were not possible, and outcomes were based on a daily diary of their symptoms |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The number of patients with lost to follow‐up was only one in each group |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Colombel 2008 | This study was not a RCT |
| NCT00152425 | Maintenance of remission study The study participants were the patients responded to CZP |
| NCT00307931 | This study was not a RCT |
| NCT00349752 | Maintenance of remission study The study participants were the patients in remission and receiving corticosteroids |
| NCT00354367 | This study was not conducted |
| NCT01024647 | This study was not a RCT |
| NCT01053559 | This study was not a RCT |
| NCT01582568 | This study was not a RCT |
| Sandborn 2008 | This study was not a RCT |
| Sandborn 2012 | This study was not a RCT |
| Vermeire 2008 | Maintenance of remission study The study participants were the patients who responded to CZP. Moreover, the participants were allocated to two different dosing schedules of CZP (every two weeks or every four weeks) |
| Winter 2004 | CZP was administered intravenously rather than subcutaneously |
CZP: certolizumab pegol; RCT: randomized controlled trial
Differences between protocol and review
A trial search coordinator made minor changes to the search strategy to meet Cochrane standards.
Ogata 2009 included CD patients aged between ≥ 16 years and < 65 years, although age of the patients in our protocol was defined as ≥ 18 years. Because there were only two patients of < 18 years in each group, we decided to include Ogata 2009.
Log scales of geometric mean CRP ratios between baseline and week 8, instead of mean changes from baseline, were evaluated for secondary outcomes because only the data of the geometric mean CRP ratios were available.
Adverse and serious adverse events were based on judgement in the primary studies because we could not obtain the detailed severity of adverse events.
Contributions of authors
Drafting the protocol: Hajime Yamazaki, Ryuhei So, Katsuyoshi Matsuoka, Taku Kobayashi, Shinichiro Shinzaki, Minoru Matsuura, Shinji Okabayashi, Yuki Kataoka, Yasushi Tsujimoto, Toshi A Furukawa, Norio Watanabe
Developing and running the search strategy: Hajime Yamazaki, Yuki Kataoka, Cochrane IBD Group Information Specialist
Obtaining copies of studies: Hajime Yamazaki, Katsuyoshi Matsuoka, Taku Kobayashi, Shinji Okabayashi
Selecting which studies to include: Hajime Yamazaki, Ryuhei So
Extracting data from studies: Hajime Yamazaki, Ryuhei So
Entering data into RevMan: Hajime Yamazaki
Carrying out the analysis: Hajime Yamazaki, Yuki Kataoka, Yasushi Tsujimoto, Toshi A Furukawa, Norio Watanabe
Interpreting the analysis: Hajime Yamazaki, Katsuyoshi Matsuoka, Taku Kobayashi, Shinichiro Shinzaki, Minoru Matsuura, Shinji Okabayashi
Drafting the final review: Hajime Yamazaki, Ryuhei So, Katsuyoshi Matsuoka, Taku Kobayashi, Shinichiro Shinzaki, Minoru Matsuura, Shinji Okabayashi, Yuki Kataoka, Yasushi Tsujimoto, Toshi A Furukawa, Norio Watanabe
Updating the review: Hajime Yamazaki, Ryuhei So, Katsuyoshi Matsuoka, Taku Kobayashi, Shinichiro Shinzaki, Minoru Matsuura, Shinji Okabayashi, Yuki Kataoka, Yasushi Tsujimoto, Toshi A Furukawa, Norio Watanabe
Sources of support
Internal sources
No sources of support supplied
External sources
-
Japanese Society for Inflammatory Bowel Disease, Japan.
Research grant
Declarations of interest
Hajime Yamazaki: None known
Ryuhei So: None known
Katsuyoshi Matsuoka received research grants from Mitsubishi Tanabe Pharma, Kyorin Pharmaceutical, Takeda Pharmaceutical, Shionogi, EA pharma, Zeria Pharmaceutical, Nippon Kayaku, Thermofisher Scientific, Sekisui Medical, Abbvie, Alfesa Pharma Corporation, Kissei, Pfizer, and Mochida Pharmaceutical outside of this work; consultant fees from Gilead, Thermofisher Scientific, Alfresa Corporation, Covidien, Abbvie, Janssen Pharmaceutical, Astellas, JIMRO, Pfizer, Sekisui Medical, Celltrion, Eli Lilly, Chugai Pharmaceutical, Allergan, EA Pharma, Takeda Pharmaceutical, Mitsubishi Tanabe Pharma, Asahi Kasei Medical, and Daiichi‐Sankyo; lecture fees from Gilead, Mitsubishi‐Tanabe Pharma, Eisai, Abbvie, EA Pharma, Janssen Pharmaceutical, Thermofisher Scientific, Kyorin Pharmaceutical, Astellas, Asahi Kasei Medical, Kyowa Hakko Kirin, Mochida Pharmaceutical, Boehringer Ingelheim, Kissei, Takeda Pharmaceutical, JIMRO, Daiichi‐Sankyo, Pfizer, Zeria Pharmaceutical, and AstraZeneca.
Taku Kobayashi received lecture fees from Abbvie Inc, Kyorin Pharmaceutical, Mitsubishi Tanabe, EA Pharma, Medtronic Co.,Ltd, Janssen, Mochida Fharmaceutical, Takeda Pharmaceutical, Gilead Sciences, Nippon Kayaku, JIMRO, ZERIA Pharmaceutical, Astellas, Asahi Kasei Medical, Thermo Fisher Scientific, Alfresa Pharma, Celltrion, Pfizer, Eli Lilly, Ferring Pharmaceuticals, Covidien, Eizai, and Ajinomoto Pharma; consulting/advisory fees from Abbvie Inc, Kyorin Pharmaceutical, Janssen, Mochida Fharmaceutical, Takeda Pharmaceutical, Gilead Sciences, Nippon Kayaku, Alfresa Pharma, Celltrion, Pfizer, and Eli Lilly; grants from Alfresa Pharma, Nippon Kayaku, EA Pharma, and Thermo Fisher Scientific.
Shinichiro Shinzaki received consulting or lecture fees from Abbvie, Aspen, EA Pharma, Kissei, Kyorin, Janssen, JIMRO, Mitsubishi‐Tanabe, Mochida, Nippon Kayaku, Takeda, Zeria and serves as an advisory board member for Janssen and Takeda.
Minoru Matsuura received lecture fees from Janssen Pharamaceutical. Co. Ltd, Takeda Pharmaceutical Co. Ltd., Kissei Pharmaceutical Co. Ltd., Mitsubishi‐Tanabe Pharma, Abbvie, EA Pharma Co., Ltd., KYORIN Pharmaceutical, ZERIA Pharmaceutical, AstraZeneka, Daiichi‐Sankyo, Mochida Pharmaceutical, JIMRO, Astellas Pharma Inc. and Nippon Kayaku Co., Ltd.
Shinji Okabayashi: None known
Yuki Kataoka: None known
Yasushi Tsujimoto: None known
Toshi A Furukawa has received lecture fees from Meiji, Mitsubishi‐Tanabe, MSD and Pfizer. He has received research support from Mitsubishi‐Tanabe.
Norio Watanabe: None known
New
References
References to studies included in this review
Ogata 2009 {published and unpublished data}
- NCT00291668. Clinical study of CDP870/Certolizumab Pegol in patients with active Crohn's disease. https://clinicaltrials.gov/ct2/show/study/NCT00291668.
- Ogata H, Ito H, Motoya S, Takazoe M, Suzuki Y, Matsumoto T, et al. Certolizumab pegol is effective at inducing and maintaining response and remission in Japanese patients with Crohn’s disease: Results from induction and maintenance studies. Gut. 2009; Vol. 58:A170.
Sandborn 2007 {published and unpublished data}
- NCT00152490. A study to test the effect of CDP870 in the treatment of Crohn's disease over 26 weeks, comparing CDP870 to a dummy drug (placebo). https://clinicaltrials.gov/ct2/show/NCT00152490.
- Sandborn WJ, Feagan BG, Honiball PJ, Rutgeerts P, Mason D, Bloomfield R, et al. Certolizumab pegol for the treatment of Crohn's disease. New England Journal of Medicine 2007;357(3):228‐38. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Feagan BG, Stoinov S, Honiball PJ, Rutgeerts P, McColm JA. Certolizumab pegol administered subcutaneously is effective and well tolerated in patients with active Crohn's disease: results from a 26‐week, placebo controlled phase III study (PRECiSE 1). Gastroenterology. 2006; Vol. 130:A107‐8.
Sandborn 2011 {published and unpublished data}
- EUCTR‐001913‐41. A phase IIIb, multinational, randomized, double‐blind, placebo‐controlled trial to assess the efficacy and safety of certolizumab pegol, a pegylated Fab' fragment of a humanized anti‐TNF‐alpha monoclonal antibody, administered subcutaneously at weeks 0, 2 and 4 in subjects with moderately to severely active Crohn’s disease. https://www.clinicaltrialsregister.eu/ctr‐search/search?query=2007‐001913‐41.
- NCT00552058. Study to evaluate efficacy and safety of certolizumab pegol for induction of remission in patients with Crohn's disease. https://ClinicalTrials.gov/show/NCT00552058.
- Sandborn W, Schreiber S, Feagan B, Rutgeerts P, Younes Z, Bloomfield R, et al. Induction therapy with certolizumab pegol in patients with moderate to severe Crohn's disease: a placebo‐controlled trial. American Journal of Gastroenterology. 2010; Vol. 105:S419.
- Sandborn WJ, Schreiber S, Feagan BG, Rutgeerts P, Younes ZH, Bloomfield R, et al. Certolizumab pegol for active Crohn's disease: a placebo‐controlled, randomized trial. Clinical Gastroenterology & Hepatology 2011;9(8):670‐8. [DOI] [PubMed] [Google Scholar]
Schreiber 2005 {published and unpublished data}
- Rutgeerts P, Schreiber S, Feagan B, Keininger DL, O'Neil L, Fedorak RN. Certolizumab pegol, a monthly subcutaneously administered Fc‐free anti‐TNFalpha, improves health‐related quality of life in patients with moderate to severe Crohn's disease. International Journal of Colorectal Disease 2008;23(3):289‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S, Rutgeerts P, Fedorak R, Khaliq‐Kareemi M, Kamm MA, Patel J, et al. CDP870, a humanized anti‐TNF antibody fragment, induces clinical response with remission in patients with active Crohn's disease (CD). Gastroenterology. 2003; Vol. 124:A61.
- Schreiber S, Rutgeerts P, Fedorak RN, Khaliq‐Kareemi M, Kamm MA, Boivin M, et al. A randomized, placebo‐controlled trial of certolizumab pegol (CDP870) for treatment of Crohn's disease. Gastroenterology 2005;129(3):807‐18. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Colombel 2008 {published data only}
- Colombel JF, Hebuterne X. Endoscopic mucosal improvement in patients with active Crohn's disease treated with certolizumab pegol: first results of the music clinical trial. American Journal of Gastroenterology. 2008; Vol. 103:S432.
- NCT00297648. Mucosal healing study in Crohn's disease (CD) (MUSIC). https://clinicaltrials.gov/ct2/show/NCT00297648.
NCT00152425 {published data only}
- NCT00152425. Study to test the effect of CDP870 in the treatment of Crohn's disease over 26 weeks, comparing CDP870 to a dummy drug (placebo), following 3 doses of active drug (CDP870). https://clinicaltrials.gov/ct2/show/NCT00152425.
NCT00307931 {published data only}
- NCT00307931. Certolizumab pegol for treatment of adult Greek patients with moderate to severe Crohn's disease who failed infliximab. https://clinicaltrials.gov/ct2/show/NCT00307931.
NCT00349752 {published data only}
- NCT00349752. Corticosteroid sparing effect of certolizumab in Crohn's disease (COSPAR1). https://clinicaltrials.gov/ct2/show/NCT00349752.
NCT00354367 {published data only}
- NCT00354367. Evaluate efficacy of certolizumab in Crohn's patients with draining fistulas. https://clinicaltrials.gov/ct2/show/NCT00354367.
NCT01024647 {published data only}
- NCT01024647. Optimizing cimzia in Crohn's patients. https://clinicaltrials.gov/ct2/show/NCT01024647.
NCT01053559 {published data only}
- NCT01053559. Assessment of small bowel healing in Crohn's disease patients treated with cimzia using wireless capsule endoscopy. https://clinicaltrials.gov/ct2/show/NCT01053559.
NCT01582568 {published data only}
- NCT01582568. EUS evaluation of perianal and peri‐rectal fistulizing Crohn's disease with CERTOLIZUMAB treatment (EUS). https://clinicaltrials.gov/ct2/show/NCT01582568.
Sandborn 2008 {published data only}
- Sandborn W J. Certolizumab pegol for moderate‐to‐severe Crohn's disease. Gastroenterology & Hepatology 2008;4(7):467‐8. [PMC free article] [PubMed] [Google Scholar]
Sandborn 2012 {published data only}
- Sandborn W.J, Younes ZH, Pierre‐Louis B, D'Haens GR. Efficacy of certolizumab pegol induction and maintenance therapy in patients with Crohn's disease not in remission following active or placebo induction. Gastroenterology. 2012; Vol. 142:S565.
Vermeire 2008 {published data only}
- NCT00308581. Certolizumab in Crohn's Disease Patients With Loss of Response or Intolerance to Infliximab. https://clinicaltrials.gov/ct2/show/NCT00308581.
- Vermeire S. Abreu MT. D'Haens G, Colombel JF, Mitchev K, Fedorak R, et al. Efficacy and safety of certolizumab pegol in patients with active Crohn’s disease who previously lost response or were intolerant to infliximab: open‐label induction preliminary results of the Welcome study. Gastroenterology. 2008; Vol. 134:A67‐68.
Winter 2004 {published data only}
- Winter TA, Wright J, Ghosh S, Jahnsen J, Innes A, Round P. Intravenous CDP870, a PEGylated Fab' fragment of a humanized antitumour necrosis factor antibody, in patients with moderate‐to‐severe Crohn's disease: an exploratory study. Alimentary Pharmacology and Therapeutics 2004;20:1337‐46. [DOI] [PubMed] [Google Scholar]
Additional references
Baumgart 2012
- Baumgart DC, Sandborn WJ. Crohn's disease. Lancet 2012;380:1590‐605. [DOI] [PubMed] [Google Scholar]
Best 1976
- Best WR, Becktel JM, Singleton JW, Kern F Jr. Development of a Crohn's disease activity index. National Cooperative Crohn's Disease Study. Gastroenterology 1976;70:439‐44. [PubMed] [Google Scholar]
Canavan 2007
- Canavan C, Abrams KR, Mayberry JF. Meta‐analysis: mortality in Crohn's disease. Alimentary Pharmacology and Therapeutics 2007;25:861‐70. [DOI] [PubMed] [Google Scholar]
Cosnes 2011
- Cosnes J, Gower‐Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011;140:1785‐94. [DOI] [PubMed] [Google Scholar]
Daperno 2004
- Daperno M, D'Haens G, Assche G, Baert F, Bulois P, Maunoury V, et al. Development and validation of a new, simplified endoscopic activity score for Crohn's disease: the SES‐CD. Gastrointestinal Endoscopy 2004;60:505‐12. [DOI] [PubMed] [Google Scholar]
Ford 2011
- Ford AC, Sandborn WJ, Khan KJ, Hanauer SB, Talley NJ, Moayyedi P. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta‐analysis. American Journal of Gastroenterology 2011;106:644‐59. [DOI] [PubMed] [Google Scholar]
Frolkis 2014
- Frolkis AD, Lipton DS, Fiest KM, Negron ME, Dykeman J, deBruyn J, et al. Cumulative incidence of second intestinal resection in Crohn's disease: a systematic review and meta‐analysis of population‐based studies. American Journal of Gastroenterology 2014;109:1739‐48. [DOI] [PubMed] [Google Scholar]
Gomollon 2017
- Gomollon F, Dignass A, Annese V, Tilg H, Assche G, Lindsay JO, et al. 3rd European evidence‐based consensus on the diagnosis and management of Crohn's Disease 2016: Part 1: diagnosis and medical management. Journal of Crohn's and Colitis 2017;11:3‐25. [DOI] [PubMed] [Google Scholar]
Guyatt 1989
- Guyatt G, Mitchell A, Irvine E J, Singer J, Williams N, Goodacre R, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology 1989;96:804‐10. [PubMed] [Google Scholar]
Harvey 1980
- Harvey RF, Bradshaw JM. A simple index of Crohn's‐disease activity. Lancet 1980;1:514. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assesing risk of bias in included studies. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Kawalec 2013
- Kawalec P, Mikrut A, Wisniewska N, Pilc A. Tumor necrosis factor‐alpha antibodies (infliximab, adalimumab and certolizumab) in Crohn's disease: systematic review and meta‐analysis. Archives of Medical Science 2013;9:765‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mary 1989
- Mary JY, Modigliani R. Development and validation of an endoscopic index of the severity for Crohn's disease: a prospective multicentre study. Groupe d'Etudes Therapeutiques des Affections Inflammatoires du Tube Digestif (GETAID). Gut 1989;30:983‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Molodecky 2012
- Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012;142:46‐54. [DOI] [PubMed] [Google Scholar]
Nesbitt 2007
- Nesbitt A, Fossati G, Bergin M, Stephens P, Stephens S, Foulkes R, et al. Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti‐tumor necrosis factor alpha agents. Inflammatory Bowel Diseases 2007;13:1323‐32. [DOI] [PubMed] [Google Scholar]
Nielsen 2013
- Nielsen OH, Ainsworth MA. Tumor necrosis factor inhibitors for inflammatory bowel disease. New England Journal of Medicine 2013;369:754‐62. [DOI] [PubMed] [Google Scholar]
Olesen 2016
- Olesen CM, Coskun M, Peyrin‐Biroulet L, Nielsen OH. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel diseases. Pharmacology and Therapeutics 2016;159:110‐9. [DOI] [PubMed] [Google Scholar]
Peyrin‐Biroulet 2010
- Peyrin‐Biroulet L, Loftus E V Jr, Colombel JF, Sandborn WJ. The natural history of adult Crohn's disease in population‐based cohorts. American Journal of Gastroenterology 2010;105:289‐97. [DOI] [PubMed] [Google Scholar]
Rutgeerts 1990
- Rutgeerts P, Geboes K, Vantrappen G, Beyls J, Kerremans R, Hiele M. Predictability of the postoperative course of Crohn's disease. Gastroenterology 1990;99:956‐63. [DOI] [PubMed] [Google Scholar]
Schreiber 2011
- Schreiber S. Certolizumab pegol for the treatment of Crohn's disease. Therapeutic Advances in Gastroenterology 2011;4(6):375‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Shao 2009
- Shao L M, Chen M Y, Cai J T. Meta‐analysis: the efficacy and safety of certolizumab pegol in Crohn's disease. Alimentary Pharmacology and Therapeutics 2009;29:605‐14. [DOI] [PubMed] [Google Scholar]
Talley 2011
- Talley N J, Abreu M T, Achkar J P, Bernstein C N, Dubinsky M C, Hanauer S B, et al. An evidence‐based systematic review on medical therapies for inflammatory bowel disease. American Journal of Gastroenterology 2011;106(Suppl 1):S2‐25; quiz S26. [DOI] [PubMed] [Google Scholar]
Terdiman 2013
- Terdiman JP, Gruss CB, Heidelbaugh JJ, Sultan S, Falck‐Ytter YT, Practice AGA Institute Clinical, et al. American Gastroenterological Association Institute guideline on the use of thiopurines, methotrexate, and anti‐TNF‐alpha biologic drugs for the induction and maintenance of remission in inflammatory Crohn's disease. Gastroenterology 2013;145:1459‐63. [DOI] [PubMed] [Google Scholar]
Torres 2016
- Torres J, Mehandru S, Colombel JF, Peyrin‐Biroulet L. Crohn's disease. Lancet 2016;389:1741‐55. [DOI] [PubMed] [Google Scholar]
Vermeire 2010
- Vermeire S, Schreiber S, Sandborn W J, Dubois C, Rutgeerts P. Correlation between the Crohn's disease activity and Harvey‐Bradshaw indices in assessing Crohn's disease severity. Clinical Gastroenterology and Hepatology 2010;8:357‐63. [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36‐item short‐form health survey (SF‐36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473‐83. [PubMed] [Google Scholar]