

Abstract
Background
Osteoporosis is a bone mineralisation disorder occurring in about one third of adults with cystic fibrosis. Bisphosphonates can increase bone mineral density and decrease the risk of new fractures in post‐menopausal women and people receiving long‐term oral corticosteroids.
Objectives
To assess the effects of bisphosphonates on the frequency of fractures, bone mineral density, quality of life, adverse events, trial withdrawals, and survival in people with cystic fibrosis.
Search methods
We searched the Cystic Fibrosis and Genetic Disorders Group Trials Register of references (identified from electronic database searches and handsearches of journals and abstract books) on 13 January 2014.
Additional searches of PubMed were performed on 13 January 2014.
Selection criteria
Randomised controlled trials of at least six months duration studying bisphosphonates in people with cystic fibrosis.
Data collection and analysis
Two authors independently selected trials and extracted data. Trial investigators were contacted to obtain missing data.
Main results
Nine trials were identified and seven (with a total of 237 adult participants) were included.
Data were combined (when available) from six included studies in participants without a lung transplant. Data showed that there was no significant reduction in fractures between treatment and control groups at 12 months, odds ratio 0.72 (95% confidence interval 0.13 to 3.80). No fractures were reported in studies with follow‐up at 24 months. However, in patients taking bisphosphonates after six months the percentage change in bone mineral density increased at the lumbar spine, mean difference 4.61 (95% confidence interval 3.90 to 5.32) and at the hip or femur, mean difference 3.35 (95% confidence interval 1.63 to 5.07); but did not significantly change at the distal forearm, mean difference ‐0.49 (95% confidence interval ‐2.42 to 1.45). In patients taking bisphosphonates, at 12 months the percentage change in bone mineral density increased at the lumbar spine, mean difference 6.10 (95% confidence interval 5.10 to 7.10) and at the hip or femur, mean difference 4.35 (95% confidence interval 2.99 to 5.70). At 24 months, in patients treated with bisphosphonates the percentage change in bone mineral density also increased at the lumbar spine, mean difference 5.49 (95% confidence interval 4.38 to 6.60) and at the hip or femur, mean difference 6.05 (95% confidence interval 3.74 to 8.36). There was clinical heterogeneity between studies and not all studies reported all outcomes. Bone pain was the most common adverse event with intravenous agents. Flu‐like symptoms were also increased in those taking bisphosphonates.
In participants with a lung transplant (one study), intravenous pamidronate did not change the number of new fractures. At axial sites, bone mineral density increased with treatment compared to controls: percentage change in bone mineral density at lumbar spine, mean difference 6.20 (95% confidence interval 4.28 to 8.12); and femur mean difference 7.90 (95% confidence interval 5.78 to 10.02).
Authors' conclusions
Oral and intravenous bisphosphonates increase bone mineral density in people with cystic fibrosis. Severe bone pain and flu‐like symptoms may occur with intravenous agents. Additional trials are needed to determine if bone pain is more common or severe (or both) with the more potent zoledronate and if corticosteroids ameliorate or prevent these adverse events. Additional trials are also required to further assess gastrointestinal adverse effects associated with oral bisphosphonates. Trials in larger populations are needed to determine effects on fracture rate and survival.
Plain language summary
Bisphosphonates for osteoporosis in people with cystic fibrosis
Cystic fibrosis is a serious genetic disorder that affects many organs (e.g. lung and pancreas). It commonly leads to reduced bone mineral density, known as osteoporosis, which increases the likelihood of fractures. The short‐term and long‐term effects of fractures (e.g. rib and vertebral) may make lung disease worse. Bisphosphonates are drugs that increase bone mineral density by slowing down bone resorption. They are used to treat osteoporosis caused by menopause or the use of corticosteroid drugs.
The evidence available was limited to six trials with participants who had not undergone lung transplants (total of 203 adults) and one trial with 34 adults who had undergone lung transplantation. Bisphosphonates consistently increased bone mineral density at the lumbar spine and hip regions. The rates of fractures (vertebral and non‐vertebral) or deaths were not reduced by bisphosphonate therapy. However, this may be related to the small numbers of participants involved and the short duration of the trials. Severe bone pain and flu‐like symptoms were commonly linked to intravenous bisphosphonates, especially in people not using corticosteroids. More research is needed to assess the effect of pre‐treatment with corticosteroids. Additional trials are needed to determine if bone pain is more common or severe (or both) with the stronger drug zoledronate and if corticosteroids lessen or prevent these adverse events. Additional trials are also required to further assess gastrointestinal adverse effects associated with oral bisphosphonates. Trials in larger populations are needed to determine effects on fracture rate and survival.
Background
Description of the condition
Cystic fibrosis (CF) is the most common serious autosomal recessive genetic disorder in the Caucasian population. It is caused by mutations in the cystic fibrosis transmembrane regulator (CFTR) gene (Riordan 1989). This genetic disorder occurs in approximately one in 3500 live births in the United States of America; the incidence varies between racial and ethnic groups, being more common in Caucasians (Cystic Fibrosis Foundation Patient Registry 2013). One in 25 individuals carry the defective gene (Yankaskas 1999). The major components of CF are lung disease and pancreatic insufficiency. In the past, only one third of individuals with CF lived to the age of 18 years (Yankaskas 1999). However, recent advances in medical science and technology have increased the life expectancy of people with CF into the fourth and fifth decades of life (Cystic Fibrosis Foundation Patient Registry 2013). In the United States of America, the predicted median survival in 1992, 2002 and 2012 was 29.4 years, 31.3 years and 41.1 years respectively. Hence, as survival improves, long‐term sequelae of the disease (Quon 2012), such as osteoporosis (Paccou 2013; Stalvey 2013a), diabetes mellitus (Kelly 2013) and liver disease (Rowland 2011) are of increasing significance.
Osteoporosis is a disorder of bone mineralisation that decreases bone mineral density (BMD) and makes bones brittle and more susceptible to fracture. Osteopenia refers to a milder degree of bone demineralisation. Bone density is currently measured using dual‐energy X‐ray absorptiometry (DXA), which measures absolute bone density in grams per centimetre squared (gm/cm2) and can be compared to a population mean. It is usual to express BMD as standard deviations from the population mean, either as a Z‐score (compared to age‐ and sex‐matched data) or a T‐score (compared to the healthy young adult mean for the participant's sex). The World Health Organization (WHO) classifies osteopenia as a T‐score of between ‐1 and ‐2.5, and osteoporosis as a T‐score of ‐2.5 or less (Kanis 1994). The International Society for Clinical Densitometry has recommendations with regard to BMD reporting (ISCD Official Positions 2013). However, BMD is only one of the factors that determine the risk of fracture. It is osteoporotic fractures which account for the morbidity and mortality associated with osteoporosis (Cummings 1995).
Decreased BMD has been observed in children with CF (Bianchi 2006; Sermet‐Gaudelus 2007), adolescents with CF (Bianchi 2006; Buntain 2004; Caldeira 2008) and adults with CF (Bianchi 2006; Buntain 2004). A longitudinal study of 151 adults with CF aged 15 to 52 years showed that 34% of participants had a DXA Z‐score of ≤ ‐2 (Haworth 2001). A recent systematic literature review with meta‐analysis reported the pooled prevalence of osteoporosis (total of 888 patients) and osteopenia (total of 697 patients) in adults with CF was 23.5% (95% confidence interval (CI) 16.6 to 31.0) and 38% (95% CI 28.2 to 48.3) respectively (Paccou 2010). Other studies have not detected differences in BMD between children with CF and those without (Buntain 2004), but inadequate bone mass accrual during childhood and adolescence has been reported (Buntain 2006). Definitions of osteoporosis and osteopenia may vary between studies.
The etiology of CF‐related bone disease is thought to be multifactorial. An imbalance of increased bone resorption and decreased bone formation has been observed (Boyle 2006; Gore 2010), even in young prepubertal patients (Ambroszkiewicz 2013). Proposed contributing factors to bone disease include malnutrition and exocrine pancreatic insufficiency; vitamin D, vitamin K or calcium deficiency; poor nutrition and growth resulting in low body weight; systemic inflammatory cytokines; use of exogenous glucocorticoids; sex hormone insufficiency, diabetes mellitus, physical inactivity resulting in decreased weight bearing activity and organ transplantation with associated immunosuppressive therapy (Aris 2005; Boyle 2006; Gore 2010; Hall 2010; Haworth 2010b; Javier 2011; Paccou 2013). Abnormalities of calcium metabolism independent of vitamin D status have been reported (Greer 2003). Many of these factors are inter‐related (e.g. pancreatic exocrine insufficiency contributing to malabsorption of vitamin D, exacerbated by decreased sun exposure if indoors due to poor health). Furthermore, BMD has been positively associated with pulmonary function, physical function, nutritional status and negatively associated with chronological age and recurrent pulmonary exacerbations (Alicandro 2013; Donadio 2013; Legroux‐Gerot 2012). Mutation of the CFTR gene itself may have a direct role in the pathogenesis of CF‐related bone disease (Dif 2004; Le Henaff 2012; Paccou 2013). Studies have indicated that the loss of CFTR activity in osteoblasts decreases the secretion of osteoprotegerin (OPG) resulting in accentuated inflammation‐driven bone resorption (Le Heron 2010; Stalvey 2013).The glycoprotein OPG acts a decoy receptor for receptor activator of nuclear factor κB ligand (RANKL), blocking the interaction of RANKL with the receptor RANK. The RANKL/RANK interaction stimulates the formation and activation of osteoclasts, hence OPG exerts an anti‐resorptive effect (Weitzmann 2013). A recent study identified that the risk of osteoporosis was related to gene polymorphisms of the gene encoding the inflammatory cytokine, tumour necrosis factor‐alpha (Shmarina 2013).
The clinical consequences of CF‐related bone disease include low bone density with potential fractures, including rib and vertebral fractures, which may be precipitated by coughing. In the systematic review by Paccou, the pooled prevalences of radiological vertebral fractures (total of 683 patients) and non‐vertebral fractures (total of 553 patients) in young adults with CF were 14% (95% CI 7.8 to 21.7) and 19.7% (95% CI 6.0 to 38.8) respectively (Paccou 2010). A study in post‐lung transplant participants with CF found an approximate two‐fold increase in the risk of non‐vertebral fractures for women aged 16 to 34 years (P = 0.015) and men aged 25 to 45 years (P = 0.04) compared with the general population (Aris 1998). Vertebral compression and rib fractures were 100‐fold and 10‐fold more common respectively than predicted (P < 0.001). Incident new vertebral fractures are commonly defined as a 15% or greater reduction in anterior, posterior, or middle vertebral height. Acutely, vertebral and rib fractures may result in pain and debilitation, resulting in diminished lung function, ineffective cough and airway clearance, limitations in respiratory physiotherapy, in addition to reduced physical activity. Chronic consequences may include kyphosis and chest wall deformities which may worsen lung function. Hence a vicious cycle of further impairment in lung function and deterioration in bone health may result (Aris 2005; Boyle 2006; Hayes 2011). Established bone disease may exclude the patient from lung transplantation, as the high‐dose corticosteroids and other immunosuppression required post‐transplantation can be expected to worsen bone disease further (Aris 1996).
Description of the intervention
Bisphosphonates are a class of drugs that inhibit bone resorption (Giger 2013; Russell 2007). Considerations in using bisphosphonate therapy include different formulations (oral, intravenous), dosage, frequency of administration, intermittent versus continuous dosing and duration of therapy (Russell 2006).
Potential side‐effects include an acute‐phase response leading to fever and 'flu'‐like symptoms (myalgia, malaise). This has been most commonly observed after the first exposure to nitrogen‐containing bisphosphonates administered intravenously, and associated with an increase in inflammatory cytokines (Sauty 1996). Osteonecrosis of the jaw has been associated with high‐dose intravenous bisphosphonate therapy in patients with malignancy (Pendrys 2008). Prolonged bisphosphonate use (exceeding five years) has been associated with atypical femoral fractures, a rare subset of subtrochanteric and femoral shaft fractures (Saleh 2013). Upper gastrointestinal side‐effects (e.g. oesophagitis) have been associated with oral agents (Cryer 2002).
How the intervention might work
Bisphosphonates are selectively taken up by bone and exert inhibitory effects on osteoclasts, cells that act to dissolve and resorb bone (Russell 2007). Individual drugs within this class have different mineral binding affinities and molecular modes of action (e.g. nitrogen‐containing pamidronate, alendronate, risedronate, ibandronate and zoledronate are more potent than the non‐nitrogen containing etidronate and clodronate) (Russell 2006).
Bisphosphonates have proven effective treatments for disorders of excessive bone resorption (Giger 2013). These disorders include Paget's disease of bone, myeloma, bone metastases (Russell 2006); postmenopausal osteoporosis (Black 1996); other forms of osteoporosis (e.g. associated with glucocorticoid administration (Saag 1998)); and children with the 'brittle bone' disorder, osteogenesis imperfecta (Glorieux 1998; Phillipi 2008). Positive outcomes have included increased bone mass, BMD and a reduction in fracture frequency.
Bone resorption has been observed to increase during CF pulmonary exacerbations due to the stimulatory effect of pro‐inflammatory cytokines on osteoclast activity (Shead 2010). Loss of the CFTR in osteoblasts may exacerbate the effect of inflammation to stimulate osteoclastogenesis (Le Heron 2010; Stalvey 2013; Weitzmann 2013). Since bisphosphonates inhibit osteoclastic bone resorption, these agents may minimise bone loss in this context.
A two‐year study of intermittent cyclical etidronate in 423 postmenopausal women demonstrated a significant increase in BMD at the spine and a decrease in new fractures (Watts 1990). In a study of 2027 women with at least one existing vertebral fracture, alendronate was shown to increase BMD at the spine and hip and to decrease fractures at the hip, wrist and spine after three years of treatment (Black 1996). Pamidronate in combination with calcium was studied over an 18‐month period in an initial cohort of 35 postmenopausal women (mean age 64.5 years) with at least one atraumatic vertebral fracture due to osteoporosis. After one year, BMD increased in the lumbar spine (P < 0.001), although there were no changes in the femoral neck (Fromm 1991). Oral risedronate has been observed to secondarily prevent vertebral and non‐vertebral fractures in postmenopausal women (Wells 2008).
A study of 477 participants with glucocorticoid‐induced osteoporosis found alendronate to be highly effective in increasing BMD at the spine and femoral neck, with a significant reduction in the number of incident fractures (Saag 1998). Another one‐year study compared two regimens of intravenous pamidronate (a single infusion or once every three months) for the primary prevention of glucocorticoid‐induced osteoporosis. The study population consisted of 32 participants who required long‐term glucocorticoid therapy with at least 10 mg of prednisone daily. A highly significant difference was observed between both pamidronate regimens and the control group at the lumbar spine (P < 0.001) and femoral neck (P < 0.01). Both pamidronate regimens effectively achieved primary prevention of glucocorticoid‐induced osteoporosis (Boutsen 2001). This evidence is particularly promising since corticosteroid use is associated with osteoporosis among people with CF.
A three‐year randomized double‐blind trial of 300 mg oral pamidronate daily compared with placebo was conducted in 105 participants with rheumatoid arthritis (Eggelmeijer 1996). Inflammation, decreased mobility and glucocorticoid use may contribute to the risk of osteoporosis in individuals with rheumatological conditions. After three years, lumbar spine and forearm BMD had increased significantly in the pamidronate‐treated group, while there were non‐significant changes in the placebo‐treated group. Changes were significantly different between the treatment and placebo groups (Eggelmeijer 1996).
Why it is important to do this review
This is an update of a previous Cochrane review (Brenckmann 2001; Conwell 2009; Conwell 2012).
A multi‐faceted approach to the prevention and treatment of low BMD has been recommended including optimising vitamin, calcium and nutritional status, encouraging weight‐bearing exercise, endocrinological assessment and management of delayed puberty or hypogonadism, aggressive treatment of pulmonary infections, minimizing exposure to corticosteroids and treatment of CF‐related diabetes (Aris 2005). A consensus statement addressing the issue of bone health and disease in CF recommends consideration of oral or intravenous bisphosphonates in individuals with DXA T‐ or Z‐scores ≤ 2.0, awaiting transplant or BMD loss more than 3% to 5% per year (Aris 2005). A caution is placed on the use of intravenous bisphosphonates due to the association with severe bone pain in this population group.
The UK Cystic Fibrosis Trust Bone Mineralisation Working Group outlined specific recommendations for consideration of bisphosphonate treatment in adults and children, justifying the importance of the previous Cochrane reviews of the available evidence assessing bisphosphonates for osteoporosis in people with CF (Cystic Fibrosis Trust Report 2007).
The more recent European Cystic Fibrosis Bone Mineralisation guidelines outline detailed recommended assessment, prevention and treatment strategies (Sermet‐Gaudelus 2011). Specifically, these guidelines recommend consideration of bisphosphonate treatment in adults who:
are taking continuous systemic oral glucocorticoids for at least three months with a BMD Z/T‐score of ‐1.5 or less;
sustain a low trauma fracture while taking systemic glucocorticoids;
have had a low trauma fracture or in whom the lumbar spine or total hip or femoral neck Z/T‐score is ‐2 or less (or both) and there is evidence of significant bone loss (more than 4% per year) on serial DXA measurements despite optimisation of clinical care;
are awaiting or have undergone solid organ transplantation and have a BMD Z/T‐score of ‐1.5 or less;
are starting a prolonged course of oral glucocorticoids (longer than three months) and have a BMT Z/T‐score of ‐1.5 or less (Sermet‐Gaudelus 2011).
These 2011 guidelines also recommend the consideration of bisphosphonate treatment in children after failure of optimal conservative treatment who:
are taking continuous systemic glucocorticoids for at least three months and have a history of low trauma fracture or BMD Z‐score of ‐2 or less (or both);
have a BMD Z‐score of ‐2 or less in the total body or lumbar spine and a history of low‐trauma extremity fractures or vertebral compression fractures;
are awaiting or have undergone solid organ transplantation and have a BMD Z‐score of ‐2 or less;
are prescribed continuous systemic glucocorticoids and have a BMD Z‐score of ‐2 or less (Sermet‐Gaudelus 2011).
In transplant recipients, the recommendations are that:
low BMD and a history of low trauma fractures are not an absolute contraindication for lung transplant listing in people with CF;
bone loss after solid organ transplantation can be lessened by the prescription of bisphosphonates in people with CF (Sermet‐Gaudelus 2011).
Hence, a review of the available evidence assessing bisphosphonates for osteoporosis in people with CF is important.
Objectives
To determine whether bisphosphonates cause the following changes in people with CF:
decrease fractures (vertebral and non‐vertebral);
improve BMD measured using DXA or, if available, using other methods of bone density measurement such as single energy X‐ray absorptiometry (SXA) and quantitative tomography (QCT);
increase quality of life;
increase adverse events, including bone pain and gastrointestinal adverse events;
change the number of withdrawals due to all causes and due to adverse events;
increase survival.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials. Published papers and abstracts were included. Trials published in all languages were considered for inclusion.
Types of participants
People of all ages and of both sexes with CF diagnosed clinically or by sweat and genetic testing, including all degrees of disease severity and bone density.
Types of interventions
All trials examining bisphosphonates compared to controls (other bisphosphonates, placebo or usual treatment) for treating or preventing osteoporosis in people with CF were considered for inclusion. All doses and routes of administration were considered. Trials of a minimum duration of six months were included to allow time to observe an effect of treatment.
Types of outcome measures
Primary outcomes
Fractures (number of participants with any fracture and number of fractures at all sites, spine, hip, wrist)
Secondary outcomes
-
Bone density as measured by dual‐energy X‐ray absorptiometry (DXA), reported as per cent change from baseline. Data reported using other methods of bone density measurement such as single energy X‐ray absorptiometry (SXA) and quantitative tomography (QCT) would be used if available but analysed separately:
lumbar spine
hip or femur
radius
total body
Quality of life (QoL) (CF‐related, osteoporosis‐related or general QoL measures)
Adverse events such as bone pain, hypocalcaemia and gastrointestinal adverse events (number of participants, number of adverse events)
-
Withdrawals
withdrawals due to adverse events
total withdrawals
Survival
Search methods for identification of studies
Electronic searches
Relevant trials were identified from the Group's Cystic Fibrosis Trials Register using the terms: CF‐related bone disease AND bisphosphonates.
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue), quarterly searches of MEDLINE, a search of Embase to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work is identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Cystic Fibrosis and Genetic Disorders Group Module.
Date of the most recent search of the Group's Cystic Fibrosis Trials Register: 13 January 2014.
Additionally LC performed personal searches of PubMed on 14 January 2014 (all years up to 14 January 2014) (Appendix 1; Appendix 2).
Searching other resources
For the original review, abstracts from major osteoporosis conferences (The American Society for Bone and Mineral Research and The Institute of Biomedical Science (ASBMR‐IBM) 2nd Joint Meeting 1998; ASBMR 21st Annual Meeting 1999) were searched and also the reference lists from the retrieved articles.
In an earlier update, the reference lists for the retrieved articles were also searched and from that, a further paper was identified (Conway 2004).
Data collection and analysis
Selection of studies
In the original review, two authors (CB, AP) independently reviewed the trials in order to assess which trials should be included. For the updated reviews from 2009, two authors (LC, AC) independently reviewed the trials included in the original review in addition to trials identified by the updated searches. If there was disagreement about whether a trial should be included, the authors planned to ask an independent author from a third centre to review the paper(s) in question. The authors documented the reasons for excluding any trial.
Data extraction and management
In the original review, each author independently extracted data for the outcome measures listed below. The review authors contacted the first authors of the included trials to verify their data and obtain unpublished data where necessary. The authors used the Cochrane Review Manager software (Version 4.1) to compile and analyse the data (Review Manager 2003). The authors compared their data and resolved differences by referring to the original article; they planned to resolve any remaining differences with a third individual.
From 2009 onwards, the authors (LC, AC) reviewed trials that satisfied the inclusion criteria and recorded the following information when available: study setting; year of study; source of funding; participant recruitment details (including number of eligible participants); inclusion and exclusion criteria; randomisation and allocation concealment method; numbers of participants randomised; blinding (masking) of participants, care providers and outcome assessors; dose and type of intervention; duration of therapy; co‐interventions; numbers of participants not followed up; reasons for withdrawals from study protocol (clinical, side‐effects, refusal and other); side‐effects of therapy; and whether intention‐to‐treat analyses were possible. The review authors requested further information from the two authors but no response was received. Data were reported at time‐points 6 months, 12 months and annually thereafter which are appropriate time‐points to assess treatment effects on fracture frequency and duration. For per cent change in BMD, the review authors combined and reported data for the end of study as it is unknown what length of study is needed for effect of treatment to be evident. The review authors used the Cochrane Review Manager software (Version 5.1) to compile and analyse the data (Review Manager 2011). Similarly, the authors compared their data and resolved differences by referring to the original article.
The review authors conducted separate analyses for participants who had received a lung transplant and for those who had not. At this stage, the number of people with CF who have received other organ transplants is small. Therefore, the authors included individuals with other organ transplants in the analysis of participants with a lung transplant, since they share a common risk factor for osteoporosis, that is the long‐term use of immunosuppressive agents which lower BMD.
Assessment of risk of bias in included studies
The two authors who performed the original review independently assessed the quality of the trials using the system as described by Jadad (Jadad 1996). If there was disagreement about whether a trial should be included, or about the quality score it should receive, the authors asked an independent author from a third centre to review the paper(s) in question.
In this review update, the authors (LC, AB) independently assessed the risk of bias in the trials using the criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011):
randomisation process i.e. the generation of allocation sequence;
allocation process i.e. quality of allocation blinding;
degree of blinding (of clinician or person delivering treatment, of participant or of outcome assessor);
is attrition bias present i.e. use of intention‐to‐treat analysis?
is selective reporting present?
For each of the criteria, the authors gave a judgement of a high, low or unclear risk of bias based on guidance from The Cochrane Collaboration (Higgins 2011) and gave the reasons for these judgements in the risk of bias tables (Characteristics of included studies).
Measures of treatment effect
For the dichotomous outcome variables (adverse events, fractures, survival) of each individual study, the authors calculated the odds ratio (OR) using a modified intention‐to‐treat analysis (where incomplete data assumes failure of treatment). They also calculated the summary ORs and 95% confidence intervals (CIs) using the Cochrane Collaboration's statistical package, RevMan 5 (Review Manager 2011). They planned to calculate the numbers needed to treat (NNT) from the pooled OR and its 95% CIs applied to a specified baseline risk, which is the sum of all the events in the control groups (in all trials) divided by the total participant numbers in control groups in all trials using an online calculator (Cates 2003).
For continuous outcomes (changes in BMD and laboratory values), the authors recorded the mean relative change from baseline for each group or mean post‐treatment or post‐intervention values and standard deviation (SD). They planned to calculate the SDs if standard errors were reported. They would then calculate a pooled estimate of treatment effect by the mean difference (MD) and 95% CIs, again using the statistical package from The Cochrane Collaboration (Review Manager 2011).
Unit of analysis issues
There were no cross‐over studies, but if there had been, the authors planned only to use data from the first arm of the study for any analysis due to the potential for a carry‐over effect. They would have analysed count data of rare events as rates (number of counts to the amount of time during which they could have happened). They planned to use the summary statistic rate ratio in meta‐analysis. For rate ratios of common events, whereby one participant may have more than one event, they planned to use the generic inverse variance (GIV) method of analysis.
Dealing with missing data
The review authors contacted primary authors of studies to obtain missing data or clarify data.
Assessment of heterogeneity
The authors planned to describe any heterogeneity between the study results and test it to see if it reached statistical significance using the I2 statistic (Higgins 2003). This measure describes the percentage of total variation across studies that are due to heterogeneity rather than by chance (Higgins 2003).
The values of I2 lie between 0% and 100%, and a simplified categorization of heterogeneity that the authors planned to use is of low (I2 value of 0% to 40%), moderate (I2 value 30% to 60%), substantial (I2 value 50% to 90%) and considerable (75% to 100%) (Higgins 2011). The P value from the chi‐squared test also indicates the strength of evidence for heterogeneity, considered to be statistically significant when the P value was less than 0.10 (Higgins 2011).
Assessment of reporting biases
In order to identify selective reporting, the review authors compared outcome measures reported within the published papers to the measures described in the methodology sections or with the study protocols if they were available.
Data synthesis
The authors included the results from studies that met the inclusion criteria and reported any of the outcomes of interest in the subsequent meta‐analyses using a fixed‐effect model. They would have included the 95% CI, estimated using a random‐effects model, if there was at least moderate heterogeneity (I2 over 30%).
If studies reported outcomes using different measurement scales, the authors planned to estimate the standardised mean difference.
Subgroup analysis and investigation of heterogeneity
The authors planned to analyse the trials in the subgroups of children (aged 18 years or less) and adults (over 18 years). There were no trials fulfilling the definition of the former subgroup, but the authors will carry out this planned subgroup analysis if they are able to include a sufficient number of relevant trials (at least 10) in a future update of this review.
Sensitivity analysis
The authors had also planned to conduct sensitivity analyses to assess the impact of the potentially important factors on the overall outcomes:
differences in the medications used in the intervention and comparison groups;
analysis using random effects model;
analysis by "treatment received"; and
analysis by "intention‐to‐treat".
Results
Description of studies
Results of the search
In the original review (Brenckmann 2001), two studies were identified. With the present update, a total of nine clinical trials were identified that were all published in English (Aris 2000; Aris 2004; Boyle 2005; Chapman 2009; Conway 2004; Hardin 2005; Haworth 2001; Haworth 2011; Papaioannou 2008). Seven trials were identified from the search of the Group's Cystic Fibrosis Trials Register (Aris 2000; Aris 2004; Boyle 2005; Chapman 2009; Hardin 2005; Haworth 2001; Haworth 2011). Additional PubMed searches as detailed in the appendices (Appendix 1; Appendix 2) identified one further trial (Papaioannou 2008) and the full paper to the previously identified abstracts to the later Haworth trial. Searching the reference lists for the retrieved articles identified a further trial (Conway 2004).
Seven clinical trials were included and two were excluded (see below).
Included studies
Seven clinical trials met the inclusion criteria for this review (Aris 2000; Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008). One trial was published only as an abstract (Boyle 2005). We contacted the lead investigator for details of the full publication, without receiving a reply. Four trials were published both as abstracts and full review papers (Aris 2000; Aris 2004; Haworth 2001; Haworth 2011). Two trials were published as full review papers (Chapman 2009; Papaioannou 2008).
All the trials had similar designs, duration of planned intervention (12 to 24 months) and outcome measures. None of the trials reported sample size or power calculations. None of the studies included children (aged 18 years or less). In the Boyle trial, enrolment was ceased after five participants were randomised and follow‐up duration was decreased from 12 months to 6 months due to adverse events (i.e. musculoskeletal pain) (Boyle 2005). The follow‐up duration of another trial was also shortened from 12 months to 6 months due to adverse events (i.e. bone pain) (Haworth 2001).
One trial assessed 24 months of intravenous pamidronate in 34 post‐transplant adults with CF (Aris 2000). The participants in the other six trials were non‐transplanted adults with CF.
Other factors that will contribute to the heterogeneity in the results of the review include differences in trial populations and concomitant general health, activity levels and medications (prednisone, cyclosporin A, azathioprine in the transplant group).
All participants in six of the trials received supplemental calcium and vitamin D (Aris 2000; Aris 2004; Boyle 2005; Chapman 2009; Haworth 2011; Papaioannou 2008). In the study by Haworth and colleagues, participants with pancreatic insufficiency were prescribed long‐term oral vitamin D supplements (all participants except one participant in the control group) (Haworth 2001). Oral prednisolone was administered to all participants of one study for three days starting on the morning of the first infusion (Chapman 2009). This was repeated with subsequent infusions if a reaction to the first infusion was thought likely.
The types of bisphosphonate assessed in the trials included intravenous pamidronate in two trials, 31 participants in the earlier Haworth trial and 34 participants in the earlier Aris trial (Aris 2000; Haworth 2001); oral alendronate in two trials, 53 participants in the later Aris trial and 56 participants in the Papaioannou trial (Aris 2004; Papaioannou 2008); oral risedronate (36 participants) (Haworth 2011); intravenous zoledronate (22 participants) (Chapman 2009) and intravenous zoledronate (40 participants planned) (Boyle 2005).
Funding for one trial was provided by grants from the CF Foundation and the Verne S. Caviness General Center for Clinical Research (Aris 2000). Another trial was funded by the U.S. Food and Drug administration, Merck and Co, Inc., the Clinical Nutrition Research Unit, the Verne S. Caviness General Center for Clinical Research at University of North Carolina, the CF Foundation and the NIDDK (Aris 2004). Novartis Pharmaceuticals Pty Ltd, Australia partly funded one trial (Chapman 2009). Study funding was also provided by Merck Frosst Canada (Papaioannou 2008). Haworth and colleagues received funding from the CF Trust in the UK (Haworth 2001; Haworth 2011). The later Haworth study was funded by unrestricted educational grants from Proctor & Gamble (Norwich, USA) and the Cystic Fibrosis Trust, with support to investigators from the UK National Institute of Health Research (Haworth 2011).
Excluded studies
Two clinical trials were excluded (Conway 2004; Hardin 2005).
One trial was a prospective open study assessing the effect of oral bisphosphonates on BMD and body composition in adults with CF (Conway 2004). It was not a randomised controlled trial. The other trial assessed the effect of growth hormone on bone mineral content in children with CF (Hardin 2005). It did not assess the use of bisphosphonates and hence did not meet the inclusion criteria for this review.
Risk of bias in included studies
For detailed information on the risk of bias of each included trial, please refer to the risk of bias tables attached to the 'Characteristics of included studies' section of this review.
Allocation
All trials stated that allocation was in accordance to randomisation. One trial reported the use of a computer‐generated randomisation code, stratified according to institution (prepared by an independent randomisation centre) with use of block allocation to ensure equitable distribution to each treatment group (Papaioannou 2008). In another trial, participants were allocated to treatment or placebo using a computer programme to minimise differences between groups in treatment centre, sex and baseline lumbar spine BMD (Haworth 2011). We judged these two trials to have a low risk of bias. Two trials described a "blocks of four" design, but the actual method of randomisation was not discussed. We judged the risk of bias due to the generation of the randomisation sequence as unclear (Aris 2000; Aris 2004). In the other trials, the method of randomisation and hence the risk of bias was unclear (Boyle 2005; Chapman 2009; Haworth 2001).
Concealment of allocation and hence risk of bias was low in two trials (Haworth 2011; Papaioannou 2008) and unclear in the remaining five trials (Aris 2000; Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001).
Blinding
Clinician or person delivering treatment
In one trial, clinicians giving the interventions were not blinded (Aris 2000) and thus risk of bias was high and in another, this was not reported (Haworth 2001). Three trials were described as "double‐blind", but the blinding of those delivering treatment was not specifically discussed (Aris 2004; Boyle 2005; Chapman 2009). In another trial, blinding was not reported (Haworth 2001). Hence, these four trials were judged to have an unclear risk of bias. Clinicians delivering treatment were blinded in two trials, leading us to judge them to have a low risk of bias (Papaioannou 2008; Haworth 2011).
Participants
In one trial, participants were not blinded to the treatment group (Aris 2000) (thus a high risk of bias) and in another trial, this was not reported leading to an unclear risk of bias (Haworth 2001). Participants were blinded to the intervention group in the other five trials, which we therefore judged to have a low risk of bias (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2011; Papaioannou 2008).
Outcome assessors
In one trial, only the radiologist who interpreted the DXA scans was blinded (Aris 2000). In the Chapman trial, it was stated that the personnel who performed and analysed the DXA scans were blinded to the treatment group, but it was not clear whether all the outcome assessors were blinded (Chapman 2009). In one trial, blinding was not reported (Haworth 2001) and two other trials were described as "double‐blind" although it was not specifically discussed whether all the outcome assessors were blinded (Aris 2004; Boyle 2005).
Hence considering these different types of participants and personnel together, there was some risk of bias in five of the trials (Aris 2000; Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001).
Person(s) responsible for participants care, participants and outcome assessors were blinded to treatment group allocation in the trial by Papaioannou (Papaioannou 2008). In the later Haworth trial, only the study pharmacist had access to the treatment allocation (Haworth 2011). Hence there was a low risk of bias in these trials (Haworth 2011; Papaioannou 2008).
Incomplete outcome data
All seven trials described withdrawals from treatment. Further details can be found in the 'Risk of bias' tables in Characteristics of included studies (Aris 2000; Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008).
In one trial, five withdrawals between commencement of protocol and outcomes measured at six months were described, but not delineated as to whether they were in the treatment or the control group (Aris 2004). In the earlier Aris trial, it was described that three participants died during the course of the study before the first primary end‐point measurement (causes of death were one each from sepsis, acute respiratory distress syndrome and obliterative bronchiolitis). These participants were excluded from the final analysis of baseline characteristics and outcome data. However, it was not reported which treatment group they were in (Aris 2000). Hence the risk of bias was unclear for both these trials. In another trial, it was unclear which specific participants had BMD measurements available at each time‐point, particularly for forearm measures (fewer measurements compared with lumbar spine and femoral neck) (Chapman 2009).
In the trial by Boyle, 40 participants were planned for enrolment but only five enrolled (three in treatment group) before the study was stopped by the Data and Safety Monitoring Board after three participants experienced adverse effects. It was assumed that the three patients with bone pain were in the treatment group although this was not stated, hence was judged to be low risk of bias (Boyle 2005). Details of withdrawals were provided by the author for one study i.e. unpublished data (Haworth 2011) thus judged as low risk of bias. Two other trials were also judged as low risk (Haworth 2001; Papaioannou 2008).
Withdrawal rates and hence risk of attrition bias varied. For example, from protocol commencement to BMD (lumbar spine) measurement at end of study, total withdrawals were 0/34 (0%) (Aris 2000),13/53 (25%) (Aris 2004), 3/22 (14%) (Chapman 2009), 3/31 (10%) (Haworth 2001), 12/36 (33.3%) (Haworth 2011), 8/56 (14.2%) (Papaioannou 2008) and 3/5 (60%) (Boyle 2005).
Selective reporting
Outcome measures described in the methodology were reported for most trials, hence we judged them to have a low risk of bias (Aris 2004; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008). The exception was the serum and urine biochemical measurements at the two‐day time‐point (only after first pamidronate infusion in intervention group) which were not reported in one trial (Aris 2000). The remaining trial was only published as an abstract with insufficient detail to allow us to make a judgement on this domain (unclear risk of bias) (Boyle 2005).
Other potential sources of bias
There were other potential sources of bias in three trials (Aris 2004; Boyle 2005; Haworth 2001). One trial was designed to be two years in length, but few participants were willing to consent to such a lengthy study, so the protocol was revised to measure the primary endpoint at 12 months (Aris 2004). Another trial duration was planned for one year, but was shortened to six months because of adverse events (Haworth 2001). In the third trial, the study was stopped by its Data and Safety Monitoring Board after three participants experienced dramatic musculoskeletal pain, two requiring emergency room assessment. Symptoms began six to eight hours after infusion, peaked at 12 to 18 hours, and were characterized by severe chest and back pain. Along with musculoskeletal pain, one participant also experienced a fever of 104°F lasting for several hours and a rise in Tumour Necrosis Factor‐α. Although the most severe symptoms resolved within 48 to 72 hours, participants reported continued arthralgias for up to a week (Boyle 2005).
Effects of interventions
Seven trials were included, six examined participants who had not undergone lung transplantation (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008); the other studied participants received lung transplants (Aris 2000). All participants were adults.
Trials of participants who have not received lung transplant
Primary outcome
1. New fractures
Combining data from three trials for vertebral fractures at the 12‐month time‐point; there were no vertebral fractures reported for either group (20 participants in each) in the trial assessing 10 mg daily of oral alendronate (Aris 2004) or for either group (10 participants in each) in the trial assessing intravenous zolendrate every three months (Chapman 2009). However, there were two new vertebral fractures in the control group (24 participants) compared to none in the 23 participants in the group receiving 70 mg weekly of oral alendronate (Papaioannou 2008). Hence, there was no statistically significant difference in the number of participants with new non‐vertebral fractures in the treatment group compared to controls after 12 months of intervention, OR 0.19 (95% CI 0.01 to 4.21) (Analysis 1.1). At the 24‐month time‐point, data from two trials did not show any vertebral fractures in either treatment or control group (Chapman 2009; Haworth 2011) (Analysis 1.1).
1.1. Analysis.

Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 1 Vertebral fractures.
Data from the two trials which reported non‐vertebral fractures at 12 months were combined (Aris 2004; Chapman 2009). There was no statistically significant difference in the number of participants with new non‐vertebral fractures in the treatment group compared to controls, OR 2.11 (95% CI 0.18 to 25.35) (Analysis 1.2). In the Aris trial, 2 out of 20 participants in the alendronate group reported fractures (arm and rib) compared to 1 out of 20 participants in the control group (toe fracture) (Aris 2004); there were no reported fractures in the Chapman trial (Chapman 2009). Neither did Chapman report any fractures in either group at 24 months (Chapman 2009). Similarly, there were no non‐vertebral fractures in either group with the intervention of 35 mg weekly of oral risedronate for 24 months (Haworth 2011).
1.2. Analysis.
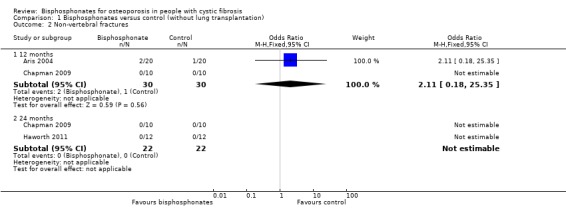
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 2 Non‐vertebral fractures.
Combining data for both vertebral and non‐vertebral fractures from the available studies, there was no significant difference between groups at 12 months, OR 0.72 (95% CI 0.13 to 3.80) and no fractures reported at 24 months (Analysis 1.3).
1.3. Analysis.
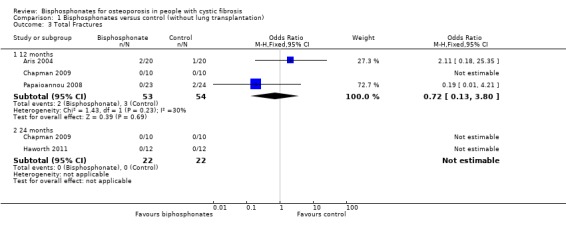
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 3 Total Fractures.
Fractures were not reported as an outcome measure in the other two trials. We contacted the authors to clarify if these had been measured in the trials, but did not receive a reply (Boyle 2005; Haworth 2001).
Secondary outcomes
1. Per cent change in BMD
a. Lumbar spine
We have presented individual time‐points (Analysis 1.4) and summary end of study data (Analysis 1.5).
1.4. Analysis.
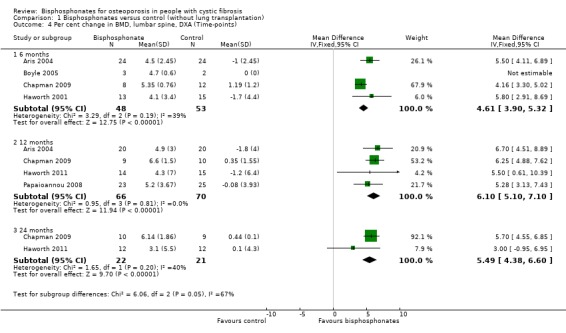
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 4 Per cent change in BMD, lumbar spine, DXA (Time‐points).
1.5. Analysis.
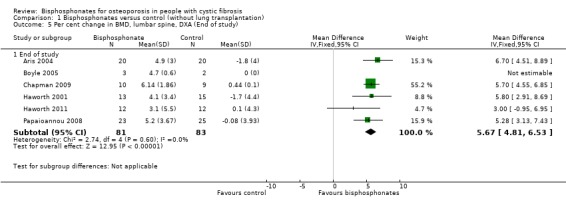
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 5 Per cent change in BMD, lumbar spine, DXA (End of study).
All six studies reported on this outcome (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008).
At the six‐month time‐point, data from the four studies showed there was a significant increase in BMD measured at the lumbar spine in the treatment group, MD 4.61 (95% CI 3.90 to 5.32) (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001) (Analysis 1.4). In the study by Aris, participants receiving oral alendronate had increased lumbar spine BMD after six months of the intervention; lumbar spine BMD decreased in control group participants at this time‐point (Aris 2004). The MD for per cent change BMD at the lumbar spine after six months was 5.50 (95% CI 4.11 to 6.89) (Aris 2004). The Boyle trial was ceased after only five participants were enrolled (three in the treatment group). The mean (SD) lumbar spine BMD had increased by mean (SD) 4.7 (0.6)% six months after the single dose of intravenous zoledronate compared to no change in the two participants in the placebo group (Boyle 2005). In the second study, intravenous zoledronate was associated with a statistically significant positive effect on BMD at the lumbar spine after six months of treatment, MD 4.16 (95% CI 3.30 to 5.02) (Chapman 2009). In the Haworth study, after six months intravenous pamidronate intervention, participants in the control group had decreased lumbar spine BMD; participants in the treatment group had gained BMD in this region, MD for per cent change lumbar spine BMD at six months was 5.80 (95% CI 2.91 to 8.69) (Haworth 2001). There was low statistical heterogeneity between studies; I2 = 39% at the six‐month time‐point.
Four studies had data available at the 12‐month time‐point (Aris 2004; Chapman 2009; Haworth 2011; Papaioannou 2008) (Analysis 1.4). These combined data showed a significant increase in BMD in favour of bisphosphonates, MD 6.10 (95% CI 5.10 to 7.10). In the study by Aris, participants in the treatment group had increased lumbar spine BMD after 12 months of the intervention. This decreased in control group participants at this time‐point, MD 6.70 (95% CI 4.51 to 8.89) (Aris 2004). In the Chapman study, the intervention showed a statistically significant positive effect on BMD at the lumbar spine after 12 months of treatment, MD 6.25 (95% CI 4.88 to 7.62) (Chapman 2009). The other studies also showed a significant increase in BMD at the lumbar spine with MD 5.50 (95% CI 0.61 to 10.39) for the oral risedronate trial (Haworth 2011) and MD 5.28 (95% CI 3.13 to 7.43) for the second oral alendronate trial (Papaioannou 2008). At this 12‐month time point, there was also low statistical heterogeneity between studies; I2 = 0% (Analysis 1.4).
Data for lumbar spine BMD at the 24‐month time‐point were reported by two trials and this result, too, was significant in favour of the treatment group, MD 5.49 (95% CI 4.38 to 6.60), although only one trial showed a statistically significant positive effect of bisphosphonates with MD 5.70 (95% CI 4.55 to 6.85) (Chapman 2009) compared with MD 3.00 (95% CI ‐0.95 to 6.95) (Haworth 2011); for this time‐point I2 = 40% (Analysis 1.4).
Pooling end of study results for trials assessing bisphosphonates, there were positive BMD effects at the lumbar spine, MD 5.67 (95% CI 4.81 to 6.53) (Analysis 1.5). There was no statistical heterogeneity between studies for lumbar spine BMD (I2 = 0%) (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008).
b. Total hip or femur
Five studies reported on this outcome (Aris 2004; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008). We have presented individual time‐points (Analysis 1.6) and summary end of study data (Analysis 1.7).
1.6. Analysis.

Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 6 Per cent change in BMD, total hip / femur, DXA (Time‐points).
1.7. Analysis.
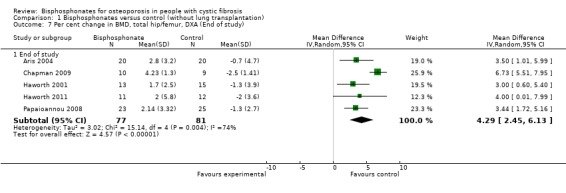
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 7 Per cent change in BMD, total hip/femur, DXA (End of study).
Combined data at six months showed a significant result in favour of bisphosphonates for total hip or femur BMD, MD 3.35 (95% CI 1.63 to 5.07) (Analysis 1.6). In the Aris trial, participants receiving oral alendronate had increased total hip or femur BMD after six months of the intervention; but this outcome was unchanged in the control group at this time, MD 2.20 (95% CI 0.81 to 3.59) (Aris 2004). Chapman reported that intravenous zoledronate was associated with a statistically significant positive effect on BMD at the total hip or femur regions after six months of treatment, MD 4.63 (95% CI 3.49 to 5.77) (Chapman 2009). In the earlier Haworth trial, after the six months intravenous pamidronate intervention, participants in the control group had decreased total hip or femur BMD; participants in the treatment group had gained BMD in this region, MD 3.00 (95% CI 0.60 to 5.40) (Haworth 2001).
Data from four trials were combined at the 12‐month time‐point and showed a significant increase in total hip or femur BMD, MD 4.35 (95% 2.99 to 5.70) (Aris 2004; Chapman 2009; Haworth 2011; Papaioannou 2008) (Analysis 1.6). At 12 months, participants in the Aris trial receiving oral alendronate had increased total hip or femur BMD but this decreased compared to baseline in the control group, MD 3.50 (95% CI 1.01 to 5.99) (Aris 2004). Chapman reported a statistically significant positive effect on BMD at the total hip or femur regions in the zolendrate group after 12 months of treatment, MD 5.71 (95% CI 4.25 to 7.17) (Chapman 2009). Papaioannou also showed a significant increase in BMD at the total hip or femoral neck with MD 3.44 (95% CI 1.72 to 5.16) (Papaioannou 2008). The trial assessing oral risedronate did not show a significant increase in BMD at this site at 12 months, MD 3.80 (95% CI ‐0.43 to 8.03) (Haworth 2011).
At the 24‐month time‐point, the two studies were also significant in favour of the intervention, MD 6.05 (CI 95% 3.74 to 8.36) (Chapman 2009; Haworth 2011) (Analysis 1.6).
Pooling end of study results for all bisphosphonates, there were positive BMD effects at the total hip or femur, MD 4.29 (95% CI 2.45 to 6.13) (Analysis 1.7).
There was substantial heterogeneity between studies for total hip or femur (I2 = 73% at six months) and (I2 = 74% for end of study). At the 12‐month and 24‐month time points, there was low statistical heterogeneity between studies; I2 = 38% and 39% respectively.
c. Radius
Please refer to Analysis 1.8, Analysis 1.9 and Analysis 1.10.
1.8. Analysis.

Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 8 Per cent change in BMD, distal radius, SXA (Time‐points).
1.9. Analysis.
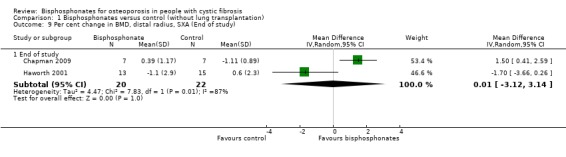
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 9 Per cent change in BMD, distal radius, SXA (End of study).
1.10. Analysis.
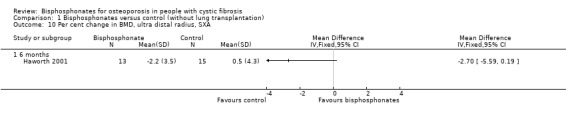
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 10 Per cent change in BMD, ultra distal radius, SXA.
Two studies reported data for distal radius (Chapman 2009; Haworth 2001) and one reported data for ultra distal radius (Haworth 2001).
Combined data for distal radius at the six‐month time‐point do not show a significant result, MD ‐0.49 (95% CI ‐2.42 to 1.45). In the Chapman study, distal radial BMD was not statistically different from the control group at six months, MD 0.32 (95% CI ‐0.50 to 1.14). In the Haworth study, there was a non‐significant decrease in BMD as measured by SXA of the distal forearm, MD ‐1.70 (95% CI ‐3.66 to 0.26) (Analysis 1.8).
Only Chapman reported data for the 12‐month and 24‐month time‐points. At 12 months, distal radial BMD was not statistically different from the control group, MD 0.32 (95% CI ‐0.30 to 0.94). However, at 24 months, this was significant in favour of bisphosphonates, MD 1.50 (95% CI 0.41 to 2.59) (Chapman 2009) (Analysis 1.8).
Pooling end of study results, there was no significant change in BMD at the distal forearm, MD 0.01 (95% CI ‐3.12 to ‐3.14) (Chapman 2009; Haworth 2001) (Analysis 1.9).
As is the case for total hip or femur, there was substantial statistical heterogeneity between studies for distal radius (I2 = 71% at 6 months).
In the Haworth study, measurements of appendicular sites showed opposite trends. There was a non‐significant decrease in BMD as measured by SXA of the ultra distal forearm in participants receiving pamidronate at six months, MD ‐2.70 (95% CI ‐5.59 to 0.19) (Haworth 2001) (Analysis 1.10).
2. Quality of life
No quality of life measurements were reported in five of the trials (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011). One trial of oral alendronate assessed mean changes over 12 months using the physical component score and mental component score of the Medical Outcomes Study 36‐item short form, version 2 (SF‐36v2) (Papaioannou 2008) (Analysis 1.11). There was no significant change in the physical component score, MD 2.51 (95%CI ‐1.38 to 6.40); mental component score, MD ‐5.93 (95% CI ‐11.73 to ‐0.13); and total score MD ‐0.11 (95% CI ‐3.35 to 3.12).
1.11. Analysis.
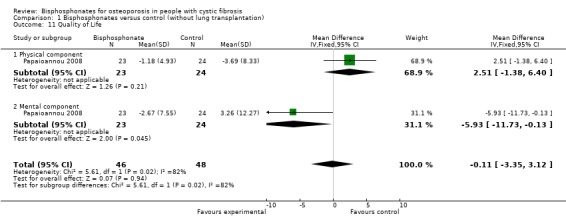
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 11 Quality of Life.
3. Adverse events
Six studies reported on adverse events (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008).
We were able to present combined data in a meta‐analysis for bone pain (Analysis 1.12), fever (Analysis 1.13) and information on gastrointestinal adverse effects. For this outcome information was not available per patient for all studies, so has been presented narratively. If the required information from the study investigators becomes available, a meta‐analysis will be performed for a future update of the review.
1.12. Analysis.
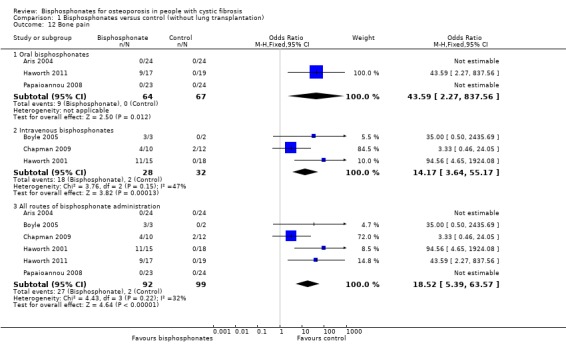
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 12 Bone pain.
1.13. Analysis.
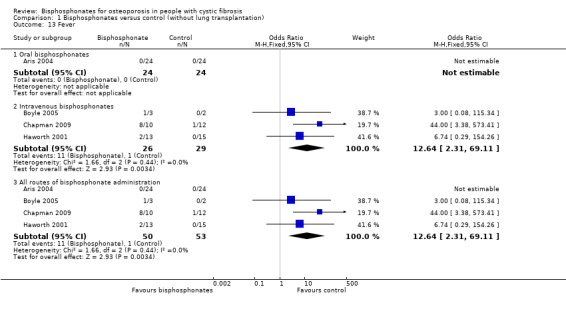
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 13 Fever.
a. Bone Pain
In one study of oral alendronate for 12 months, none of the participants in either the intervention or the control group experienced bone pain (Aris 2004). The other study also assessing oral alendronate for 12 months did not report bone pain in any participants; three of the participants in the treatment group used oral corticosteroids with the mean yearly cumulative dose 49.32 mg (Papaioannou 2008). In the 24‐month trial of oral risedronate, four participants withdrew completely due to bone pain (three in the first 12 months) and one participant discontinued the study medication due to bone pain. The Haworth trial of oral risedronate reported that bone pain was recorded 19 times by nine different risedronate patients in the first year after randomisation, while there were no episodes of bone pain in the placebo group, OR 43.59 (95% CI 2.27 to 837.56). Fourteen of 19 (74%) episodes occurred within the first 56 days after randomisation. Five events (26%) were classified as severe and led to the discontinuation of the study medication. Of the other events, ten were classified as moderate and four as mild. Only two (11%) events were considered to be unrelated to the study medication. Bone pain usually started within 24 hours of the first dose of risedronate and lasted for approximately 72 hours (Haworth 2011).
Pooling the results for intravenous bisphosphonates, including the study in which participants received oral prednisone daily for three days on the morning of the first infusion (repeated with subsequent infusions if a reaction to the first infusion was thought likely), the occurrence of bone pain was significantly higher in participants in the treatment groups (18 out of 28) compared to control groups (2 out of 32), OR 14.17 (95% CI 3.64 to 55.17) (Boyle 2005; Chapman 2009; Haworth 2001). Boyle reported that three participants experienced dramatic musculoskeletal pain, two requiring emergency room assessment. Symptoms began six to eight hours after infusion, peaked at 12 to 18 hours, and were characterized by severe chest and back pain. Although the most severe symptoms resolved within 48 to 72 hours, participants reported continued arthralgias for up to a week. It was presumed that these three participants were those in the treatment group (no reply was received to an attempt to contact the author) (Boyle 2005). Chapman reported that there was no significant difference between treatment and control groups with respect to musculoskeletal pain (4 out of 10 in the zoledronate group, 2 out of 12 in the control group), OR 3.33 (95% CI 0.46 to 24.05) (Chapman 2009). In the Haworth 2001 trial, no participants in the control group experienced bone pain; but 11 out of 15 participants in the treatment group experienced moderate to severe pain following the first dose of medication, OR 94.56 (95% CI 4.65 to 1924.08). Nine participants reported severe bone pain. The pain was reported to be excruciating in seven participants rendering them bed bound and making sputum expectoration and physiotherapy difficult. None of the participants experiencing bone pain were taking corticosteroids, while three of the four participants without bone pain in the treatment group did (Haworth 2001).
Combining study results for all routes of administration, bisphosphonates were associated with significantly higher occurrence of bone pain, OR 18.52 (95% CI 5.39 to 63.57) (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008).
b. Fever
In the 12‐month study of oral alendronate, none of the participants in either the intervention or the control group experienced fever (Aris 2004). In the other trials of oral bisphosphonates, it was not stated that fever was one of the adverse events that were reported (Haworth 2011; Papaioannou 2008).
Combined data from the trials of intravenous bisphosphonates show participants in the treatment groups were significantly more likely to experience fever, OR 12.64 (95% CI 2.31 to 69.11) (Boyle 2005; Chapman 2009; Haworth 2001). Boyle reported that one participant experienced a fever of 104°F lasting for several hours and a rise in Tumour Necrosis Factor‐α, OR 3.00 (95% CI 0.08 to 115.34) (Boyle 2005). In the Chapman trial the number of participants experiencing fever was significantly higher in the treatment group (8 out of 10 in the zoledronate group, 1 out of 12 in the control group), OR 44.00 (95% CI 3.38 to 573.41) (Chapman 2009). Haworth reported that two of the nine participants in the treatment group had febrile reactions, OR 6.74 (95% CI 0.29 to 154.26) (Haworth 2001).
c. Gastrointestinal adverse effects
Gastrointestinal adverse effects were described in the trials assessing oral bisphosphonates (Aris 2004; Haworth 2011; Papaioannou 2008). In the Aris study, three cases of diarrhoea were reported; one participant was receiving alendronate and two were receiving placebo. This was accompanied by abdominal cramping and loss of appetite. These participants continued to experience diarrhoea after discontinuing the study medication. One participant reported dysphagia but it was not clear which study group they were in (Aris 2004). In Papaioannau's trial of oral alendronate, there were 10 gastrointestinal‐related adverse events in the treatment group (three events of nausea or vomiting or both and one event of each of reflux, difficulty swallowing, oesophagitis, constipation, gastrointestinal upset, intestinal obstruction and stomach pain or burn). There were seven gastrointestinal‐related adverse events in the control group (four events of nausea or vomiting or both and one event of each of constipation, intestinal obstruction and stomach pain or burn) (Papaioannou 2008). Gastrointestinal symptoms were also reported in the oral risedronate trial (26 in the treatment group and 16 in the placebo group). These events corresponded to the same number of participants in each study group (nine in risedronate, nine in placebo) (Haworth 2011).
d. Other adverse effects
Chapman reported that musculoskeletal side effects were experienced following 27 of 63 zoledronate infusions, but after only 4 of 73 placebo infusions. This was despite administering prednisolone to all participants with at least the first infusion, and these side effects were more common after the first than the subsequent infusions. In six participants, the reactions were described as severe, in two cases resulting in withdrawal. One of these participants required admission to hospital with severe pain and fever restricting movement, with onset 12 hours after the first infusion; this resolved after four days, but led to subsequent withdrawal from the study. The other participant experienced fever, rigor, bone pain and headache 24 hours after the first infusion with resolution in five days. Similar effects were experienced after the second infusion followed by study withdrawal (Chapman 2009).
In the 24‐month trial of oral risedronate, two participants discontinued the study medication in the first 12 months due to muscle aches or generalised pain. Within two years of randomisation 346 adverse events were recorded; most events were classed as respiratory exacerbations (n = 230) and it was stated that there was no difference in incidence between the two groups (Haworth 2011).
In the earlier Haworth trial, it was reported that one participant developed phlebitis around the infusion site (Haworth 2001).
Papaionnou reported serious adverse events in the alendronate group as exacerbation of CF (n = 3), bronchial superinfection (n = 1), hypoglycaemic seizure (n = 1), gastrointestinal obstruction (n = 1) and intestinal obstruction (n = 1). Serious adverse events reported for the control group included an exacerbation of CF (n = 3), with two of these participants experiencing additional gastrointestinal complaints (Papaioannou 2008).
None of the studies assessed calcium levels post‐intravenous bisphosphonate infusion (Boyle 2005; Chapman 2009; Haworth 2001).
4. Withdrawals
We have presented data for withdrawals due to adverse events (Analysis 1.14) and total withdrawals (Analysis 1.15).
1.14. Analysis.
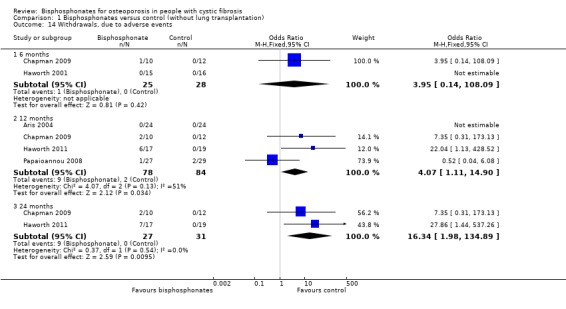
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 14 Withdrawals, due to adverse events.
1.15. Analysis.
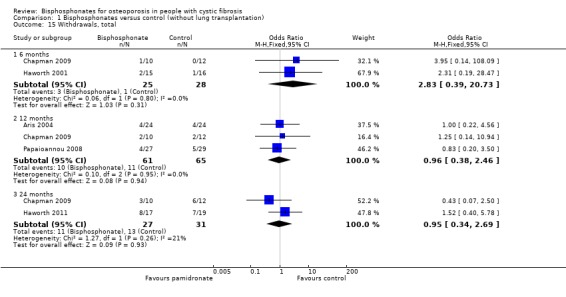
Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 15 Withdrawals, total.
Five studies described withdrawals from the study. At 12 and 24 months, there were significantly more withdrawals due to adverse events in the treatment group compared to the control group, OR 4.07 (95% CI 1.11 to 14.90) and 16.34 (95% CI 1.98 to 134.89) respectively. At 6, 12 and 24 months, data from these trials do not show any significant difference between treatment or control groups for total withdrawals for any reason (Aris 2004; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008).
In the Aris study, of the 53 participants who started the protocol, five dropped out in the first six months. The reasons included pregnancy (n = 1, experienced a spontaneous abortion in her first trimester), dysphagia (n = 1, experienced difficulty swallowing the first tablet and had similar difficulties swallowing other medications), and diarrhoea (n = 3). For the first two participants, it was unclear which group they were allocated to. One of the participants experiencing diarrhoea was receiving alendronate and two were receiving placebo. No participants withdrew due to gastrointestinal reflux symptoms. Of the 48 participants who completed DXA assessment at six months beyond baseline, 4 out of 24 in each group withdrew before the DXA assessment at 12 months beyond baseline. In the treatment group, the withdrawals were due to lung transplantation (n = 1), moving (n = 2) and non‐compliance (n = 1); in the control group these were due to lung transplantation (n = 2), death (n = 1) and moving (n = 1) (Aris 2004).
In the first 12 months of the later Haworth trial, 3 out of 17 in the oral risedronate group withdrew from the study completely (due to bone pain) and three patients discontinued the study medication (one citing bone pain and the other two participants citing muscle aches or generalised pain) but remained in the study for follow up. Between 12 and 24 months one further participant withdrew from the intervention group (citing bone pain). At 24 months, 12 participants remained in study with nine still taking the study drug. Immediately after randomisation one participant in the placebo group withdrew consent before taking the study medication. Therefore, only 18 participants were commenced on placebo. By 12 months, two participants in the placebo group withdrew consent and one participant had died. By 24 months, three participants in the placebo group had withdrawn consent and four patients had died, hence at 24 months, 12 participants remained in the control group of the study. In the 24 months there was no significant difference between groups with respect to total withdrawals (8 of 17 in the risedronate group ceased the medication, 7 out of 19 in the control group), OR 1.52 (95% CI 0.40 to 5.78) (Haworth 2011).
In the 12‐month trial by Papaioannou, 4 of 27 in the oral alendronate group withdrew (one due to an adverse event, one withdrew consent and two due to non‐compliance). In the placebo group, 5 of 29 withdrew (two due to non‐compliance, two due to an adverse event and one was lost to follow‐up) (Papaioannou 2008).
At 24 months, Chapman reports there was no significant difference between intravenous zoledronate and control with respect to total withdrawals (3 out of 10 in the treatment group, 6 out of 12 in the control group), OR 0.43 (95% CI 0.07 to 2.50) or withdrawals due to adverse events (2 out of 10 in the treatment group, 0 of 12 in the treatment group), OR 7.35 (0.31 to 173.13). The adverse events have been described previously. The withdrawal that was unrelated to adverse events in the treatment group was due to loss to follow‐up. The six withdrawals in the control group were due to loss to follow‐up (n = 1), decreased BMD to withdrawal threshold (n = 3), poor compliance to study requirements (n = 1) and a combination of the latter two reasons (n = 1) (Chapman 2009).
Haworth also reports no significant difference between intravenous pamidronate and control groups with respect to total withdrawals over the six months (2 out of 15 in the pamidronate group, 1 out of 16 in the control group), OR 2.31 (95% CI 0.19 to 28.47). One participant from each group died, while a second participant in the pamidronate group withdrew in order to receive a double lung transplant. No participant withdrew due to other adverse events (Haworth 2001).
5. Survival
We have presented data on this outcome in the graphs (Analysis 1.16).
1.16. Analysis.

Comparison 1 Bisphosphonates versus control (without lung transplantation), Outcome 16 Survival.
Of the 48 participants in the oral alendronate trial who completed DXA assessment at six months beyond baseline, one participant out of 24 in the control group died during the latter six months of the study compared to none in the treatment group. This difference was not statistically significant, OR 3.13 (95% CI 0.12 to 80.68) (Aris 2004). Combining this survival data with the 12‐month outcome data of the oral risedronate trial did not show a statistically significant difference, OR 2.98 (95% CI 0.30 to 29.84) (Aris 2004; Haworth 2011).
Three of the trials reported there were no deaths in either the treatment or control group (Boyle 2005; Chapman 2009; Papaioannou 2008).
Haworth reported that intravenous pamidronate treatment for a duration of six months did not significantly affect survival compared with the control group (14 out of 15 survivors in the pamidronate group, 15 out of 16 in the control group), OR 0.93 (95% CI 0.05 to 16.39) (Haworth 2001).
Haworth also reported that oral risedronate treatment for 24 months did not significantly affect survival compared with the control group (16 out of 17 survivors in the risedronate group, 15 out of 19 in the control group), 4.27 (95% CI 0.43 to 42.63) (Haworth 2011).
Trial of participants who have received lung transplant
Only one trial reported on this comparison (Aris 2000).
Primary outcome
1. New fractures
There was no statistically significant difference in the number of participants with new vertebral fractures in the bisphosphonate (pamidronate) group (3 out of 16 participants) versus the control group (1 out of 18 participants), OR 3.92 (95% CI 0.36 to 42.20) (Analysis 2.1). There was also no statistically significant difference in the number of participants with new non‐vertebral fractures in the treated (3 out of 16 participants) versus the untreated group (6 out of 18 participants), OR 0.46 (95% CI 0.09 to 2.27) (Analysis 2.2). Considering any fractures, there was also a non‐significant result, OR 0.94 (95% CI 0.24 to 3.77) (Analysis 2.3).
2.1. Analysis.
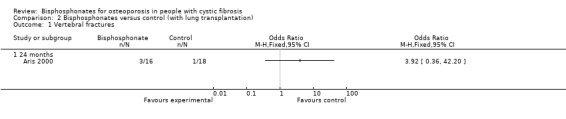
Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 1 Vertebral fractures.
2.2. Analysis.
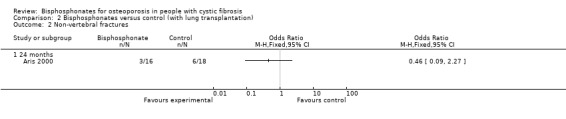
Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 2 Non‐vertebral fractures.
2.3. Analysis.

Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 3 Total Fractures.
Secondary outcomes
1. Per cent change in BMD
a. Lumbar spine
At the end of two years, the per cent change in BMD was significantly higher in the treatment group than in the control group at the lumbar spine, MD 6.20 (95% CI 4.28 to 8.12) (Analysis 2.4).
2.4. Analysis.
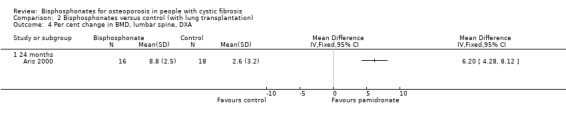
Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 4 Per cent change in BMD, lumbar spine, DXA.
b. Hip or femur
Likewise, at the end of two years, the per cent change in BMD was significantly higher in the treatment group than in the control group at the femur, MD 7.90 (95% CI 5.78 to 10.02) (Analysis 2.5).
2.5. Analysis.

Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 5 Per cent change in BMD, femur, DXA.
2. Quality of life
No quality of life measurements were reported (Aris 2000).
3. Adverse events
None of these participants, all of whom were receiving corticosteroids, experienced any bone pain (Analysis 2.6). Participants were assessed 24‐hours post‐infusion for cellulitis, thrombophlebitis, or fever; no cases were detected. Twenty‐four hours later, serum calcium, phosphorus, and magnesium and a complete blood count were analysed. The study authors reported that there were no significant differences compared with pre‐infusion data, with no episodes of hypocalcaemia and three episodes of mild hypervitaminosis D that resolved spontaneously. The trial in post‐transplant participants also reported that there was no significant difference in the degree of immunosuppression between the treatment and control groups (Aris 2000).
2.6. Analysis.
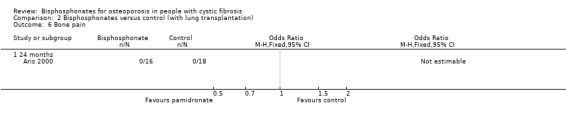
Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 6 Bone pain.
4. Withdrawals
There were no withdrawals other than those due to death in either treatment group (Analysis 2.7; Analysis 2.8).
2.7. Analysis.
Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 7 Withdrawals, due to adverse events.
2.8. Analysis.

Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 8 Withdrawals, total.
5. Survival
Three participants died before the first BMD data could be collected at six months; these participants were excluded from further analysis and thus cannot be included as data in this review. The authors stated that the exclusion of these three participants did not significantly affect the subsequent analysis (Aris 2000). No other participants died during the course of the trial (Analysis 2.9).
2.9. Analysis.
Comparison 2 Bisphosphonates versus control (with lung transplantation), Outcome 9 Survival.
Sensitivity analysis
a. Differences in the medications used in the intervention and comparison groups
Considering non‐transplanted participants, the differences in adverse events between oral and intravenous bisphosphonates have been described. The per cent increase in BMD at the lumbar spine still increased significantly at six months when oral bisphosphonates were analysed separately to intravenous bisphosphonates; oral bisphosphonates, MD for per cent change was 5.50 (95% CI 4.11 to 6.89) (Aris 2004), intravenous bisphosphonates, MD 4.29 (95% CI 3.47 to 5.12) (Boyle 2005; Chapman 2009; Haworth 2001). This was also observed for the effect on lumbar spine BMD (12 months and end of study) and hip or femur BMD (6 months, 12 months, 24 months and for outcomes at the end of study). At 24 months, only intravenous bisphosphonates significant increased BMD at the lumbar spine.
b. Analysis using random‐effects model
Combining data for both vertebral and non‐vertebral fractures from the available studies, there was still no significant difference between groups using the random‐effects model, OR 0.76 (95% CI 0.07 to 7.92) (Analysis 1.3).
Using the random‐effects model, the MD results for per cent increases in BMD for lumbar spine were still significant at the six‐month time‐point, MD 4.81 (95% CI 3.75 to 5.88); the 12‐month time‐point, MD 6.10 (95% CI 5.10 to 7.10); the 24‐month time‐point, MD 5.04 (95% CI 2.76 to 7.31) and end of study, MD 5.67 (95% CI 4.81 to 6.53) (Analysis 1.4; Analysis 1.5).
Using the random‐effects model, the MD results for per cent increases in BMD for total hip or femur were also still significant at the six‐month time‐point, MD 3.35 (95% CI 1.63 to 5.07); the 12‐month time‐point, MD 4.35 (95% CI 2.99 to 5.70); the 24‐month time‐point, MD 6.05 (95% CI 3.74‐8.36) and end of study, MD 4.29 (95% CI 2.45 to 6.13) (Analysis 1.6; Analysis 1.7).
c. Analysis by "treatment received"
Due to the lack of data, this was not possible.
d. Analysis by "intention‐to‐treat"
Due to the lack of data, this was not possible.
Discussion
Six studies investigated the effect of bisphosphonates in adults with CF who had not undergone lung transplantation (Aris 2004; Boyle 2005; Chapman 2009; Haworth 2001; Haworth 2011; Papaioannou 2008). One study assessed this in adults with CF who had undergone lung transplantation (Aris 2000). Oral and intravenous bisphosphonates increased BMD in adults with CF. However, severe bone pain and flu‐like symptoms were common with intravenous agents, especially in participants not taking glucocorticoids. A reduction in fracture rate was not observed.
Oral bisphosphonates (alendronate and risedronate) were associated with increased BMD at the lumbar spine and hip or femur regions in adults with CF who had not undergone lung transplantation in the meta‐analysis from three studies (Aris 2004; Haworth 2011; Papaioannou 2008). Three trials assessing different intravenous bisphosphonate regimens (agent, duration and frequency) in this population group also showed improved lumbar spine BMD (Boyle 2005; Chapman 2009; Haworth 2001). Both trials with hip or femur BMD as a measure showed positive effects on this outcome (Chapman 2009; Haworth 2001). The two studies that assessed radial BMD had conflicting results. Radial BMD was significantly improved in the zoledronate study of 24‐months duration (Chapman 2009), but not in the pamidronate study with six‐months follow‐up (Haworth 2001).
There was significant clinical heterogeneity between the studies but in the majority, only low statistical heterogeneity.
Lumbar spine and hip BMD were also improved by intravenous pamidronate in a single trial in adult CF participants post‐lung transplantation (Aris 2000).
These trials provide valuable data on two different populations; adults with CF who have received a transplant and those who have not. Although the inclusion criteria, duration of follow up and the magnitude of effect were different for the trials, similar trends for BMD effect were seen, suggesting that the beneficial effects of oral and intravenous bisphosphonates might be generalisable to a fairly broad population of people living with CF.
Bone mineral density is only an intermediate outcome. The more clinically important endpoint is the occurrence of new fractures. There was no significant effect of treatment on fractures (total, vertebral or non‐vertebral) in participants with or without lung transplantation (Aris 2000; Aris 2004; Chapman 2009; Haworth 2011; Papaioannou 2008).
Only one trial assessed quality of life, with no significant effect of intervention on physical or mental components of the score (Papaioannou 2008).
Fever was not reported in association with oral bisphosphonates (Aris 2004). Gastro‐oesophageal symptoms were reported, but not significantly increased, in the trials of oral bisphosphonates (Aris 2004; Haworth 2011; Papaioannou 2008).
Bone pain was reported in one of the three trials of oral bisphosphonates (Haworth 2011). Participants who did not receive corticosteroids during the clinical trial of intravenous pamidronate were more likely to experience bone pain (Haworth 2001). The observed relationship between the regular use of glucocorticoid therapy and lack of bone pain may be explained by evidence that corticosteroids suppress the release of TNF‐alpha (Steer 1997), an inflammatory cytokine known to increase bone resorption. Haworth suggests that bone pain may be avoided by prescribing a short course of oral corticosteroids before and at the time of pamidronate infusion (Haworth 2001). Zoledronate, a more potent bisphosphonate than pamidronate was associated with severe musculoskeletal pain resulting in discontinuation of one trial early in enrolment (Boyle 2005). In a subsequent study of intravenous zoledronate, participants were prescribed daily prednisolone for three days commencing on the day of the first infusion (and repeated if thought necessary with subsequent infusions). Musculoskeletal pain was not significantly increased in the treatment group. However, fever and other flu‐like effects were more commonly reported, severe in some participants, although the majority continued treatment. The authors proposed that a longer period of treatment with corticosteroids, including pre‐treatment dosing may diminish such adverse effects (Chapman 2009). A consensus statement has recommended pre‐treatment with oral corticosteroids for three to five days prior to bisphosphonate infusion, but this has not been studied in an RCT (Aris 2005). None of the participants in the lung‐transplanted group, all of whom were receiving corticosteroids, experienced any bone pain.
For treatment duration of 12 to 24 months, there were significantly more withdrawals due to adverse events than in those not receiving bisphopsphonates. There was no significant effect on survival. The lack of significant differences in the rate of fractures, gastrointestinal adverse effects, withdrawals or survival may relate to the small numbers of participants involved and the short duration of the trials
The risk of bias in the trials due to blinding was judged as present (Aris 2000; Haworth 2001), unclear (Aris 2004; Boyle 2005; Chapman 2009) or low (Haworth 2011; Papaioannou 2008). When participants are aware of the treatment they are receiving, they may be more or less likely to report adverse events. The judgment of individuals who collect and interpret patient data may be affected when the assessor is aware of the treatment a participant is receiving. Lack of blinding may result in biased results.
Authors' conclusions
Implications for practice.
Based on seven small trials, oral and intravenous bisphosphonates cause a rapid increase in spine and hip or femoral BMD in adults with CF. Only two trials assessed the effect on distal radial BMD, with only the study of longer duration reporting an improvement. Severe bone pain is common with the use of intravenous bisphosphonates in participants with CF not receiving oral corticosteroids, and may limit tolerability. Severe bone pain was also observed in one of three trials of oral bisphosphonates. Oral bisphosphonate treatment did not cause increased gastrointestinal symptoms. Only one trial assessed quality of life and did not show a benefit. These short‐term trials (maximum of two years) did not show fracture reduction or survival benefit. Currently, no other options for the treatment of CF‐related osteoporosis have been reported in the public domain. Thus, although bisphosphonates increase BMD, no recommendation can be made concerning the use of bisphosphonates in participants with CF as an effect on fractures could not be demonstrated.
Implications for research.
This area of research would benefit from a large multicentre RCT of bisphosphonates (oral and intravenous) with separate analyses for participants with and without lung transplantation to measure the effectiveness of these therapies on outcomes important to people with CF, such as fractures and survival. These outcomes would require long‐term studies. Data on adverse events such as gastrointestinal events, bone pain and fever, in addition to the use of corticosteroids should also be recorded. Studies assessing pre‐treatment with corticosteroids are required.
What's new
| Date | Event | Description |
|---|---|---|
| 13 April 2015 | Amended | Contact details updated. |
History
Protocol first published: Issue 2, 2000 Review first published: Issue 4, 2001
| Date | Event | Description |
|---|---|---|
| 13 March 2014 | New citation required but conclusions have not changed | Text updated with new references. No new studies added, hence conclusions remain the same. |
| 13 March 2014 | New search has been performed | A search of the Cystic Fibrosis & Genetic Disorders Group's Cystic Fibrosis Trials Register did not identify any new studies potentially eligible for inclusion in this review. |
| 15 February 2012 | New citation required but conclusions have not changed | One of the new references identified was the full paper publication of a study previously identified as an e‐publication ahead of print and included (Chapman 2009). The other study (two references) has been included in this update (Haworth 2011). A study previously listed as 'Awaiting classification' has also now been included (Papaioannou 2008). |
| 15 February 2012 | New search has been performed | A search of the Group's Cystic Fibrosis Trials Register identified three new references to two separate studies. |
| 26 April 2010 | Amended | Contact details updated. |
| 18 July 2009 | New citation required and conclusions have changed | A new review team has updated the review. As part of this update the sections 'Methods' and 'Risk of Bias in included studies' have been substantively revised and updated. |
| 18 July 2009 | New search has been performed | A search of the Group's Cystic Fibrosis Trials Register was run in October 2008. Five references to four new trials were identified; two trials have been included (Aris 2004; Chapman 2009) and two trials have been excluded (Conway 2004; Hardin 2005). One newly identified trial has been listed as 'Awaiting classification' until the authors have been able to contact the trial investigators (Papaioannou 2008a). |
| 10 November 2004 | New search has been performed | A search of the Group's Cystic Fibrosis Trials Register was run in August 2004, but no new references were identified. |
| 12 November 2003 | New search has been performed | The searches identified no new studies to be included in the review. Minor style changes have been made. |
| 13 November 2002 | New search has been performed | An additional reference [abstract] to the Haworth 2001 trial has been incorporated into the review. |
| 20 August 2001 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We acknowledge the efforts of Dr Christine Brenkmann and Dr Alexandra Papaioannou who performed the original review and to the Cochrane CF and Genetic Disorders Review Group (Olwen Beaven, Rosalind Smyth, Ruaraidh Hill, Tracey Remmington and Gerard Ryan) and the external peer reviewers for their time and valuable comments during the preparation of the original review. The current authors similarly acknowledge the assistance of the Cochrane CF and Genetic Disorders Review Group (Nikki Jahnke, Natalie Hall) and associated referees during the update of this review.
Dr. Charles Haworth and Dr. Robert Aris generously provided unpublished data for the analyses for the original review. That data is now published. Dr Haworth also provided unpublished data related to his later trial for inclusion in this update (Haworth 2011).
Appendices
Appendix 1. PubMed search strategy
| Search terms |
| zoledronate AND cystic fibrosis |
Appendix 2. PubMed search strategy
| Search terms |
| "Diphosphonates" [Mesh] AND "Cystic Fibrosis" [Mesh] |
Data and analyses
Comparison 1. Bisphosphonates versus control (without lung transplantation).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Vertebral fractures | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 12 months | 3 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.01, 4.21] |
| 1.2 24 months | 2 | 44 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Non‐vertebral fractures | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 12 months | 2 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.11 [0.18, 25.35] |
| 2.2 24 months | 2 | 44 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Total Fractures | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 12 months | 3 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.13, 3.80] |
| 3.2 24 months | 2 | 44 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Per cent change in BMD, lumbar spine, DXA (Time‐points) | 6 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 6 months | 4 | 101 | Mean Difference (IV, Fixed, 95% CI) | 4.61 [3.90, 5.32] |
| 4.2 12 months | 4 | 136 | Mean Difference (IV, Fixed, 95% CI) | 6.10 [5.10, 7.10] |
| 4.3 24 months | 2 | 43 | Mean Difference (IV, Fixed, 95% CI) | 5.49 [4.38, 6.60] |
| 5 Per cent change in BMD, lumbar spine, DXA (End of study) | 6 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 End of study | 6 | 164 | Mean Difference (IV, Fixed, 95% CI) | 5.67 [4.81, 6.53] |
| 6 Per cent change in BMD, total hip / femur, DXA (Time‐points) | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 6 months | 3 | 96 | Mean Difference (IV, Random, 95% CI) | 3.35 [1.63, 5.07] |
| 6.2 12 months | 4 | 135 | Mean Difference (IV, Random, 95% CI) | 4.35 [2.99, 5.70] |
| 6.3 24 months | 2 | 42 | Mean Difference (IV, Random, 95% CI) | 6.05 [3.74, 8.36] |
| 7 Per cent change in BMD, total hip/femur, DXA (End of study) | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 End of study | 5 | 158 | Mean Difference (IV, Random, 95% CI) | 4.29 [2.45, 6.13] |
| 8 Per cent change in BMD, distal radius, SXA (Time‐points) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 6 months | 2 | 48 | Mean Difference (IV, Random, 95% CI) | ‐0.49 [‐2.42, 1.45] |
| 8.2 12 months | 1 | 18 | Mean Difference (IV, Random, 95% CI) | 0.32 [‐0.30, 0.94] |
| 8.3 24 months | 1 | 14 | Mean Difference (IV, Random, 95% CI) | 1.5 [0.41, 2.59] |
| 9 Per cent change in BMD, distal radius, SXA (End of study) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1 End of study | 2 | 42 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐3.12, 3.14] |
| 10 Per cent change in BMD, ultra distal radius, SXA | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 10.1 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Quality of Life | 1 | 94 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐3.35, 3.12] |
| 11.1 Physical component | 1 | 47 | Mean Difference (IV, Fixed, 95% CI) | 2.51 [‐1.38, 6.40] |
| 11.2 Mental component | 1 | 47 | Mean Difference (IV, Fixed, 95% CI) | ‐5.93 [‐11.73, ‐0.13] |
| 12 Bone pain | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 Oral bisphosphonates | 3 | 131 | Odds Ratio (M‐H, Fixed, 95% CI) | 43.59 [2.27, 837.56] |
| 12.2 Intravenous bisphosphonates | 3 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 14.17 [3.64, 55.17] |
| 12.3 All routes of bisphosphonate administration | 6 | 191 | Odds Ratio (M‐H, Fixed, 95% CI) | 18.52 [5.39, 63.57] |
| 13 Fever | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 Oral bisphosphonates | 1 | 48 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.2 Intravenous bisphosphonates | 3 | 55 | Odds Ratio (M‐H, Fixed, 95% CI) | 12.64 [2.31, 69.11] |
| 13.3 All routes of bisphosphonate administration | 4 | 103 | Odds Ratio (M‐H, Fixed, 95% CI) | 12.64 [2.31, 69.11] |
| 14 Withdrawals, due to adverse events | 5 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 6 months | 2 | 53 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.95 [0.14, 108.09] |
| 14.2 12 months | 4 | 162 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.07 [1.11, 14.90] |
| 14.3 24 months | 2 | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 16.34 [1.98, 134.89] |
| 15 Withdrawals, total | 5 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 6 months | 2 | 53 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.83 [0.39, 20.73] |
| 15.2 12 months | 3 | 126 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.38, 2.46] |
| 15.3 24 months | 2 | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.34, 2.69] |
| 16 Survival | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 6 months | 2 | 36 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.05, 16.39] |
| 16.2 12 months | 3 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.98 [0.30, 29.84] |
| 16.3 24 months | 2 | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.27 [0.43, 42.63] |
Comparison 2. Bisphosphonates versus control (with lung transplantation).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Vertebral fractures | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 24 months | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Non‐vertebral fractures | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 24 months | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Total Fractures | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 24 months | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Per cent change in BMD, lumbar spine, DXA | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 24 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Per cent change in BMD, femur, DXA | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 24 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Bone pain | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1 24 months | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Withdrawals, due to adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8 Withdrawals, total | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1 24 months | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Survival | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1 24 months | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Aris 2000.
| Methods | Randomised controlled trial, parallel design. Trial duration 2 years. Single centre, university hospital, USA. |
|
| Participants | Inclusion criteria: CF; 1 to 12 months post‐lung transplantation; ambulatory.
Exclusion criteria: primary graft failure or other post‐operative morbidities that precluded long‐term survival; renal insufficiency (serum creatinine > 3.0 mg/dl); or pregnancy. Total participants: n = 34 (17 female). Treatment group: n = 16 (7 female); mean (SD) age 27.5 years (6.6 years); Control group: n = 18 (10 female); mean (SD) age 29.1 years (6.4 years). Groups similar in age, gender, baseline T‐scores, renal function, hospitalisation rates, immunosuppressant levels, change in lung function and BMI over study period. 13 in treatment group and 12 controls had baseline T‐scores < ‐2.5 at a minimum of one site; all others ‐1 < T < ‐2.5 at a minimum of one site. |
|
| Interventions | Treatment group: intravenous pamidronate (30 mg every 3 months) plus oral vitamin D (800 IU/day) and oral calcium (1 g/day) Control group: oral vitamin D (800 IU/day) and oral calcium (1 g/day) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | 44 people with CF were eligible during the course of this study, 7 died immediately post‐operatively and were therefore not eligible for this trial. As outlined above, 3 people died during the course of the study before the first primary end point measurement. 34 people were included in the final analyses. Funding was provided by grants from the CF Foundation and the Verne S. Caviness General Center for Clinical Research. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Blocks of four" design stated (stratified on basis of gender and severity of osteoporosis using spine z score of ‐3.0), but actual method of randomisation is not discussed. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Person(s) responsible for participants care and participants were not blinded. Of outcome assessors, only the radiologist who interpreted the DXA scans was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It was described that 3 participants died during the course of the study before the first primary end‐point measurement (causes of death were one each from sepsis, acute respiratory distress syndrome and obliterative bronchiolitis). These participants were excluded from the final analysis of baseline characteristics and outcome data. However, it was not reported which treatment group they were in. |
| Selective reporting (reporting bias) | High risk | Serum and urine biochemical measurements that were measured at 2 days (only after first pamidronate infusion in intervention group) were not reported. |
| Other bias | Unclear risk | None identified. |
Aris 2004.
| Methods | Randomised controlled trial; parallel design; double‐blind placebo‐controlled. Trial duration 1 year for primary outcome measure (trial was intended to be 2 years duration). Single centre, adult CF centre, USA. |
|
| Participants | Inclusion criteria: CF; ambulatory; DXA showed a spine or femur T‐score of ‐1 or less.
Exclusion criteria: primary graft failure or other post‐operative morbidities that precluded long‐term survival; renal insufficiency (serum creatinine >3.0 mg/dl); active upper gastrointestinal disease; chronic oral glucocorticoid usage (>10 mg every day); organ transplantation; a history of bisphosphonate intolerance or use; and pregnancy. 101 participants consented to be screened, 86 qualified and 53 started protocol and were randomised. Total participants: n = 48 (23 female). Treatment group: n = 24 (9 female); mean (SD) age 28 years (7 years). Control group: n = 24 (14 female); mean (SD) age 27 years (9 years). At baseline, osteoporosis was found in 3 participants and osteopenia was present in 20 participants in both the treatment and control groups. |
|
| Interventions | Treatment group: oral alendronate (10 mg daily) plus oral vitamin D (800 IU/day) and oral calcium carbonate (1000 mg/day) Control group: oral vitamin D (800 IU/day) and oral calcium carbonate (1000 mg/day) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Funded by the U.S. Food and Drug administration, Merck and Co, Inc., the Clinical Nutrition Research Unit, the Verne S. Caviness General Center for Clinical Research at University of North Carolina, the CF Foundation and the NIDDK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Blocks of four" design stated, but actual method of randomisation is not discussed. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as "double‐blind". Participants: blinded. Clinicians or persons delivering treatment: unclear if clinicians involved in the study and clinicians managing the medical problems of the participants were all blinded. Outcome assessors: stated that the musculoskeletal radiologist who analysed baseline and end‐of‐study chest radiographs for fracture was blinded, not specifically stated that other outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals up to 6 months: 5 withdrawals in total, but not stated whether they were in treatment or control group, so 48 participants were evaluable. Reasons for dropping out: pregnancy (n=1); diarrhoea and weight loss (n=3); dysphagia (n=1). The three participants with diarrhoea reported abdominal cramping, loss of appetite, and diarrhoea before the medications began that worsened during the study but persisted after the study medications were discontinued, one participant was on alendronate and two on placebo. Withdrawals between 6 and 12 months: 4 withdrawals from each group were described. Treatment group: 4 drop‐outs; reasons were: transplanted (n = 1); moved (n = 2); non‐compliance (n = 1). Placebo group: 4 drop‐outs; reasons were: transplanted (n = 2); died (n = 1); moved (n = 1). Withdrawals between 1 and 2 years Treatment group: 9 drop‐outs; reasons were: moved (n = 2); committed to only 1 year (n = 7). Placebo group: 7 drop‐outs; reasons were: moved (n = 1); committed to only 1 year (n = 6). The primary end‐point measure was analysed in 40/53 (75%) participants, hence there is a risk of attrition bias. Stated that an intention‐to‐treat principle was used in the analyses of the treatment endpoints. |
| Selective reporting (reporting bias) | Low risk | Outcome measures that were described in the methods section were reported in the results section. |
| Other bias | High risk | Described that protocol was originally designed to be 2 years in length, but few participants were willing to consent to such a lengthy study, so protocol was revised to measure the primary endpoint at 12 months. |
Boyle 2005.
| Methods | Randomised, double‐blinded, placebo‐controlled trial; parallel design. Trial duration 6 months (originally intended for 12 months). |
|
| Participants | Inclusion criteria: CF; osteopenia of the lumbar spine (T‐scores ‐1.0 to ‐2.5); serum 25‐hydroxyvitamin D levels ≥ 20ng/ml prior to infusion.
Exclusion criteria: existing osteoporosis, prior treatment with bisphosphonates or previous lung transplant. N = 40 planned for enrolment but only 5 enrolled (3 in treatment group) before study stopped by Data and Safety Monitoring Board (see notes). |
|
| Interventions | Treatment group: intravenous zoledronate, 5 mg infusion administered on a single occasion over 20 minutes plus supplemental oral vitamin D (800 IU) and oral calcium (1000 mg) daily Control group: supplemental oral vitamin D (800 IU) and oral calcium (1000 mg) daily |
|
| Outcomes |
|
|
| Notes | The study was stopped by its Data and Safety Monitoring Board after 3 participants experienced dramatic musculoskeletal pain, 2 requiring emergency room assessment. Symptoms began 6 to 8 hours after infusion, peaked at 12 to 18 hours, and were characterized by severe chest and back pain. Along with musculoskeletal pain, one participant also experienced a fever of 104°F lasting for several hours and a rise in tumour necrosis factor‐α. Although the most severe symptoms resolved within 48 to 72 hours, participants reported continued arthralgias for up to a week. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, but process not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as 'double‐blind'. Participants: blinded. Not discussed if clinicians or persons delivering treatment and outcome assessors were both blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Based on interpretation of data, we have presumed that the 3 participants who had severe bone pain were the 3 in the treatment group. Clarification from the author was requested but not received. |
| Selective reporting (reporting bias) | Unclear risk | Abstract only but outcome measures were described in the results. |
| Other bias | High risk | The study was stopped by its Data and Safety Monitoring Board after 3 participants experienced dramatic musculoskeletal pain, 2 requiring emergency room assessment. Symptoms began 6 to 8 hours after infusion, peaked at 12 to 18 hours, and were characterized by severe chest and back pain. Along with musculoskeletal pain, one participant also experienced a fever of 104oF lasting for several hours and a rise in Tumour Necrosis Factor‐α. Although the most severe symptoms resolved within 48 to 72 hours, participants reported continued arthralgias for up to a week. |
Chapman 2009.
| Methods | Randomised, double‐blinded, placebo‐controlled trial; parallel design. Trial duration 24 months. Multicentre, 2 sites, CF clinics, Australia. |
|
| Participants | Inclusion criteria: CF (diagnosis previously made by sweat chloride test and an appropriate CF phenotype); ≥ 18 years; bone density T‐score <‐1.5 in at least one of three sites (hip (femoral neck), lumbar spine 2 to 4 (L2 to L4) and distal forearm) in the month before study commencement. Exclusion criteria: pre‐existing, symptomatic, fragility fractures; untreated hyperthyroidism, primary hyperparathyroidism or hypogonadism; bisphosphonate treatment in the three months before starting the study; serum calcium concentration below the lower limit of the laboratory normal range; serum creatinine concentration more than 1.5 times the upper limit of the laboratory normal range; serum ALT, ALP or bilirubin more than 3 times the upper limit of the laboratory normal range; on the waiting list for lung transplantation; pregnant or lactating; considered unlikely to complete the study. Total participants: n = 22 (5 females). Age range over all: males 21 to 47 years, females 19 to 28 years. Treatment group: n = 10 (3 female); mean (SD) age 30.1 (2.2) years. Control group: n = 12 (2 females) mean (SD) age 28.6 (2.4) years. |
|
| Interventions | Treatment group: intravenous zoledronic acid (zoledronate) in 100 ml of normal saline infused over 15 minutes every 3 months for 21 months (8 infusions in total). For 5 out of 63 doses, 4 mg zoledronate was administered, then dose reduced to 2 mg for subsequent doses (due to febrile reactions to the higher dose in several participants). Placebo group: 100 ml normal saline as above. All participants were prescribed calcium carbonate 600 mg and vitamin D2 1000 IU each twice daily at least 3 days before the first treatment infusion and continued throughout the study. All participants were prescribed oral prednisolone 25 mg/day for 3 days starting on the morning of the first infusion; repeated with subsequent infusions if a reaction to the first infusion was thought likely. If there were side effects of the study infusion that were considered to be possibly due to the infusion during the first or any subsequent infusion, at the discretion of the investigator and participant, oral analgesia (paracetamol) was also administered for subsequent infusions. |
|
| Outcomes |
|
|
| Notes | Novartis Pharmaceuticals Pty Ltd, Australia partly funded this trial. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, but process not discussed. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as 'double‐blind'. Participants: blinded. Outcome assessors: DXA scans were performed and analysed by personnel blinded to treatment assignment. Not specifically discussed if clinicians or persons delivering treatment and other outcome assessors were all blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawals described and equal across groups ‐ there were 3/10 in treatment group and 5/12 in control group. In the treatment group, 2 participants withdrew due to side‐effects, 1 due to psychiatric illness. In the placebo group, one participant was lost to follow‐up, one participant's BMD decreased to withdrawal threshold, 2 participants were poorly compliant to study protocols and in one participant, both of the latter two reasons were applicable. However, it was unclear which specific participants had BMD measurements available at each time‐point, particularly for forearm measures (fewer measurements compared with lumbar spine and femoral neck). |
| Selective reporting (reporting bias) | Low risk | Outcome measures that were described in the methods section were reported in the results section. |
| Other bias | Low risk | None identified. |
Haworth 2001.
| Methods | Randomised controlled trial; parallel design. Trial duration planned for 1 year, but was shortened to 6 months because of adverse events. Single centre, UK. |
|
| Participants | Inclusion criteria: CF; no organ transplantation; 70% of all eligible participants in a longitudinal BMD study recruited after one year of follow‐up; no prior treatment with bone‐sparing agents; BMD z score of ≦ ‐2 at lumbar spine, proximal femur or distal forearm. Total participants: n = 31 (9 female); mean (SD) age 26.1 (5.8) years; mean (SD) BMI 21.1 (2.7) kg/m2; mean (SD) FEV1 50.9 (20.3) % of predicted treatment. Groups similar with respect to age, initial BMD, bone biochemistry and respiratory disease severity. Treatment group: n = 15 (more females in this group but exact number not reported) control group: n = 16 3 participants did not complete the study (1 participant in the treatment group received a double lung transplant and 1 participant in each group died of respiratory failure). |
|
| Interventions | Treatment group: intravenous pamidronate 30 mg every 3 months for 6 months (2 doses) plus oral calcium (1 g daily) Control group: oral calcium (1 g daily) All participants with pancreatic insufficiency (relevant to all except one in control group) continued long‐term oral vitamin D (900 IU/day). |
|
| Outcomes |
|
|
| Notes | Received funding from the CF Trust in the UK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, but process not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not possible to blind as they did not give placebo infusions, so patients and clinicians would know who was in the treatment group and who in the control group. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals were reported; 1 participant in each group died of respiratory failure and 1 participant in the treatment group underwent a double lung transplant. |
| Selective reporting (reporting bias) | Low risk | Outcome measures that were described in the methods section were reported in the results section. |
| Other bias | High risk | Trial duration planned for 1 year, but was shortened to 6 months because of adverse events. |
Haworth 2011.
| Methods | Randomised, double‐blinded, placebo‐controlled trial; parallel design. Trial duration 24 months. Multicentre, 4 sites in United Kingdom, 1 site in Ireland. |
|
| Participants | Inclusion criteria: CF (diagnosis on the basis of a positive sweat test or gene analysis and a consistent CF phenotype); > 18 years, low BMD defined as lumbar spine, total hip or femoral neck BMD z score < 1. Exclusion criteria: prescription of daily oral glucocorticoids for 6 weeks or more in the 12 months preceding the study; breast feeding, pregnancy, desire to become pregnant within 3 years; listed for, or recipient of solid organ transplant; history of gastroscopy proven oesophageal abnormalities; renal impairment (elevated serum creatinine and an estimated creatinine clearance of 30 ml/min or less); hypocalcaemia; previous prescription of bone active drugs (bisphosphonates, hormone replacement therapy, raloxifene, calcitriol, calcitonin, teriparatide); biochemical evidence of vitamin D deficiency in the 12 months prior to the screening visit (25‐hydroxyvitamin D level < 10 ng/ml and PTH > 45 pg/ml); previous poor clinic attendance; previous poor adherence; pre‐terminal illness or other serious concomitant illness. Female participants of reproductive age were advised not to become pregnant for at least 12 months after study completion. Total participants: n = 36 (9 females). Treatment group: n = 17 (4 females); mean (SD) age 30.2 (12) years. Control group: n = 19 (5 females); mean (SD) age 27.8 (8.0) years. |
|
| Interventions | Treatment group: once weekly oral risedronate 35 mg. Control group: once weekly identical placebo. Both groups were both prescribed Calcichew D3 Forte 2 tablets daily which provides 1000 mg calcium + 800 IU vitamin D3/day. Patients were advised to continue their standard multivitamin supplements. |
|
| Outcomes |
|
|
| Notes | Concomitant medications were recorded at study visits (3, 6, 12, 18 and 24 months). Pregnancy tests were performed in females at study visits (3, 6, 12, 18 and 24 months). Funded by unrestricted educational grants from Proctor & Gamble (Norwich, USA) and the Cystic Fibrosis Trust, with support to investigators from the UK National Institute of Health Research. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were allocated to risedronate or placebo using a computer programme to minimise differences between groups in treatment centre, sex and baseline lumbar spine BMD. |
| Allocation concealment (selection bias) | Low risk | Only the study pharmacist had access to the treatment allocation. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical placebo used. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals are described in full and fairly equally spread across groups. Intervention group: in the first 12 months, 3 out of 17 in the oral risedronate group withdrew from the study completely (due to bone pain) and 3 patients discontinued the study medication (one citing bone pain and the other 2 participants citing muscle aches or generalised pain) but remained in the study for follow up. Between 12 and 24 months one further participant withdrew, citing bone pain. At 24 months, 12 participants in the intervention group remained in study with 9 still taking the study drug. Placebo group: immediately after randomisation 1 participant in the placebo group withdrew consent before taking the study medication. Therefore, only 18 participants were commenced on placebo. By 12 months, 2 participants in the placebo group withdrew consent and 1 participant had died. By 24 months, 3 participants in the placebo group had withdrawn consent and 4 patients had died, hence at 24 months, 12 participants remained in the control group of the study. Primary outcome data included 12/17 of risedronate group (although only 9 still on drug); 12/19 placebo group. In the published abstract, it was not clear if the analysis included the 3 participants. |
| Selective reporting (reporting bias) | Low risk | Outcome measures that were described in the methods section were reported in the results section. |
| Other bias | Low risk | None identified. |
Papaioannou 2008.
| Methods | Randomised, placebo‐controlled trial. Trial duration: 12 months. Multi‐centre, 6 Canadian CF specialty clinics. |
|
| Participants | Inclusion criteria: participants had CF confirmed by positive sweat test result or DNA acid analysis and a BMD T score of 1.0, as determined by dual‐energy radiograph absorptiometry. Exclusion criteria: organ transplantation; endoscopy‐proven oesophagitis, gastritis, and ulceration; metabolic bone disorders; severe renal disease; use of systemic corticosteroids (dose 7.5 mg/day) or other drugs known to influence bone metabolism in the previous 6 months; osteomalacia and other documented contraindications. Total adults randomised: n = 56 (22 female). 9 withdrew, 47 completed study. Treatment group: n = 27 (10 female); mean (SD) age 28.1 (7.7) years. 4 withdrew, 23 completed study. Control group: n = 29 (12 female); mean (SD) age 30.9 (9.7) years. 5 withdrew, 24 completed study. |
|
| Interventions | Treatment group: oral alendronate (70 mg) once weekly for 12 months. Control group: placebo. Medication was taken while sitting upright and with water only on an empty stomach at least 30 min before first food or beverage of the day. In addition, all participants received 800 IU of vitamin D and 1000 mg of calcium (500 mg supplementation, 500 mg from diet) daily. |
|
| Outcomes | In‐clinic assessments at baseline, 6 and 12 months; telephone follow‐up conducted by study staff at months 3 and 9.
Safety analyses included all vertebral fractures, osteoporosis‐related fractures, adverse reactions, and abnormal findings that had been detected through laboratory tests and physical examinations. Documentation for all adverse events were blinded and adjudicated by the external Data Safety Monitoring Committee. All adverse events were reported regardless of attribution to study medication. |
|
| Notes | Participants who received at least 80% of the study drug were classified as being adherent to the protocol. 5 participants completed the study protocol but received sub‐optimal dosing (< 80% adherence; treatment group, 3 participants; control group, 2 participants). 1 of the participants in the treatment group missed > 50% of doses. Stopping and study withdrawal rules were monitored by an external Data Safety Monitoring Committee. During the study, 3 participants in the treatment group used oral corticosteroids compared to none in the control group. Study funding provided by Merck Frosst Canada. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation code, stratified according to institution was prepared by an independent randomisation centre and block allocation was employed to ensure equitable distribution to each treatment group. |
| Allocation concealment (selection bias) | Low risk | The randomisation code was prepared by an independent randomisation centre and the medication treatment arm was concealed from all participants, central and local site coordinators, physicians, staff, and caregivers. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Person(s) responsible for participants care, participants and outcome assessors were blinded to treatment group allocation. A medical physicist, who was blinded to the study treatment arm and study status, reviewed all DXA scans. Radiographs were sent to the central methods centre, and read independently by two radiologists who were blinded to the study treatment arm. Differences between radiologists were resolved by consensus. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All analyses were performed as intention‐to‐treat and included all available data. Withdrawals were described. Treatment group: 27 randomised, 4 withdrew (2 non‐compliance, 1 due to adverse event, 1 withdrew consent). 23 completed study. Control group: 29 randomised, 5 withdrew (2 non‐compliance, 2 due to adverse event, 1 lost to follow‐up). 24 completed study. |
| Selective reporting (reporting bias) | Unclear risk | Outcome measures that were described in the methods section were reported in the results section. It was reported that there were no differences in baseline CRP, 25‐hydroxyvitamin D, PTH or CTX levels between the risedronate patients who experienced bone pain and those that did not (but data not shown). |
| Other bias | Low risk | None identified. |
ALP: alkaline phosphatase ALT: alanine aminotransferase BMD: bone mineral density BMI: body mass index CF: cystic fibrosis DXA: dual‐energy x‐ray absorptiometry FEV1: forced expiratory volume in one second IU: international units PTH: parathyroid hormone SD: standard deviation SF‐36v2: Medical Outcomes Study 36‐item short form, version 2 SXA: single energy x‐ray absorptiometry
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Conway 2004 | This study was a prospective open design, not a randomised controlled trial. |
| Hardin 2005 | This study assessed the effect of growth hormone on total‐body bone mineral content in pre‐pubertal children with CF. It did not assess the effect of bisphosphonates. |
CF: cystic fibrosis
Contributions of authors
Dr Christine Brenckmann and Dr. Alexandra Papaioannou conceived and designed the original review. Until Issue 1, 2005 of The Cochrane Library, they performed the data collection and interpretation. Christine Brenckmann performed the data management and analysis, and wrote the review. Christine Brenckmann acted as guarantor of the review until the review team stepped down in 2007.
From Issue 4, 2009 of The Cochrane Library a new review team has taken on this review. The lead author and guarantor of the review is Dr Louise Conwell. Co‐author is Professor Anne Chang.
Sources of support
Internal sources
-
Royal Children's Hospital Foundation, Australia.
Research support for AC
External sources
-
Australia National Health and Medical Research Council, Australia.
Provision of Practitioner Fellowship for AC
-
Queensland Smart State, Australia.
Provision of Clinical Fellowship for AC
Declarations of interest
None known.
Edited (no change to conclusions)
References
References to studies included in this review
Aris 2000 {published data only}
- Aris RM, Lester GE, Neuringer IP, Winders AW, Gott KK, Rea J, et al. Efficacy of pamidronate for osteoporosis in cystic fibrosis patients following lung transplantation [abstract]. Pediatric Pulmonology 1998;Suppl 17:365. [Google Scholar]
- Aris RM, Lester GE, Renner JB, Winders A, Denene Blackwood A, Lark RK, et al. Efficacy of pamidronate for osteoporosis in patients with cystic fibrosis following lung transplantation. American Journal of Respiratory and Critical Care Medicine 2000;162(3 part 1):941‐6. [DOI] [PubMed] [Google Scholar]
- Aris RM, Ontjes DA, Winders AW, Blackwood D, Lester GE. Effect of pamidronate on bone biomarkers in post‐transplant osteoporotic cystic fibrosis patients [abstract]. Pediatric Pulmonology 1998;Suppl 17:364‐5. [Google Scholar]
Aris 2004 {published data only}
- Aris RM, Lester GE, Camaniti M, Hensler M, Lark RK, Hecker TM, et al. Efficacy of alendronate in CF adults with low bone density. Pediatric Pulmonology 2003;S25:351. [Google Scholar]
- Aris RM, Lester GE, Caminiti M, Blackwood AD, Hensler M, Lark RK, Hecker TM, Renner JB, Guillen U, Brown SA, Neuringer IP, Chalermskulrat W, Ontjes DA. Efficacy of alendronate in adults with cystic fibrosis with low bone density. American journal of respiratory and critical care medicine 2004;169(1):77‐82. [DOI] [PubMed] [Google Scholar]
Boyle 2005 {published data only}
- Boyle MP, Lechtzin N, Watts S. Zoledronate therapy for decreased bone density in adults with cystic fibrosis [abstract]. Pediatric Pulmonology 2005;40(S28):353. [Google Scholar]
Chapman 2009 {published data only}
Haworth 2001 {published data only}
- Haworth CS, Selby PL, Adams JE, Mawer EB, Horrocks AW, Webb AK. Effect of intravenous pamidronate on bone mineral density in adults with cystic fibrosis. Thorax 2001;56(4):314‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth CS, Selby PL, Mawer EB, Adams JE, Verma A, Phillips A, et al. Pamidronate increases axial bone density in cystic fibrosis patients [abstract]. Pediatric Pulmonology 1999;Suppl 19:295. [Google Scholar]
- Haworth CS, Selby PL, Mawer EB, Adams JE, Webb AK. Intravenous pamidronate increases axial bone density in cystic fibrosis adults [abstract]. Thorax 1999;54(Suppl 3):A67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth CS, Selby PL, Webb AK, Adams JE, Freemont TJ. Oral corticosteroids and bone pain after pamidronate in adults with cystic fibrosis [letter]. Lancet 1999;353(9167):1886. [DOI] [PubMed] [Google Scholar]
- Haworth CS, Selby PL, Webb AK, Mawer EB, Adams JE, Freemont TJ. Severe bone pain after intravenous pamidronate in adult patients with cystic fibrosis [letter]. Lancet 1998;352(9142):1753‐4. [DOI] [PubMed] [Google Scholar]
Haworth 2011 {published and unpublished data}
- Haworth CS, Sharples L, Hughes V, Elkin SL, Hodson M, Conway S, Etherington C, Elborn JS, Rendall J, Wheaton E, Kadri E, Elliott J, Hlaing T, Barker H, Compston J. Two‐year multicenter, randomised, double‐blind, placebo‐controlled trial assessing the effect of weekly Risedronate on bone mineral density in adults with CF [abstract]. Pediatric Pulmonology 2010;45(Suppl 33):423. [Google Scholar]
- Haworth CS, Sharples L, Hughes V, Elkin SL, Hodson ME, Conway SP, et al. Multicentre trial of weekly risedronate on bone density in adults with cystic fibrosis. Journal of Cystic Fibrosis 2011;10(6):470‐6. [DOI: 10.1016/j.jcf.2011.07.007] [DOI] [PubMed] [Google Scholar]
- Haworth CS, Sharples L, Hughes V, Elsin SL, Hodson ME, Conway SP, Etherington C, Elborn JS, Rendall J, Wheaton E, Kadri E, Elliott J, Barker HC, Compston J. Multicentre randomised double blind placebo controlled trial assessing the effect of weekly risedronate on bone mineral density in adults with cystic fibrosis [abstract]. Journal of Cystic Fibrosis 2010;9(Suppl 1):S85. [DOI] [PubMed] [Google Scholar]
Papaioannou 2008 {published data only}
- Papaioannou A, Kennedy CC, Freitag A, Ioannidis G, O'Neill J, Webber C, et al. Alendronate once weekly for the prevention and treatment of bone loss in Canadian adult cystic fibrosis patients (CFOS trial). Chest 2008;134(4):794‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Conway 2004 {published data only}
- Conway SP, Oldroyd B, Morton A, Truscott JG, Peckham DG. Effect of oral bisphosphonates on bone mineral density and body composition in adult patients with cystic fibrosis: a pilot study. Thorax 2004;59(8):699‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hardin 2005 {published data only}
- Hardin DS, Ahn C, Prestidge C, Seilheimer DK, Ellis KJ. Growth hormone improves bone mineral content in children with cystic fibrosis. Journal of paediatric endocrinology & metabolism: JPEM 2005;18(6):589‐95. [DOI] [PubMed] [Google Scholar]
Additional references
Alicandro 2013
- Alicandro G, Bisogno A, Battezzati A, Bianchi ML, Corti F, Colombo C. Recurrent pulmonary exacerbations are associated with low fat free mass and low bone mineral density in young adults with cystic fibrosis. Journal of Cystic Fibrosis 2013 Nov 28 [Epub ahead of print]:S1569‐993. [PUBMED: 24291530] [DOI] [PubMed]
Ambroszkiewicz 2013
Aris 1996
- Aris RM, Neuringer IP, Weiner MA, et al. Severe osteoporosis before and after lung transplantation. Chest 1996;109(5):1176‐83. [DOI] [PubMed] [Google Scholar]
Aris 1998
- Aris RM, Renner JB, Winders AD, Buell HE, Riggs DB, Lester GE, et al. Increased rate of fractures and severe kyphosis: sequelae of living into adulthood with cystic fibrosis. Annals of Internal Medicine 1998;128(3):186‐93. [DOI] [PubMed] [Google Scholar]
Aris 2005
- Aris RM, Merkel PA, Bachrach LK, Borowitz DS, Boyle MP, et al. Consensus Statement: Guide to Bone Health and Disease in Cystic Fibrosis. The Journal of Clinical Endocrinology and Metabolism 2005;90(3):1888‐96. [DOI] [PubMed] [Google Scholar]
Bianchi 2006
- Bianchi ML, Romano G, Saraifoger S, Costantini D, Limonta C, Colombo C. BMD and body composition in children and young patients affected by cystic fibrosis. Journal of Bone and Mineral Research 2006;21(3):388‐96. [DOI] [PubMed] [Google Scholar]
Black 1996
- Black DM, Cummings SR, Karpf DB, Cauley JA, Thompson DE, Nevitt MC, et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Lancet 1996;348(9041):1535‐41. [DOI] [PubMed] [Google Scholar]
Boutsen 2001
- Boutsen Y, Jamart J, Esselinckx W, Devogelaer JP. Primary prevention of glucocorticoid‐induced osteoporosis with intravenous pamidronate and calcium: a prospective controlled 1‐year study comparing a single infusion, an infusion given once every 3 months, and calcium alone. Journal of Bone Mineral Research 2001;16(1):104‐12. [DOI] [PubMed] [Google Scholar]
Boyle 2006
- Boyle MP. Update on maintaining bone health in cystic fibrosis. Current Opinion in Pulmonary Medicine 2006;12(6):453‐8. [DOI] [PubMed] [Google Scholar]
Buntain 2004
- Buntain HM, Greer RM, Schluter PJ, Wong JC, Batch JA, Potter JM, et al. Bone mineral density in Australian children, adolescents and adults with cystic fibrosis: a controlled cross sectional study. Thorax 2004;59(2):149‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Buntain 2006
- Buntain HM, Schluter PJ, Bell SC, Greer RM, Wong JC, Batch J, et al. Controlled longitudinal study of bone mass accrual in children and adolescents with cystic fibrosis. Thorax 2006;61(2):146‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Caldeira 2008
- Caldeira RJ, Fonseca VD, Gomes Junio SC, Miranda Chaves CR. Prevalence of bone mineral disease among adolescents with cystic fibrosis. Journal of Pediatrics 2008;84(1):18‐25. [DOI] [PubMed] [Google Scholar]
Cates 2003 [Computer program]
- Cates C. Visual Rx. Online NNT Calculator. http://www.nntonline.net/: Cates C, 2003.
Cryer 2002
- Cryer B, Bauer DC. Oral bisphosphonates and upper gastrointestinal tract problems: what is the evidence?. Mayo Clinic proceedings. Mayo Clinic 2002;77(10):1031‐43. [DOI] [PubMed] [Google Scholar]
Cummings 1995
- Cummings SR, Nevitt MC, Browner WS, Stone K, Fox KM, Ensrud KE, et al. Risk factors for hip fracture in white women: Study of Osteoporotic Fractures Research Group. New England Journal of Medicine 1995;332(12):767‐73. [DOI] [PubMed] [Google Scholar]
Cystic Fibrosis Foundation Patient Registry 2013
- Cystic Fibrosis Foundation. Patient Registry 2012 Annual Report. Bethesda, Maryland: Cystic Fibrosis Foundation, 2013. [Google Scholar]
Cystic Fibrosis Trust Report 2007
- Cystic Fibrosis Trust. Report of the UK Cystic Fibrosis Trust Bone Mineralisation Working Group. Report of the UK Cystic Fibrosis Trust Bone Mineralisation Working Group. Kent: Cystic Fibrosis Trust, February 2007. [Google Scholar]
Dif 2004
- Dif F, Marty C, Baudoin C, Vernejoul MC, Levi G. Severe osteopenia in CFTR‐null mice. Bone 2004;35(3):595‐603. [PUBMED: 15336594] [DOI] [PubMed] [Google Scholar]
Donadio 2013
- Donadio MV, Souza GC, Tiecher G, Heinzmann‐Filho JP, Paim TF, Hommerding PX, et al. Bone mineral density, pulmonary function, chronological age, and age at diagnosis in children and adolescents with cystic fibrosis. Jornal de Pediatria 2013;89(2):151‐7. [PUBMED: 23642425] [DOI] [PubMed] [Google Scholar]
Eggelmeijer 1996
- Eggelmeijer F, Papapoulos SE, Paassen HC, Dijkmans BA, Valkema R, Westedt ML, et al. Increased bone mass with pamidronate treatment in rheumatoid arthritis. Results of a three‐year randomized, double‐blind trial. Arthritis and Rheumatism 1996;39(3):396‐402. [DOI] [PubMed] [Google Scholar]
Fromm 1991
- Fromm G, Vega E, Plantalech L, Galich AM, Mautalen CA. Differential action of pamidronate on trabecular and cortical bone in women with involutional osteoporosis. Osteoporosis International 1991;1(3):129‐33. [DOI] [PubMed] [Google Scholar]
Giger 2013
- Giger EV, Castagner B, Leroux JC. Biomedical applications of bisphosphonates. Journal of Controlled Release: official journal of the Controlled Release Society 2013;167(2):175‐88. [PUBMED: 23395668] [DOI] [PubMed] [Google Scholar]
Glorieux 1998
- Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. The New England Journal of Medicine 1998;339(14):947‐52. [DOI] [PubMed] [Google Scholar]
Gore 2010
- Gore AP, Kwon SH, Stenbit AE. A roadmap to the brittle bones of cystic fibrosis. Journal of Osteoporosis 2010;2011:926045. [PUBMED: 21209785] [DOI] [PMC free article] [PubMed] [Google Scholar]
Greer 2003
- Greer RM, Buntain HM, Potter JM, Wainwright CE, Wong JC, O'Rourke PK, et al. Abnormalities of the PTH‐vitamin D axis and bone turnover markers in children, adolescents and adults with cystic fibrosis: comparison with healthy controls. Osteoporosis International 2003;14(5):404‐11. [DOI] [PubMed] [Google Scholar]
Hall 2010
- Hall WB, Sparks AA, Aris RM. Vitamin D deficiency in cystic fibrosis. International Journal of Endocrinology 2010;2010:218691. [PUBMED: 20148079] [DOI] [PMC free article] [PubMed] [Google Scholar]
Haworth 2010b
- Haworth CS. Impact of cystic fibrosis on bone health. Current Opinion in Pulmonary Medicine 2010;16(6):616‐22. [PUBMED: 20739891] [DOI] [PubMed] [Google Scholar]
Hayes 2011
- Hayes M, Yaster M, Haythornthwaite JA, Riekert K, Nelson K, White E, et al. Pain is a common problem affecting clinical outcomes in adults with cystic fibrosis. Chest 2011;140(6):1598‐603. [PUBMED: 21659431] [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
ISCD Official Positions 2013
- International Society for Clinical Densitometry. 2013 ISCD Official Positions [Updated August 2013]. www.iscd.org (accessed 27 January 2014).
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, McQuay HJ. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled clinical trials 1996;17(1):1‐12. [DOI] [PubMed] [Google Scholar]
Javier 2011
- Javier RM, Jacquot J. Bone disease in cystic fibrosis: What's new?. Joint, Bone, Spine: Revue du Rhumatisme 2011;78(5):445‐50. [PUBMED: 21233000] [DOI] [PubMed] [Google Scholar]
Kanis 1994
- Kanis JA. Osteoporosis. Philadelphia: Blackwell Science, 1994. [Google Scholar]
Kelly 2013
- Kelly A, Moran A. Update on cystic fibrosis‐related diabetes. Journal of Cystic Fibrosis 2013;12(4):318‐31. [PUBMED: 23562217] [DOI] [PubMed] [Google Scholar]
Le Henaff 2012
- Henaff C, Gimenez A, Hay E, Marty C, Marie P, Jacquot J. The F508del mutation in cystic fibrosis transmembrane conductance regulator gene impacts bone formation. The American Journal of Pathology 2012;180(5):2068‐75. [PUBMED: 22449949] [DOI] [PubMed] [Google Scholar]
Le Heron 2010
- Heron L, Guillaume C, Velard F, Braux J, Touqui L, Moriceau S, et al. Cystic fibrosis transmembrane conductance regulator (CFTR) regulates the production of osteoprotegerin (OPG) and prostaglandin (PG) E2 in human bone. Journal of Cystic Fibrosis 2010;9(1):69‐72. [PUBMED: 20005786] [DOI] [PubMed] [Google Scholar]
Legroux‐Gerot 2012
- Legroux‐Gerot I, Leroy S, Prudhomme C, Perez T, Flipo RM, Wallaert B, et al. Bone loss in adults with cystic fibrosis: prevalence, associated factors, and usefulness of biological markers. Joint, bone, spine : revue du rhumatisme 2012;79(1):73‐7. [PUBMED: 21733729] [DOI] [PubMed] [Google Scholar]
Paccou 2010
- Paccou J, Zeboulon N, Combescure C, Gossec L, Cortet B. The prevalence of osteoporosis, osteopenia, and fractures among adults with cystic fibrosis: a systematic literature review with meta‐analysis. Calcified Tissue International 2010;86(1):1‐7. [PUBMED: 19949942] [DOI] [PubMed] [Google Scholar]
Paccou 2013
- Paccou J, Fardellone P, Cortet B. Cystic fibrosis‐related bone disease. Current Opinion in Pulmonary Medicine 2013;19(6):681‐6. [PUBMED: 24060979] [DOI] [PubMed] [Google Scholar]
Pendrys 2008
- Pendrys DG, Silverman SL. Osteonecrosis of the jaws and bisphosphonates. Current osteoporosis reports 2008;6(1):31‐8. [DOI] [PubMed] [Google Scholar]
Phillipi 2008
- Phillipi CA, Remmington T, Steiner RD. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD005088.pub2] [DOI] [PubMed] [Google Scholar]
Quon 2012
- Quon BS, Aitken ML. Cystic fibrosis: what to expect now in the early adult years. Paediatric Respiratory Reviews 2012;13(4):206‐14. [PUBMED: 23069117] [DOI] [PubMed] [Google Scholar]
Review Manager 2003 [Computer program]
- The Cochrane Collaboration. Review Manager (RevMan). Version 4.2 for Windows. Oxford, England: The Cochrane Collaboration, 2003.
Review Manager 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Riordan 1989
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characteristics of complementary DNA. Science 1989;245(4922):1066‐73. [DOI] [PubMed] [Google Scholar]
Rowland 2011
- Rowland M, Bourke B. Liver disease in cystic fibrosis. Current Opinion in Pulmonary Medicine 2011;17(6):461‐6. [PUBMED: 21881510] [DOI] [PubMed] [Google Scholar]
Russell 2006
- Russell RG. Bisphosphonates from bench to bedside. Annals New York Academy of Sciences 2006;1068:367‐401. [DOI] [PubMed] [Google Scholar]
Russell 2007
- Russell RG, Xia Z, Dunford JE, Oppermann U, Kwaasi A, Hulley PA, et al. Bisphosphonates: An update on mechanisms of action and how these relate to clinical efficacy. Annals of the New York Academy of Sciences 2007;1117:209‐57. [DOI] [PubMed] [Google Scholar]
Saag 1998
- Saag KG, Emkey R, Schnitzer TJ, Brown JP, Hawkins F, Goemaere S, et al. Alendronate for the prevention and treatment of glucocorticoid‐induced osteoporosis. New England Journal of Medicine 1998;339(5):292‐9. [DOI] [PubMed] [Google Scholar]
Saleh 2013
- Saleh A, Hegde VV, Potty AG, Lane JM. Bisphosphonate therapy and atypical fractures. The Orthopedic Clinics of North America 2013;44(2):137‐51. [PUBMED: 23544820] [DOI] [PubMed] [Google Scholar]
Sauty 1996
- Sauty A, Pecherstorfer M, Zimmer‐Roth I, Fioroni P, Juillerat L, Markert M, et al. Interleukin‐6 and tumor necrosis factor alpha levels after bisphosphonates treatment in vitro and in patients with malignancy. Bone 1996;18(2):133‐9. [DOI] [PubMed] [Google Scholar]
Sermet‐Gaudelus 2007
- Sermet‐Gaudelus I, Souberbielle JC, Ruiz JC, Vrielynck S, Heullion B, Azhar I, et al. Low bone mineral density in young children with cystic fibrosis. American Journal of Respiratory & Critical Care Medicine 2007;175(9):951‐7. [DOI] [PubMed] [Google Scholar]
Sermet‐Gaudelus 2011
- Sermet‐Gaudelus I, Bianchi ML, Garabedian M, Aris RM, Morton A, Hardin DS, et al. European cystic fibrosis bone mineralisation guidelines. Journal of Cystic Fibrosis 2011;10 Suppl 2:S16‐23. [PUBMED: 21658635] [DOI] [PubMed] [Google Scholar]
Shead 2010
- Shead EF, Haworth CS, Barker H, Bilton D, Compston JE. Osteoclast function, bone turnover and inflammatory cytokines during infective exacerbations of cystic fibrosis. Journal of Cystic Fibrosis 2010;9(2):93‐8. [PUBMED: 20006563] [DOI] [PubMed] [Google Scholar]
Shmarina 2013
- Shmarina G, Pukhalsky A, Petrova N, Zakharova E, Avakian L, Kapranov N, et al. TNF gene polymorphisms in cystic fibrosis patients: contribution to the disease progression. Journal of Translational Medicine 2013;11:19. [PUBMED: 23343370] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stalvey 2013
- Stalvey MS, Clines KL, Havasi V, McKibbin CR, Dunn LK, Chung WJ, et al. Osteoblast CFTR inactivation reduces differentiation and osteoprotegerin expression in a mouse model of cystic fibrosis‐related bone disease. PloS One 2013;8(11):e80098. [PUBMED: 24236172] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stalvey 2013a
- Stalvey MS, Clines GA. Cystic fibrosis‐related bone disease: insights into a growing problem. Current Opinion in Endocrinology, Diabetes and Obesity 2013;20(6):547‐52. [PUBMED: 24468756] [DOI] [PMC free article] [PubMed] [Google Scholar]
Steer 1997
- Steer JH, Vuong Q, Joyce DA. Suppression of human monocyte tumour necrosis factor‐alpha release by glucocorticoid therapy: relationship to systemic monocytopaenia and cortisol suppression. British Journal of Clinical Pharmacology 1997;43(4):383‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Watts 1990
- Watts NB, Harris ST, Genant HK, Wasnich RD, Miller PD, Jackson RD, et al. Intermittent cyclical etidronate treatment of postmenopausal osteoporosis. New England Journal of Medicine 1990;323(2):73‐9. [DOI] [PubMed] [Google Scholar]
Weitzmann 2013
- Weitzmann MN. The role of inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica 2013;2013:125705. [PUBMED: 24278766] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wells 2008
- Wells GA, Cranney A, Peterson J, Boucher M, Shea B, Welch V, et al. Risedronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database of Systematic Reviews 2008, Issue 1. [DOI: 10.1002/14651858.CD004523.pub3] [DOI] [PubMed] [Google Scholar]
Yankaskas 1999
- Yankaskas JR, Knowles MR. Cystic Fibrosis in Adults. Philadelphia: Lippincott‐Raven, 1999. [Google Scholar]
References to other published versions of this review
Brenckmann 2001
- Brenckmann C, Papaioannou A. Bisphosphonates for osteoporosis in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD004523.pub2] [DOI] [PubMed] [Google Scholar]
Conwell 2009
- Conwell LS, Chang AB. Bisphosphonates for osteoporosis in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD002010.pub2] [DOI] [PubMed] [Google Scholar]
Conwell 2012
- Conwell LS, Chang AB. Bisphosphonates for osteoporosis in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD002010.pub3] [DOI] [PubMed] [Google Scholar]