

Abstract
Background
Trifluoperazine is a long‐established high potency typical antipsychotic drug used in the treatment of schizophrenia and schizophrenia‐like illnesses.
Objectives
To determine absolute effects of trifluoperazine for schizophrenia and schizophrenia‐like illnesses compared with placebo.
To critically appraise and summarise current evidence on the resource use, cost and economic evaluation of trifluoperazine compared with placebo for schizophrenia.
Search methods
Searches of the Cochrane Schizophrenia Group's register of trials (July 2012), supplemented with handsearching, reference searching, personal communication and contact with industry. Two review authors undertook a search for economic studies using the Cochrane Schizophrenia Group's Health Economic Database (CSzGHED) on the 9th April 2013.
Selection criteria
All available clinical randomised trials involving people with schizophrenia and schizophrenia‐like illnesses that compare trifluoperazine with placebo.
Data collection and analysis
Studies for the effects of interventions were reliably selected by a review team and data were doubly independently extracted to reduce bias. We only used dichotomous data, using intention‐to‐treat analysis when possible. Data were estimated using risk ratio (RR) with 95% confidence intervals (CI). A 'Summary of findings' table was produced, where possible, for each primary outcome using GRADE. Economic studies were searched and reliably selected by review authors (VF and SS) to provide an economic summary of available data. Where no relevant economic studies were eligible for inclusion, the economic review team valued the already‐included effectiveness outcome data to provide a rudimentary economic summary.
Main results
This review included 10 studies with a total number of 686 participants featuring in 20 different outcomes of interest. Overall, there was significant clinical improvement in clinical global state at medium term amongst people receiving trifluoperazine (3 RCTs, n = 417, RR 4.61, CI 1.54 to 13.84, low quality evidence) and significantly fewer people receiving trifluoperazine left the studies early due to relapse or worsening at medium term (2 RCTs, n = 381, RR 0.34, CI 0.23 to 0.49, low quality evidence). However, results were equivocal for leaving the study early at medium term for any reason (2 RCTs, n = 391, RR 0.80, CI 0.17 to 3.81, very low quality evidence) and due to severe adverse effects (2 RCTs, n = 391, RR 1.54, CI 0.56 to 4.24, very low quality evidence). Equivocal data were also found for intensified symptoms at medium term (2 RCTs, n = 80, RR 1.05, CI 0.54 to 2.05, very low quality evidence) and rates of agitation or distress again at medium term (1 RCT, n = 52, RR 2.00, CI 0.19 to 20.72, very low quality evidence). Comparison between low and high‐dose trifluoperazine with placebo from a single study provided equivocal evidence of effects. For economic outcomes, we valued outcomes in GBP terms and presented them in additional tables; there was an estimated saving of £3488.3 in favour of trifluoperazine. However, numerous assumptions were made and these savings need to be interpreted in light of those assumptions.
Authors' conclusions
Our results agree with existing evidence that compared to placebo, trifluoperazine is an effective antipsychotic for people with schizophrenia. Furthermore, our review provides supportive evidence that trifluoperazine increases the risk of extrapyramidal adverse effects. Although the effect sizes against placebo are similar to those observed with other agents, they are based on data from many small, pre‐CONSORT trials with generally either a low or very low GRADE evidence that has limited implication for clinical practice. Large, independent trials are needed that adhere to the CONSORT statement to compare trifluoperazine with placebo used in the treatment of schizophrenia and schizophrenia‐like illnesses.
Keywords: Humans; Antipsychotic Agents; Antipsychotic Agents/adverse effects; Antipsychotic Agents/therapeutic use; Dyskinesia, Drug‐Induced; Dyskinesia, Drug‐Induced/etiology; Randomized Controlled Trials as Topic; Schizophrenia; Schizophrenia/drug therapy; Trifluoperazine; Trifluoperazine/adverse effects; Trifluoperazine/therapeutic use
Plain language summary
Trifluoperazine versus placebo for schizophrenia
Trifluoperazine (trade name Stelazine) is a long‐established antipsychotic drug that has been used since the 1950s to treat schizophrenia. It is one of the first generation (typical) drugs that have proven very effective for treating the ‘positive symptoms’ of schizophrenia, such as hearing voices, seeing things and having strange beliefs. These drugs may cause side effects such as involuntary shaking, restlessness and movement disorders such as having a strange posture.
There are also more modern drugs (second generation and atypical antipsychotic drugs). These are effective with the ‘positive symptoms’ of mental illness but also help treat ‘negative symptoms’ such as apathy, weight gain and loss of emotion in people with schizophrenia. These more modern drugs are much more expensive.
This review is based on a search for trials carried out in July 2012, and includes 10 studies with 686 participants. The aim was to determine the effects of trifluoperazine for schizophrenia when compared with placebo (a ‘dummy’ treatment). As expected, people given trifluoperazine showed a significant improvement compared to placebo in both the short and medium term, reinforcing the use of this well‐established typical antipsychotic for people with schizophrenia. However, trifluoperazine can cause side effects such as confusion, agitation, having a dry mouth and blurred vision, but causes less sedation and dizzy spells, so is generally well tolerated by people with schizophrenia.
The authors of the review conclude that trifluoperazine has similar effectiveness to other common antipsychotic drugs, although it may cause more side effects. Evidence used in the review was also graded as low or very low quality. In the light of this, use of other antipsychotic drugs should be considered before starting on trifluoperazine. Most of the included studies were conducted roughly 40 years ago so new, large, comprehensive and independent research trials are needed.
This plain language summary has been written by a consumer Ben Gray from RETHINK.
Summary of findings
Summary of findings for the main comparison. TRIFLUOPERAZINE versus PLACEBO for schizophrenia.
| TRIFLUOPERAZINE versus PLACEBO for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: inpatient and outpatient Intervention: TRIFLUOPERAZINE versus PLACEBO | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | TRIFLUOPERAZINE versus PLACEBO | |||||
| Global state ‐ clinical improvement ‐ medium term As defined by each study Follow‐up: mean 19 weeks | Study population | RR 4.61 (1.54 to 13.84) | 417 (3 studies) | ⊕⊕⊝⊝ low4,5 | ||
| 20 per 10001,2 | 94 per 1000 (31 to 282)3 | |||||
| Moderate | ||||||
| 19 per 10001,2 | 88 per 1000 (29 to 263)3 | |||||
| Global state ‐ relapse or worsening ‐ medium term Numbers of participants experiencing relapse/worsening Follow‐up: mean 5 months | Study population | RR 0.34 (0.23 to 0.49) | 381 (2 studies) | ⊕⊕⊝⊝ low6,7 | ||
| 389 per 10001,2 | 132 per 1000 (90 to 191) | |||||
| Moderate | ||||||
| 250 per 10001,2 | 85 per 1000 (58 to 123) | |||||
| Mental state ‐ any clinically significant response in psychotic symptoms (as defined by each study) ‐ medium term Numbers of participants experiencing 'intensified symptoms' Follow‐up: mean 16 weeks | Study population | RR 1.05 (0.54 to 2.05) | 80 (2 studies) | ⊕⊝⊝⊝ very low6,8,9 | ||
| 225 per 10002 | 236 per 1000 (122 to 461) | |||||
| Moderate | ||||||
| 225 per 10002 | 236 per 1000 (122 to 461) | |||||
| Leaving the study early ‐ any reason ‐ medium term Number of participants leaving the studies early Follow‐up: mean 5 months | Study population | RR 0.67 (0.38 to 1.19) | 523 (5 studies) | ⊕⊝⊝⊝ very low6,8,11,12 | ||
| 336 per 100010 | 225 per 1000 (128 to 400) | |||||
| Moderate | ||||||
| 115 per 100010 | 77 per 1000 (44 to 137) | |||||
| Severe adverse effects ‐ short term Numbers of participants leaving the studies due to severe adverse effects Follow‐up: mean 2 months | Study population | RR 1.31 (0.22 to 7.8) | 67 (2 studies) | ⊕⊝⊝⊝ very low8,9,13 | ||
| 77 per 10001,2 | 101 per 1000 (17 to 600) | |||||
| Moderate | ||||||
| 71 per 10001,2 | 93 per 1000 (16 to 554) | |||||
| Behaviour ‐ any clinically significant agitation or distress ‐ medium term As defined by each study Follow‐up: 4 months | 38 per 1000 | 77 per 1000 (7 to 797) | RR 2 (0.19 to 20.72) | 52 (1 study) | ⊕⊝⊝⊝ very low6,8,9 | |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | No studies reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk relates to the population percentage in the control group. 2 Median control group risk presented. 3 Data were presented as the positive outcome of 'clinical improvement' so the higher value indicates a favourable outcome. 4 Risk of bias: 'serious' ‐ 33% of the studies rated as a 'high' risk or bias over one or more of the domains; 100% of the included studies did not adequately describe randomisation methods. 5 Indirectness: 'serious' ‐ only 33% of the studies directly compared trifluoperazine to a placebo whereas 67% had other drug interventions in their respective trials. 6 Risk of bias: 'serious' ‐ 100% of the studies rated as a 'high' risk or bias over one or more of the domains; 100% of the included studies did not adequately describe randomisation methods. 7 Indirectness: 'serious' ‐ 50% of the studies directly compared trifluoperazine to a placebo whereas the remaining 50% had other drug interventions in their respective trials. 8 Imprecision: 'serious' ‐ 95% confidence intervals for best estimate of effect include both 'no effect' and appreciable benefit/harm. 9 Indirectness: 'serious' ‐ 100% had other drug interventions in their respective trials. 10 Note: moderate heterogeneity between studies. 11 Inconsistency: 'serious' ‐ moderate heterogeneity evident (I2 = 47%). 12 Indirectness: 'serious' ‐ 20% of the studies directly compared trifluoperazine to a placebo whereas the remaining 80% had other drug interventions in their respective trials. 13 Risk of bias: 'serious' ‐ 50% of the studies rated as a 'high' risk or bias over one or more of the domains; 50% of the included studies did not adequately describe randomisation methods.
Background
Description of the condition
Schizophrenia is a term used for the most common form of psychiatric disorder characterised by psychotic symptoms, involving a change in a person’s thoughts, emotions, behaviour and perception of reality. It has an estimated mean incidence of 0.11 per 1000 population (range 0.07 to 0.17 per 1000) with a lifetime prevalence between 0.4% and 1.4% (NICE 2010). Onset is commonly during adolescence or young adulthood (Saha 2005) but can occur at any age, with mean age of onset about five years greater in women (NICE 2010). Symptoms are divided into positive and negative; positive symptoms encompass hallucinations, delusions and disordered thinking, whilst negative symptoms consist of social withdrawal and a loss of interest, energy and emotion (NICE 2010; RCPSYCH 2010). Although it is common to have a negative prodrome preceding positive symptoms, the course, duration and severity of schizophrenia varies considerably and is usually unique to each person (Lankappa 2012; NICE 2010). People with schizophrenia have higher risk mortality than the general population due to increased rates of death through suicide and accidents as well as organic diseases such as cardiovascular, renal and respiratory disease (BNF 2012; Saha 2007; Tiihonen 2009). Furthermore, people with schizophrenia experience social problems including social exclusion, reduced employment opportunities , problems with relationships and a lack of public understanding of the disorder creating a harmful stigma (NICE 2010). Key treatment for this illness is medication. This has been shown to be of benefit for at least 'positive symptoms', but effectiveness relies largely on adherence. Unfortunately, these medications are not without their adverse effects, which highlights the importance of selecting the right medication and thoughtfully involving the recipient of care in the process.
Description of the intervention
Trifluoperazine, trade name Stelazine, is a long‐established antipsychotic that has been used since the 1950s to treat schizophrenia. It is one of the first generation (typical) drugs that has a high potency, having a greater bind to the D2 receptor (Turner 2007). As with most first generation drugs, trifluoperazine is known to cause extrapyramidal side effects (EPS), including pseudo‐Parkinsonism, dystonia, akathisia and tardive dyskinesia. There is no clear choice of first‐line antipsychotics for schizophrenia as the efficacy of the drugs available is not too dissimilar and the choice of medication is usually made based on availability, cost, side‐effect profile and individual patient circumstances (BNF 2012). Newer generation (atypical) drugs have become available and claim to be more effective in treating negative symptoms with fewer EPS, however systematic reviews have demonstrated that this claim by the pharmaceutical industry is not always accurate (Leucht 2003).
How the intervention might work
Pharmacodynamics of trifluoperazine: it is a high potency derivative of phenothiazine and is chemically related to chlorpromazine. It causes a post‐synaptic D2 dopamine receptor blockade in the brain, specifically the mesolimbic, mesocortical centres and the striatum, the latter of which is responsible for EPS (Arana 2000). Because of a decrease in homovanillic acid levels (primary dopamine metabolite), clinical effects of trifluoperazine normally take weeks to occur (Bazire 2000). It has weak anticholinergic and sedative effects whilst having strong extrapyramidal and antiemetic effects. Trifluoperazine is readily absorbed by the gastrointestinal tract (GI) and will peak in the plasma after one and a half to six hours. It is a protein‐binding drug and so will influence secretion into breast milk (care must be taken for pregnant or breast feeding women). Trifluoperazine has a low potency of cholinergic blockade and causes parasympatholytic side effects such as confusion, agitation, dry mouth and blurred vision. It weakly acts at histamine and alpha‐adrenergic receptors relative to the other typical antipsychotics causing less sedation and orthostatic hypotension, hence is generally well tolerated (Kaplan 1998).
Why it is important to do this review
Trifluoperazine is a well‐established antipsychotic drug used to treat schizophrenia. The previous trifluoperazine Cochrane review was undertaken nearly a decade ago (Marques 2004) and does include a placebo comparison. Although relative effects of trifluoperazine against other antipsychotic drugs are important, establishing an up‐to‐date absolute measure of clinical outcomes, efficacy and effects of this less expensive drug is needed. More recently, there has been a shortage of supplies of oral trifluoperazine tablets in the UK and there is emerging evidence that patients receiving this medication who were under the care of their GPs for schizophrenia are beginning to relapse and seek input from secondary care (see: Mental Health Care and PJ Online).
In terms of the costs of schizophrenia, this was estimated at about £6.7 billion in England in 2004/2005, of which the direct costs were £2 million, while the indirect costs accounted for the rest (Mangalore 2007). The cost of trifluoperazine itself is inexpensive, at £5.87 for 112 5 mg tablets. Every person who needs trifluoperazine, in general, requires about 15 mg a day (or 450 mg per month) costing £4.72 (BNF 2012). The newer, atypical antipsychotics in comparison are more expensive than typical antipsychotics, with olanzapine available at £13.11 for 28 5 mg tablets, and clozapine (Clozaril) at £21.56 for 28 100 mg tablets.
It is essential to complement the clinical effectiveness of trifluoperazine with its cost‐effectiveness. Davies et al. (Davies 2007) conducted a study on cost‐effectiveness of the first generation antipsychotics (i.e. flupenthixol, trifluoperazine, chlorpromazine) and the second generation antipsychotics (i.e. risperidone, olanzapine, amisulpiride). The study findings argue that there is no evidence to suggest that atypical (second generation) antipsychotics are more cost‐effective than typical (first generation) antipsychotics. Recommended first‐line treatment for schizophrenia are second‐generation antipsychotic medications, which tend to be more expensive than first‐generation antipsychotics; it is therefore important to assess the effectiveness and cost‐effectiveness of the older medications.
Objectives
To determine absolute effects of trifluoperazine for schizophrenia and schizophrenia‐like illnesses when compared with placebo.
To critically appraise and summarise current evidence on the resource use, cost and economic evaluations of trifluoperazine for schizophrenia.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. If a trial was described as 'double blind' but implied randomisation, we included such trials in a sensitivity analysis (see Sensitivity analysis). If their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion did result in important clinically significant, but not necessarily statistically significant differences, we did not add the data from these lower quality studies to the results of the better trials, but presented such data within a subcategory. We excluded quasi‐randomised studies, such as those allocating by alternate days of the week. Where people were given additional treatments within trifluoperazine, we only included data if the adjunct treatment was evenly distributed between groups and it was only the trifluoperazine that was randomised.
With regards to selecting studies for economic evaluations, review authors (SS and VF) categorised studies as per the following: Type A ‐ Full economic evaluation (within the framework of RCT): studies that focus on cost‐effectiveness analysis, cost‐utility analysis and cost‐benefit analysis. Type B ‐ Partial economic evaluation (within the framework of RCT): studies that focus on cost‐analysis and cost‐minimisation studies of trifluoperazine. Type C ‐ Randomised trials that reported limited information, such as estimates of resources use or costs associated with trifluoperazine.
Types of participants
Participants with schizophrenia or related disorders, including schizophreniform disorder, schizoaffective disorder and delusional disorder, again, by any means of diagnosis.
We were interested in making sure that information was as relevant to the current care of people with schizophrenia as possible. We therefore sought to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses).
Types of interventions
1. Trifluoperazine
Any dose administered by any means. We sought to keep to BNF 2012 doses ‐ "initially 5 mg twice daily, increased by 5 mg daily after 1 week, then at intervals of 3 days, according to the response; elderly reduce initial dose by at least half" ‐ and to consider any dose over 30 mg as very high. Doses outside this range were further investigated using sensitivity analysis.
2. Placebo
Any form of placebo or no treatment alternative.
Types of outcome measures
We divided all outcomes into short term (less than three months), medium term (three to six months) and long term (over six months).
Primary outcomes
1. Global state 1.1 Any clinically significant response in medium term global state (as defined by each study).
2. Behaviour 2.1 Any clinically significant agitation or distress (as defined by each study).
3. Relapse +/‐ hospitalisation 3.1 Relapse including any hospitalisation of a participant within a study.
4. Severe adverse effects 4.1 Any clinically significant severe short term adverse effects based on relevant rating scales.
Secondary outcomes
1. Global state 1.1 Average score/change in global state ‐ short and long term. 1.2 Relapse.
2. Behaviour 2.1 Use of adjunctive medication for sedation. 2.2 Aggression to self or others.
3. Mental state 3.1 Any clinically significant response in psychotic symptoms. 3.2 Average score/change in psychotic symptoms. 3.3 Any clinically significant response in positive symptoms. 3.4 Average score/change in positive symptoms. 3.5 Any clinically significant response in negative symptoms. 3.6 Average score/change in negative symptoms.
4. Leaving the study early 4.1 Any reason. 4.2 Due to adverse effects. 4.3 Due to relapse.
5. Extrapyramidal adverse effects 5.1 Use of any anti‐Parkinsonism drugs. 5.2 Average score/change in extrapyramidal adverse effects. 5.3 Tardive dyskinesia. 5.4 Acute dystonia. 5.5 Akathisia. 5.6 Pseudo‐Parkinsonism.
6. Other adverse effects/event, general and specific 6.1 Death.
7. Hospital and service utilisation outcomes 7.1 Hospital admission. 7.2 Average change in days in hospital. 7.3 Improvement in hospital status (for example: change from formal to informal admission status, use of seclusion, level of observation).
8. Economic outcomes
8.1 Average change in total cost of medical and mental health care. 8.2 Total indirect and direct costs. 8.3 Direct resource use: 8.3.1 Outpatients ‐ number of contacts (GP consultation, psychiatrist, psychologists, psychiatric nurse, counsellor, social worker). 8.3.2 Hospitalisation (taking battery of tests, patients’ physical, psychiatric and psychological profile and psychological assessment, number of days, relapse). 8.3.3 Medication (different types of antipsychotics to include dose and frequency, treatment of side‐effects). 8.3.4 Psychological therapies (different types of psychological therapies to include session numbers and frequency) 8.3.5Other resources (day centres, night shelter) and transportation for medical care visits. 8.4 Indirect resource use: 8.4.1 Family, relative and friends resources. 8.4.2 Police, criminal justice system. 8.4.3 Benefits paid, social security payments. 8.4.4 Employment agency workers, absence from work, loss of productivity. 8.5 Cost‐effectiveness ratios represented by ICER. 8.6 Cost‐utilities represented by incremental costs per QALY or DALYs 8.7 Cost benefit represented by net Benefit Ratio, others.
9. Quality of life/satisfaction with care for either recipients of care or caregivers 9.1 Significant change in quality of life/satisfaction. 9.2 Average score/change in quality of life/satisfaction.
10. Cognitive response 10.1 Any clinically important change. 10.2 Any change, general and specific.
11. 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used GRADE profiler (GRADEPRO) to import data from RevMan 5.1 (Review Manager) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
1. Global state ‐ any clinically significant response in global state ‐ medium term. 2. Global state ‐ relapse +/‐ hospitalisation ‐ medium term. 3. Mental state ‐ any clinically significant response in psychotic symptoms ‐ medium term. 4. Leaving the study early ‐ medium term. 5. Severe adverse side effects ‐ short term. 6. Behaviour ‐ any clinically significant response in behaviour ‐ medium term. 7. Economic outcomes.
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia Group Trial Register
We searched the Cochrane Schizophrenia Group's register (May 2012), which is based on regular searches of CINAHL, EMBASE, MEDLINE and PsycINFO. Trifluoperazine is known by many names, so we constructed the following search phrase to assist identification using the following search strategy:
Trifluoperazine‐phrase = *10‐[3‐(4‐methyl‐1‐piperazinyl)propyl]‐2‐trifluoromethylpheno thiazine (hydrochloride)* or *terfluzine* or *terfluzinor discimer* or *eskazine foille* or *iremo* or *piero* or *jatroneural* or *modalina* or *oxyperazine* or *sedofren* or *sporalon* or *stelazine* or *stelazina* or *stelium* or *terflurazine* or *terfluoperazine* or *SKF 5019* or *7623 RP* or *trifluoperazine* or *Solazine*.
See Appendices for details of the original search.
2. Cochrane Schizophrenia Group Health Economic Database
For the economic search, the economic review team replicated the above strategy in the Cochrane Schizophrenia Group Health Economic Database (CSzGHED), 9th April 2013. The database of studies relates to cost‐effectiveness of schizophrenia treatments. This database was constructed from systematic searches of four databases: Health Economic Evaluation Database (HEED), National Health Services Health Economic Database (NHS EED), Cost‐Effectiveness Analysis Registry (CEA) and EconLit as well as Cochrane Registry.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Selection of studies
Review author KM independently inspected citations from the searches and identify relevant abstracts. A random 20% sample was independently re‐inspected by review authors KK and EH to ensure reliability. Where disputes arose, the full report was acquired for more detailed scrutiny. Full reports of the abstracts meeting the review criteria were obtained and inspected by KM. Again, a random 20% of reports were re‐inspected by KK and EH in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we contacted the authors of the study for clarification.
For the selection of economic studies, two authors (VF and SS) inspected all retrieved citations identified by the economic database search, and where disputes arose, the full report was acquired for further inspection.
Data extraction and management
1. Extraction
Review author KM extracted data from all included studies. In addition, to ensure reliability, KK and EH independently extracted data from a random sample of these studies, comprising 10% of the total. Again, any disagreements were discussed, decisions documented and, if necessary, authors of studies contacted for clarification. With remaining problems KM, KK and EH helped to clarify issues and these final decisions were documented. Data presented only in graphs and figures were extracted whenever possible, but included only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. Had we identified multi‐centre studies, we would have extracted data relevant to each component centre separately; however, we found no multi‐centre studies to include.
For the economic analysis, studies of Type A and B (see Types of studies), were investigated by VF and SS, investigated whether appraisal had already been undertaken by the National Health Service's Economic Evaluation Database (NHS EED) using their search tool derived for this purpose. If appraisal had not been undertaken, we applied this tool to the data. In this current review, there were only Type C studies available; therefore, we extracted outcome data directly from the already‐included effectiveness studies. We recognised that much information would be lacking to get results that are both valid and reliable (this is a pilot economic study and there is a risk that the results may be incorrect).
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We would have included continuous data from rating scales only if: a) the psychometric properties of the measuring instrument were described in a peer‐reviewed journal (Marshall 2000); and b) the measuring instrument was not written or modified by one of the trialists for that particular trial. Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, in Description of studies we would have noted if this was the case or not. However, no scale‐derived data were found to include in this review.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint) which can be difficult in unstable and difficult to measure conditions such as schizophrenia. Had we found any scale data to include, we had decided that we would primarily use endpoint data, and only use change data if the former were not available. Endpoint and change data would have been combined in the analysis, with mean differences (MD) rather than standardised mean differences (Higgins 2011). Again, we found no such data to be included.
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we would have applied the following standards to all data before inclusion: a) standard deviations (SDs) and means are reported in the paper or obtainable from the authors; b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution, (Altman 1996); c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), (Kay 1986)), which can have values from 30 to 210), the calculation described above is modified to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and 'S min' is the minimum score.
Endpoint scores on scales often have a finite start and end point and these rules can be applied. Skewed data pose less of a problem when looking at means if the sample size is large (> 200) and we would have entered these into the syntheses. We would have presented skewed endpoint data from studies of less than 200 participants in ‘Additional tables’ rather than enter such data in analyses. When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We would have presented and entered change data into analyses. However, we found no such data.
2.5 Common measure
To facilitate comparison between trials, we would have converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month). However, we found no such variables.
2.6 Conversion of continuous to binary
Had we found any continuous data, we would have made the effort to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a).
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for trifluoperazine. Where keeping to this makes it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not improved') we reported data where the left of the line indicates an unfavourable outcome. This is noted in the relevant graphs, and is the case for the outcome of 'clinical improvement'.
Assessment of risk of bias in included studies
Again, review authors KM, KK and EH worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the raters disagreed, the final rating was made by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted the authors of the studies in order to obtain further information. Non‐concurrence in quality assessment was reported, but when disputes arose as to which category a trial is to be allocated, again, we resolved this by discussion.
The level of risk of bias is noted in both the text of the review and in the Table 1.
This review also aimed to assess the overall methodological quality of each study included in the economic evaluation. Assessment of risk bias was carried out using the checklist developed by Drummond 1996 and the CHEC criteria list (Evers 2005) for Type A and B studies. Had we found any economic studies of Type A or B level, this would have been noted in the summary as well as in Table 2. In this current review, only Type C level studies were used, and therefore the same judgement for risk of bias was employed as for the effectiveness studies.
1. Economic summary.
| Study | Country | Participants | Perspective | Type of Economic Evaluation | Resource Use provided | Unit Costs Provided | ICER | QALY/DALY | Net Benefit Ratio | Grading |
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). For statistically significant results, we used 'Summary of findings' tables to calculate the number needed to treat to provide benefit /to induce harm (NNTB/H) statistic and its 95% CI.
2. Continuous data
Had we encountered continuous data, we would have estimated mean difference (MD) between groups. We would prefer not to calculate effect size measures (standardised mean difference(SMD)). However, if scales of very considerable similarity had been used, we would have presumed there was a small difference in measurement, and would have calculated the effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
We did not find any cluster‐randomised trials. If clustering had not been accounted for in primary studies, we planned to present the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation coefficients for their clustered data and to adjust for this by using accepted methods (Gulliford 1999).
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC is not reported, it will be assumed to be 0.1 (Ukoumunne 1999).
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we would have only used data of the first phase of cross‐over studies. However, no such studies were found.
3. Studies with multiple treatment groups
There were no studies incorporating multiple treatment groups; however, for future versions of this review, where a study involves more than two treatment arms, if relevant, the additional treatment arms would be presented in comparisons. If data are binary these would be simply added and combined within the two‐by‐two table. If data are continuous we would combine the data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011). Where the additional treatment arms are not relevant, we will not use these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, where more than 50% of data were unaccounted for, we did not reproduce these data or use them within analyses, (except for the outcome 'leaving the study early'). When, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we marked such data with (*) to indicate that such a result may well be prone to bias. This was the case in Prien 1969*.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). Those leaving the study early were all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. For these outcomes the rate of those who stayed in the study ‐ in that particular arm of the trial ‐ were used for those who did not. We undertook a sensitivity analysis to test how prone the primary outcomes were to change when data only from people who complete the study to that point are compared to the ITT analysis using the above assumptions.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported, we would have reproduced these. However, no continuous data were found.
3.2 Standard deviations
If standard deviations (SDs) were not reported, we would first have tried to obtain the missing values from the authors. If not available, where there are missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals (CIs) available for group means, and either a 'P' value or 't' value available for differences in mean, we would calculate them according to the rules described in the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011): When only the SE is reported, SDs are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, t or F values, CIs, ranges or other statistics. If these formulae do not apply, we would calculate the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless would examine the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, if LOCF data were used in the trial, if less than 50% of the data have been assumed, we would have presented and used these data and indicated that they were the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, these were fully discussed.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose, these were fully discussed.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
Heterogeneity between studies was investigated by considering the I2 method alongside the Chi2 'P' value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. 'P' value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, was interpreted as evidence of substantial levels of heterogeneity (Section 9.5.2 ‐ Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in section 10.1 of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011). We sought to locate protocols of included randomised trials. If the protocol had been available, outcomes in the protocol and in the published report would have been compared. However, no protocols were available, so outcomes listed in the methods section of the trial report were compared with actually reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are again described in Section 10 of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We intended not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In this version of the review, no funnel plots were used, because no single outcome included 10 or more studies.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose a random‐effects model for all analyses.
Pilot Economic Summary
“It has been argued for many years that promoting effective care without taking into account the cost of care and the value of any health gain can lead to inefficient use of public and private funds allocated to health care, which may indirectly result in harm for individuals and the public” (Williams 1987).
We intended to summarise data from type A and type B studies. Data were summarised according to the Cochrane Campbell Economic Methods Group (Higgins 2011) and if information had been available, a narrative abstract would have been presented for each included study in (see Table 2).
We anticipated that most studies would be Type C level of economic evidence and that we would use data from such studies to calculate a GBP value associated with the outcomes. These approximate values were calculated by (a) using the Personal Social Services Research Unit (PSSRU ‐ NHS reference costs for mental health services) calculation of £338 (weighted mean average of all adult mental health inpatient bed days) per hospital bed day based in a UK NHS setting (PSSRU 2012), and (b) assuming that one relapse equals one hospital admission, a median length of stay as 16 days, as per Hospital Episode Statistics 2012 (HES 2012; main speciality ‘adult mental illness’), we utilised results of the effects of the intervention that presented service use data for an adult ward as well as for relapse rates (HES is a data warehouse containing details of all admissions, outpatient appointments and A&E attendances at NHS hospitals in England) (c) assuming that the adjunctive medication used was phenobarbital and that it would be prescribed for no longer than 14 days at an average dose of 120 mgs per day. The cost for this was obtained from the BNF which provides unit costs for the medication (d) assuming that for the treatment of extra‐pyramidal side‐effects, procyclidine was used at a dose of 10 mgs three times a day for 14 days. The cost for this was obtained from the BNF which provides unit costs for the medication (e) assuming that for the treatment of akathesia, propranolol was prescribed at a dose of 80 mgs twice a day for 14 days. The cost for this was obtained from the BNF which provides unit costs for the medication.
We have not factored any associated costs (including cost and resource use of treatment) prior to the relevant measured outcomes being considered. We are using UK NHS PSSRU reference costs of 2012 as well as BNF costs from 2013 and therefore present the outcomes in terms of a GBP saving using relative risks obtained from the effectiveness part of the review, which we have considered to be a proxy for resource use.
The authors wish to emphasise the numerous assumptions that have been made for the purposes of presenting this economic data, specifically at Type C studies:
The current included studies contributing to the Type C studies were undertaken between the years of 1961 to 1975; and, taking this into account ‐
The average length of stay and costs have been calculated from current available data, that is, according to 2012 HES costs, from most primarily a UK NHS perspective; and
The GBP value data that are presented reflect a proxy measure only; that is, the GBP value of the intervention effect on the measured outcome, and not taking into account any costs or resource use that may likely have been incurred prior to the actual outcome (which includes, but is not limited to, costs and resource use prior to intervention, the intervention itself and post‐intervention up to outcome)..
We are aware that Cochrane systematic reviews are international in context and in reception; however, we have adopted a UK NHS perspective for the purposes of this review – partly because we have been funded by the National Institute of Health Research (NIHR) (NIHR Cochrane Programme Grant 2011, UK Reference number: 10/4001/15) to undertake a series of economic evaluations within systematic reviews.
“…[I]n the face of scarce resources, decision makers often need to consider not only whether an intervention works, but also whether its adoption will lead to a more efficient use of resources” (Higgins 2011).
The comparisons considered in this review involve trifluoperazine (treatment) versus placebo. The value of incorporating consideration of the economics of a treatment versus placebo comparison is extremely limited, since in practice patients are not treated with placebo. We are aware that any economic analysis of a treatment versus placebo comparison, any apparent differences between comparison groups in terms of resource use or costs are likely to be overestimated (relative to a treatment versus alternative treatment comparison) and are therefore unlikely to be applicable to any target setting (assuming that an alternative treatment is more effective than placebo), thus limiting the value of such an analysis for end users. However, we believe, at least for schizophrenia, placebo (or nothing) is often the clinical option chosen by the person with the illness, and to consider the economic issues surrounding this seems sensible (Bartko 1988).
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.2 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of trifluoperazine for people with schizophrenia in general. In addition, however, we wanted to report data on subgroups of people in the same clinical state, stage and with similar problems. All participants in the included studies were classified as chronic schizophrenia; however, perhaps owing to the age of the included studies and low‐quality reporting standards, details of individual participants were not provided, meaning that subgroup analyses were not possible. We have, however, presented details for trifluoperazine high dose versus placebo and low dose versus placebo separately.
2. Investigation of heterogeneity
If inconsistency was high, this was reported. First, we investigated whether data were entered correctly. Second, if data were correct, we visually inspected the graph and outlying studies were successively removed to see if homogeneity was restored. For this review we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, data would be presented. If not, data were not pooled and issues were discussed. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We did not undertake analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes, we included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then all data were employed from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. If there was a substantial difference, we reported results and discussed them, but continued to employ our assumption.
Had we found continuous data, and made assumptions regarding missing SDs data (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. A sensitivity analysis would have been undertaken to test how prone results were to change when completer‐only data only were compared to the imputed data using the above assumption. If there was a substantial difference, we would have reported results and discussed them but would have continued to employ our assumption. However, no such data were found.
3. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available), allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then data from these trials were included in the analysis.
4. Imputed values
We would have also undertaken a sensitivity analysis to assess the effects of including data from trials where we would have used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If substantial differences were noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not have pooled data from the excluded trials with the other trials contributing to the outcome, but would have presented them separately. Again, no such data were imputed.
5. Fixed and random effects
All data were synthesised using a random‐effects model, however, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether this altered the significance of the results.
6. Unusual doses of trifluoperazine
Again, only working with primary outcomes, we investigated whether doses over 30 mg of trifluoperazine had any different effects than more modest doses.
7. Economic summary
We undertook a sensitivity analysis taking into account both the upper and lower confidence intervals for the risk ratios, of the outcomes of interest, and calculated a saving based on these values to investigate how far this affects the direction of the estimated value.
Results
Description of studies
For in‐depth descriptions of the studies please see Characteristics of included studiesCharacteristics of excluded studies, and Characteristics of studies awaiting classification.
Results of the search
1. Effects of intervention
The electronic search (9 July 2012) identified more than 500 empirical clinical studies which were potentially eligible for inclusion. We screened all results initially, excluding over 313 records that were not relevant; with our second screening, 187 articles were assessed resulting in 24 full‐text articles fully assessed for eligibility. After further assessment, we found 12 potential studies eligible for inclusion. During the cross‐checking process however, two further studies were excluded; Hamilton 1963 and Hunt 1967 did not provide relevant data rendering them unusable, thus we were able to include only 10 studies (Figure 1).
1.
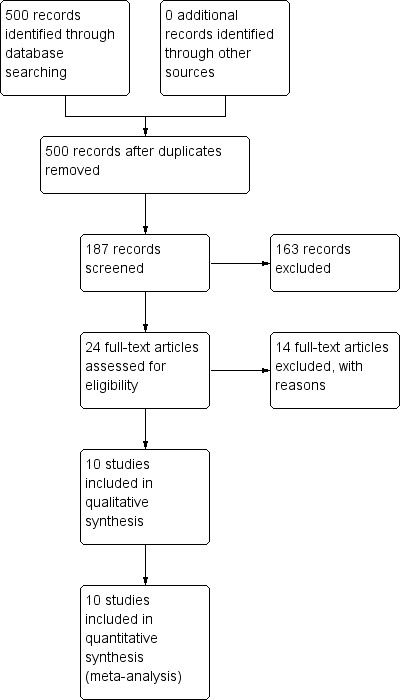
Study flow diagram.
2. Economic
We identified 15 potential studies in our economic evaluation search, none of which met our inclusion criteria for Type A and B. Twelve studies were excluded (See Table 3) and three are awaiting classification (See Table 4). See also Figure 2. Had we found any studies of Type A or B quality, they would have been presented in Table 2. We therefore present data for Type C economic evaluation only.
2. Economic studies: excluded.
| Study ID | Status | Reasons for exclusion | Study type |
| Davies 2007 | Excluded | Allocation: randomised. Particiapants: schizophrenia. Interventions: first vs second generation antipsychotics. Outcomes: no specific outcome measures for trifluoperazine. |
Type A |
| Filippelli 2005 | Excluded | Allocation: randomised. Particiapants: schizophrenia. Interventions: atypical vs typical antipsychotics. Outcomes: no specific outcome measures for trifluoperazine. |
Type A |
| Galvin 1999 | Excluded | Allocation: not randomised. | N/A |
| Ghaemi 2001 | Excluded | Allocation: not randomised. | N/A |
| Hanrahan 2006 | Excluded | Allocation: randomised. Particiapants: schizophrenia. Interventions: atypical vs conventional antipsychotics. Outcomes: no specific outcome measures for trifluoperazine. |
Type A |
| Knapp 2008 | Excluded | Allocation: randomised. Particiapants: schizophrenia. Interventions: olanzapine vs other antipsychotics. Outcomes: no specific outcome measures for trifluoperazine. |
Type A |
| Lewis 1998 | Excluded | Allocation: randomised (systematic review). | N/A |
| Lewis 2006 | Excluded | Allocation: randomised (systematic review). | N/A |
| Martin 2006 | Excluded | Allocation: randomised (systematic review). | N/A |
| Mould 2009 | Excluded | Allocation: randomised. Particiapants: schizophrenia. Interventions: cost and effectiveness of ziprasidone, olanzapine, risperidone, haloperidol and clozapine (not trifluoperazine). |
Type A |
| Stargardt 2008 | Excluded | Allocation: not randomised. | N/A |
| Suttajit 2009 | Excluded | Allocation: randomised (systematic review). | N/A |
3. Economic studies: awaiting classification.
| Study ID | Status | Notes |
| Mapelli 2004 | Awaiting classification. | Full article not obtained. |
| Mauskopf 1999 | Awaiting classification. | Full article not obtained. |
| Percudani 2003 | Awaiting classification. | Full article not obtained. |
2.
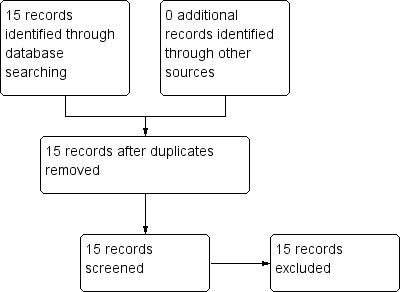
Study flow diagram: economic summary (2013)
Included studies
Further details of the 10 included studies in this review are provided in the Characteristics of included studies.
We found no studies meeting our inclusion criteria for economic evaluation Types A and B. However, for Type C‐level evidence, we utilised the data from relevant economic outcomes of interest (relapse and hospital discharge) from three trials included in the effectiveness section of the systematic review (Prien 1969*; Reardon 1966; Schiele 1961).
1. Length of studies
The duration of the studies included ranged from four weeks in Clark 1975, through to seven months in Marjerrison 1964. The majority of the trials were between two and four months long, and in one study there was an additional observation trial period. In Schiele 1961 the study lasted for 16 weeks with an additional 22‐week trial period that included some of the originally randomised participants.
2. Clinical state
Participants in nine of the included studies were described as having chronic schizophrenia; however there were no diagnostic criteria described with this judgement (Bishop 1964; Clark 1975; Gross 1974; Gwynne 1962; Marjerrison 1964;Menon 1972; Pinard 1972; Prien 1969* and Schiele 1961) and one study described participants as having acute paranoid schizophrenia using the Bleuler Criteria (Reardon 1966).
3. Diagnosis
In nine of the included studies the diagnosis of all participants was schizophrenia (Bishop 1964; Clark 1975; Gross 1974; Gwynne 1962; Menon 1972; Pinard 1972; Prien 1969*; Schiele 1961; Reardon 1966). Diagnosis of 14% of participants in Marjerrison 1964, however, was described as 'chronic psychotic'. We decided to include this study, as the vast majority of participants had schizophrenia, and the study investigated the effects of trifluoperazine on schizophrenia as its primary focus.
4. Exclusions
Where exclusion criteria were listed, these often included physical or neurological disease, mental deficiency, epilepsy, organic brain disease and those who had been hospitalised for under two years (Bishop 1964; Clark 1975; Gross 1974). Clark 1975 excluded any patients who had history of renal or metabolic disease, people under the age of 18, and those suffering from central nervous system (CNS) syphilis. Additional exclusion criteria used in Gross 1974 were drug addictions and severe depression. Schiele 1961 excluded any patients over the age of 55 years 'with a history of complicating organic factors'.
5. Age
The range of ages of participants ranged from 18 years (Prien 1969*), to 67 years (Gross 1974). Eight of the studies (Bishop 1964; Clark 1975; Gross 1974; Gwynne 1962; Marjerrison 1964; Menon 1972; Prien 1969*; Schiele 1961) gave a mean value for the ages of participants with this ranging from 33.25 (Menon 1972) to 49 years (Gwynne 1962). However, the majority of the mean values were between 40 and 49 years. Only Reardon 1966 gave no data relating to age.
6. Gender
In total there were n = 508 men and n = 402 women in the included studies. However, out of this number, there were only data available for n = 159 men and n = 108 women (Bishop 1964; Clark 1975; Gross 1974; Gwynne 1962; Marjerrison 1964; Menon 1972; Schiele 1961). Pinard 1972 stated that men and women were "equally represented", and Reardon 1966 described male and female distribution as "comparable". Prien 1969* did not provide details of male/female distribution amongst groups. Schiele 1961 was the only study to target only male participants.
7. Study size
There were a total of n = 910 participants in the included studies; however, only n = 686 were relevant to this review after selecting data concerning only trifluoperazine or placebo, as some studies had additional treatment arms comparing other antipsychotic drugs. The study sizes varied from 34 participants (Reardon 1966) to "approximately 360" participants (Prien 1969*) with a mean of n = 90 participants between studies. Again, after selecting data from participants only relevant to our comparison, the range of study sizes varied from n = 23 to "approximately 360" participants, with a mean of n = 69 participants.
8. Setting
Nine of the 10 included studies were conducted in a hospital setting, with the majority completed in the USA (Bishop 1964; Clark 1975; Gwynne 1962; Prien 1969*; Reardon 1966; Schiele 1961), followed by Canada (Marjerrison 1964; Pinard 1972) and India (Menon 1972). While Gross 1974 described the setting as a "rehabilitative half‐way house", again in the USA.
9. Interventions
We were interested only in the populations that included trifluoperazine and placebo interventions. In most of the included studies there were comparisons to other drugs, which we did not utilise. The dose given of trifluoperazine ranged from a minimum of 5 mg a day (Gross 1974) through to a maximum high dose of 80 mg a day (Prien 1969*). The mean of dose of all included studies was 30 mg/day. All included studies administered trifluoperazine and placebo in oral capsule form. Two studies implemented a fixed dose of 15 mg/day (Menon 1972; Pinard 1972); two studies increased the dose, starting from 5 mg/day increasing to 40 mg/day (Bishop 1964; Gwynne 1962. Two studies increased dosage over the duration of the study, from 10 mg/day to 50 mg/day (Clark 1975) and 20 mg/day to 40 mg/day (Reardon 1966). One study reported a range of 10 mg/day to 50 mg/day (Schiele 1961), while Gross 1974 reported a mean of 17.5 mg/day and Marjerrison 1964 a mean of 28 mg/day. Prien 1969* compared high‐dose trifluoperazine (80 mg/day) to low‐dose trifluoperazine (15 mg/day) with placebo ‐ the results of this particular study have been pooled and presented in the main comparison, as well as two separate comparisons in the data and analysis section. This was the only study to compare high and low doses, therefore meta‐analysis was not possible.
10. Outcomes
Only binary data were available for outcomes, including: clinical improvement; severe short terms adverse effects; relapse; leaving the study early; use of anti‐Parkinson drugs. The majority of the included studies used a continuous rating scale to measure improvement; mental state; EPS and behaviour. However, with no statistical data available for use in meta‐analysis (often with only P values available), data were rendered usable.
Rating scales used in included studies that provided dichotomised outcomes are listed below:
10.1 Global state
10.1.1 Clinical global impression (CGI) (Guy 1970)
The CGl enables clinicians to quantify the severity of symptoms of any mental health problem at one point in time. Clinicians are then able to measure any improvement or worsening of symptoms over time. A seven‐point scale is used, scoring from one (= very much improved) to seven (= very much worse). Clark 1975; Gross 1974 and Prien 1969* used this scale to measure improvement, presented as a dichotomous outcome.
10.1.2 Minnesota Multiphasic Personality Inventory (MMPI) (Hathaway 1940)
The MMPI was originally developed in 1939, to assess personality traits and psychopathology, administered only by a psychologist specifically trained to do so. It has since been revised ‐ in 1989 (MMPI‐2), 1992 (MMPI‐2) and 2003 (MMPI‐2 RF). The 1939 scale used an empirical keying approach, which derived clinical scales by selecting items endorsed by patients known to have been diagnosed with certain pathologies. The later versions of this scale developed the use of sub scales, to allow for more accessible interpretation of results. The most recent scale consists of two sub scales with a total of 567 items; a clinical sub scale, which assesses 10 traits (including depression, hysteria, paranoia, schizophrenia, hypomania), as well as a validity sub scale, designed to test for inter‐rater reliability. Schiele 1961 used this scale to measure improvement, presented as a dichotomous outcome.
10.1.3 Manifest Behvaiour Scale (MBS) (Mendelsohn 1959)
This scale was designed to measure behavioural changes and consists of 90 items. The MBS subjectively measures the frequency of particular manifest behaviour items, such as 'does he talk to, or answer, what might be hallucinations?'; 'has frequent changes in mood'; 'combs his hair'; 'frequently has tantrums', with a higher score equalling a worsening in behaviour. Schiele 1961 used this scale to measure improvement, presented as a dichotomous outcome.
10.1.4 Quantification of Psychotic Symptom Severity (QPSS) (Goodrich 1953)
The QPSS is an observer‐rated scale, administered within a 15‐minute session, and intended to provide a simple, concise method of rating symptoms to aid hospital psychiatrists when rating severity of a psychotic illness. The scale, however, is not intended to "assess highly complex changes" or evaluate symptoms of non‐hospitalised people with a psychiatric illness. The scale consists of 28 items, dispersed into one of five categories, including: physical state; psychosomatic symptoms; behaviour; emotional state; and mental content items. The rater uses the accompanying 'criteria for quantitation of psychotic symptom severity' to measure the symptom between one (= most extreme degree of symptom) and four (= absence of the symptom), with a higher overall result indicating lower degree of behavioural disturbance and incapacity. Menon 1972 used this scale to measure clinical improvement in global state, presented as a dichotomous outcome.
10.2 Mental state
10.2.1 Psychotic Reaction Profile (PRP) (Lorr 1960)
The PRP was developed to document observable psychotic behaviour in a hospital setting, to be developed into a behaviour inventory for use by nurses and aides who have greater exposure and therefore greater opportunity to observe patients. The scale consists of 85 items, each categorised into one of four scales including withdrawal; thinking disorganisation; paranoid belligerence; and agitated depression. Items are answered with either 'true', 'false' or 'doesn't apply', with greater score indicating a worse outcome. Bishop 1964 used this scale to measure improvement, presented as a dichotomous outcome.
10.3 Behaviour
10.3.1 Miminal Social Behaviour Scale (MSBS) (Farina 1957)
The MSBS measures 32‐items using an environmental, subjective method; the scale is administered "in a room containing a desk, two chairs, a waste paper basket and nothing more". The rater and patient sit in the room; a set of dialogue and actions are then performed by the rater, in order to gauge the response of the patient ‐ for example, "5. The examiner says: 'won't you have a seat'" (to score one point if the patient is seated without further urging), with a higher score indicating a more favourable outcome. Bishop 1964 used this scale to measure improvement, presented as a dichotomous outcome.
10.3.2 Wings Behaviour Rating Scale (Wings) (Wing 1961)
This scale consists of two sub scales; the first measures the mental state and four typical symptoms associated with schizophrenia in a brief interview on a five‐point scale. The second sub scale measures 12 behaviour items on a three‐point scale, with a higher score indicating a more acute state. Menon 1972 used this scale to measure improvement in behaviour, presented as a dichotomous outcome.
11. Missing outcomes
None of the included studies assessed economic outcomes or quality of life/satisfaction with care for either recipients or caregivers. Nor were there any deaths reported in any of the included studies.
12. Funding
Bishop 1964 was supported by a Public Health Grant from the National Institute of Mental Health (USA), and Prien 1969* received various Public Health Service grants from the National Institute of Mental Health. Six of the 10 included studies were funded, at least in part, by pharmaceutical companies. Clark 1975 received a grant from USPHS and a grant in‐aid from Lederle laboratories. Gross 1974 trial drugs provided by McNeil Laboratories. Gwynne 1962 received study drugs from Smith, Kline & French; and Merck, Sharp & Dohme. Marjerrison 1964 received study drugs from Smith, Kline & French; and Montreal & Hoffman LaRoche. Menon 1972 received trial drugs and 'financial assistance' from Ethnor Limited. Smith, Kline & French also provided the study drugs and placebo in Reardon 1966. Funding was not stated in Pinard 1972 or Schiele 1961.
Excluded studies
In total we excluded 14 studies. Of these, six were not randomised (Cahan 1960; Leff 1971; Morton 1968; Stanley 1961; Weckowicz 1960; Weston 1961). A further six did not yield any usable data ‐ one of these was a cross‐over design that did not present any data pre‐cross‐over (Barron 1961) while another was a withdrawal study, not yielding results for placebo (Holden 1971); the other four studies did not present any extractable data (Abuzzahab 1977; Hamilton 1963; Hunt 1967; Madgwick 1958). Leff 1973 pooled results from two RCTs, rendering data unusable. Due to the length of time that has elapsed since these studies were undertaken, the review authors were unable to contact trial authors in the pursuit of attaining further information where we classified data as 'unusable'. Coons 1962 was excluded as participants were of mixed diagnosis, including participants who had received pre‐frontal lobotomy.
Twelve economic studies were excluded altogether; this was due to no randomisation in three studies (Galvin 1999; Ghaemi 2001; Stargardt 2008), four results were in fact systematic reviews (Lewis 1998; Lewis 2006; Martin 2006; Suttajit 2009) and five studies did not present specific outcome measures for trifluoperazine (Davies 2007; Filippelli 2005; Hanrahan 2006; Knapp 2008; Mould 2009). See Table 3.
Studies awaiting classification
Ortega‐Soto 1996 is available only as an abstract and we await the full paper.
Three economic studies await classification, due to full reports not being obtainable at the time of writing (See Table 4).
Risk of bias in included studies
See Figure 3 and Figure 4 for a graphical overview.
3.
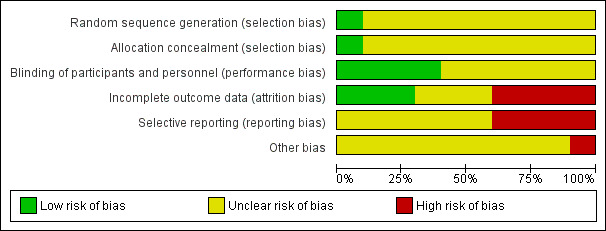
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
We excluded any studies that involved no random element as we felt that these would introduce potential for bias in our outcome results. All of the included studies were either described as randomised or implied randomisation in their allocation. Only one of the included trials gave a method of randomisation, with Lederle Laboratories providing a random block for the allocation of the drugs (Clark 1975); this was the only study to be rated as a 'low' risk of bias. None of the other included studies mentioned a method of randomisation but instead described 'random allocation', 'random assignment', or 'random division' to groups. For this reason, the remaining nine studies were rated as an 'unclear' risk of bias.
The majority of studies gave no details as to allocation concealment and were therefore rated as an 'unclear' risk; only one study provided details, in which allocation was controlled by the pharmacy, withholding allocation from investigators (Reardon 1966). This is the only study to be rated as a 'low' risk of bias.
Blinding
All but one of the 10 included studies were described as 'double blind', with a single study implied using a single (assessor) blind method (Menon 1972), with no details of participant blinding. This study was rated as an 'unclear risk'. Of the remaining nine studies, a double blind code was mentioned but not described (Pinard 1972; Prien 1969*), or identical capsules for all of the groups to prevent appearance of the drugs weakening the blinding, with no further detail as to assessor blinding (Bishop 1964; Clark 1975; Gross 1974; Gwynne 1962). Although, in a trial of this nature, it can be said that blinding would have been difficult to maintain and more prone to being broken due to the extrapyramidal side effects that the trifluoperazine manifests. In longer trials, the patients in the placebo group would also have a greater likelihood of global worsening as they were not receiving any active medication. Distribution of medication was controlled by the pharmacy in Reardon 1966, stating that ward personnel and participants were unaware of the medication they received. Marjerrison 1964 described a large effort for blinding throughout the course of the study, however blinding seemed to have been broken in the second phase of the study. For these reasons, these studies were rated with an 'unclear' risk of bias. Only Schiele 1961 described strict double blind conditions in which capsules identical in appearance were used, and only the hospital pharmacist had the code ‐ for this reason, we decided to rate this study as a 'low' risk of bias.
Incomplete outcome data
Three studies were rated as 'low' risk by the review authors, who reported either no loss to follow‐up or drop‐outs at any point (Bishop 1964), or participant drop‐outs due to adverse effects, but making use of intention‐to‐treat (ITT) (Gross 1974; Gwynne 1962). For the one of the three studies rated as 'unclear', no drop‐outs were reported throughout the trial period (Menon 1972), but due to no explicit mention of all participants completing the entire study duration, this study was rated as an 'unclear' risk. For the remaining, drop‐outs were reported either due to 'strong drug reactions', going 'AWOL', or other adverse effects, but it remains unclear how participant data were handled and to what extent ITT were used, if at all (Clark 1975; Pinard 1972). The remaining studies were all rated as a 'high' risk; either because drop‐out rates were reported with reasons but without use of ITT (Marjerrison 1964); or participants were excluded from the investigation after being assigned to electroconvulsive therapy (ECT) or transferring/home leave (Reardon 1966). Only half of the participants in each intervention group in Schiele 1961 were tested using specified rating scales throughout the course of the study, while the remaining participants were termed "untestable" and not included in analysis. Finally, Prien 1969* has been starred because the trial authors gave only an approximation of randomised participants included in the study, with group numbers varying between outcomes ‐ therefore, we found it hard to identify the true number of participants that left early or that were included in the final analysis. This particular study was subject to a sensitivity analysis.
Selective reporting
None of the included studies provided protocol information, making it difficult to ascertain any explicit selective reporting. With the data available to us, however, we found that each included study used a continuous rating scale to measure either mental state (including the BPRS or PANSS), global state (CGI), behaviour (NOSIE) as well as other scales to rate EPS and social functioning. However, none of the included studies provided usable continuous data; for instance, only providing only P values; means with no standard deviations and, in the majority of studies, only graphs and visual representations were available for inspection. For this reason, six of the included studies were rated as a 'unclear' risk under this category (Clark 1975; Gross 1974; Marjerrison 1964; Prien 1969*; Reardon 1966; Schiele 1961), while the remaining four studies were rated as a 'high' risk where outcomes expressed in the methods sections were not reported (Bishop 1964; Gwynne 1962; Menon 1972), or where methods of analysis were changed retrospectively upon completion of the study (Pinard 1972).
Other potential sources of bias
Six of the 10 included studies were funded, at least in part, by pharmaceutical companies. Clark 1975 received a grant from USPHS and a grant in‐aid from Lederle laboratories. Gross 1974 trial drugs provided by McNeil Laboratories. Gwynne 1962 received study drugs from Smith, Kline & French; and Merck, Sharp & Dohme. Marjerrison 1964 received study drugs from Smith, Kline & French; and Montreal & Hoffman LaRoche. Menon 1972 received trial drugs and 'financial assistance' from Ethnor Limited. Smith, Kline & French also provided the study drugs and placebo in Reardon 1966. Four of the included studies explicitly stated that raters were independent of treatment (Gwynne 1962; Marjerrison 1964; Menon 1972; Schiele 1961), however, we decided to keep these ratings as an 'unclear' risk because three of these studies were funded, at least in part, by a pharmaceutical company, and the primary investigator in Schiele 1961 was also the author of the MBS scale, administered to measure their primary outcome of improvement. We rated only one study (Gross 1974) as a 'high' risk of bias, in which the trials authors acknowledged that the social worker administering the social rating scale was “prejudiced by the nature of her job” by working at the 'half‐way house' in which the study was undertaken. This study also received trial drugs from McNeil Laboratories Inc. Another potential source of bias in these studies was the general small sample size; for instance, six studies have 40 or less participants relevant to our review, these include Bishop 1964; Clark 1975; Gross 1974; Menon 1972; Reardon 1966 and Schiele 1961.
Effects of interventions
See: Table 1
Only dichotomous data were found; we used risk ratios (RR) with 95% confidence intervals (CIs) throughout.
1. TRIFLUOPERAZINE versus PLACEBO
All studies provided data for this comparison (Bishop 1964; Clark 1975; Gross 1974; Gwynne 1962; Marjerrison 1964; Menon 1972; Pinard 1972; Prien 1969*; Reardon 1966; Schiele 1961) with a total of n = 910.
1.1 Global state: clinical improvement
Overall, there was a highly significant improvement in clinical state in the trifluoperazine group (6 RCTs, n = 509, RR 6.44, CI 2.72 to 15.22) as well as significant clinical improvements in both sub‐categories in the short term (zero to three months) (3 RCTs, n = 92, RR 10.93, CI 2.74 to 43.60) and medium term (three to six months) favouring trifluoperazine (3 RCTs, n = 417, RR 4.61, CI 1.54 to 13.84, Analysis 1.1).
1.1. Analysis.
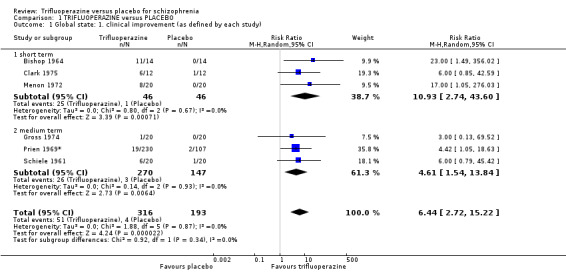
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 1 Global state: 1. clinical improvement (as defined by each study).
1.2 Behaviour: clinically significant agitation or distress (as defined by each study)
Gwynne 1962 was the only study to give usable data on agitation and found no significant difference between the two groups in the short/medium term (1 RCT, n = 52, RR 2.00, CI 0.19 to 20.72, Analysis 1.2).
1.2. Analysis.
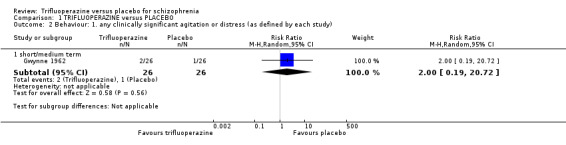
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 2 Behaviour: 1. any clinically significant agitation or distress (as defined by each study).
1.3 Behaviour: use of adjunctive medication for sedation
Data were equivocal at medium term (1 RCT, n = 50, RR 0.94, CI 0.34 to 2.61) and long term (1 RCT, n = 50, RR 0.80, CI 0.24 to 2.61, Analysis 1.3) in the one study that reported this outcome.
1.3. Analysis.
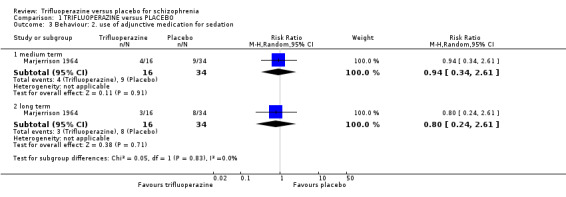
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 3 Behaviour: 2. use of adjunctive medication for sedation.
1.4 Behaviour: clinical improvement
One small study found significant clinical improvement in the short term in the trifluoperazine group compared with placebo (1 RCT, n = 40, RR 27.00, CI 1.71 to 425.36, Analysis 1.4).
1.4. Analysis.
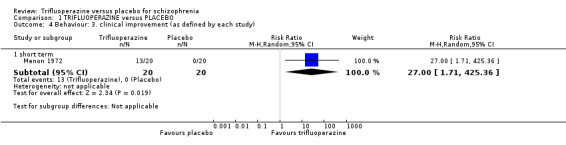
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 4 Behaviour: 3. clinical improvement (as defined by each study).
1.5 Mental state: clinically significant response in psychotic symptoms
Meta‐analysis revealed no significant differences in 'intensified' psychotic symptoms between the two groups (4 RCTs, n = 139, RR 0.75, CI 0.32 to 1.74); however, there was slight heterogeneity present (P = 0.27; I2 = 24%). No significance was shown in either the short term (2 RCTs, n = 59, RR 0.37, CI 0.09 to 1.58, again displaying slight heterogeneity; P = 0.25; I2= 25%), or by short/medium term (2 RCTs, n = 80, RR 1.05, CI 0.54 to 2.05, Analysis 1.5).
1.5. Analysis.
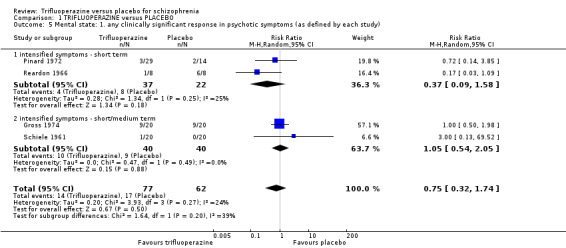
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 5 Mental state: 1. any clinically significant response in psychotic symptoms (as defined by each study).
1.6 Mental state: clinically significant response in positive symptoms
One small study showed no difference in significant clinical response (defined as 'delusions and hallucinations' in the particular study) in favour of the trifluoperazine group (1 RCT, n = 16, RR 0.17, CI 0.03 to 1.09, Analysis 1.6).
1.6. Analysis.
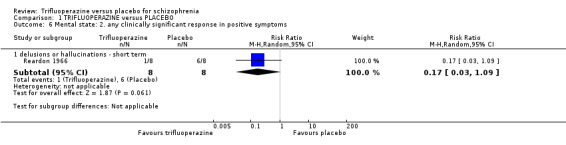
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 6 Mental state: 2. any clinically significant response in positive symptoms.
1.7 Leaving the study early: any reason
No significant difference was shown between the two groups in terms of leaving the study early for any reason (8 RCTs, n = 613, RR 0.72, CI 0.45 to 1.16, Analysis 1.7), over any time frame. There was considerable heterogeneity evident at medium term however, (2 RCTs, n = 391, RR 0.80, CI 0.17 to 3.81; P = 0.04; I2 = 77%). It must be noted that leaving the study early for any reason across all time frames carried moderate heterogeneity (P = 0.15; I2 = 35%).
1.7. Analysis.
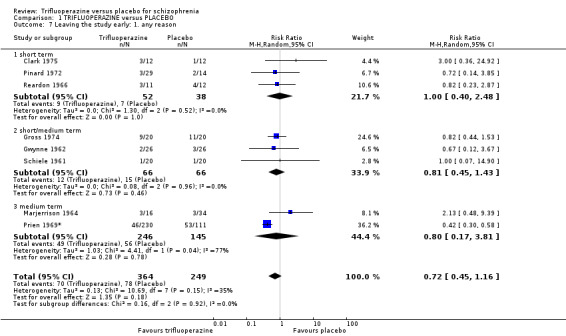
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 7 Leaving the study early: 1. any reason.
1.8 Leaving the study early: severe adverse effects
No difference was found overall (7 RCTs, n = 590, RR 1.00, CI 0.62 to 1.62), nor between groups in either short term (2 RCTs, n = 67, RR 1.31, CI 0.22 to 7.80), short/medium term (3 RCTs, n = 132, RR 0.84, CI 0.46 to 1.52) and medium term (2 RCTs, n = 391, RR 1.54, CI 0.56 to 4.24, Analysis 1.8).
1.8. Analysis.
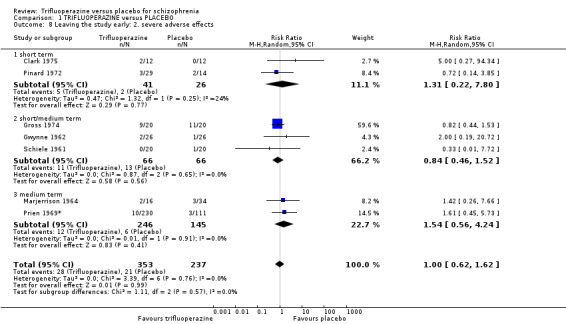
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 8 Leaving the study early: 2. severe adverse effects.
1.9 Leaving the study early: due to relapse or worsening
In the meta‐analysis, there was a significant favour for trifluoperazine, with higher numbers of people leaving the study in the placebo groups (3 RCTs, n = 404, RR 0.35, CI 0.25 to 0.50), with significant difference shown at medium term (2 RCTs, n = 381, RR 0.34, CI 0.23 to 0.49) but no difference by short term (1 RCT, n = 23, RR 0.73, CI 0.15 to 3.57, Analysis 1.9).
1.9. Analysis.
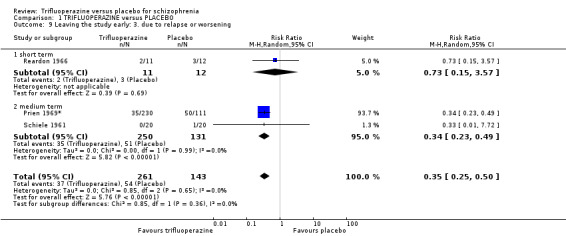
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 9 Leaving the study early: 3. due to relapse or worsening.
1.10 Extrapyramidal adverse effects: general
Overall, there were significantly fewer general extrapyramidal adverse effects in the placebo group compared with trifluoperazine (5 RCTs, n = 184, RR 2.93, CI 1.28 to 6.70), however with slight heterogeneity (P = 0.14; I2= 48%). This high significance was shown to be present in the sub category of short term (3 RCTs, n = 92, RR 4.89, CI 1.36 to 17.59), but not in short/medium term (2 RCTs, n = 92, RR 2.08, CI 0.86 to 5.02, Analysis 1.10), which also demonstrated slight heterogeneity (P = 0.17; I2= 48%).
1.10. Analysis.
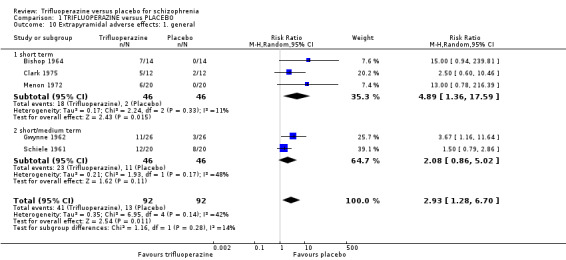
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 10 Extrapyramidal adverse effects: 1. general.
1.11 Extrapyramidal adverse effects: use of antiParkinson drugs
Overall, significantly more patients required anti‐Parkinson drugs in the trifluoperazine group compared with the placebo group (3 RCTs, n = 114, RR 5.91, CI 2.64 to 13.26). This significance was demonstrated in the short/medium term (1 RCT, n = 40, RR 4.50, CI 1.11 to 18.27) and long term (1 RCT, n = 50, RR 8.50, CI 2.78 to 25.97) but not in the short term alone (1 RCT, n = 24, RR 3.00, CI 0.36 to 24.92, Analysis 1.11).
1.11. Analysis.
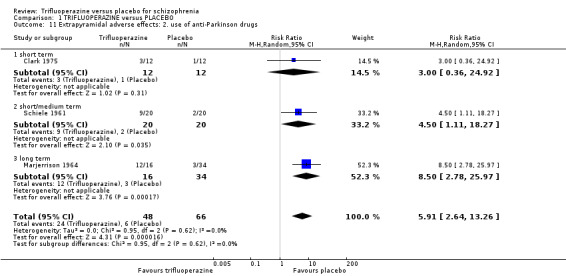
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 11 Extrapyramidal adverse effects: 2. use of anti‐Parkinson drugs.
1.12 Extrapyramidal adverse effects: dyskinesia
Meta‐analysis revealed no difference in instances of dyskinesia in the short term (2 RCTs, n = 52, RR 3.00, CI 0.33 to 27.11, Analysis 1.12).
1.12. Analysis.
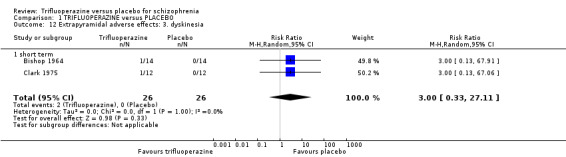
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 12 Extrapyramidal adverse effects: 3. dyskinesia.
1.13 Extrapyramidal adverse effects: akathisia
Akathisia was shown to be highly significant with more prevalence in the trifluoperazine group, consequently favouring placebo (2 RCTs, n = 369, RR 10.78, CI 3.06 to 37.99). Short‐term data were not significant (1 RCT, n = 28, RR 5.00, CI 0.26 to 95.61), but slightly favouring the placebo group. Short/medium term data (zero to six months) were significant (1 RCT, n = 341, RR 12.79, CI 3.17 to 51.53, Analysis 1.13).
1.13. Analysis.
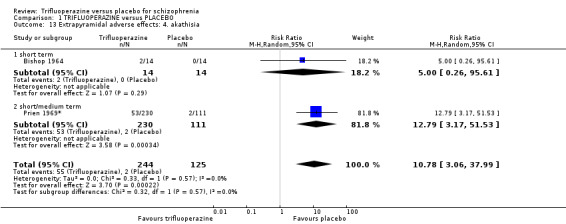
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 13 Extrapyramidal adverse effects: 4. akathisia.
1.14 Extrapyramidal adverse effects: Parkinsonism
Instances of Parkinsonism were generally higher with those receiving trifluoperazine, however significance was found only by the short/medium term (1 RCT, n = 341, RR 1.93, CI 1.19 to 3.12). The finding was not significant at short term (2 RCTs, n = 44, RR 15.00, CI 0.94 to 239.81), nor in meta‐analysis of subgroups (3 RCTs, n = 385, RR 3.43, CI 0.54 to 21.69, Analysis 1.14).
1.14. Analysis.
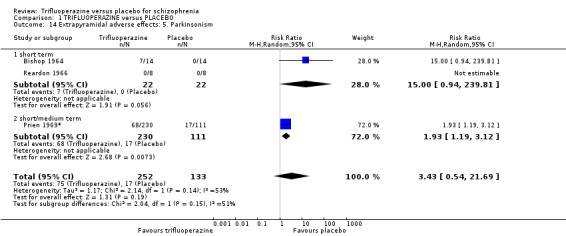
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 14 Extrapyramidal adverse effects: 5. Parkinsonism.
1.15 Extrapyramidal adverse effects: dystonia
There was no difference by the short/medium term (1 RCT, n = 341, RR 1.75, CI 0.94 to 3.29, Analysis 1.15).
1.15. Analysis.
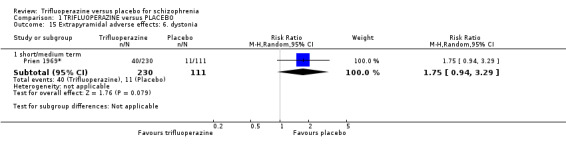
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 15 Extrapyramidal adverse effects: 6. dystonia.
1.16 Other adverse effects: general
Overall, all other adverse effects showed to be just non‐significant and slightly in favour of the placebo group, with more people receiving trifluoperazine showing more adverse effects (5 RCTs, n = 192, RR 1.90, CI 0.77 to 4.70), however, heterogeneity was considerable (P = 0.03; I2 = 62%). Results were homogenous at short term, favouring placebo (2 RCTs, n = 68, RR 13.98, CI 1.94 to 100.64), and again by medium term (1 RCT, n = 44, RR 2.38, CI 0.37 to 15.16). However, in the short/medium term group, there was no difference (2 RCTs, n = 80, RR 1.10, CI 0.61 to 2.00, Analysis 1.16), with heterogeneity evident again (P = 0.19; I2 = 43%).
1.16. Analysis.
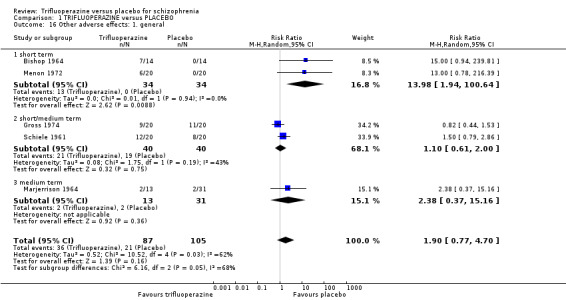
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 16 Other adverse effects: 1. general.
1.17 Other adverse effects: specific
It was found that more participants receiving placebo experienced decreased appetite in the short/medium term in the placebo group (2 RCTs, n = 381, RR 0.59, CI 0.39 to 0.89). Results were also significant for rigidity, with more participants receiving trifluoperazine experiencing rigidity in the short/medium term (1 RCT, n = 40, RR 9.00, CI 1.25 to 64.59).
Although not significant, dizziness (1 RCT, n = 341, RR 9.21, CI 0.54 to 156.86) and drowsiness (1 RCT, n = 24, RR 7.00, CI 0.40 to 122.44) appeared more frequently in the trifluoperazine group between zero to 6 months. Higher instances of oculogyric crisis were shown in the trifluoperazine group at short term (1 RCT, n = 24, RR 3.00, CI 0.13 to 67.06) and blurred vision in the short/medium term (2 RCTs, n = 92, RR 5.00, CI 0.26 to 98.00); furthermore, incoordination occurred more frequently in the trifluoperazine group (1 RCT, n = 40, RR 3.50, CI 0.83 to 14.83, Analysis 1.17).
1.17. Analysis.
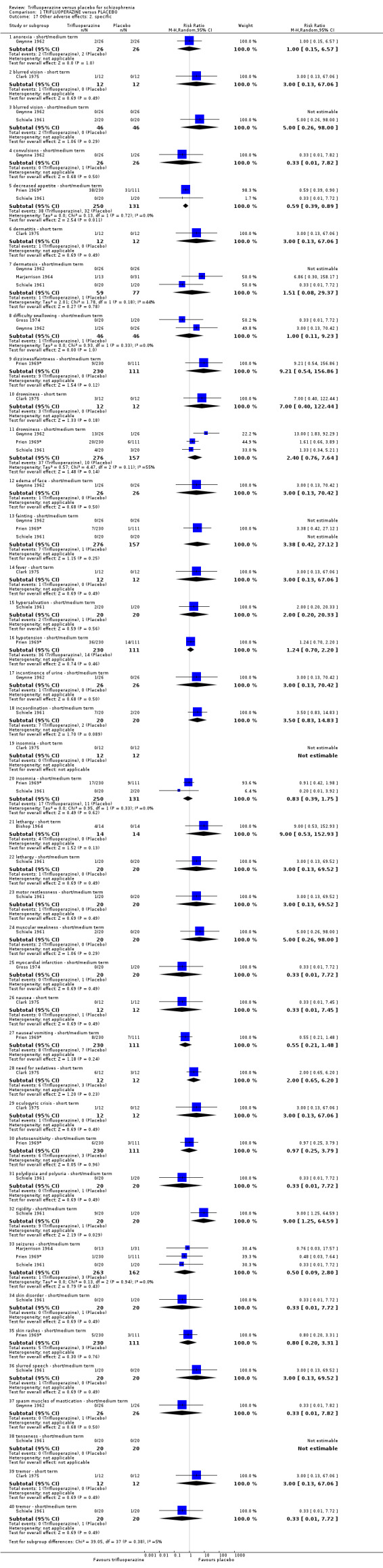
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 17 Other adverse effects: 2. specific.
1.18 Other adverse effects: laboratory data
All data for laboratory tests were equivocal, for instance, with only slightly more people receiving trifluoperazine reported as either having lost weight (1 RCT, n = 24, RR 2.00, CI 0.45 to 8.94) or gained weight (1 RCT, n = 24, RR 2.00, CI 0.21 to 19.23, Analysis 1.18) at short term.
1.18. Analysis.
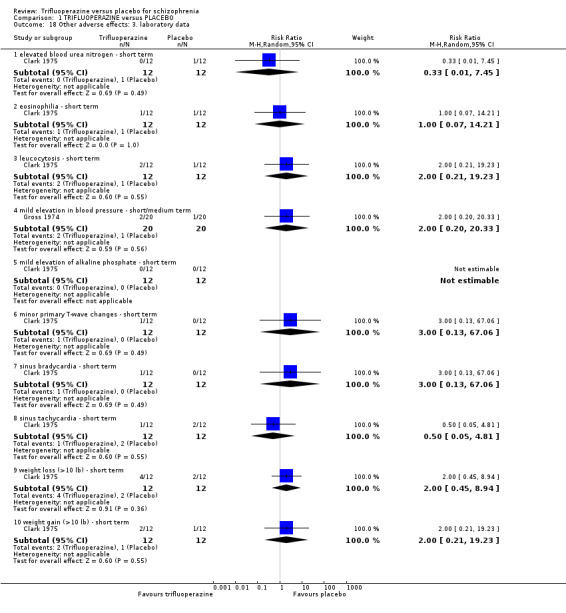
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 18 Other adverse effects: 3. laboratory data.
1.19 Hospital and service utilisation outcomes: hospital transfer/home leave
There was no difference in the number of people either transferring hospitals or experiencing home leave (1 RCT, n = 23, RR 1.09, CI 0.08 to 15.41, Analysis 1.19).
1.19. Analysis.
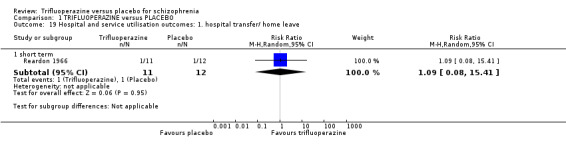
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 19 Hospital and service utilisation outcomes: 1. hospital transfer/ home leave.
1.20 Hospital and service utilisation outcomes: hospital discharge
There was a slight favouring for trifluoperazine with this outcome, with more persons discharged compared with people receiving placebo in the medium term (1 RCT, n = 40, RR 3.00, CI 0.13 to 69.52, Analysis 1.20).
1.20. Analysis.
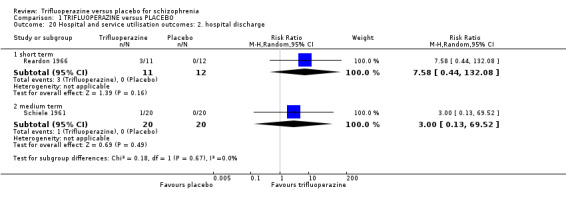
Comparison 1 TRIFLUOPERAZINE versus PLACEBO, Outcome 20 Hospital and service utilisation outcomes: 2. hospital discharge.
1.21 Economic outcomes
Cost of adjunctive medication for sedation
The clinical assumption used was based on unit costs of phenobarbital (60 mg, 28‐tab pack = £5.75, BNF) and a 14‐day treatment period at 120 mg at night‐time per patient, per day. We synthesised estimates of differences in resource use by using relative risk as a proxy measure (see Table 5). The relative risk was 0.8(0.24,2.61) and using this the GBP value obtained for trifluoperazine was £4.6(1.38,15.0) as compared to £5.75 for placebo
4. Economic: Differences in resource use using Relative Risk (RR).
| Resource | Base case | Favouring trifluoperazine (CI) | Favouring placebo (CI) | ||||||
| RR | Trifluoperazine | Placebo | RR | Trifluoperazine | Placebo | RR | Trifluoperazine | Placebo | |
| Adjunctive medication | 0.8 | £4.6 | £5.75 | 0.24 | £1.38 | £5.75 | 2.61 | £15 | £5.75 |
| Use of anti‐parkinson drugs | 5.91 | £25.53 | £4.32 | 2.64 | £11.40 | £4.32 | 13.25 | £57.24 | £4.32 |
| Treatment for akathisia | 10.78 | £8.46 | £0.79 | 3.06 | £2.42 | £0.79 | 37.99 | £30.01 | £0.79 |
| Relapse | 0.35 | £1892 | £5408 | 0.25 | £1352 | £5408 | 0.50 | £2704 | £5408 |
| Total: | £1930.59 | £5418.86 | £1367.2 | £5418.86 | £2806.25 | £5418.86 | |||
Cost of anti‐parkinson medication
The clinical assumption used was based on unit costs (5 mg, net price 28‐tab pack = £1.44, BNF) and a 14‐day treatment period at 10 mg three times a day, per patient. We synthesised estimates of differences in resource use by using relative risk as a proxy measure. The relative risk was 5.91(2.64,13.25) and using this the GBP value obtained was £25.53(11.40,57.24) as compared to £4.32 for placebo
Cost for treatment of akathisia
The clinical assumption used was based on unit costs (80 mg, 56‐tab pack = £1.57, BNF) and a 14‐day treatment period at 80 mg twice a day, per patient. We synthesised estimates of differences in resource use by using relative risk as a proxy measure. The relative risk was 10.78(3.06,37.99) and using this the GBP value obtained was £4.6(1.38,15.0) as compared to £0.79 for placebo
Cost of relapse
This assumption was based on the HES‐calculated median length of hospital stay of 16 days (HES 2012) multiplied by the PSSRU‐calculated mean daily cost of hospitalisation of £338 (PSSRU 2012). We synthesised estimates of differences in resource use by using relative risk as a proxy measure. The relative risk was 0.35(0.25,0.50) and using this the GBP value obtained was £1892(1352,2704) as compared to £5408 for placebo.
Total: comparative savings
Our calculations show that there is a cost‐saving of £3488.3, when comparing placebo as against trifluoperazine, and most of these savings are due to an increase in relapse costs. Even using confidence intervals (95% CI) of effectiveness data, we still have savings of at least £2,612.6 in favour of trifluoperazine when using the upper‐end of the CI, which could go up to £4051.9 if using the lower‐end of the CI.
2. TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO
Only one study provided data for this comparison (Prien 1969*), therefore meta‐analysis was not possible (n = 224). The low dose of trifluoperazine employed in this study was 15 mg/day. We decided to present the data for this study separately due to the incredibly high dose of trifluoperazine employed in the high dose group (80 mg a day).
2.1 Global state: clinical improvement
There was significantly greater (P = 0.04) improvement amongst people receiving low dose trifluoperazine at medium term than placebo (1 RCT, n = 220, RR 4.73, CI 1.06 to 21.11, Analysis 2.1).
2.1. Analysis.
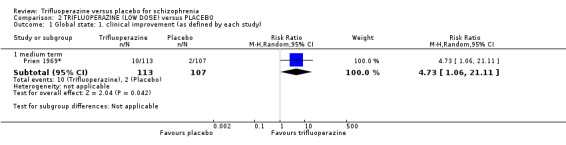
Comparison 2 TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO, Outcome 1 Global state: 1. clinical improvement (as defined by each study).
2.2 Leaving the study early: any reason
Significantly more people receiving placebo left the study early due to any reason by medium term (1 RCT, n = 224, RR 0.39, CI 0.25 to 0.60, Analysis 2.2).
2.2. Analysis.
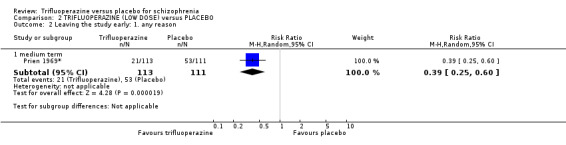
Comparison 2 TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO, Outcome 2 Leaving the study early: 1. any reason.
2.3 Leaving the study early: severe adverse effects
There was no difference in the amount of people leaving the study due to severe adverse effects by medium term (1 RCT, n = 224, RR 0.33, CI 0.03 to 3.10, Analysis 2.3).
2.3. Analysis.
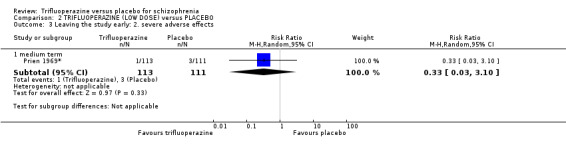
Comparison 2 TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO, Outcome 3 Leaving the study early: 2. severe adverse effects.
2.4 Leaving the study early: due to relapse or worsening
Significantly more people receiving placebo left the study early due to relapse or worsening by medium term (1 RCT, n = 224, RR 0.39, CI 0.25 to 0.61, Analysis 2.4).
2.4. Analysis.
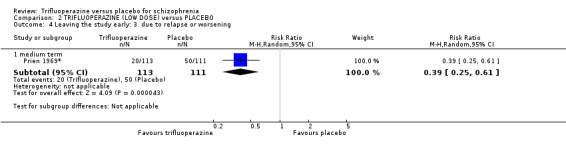
Comparison 2 TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO, Outcome 4 Leaving the study early: 3. due to relapse or worsening.
2.5 Extrapyrimidal adverse effects: akathisia
There were significantly greater instances of akathisia experienced by people receiving the low dose trifluoperazine (1 RCT, n = 224, RR 6.88, CI 1.60 to 29.56, Analysis 2.5).
2.5. Analysis.
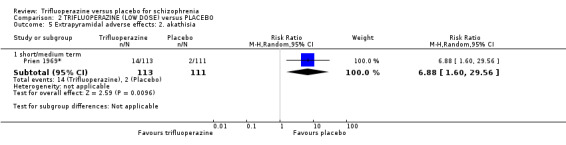
Comparison 2 TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO, Outcome 5 Extrapyramidal adverse effects: 2. akathisia.
2.6 Extrapyrimidal adverse effects: Parkinsonism
There was no difference in Parkinsonism events between groups (1 RCT, n = 224, RR 0.69, CI 0.35 to 1.38, Analysis 2.6).
2.6. Analysis.
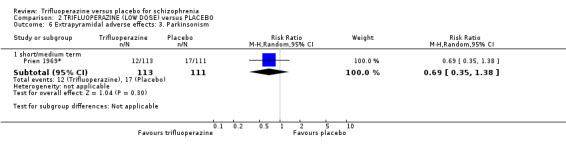
Comparison 2 TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO, Outcome 6 Extrapyramidal adverse effects: 3. Parkinsonism.
2.7 Extrapyrimidal adverse effects: dystonia
There was no difference in dystonia events between groups (1 RCT, n = 224, RR 0.98, CI 0.44 to 2.17, Analysis 2.7).
2.7. Analysis.
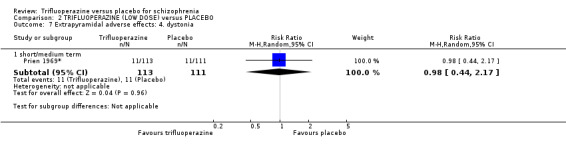
Comparison 2 TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO, Outcome 7 Extrapyramidal adverse effects: 4. dystonia.
2.8 Other adverse effects: specific
Again, there was little difference in levels of specific adverse effects between groups; with slightly higher instances amongst people receiving trifluoperazine of effects such as fainting (1 RCT, n = 224, RR 4.91, CI 0.58 to 41.37); dizziness and fainting (1 RCT, n = 224, RR 6.88, CI 0.36 to 131.62) and hypotension (1 RCT, n = 224, RR 1.47, CI 0.79 to 2.75), however, this was not significant. Only higher instances of decreased appetite were shown in the placebo group, demonstrating statistical significance (1 RCT, n = 224, RR 0.60, CI 0.36 to 1.00, Analysis 2.8).
2.8. Analysis.
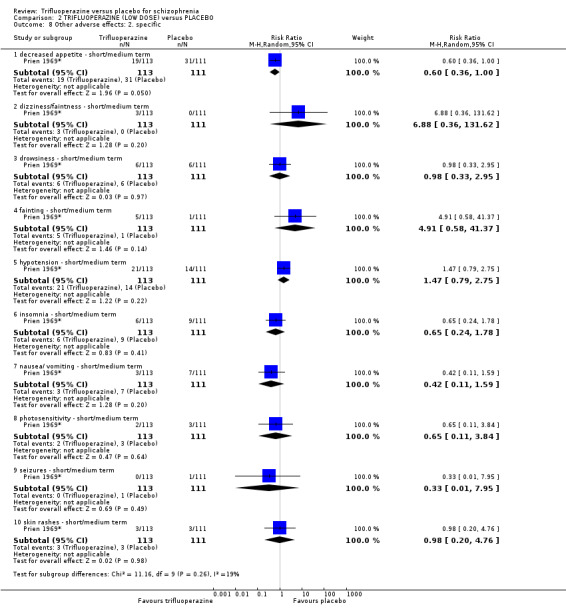
Comparison 2 TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO, Outcome 8 Other adverse effects: 2. specific.
3. TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO
Only one study used a fixed high dose throughout the duration of the entire study (Prien 1969*), therefore meta‐analysis was not possible (n = 228). The high dose of trifluoperazine employed in this study was 80 mg/day.
3.1 Global state: clinical improvement
Slightly more people receiving high‐dose trifluoperazine demonstrated improvement, however this was not a statistically significant finding (1 RCT, n = 228, RR 1.42, CI 0.52 to 3.87, Analysis 3.1).
3.1. Analysis.
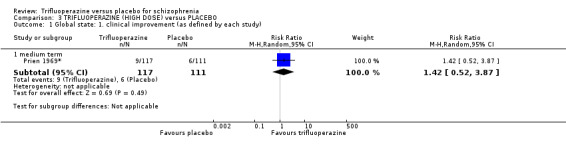
Comparison 3 TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO, Outcome 1 Global state: 1. clinical improvement (as defined by each study).
3.2 Leaving the study early: any reason
Significantly more people from the placebo group left the study early for 'any reason' (1 RCT, n = 228, RR 0.45, CI 0.30 to 0.67, Analysis 3.2).
3.2. Analysis.
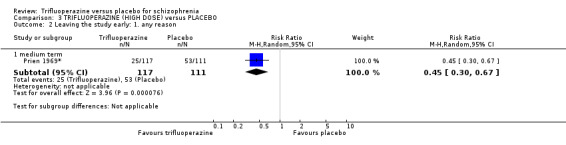
Comparison 3 TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO, Outcome 2 Leaving the study early: 1. any reason.
3.3 Leaving the study early: severe adverse effects
More people from the trifluoperazine group left the study early owing to severe adverse effects; however this was not a statistically significant finding (1 RCT, n = 228, RR 2.85, CI 0.79 to 10.24, Analysis 3.3).
3.3. Analysis.
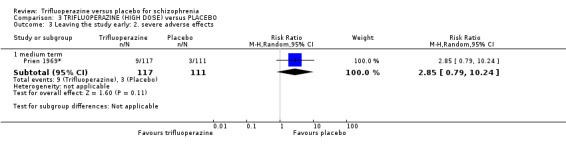
Comparison 3 TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO, Outcome 3 Leaving the study early: 2. severe adverse effects.
3.4 Leaving the study early: due to relapse or worsening
Significantly more people receiving placebo left the study early due to 'deteriorated behaviour' (1 RCT, n = 228, RR 0.28, CI 0.17 to 0.48, Analysis 3.4).
3.4. Analysis.
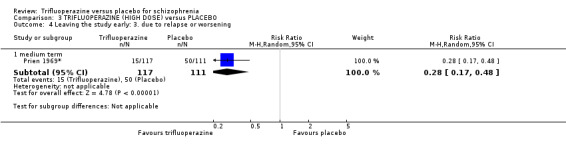
Comparison 3 TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO, Outcome 4 Leaving the study early: 3. due to relapse or worsening.
3.5 Extrapyrimidal adverse effects: akathisia
Significantly more people who received trifluoperazine experienced akathisia than people receiving placebo (1 RCT, n = 228, RR 18.50, CI 4.58 to 74.80, Analysis 3.5).
3.5. Analysis.
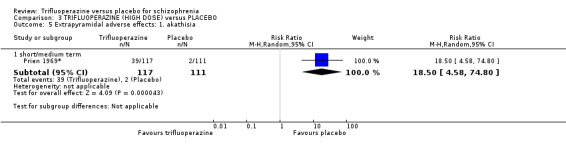
Comparison 3 TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO, Outcome 5 Extrapyramidal adverse effects: 1. akathisia.
3.6 Extrapyrimidal adverse effects: Parkinsonism
There was slight favour for placebo by short/medium term, however this was not statistically significant (1 RCT, n = 228, RR 3.13, CI 1.94 to 5.03, Analysis 3.6).
3.6. Analysis.
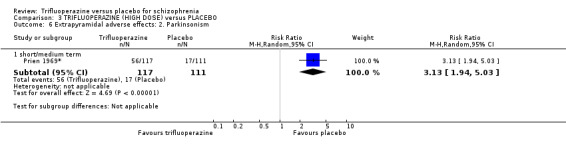
Comparison 3 TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO, Outcome 6 Extrapyramidal adverse effects: 2. Parkinsonism.
3.7 Extrapyrimidal adverse effects: dystonia
There were significantly higher instances of dystonia amongst people receiving high dose trifluoperazine by short/medium term (1 RCT, n = 228, RR 2.50, CI 1.31 to 4.76, Analysis 3.7).
3.7. Analysis.
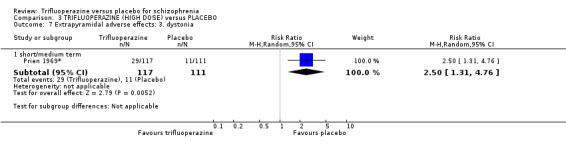
Comparison 3 TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO, Outcome 7 Extrapyramidal adverse effects: 3. dystonia.
3.8 Other adverse effects: specific
A greater number of people taking placebo experienced decreased appetite by short/medium term (1 RCT, n = 228, RR 0.58, CI 0.35 to 0.97) with only slight statistical significance (P = 0.04).There were slightly higher instances of dizziness/faintness (1 RCT, n = 228, RR 12.34, CI 0.70 to 216.49); drowsiness (1 RCT, n = 228, RR 2.21, CI 0.88 to 5.56) and fainting (1 RCT, n = 228, RR 1.90, CI 0.17 to 20.63, Analysis 3.8).
3.8. Analysis.
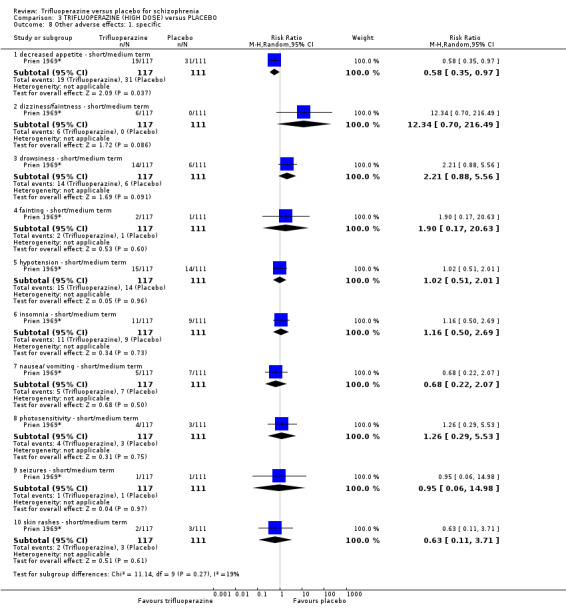
Comparison 3 TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO, Outcome 8 Other adverse effects: 1. specific.
4. Sensitivity analysis
4.1 Implication of randomisation
All included studies at least stated that they used random allocation of participants to treatment. However, no study mentions the procedure or type of randomisation apart from Clark 1975. When all studies that did not provide adequate details of randomisation techniques were excluded from the analysis of our primary outcomes, the following changes were noted.
4.1.1 Global state: any clinically significant response in medium term global state (as defined by each study)
There were no longer significant levels of improvement among people taking trifluoperazine, with only the one study providing any data at the short term (1 RCT, n = 24, RR 6.00, CI 0.85 to 42.59).
4.1.2 Behaviour: any clinically significant agitation or distress as defined by each study
As a result of removing the single study that measured this outcome with a rating of 'unclear' risk of bias associated with unclear randomisation techniques, there were no data left to compare.
4.3.3 Severe adverse effects: any clinically significant severe short term adverse effects based on relevant rating scales
Data reported for people leaving the study early due to severe adverse effects at short term were only available after removing studies that were unclear as to randomisation. The results remain largely equivocal, but removed the slight heterogeneity that was found in the meta‐analysis (1 RCT, n = 24, RR 5.00, CI 0.27 to 94.34).
4.1.4 Relapse +/‐ hospitalisation: relapse including any hospitalisation of a participant within a study
Again, data were reported for people leaving the study early due to relapse; after 'unclear' risk studies were removed from the analysis, there were no data left to compare.
4.2 Assumptions for lost binary data
We undertook a sensitivity analysis to test how prone the primary outcomes were to change when data only from people who completed the study to that point were compared to the intention‐to‐treat (ITT) analysis using the above assumptions.
4.2.1 Global state: any clinically significant response in medium term global state (as defined by each study)
At short term, when comparing completer‐only data, data remain extremely significant, but with greater favour for trifluoperazine than when using ITT data (3 RCTs, n = 88, RR 11.97, CI 3.04 to 47.16). Data by medium term remain significant, but with a lower P value (P = 0.02 instead of P = 0.006) (3 RCTs, n = 300, RR 3.59, CI 1.21 to 10.67), with meta‐analysis of both short‐ and medium‐term subgroups still in significant favour of trifluoperazine (6 RCTs, n = 388, RR 5.72, CI 2.44 to 13.43).
4.2.2 Behaviour: any clinically significant agitation or distress as defined by each study
Completer‐only data remain equivocal (1 RCT, n = 47, RR 1.92, CI 0.19 to 19.73).
4.2.3 Severe adverse effects: any clinically significant severe short term adverse effects based on relevant rating scales
As per our protocol, those leaving the study early were all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects, therefore we did not complete a sensitivity analysis comparing how likely the results are to change when compared with completer‐only data, as this would have the effect of unfairly over‐estimating the amount of severe adverse effects witnessed in any one arm of the study.
4.2.4 Relapse +/‐ hospitalisation: relapse including any hospitalisation of a participant within a study
There is little difference in numbers of people leaving the studies early due to relapse when completer‐only data are used, however results display greater significance for trifluoperazine at short term (1 RCT, n = 16, RR 0.67, CI 0.15 to 2.98), medium term (2 RCTs, n = 280, RR 0.22, CI 0.16 to 0.30) and at both short and medium term together (3 RCTs, n = 296, RR 0.24, CI 0.16 to 0.37).
4.3 Risk of bias
Nine out of 10 studies were rated as a high risk of bias across one of the risk of bias domains for any of the primary outcomes (with Clark 1975 as the only exception), including incomplete outcome data (attrition bias); selective reporting (reporting bias); and other bias. Nine out of 10 studies were also rated as an 'unclear' risk of bias under the randomisation factor (again, with Clark 1975 as the only exception) and, no study was rated as a 'high' risk of bias for either allocation concealment (selection bias), nor blinding of participants or personnel (performance bias) when assessing our primary outcomes, which are listed as follows:
4.3.1 Global state: any clinically significant response in medium term global state (as defined by each study)
After removing studies rated as a 'high' risk of bias for incomplete outcome data (attrition bias) from the meta‐analysis, the results still favoured trifluoperazine at medium term for greater levels of improvement, however the results are no longer statistically significant, with much wider confidence intervals and far less power resulting from excluding the larger studies (1 RCT, n = 40, RR 3.00, CI 0.13 to 69.52).
4.3.2 Behaviour: any clinically significant agitation or distress as defined by each study
As a result of removing the single study that measured this outcome with a rating of 'high' risk of bias under the selective reporting domain, there were no data left to compare.
4.3.3 Severe adverse effects: any clinically significant severe short term adverse effects based on relevant rating scales
There were data reported for people leaving the study early due to severe adverse effects at short term; there was little difference in the results after removing 'high' risk studies, with placebo still favoured (non‐significantly), however heterogeneity was removed as a consequence (1 RCT, n = 24, RR 5.00, CI 0.27 to 94.34).
4.3.4 Relapse +/‐ hospitalisation: relapse including any hospitalisation of a participant within a study
Again, data were reported for people leaving the study early due to relapse; after 'high' risk studies were removed from analysis, there were no data left to compare.
4.4 Imputed values
There were no usable continuous data and we did not impute any statistical values.
4.5 Fixed‐effect and random‐effects
Random‐effects were used throughout. Within the primary outcomes, no outcomes were affected when switching to a fixed‐effect model.
4.6 Unusual doses of trifluoperazine
Seven out of the 10 studies that provided data for the primary outcomes at some point, used doses of trifluoperazine above the current BNF 2012 guidelines of 30 mg per day (Bishop 1964; Clark 1975; Gross 1974; Gwynne 1962; Prien 1969*; Reardon 1966; Schiele 1961). The range was from as low as 5 mg a day (Gross 1974) to the highest dose of 80 mg a day for one of the trifluoperazine groups in Prien 1969*. Two studies used fixed doses of 15 mg a day (Menon 1972; Pinard 1972) and one of the trifluoperazine groups in Prien 1969* received 'low dose' of 15 mg a day. Marjerrison 1964 used just below 30 mg a day, with a mean of 27 mg a day. We removed the seven studies that used more than 30 mg a day from the analysis of the primary outcomes to see if this changes the estimate of the effect:
When removing relevant studies that used more than 30 mg a day, only data for short term remained, which still demonstrated statistically significant favour for global state improvement amongst the trifluoperazine group (1 RCT, n = 40, RR 17.00 95% CI 1.05 to 276.03, Analysis 1.1). Data, after removing Bishop 1964 and Clark 1975 still show favour for trifluoperazine, although this is not statistically significant (1 RCT, n = 24, RR 17.00 95% CI 1.05 to 276.03). Due to the greater power of Prien 1969*, if this study alone is removed from medium term, the data are rendered just insignificant (2 RCTs, n = 80, RR 4.90 95% CI 0.89 to 26.85, Analysis 1.1). These data do suggest that, apart from short term clinical global improvement, studies with more power and larger doses in the medium term seem to show higher levels of clinical global improvement. No other changes worthy of note occured in the other primary outcomes when looking at study doses.
There is little difference when comparing the high dose (80 mg a day) and low dose (15 mg a day) groups with placebo in Prien 1969* when assessing levels of global improvement, with statistical significance (P = 0.04) just shown in the low‐dose group (1 RCT, n = 220, RR 4.73, CI 1.06 to 21.11, Analysis 2.1). When using higher doses of 80 mg a day, Prien 1969* records higher levels of leaving the study early due to severe adverse effects (1 RCT, n = 228, RR, 2.85, CI 0.79 to 10.24, Analysis 3.3), although not significant. Prien 1969* shows significantly more extrapyramidal adverse effects with a high dose of 80 mg a day, including akathisia (1 RCT, n = 228, RR 18.50, CI 4.58 to 74.80, Analysis 3.5) and Parkinsonism (1 RCT, n = 228, RR 3.13, CI 1.94 to 5.03, Analysis 3.6). From this relatively large study, it is shown that this higher dose seems to promote global improvement, but also cause significantly more adverse effects, especially extrapyramidal phenomenon.
4.7 Economic outcomes
The effectiveness data that these values are based on, however, are based on small sample sizes and numbers of events, as well as imprecise effect estimates, as the 95% confidence interval for best estimate of effect include both no effect and appreciable benefit/harm. This calls into question the reliability of any estimated 'savings' that may be seen between groups; particularly so, as the results are merely placing a value on the effectiveness data, which takes into account very limited associated direct or indirect costs or resources associated with treatment. However, the same caveats remain for these assumption ‐ particularly that, even when taking into consideration the upper and lower confidence intervals of the risk ratios, one cannot ignore the unaccounted‐for costs of, for example, treatment for some of the associated adverse effects of trifluoperazine, which may‐well offset any savings.
Discussion
Summary of main results
1. TRIFLUOPERAZINE versus PLACEBO
As can be seen from the Table 1 that, despite seven trials providing data (total n = 576), the overall quality of evidence was either low or very low, reiterating that further studies are needed.
1.1 Global state
As expected, patients with the trifluoperazine intervention showed a significant clinical improvement compared with placebo in both short and medium term, reinforcing the use of this well‐established typical antipsychotic in people with schizophrenia.
1.2 Behaviour
Only one small study was usable so more studies are needed to attain a realistic view of this outcome. Nonetheless, no significant agitation was found between the two interventions. The results did demonstrate an extra patient in the trifluoperazine group classed as agitated, which could well be a reflection of the side effects of trifluoperazine, even though this drug is primarily known for its extrapyramidal side effects (BNF 2012). Even though insignificant, it is expected that adjunct sedative medication would be used more frequently in the placebo group due to the better control of symptoms and possible sedatory effects of trifluoperazine (BNF 2012, RCPSYCH 2009). Further studies reporting this are needed. Only one small study (Menon 1972) reported clinical improvement in behaviour as an outcome and as expected, more patients in the trifluoperazine group showed improved behaviour, again due to the antipsychotic effects of the drug and its possible sedatory effects (BNF 2012, RCPSYCH 2009).
1.3 Mental state
Even though overall no significance was shown, short‐term response in psychotic symptoms demonstrated greater intensified symptoms for people receiving placebo, favouring the trifluoperazine group; a result that is generally expected from this antipsychotic drug. In the one study that reported this outcome, trifluoperazine was found to be more effective at treating positive symptoms of delusions and hallucinations. This is again expected as typical antipsychotics, are known to have a greater effect on treating positive symptoms of schizophrenia than the negative symptoms. Newer generation atypical antipsychotics are said to be more effective than typical at treating the negative symptoms (BNF 2012; RCPSYCH 2009).
1.4 Leaving the study early
All sub‐categories, although insignificant, did generally show more patients receiving placebo leaving the study early, with the relatively large study of Prien 1969* having significant data. Gross 1974 mentions two patients in the placebo intervention leaving the study early, one due to dysphagia and one due to a myocardial infarction; these are unlikely to be due to the placebo intervention and more likely to be due to chance and random allocation. For example, the maximum age range was up to 67 years, dysphagia and a myocardial infarction could be due to other events, even though an effort was made to exclude patients with other underlying co‐morbidities. Results were moderately heterogeneous (35%). This could well be due to the different study designs that included comparisons of other antipsychotics, as well as the setting (Gross 1974 was the only outpatient study), patient population and demographics and the vast number of possible adverse effects that can be experienced on or off of trifluoperazine.
For those leaving the study early due to severe adverse effects, there was no difference in the results of the meta‐analysis. To clarify, here, we defined 'severe' as any adverse effect that necessitated a patient to leave the study early. Defining a 'severe' adverse event is very subjective; any adverse effect could lead to the termination of a patient from a study if severe enough, occurring at the right time in the right individual. More severe adverse effects could potentially have been included, but due to lack of information from the trials, these data could not be determined and are represented in the outcome 'other adverse effects'.
1.5 Extrapyramidal adverse effects
Our results agree with what is already well known about trifluoperazine; that it has a greater incidence of extrapyramidal effects. It was expected that more anti‐Parkinson drugs would be administered as significantly more people in the trifluoperazine group suffered general extrapyramidal adverse effects. Our results only show significance in the combined short/medium term and long term sub categories, suggesting that anti‐Parkinson drugs are not needed as often in the short term. To reliably suggest this, however, a larger study with greater power is needed. On the other hand, this result could suggest that the onset of these extrapyramidal side effects may take longer than three months. However, due to the lack of information, as the short/medium term sub category was significant, this suggests extrapyramidal results could well have occurred in the short term. The BNF 2012 recommends that 'patients should receive an antipsychotic drug for 4–6 weeks before it is deemed ineffective', suggesting that effects could well happen in the short term. Dyskinesia occurred more frequently in the trifluoperazine group; however, between the two small studies, there is not enough power to state this as a significant result. As stated above, this may reflect dyskinesia as a later‐developing side effect of trifluoperazine use, but specific data in all three stated time frames would be needed as well as more studies to increase the power of the evidence. The larger study of Prien 1969* was the driving‐force behind the significance of the results showing higher instances of akathisia with people receiving trifluoperazine. These results suggest that akathisia is more prevalent in those taking trifluoperazine when compared with placebo and also that akathisia is more prevalent than dyskinesia. The original review by Marques 2004 found that dyskinesia was more prevalent than akathisia and it is difficult to find statistical evidence elsewhere supporting this. More studies are needed.
1.6 Other adverse effects
Results suggest that more patients receiving trifluoperazine experienced general adverse effects than on placebo. However, there were instances of specific adverse effects experienced with placebo, including convulsions; decreased appetite; insomnia; nausea; skin disorders and tremors. We also found that three studies each reported higher instances of seizures in the placebo group. Again, it must be noted that there was significant heterogeneity between groups for general adverse effects; more studies, with particular focus on adverse effects are needed, especially with a drug like trifluoperazine that is well known to cause them. Of note, Schiele 1961 reported a highly significant prevalence of rigidity in the trifluoperazine group, which could be linked to extrapyramidal phenomenon. Hypotension and dizziness/faintness could be linked to the use of trifluoperazine and this is supported by evidence used in current guidelines (BNF 2012). Other adverse effects found in this review such as blurred vision, which could imply anticholinergic effects (Marques 2004) and oculogyric crisis are also stated amongst the many possible adverse effects in the BNF 2012 and the original review of Marques 2004.
1.7 Hospital and service utilisation outcomes
If any future studies are carried out using already hospitalised patients, this outcome could be of importance as release from hospital implies an overall clinical global improved state. Although only a small single study provided these data, results suggested release from hospital occurred more frequently in the trifluoperazine group, showing the benefit of the drug against placebo. This is an area of interest for future studies and the concept could be further transferred for potential outpatient studies under outcomes such as discharge or relief from medication.
1.8 Economic
As we have stated, the current economic evidence we present uses up‐to‐date costs calculated using GBP PSSRU unit costs of health and social care; we have used BNF costs of 2013, and clinical judgement and opinions of a UK‐based mental health professional; baring this in mind, the Type C level of economic evidence used was taken from the effect of intervention data from studies that were conducted between 1961 and 1975 in the USA, Canada or India. Therefore, the international applicability of the data presented is questionable, as is the accuracy of the potential costs that may be incurred and the savings that we state have occurred due to the intervention. What the results do show us, however, is what is already known ‐ that there is a price attached to outcome measures commonly addressed in systematic reviews. This pilot economic summary is intended to highlight and promote discussion of including an economic perspective within the Cochrane Schizophrenia Group’s systematic reviews, and to encourage review authors to incorporate a user‐friendly economic analysis in reviews. It must be borne in mind that it is likely that, in clinical practice, organisation of care and treatment protocols for schizophrenia patients would all impact on care, treatment, management of side‐effects, and hospital discharge ‐ including aftercare ‐ and have very likely changed considerably since 1961‐75, which may undermine the present day applicability of the outcomes that underpin the economic analysis presented.
2. TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO
The only study to include specific low‐dose and high‐dose trifluoperazine was the largest study of Prien 1969*, which used a fixed dose of 15 mg a day. Even with such a large study, due to the high risk of bias associated with this study, it is difficult to have confidence in any results ‐ these should be interpreted with caution.
2.1 Global state; leaving the study early; adverse effects
Clinical improvement was significant for people receiving low‐dose trifluoperazine than for people receiving placebo. Results of the two forest plots comparing low‐dose trifluoperazine versus placebo and high‐dose trifluoperazine versus placebo also demonstrate that more people receiving a low dose improved than those receiving a high dose in the medium term. The evidence taken from this single study suggests that a lower dose leads to more effective clinical improvement than such a high dose. With a significant amount of participants leaving the study early from the placebo group, this could well be an indirect measure of acceptability of treatment, with a greater number of people receiving placebo leaving due to any reason, and due to relapse of worsening. However, results show no difference between low‐dose trifluoperazine or placebo with extrapyramidal adverse effects such as Parkinsonism and dystonia.
3. TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO
Again, the only study to report data for such a high dose of trifluoperazine (80 mg a day) was Prien 1969*.
3.1 Global state; leaving the study early; adverse effects
It would be considered unethical nowadays to undertake a study that directly compares such high doses of trifluoperazine to either placebo or a low dose. The results for clinical improvement from all pooled trifluoperazine versus placebo studies (with a total n = 513) employ a mean of 33 mg/day ‐ a dose that we now consider high based on the BNF 2012 recommendations, but which at the time of the studies (between 37 to 52 years ago) showed clinical improvement. This single study, with a high dose of 80 mg/day, did not yield significant results as regards clinical improvement and showed that a significantly greater amount of extrapyramidal symptoms (akathisia; Parkinsonism; dystonia) were experienced by people in the high‐dose group compared to placebo. These results indicate that exceptionally high doses will not serve to improve global state, nor limit risk of extrapyramidal side effects (EPS). However, greater numbers of people taking placebo left the study early due to any reason, including relapse or worsening. This indirect measure of acceptability of treatment is more in favour of high‐dose trifluoperazine, but at the cost of greater losses of trifluoperazine participants due to severe adverse effects.
Again, due to the high risk of bias associated with this study, results need interpreting with caution.
Overall completeness and applicability of evidence
1. Completeness
Apart from Clark 1975, which mentioned obtaining consent from patients' families, no other study gave any insight into how and even if consent was obtained from the patients or their relatives/next of kin to take part in the trial, which leaves a question mark over the ethics of these trials. Futhermore, Clark 1975 was the only study to mention how they randomly allocated the patients in the trial (block randomisation). This leaves an unclear risk of allocation bias for all of the remaining studies. There were a large number of small studies included from many years ago (Marques 2004). So not only were all the trials pre‐CONSORT (Moher 2001), but the data provided were very limited and, of the data that were provided, only a portion could be used due to incompleteness. No contact with the original authors was possible due to the length of time that has lapsed, therefore further data could not be obtained for use in the review. Future studies, if any, must follow CONSORT (Moher 2001) and include or make all data freely accessible whether used or not. Data must be clearly presented in visual tables and written format and state exactly what is being measured as well as how and when it is being measured. For example, number of adverse effects experienced by a patient; time frame; what they experienced and how often. Other outcomes such as patient satisfaction; quality of life and economic outcomes were not assessed.
1. Applicability
All studies apart from Gross 1974 were conducted in a hospital setting. Nowadays, with schizophrenia being diagnosed earlier and treatment provided earlier (RCPSYCH 2011), prognosis is better, with more care provided in the community, so these older studies do not reflect this transition. Many studies excluded participants with co‐morbidities to reduce the risk of bias of the results and outcomes. Many studies also used higher than BNF 2012 recommended daily doses of trifluoperazine. This does occur in practice; if higher doses are needed to control symptoms then anti‐Parkinson drugs are often administered to try and limit, in particular, extrapyramidal effects (RCPSYCH 2009).
There are no Type A or B economic evaluations conducted on trifluoperazine. With Type C data, which report outcome measures, we have attempted to value the outcomes in GBP terms. This may not be an accurate measure of costs incurred or saved and as such we would not recommend that these results be used. This is a pilot study and we intended to encourage debate on how best to use such reported data.
Quality of the evidence
1. General
Of the 10 included studies, all are pre‐CONSORT (Moher 2001) and are graded either low or very low quality.To a large extent, these studies have not mentioned that they followed a specific method of randomisation and much of the data was unusable.
2. Specific
Ten studies with a total number 686 relevant participants featured in 20 outcomes in this review. The results showed that there was a global improvement in patients’ mental health status. Although these studies have shown improvement in patients’ mental health status, they have not methodologically followed randomised clinical trials designs.
2.1 Economic evaluation
We did not come across any studies that were of Type A or B economic evaluation (which includes but not limited to cost‐effectiveness analysis, cost‐utility analysis and cost‐benefit analysis).
Potential biases in the review process
The data extraction was conducted by researchers KM and EH, with review author KK independently extracting data for a second time from all included 10 studies. The reviews were cross‐checked by a fourth independent review author. Bias may be introduced through the extraction process but efforts beyond stated in the protocol have been employed to try reduce this.
Agreements and disagreements with other studies or reviews
This review focused on trifluoperazine versus placebo. In general, the results of this review come in line with the review conducted by Marques 2004 who indicate that trifluoperazine is more effective in comparison to placebo (as well as other treatments).
Limitations of the review
The study authors appreciate the limitations of the methods used in the economic summary. It is fair to say that the data provided display a rudimentary estimate of the value in GBP terms associated with the outcomes of relapse and hospital discharge. As proxy measures are used for total costs, it is acknowledged that the cost of treatment may well change the direction of the estimated result. The aim of piloting this economic summary is to generate debate and discussion; to not take effectiveness data at face value without considering potential economic consequences; and to utilise the data in a way that will be of greater use to decision/policy‐makers. We will welcome discussion on the methods used for Type C level economic evidence and the planned methods described for Type A and B level evidence should future reviews find those studies. We are aware that policy‐makers and commissioners often consider these reviews while deciding on service provision and valuing outcomes in GBP terms may make it easier to review evidence such as relapse.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
Trifluoperazine is a well‐recognised typical antipsychotic and this review has shown it does have a benefit over placebo, although adverse effects, particularly extrapyramidal side effects, are a major factor to consider. With the wide range of antipsychotics now available and the development of atypical antipsychotics, although personal preference, others may be more beneficial to try first which have shown to be more effective and have a reduced adverse‐effect profile.
2. For clinicians
Clinicians and patients must collaborate to use the drug that is most suitable for the patient, with all relevant circumstances considered. In agreement with Marques 2004, trifluoperazine has similar efficacy to other common antipsychotics although may contribute to more extrapyramidal events. In this light, use of potential other antipsychotics should be considered before starting on trifluoperazine.
3. For managers/policy‐makers
Newer, larger and more comprehensive independent trials, to also include economic evaluation, are needed to compare trifluoperazine with other available antipsychotics rather than placebo, as it is well documented that trifluoperazine is an effective antipsychotic. If future evidence supports the existing evidence, that many typical antipsychotics are as effective as atypical antipsychotics, but also cost‐effective or cost‐beneficial, then a trend could be set to revert back to use of these less expensive drugs, particularly in the current economic climate.
Implications for research.
1. General
Adherence to the CONSORT statement (Moher 2001) would probably have resulted in this review being more conclusive. Clear descriptions of randomisation would have reassured users of these trials that selection bias had been minimised and well described and tested blinding could have encouraged greater confidence in the control of performance and detection bias. As mentioned earlier in this review, studies did not report on design, methodology or analysis of their clinical trials. Therefore, it was not possible to assess the internal validity of the targeted studies. It was found that all studies used binary data, but none mentioned how they converted continuous data into binary and what methods they employed to do so.
2. Specific
Most of the included studies were conducted roughly 40 years ago. Although not a priority question for funders to address, any future studies should adhere to the CONSORT statement in order to improve reporting standards (as described in Overall completeness and applicability of evidence). Any further trials with the sole comparators of trifluoperazine versus placebo are unlikely, based on the available evidence and current accepted practice. The review authors have constructed a suggested design of future study with additional comparators should any new independent clinical trials using trifluoperazine ever be considered (See Table 6).
5. Suggested design for future research.
| Allocation: randomised, clearly described, concealed.
Blindness: double, described and tested.
Duration: 12 months. Setting: community. |
| Diagnosis: schizophrenia or schizophrenia‐like illness, clearly described and documented. N = 600.* Age: any. Sex: both. Exclusion: none but full medical history must be taken into account as well as thorough health state evaluation to reduce potential confounders. |
| 1. Oral trifluoperazine: dose flexible within current guideline recommended limits BNF 2012, N = 150.
2. Oral clozapine: dose flexible within current guideline recommended limits BNF 2012, N = 150. 3. Oral atypical antipsychotic: dose flexible current guideline within recommended limits BNF 2012, N = 150. **4. Oral placebo: N = 150. |
| All outcomes are grouped by time measured at: one month, three months, six months, nine months and 12 months. Mortality. Specific behaviours ‐ self‐harm, including suicide, injury to others, aggression. Global outcomes ‐ overall improvement, use of any relevant additional medication, relapse. Service outcomes ‐ hospital admission and duration of stay, for any reason. Mental state ‐ no clinically important change in general mental state, no clinically important change in psychotic symptoms, broken down into positive and negative symptoms. Adverse effects ‐ clinically important adverse effects, defining severe adverse effects and including all extrapyramidal phenomena. Leaving the study early ‐ any reason; severe adverse effects. Economic outcomes. |
| * Powered to be able to identify a difference of ˜20% between groups for primary outcome with adequate degree of certainty. ** Issues about how ethical it is to give placebo to patients suffering with schizophrenia may arise, especially when all these drugs have been shown to be more beneficial than placebo in past RCTs. |
RCT ‐ randomised controlled trial
Acknowledgements
The Cochrane Schizophrenia Group Editorial Base in Nottingham produces and maintains standard text for use in the Methods section of their reviews. We have used this text as the basis of what appears here and adapted it as required.
The search terms were developed by the Trial Search Co‐ordinator of the Cochrane Schizophrenia Group (who, at the time of searching was Samantha Roberts) and the contact author of this protocol.
Thank you also goes to Stephanie Sampson and Hannah Jones from the Cochrane Schizophrenia Group for all their invaluable knowledge and support given to the authors in writing this review.
We would like to thank the contribution, help and advice received from Toby Lasserson (Cochrane Editorial Unit), Luke Vale and Ian Shemilt (Campbell & Cochrane Economics Methods Group) during the development of the economic evaluation.
Appendices
Appendix 1. Original search (2002)
1. The Cochrane Schizophrenia Group's Register (March 2002) was searched using the phrase:
{[(trifluoperazine‐phrase) in title, abstract or index terms of REFERENCE] or [*trifluoperazine* in interventions of STUDY]}
Data and analyses
Comparison 1. TRIFLUOPERAZINE versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. clinical improvement (as defined by each study) | 6 | 509 | Risk Ratio (M‐H, Random, 95% CI) | 6.44 [2.72, 15.22] |
| 1.1 short term | 3 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 10.93 [2.74, 43.60] |
| 1.2 medium term | 3 | 417 | Risk Ratio (M‐H, Random, 95% CI) | 4.61 [1.54, 13.84] |
| 2 Behaviour: 1. any clinically significant agitation or distress (as defined by each study) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short/medium term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.72] |
| 3 Behaviour: 2. use of adjunctive medication for sedation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 medium term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.34, 2.61] |
| 3.2 long term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.24, 2.61] |
| 4 Behaviour: 3. clinical improvement (as defined by each study) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 short term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 27.0 [1.71, 425.36] |
| 5 Mental state: 1. any clinically significant response in psychotic symptoms (as defined by each study) | 4 | 139 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.32, 1.74] |
| 5.1 intensified symptoms ‐ short term | 2 | 59 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.09, 1.58] |
| 5.2 intensified symptoms ‐ short/medium term | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.54, 2.05] |
| 6 Mental state: 2. any clinically significant response in positive symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 delusions or hallucinations ‐ short term | 1 | 16 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.03, 1.09] |
| 7 Leaving the study early: 1. any reason | 8 | 613 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.45, 1.16] |
| 7.1 short term | 3 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.40, 2.48] |
| 7.2 short/medium term | 3 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.45, 1.43] |
| 7.3 medium term | 2 | 391 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.17, 3.81] |
| 8 Leaving the study early: 2. severe adverse effects | 7 | 590 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.62, 1.62] |
| 8.1 short term | 2 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.22, 7.80] |
| 8.2 short/medium term | 3 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.46, 1.52] |
| 8.3 medium term | 2 | 391 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [0.56, 4.24] |
| 9 Leaving the study early: 3. due to relapse or worsening | 3 | 404 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.25, 0.50] |
| 9.1 short term | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.15, 3.57] |
| 9.2 medium term | 2 | 381 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.23, 0.49] |
| 10 Extrapyramidal adverse effects: 1. general | 5 | 184 | Risk Ratio (M‐H, Random, 95% CI) | 2.93 [1.28, 6.70] |
| 10.1 short term | 3 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 4.89 [1.36, 17.59] |
| 10.2 short/medium term | 2 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 2.08 [0.86, 5.02] |
| 11 Extrapyramidal adverse effects: 2. use of anti‐Parkinson drugs | 3 | 114 | Risk Ratio (M‐H, Random, 95% CI) | 5.91 [2.64, 13.26] |
| 11.1 short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.36, 24.92] |
| 11.2 short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 4.5 [1.11, 18.27] |
| 11.3 long term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 8.5 [2.78, 25.97] |
| 12 Extrapyramidal adverse effects: 3. dyskinesia | 2 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.33, 27.11] |
| 12.1 short term | 2 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.33, 27.11] |
| 13 Extrapyramidal adverse effects: 4. akathisia | 2 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 10.78 [3.06, 37.99] |
| 13.1 short term | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.26, 95.61] |
| 13.2 short/medium term | 1 | 341 | Risk Ratio (M‐H, Random, 95% CI) | 12.79 [3.17, 51.53] |
| 14 Extrapyramidal adverse effects: 5. Parkinsonism | 3 | 385 | Risk Ratio (M‐H, Random, 95% CI) | 3.43 [0.54, 21.69] |
| 14.1 short term | 2 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 15.0 [0.94, 239.81] |
| 14.2 short/medium term | 1 | 341 | Risk Ratio (M‐H, Random, 95% CI) | 1.93 [1.19, 3.12] |
| 15 Extrapyramidal adverse effects: 6. dystonia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 short/medium term | 1 | 341 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.94, 3.29] |
| 16 Other adverse effects: 1. general | 5 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.90 [0.77, 4.70] |
| 16.1 short term | 2 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 13.98 [1.94, 100.64] |
| 16.2 short/medium term | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.61, 2.00] |
| 16.3 medium term | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 2.38 [0.37, 15.16] |
| 17 Other adverse effects: 2. specific | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 anorexia ‐ short/medium term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.57] |
| 17.2 blurred vision ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 67.06] |
| 17.3 blurred vision ‐ short/medium term | 2 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.26, 98.00] |
| 17.4 convulsions ‐ short/medium term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.82] |
| 17.5 decreased appetite ‐ short/medium term | 2 | 381 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.39, 0.89] |
| 17.6 dermatitis ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 67.06] |
| 17.7 dermatosis ‐ short/medium term | 3 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.08, 29.37] |
| 17.8 difficulty swallowing ‐ short/medium term | 2 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.11, 9.23] |
| 17.9 dizziness/faintness ‐ short/medium term | 1 | 341 | Risk Ratio (M‐H, Random, 95% CI) | 9.21 [0.54, 156.86] |
| 17.10 drowsiness ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.40, 122.44] |
| 17.11 drowsiness ‐ short/medium term | 3 | 433 | Risk Ratio (M‐H, Random, 95% CI) | 2.40 [0.76, 7.64] |
| 17.12 edema of face ‐ short/medium term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.42] |
| 17.13 fainting ‐ short/medium term | 3 | 433 | Risk Ratio (M‐H, Random, 95% CI) | 3.38 [0.42, 27.12] |
| 17.14 fever ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 67.06] |
| 17.15 hypersalivation ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.20, 20.33] |
| 17.16 hypotension ‐ short/medium term | 1 | 341 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.70, 2.20] |
| 17.17 incontinence of urine ‐ short/medium term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.42] |
| 17.18 incoordination ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 3.5 [0.83, 14.83] |
| 17.19 insomnia ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 17.20 insomnia ‐ short/medium term | 2 | 381 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.39, 1.75] |
| 17.21 lethargy ‐ short term | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 9.00 [0.53, 152.93] |
| 17.22 lethargy ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 69.52] |
| 17.23 motor restlessness ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 69.52] |
| 17.24 muscular weakness ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.26, 98.00] |
| 17.25 myocardial infarction ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.72] |
| 17.26 nausea ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.45] |
| 17.27 nausea/ vomiting ‐ short/medium term | 1 | 341 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.21, 1.48] |
| 17.28 need for sedatives ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.65, 6.20] |
| 17.29 oculogyric crisis ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 67.06] |
| 17.30 photosensitivity ‐ short/medium term | 1 | 341 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.25, 3.79] |
| 17.31 polydipsia and polyuria ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.72] |
| 17.32 rigidity ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 9.00 [1.25, 64.59] |
| 17.33 seizures ‐ short/medium term | 3 | 425 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.09, 2.80] |
| 17.34 skin disorder ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.72] |
| 17.35 skin rashes ‐ short/medium term | 1 | 341 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.20, 3.31] |
| 17.36 slurred speech ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 69.52] |
| 17.37 spasm muscles of mastication ‐ short/medium term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.82] |
| 17.38 tenseness ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 17.39 tremor ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 67.06] |
| 17.40 tremor ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.72] |
| 18 Other adverse effects: 3. laboratory data | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 18.1 elevated blood urea nitrogen ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.45] |
| 18.2 eosinophilia ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.21] |
| 18.3 leucocytosis ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.21, 19.23] |
| 18.4 mild elevation in blood pressure ‐ short/medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.20, 20.33] |
| 18.5 mild elevation of alkaline phosphate ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 18.6 minor primary T‐wave changes ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 67.06] |
| 18.7 sinus bradycardia ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 67.06] |
| 18.8 sinus tachycardia ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.05, 4.81] |
| 18.9 weight loss (>10 lb) ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.45, 8.94] |
| 18.10 weight gain (>10 lb) ‐ short term | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.21, 19.23] |
| 19 Hospital and service utilisation outcomes: 1. hospital transfer/ home leave | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 19.1 short term | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.08, 15.41] |
| 20 Hospital and service utilisation outcomes: 2. hospital discharge | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 20.1 short term | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 7.58 [0.44, 132.08] |
| 20.2 medium term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 69.52] |
Comparison 2. TRIFLUOPERAZINE (LOW DOSE) versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. clinical improvement (as defined by each study) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 medium term | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 4.73 [1.06, 21.11] |
| 2 Leaving the study early: 1. any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.25, 0.60] |
| 3 Leaving the study early: 2. severe adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.03, 3.10] |
| 4 Leaving the study early: 3. due to relapse or worsening | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.25, 0.61] |
| 5 Extrapyramidal adverse effects: 2. akathisia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 6.88 [1.60, 29.56] |
| 6 Extrapyramidal adverse effects: 3. Parkinsonism | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.35, 1.38] |
| 7 Extrapyramidal adverse effects: 4. dystonia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.44, 2.17] |
| 8 Other adverse effects: 2. specific | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 decreased appetite ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.36, 1.00] |
| 8.2 dizziness/faintness ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 6.88 [0.36, 131.62] |
| 8.3 drowsiness ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.33, 2.95] |
| 8.4 fainting ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 4.91 [0.58, 41.37] |
| 8.5 hypotension ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.79, 2.75] |
| 8.6 insomnia ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.24, 1.78] |
| 8.7 nausea/ vomiting ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.11, 1.59] |
| 8.8 photosensitivity ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.11, 3.84] |
| 8.9 seizures ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.95] |
| 8.10 skin rashes ‐ short/medium term | 1 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.20, 4.76] |
Comparison 3. TRIFLUOPERAZINE (HIGH DOSE) versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. clinical improvement (as defined by each study) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.52, 3.87] |
| 2 Leaving the study early: 1. any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.30, 0.67] |
| 3 Leaving the study early: 2. severe adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 2.85 [0.79, 10.24] |
| 4 Leaving the study early: 3. due to relapse or worsening | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.28 [0.17, 0.48] |
| 5 Extrapyramidal adverse effects: 1. akathisia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 18.50 [4.58, 74.80] |
| 6 Extrapyramidal adverse effects: 2. Parkinsonism | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 3.13 [1.94, 5.03] |
| 7 Extrapyramidal adverse effects: 3. dystonia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 2.50 [1.31, 4.76] |
| 8 Other adverse effects: 1. specific | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 decreased appetite ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.35, 0.97] |
| 8.2 dizziness/faintness ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 12.34 [0.70, 216.49] |
| 8.3 drowsiness ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 2.21 [0.88, 5.56] |
| 8.4 fainting ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 1.90 [0.17, 20.63] |
| 8.5 hypotension ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.51, 2.01] |
| 8.6 insomnia ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.50, 2.69] |
| 8.7 nausea/ vomiting ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.22, 2.07] |
| 8.8 photosensitivity ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.29, 5.53] |
| 8.9 seizures ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.06, 14.98] |
| 8.10 skin rashes ‐ short/medium term | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.11, 3.71] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bishop 1964.
| Methods | Allocation: random. Blinding: double. Duration: 10 weeks. Design: parallel (3 groups), single centre. Setting: inpatient, Toulane research wards, East Louisiana State Hospital, Jackson, Louisisana (USA). |
|
| Participants | Diagnosis: chronic schizophrenia. N = 42 *(n = 28 included in the analysis ‐ see interventions). Age: trifluoperazine ‐ mean 42.9 years; placebo ‐ mean 40.4 years; range 21‐53 years across both groups. Sex: 21M, 21F *(14M, 14F included in the analysis ‐ 7M, 7F in each group). Ethnicity: not stated. Consent: not stated. History: time hospitalised ranges from 3‐27 years, with a mean of 12.4 years for trifluoperazine and 11.7 in placebo groups. Included: off medication for at least 60 days and no committed to other projects. Excluded: concomitant physical or neurological disorder. |
|
| Interventions | 1. Trifluoperazine: maximum dose 40 mg/day (week one: 5 mg/day; week two: 10 mg/day; week three: 20 mg/day; week four: 30 mg/day; weeks five‐10: 40 mg/day), n = 14. 2. Placebo: n = 14. *(3. Butaperazine: maximum dose 200 mg/day, n = 14 ‐ this group was not included in the analysis). |
|
| Outcomes | Global state: improvement (given as a single rating by averaging the final scores of four raters, using the Beckombergo Rating Scale; Psychotic Reactive Profile; Tulane Test Battery; and Minimal Social Behaviour scales). Extrapyramidal adverse effects: akathisia; dyskinesia; Parkinsonism‐like symptoms. Other adverse effects: lethargy; general adverse effects. Unable to use ‐ Global state: individual scores for BRS; PRP; TTB; and MSBS ‐ no mean or SD. Use of anti‐Parkinson drugs: half of the study population (n = 7 in each of the three groups of n = 14) received anti‐Parkinson medication as part of the treatment regimen from the beginning of the study until completion, therefore data are not presented as an effect of the study medication. Furthermore, no data were presented for the placebo group (selective reporting). |
|
| Notes | This study had an 'additional variable' (p675) of assessing the effect of the anti‐Parkinson medication Artane on half of the participants in each group (n = 7 from each group) with the remaining participants receiving an Artane placebo. No data were reported for the placebo group, only for the active drug groups. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ quote “the 42 patients were divided randomly into three groups of 14, each containing 7 males and 7 females” (p674), no further details of randomisation methods. |
| Allocation concealment (selection bias) | Unclear risk | No description. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double ‐ quote, "all personnel involved in the project [remained] blind as to the medication a patient was receiving" (p675). Quote, “both drugs and placebo were supplied in capsules of identical appearance (Parke‐Davis No.2 pink) and were dispensed from individual medicine bottles” (p675). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None detected. |
| Selective reporting (reporting bias) | High risk | For the 'additional variable' to this study, no data were reported for the placebo group regarding numbers of participants who experienced 'extrapyramidal reactions', only data for the active drug groups were presented. No statistical data reported. |
| Other bias | Unclear risk | Funding: supported by Public Health Grant 5 TI‐MH‐03701‐04 (Psychopharmacology Service Center, National Institute of Mental Health). Rating scales: raters not stated to be independent of treatment. Prinicpal investigator created the Tulane Test Battery scale utilised in the study. |
Clark 1975.
| Methods | Allocation: random. Blinding: double. Duration: 4 weeks. Design: parallel (3 groups), single centre. Setting: inpatient, special research ward (USA). |
|
| Participants | Diagnosis: chronic schizophrenia (newly admitted, including ‐ paranoid n = 7; undifferentiated n = 10; hebephrenic n = 1; n = 2 schizoaffective). N = 43 *(n = 24 included in the analysis ‐ see interventions). Age: trifluoperazine ‐ mean 41.4 years; placebo ‐ mean 36.9 years. Sex: 21M, 16F *(13M, 11F included in the analysis ‐ 6M, 6F trifluoperazine; 7M, 5F placebo). Ethnicity: not stated. Consent: family permission. History: newly admitted; mean age of first hospitalisation in placebo 27.4 years; trifluoperazine 31.5 years. Included: diagnosis of schizophrenia confirmed by research scientists; minimum of two years duration of illness; over 18 years old; no evidence of mental deficiency, epilepsy, CNS syphilis, or other types of organic brain disease or significant metabolic disease, liver disease, cardiovascular disease, or renal disease. Excluded: females of child‐bearing potential excluded. |
|
| Interventions | 1. Trifluoperazine: initial dose 5 mg capsules twice daily, increased by 2 capsules twice weekly until maximum of 10 capsules (50 mg) was reached on 15th day, n = 12. 2. Placebo: initial dose 2 capsules, increased by 2 capsules twice weekly until maximum of 10 capsules was reached on 15th day, n = 12. *(3. Loxapine: initial dose 2 capsules, increased by 2 capsules twice weekly until maximum of 10 capsules (100 mg) was reached on 15th day, n = 13 ‐ this group was not included in the analysis). |
|
| Outcomes | Global state: improvement (a rating of 'marked' or 'moderate' improvement according to the psychiatrists' CGI‐I scale). Leaving study early: any reason; due to adverse effects. Adverse effects: sedation; drowsiness; oculogyric crisis; dermatitis; blurred vision; insomnia; nausea; fever; tremor. Laboratory tests: leucocytosis; eosinophilia; elevated blood nitrogen urea nitrogen; mild elevation of alkaline phosphate; sinus tachycardia; sinus bradycardia; minor primary t‐wave changes; weight‐gain; weight‐loss. Extrapyramidal adverse effects: use of anti‐Parkinsonian drugs; EPS symptoms; dyskinesia. Unable to use: Mental state: BPRS – no SD. Behaviour: NOSIE rating scale ‐ only adjusted final means and P values reported. Adverse effects – not all reported according to intervention allocation. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised – quote, “subjects were assigned sequentially to treatment by means of a pre‐randomised list blocked on 3 provided by Lederle Laboratories” (p287). |
| Allocation concealment (selection bias) | Unclear risk | Allocation determined by Lederle Laboratories. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double – quote, “medication was dispensed as identically‐appearing capsules… in bottles labelled only with the patient’s name. The double‐blind technique was followed throughout the study” (p287). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Loss to follow‐up: n = 6 ‐ three participants went AWOL with no final measurements made; n = 1 trifluoperazine‐treated participant was dropped after 12 days because the lymphadenopathy and lymphocytosis noted at baseline had not resolved; another trifluoperazine‐treated participant was terminated after three weeks because of a severe drug reaction characterised by tremor and an elevated temperature; one loxapine‐treated participant was terminated after 10 days when family withdrew consent without reason (p288). ITT used for some outcomes. Quote, “five additional subjects were admitted to the study but were dropped before adequate measures could be obtained” (p288). |
| Selective reporting (reporting bias) | Unclear risk | Not all outcomes reported – SD’s not reported for scales including BRPS, CGI‐I, PGI‐S.ILL, NGI‐IMP, NGI‐S.ILL. Not all adverse effects reported according to treatment group. |
| Other bias | Unclear risk | Funding: USPHS Grants MH 11666 and MH 21409, and a grant in‐aid from Lederle laboratories. Rating scales: one psychiatrist carried out all psychiatric ratings in 32 participants; a second psychiatrist carried out all psychiatric ratings in 5 participants. A psychiatric research nurse observed the daily behaviour of participants and rated them using the NOISE ‐ it is not clear whether raters were independent of treatment. |
Gross 1974.
| Methods | Allocation: random.
Blinding: double.
Duration: 16 weeks (with a further 36‐week open evaluation). Design: parallel (3 groups), single centre. Setting: rehabilitative 'half‐way house', offering 24hr supervision, Harbor View House (USA). |
|
| Participants | Diagnosis: chronic schizophrenia. N = 61 *(n = 40 included in the analysis ‐ see interventions). Age: pimozide: mean = 44.8, range 21‐66 years; trifluoperazine; mean = 47.5, range 20‐67 years; placebo; mean = 44.8, range 24‐64 years. Sex: 37M, 24F *(25M, 14F included in the analysis ‐ 14M, 6F trifluoperazine; 12M, 8F placebo). Ethnicity: not stated. Consent: not stated. History: diagnosis of chronic schizophrenia for at least two years/ previous psychiatric hospitalisation for schizophrenia, with symptoms severe enough to have required continuous treatment with antipsychotic medication within the past three months. Included: demonstrated ‘key schizophrenic symptoms’ ‐ including conceptual disorganisation; emotional withdrawal; blunted affect bizarre mannerisms; unusual thought content; hallucinations. Demonstrable capacity to respond to psychotropic drug treatment as evidenced by improvement in the ‘manifestations of his psychosis’. Excluded: epilepsy; drug addiction; severe depression; mental retardation [sic]; organic brain disease; significant physical disease, or those who require heavy sedation or chemical restraint to control symptomology. |
|
| Interventions | 1. Trifluoperazine: dose range 5 to 30 mg/day, mean 17.5 mg/day, n = 20. 2. Placebo: once daily, n = 20. *(3. Pimozide: dose range 2 to 12 mg/day, mean 6.3 mg/day, n = 21 ‐ this group was not included in the analysis).3 |
|
| Outcomes | Global state: improvement (measured using the CGI1). Leaving the study early: due to adverse effects; any reason2. Mental state ‐ clinically significant response in psychotic symptoms (defined as 'intensified symptoms'). Unable to use ‐ Global state: CGI ‐ therapeutic effect (improvement data more meaningful). Mental state: BPRS (no usable data). Adverse effects (no usable data). Social functioning: Family Rating Form (no usable data), Harbor View House Resident Rating Report (unpublished scale). |
|
| Notes |
1Global state: for CGI improvement, 'very much improved' and 'much improved' data was used. 2Specific side effects under 'intensified symptoms' not elaborated on but included under mental state as 'Clinically significant response in psychotic symptoms'. 3Prior to transfer to study medication, participants had received a neuroleptic for at least four weeks, the last two weeks of which were stabilised at a fixed daily dose, not exceeding ‐ chlorpromazine 500 mg; thioridazine 500 mg; trifluoperazine 30 mg; fluphenazine 30 mg. Drugs administered once daily prior to breakfast in identical capsules containing either pimozide 2 mg; trifluoperazine 5 mg; or placebo. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ quote, "randomly assigned" (p698). No further details. |
| Allocation concealment (selection bias) | Unclear risk | No description. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double ‐ "identical appearing capsules" provided (p698) ‐ no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Follow‐up: 59% ‐ 24% of pimozide‐treated patients, 45% of trifluoperazine treated patients and 55% of placebo‐treated patients failed to complete the study due to intensified symptoms (n = 4 receiving pimozide; n = 9 receiving trifluoperazine; n = 9 receiving placebo); discharge, "to live independently" (n = 1 receiving pimozide); difficulty swallowing (n = 1 receiving placebo) and myocardial infarction (n = 1 receiving placebo). ITT used. |
| Selective reporting (reporting bias) | Unclear risk | Statistical reporting was incomplete for all scale data (no SDs). |
| Other bias | High risk | Funding: Pimozide provided by McNeil Laboratories Inc. Rating scales: not stated to be independent of treatment. The social rating scale was administered by a social worker who worked at Harbour View House and was “prejudiced by the nature of her job” (p700). She reviewed nearly all the patients as being improved or remaining at pre‐trial levels even when taking the placebo. |
Gwynne 1962.
| Methods | Allocation: random. Blinding: double. Duration: 4 months. Design: parallel (3 groups), single centre. Setting: inpatient, closed wards, Athens State Hospital, Columbus, Ohio (USA). |
|
| Participants | Diagnosis: schizophrenia (including hebephrenic; catatonic; paranoid; chronic undifferentiated). N = 78 *(n = 52 included in the analysis ‐ see interventions). Age: < 60, mean age 49 years. Sex: 39M, 39F *(26M, 26F included in the analysis ‐ x3 groups, with 13M and 13F in each). Ethnicity: not stated. Consent: not stated. History: average time in hospital of 20 years; all participants had previously responded poorly to somatic therapy; no other type of therapy given for at least one month; none of the participants had received trifluoperazine before. Included: diagnosis of schizophrenia for a period of 5 years or more; a history of withdrawal for one year or more. Excluded: not stated. |
|
| Interventions | 1.Trifluoperazine: week one: 5 mg twice daily (total 10 mg/day); week two: 10 mg twice daily (total 20 mg/day); week three: 15 mg twice daily (total 30 mg/day); after this time, 20 mg twice daily (total 40 mg/day) until 'maximum improvement or side‐effects intervened', n = 26. 2. Placebo: twice daily, n = 26. *(3. Chlorpromazine: set dose schedule over a four‐week period, week 1: 50 mg twice daily (total 100 mg/day); week 2: 100 mg twice daily (total 200 mg/day); week 3: 150 mg twice daily (total 300 mg/day); after this time, 200 mg twice daily (total 400 mg/day) until 'maximum improvement or side‐effects intervened', n = 26 ‐ this group was not included in the analysis).1 |
|
| Outcomes | Behaviour: agitation (undefined). Leaving the study early: any reason; due to adverse effects. Extrapyramidal adverse effects: general. Other adverse effects: general; specific ‐ difficulty swallowing; spasm muscles of mastication; drowsiness; blurred vision; anorexia; dermatitis; oedema of the face; incontinence of urine; fainting; convulsions. Unable to use ‐ Global state: The Lorr Multidimensional Scale for Rating Psychiatric Patients (MSRPP) – P values only. Need for additional medication: for side effects of study medication ‐ no usable data. |
|
| Notes |
1Dosages were reduced when maximum improvement appeared to have been achieved. Where reduction of dosage resulted in exacerbation of symptoms, the dosage was again raised. Benztropine methanesulfonate was the only other drug administered to participants who developed adverse effects relating to the medication. This was done by initially lowering the dosage and adding 2 mg benztropine methanesulfonate daily. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ quote, "three groups of 26 patients… were formed by random selection” (p451). |
| Allocation concealment (selection bias) | Unclear risk | No description. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double (implied) ‐ "[A]ll drugs were identical in appearance and taste...[N]one of the evaluators had any knowledge of the drug groups and the code remained unbroken until the completion of the study” (p452‐3). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | n = 2/26 left in trifluoperazine and n = 1/26 in the placebo group due to severe EPS; a further n = 2 left the placebo group as they were judged by the investigator (not acting as an evaluator) to need 'active medication.' ITT used. |
| Selective reporting (reporting bias) | High risk | MSRPP scale was used; evaluations had different results; no means or SDs reported for outcome data. |
| Other bias | Unclear risk | Funding: Smith, Kline and French provided chlorpromazine and trifluoperazine; Merck, Sharp & Dohme provided benztropine. Rating scales: three independent evaluations for each participant were obtained from the ward attendants. In addition, two psychiatric residents independently evaluated each participant using the MSRPP. Other: quote, "a legitimate criticism of is that the five patient lost to the study before the first post‐medication evaluation should have been rated as failures. As far as bias is concerned the weighting is against the trifluoperazine group and in favour of the placebo, but the chlorpromazine group might have shown better advantage if the latter course had been taken" (p454). |
Marjerrison 1964.
| Methods | Allocation: random. Blinding: double. Duration: 7 months, two phases: (i) 5 months; (ii) 2 months. Design: parallel (4 groups); single centre. Setting: inpatient, Saskatchewan Hospital, North Battleford (Canada). |
|
| Participants | Diagnosis: n = 76 schizophrenia (remaining n = 12 with other ‘chronic psychotic’ diagnosis)1. N = 88 *(n = 50 included in the analysis ‐ see interventions). Age: mean 48 years. Sex: 38M, 40F *(21M, 23F included in the analysis ‐ 6M, 7F trifluoperazine; 15M, 16F placebo). Ethnicity: not stated. Consent: not stated. History: mean length of illness – placebo: 20 years; trifluoperazine: 13 years. Mean length of current hospitalisation ‐ placebo: 19 years; trifluoperazine: 13 years. All participants had received phenothiazines continuously for at least one year. Included: ‘highly treatment‐resistive’; long‐term inpatients from two male and two female continued‐treatment wards. Excluded: history of epileptic seizures; those receiving ‘psychotic energizers’ (anti‐depressive compounds); those who were considered likely candidates for imminent discharge. |
|
| Interventions | 1. Trifluoperazine: 10 mg capsules, n = 16. i) phase 1: mean dose 2.9 capsules = 29 mg; ii) phase 2: mean dose 2.7 capsules = 27 mg. 2. Placebo: n = 34. i) phase 1: mean = 4.1 capsules; ii) phase 2: mean = 5.7 capsules.2 *(3. Usual phenothiazine: varying doses determined clinically in both phase 1 and 2, n = 30 ‐ this group was not included in the analysis). *(4. Chlorprothixene: 50 mg capsules, n = 8 ‐ this group was not included in the analysis: i) phase 1: mean dose 4.0 capsules = 200 mg; ii) phase 2: mean dose 5.4 capsules = 270 mg). |
|
| Outcomes | Leaving the study early: any reason; due to adverse effects. Adverse effects: dermatosis; seizure. Behaviour: use of adjunctive medication for sedation (barbiturate). Extrapyramidal adverse effects: use of anti‐Parkinson drugs. Unable to use ‐ Global and mental state: clinical worsening in one patient in each the trifluoperazine and placebo group ‐ no usable data. Behaviour: PRP ‐ no means or SD. |
|
| Notes |
1The n = 12 of mixed diagnosis were, quote, “evenly distributed among the groups” (p293). 2Anti‐Parkinsonian drugs used 'when necessary' (p292). Where a participants' behaviour were not adequately controlled by a higher‐dose of either treatment prescription, barbiturate sedatives were permitted for use alongside study medication. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ randomly assigned ‐ no further details. |
| Allocation concealment (selection bias) | Unclear risk | No description. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double – participants kept on their original wards throughout study duration, in order to enable "continuity of observation by the behaviour‐rating nurses in charge" (p291) in addition to other psychosocial treatments continued 'as usual'; no changes to recreational leave or discharge policy for the study population. Drugs given in individually‐assigned colour‐coded bottles with variations of dosage determined by clinical staff, with doses administered in a "multiplicity of forms" to minimise observer bias towards a particular prescribed drug (p292). Large effort described to keep patients and study personal blinded. However, 16/31 patients in the placebo group phase II were given 'no medication' whilst 15/31 were given a 'second placebo', implying no blinding of half the placebo group in phase II. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Follow‐up: 89% ‐ n = 10 participants left the study early. From the trifluoperazine group: n = 1 was dropped due to 'clinical worsening', n = 1 was transferred to another hospital for administrative reasons, and n = 1 was discontinued due to adverse reaction (dermatosis). From the placebo group: n = 2 were dropped due to 'marked worsening' and n = 1 left due to 'idiopathic seizures'. From the usual phenothiazine group: n = 3 were dropped due to 'clinical worsening', and n = 1 was dropped due to improvement leading to ward transfer for discharge planning (p294). No ITT used in the trial, but used for meta‐analysis. |
| Selective reporting (reporting bias) | Unclear risk | No means or SD reported for scales. |
| Other bias | Unclear risk | Funding: trifluoperazine and chlorprothixene supplied by Smith, Kline and French; Montreal and Hoffman LaRoche. Rating scales: raters independent of treatment. |
Menon 1972.
| Methods | Allocation: random. Blinding: unclear. Duration: 16 weeks (6‐week observation period; 10 weeks treatment period). Design: parallel (3 groups), single centre. Setting: inpatient, Government Mental Hospital, Madras, India. |
|
| Participants | Diagnosis: chronic schizophrenia (clinical diagnosis, when evidence of thought disorder; poverty of ideas; fixility of attitudes; narrowing of interest; apathy; lack of initiative; catatonic mannerisms; delusions and hallucinations). N = 60 *(n = 40 included in the analysis ‐ see interventions). Age: range 20‐52 years old (trifluoperazine group mean 37.50 years; placebo group 34.60 years). Sex: 30M, 30F *(20M, 20F included in analysis ‐ x3 groups, with 10M and 10F in each). Ethnicity: not stated. Consent: not stated. History: length of hospitalisation range 1.5‐9 years (trifluoperazine group mean 3.75 years; placebo group 4.13 years). Included: continuous hospitalisation for minimum of 1 year; ‘normal intelligence’. Excluded: presence of physical complications (e.g. pulmonary tuberculosis, liver disorders, diabetes, hypertension and other organic involvement). |
|
| Interventions | 1. Trifluoperazine: fixed dose 5.0 mg tablet tds, = 15 mg daily, n = 20. 2. Placebo: fixed dose 1 tds, n = 20. *(3. Trifluperidol: fixed dose 0.5 mg tablet tds, = 1.5 mg daily, n = 20 ‐ this group was not included in the analysis). |
|
| Outcomes | Global state: clinical improvement (QPSS rating) (defined as ‘marked’ or ‘moderate’ improvement). Behaviour: clinical improvement (Wings rating) (defined as ‘marked’ or ‘moderate’ improvement). Extrapyramidal adverse effects ‐ general. 1Adverse effects: EPS. Unable to use ‐ Anti‐Parkinsonism drugs: administered as needed (no data). Laboratory data: blood count, urine analysis, liver profiles (no data reported). |
|
| Notes | 1Adverse effects were calculated from a percentage. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ quote, "random allocation of patients to the three trial groups" (p20) ‐ no further details. |
| Allocation concealment (selection bias) | Unclear risk | Quote ‐ "random allocation of patients to the three trial groups ensured against any bias entering in the allocation of patients to the particular treatment" (p20) ‐ no further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Single (assessor) – "bias in the evaluation of treatment was avoided by keeping the research workers blind" (p20). No details of participant blinding. Quote: "the research workers evaluating the effects of the drugs were kept "blind" as to the medication each patient received" (p18). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details. |
| Selective reporting (reporting bias) | High risk | No data reported for laboratory investigations. No statistical data reported for rating scales QPSS and Wings. |
| Other bias | Unclear risk | Funding: Ethnor Limited (India) supplied drugs and ‘financial assistance’. Raters: psychiatric assessment made independently by two psychiatrists (QPSS and Wings). |
Pinard 1972.
| Methods | Allocation: random.
Blinding: double.
Duration: 70 days (preceded by 21 days where all had chlorpromazine 100 mg/day). Setting: inpatient, St‐Jean‐de‐Dieu Hospital, Research Unit, Montréal, Canada. Design: parallel (5 groups), single centre. |
|
| Participants | Diagnosis: chronic schizophrenia, BPRS average ˜45.
N = 80 *(n = 48 included in the analysis ‐ see interventions). Age: range 20‐60 years. Sex: "equally represented". Ethnicity: not stated. History: not stated. Included: hospitalised > 2 years; no exacerbation in last year. Excluded: not stated. Consent: not stated. |
|
| Interventions | 1. Trifluoperazine: dose 5 mg/three times daily, n = 14.
2. Trifluoperazine: dose 15 mg/day, n = 15.
3. Placebo: n = 14. *(4. Pimozide: dose 3 mg/day, n = 16 ‐ this group was not included in the analysis). *(5. Pimozide: dose 6 mg/day, n = 15 ‐ this group was not included in the analysis). Chlorpromazine, methyprylon, benztropine as required. |
|
| Outcomes | Mental state: clinically significant response in psychotic symptoms (defined as 'psychotic set‐backs and suicidal thoughts'). Leaving the study early: for any reason; due to adverse effects (including psychotic setback and suicidal thoughts). Unable to use ‐ Mental state: BPRS ‐ no usable ('P' values only). Extrapyramidal adverse effects: BPS ‐ treatment effect on EPS symptoms rating scale ‐ no usable data (graph only). Behaviour: NOSIE ‐ no usable data (graph only). Insight scale: Echelle D'Autocritique (only correlations). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ matched for symptom severity (BPRS), ward, attending physician and their evaluator (p22). |
| Allocation concealment (selection bias) | Unclear risk | No description. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double ‐ quote, “at all times, the double blind technique was respected” (p23). It is unclear who administered the rating scales used at different intervals throughout the study period. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Follow‐up ‐ 93%. Six participants left the study early due to psychotic set‐backs, suicidal thoughts and acute cholecystitis. Unsure if included in analysis ‐ ITT unclear. |
| Selective reporting (reporting bias) | High risk | Statistical reporting was incomplete for all scale data (no SDs). Covariance analysis was used rather than the BPRS as originally stated in the protocol, which was said to be 'ineffective in revealing significant differences' due to drop‐outs. |
| Other bias | Unclear risk | Funding: not stated. Rating scales: not stated to be independent of treatment. |
Prien 1969*.
| Methods | Allocation: random. Blinding: double. Duration: 24 weeks, 4 week observation period pre‐trial. Setting: inpatient, 6 hospitals ‐ Broughton State Hospital, NC, Dorothea Dix State Hospital, NC, Kentucky State Hospital, KY, Manhattan State Hospital, NY, St. Louis Hospital, MO, Springfield State Hospital, MD (USA). Design: parallel (3 groups), single centre. |
|
| Participants | Diagnosis: chronic schizophrenia. N = 3411. Age: 18‐55 (mean age 41.8 years, 60% were 45+ years old). Sex: 180M, 180F (30M, 30F from each hospital). Ethnicity: not stated. History: chronic schizophrenics with length of hospitalisation 2‐33 years with mean 15 years. 55% of patients were hospitalised 10+ years. Inclusion: a primary diagnosis of schizophrenia, age between 18‐55, continuous hospitalisation for at least 2 years. Exclusion: organic brain disease, mental deficiency or medical conditions that would otherwise put the patient at increased risk when taking high dose drugs. Consent: not stated. |
|
| Interventions | 1. Trifluoperazine: (high dose) 80 mg/day, gradual increase from previous dose to 80 mg/day after 35 days, n = 117. 2. Trifluoperazine: (low dose) 15 mg/day, n = 113. 3. Placebo: n = 111. |
|
| Outcomes | Global state: clinical improvement (defined a 'markedly improved' using the Doctor's Global Improvement Scale) ‐ together and by high dose and low dose. Extrapyramidal adverse effects: akathisia; Parkinsonian reaction; dystonia ‐ together and by high dose and low dose. 2Other adverse effects: specific ‐ seizures; hypotension; dizziness; fainting; nausea/vomiting; skin rashes; photosensitivity; insomnia; drowsiness; decreased appetite ‐ together and by high dose and low dose. Leaving the study early: any reason; due to severe adverse effects; due to relapse or worsening ‐ together and by high dose and low dose. Unable to use: Mental state: BPRS; IMPS ‐ no usable data. Behaviour: NOSIE ‐ no usable data. Hospital and service utilisation outcomes: follow‐up ‐ discharge ‐ not enough data. |
|
| Notes |
1Study paper reads: "approximately 60 chronic schizophrenics...were selected at each hospital [and] each treatment group consisted of approximately 120 patients" (p54). No concrete data were found for true participant numbers; however, we used the data available to us that represented n = 117 in high‐dose trifluoperazine; n = 113 in low‐dose trifluoperazine; and n = 111 in placebo groups (N = 341). 2All adverse effect data and numbers leaving the study early were calculated from percentages. High and low doses were combined as well as reported separately versus placebo. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ quote, "patients were randomly assigned to one of three groups” (p306). |
| Allocation concealment (selection bias) | Unclear risk | No description. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double ‐ “all medication was administered in capsule form under double‐blind conditions for 24 weeks” (p306). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Where the study only gives an approximation as to the original number of randomised participants (N = 360), it is hard to identify the true number of participants that dropped out or that were included in the final analysis. |
| Selective reporting (reporting bias) | Unclear risk | No data provided for IMPS, BPRS, NOSIE, Global Improvement Scale, or Discharge‐Readiness Inventory (DRI). |
| Other bias | Unclear risk | Funding: Public Health Service grants: MH‐10292, MH‐10332, MH‐11384, MH‐10989, MH‐11046, MH‐11047 and contract SA‐43‐ph‐3064 all from the National Institute of Mental Health. Rating Scales: scales were administered by the research physician; unclear whether they were independent of treatment. |
Reardon 1966.
| Methods | Allocation: random. Blinding: double. Duration: 4 ‐ 12 weeks. Setting: inpatient (USA). Design: parallel (3 groups), single centre. |
|
| Participants | Diagnosis: acute paranoid schizophrenia (Bleuler criteria). N = 34 *(n = 23 included in the analysis ‐ see interventions). Age: no data. Sex: 22M,12F ("number of males and females in each group were comparable"). Ethnicity: not stated. History: not stated. Included: those who demonstrated a thinking and affect disturbance, and who admitted the presence of persecutory delusions and hallucinations within 10 days prior to admission were selected. Exclusion: not stated. Consent: not stated. |
|
| Interventions | 1. Trifluoperazine: 20 mg/day for first week, increased to 40 mg/day for the remainder of the study, n = 11. 2. Placebo: either 2 to 4 cc. or 5 to 10 cc 'as though it were one of the active drugs', n = 12. *(3. Chlorpromazine: 300 mg/day for first week, increased to 600 mg until completion of study, n = 11 ‐ this group was not included in the analysis). (IM barbiturates were administered 'on occasion' to deal with aggressive or assaultive behaviour). |
|
| Outcomes | Leaving the study early: any reason; due to adverse effects (relapse/ worsening ‐ ECT). Extrapyramindal adverse effects: Parkinsonism. Mental state: clinically significant response in positive symptoms (exhibited delusions and hallucinations); clinically significant response in psychotic symptoms (defined as exhibiting delusions and hallucinations). Hospital and service utilisation outcomes: hospital transfer/ home leave. Unable to use ‐ Global state and cognitive response: MMPI, Shipley Hartford and four sub tests of WAIS rating scores ‐ only P values given. Use of anti‐Parkinson drugs: Artane (10 mg) daily was given to all patients. No data and no Parkinsonian symptoms were observed. Behaviour: no usable data. |
|
| Notes | ITT used for outcomes not relating to adverse effects (leaving the study early and service utilisation outcomes only). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ the pharmacy controlled the allocation ‐ quote, “each subject was placed on a ward and randomly assigned trifluoperazine, chlorpromazine or placebo by the pharmacy” (p266) ‐ no further details. |
| Allocation concealment (selection bias) | Low risk | Allocation was controlled by the pharmacy; ward personnel and investigators did not know which drug each participant received. Participants were placed in active treatment wards in order to quote: "avoid the therapeutic milieu effect that might occur with placement on a special research unit" (p266). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double ‐ allocation was controlled by the pharmacy; ward personnel and investigators did not know which drug each participant received. Placebo administered quote: "as though it were one of the active drugs" (p266). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Follow‐up: 74%. Six participants were excluded from the investigation because they were given ECT (n = 2 receiving trifluoperazine; n = 1 receiving chlorpromazine; n = 3 receiving placebo) with an addition n = 3 (one from each group) removed from the study after transfer or home leave. Limited data provided and subjective clinical observations used. |
| Selective reporting (reporting bias) | Unclear risk | MMPI, Shipley Hartford WAIS ‐ no scale data or SDs reported. |
| Other bias | Unclear risk | Funding: Smith, Kline and French supplied the drugs and placebo. Rating scales: not clear whether nurses or other raters were independent of treatment. |
Schiele 1961.
| Methods | Allocation: random. Blinding: double. Duration: 16 weeks (22‐week 'additional trial period'). Setting: inpatient, St Cloud, Minnesota (USA). Design: parallel (four groups), single centre. |
|
| Participants | Diagnosis: chronic schizophrenia. N = 80 *(n = 40 included in the analysis ‐ see interventions). Age: average 40.6 years. Sex: 80M. Ethnicity: not stated. History: participants were either "withdrawn or subject to periodic disturbances, and they were generally ineffective. All needed supervision and management" and most needed closed‐ward care. Average continuous hospitalisation for 10.0 years. Medication received prior to study included chlorpromazine (n = 30), mepazine (n = 35), trifluoperazine (n = 6), prochlorperazine (n = 2), various combinations (n = 7). Inclusion: diagnosis of schizophrenia; without history or evidence of complicating organic factors. Exclusion: age greater than 55 years; lobotomy. Consent: not stated. |
|
| Interventions | 1. Trifluoperazine: 5 mg capsules (10 to 50 mg/day), n = 20. 2. Placebo: n = 20. *(3. Chlorpromazine: 100 mg capsules (200 to 1000 mg/day), n = 20. *(4. Thioridazine: 100 mg capsules (200 to 1000 mg/day), n = 20. (Medication varied between 2 to 10 capsules a day given 2/4 times a day; anti‐Parkinsonian medication benztropine methanesulfonate used as needed to control EPS; phenobarbital used temporarily for sedation). |
|
| Outcomes | Global state: clinical improvement (defined as a global estimate of the amount of change in clinical condition using scores from the MBS and MMPI, including 'considerable improvement' and 'moderate improvement' ‐ judgement made by the investigators with the ward physician as chairman). Mental state: any clinically significant response in psychotic symptoms (defined as 'psychiatric condition becoming and remaining worse'). Hospital and service utilisation outcomes: discharge. Extrapyrimidal adverse effects: general side effects. Extrapyrimidal adverse effects: use of anti‐Parkinson drugs. Adverse effects: drowsiness; motor restlessness; rigidity; tremors; hypersalivation; slurred speech; incoordination; insomnia; skin disorder; fainting; blurred vision; lethargy; muscle weakness; tenseness; seizure; polydipsia and polyuria; decreased appetite. Leaving the study early: any reason; due to relapse or worsening. Unable to use ‐ Behaviour: MBS and MMPI ‐ no SD (adjusted means only). Improvement from 22‐week 'additional treatment period': blinding broken; only 71% participants from original sample. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random ‐ no further description. |
| Allocation concealment (selection bias) | Unclear risk | No description. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double ‐ quote, "strict double blind conditions...individual bottle of medication...capsules were identical in appearance..only hospital pharmacist had the code...". In the additional 22‐week trial period the double blind procedure was modified, these results were handled separately in the study. We have only used data from the first 16 weeks of the study. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 43 participants (n = 13 in the thioridazine group, and n = 10 from each of the other three groups) were tested at each specified point during the study using the MMPI, with the remainder of participants termed "untestable" (p155). |
| Selective reporting (reporting bias) | Unclear risk | No statistical data reported for the MMPI and MBS scales. |
| Other bias | Unclear risk | Funding: not stated. Rating scales: Manifest Behaviour Scale (MBS) completed twice on each participant by two nursing assistants working independently. Primary investigator was author of the MBS. Exclusion criteria: participants who had lobotomies, meeting the exclusion criteria, were "inadvertently included", with n = 2 in the thioridazine group and n = 1 chlorpromazine group. |
Rating scales
BPRS ‐ Brief Psychiatric Rating Scale BRS ‐ Beckombergo Rating Scale CGI ‐ Clinical Global Impressions Scale IMPS ‐ Inpatient Multidimensional Psychiatric Scale MMPI ‐ Minnesota Multiphasic Personality Inventory MSBS ‐ Minimal Social Behaviour Scale NOSIE ‐ Nurses' Observation Scale for Inpatient Evaluation PRP ‐ Psychotic Reactive Profile QPSS ‐ Quantification of Psychotic Symptom Severity TTB ‐ Tulane Test Battery WAIS ‐ Welchsler Adult Intelligence Scale
Other CNS ‐ central nervous system ECT ‐ electroconvulsive therapy EPS ‐ extrapyramidal symptoms IM ‐ intramuscular ITT ‐ intention‐to‐treat SD ‐ standard deviation tds ‐ three times daily
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abuzzahab 1977 | Allocation: not clear. Participants: chronic schizophrenia. Interventions: trifluoperazine versus penfluridol versus placebo. Outcomes: no usable data (literature review). |
| Barron 1961 | Allocation: random. Participants: 'chronically mentally ill'. Interventions: trifluoperazine versus chlorprothixene versus placebo. Outcomes: no usable data (cross‐over study ‐ no results available pre‐cross‐over). |
| Cahan 1960 | Allocation: not randomised. |
| Coons 1962 | Allocation: randomised. Participants: mixed diagnoses. |
| Hamilton 1963 | Allocation: randomised. Participants: chronic schizophrenia. Interventions: trifluoperazine versus prochlorperazine versus placebo. Outcomes: no usable data. |
| Holden 1971 | Allocation: not clear. Participants: schizophrenia. Interventions: trifluoperazine versus thiothixene. |
| Hunt 1967 | Allocation: random. Participants: schizophrenia. Interventions: trifluoperazine versus oxypertine. |
| Leff 1971 | Allocation: not randomised. |
| Leff 1973 | Allocation: randomised. Participants: schizophrenia. Interventions: pooled data from two RCTs ‐ fluphenazine decanoate versus placebo and trifluoperazine versus placebo. |
| Madgwick 1958 | Allocation: not stated. Participants: chronic schizophrenia. Interventions: trifluoperazine ‐ once stabilised, half continued trifluoperazine, half withdrawn using placebo. Outcomes: no usable data (withdrawal study). |
| Morton 1968 | Allocation: not randomised. |
| Stanley 1961 | Allocation: not randomised. |
| Weckowicz 1960 | Allocation: not randomised |
| Weston 1961 | Allocation: not randomised. |
RCT ‐ randomised controlled trial
Characteristics of studies awaiting assessment [ordered by study ID]
Ortega‐Soto 1996.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | To be assessed |
Differences between protocol and review
Under outcome 1.13 Hospital and service utilisation outcomes (Analysis 1.19), follow‐up data were added as they were available and thought to be of relevance and use.
Primary outcome 4. Relapse +/‐ hospitalisation was added under primary outcome 1. Global state (Analysis 1.2).
In the Sensitivity analysis, 'Usual doses of trifluoperazine' date were included from secondary outcomes as they were deemed relevant and important.
An economic review team added and carried out an economic studies search in order to identify any high‐quality economic analyses relating to the intervention. Where this was not possible, the economic review authors would have used a lower‐grade of 'economic summary', using the calculations described in the Methods section of this review.
Contributions of authors
Kai Koch ‐ wrote the protocol, independently re‐extracted data from all included studies, extracted and input all dichotomous data, and wrote text for: effects of intervention, sensitivity analysis, summary of results, overall completeness and applicability of evidence, implications for practice and the abstract. Produced 'Summary of findings' table and Risk of bias' graphs. Checked the final version.
Kamel Mansi ‐ checked the protocol, helped search and selected relevant studies, excluding studies and extracted data from included studies. Wrote text for: quality of the evidence, potential biases in the review process and implications for research.
Euan Haynes ‐ checked the protocol, helped KM to exclude studies and extract data from included studies. Assisted with input of the data and wrote text for: description of the studies and risk of bias in included studies.
Clive E Adams ‐ checked the protocol and review.
Stephanie Sampson ‐ support and advisor for authors in data extraction and write up of results, carried out and completed economic evaluation of studies, .
Vivek Futardo ‐ developed economic evaluation, carried out completed economic evaluation of studies.
Sources of support
Internal sources
Cochrane Schizophrenia Group, UK.
University of Nottingham, UK.
External sources
-
NIHR Cochrane Programme Grant 2011, UK.
Reference number: 10/4001/15
Declarations of interest
Kai Koch ‐ none. Kamel Mansi ‐ none. Euan Haynes ‐ none. Clive E Adams ‐ none. Stephanie Sampson ‐ none. Vivek Furtado ‐ none.
New
References
References to studies included in this review
Bishop 1964 {published data only}
- Bishop MP, Gallant DM, Nesselhof W, Sprehe DJ. A controlled evaluation of butaperazine in chronic schizophrenic patients. Diseases of the Nervous System 1964;25:674‐83. [PubMed] [Google Scholar]
Clark 1975 {published data only}
- Clark ML, Paredes A, Costiloe JP, Wood F, Barrett A. Loxapine in newly admitted chronic schizophrenic patients. Journal of Clinical Pharmacology 1975;15(4 Pt 1):286‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gross 1974 {published data only}
- Gross HS. A double‐blind comparison of once‐a‐day pimozide, trifluoperazine, and placebo in the maintenance care of chronic schizophrenic outpatients. Current Therapeutic Research, Clinical and Experimental 1974;16(7):696‐705. [PubMed] [Google Scholar]
Gwynne 1962 {published data only}
- Gwynne P, Hundziak M, Kavtschitsch J, Lefton M, Pasamanick B. Efficacy of trifluoperazine on withdrawal in chronic schizophrenia. Journal of Nervous and Mental Disease 1962;134:451‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Marjerrison 1964 {published data only}
- Marjerrison G, Irvine D, Stewart CN, Williams R, Matheu H, Demay M. Withdrawal of long term phenothiazines from chronically hospitalized psychiatric patients. Canadian Psychiatric Association Journal 1964;60:290‐8. [DOI] [PubMed] [Google Scholar]
Menon 1972 {published data only}
- Menon MS, Ramachandran V. A controlled clinical trial of trifluperidol on a group of chronic schizophrenic patients. Current Therapeutic Research 1972;14(1 January):17‐21. [PubMed] [Google Scholar]
Pinard 1972 {published data only}
- Pinard G, Prenoveau Y, Fliesen W, Elie R, Bielmann P, Lamontagne Y, et al. Pimozide and social rehabilitation of chronic schizophrenic patients [Le Pimozide et la reintegration sociale des schizophrenes chroniques]. L'Encephale 1972;61(1):53‐66. [MEDLINE: ] [PubMed] [Google Scholar]
Prien 1969* {published data only}
- Prien RF, Levine J, Cole JO. High dose trifluoperazine therapy in chronic schizophrenia. American Journal of Psychiatry 1969;126(3):305‐13. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Reardon 1966 {published data only}
- Reardon JD, Abrams S. Acute paranoid schizophrenia (treatment with chlorpromazine, trifluoperazine and placebo). Diseases of the Nervous System 1966;27:265‐70. [PubMed] [Google Scholar]
Schiele 1961 {published data only}
- Schiele BC, Vestre ND, Stein KE. A comparison of thioridazine, trifluoperazine, chlorpromazine, and placebo: a double‐blind controlled study on the treatment of chronic hospitalized, schizophrenic patients. Journal of Clinical and Experimental Psychopathology 1961;22(3):151‐62. [MEDLINE: ] [PubMed] [Google Scholar]
References to studies excluded from this review
Abuzzahab 1977 {published data only}
- Abuzzahab FS. The treatment of schizophrenia with long‐acting oral neuroleptics: a six‐month double‐blind investigation of penfluridol versus trifluoperazine. Psychopharmacology Bulletin 1977;13(3):26‐7. [MEDLINE: ] [PubMed] [Google Scholar]
Barron 1961 {published data only}
- Barron A, Beckering B, Rudy LH, Smith JA. A "double‐blind" study comparing RO 4‐0403, trifluoperazine and a placebo in chronically ill mental patients. American Journal of Psychiatry 1961;118:347‐8. [Google Scholar]
Cahan 1960 {published data only}
- Cahan RB. Efficacy of trifluoperazine in chronic mental illness. American Journal of Psychiatry 1960;116:838. [DOI] [PubMed] [Google Scholar]
Coons 1962 {published data only}
- Coons WH, Boyd BA, White JG. Chlorpromazine, trifluoperazine and placebo with long term mental hospital patients. Canadian Psychiatric Association Journal 1962;7:159‐63. [DOI] [PubMed] [Google Scholar]
Hamilton 1963 {published data only}
- Hamilton M, Hordern A, Waldrop FN, Lofft J. A controlled trial on the value of prochlorperazine, trifluoperazine and intensive group treatment. British Journal of Psychiatry 1963;109:510‐22. [DOI] [PubMed] [Google Scholar]
Holden 1971 {published data only}
- Holden J, Itil T, Gannon P, Keskiner A. The clinical effects of intramuscular thiothixene and trifluoperazine in chronic schizophrenia: a comparative study. Current Therapeutic Research, Clinical and Experimental 1971;13(5):298‐310. [MEDLINE: ] [PubMed] [Google Scholar]
Hunt 1967 {published data only}
- Hunt PV. A comparison of the effects of oxypertine and trifluoperazine in withdrawn schizophrenics. British Journal of Psychiatry 1967;113(505):1419‐24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Leff 1971 {published data only}
- Leff JP, Wing JK. Trial of maintenance therapy in schizophrenia. British Medical Journal 1971;3(775):599‐604. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leff 1973 {published data only}
- Leff JP. Influence of selection of patients on results of clinical trials. British Medical Journal 1973;4(5885):156‐8. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Madgwick 1958 {published data only}
- Madgwick JRA, McNeill DLM, Driver M, Preston GC. Stelazine (trifluoperazine). A preliminary report on a clinical trial. Journal of Mental Science 1958;104:1195‐8. [DOI] [PubMed] [Google Scholar]
Morton 1968 {published data only}
- Morton MR. A study of the withdrawal of chlorpromazine or trifluoperazine in chronic schizophrenia. American Journal of Psychiatry 1968;124(11):1585‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stanley 1961 {published data only}
- Stanley WJ, Walton D. Trifluoperazine (stelazine). A controlled clinical trial in chronic schizophrenia. Journal of Mental Science 1961;107:250‐7. [Google Scholar]
Weckowicz 1960 {published data only}
- Weckowicz TE, Ward TF. Clinical trial of 'stelazine' on apathetic chronic schizophrenics. Journal of Mental Science 1960;106:1008‐15. [DOI] [PubMed] [Google Scholar]
Weston 1961 {published data only}
- Weston FK, Loftus AP. A terminal double‐blind trial of tri‐fluoperazine ("stelazine") in chronic schizophrenia. Medical Journal of Australia 1961;48(1):776‐80. [MEDLINE: ] [PubMed] [Google Scholar]
References to studies awaiting assessment
Ortega‐Soto 1996 {published data only}
- Ortega‐Soto HA, Brunner E, Apiquian R, Torre MP, Ulloa RE. Typical antipsychotics: the threshold doses strategy. Proceedings of the 20th Collegium Internationale Neuro‐Psychopharmacologicum Congress; 1996 Jun 23‐27; Melbourne, Australia. 1996.
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Arana 2000
- Arana G, Rosenbaum J. Handbook of Psychiatric Drug Therapy. USA. 4. Philadelphia: Lippincott Williams and Wilkins, 2000. [Google Scholar]
Bartko 1988
- Bartko G, Herczeg I, Zador G. Clinical symptomatology and drug compliance in schizophrenic patients. Acta Psychiatrica Scandinavica 1988;77(1):74‐6. [DOI] [PubMed] [Google Scholar]
Bazire 2000
- Bazire. Psychotropic Drug Directory. USA. New York: Butler and Tanner Limited, 2000. [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
BNF 2012
- BMJ Group and Pharmaceautical Press. British National Formulary No. 63. Antipsychotic drugs. BNF 2012.
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
CEA
- Cost‐Effectiveness Analysis Registry (CEA). https://research.tufts‐nemc.org/cear4/ accessed 11/09/13.
Davies 2007
- Davies LM, Lewis S, Jones PB, Barnes TR, Gaughran F, Hayhurst K, et al. Cost‐effectiveness of first‐ v. second‐generation antipsychotic drugs: results from a randomised controlled trial in schizophrenia responding poorly to previous therapy. British Journal of Psychiatry 2007;191:14‐22. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Drummond 1996
- Drummond MF, Jefferson TO. Guidelines for authors and peer reviewers of economic submissions to the BMJ. BMJ 1996;313(3 August):275‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Evers 2005
- Evers S, Goossens M, Vet H, Tulder M, Ament A. Criteria list for assessment of methodological quality of economic evaluations: Consensus on Health Economic Criteria. International Journal of Technology Assessment in Health Care 2005;21(2 Spring):240‐5. [PubMed] [Google Scholar]
Farina 1957
- Farina A, David A, Guskin S. A scale for measuring minimal social behaviour. Journal of Consultant Psychology 1957;21:265‐8. [DOI] [PubMed] [Google Scholar]
Filippelli 2005
- Filippelli E, Biricolti G, Scarano C, Russo F, Luciano L. Treatment of psychotic disorders with olanzapine, risperidone and typical neuroleptics: a comparative cost‐effectiveness evaluation in a local psychiatric setting. Farmeconomia e Percorsi Terapeutici 2005;6(3):161‐8. [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Galvin 1999
- Galvin PM, Knezek LD, Rush AJ, Toprac MG, Johnson B. Clinical and economic impact of newer versus older antipsychotic medications in a community mental health center. Clinical Therapeutics 1999;21(6):1105‐16. [DOI] [PubMed] [Google Scholar]
Ghaemi 2001
- Ghaemi SN, Kirkwood CK, Sambur MR, Ko JY, Howden KL, Duong Q, et al. Economic outcomes of risperidone in comparison to typical neuroleptic agents for treatment‐resistant psychosis: a community‐based study. Journal of Pharmacy Technology 2001;17:273‐8. [Google Scholar]
Goodrich 1953
- Goodrich DW. Quantification of the severity of overt psychotic symptoms. American Journal of Psychiatry 1953;110:334‐41. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1970
- Guy W, Bonato RR. Clinical Global Impressions. In: Guy W, Bonato RR editor(s). Manual for the ECDEU Assessment Battery. 2 Rev. National Institute of Mental Health, 1970:12‐1‐12‐6. [Google Scholar]
Hanrahan 2006
- Hanrahan P, Luchins DJ, Fabian R, Tolley G. Cost‐effectiveness of atypical antipsychotic medications versus conventional medication. Expert Opinion in Pharmacotherapy 2006;7(13):1749‐58. [DOI] [PubMed] [Google Scholar]
Hathaway 1940
- Hathaway SR, McKinley JC. A multiphasic personality schedule (Minnesota): I. Construction of the schedule. Journal of Psychology 1940;10:249‐54. [Google Scholar]
HEED
- Health Economic Evaluation Database (HEED). Online ISBN: 9780470510933. [DOI: 10.1002/9780470510933] [DOI]
HES 2012
- Hospital Episode Statistics, Admitted Patient Care ‐ England 2011‐12: Main Specialties (.xls). http://www.hscic.gov.uk/searchcatalogue?productid=9161&q=title%3a%22hospital+episode+statistics%22&sort=Relevance&size=10&page=1#top (accessed May 2013) 2011‐12.
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Kaplan 1998
- Kaplan H, Sadock B. Pocket Handbook of Clinical Psychiatry. USA. 2. New York: Williams and Wilkins, 1998. [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Knapp 2008
- Knapp M, Windmeijer F, Brown J, Kontodimas S, Tzivelekis S, Haro JM, et al. Cost‐utility analysis of treatment with olanzapine compared with other antipsychotic treatments in patients with schizophrenia in the pan‐European SOHO study. PharmacoEconomics 2008;26(4):341‐58. [DOI] [PubMed] [Google Scholar]
Lankappa 2012
- Lankappa S, Gandhi R. Quetiapine versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2012, Issue 7. [DOI: 10.1002/14651858.CD009935] [DOI] [Google Scholar]
Leucht 2003
- Leucht S, Wahlbeck K, Hamann J, Kissling W. New generation antipsychotics versus low‐potency conventional antipsychotics: a systematic review and meta‐analysis. Lancet 2003;361(May 10):1581‐9. [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lewis 1998
- Lewis R. Typical and atypical antipsychotics in adolescent schizophrenia: efficacy, tolerability, and differential sensitivity to extrapyramidal symptoms. Canadian Journal of Psychiatry 1998;43(6):596‐604. [DOI] [PubMed] [Google Scholar]
Lewis 2006
- Lewis SW, Davies L, Jones PB, Barnes TRE, Murray RM, Kerwin R, et al. Randomised controlled trials of conventional antipsychotic versus new atypical drugs, and new atypical drugs versus clozapine, in people with schizophrenia responding poorly to, or intolerant of, current drug treatment. Health Technology Assessment 2006;10(17):1‐182. [DOI] [PubMed] [Google Scholar]
Lorr 1960
- Lorr MO, O'Connor JP, Stafford JW. The psychotic reaction profile. Journal of Clinical Psychology 1960;16:241‐5. [PubMed] [Google Scholar]
Mangalore 2007
- Mangalore R, Knapp M. Cost of schizophrenia in England. Journal of Mental Health Policy and Economics 2007;10(1):23‐41. [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Martin 2006
- Martin JL, Perez V, Sacristan M, Rodriguez‐Artalejo F, Martinez C, Alvarez E. Meta‐analysis of drop‐out rates in randomised clinical trials, comparing typical and atypical antipsychotics in the treatment of schizophrenia. European Psychiatry 2006;21(1):11‐20. [DOI] [PubMed] [Google Scholar]
Mendelsohn 1959
- Mendelsohn RM, Penman AS, Schiele BC. Massive chlorpromazine therapy: the nature of behavioural changes. Psychiatric Quarterly 1959;33(Jan):1‐22. [DOI] [PubMed] [Google Scholar]
Mental Health Care
- Mental Health Care: reliable and up‐to‐date information about psychosis for family members and friends. http://www.mentalhealthcare.org.uk/pharmacist_answers_september_august_2011 accessed September 2013.
Moher 2001
- Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. JAMA 2001;285(12):1987‐91. [DOI] [PubMed] [Google Scholar]
Mould 2009
- Mould QJ, Contreras HI, Verduzco W, Mejia AJM, Garduno EJ. Cost‐effectiveness simulation analysis of schizophrenia at the Instituto Mexicano del Seguro Social: Assessment of typical and atypical antipsychotics. Revista de Psiquiatrí́a y Salud Mental 2009;2(3):108‐18. [DOI] [PubMed] [Google Scholar]
NICE 2010
- NICE. Schizophrenia: Core interventions in the treatment and management in adults in primary and secondary care ‐ National Clinical Guideline Number 82. http://www.nice.org.uk/nicemedia/pdf/CG82FullGuideline.pdf. The British Psychological Society and The Royal College of Psychiatrists, 2010:4‐41.
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
PJ Online
- PJ Online: Do you accept that medicine shortages have not harmed patients?. http://www.pjonline.com/poll/do_you_accept_that_medicine_shortages_have_not_harmed_patients Accessed September 2013.
PSSRU 2012
- Compiled by Lesley Curtis. Unit costs of health and social care 2012. http://www.pssru.ac.uk/project‐pages/unit‐costs/2012/ (accessed May 2013) 2012:47.
RCPSYCH 2009
- Royal College of Psychiatrists. Antipsychotics. RCPSYCH 2009.
RCPSYCH 2010
- Royal College of Psychiatrists. Schizophrenia. RCPSYCH 2010.
RCPSYCH 2011
- Royal College of Psychiatrists. Schizophrenia. RCPSYCH 2011.
Saha 2005
- Saha S, Chant D, Welham J, McGrath J. Systematic review of the prevalence of schizophrenia. PLoS Medicine 2005;2(5):0413‐33. [DOI: 10.137/journal.pmed.0020141] [DOI] [PMC free article] [PubMed] [Google Scholar]
Saha 2007
- Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia. Is the differential mortality gap worsening over time?. Archives of General Psychiarty 2007;64(10):1123‐31. [DOI: 10.1001/archpsyc.64.10.1123] [DOI] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Stargardt 2008
- Stargardt T, Weinbrenner S, Busse R, Juckel G, Gericke CA. Effectiveness and cost of atypical versus typical antipsychotic treatment for schizophrenia in routine care. Journal of Mental Health Policy and Economics 2008;11(2):89‐97. [PubMed] [Google Scholar]
Suttajit 2009
- Suttajit S, Srisurapanont M, Maneeton B, Maneeton N, Suttajit S. Quetiapine versus typical antipsychotic medications for schizophrenia. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD007815] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tiihonen 2009
- Tiihonen J, Lonnqvist J, Wahlbeck K, Klaukka T, Niskanen L, Tanskanen A, et al. 11‐year follow‐up of mortality in patients with schizophrenia: a population‐based cohort study (FIN11 study). Lancet 2009;374(9690):620‐7. [DOI] [PubMed] [Google Scholar]
Turner 2007
- Turner T. Chlorpromazine: unlocking psychosis. BMJ 2007;334(Suppl 1):s7. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Williams 1987
- Williams, A (editor). Health economics: the cheerful face of dismal science?. Health and economics. London (UK): Macmillan, 1987. [Google Scholar]
Wing 1961
- Wing JK. A simple and reliable subclassification of chronic schizophrenia. Journal of Mental Science 1961;107:862‐75. [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
Marques 2004
- Marques LO, Lima MS, Soares BG. Trifluoperazine for schizophrenia. Cochrane Database of Systematic Reviews 2004, Issue 1. [DOI: 10.1002/14651858.CD003545; PUBMED: 14974020] [DOI] [PMC free article] [PubMed] [Google Scholar]