

Abstract
Colorectal cancer (CRC) is one of the most frequent, malignant gastrointestinal tumors, and strategies and effectiveness of current therapy are limited. A series of benzimidazole-isoquinolinone derivatives (BIDs) was synthesized and screened to identify novel scaffolds for CRC. Of the compounds evaluated, 7g exhibited the most promising anti-cancer properties. Employing two CRC cell lines, SW620 and HT29, 7g was found to suppress growth and proliferation of the cell lines at a concentration of ~ 20 μM. Treatment followed an increase in G2/M cell cycle arrest, which was attributed to cyclin B1 and cyclin-dependent kinase 1 (CDK1) signaling deficiencies with simultaneous enhancement in p21 and p53 activity. In addition, mitochondrial-mediated apoptosis was induced in CRC cells. Interestingly, 7g decreased phosphorylated AKT, mTOR and 4E-BP1 levels, while promoting the expression/stability of PTEN. Since PTEN controls input into the PI3K/AKT/mTOR pathway, anti-proliferative effects can be attributed to PTEN-mediated tumor suppression. Collectively, these results suggest that BIDs exert antitumor activity in CRC by impairing PI3K/AKT/mTOR signaling. Against a small kinase panel, 7g exhibited low affinity at 5 μM suggesting anticancer properties likely stem through a non-kinase mechanism. Because of the novelty of BIDs, the structure can serve as a lead scaffold to design new CRC therapies.
Keywords: Colorectal cancer, Benzimidazole, Isoquinolinones, Cell-cycle arrest, Apoptosis
1. Introduction
Human colorectal cancer (CRC) is the most predominant digestive malignant tumor in the world and accounts for approximately 10% of all tumors.1 Metastatic CRC is the fourth leading cause of cancer-related mortality, which exceeds 600,000 deaths annually.2 Although mortality has decreased from advances in screening, early detection, and innovative treatment strategies, relapses from treatment-resistant CRC remain frequent.3,4 Therefore, it is imperative to further study the mechanisms governing CRC progression and resistance and identify tractable therapies that can further reduce CRC morbidity and mortality.
Nitrogenous heterocycles are prolific in medicinal and organic chemistry.5–7 Exploring fast and expeditious routes to generate diverse, nitrogenous heterocycles is paramount in drug discovery. Due to distinct and compelling therapeutic properties, natural and synthetic benzimidazole and isoquinolinone derivatives have been widely investigated in medicinal and organic chemistry.8–10 Benzimidazole-isoquinolinone derivatives have been synthesized as potential inhibitors of phosphodiesterase 4 (PDE4) employing an innovative amberlyst-15 catalyst.11 Similar derivatives were developed with antimicrobial activity against Trypanosoma cruzi (T. cruzi) epimastigotes (GI50 value is 0.5 μM).12 These compounds also exhibited anti-inflammatory properties and anticancer activity against ovary (IGROV-1), breast (MCF-7) and CNS (SF-295) human cancer cell lines.13
Multicomponent reactions (MCRs) have attracted considerable attention as a powerful tool to access highly functionalized heterocyclic skeletons of chemical and biomedical importance.14–16 Particularly, the Ugi reaction is well known as a versatile and highly efficient synthetic tool with extremely green synthetic methods, such as mild reaction conditions, no metal catalysts, short reaction times, high yields, and atom economy.17 We have an interest in employing the Ugi cascade reaction for the formation of new C—N/C—C bonds to construct heterocycles for drug discovery efforts.18,19 In continuation of our work in search of potent anticancer scaffolds, a series of fused benzimidazole-isoquinolinone derivatives (BIDs) were synthesized and screened for CRC anticancer activity, which we report herein.
The PI3K/AKT/mTOR signaling pathway is a very important signal transduction cascade engaged in a wide range of biological processes, such as cell survival, proliferation, apoptosis, cellular metabolism and the stress response.20,21 This signaling pathway is of particular importance to CRC because 20% of all CRCs harbor a mutation in the PIK3CA gene. The mutation constitutively activates the PI3K/AKT/mTOR pathway causing an increase in pro-survival gene expression.22 Previous studies discovered that activation of the PI3K/AKT signaling-axis inhibits colorectal cancer cell apoptosis.23 On the contrary, inhibiting the activation of PI3K/AKT and its downstream target mTOR results in the initiation of apoptosis and autophagy. Furthermore, previous studies suggest that small molecule inhibition of the PI3K/AKT/mTOR signaling pathway could be utilized as a therapeutic strategy to treat CRC.24,25
In this present study, we first determined the effect of BIDs on the growth of two colorectal cancer cell lines, SW620 and HT29. We found that compound 7g could inhibit the growth of SW620 and HT29 cells in a time and concentration dependent manner. Furthermore, we found compound 7g induced G2/M cell cycle arrest and apoptosis in both SW620 and HT29 cells. To understand the mechanism behind growth inhibition in CRC, we examined PI3K/AKT/mTOR signaling and found that activity of AKT was inhibited in both SW620 and HT29 cells. In addition, the phosphorylation level of mTOR decreased as well as activity of downstream signaling partners, such as 4E-BP, in a concentration-dependent manner. Taken together, this data suggests compound 7g inhibited the growth of CRC and induced apoptosis through a PI3K/AKT/mTOR dependent mechanism.
2. Results and discussion
2.1. Results
2.1.1. Chemistry
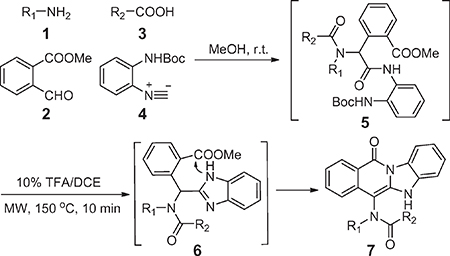
We conducted an Ugi four component reaction (U-4CR) using an amine 1, methyl 2-formylbenzoate 2, carboxylic acid 3 and isonitrile 4 in methanol. After stirring overnight at room temperature, the solvent was removed under a stream of nitrogen, and the crude Ugi product 5 was deprotected and cyclized. Intermediate 6 was used without purification and reacted under microwave irradiation using 10% TFA/DCE at 150 °C for 15 min.19 A series of fused benzimidazole-isoquinolinones (BIDs) were obtained using these conditions. The results indicate that the Ugi product can be reacted crude without significantly impacting deprotection and cyclization yields. This two-step, one-pot procedure is mild and convenient and affords the desired BIDs in yields of 52–82% (Table 1).
Table 1.
Structures, yields and chemical properties of fused benzoimidazole-isoquinolines 7a–n.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Comp. | Yield (%)a | CLogP | tPSA |
| 1 | Cyclopropyl | 2-thienyl | 7a | 82 | 3.799 | 52.65 |
| 2 | iBu | 2-thienyl | 7b | 52 | 4.8955 | 52.65 |
| 3 | iBu | 2-furanyl | 7c | 72 | 4.0715 | 61.88 |
| 4 | 2-BrC6H4 | 2-furanyl | 7d | 63 | 7.5791 | 52.65 |
| 5 | 2-BrC6H4 | Acetyl | 7e | 75 | 4.672 | 52.65 |
| 6 | 2-BrC6H4 | 3-pyridyl | 7f | 71 | 5.5395 | 65.01 |
| 7 | iBu | 3-pyridyl | 7g | 72 | 3.7945 | 65.01 |
| 8 | iBu | 2-COOHC6H4 | 7h | 76 | 4.9034 | 89.95 |
| 9 | Bzl | Ph | 7i | 62 | 6.0965 | 52.65 |
| 10 | Ph | Ph | 7j | 71 | 6.2375 | 52.65 |
| 11 | Phenethyl | 4-pyridyl | 7k | 67 | 4.4345 | 65.01 |
| 12 | H | Ph | 7l | 66 | 3.8747 | 61.44 |
| 13 | 2-BrC6H4 | 2-furanyl | 7m | 76 | 5.8165 | 61.88 |
| 14 | 2-BrC6H4 | methyl | 7n | 78 | 3.5115 | 52.65 |
Isolated yield (%) from the one-pot procedure.
2.1.2. Compound 7g inhibits proliferation and viability of colorectal cancer cells in time and concentration dependent manners
In order to evaluate the potential of BIDs to inhibit CRC growth, growth inhibition (GI50) values were determined in two CRC cell lines. As shown in Table 2, compound 7g obtained the best growth inhibition (SW620 and HT29 are 23.78 and 24.13 μM, respectively) and was selected for further studies. SW620 and HT29 cells were treated with compound 7g at different concentrations (0, 12.5, 25, 50, 100, 200 μM) and evaluated for viability at different time points (12, 24, 48 and 72 h). The results indicate that 7g inhibits the growth of SW620 and HT29 cells in a time and concentration dependent manner (Fig. 1A).
Table 2.
GI50 of BIDs against colorectal cancer cell lines.
| Entry | Comp. | GI50 (μM)a |
|
|---|---|---|---|
| SW620 | HT29 | ||
| 1 | 7a | 47.60 ± 3.76 | 51.30 ± 5.29 |
| 2 | 7b | 44.05 ± 7.37 | 49.52 ± 6.86 |
| 3 | 7c | 53.46 ± 4.63 | 52.80 ± 5.90 |
| 4 | 7d | 46.03 ± 4.61 | 43.73 ± 7.59 |
| 5 | 7e | 46.89 ± 7.16 | 57.20 ± 5.25 |
| 6 | 7f | 57.87 ± 5.25 | 61.27 ± 7.42 |
| 7 | 7g | 23.78 ± 4.06 | 24.13 ± 3.88 |
| 8 | 7h | 52.70 ± 5.94 | 59.80 ± 6.65 |
| 9 | 7i | 69.70 ± 7.14 | 74.30 ± 4.32 |
| 10 | 7j | > 100 | > 100 |
| 11 | 7k | 48.90 ± 6.06 | 52.30 ± 5.82 |
| 12 | 7l | > 100 | > 100 |
| 13 | 7m | > 100 | > 100 |
| 14 | 7n | > 100 | > 100 |
Each GI50 value was calculated from 3 independent experiments conducted in sextuplicate.
Fig. 1.

Compound 7g decreased proliferation and viability of colorectal cancer cell lines SW620 and HT29. (A) Cell proliferation and GI50 were assessed by MTT. SW620 and HT29 cells were treated with compound 7g at different concentrations (0, 12.5, 25, 50, 100, 200 μM) for 12 h, 24 h, 48 h, 72 h respectively, and then cell viability and GI50 were measured and calculated. Data are the mean ± SD of three independent experiments, and each experiment was conducted in sextuplicate. (B, C) BrdU immunofluorescence staining was conducted in SW620 and HT29 cell lines after treatment with compound 7g. The nuclei of SW620 and HT29 were exposed to compound 7g at 0, 25, 50, and 75 μM for 48 h, and then stained with BrdU (red fluorescence), primary mouse antibody against BrdU and anti-mouse IgG secondary antibody. Fluorescence microscopy was conducted after incubating with DAPI (blue fluorescence). The bar length for the group as shown in the panels is 100 μm. (D) The percentage of positive BrdU from panel B and C were analyzed, respectively. Every value was the average of three independent experiments. Data were shown as the mean ± SD. * = p < 0.05; ** = p < 0.01. (E) The effects of compound 7g on the clonogenicity of SW620 and HT29 were analyzed using the colony formation assay.
To investigate the effect of proliferative inhibition of compound 7g on SW620 and HT29, immunofluorescent analysis was conducted. As shown in Fig. 1B–D, immunofluorescence confirmed that 7g inhibited the proliferation of SW620 and HT29 in a concentration-dependent manner. Subsequently, 7g was evaluated in a colony formation assay to determine effects on clonogenicity of SW620 and HT29 cells. Colony size and numbers were dramatically reduced after exposure to compound 7g for two weeks (Fig. 1E).
2.1.3. Compound 7g induces G2/M cell cycle arrest of human colorectal cancer cells by decreasing cyclin B1 and CDK1
In order to explore the effects on cell cycle progression, SW620 and HT29 cells were treated with compound 7g at four concentrations (0, 25, 50, 75 μM) for 48 h and then analyzed by flow cytometry. The average G2/M proportions of SW620 and HT29 increased in a concentration-dependent manner. However, there was no significant difference in either cell line at lower concentrations. These results indicate that compound 7g can induce cell cycle arrest of CRC cells at the G2/M checkpoint.
To investigate the relationship between 7g-mediated cell cycle arrest and cycle-related proteins, levels of cyclin B1, CDK1, p21 and p53 were analyzed by western blot. As shown in Fig. 2C, levels of cyclin B1 and CDK1 decreased in both SW620 and HT29 cells, while p21 and p53 increased. Furthermore, expression levels of cyclin B1, CDK1, p21 and p53 changed in a concentration-dependent manner. Collectively, our data suggests that compound 7g induces cell cycle arrest at the G2/M checkpoint by altering expression or stability of cell cycle-related proteins.
Fig. 2.

Compound 7g induces SW620 and HT29 cell cycle arrest at the G2/M checkpoint. (A) Cell cycle distribution was detected using flow cytometry in SW620 and HT29 cells. Both lines were treated with compound 7g for 48 h at different concentrations (0, 25, 50, and 75 μM). Cells were collected and treated with PI and RNase. (B) Quantification of SW620 and HT29 cells at various phases of the cell cycle. Each point represents the average of three independent experiments. (C) Western blotting was used to analyze the effect of compound 7g on expression of cell cycle-related proteins. SW620 and HT29 cells were treated with compound 7g for 48 h at different concentrations (0, 25, 50, and 75 μM) and were harvested, and whole protein lysates were extracted. The expression of cell cycle-related proteins (p53, p21, cyclin B1 and CDK1) was analyzed by western blot. Tubulin was used as a loading control. The relative density of cell cycle related-proteins was analyzed by the Tanon Gis Software. Data is shown as the mean ± SD. * = p < 0.05; ** = p < 0.01.
2.1.4. Compound 7g induces apoptosis by regulating PARP and BCL-2 in human CRC cells
High expression of p53 induces apoptosis of tumor cells, and expression levels of p53 increased after treatment with 7g. Hoechst staining was utilized to observe apoptotic cells in CRC cell lines treated with compound 7g. As shown in Fig. 3A, apoptotic cells were detected and the number of apoptosis bodies increased in a concentration-dependent manner.
Fig. 3.

Effect of compound 7g on apoptosis of SW620 and HT29 cells. (A) Fluorescence microscopy images of Hoechst 33342. SW620 and HT29 cells were treated with compound 7g for 48 h at 0, 25, 50, and 75 μM, and then analyzed by Hoechst 33,342 staining and fluorescence microscopy. The red arrow indicates an apoptotic cell and chromatin condensation. (B) SW620 and HT29 cells treated with compound 7g were stained with annexin V/PI. The apoptosis rate of cells treated with compound 7g for 48 h at 0, 25, 50, and 75 μM was detected by flow cytometry. Each point represents the average of three independent experiments. Data were shown as the mean ± SD. (C) The effect of compound 7g on the expression level of apoptosis-related proteins. SW620 and HT29 cells were treated with compound 7g for 48 h at four concentrations (0, 25, 50, and 75 μM). Then, the contents of apoptosis-related proteins were measured using western blot. In addition, the relative density of apoptosis related-proteins was also analyzed by the Tanon Gis Software. Every value is the average of three independent experiments. Data are shown as the mean ± SD. * = p < 0.05; ** = p < 0.01.
The ratio of apoptosis was determined in SW620 and HT29 cells using flow cytometry after treatment with compound 7g (0, 25, 50, 75 μM) for 48 h. As shown in Fig. 3B, the percentage of SW620 and HT29 cells undergoing apoptosis increased in a concentration-dependent manner.
To further investigate the effect of compound 7g on the levels of apoptosis-related proteins, protein levels of PARP, BCL-2, and MCL-1 were determined by western blot. As shown in Fig. 3C, protein levels of BCL-2 and MCL-1 decreased in both SW620 and HT29 cells after treatment with compound 7g in a concentration-dependent manner. Furthermore, PARP activation significantly increased in a concentration-dependent manner in both SW620 and HT29 cells. This data suggests that compound 7g can regulate activity of apoptosis-related proteins to induce apoptosis in human CRC cells.
2.1.5. Compound 7g suppresses SW620 and HT29 cell growth via inhibition of the PI3K/AKT/mTOR pathway
The classical PI3K/AKT signaling pathway is ubiquitous, playing a critical role in the regulation of cell proliferation, differentiation, survival, cell-cycle, apoptosis, metabolism and the stress response.26 In order to analyze the potential antiproliferative mechanism of 7g in CRC, we investigated the PI3K/AKT/mTOR pathway, which is frequently hijacked by CRC. SW620 and HT29 cells were treated with different concentrations of compound 7g (0, 25, 50, 75 μM) for 48 h. Then, levels of PTEN, AKT, phospho-AKT, mTOR, phospho-mTOR, 4E-BP1 and phospho-4E-BP1 in whole cell lysates were examined by western blot. As show in Fig. 4, treatment with compound 7g decreased p-AKT, p-mTOR and p-4E-BP1 levels in SW620 and HT29 cells but hadn’t effect on total expression levels of AKT, mTOR and 4E-BP1.
Fig. 4.

Effect of compound 7g on the PI3K/AKT/mTOR signaling pathway. (A) SW620 and HT29 cells were treated with compound 7g for 48 h at different concentrations (0, 25, 50, and 75 μM). Cells were collected and harvested for protein lysates. The expression levels of PI3K/AKT/mTOR signaling pathway related proteins (PTEN, p-AKT, AKT, p-mTOR, mTOR, 4E-BP1, p-4E-BP1) were analyzed by western blot. Tubulin was used as a loading control. (B) The relative density of PI3K/AKT/mTOR signing pathway related-proteins was analyzed by the Tannon Gis Software. Each data point is the average of three independent experiments. Data were shown as the mean ± SD. * = p < 0.05; ** = p < 0.1.
In order to confirm the specificity of compound 7g in the CRC cell lines which is dependent on the mTOR pathway, the immortalized colorectal FHC cell lines were explored. It was found that GI50 for compound 7g was > 200 μM (Supplemental Fig. 1). The result suggested that compound 7g exhibited anti-proliferation activity in the CRC cell lines likely through the inhibition of the mTOR pathway.
Interestingly, PTEN levels increased in treated cells when compared to control, which directly impacts the PI3K/AKT/mTOR pathway.19 The results suggest that PTEN levels increase in a concentration-dependent manner in both SW620 and HT29 cell lines (Fig. 4). Further, both SW620 and HT29 CRC lines contain wild-type PTEN with low endogenous expression. This suggests that 7g may impact input into the PI3K/AKT/mTOR pathway by increasing PTEN stability or expression.
PTEN activity and stability can also be impacted by protein kinase activity. We therefore screened 7g against a 97-kinase panel representative of major kinase clusters to determine kinome activity. It was found that 7g had low affinity for kinase proteins with Kd values > 5.0 μM for all tested kinases (Supplemental Table 1). Therefore, it is unlikely 7g elicits anticancer effects through a kinase mechanism.
2.2. Discussion
Benzimidazole scaffolds have attracted significant attention for their anticancer properties. Recent studies found that benzimidazole molecules can cause cell-cycle arrest or promote apoptosis by modulating kinase signaling cascades. Hence, a novel methodology was developed for the construction of a diverse suite of fused benzimidazole-isoquinolinones using a unique one-pot Ugi/deprotection/cyclization (UDC) protocol. With mild reaction conditions and a simple protocol, commercially available starting materials can be transformed into diverse, drug-like fused benzimidazole-isoquinolinones that have broad applications for synthesis and medicinal chemistry. In our study, we synthesized 14 novel benzimidazole-isoquinolinone derivatives, with compound 7g exhibiting the most promising activity against CRC lines with GI50 values of 23.78 μM (SW620) and 24.13 μM (HT29), respectively. Comparing compounds 7g to 7b and 7c, a 5 membered ring system at the R2 position did not improve activity. When comparing compound 7g to 7f, the bulky R1 group on 7f also hindered activity. Therefore, compound 7g was selected for further studies because it obtained greatest antiproliferative activity.
Treatment with 7g halted cell cycle progression at the G2/M checkpoint, which ensures only healthy cells enter replication.27,28 G2/M arrest is an important cell cycle checkpoint to stop cells with DNA damage from entering mitosis (M-phase), which triggers DNA repair or apoptosis.29 Thus, G2/M arrest is a frequent consequence of anti-cancer drugs that interfere with DNA replication. In the S phase of the cell cycle, cyclin B1 is generated and subsequently degraded in mitosis, and the cyclin B1-CDK1 complex controls the S phase transition into mitosis.30 Anti-cancer compounds can cause G2/M arrest by interfering with the cyclin B1-CDK1 complex.31 Consistent with these findings, immunoblot results indicated protein levels of cyclin B1 and CDK1 decreased in SW620 and HT29 cells treated with 7g. This data is consistent with flow cytometry and reveal that CRC cells fail to progress from G2 to mitosis after treatment, suggesting inference with the cyclin B1-CDK1 complex as a mechanism governing G2/M arrest.
Apoptosis is the most vital mechanism of anticancer drug-mediated cell death and can be induced by mitochondria signaling. The mitochondrial pathway is regulated by a diverse set of proteins, which are divided into pro-apoptotic and anti-apoptotic effectors.32–34 The pro-apoptotic effectors include Bax and Bak, while the anti-apoptotic effectors include BCL-2 and MCL-1.35 Activation is induced by the release of pro-apoptotic factors, such as cytochrome c, from the mitochondria into the cytosol. The BCL-2 family of proteins, which regulate cytochrome c release and caspases activation, are thought to bind to the mitochondrial outer membrane and regulate permeability.36 In our study, we found that protein levels of BCL-2 and MCL-1 decreased in SW620 and HT29 cells treated with compound 7g. Variation in BCL-2 and MCL-1 protein levels account for the increase in apoptotic cells. Caspases are the major proteins involved in the stimulation of the apoptotic process. The activation of caspases is considered a vital step in the induction of drug induced apoptosis as well as cleavage of poly ADP-ribose polymerase (PARP) by caspase-3.37 To determine if the activation of PARP was a result of 7g treatment, activity of PARP was determined as a function of 7g treatment using western blot. Our data demonstrate that treatment of SW620 and HT29 cells induced significant cleavage of PARP. Apoptosis also induces various morphological and biochemical characteristics. One of the remarkable hallmarks of apoptosis is that phospholipid phosphatidylserine (PS) externalizes from the inner to the outer layer of the plasma membrane to induce phagocytes.38 Analysis of treated SW620 and HT29 cells by annexin V/PI double-dyeing indicated that compound 7g induced a considerable level of cell death through the apoptotic pathway. These results suggest that 7g induces apoptosis through the mitochondria to reduce CRC proliferation.
Previous research has reported that the pathogenesis of CRC is a complex, multiple-step process.39 The PI3K/AKT/mTOR pathway is crucial for signal transduction and controls proliferation, apoptosis, migration, cellular metabolism and the stress response.20,21 Activation of the PI3K/AKT signaling axis requires PI3K to catalyze the phosphorylation of phosphatidylinositol diphosphate (PIP2) to phosphatidylinositol triphosphate (PIP3). Then, PIP3 recruits AKT to the lipid raft compartments of the plasma membrane. This process accelerates the subsequent phosphorylation of AKT by phosphoinositide-dependent kinase (PDK) 1 and PDK2, respectively.40 PTEN, a vital tumor suppressor gene and phosphatase, is a negative regulator of AKT implicated in a wide range of human malignancies. The PI3K-AKT signaling pathway is deactivated by PTEN through dephosphorylation of its main substrates.41,42 Furthermore, the PI3K/AKT pathway is constitutively activated in a large percentage of CRCs.22 Thus, in order to determine if compound 7g inhibited SW620 and HT29 proliferation through the PI3K/AKT pathway, we analyzed phosphorylation and protein levels of AKT and PTEN (Guri et al., 2016).43 The results showed that the PTEN levels increased in SW620 and HT29 cells treated with compound 7g, whereas activation of AKT, based on phosphorylation, decreased correspondingly. The mTOR oncogene is a central promotor of cell proliferation, growth, and survival in malignancies.44,45 mTOR is a pre-dominate downstream effector of PI3K/AKT and can be phosphorylated by the PI3K/AKT signaling pathway.46 Thus, we determined the phosphorylation status of mTOR and found phosphorylation declined in both SW620 and HT29 cells treated with compound 7g. In addition, we also analyzed activation of 4E-BP1, which is a downstream effector of mTOR.46 The results indicate that compound 7g inhibited 4E-BP1 phosphorylation in both CRC lines. Collectively, this data suggests compound 7g increases protein levels of PTEN, which likely decrease input into the PI3K/AKT/mTOR signaling pathway.
3. Conclusion
In conclusion, we synthesized 14 novel BIDs using MCR chemistry and 7g was selected for further studies because of activity in SW620 and HT29 CRC lines. The anti-cancer effects of 7g, as well as the possible molecular mechanism, were investigated. The results indicate that compound 7g could inhibit CRC proliferation, growth and survival. Further, 7g halted cell cycle progression at the G2/M checkpoint and induced mitochondrial-mediated apoptosis by suppressing the PI3K/AKT/mTOR signaling cascade. Based on mechanistic studies, 7g increased levels of PTEN, which likely contributes to its antiproliferative effects. Since compound 7g does not strongly inhibit kinases, its antiproliferative activity likely stems through a non-kinase mechanism. From this study, BIDs were identified as potential anticancer drugs that can be employed in the development of novel treatments for CRC.
4. Materials and methods
4.1. General procedure for compound 7 preparation
A solution of aldehyde (1.0 mmol) and amine (1.0 mmol) in MeOH (2 mL) was stirred at room temperature for 10 min in a 10 mL microwave vial. Acid (1.0 mmol) and isonitrile (1.0 mmol) were added separately. The mixture was stirred at room temperature overnight and monitored by TLC. When no isonitrile was present, the solvent was removed under a nitrogen stream. The residue was dissolved in 10% TFA/DCE, sealed and heated in a microwave at 150 °C for 15 min. After, the reaction was cooled to room temperature, the solvent was removed under reduced pressure, diluted with EtOAc (15 mL) and washed with sat. Na2CO3 and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography using a gradient of ethyl acetate/hexane (0–60%) to afford the relative fused benzimidazole-isoquinolinone 7.
4.2. Cell lines and culture
The human colon carcinoma cell lines SW620 and HT29, and normal human colon cell line FHC were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). HT29 cells were cultured in McCoy’s 5a (Gibco, 16600108, USA) medium Supplemented with 10% fetal bovine serum (FBS, Gibco, 10100147, Australia origin), SW620 cells were cultured in high-glucose DMEM (Hyclone, SH30022.01, USA) medium with 10% FBS and FHC were cultured in DMEM:F12 (Gibco, 11330032, USA) medium with 10% FBS, 10 ng/ml cholera toxin, 0.005 mg/ml insulin, 0.005 mg/ml transferrin and 100 ng/ml hydrocortisone. All cells were cultured in an incubator at 37 °C under a humidified atmosphere of 5% CO2.
4.3. MTT assay
The cell proliferation and GI50 value of compound 7g were determined by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT, Beyotime, ST316, Shanghai, China) assay. Firstly, 1 mg compound 7g was dissolved in 60 μL DMSO (Dimethylsulfoxide) to acquire the 40 mM/L mother liquor. Then, the 40 mM/L mother liquor was further diluted into 20 mM, 10 mM, 5 mM and 2.5 mM concentrations of compound 7g which were used for treating cells. 10 μL 40, 20, 10, 5 and 2.5 mM/L compound 7g were taken out and added into 2 mL complete medium to obtain final concentrations of 200, 100, 50, 25 and 12.5 μM, respectively. Furthermore, cells were counted and seeded into 96-well plates at a density of 3 × 103 cells per well containing 200 μL complete medium. After 24 h of pre-incubation, cells were changed into the medium containing compound 7g at concentrations of 0, 12.5, 25, 50, 100, 200 μM and treated for 12, 24, 48 and 72 h. To assess viability, MTT (5 mg/mL) was added to each well to a final concentration of 0.5 mg/mL and was incubated for 4 h. The medium was removed and 200 μL of DMSO was added to each well to dissolve the formazan product. The plate was agitated at 50 rpm/min for 10 min, and then absorbance was measured at 570 nm (Bio-Tek, Winooski, VT, USA). Experiments were completed in triplicate, and growth inhibition curves and GI50 values were prepared using GraphPad Prism 6.
4.4. BrdU assay
The effect of compound 7g on the proliferation of colon cancer cells was also determined by 5-bromo-2′-deoxyuridine (BrdU). Cells were harvested when SW620 and HT29 cells were at 80% confluence and seeded onto 24-well plates at 20,000 cells per well. Cells were treated with compound 7g for 48 h at 0, 25, 50, and 75 μM, and then incubated with 10 μM BrdU (Sigma, B5002–100MG, USA) for 2 h. Cells were washed with phosphate buffered saline (PBS) three times and fixed with paraformaldehyde. Cells were treated with 2 M HCl for 30 min, washed with PBS three times and treated with 10% goat serum for 30 min at 37 °C. Subsequently, cells were incubated overnight at 4 °C with monoclonal rat primary antibody against BrdU (1:2000, ab6326, Abcam, Cabridge, MA, USA). Following, cells were washed with PBS three times and then incubated with anti-mouse IgG secondary antibody (1:2000, Invitrogen, 1606260, USA) for 120 min at room temperature. The nuclear membrane was stained by DAPI (300 nM), washed three times and then analyzed by inverted fluorescence microscopy (OLYMPUS, IX73).
4.5. Colony formation assay
Colorectal cancer cell lines (SW620 and HT29) were seeded onto 6-well plates at a density of 500 cells per well containing 2 mL medium. SW620 and HT29 cells were treated with compound 7g at concentrations of 0, 25, 50, and 75 μM for 48 h, and then incubated for another 14 h without compound 7g. After, cells were washed with PBS twice and fixed with 4% paraformaldehyde for 30 min. The cells were washed twice again, stained with 0.5% crystal violet, and analyzed for formation of colonies (Beyotime, C0121, Shanghai, China).
4.6. Cell cycle analysis
Cell cycle impact of 7g on SW620 and HT29 cells was assessed by flow cytometry. SW620 and HT29 cells grew up to 80% confluence, and then were seeded onto 6-well plates and treated with various concentrations of compound 7g (0, 25, 50, 75 μM). After 48 h treatment, the cells were harvested and washed twice with clod PBS, then centrifuged at 1000 rpm for 5 min. The sample was fixed with 70% ethanol at 4 °C for 24 h. In addition, the tumor cells were washed with cold PBS, resuspended in 200 μL PBS containing 1 μL propidium iodide (PI, 5 mg/mL, Shanghai, ST512, Shanghai, China) and 5 μL RNase A (10 mg/mL, Beyotime, ST578, Shanghai, China), protected from light, and incubated for 30 min at 37 °C. The samples were analyzed by flow cytometry (Becton Dickinson, San Jose, CA).
4.7. Hoechst 33342 staining
Chromatin analysis was completed using Hoechst 33,342 staining (Invitrogen, H1399, USA). SW620 and HT29 cells were harvested and seeded onto 24-well plates, and treated with compound 7g for 48 h. Cells were fixed for 20 min with 4% paraformaldehyde, washed three times with PBS, and then treated with Hoechst 33342. After 15 min incubation at 37 °C, cells were washed three times with PBS and then analyzed via fluorescence microscopy (OLYMPUS, IX73).
4.8. Apoptosis assessment
To evaluate apoptosis of colorectal tumor cells, both SW620 and HT29 cells were treated with PI/annexin V-FITC (annexin-V-fluorescein isothiocyanate) apoptosis kit (Beyotime, C1063, Shanghai, China) according to the manufacturer’s protocol, and then analyzed by flow cytometry. Briefly, the cells were treated with compound 7g at different concentrations (0, 25, 50, 75 μM) for 48 h. Attached cells as well as floating cells were harvested, washed twice with cold PBS, and then stained with PI and annexin V-FITC for 15 min at room temperature. The PI/annexin V-FITC signal was assessed by flow cytometry (Becton Dickinson, Accuri™ C6, USA).
4.9. Western blotting analysis
Protein levels were analyzed by western blotting. Human colorectal cancer cell lines SW620 and HT29 treated with compound 7g for 48 h were collected and lysed in the lysis buffer containing RIPA (Radioimmunoprecipitation, Beyotime, Shanghai, China), a protease inhibitor and a phosphatase inhibitor (Roche, Mannheim, Germany). The concentration of proteins was measured by a BCA (Bicinchoninic acid) kit (Beyotime, P0010, Shanghai, China). Cell lysates (50 μg) were separated by 8–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore Corporation, MA, USA). The membranes were blocked in TBST containing 5% BSA (Beyotime, ST023, Shanghai, China) for 2 h at room temperature and then incubated with primary antibodies (diluted by product specification, CST, USA) overnight at 4 °C. After that, the membranes were washed five times with TBST and then incubated with horseradish peroxidase (HRP)-labeled secondary antibodies (diluted 1:10000, Sigma, USA) for 1 h at room temperature. Subsequently, membranes were washed five times with TBST and then visualized with the ECL western blot detection kit (GE Healthcare, RPN3244, USA) on a Tanon 5200 Imaging System (Tanon Science & Technology Co., Ltd, Shanghai, China).
4.10. Statistical analysis
All data were analyzed with GraphPad Prism 6.0 and expressed as the mean ± standard deviation (SD). Statistical significance of the data were analyzed by the two-tailed independent Student’s t-test, and a p < 0.05 was considered statistically significant. Each experiment was performed in triplicate.
Supplementary Material
Acknowledgments
This work was supported by Chongqing Science and Technology Committee Project of China under grant No. cstc2016jcyjA0534 and cstc2015zdcy-ztzx120003; The grants from Chongqing Education commission Project of China under Grant No. KJZH17129, KJ1501130, KJ1711284 and KJ1711287; Chongqing University of Arts and Science R2015BX03. HL and BF were supported by the grants (NIH 1R01CA194094-010 and 1R01CA197178-01A1). HKL was supported by the grants (NIH R01 CA182424 and R01 CA193813). This work was also supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number “P20 GM109005”.
Footnotes
A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2018.06.010.
References
- [1].Ferlay J, Shin H, Bray F, Forman D, Mathers C, Parkin DM. Int J Cancer. 2010;127:2893–2917. [DOI] [PubMed] [Google Scholar]
- [2].Cao Z-X, Yang Y-T, Yu S, et al. J Ethnopharmacol 2017;202:20–27. [DOI] [PubMed] [Google Scholar]
- [3].Van Custsem E, Nordlinger B, Cervantes A, Group EGW. Ann Oncol. 2010;21:v93–97. [DOI] [PubMed] [Google Scholar]
- [4].Terme M, Pernot S, Marcheteau E, et al. Cancer Res. 2013;73:539–549. [DOI] [PubMed] [Google Scholar]
- [5].Wen L, Sun Q, Zhang H, Li M. Org Biomol Chem. 2013;11:781–786. [DOI] [PubMed] [Google Scholar]
- [6].Athri P, Wilson WD. J Am Chem Soc. 2009;131:7618–7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].George Rosenker KM, Paquette WD, Johnston PA, et al. Bioorg Med Chem. 2015;23:2810–2818. [DOI] [PubMed] [Google Scholar]
- [8].Guillaumel J, Léonce S, Pierré A, et al. Eur J Med Chem. 2006;41:379–386. [DOI] [PubMed] [Google Scholar]
- [9].Tsou H-R, Liu X, Birnberg G, et al. J Med Chem. 2009;52:2289–2310. [DOI] [PubMed] [Google Scholar]
- [10].Lahue BR, Ma Y, Shipps GW Jr, Seghezzi W, Herbst R. Bioorg Med Chem Lett. 2009;19:3405–3409. [DOI] [PubMed] [Google Scholar]
- [11].Sunke R, Adepu R, Kapavarapu R, et al. Chem Commun. 2013;49:3570–3572. [DOI] [PubMed] [Google Scholar]
- [12].Bollini M, José Casal J, Alvarez DE, et al. Bioorg Med Chem. 2009;17:1437–1444. [DOI] [PubMed] [Google Scholar]
- [13].Sondhi SM, Rani R, Singh J, Roy P, Agrawal SK, Saxena AK. Bioorg Med Chem Lett. 2010;20:2306–2310. [DOI] [PubMed] [Google Scholar]
- [14].Tong S, Wang Q, Wang M-X, Zhu J. Angew Chem Int Ed. 2015;54:1293–1297. [DOI] [PubMed] [Google Scholar]
- [15].Zajdlik A, Wang Z, Hickey JL, Aman A, Schimmer AD, Yudin AK. Angew Chem Int Ed. 2013;52:8411–8415. [DOI] [PubMed] [Google Scholar]
- [16].Dömling A, Wang W, Wang K. Chem Rev. 2012;112:3083–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhao W, Huang L, Guan Y, Wulff WD. Angew Chem Int Ed. 2014;53:3436–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen Z-Z, Zhang J, Tang D-Y, Xu Z-G. Tetrahedron Lett. 2015;45:2742–2744. [Google Scholar]
- [19].Liao W-L, Li S-Q, Wang J, et al. ACS Comb Sci. 2016;18:65–69. [DOI] [PubMed] [Google Scholar]
- [20].Shimizu T, Tolcher AW, Papadopoulos KP, et al. Clin Cancer Res. 2012;18:2316–2325. [DOI] [PubMed] [Google Scholar]
- [21].Ma C, Zhu L, Wang J, et al. J Ethnopharmacol. 2015;168:349–355. [DOI] [PubMed] [Google Scholar]
- [22].Ciruelos Gil EM Cancer Treat Rev. 2014;40:862–871. [DOI] [PubMed] [Google Scholar]
- [23].Zhirnov OP, Klenk H-D. Apoptosis. 2007;12:1419–1432. [DOI] [PubMed] [Google Scholar]
- [24].Roper J, Richardson MP, Wang WV, et al. PLoS ONE. 2011;6:e25132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kumar Pandurangan A Asian Pacific J Cancer Prev. 2013;14:2201–2205. [DOI] [PubMed] [Google Scholar]
- [26].Manning BD, Toker A. Cell. 2017;169:381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kastan MB, Bartek J. Nature. 2004;432:316–323. [DOI] [PubMed] [Google Scholar]
- [28].Malumbres M, Barbacid M. Nat Rev Cancer. 2009;9:153–166. [DOI] [PubMed] [Google Scholar]
- [29].Wang Y, Ji P, Liu J, Broaddus RR, Xue F, Zhang W. Mol Cancer. 2009;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dash BC, El-Deiry WS. Mol Cell Biol 2005;25:3364–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang C-J, Wang C-S, Hung J-Y, et al. Lung Cancer. 2009;66:162–168. [DOI] [PubMed] [Google Scholar]
- [32].Oltersdorf T, Elmore SW, Shoemaker AR, et al. Nature. 2005;435:677–681. [DOI] [PubMed] [Google Scholar]
- [33].Xu X, Yi Z, Dan Q, Jiang T, Li S. J Exp Clin Cancer Res. 2011;30:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thees S, Hubbard GB, Winckler J, Schultz C, Rami A. Restor Neurol Neurosci. 2005;23:1–9. [PubMed] [Google Scholar]
- [35].Gupta S, Afaq F, Mukhtar H. Oncogene. 2002;21:3727–3738. [DOI] [PubMed] [Google Scholar]
- [36].Martinou J-C, Youle RJ. Dev Cell. 2011;21:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Walters J, Pop C, Scott FL, et al. Biochem J. 2009;424:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- [39].Hammoud SS, Cairns BR, Jones DA. Curr Opin Cell Biol. 2013;25:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mayer IA, Arteaga CL. Annu Rev Med. 2016;67:11–28. [DOI] [PubMed] [Google Scholar]
- [41].Song MS, Salmena L, Pandolfi PP. Nat Rev Mol Cell Biol. 2012;13:283–296. [DOI] [PubMed] [Google Scholar]
- [42].Hopkins BD, Parsons RE. Clin Cancer Res. 2014;20:5379–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Guri Y, Hall MN. Trends in Cancer. 2016;2:688–697. [DOI] [PubMed] [Google Scholar]
- [44].Cornu M, Albert V, Hall MN. Curr Opin Genet Dev. 2013;23:53–62. [DOI] [PubMed] [Google Scholar]
- [45].Laplante M, Sabatini DM. Cell. 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim LC, Cook RS, Chen J. Oncogene. 2017;36:2191–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.