

Abstract
Background
Thioridazine is an antipsychotic that can still be used for schizophrenia although it is associated with the cardiac arrhythmia, torsades de pointe.
Objectives
To review the effects of thioridazine for people with schizophrenia.
Search methods
For this 2006 update, we searched the Cochrane Schizophrenia Group's Register (June 2006).
Selection criteria
We included all randomised clinical trials comparing thioridazine with other treatments for people with schizophrenia or other psychoses.
Data collection and analysis
We reliably selected, quality rated and extracted data from relevant studies. For dichotomous data, we estimated relative risks (RR), with the 95% confidence intervals (CI). Where possible, we calculated the number needed to treat/harm statistic (NNT/H) on an intention‐to‐treat basis.
Main results
This review currently includes 42 RCTs with 3498 participants. When thioridazine was compared with placebo (total n=668, 14 RCTs) we found global state outcomes favoured thioridazine (n=105, 3 RCTs, RR 'no change or worse' by 6 months 0.33 CI 0.2 to 0.5, NNT of 2 CI 2 to 3). Thioridazine is sedating (n=324, 3 RCTs, RR 5.37 CI 3.2 to 9.1, NNH 4 CI 2 to 74). Generally, thioridazine did not cause more movement disorders than placebo.
Twenty‐seven studies (total n=2598) compared thioridazine with typical antipsychotics. We found no significant difference in global state (n=743, 11 RCTs, RR no short‐term change or worse 0.98 CI 0.8 to 1.2) and medium‐term assessments (n=142, 3 RCTs, RR 0.99, CI 0.6 to 1.6). We found no significant differences in the number of people leaving the study early 'for any reason' (short‐term, n=1587, 19 RCTs, RR 1.07 CI 0.9 to 1.3). Extrapyramidal adverse events lower for those allocated to thioridazine (n=1082, 7 RCTs, RR use of antiparkinsonian drugs 0.45 CI 0.4 to 0.6). Thioridazine did seem associated with cardiac adverse effects (n=74, 1 RCT, RR 'any cardiovascular adverse event' 3.17 CI 1.4 to 7.0, NNH 3 CI 2 to 5). Electrocardiogram changes were significantly more frequent in the thioridazine group (n=254, 2 RCTs, RR 2.38, CI 1.6 to 3.6, NNH 4 CI 3 to 10).
Six RCTs (total n=344) randomised thioridazine against atypical antipsychotics. Global state rating did not reveal any short‐term difference between thioridazine and remoxipride and sulpiride (n=203, RR not improved or worse 1.00 CI 0.8 to 1.3). Limited data did not highlight differences in adverse event profiles.
Authors' conclusions
Although there are shortcomings, there appears to be enough consistency over different outcomes and periods to confirm that thioridazine is an antipsychotic of similar efficacy to other commonly used antipsychotics for people with schizophrenia. Its adverse events profile is similar to that of other drugs, but it may have a lower level of extrapyramidal problems and higher level of ECG changes. We would advocate the use of alternative drugs, but if its use in unavoidable, cardiac monitoring is justified.
Keywords: Humans; Antipsychotic Agents; Antipsychotic Agents/adverse effects; Antipsychotic Agents/therapeutic use; Arrhythmias, Cardiac; Arrhythmias, Cardiac/chemically induced; Outcome Assessment (Health Care); Randomized Controlled Trials as Topic; Schizophrenia; Schizophrenia/drug therapy; Thioridazine; Thioridazine/adverse effects; Thioridazine/therapeutic use; Treatment Outcome
Thioridazine for schizophrenia
About 1% of people will get schizophrenia and it often begins early in life. Schizophrenia is typically characterised by hallucinations (perceptions without a cause), delusions (fixed and false beliefs), disordered thinking, and emotional withdrawal. The outcomes vary, but antipsychotic drugs generally help; thioridazine is one such drug. It had been thought to be effective and less prone to cause the movement disorders that can happen particularly with the older generation antipsychotics. Largely thioridazine has been withdrawn due to its links with abnormal heart rhythm but is still used in special circumstances.
We reviewed the effects of thioridazine and found many trials suggesting that it seems to be as effective as other commonly used antipsychotics for people with schizophrenia, but also justification for guidelines encouraging heart monitoring for people prescribed this drug. Where possible, we would advocate choosing other drugs in place of thioridizine.
Background
Thioridazine (Melleril/Mellaril) is a piperidine phenothiazine similar to chlorpromazine that is taken by mouth and was developed and tested soon after chlorpromazine in the 1950s (Bain 1998). In an early, important study thioridazine was found to have similar efficacy to chlorpromazine for treating people with schizophrenia (NIMH 1964) at least in terms of the 'positive' symptoms (delusions, strongly held abnormal beliefs not explainable by the person's culture, and hallucinations, abnormal perceptions).
Thioridazine has often been considered the drug of choice in the elderly because of its lower level of extrapyramidal adverse events (such as tremor, muscle stiffness, and slow body movements) and sedation (BNF 1998). However, it may be more likely to cause cognitive adverse events in the elderly, such as delirium or worsening of memory (Moreau 1986). There is also a risk of cardiotoxicity especially in combination with a tricyclic antidepressant (BNF 1998, Heiman 1977, Lipscomb 1980) and it is also more likely to cause a fall in blood pressure than other drugs. On rare occasions, thioridazine has caused pigmentary retinopathy (leading to seeing the colour brown, blurring and loss of acuity) with doses above 1000 mg/day (Rennie 1993). A dose maximum of 800 mg/day was previously recommended (BNF 1998) together with eye examination during prolonged use. In 2000, the Committee on the Safety of Medicines advised that thioridazine's use should be restricted to second‐line treatment of schizophrenia because of rare but serious cardiotoxicity; in particular, QTc prolongation and potentially life threatening ventricular arrhythmias (MHRA). In 2005, Novartis voluntarily withdrew thioridazine from the market following safety concerns. Following this, the MHRA withdrew the UK license for thioridazine, but the drug may still be imported on an unlicensed basis under the generic name Thioridazine (Neuraxpharma).
Technical background (+/‐)‐10‐[2‐(1‐methyl‐2‐piperidyl)ethyl]‐2‐(methylthio) phenothiazine (hydrochloride) or thioridazine has a higher level of cholinergic receptor binding action than chlorpromazine which may account for its higher level of cognitive and cardiac adverse events. This is also probably the reason for its lower level of extrapyramidal adverse events. Its cardiac adverse event profile is also related to a prolongation of the QT interval of the ECG (Hollister 1995, Drolet 1999) and torsades de pointes which is a ventricular arrhythmia (Roden 1993). Because of this thioridazine has been associated with mortality in overdose (Annane 1996, Buckley 1995).
Thioridazine is claimed to have a higher degree of limbic selectivity (Borison 1983, King 1995, Seeman 1983). This means it may be more selective for binding receptors in the mesolimbic dopamine system at the base of the brain. A receptor is a protein that binds a chemical messenger (neurotransmitter) such as dopamine or acetylcholine. Drugs also bind to receptors when exerting their action. These limbic dopamine receptors may be more closely involved in the symptom development of schizophrenia. Like chlorpromazine it binds to dopamine receptors, responsible for its therapeutic effect, but is not selective for D2 receptors like haloperidol (Assie 1993, Bowers 1975, Sedvall 1995). It similarly binds to different receptors such as those for serotonin, noradrenaline and histamine neurotransmitters (King 1995). This may give it a broad range of effects including adverse events, for example, binding to histamine receptors causes sedation.
Thioridazine is usually considered a 'typical' antipsychotic, i.e. the older generation of antipsychotic first developed in the 1950s. However, because it is reputed to cause fewer extrapyramidal adverse events, some authorities have classified it as an 'atypical' i.e. akin to the newer generation of antipsychotics developed in the 1990's which are also thought to have a lower propensity to cause extrapyramidal adverse events (King 1995, Trevitt 1998)
Objectives
To review the effects of thioridazine for people with schizophrenia in comparison with antipsychotics, placebo, or no treatment.
A secondary objective was to examine the effects of thioridazine for elderly people with schizophrenia.
Methods
Criteria for considering studies for this review
Types of studies
We sought all relevant randomised controlled trials. Where trials were described as 'double‐blind', but only implied that they were randomised, they were included in a sensitivity analysis. If we found no substantive difference within primary outcomes (see types of outcome measures) when these 'implied randomisation' studies were added, then we included these in the final analysis. If a substantive difference was found we only used trials that were clearly randomised and the results of the sensitivity analysis were described. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week.
Types of participants
We included people with schizophrenia, however diagnosed. Those with 'serious/chronic mental illness' or 'psychotic illness' were also included. If possible, we excluded people with schizoaffective disorder, dementing illnesses, depression and primarily problems associated with substance misuse.
Types of interventions
1. Thioridazine: any dose 2. Placebo. 3. Any other antipsychotic agent, divided into the atypical (amisulpiride, clozapine, loxapine, molindone, olanzapine, quetiapine, risperidone, sertindole, zotepine) and the typical antipsychotics (chlorpromazine, haloperidol etc).
We excluded unlicensed compounds where they did not appear to be of established efficacy.
Types of outcome measures
As schizophrenia is often a life‐long illness and thioridazine is used as an ongoing treatment, we grouped outcomes according to time periods: short‐term (up to 12 weeks), medium‐term (13 weeks up to one year) and long‐term (more than one year).
Primary outcomes
1. Service utilisation outcomes 1.1 Hospital admission
2. Clinical response 2.1 Relapse
2.2 Clinically significant response in global state ‐ as defined by each of the studies
3. Extrapyramidal side effects 3.1 Incidence of use of antiparkinson drugs
4. Other adverse effects, general and specific 4.1 Cardiac effects
Secondary outcomes
1. Death: suicide or natural causes
2. Service utilisation outcomes 2.1 Days in hospital 2.2 Change in hospital status
3. Clinical response 3.1 Average score/change in global state 3.2 Clinically significant response in mental state ‐ as defined by each of the studies 3.3 Average score/change in mental state 3.4 Clinically significant response on positive symptoms ‐ as defined by each of the studies 3.5 Average score/change in positive symptoms 3.6 Clinically significant response on negative symptoms‐ as defined by each of the studies 3.7 Average score/change in negative symptoms
4. Leaving the study early.
5. Behaviour 5.1 Clinically significant response in behaviour ‐ as defined by each of the studies 5.2 Average score/change in behaviour
6. Extrapyramidal side effects 6.1 Clinically significant extrapyramidal side effects ‐ as defined by each of the studies 6.2 Average score/change in extrapyramidal side effects
7. Other adverse effects, general and specific 7.1 Number of people dropping out due to adverse affects 7.2 Anticholinergic effects 7.3 Antihistamine effects 7.4 Prolactin related symptoms
8. Social functioning 8.1 Clinically significant response in social functioning ‐ as defined by each of the studies 8.2 Average score/change in social functioning
9. Economic outcomes
10. Quality of life/satisfaction with care for either recipients of care or carers 10.1 Significant change in quality of life/satisfaction ‐ as defined by each of the studies 10.2 Average score/change in quality of life/ satisfaction 10.3 Employment status
11. Cognitive functioning
Search methods for identification of studies
Electronic searches
1. We searched the Cochrane Schizophrenia Group's Register (June 2006) using the phrase:
[(thioridazin* or tioridazin* or thioridazide* or thioridacin* or sonapax* or mallorol* or malloryl* or meleril* or mellaril* or melleril* or melleret* or melleryl* in REFERENCES) Title, Abstract and Index term fields OR (thioridazin* in STUDY interventions field)]
This register is compiled by systematic searches of major databases, hand searches and conference proceedings (see Group Module).
2. Details of previous electronic searches 2.1. We searched the Cochrane Schizophrenia Group's Register (January 2002) using the phrase:
(thioridazine‐phrase) or #42=571 or #42=50 or#42=227]
(#42 is the field in the Register where each intervention is coded. 571 is thioridazine and 50 and 227 are Melleril)
2.2. We searched the Cochrane Schizophrenia Group's Register (September 2002) using the phrase:
[((*Meleril* or *Mellaril* or *Melleril* or *Melleryl* or *Melleretten* or *Mallorol* or *Elperil* or *Flaracantyl* or *Mefurine* or *Orsanil* or *Ridazine* or *Sonapax* or *Stalleril* or *Tirodil* or *Visergil*) in title, abstract or index terms of REFERENCE) or (Thioridazine in interventions of STUDY)]
2.3. We searched Biological Abstracts (January 1982 to September 2002) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and for schizophrenia (see Group search strategy) combined with:
[and (thioridazine‐phrase)]
2.4. We searched CINAHL (January 1982 to September 2002) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and for schizophrenia (see Group search strategy) combined with:
[and (thioridazine‐phrase)]
2.5. We searched the Cochrane Library (Issue 3, 2002) using the phrase:
[(thioridazine‐phrase) or THIORIDAZINE/explode in MeSH]
2.6. We searched EMBASE (January 1980 to September 2002) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and for schizophrenia (see Group search strategy) combined with:
[and ((thioridazine‐phrase) or explode THIORIDAZINE / all)]
2.7. We searched MEDLINE (January 1966 to September 2002) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and for schizophrenia (see Group search strategy) combined with:
[and ((thioridazine‐phrase) or THIORIDAZINE / explode in MeSH)]
2.8. We searched PsycLIT (January 1974 to September 1999) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and for schizophrenia (see Group search strategy) combined with:
[and ((thioridazine‐phrase) or THIORIDAZINE / explode in MeSH)]
2.9. We searched Sociofile (January 1974 to September 2002) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and for schizophrenia (see Group search strategy) combined with:
[and (thioridazine‐phrase)]
Searching other resources
1. Reference searching We also inspected the references of all identified studies for more studies.
2. Personal contact We contacted the first author of each included study for information regarding unpublished trials.
3. Drug company We contacted the manufacturers of proprietary thioridazine (Novartis) for additional data.
Data collection and analysis
1. Study selection For the earlier version of the review (AS) inspected all citations from the search results. MF re‐inspected a random sample (10%) of reports in order to ensure selection reliability. Potentially relevant abstracts were identified and full papers ordered and reassessed for inclusion and methodological quality. Where disagreements arose we attempted resolution by discussion, or acquired further information from the authors of trials. If doubt remained we did not include the study and added it to the list of those awaiting assessment, pending further information. For the update (2006) we (MF and JR) inspected and selected all study citations identified by the search. Where disagreement arose, this was resolved by discussion, or where doubt remained, we acquired the full article for further inspection.
2. Assessment of methodological quality We assessed the methodological quality of the included studies using criteria described in the Cochrane Handbook (Higgins 2005) and the Jadad Scale (Jadad 1996). The former is based on the evidence of a strong relationship between allocation concealment and direction of effect (Schulz 1995). The categories are defined below:
A. Low risk of bias (adequate allocation concealment) B. Moderate risk of bias (some doubt about the results) C. High risk of bias (inadequate allocation concealment). For the purpose of the analysis in this review, trials were included if they met the Cochrane Handbook criteria A or B.
The Jadad Scale measures a wider range of factors that impact on the quality of a trial. The scale includes three items:
1. Was the study described as randomised? 2. Was the study described as double‐blind? 3. Was there a description of withdrawals and drop outs?
Each item receives one point if the answer is positive. In addition, a point can be deducted if either the randomisation or the blinding/masking procedures described are inadequate. For this review we used a cut‐off of two points on the Jadad scale to check the assessment made by the Handbook criteria. However, we did not use the Jadad Scale to exclude trials.
3. Data management 3.1 Data extraction Originally (AS) independently extracted data from the included trials and a random 10% sample was checked by (JR) for accuracy. We discussed any disagreements, documented decisions and, where necessary, we contacted authors of trials for clarification. When this was not possible, we did not enter data and added the studies to the list of those awaiting assessment. For the 2006 update we (MF and JR) independently extracted data and any disagreements were resolved through discussion, where this was not possible we contacted authors for further information.
4. Data synthesis Data types: Outcomes are assessed using continuous (for example, average changes on a behaviour scale), or dichotomous measures (for example, either 'no important changes' or 'important changes' in a person's behaviour). Categorical data (for example, one of three categories on a behaviour scale, such as 'little change', 'moderate change' or 'much change' are currently not supported by RevMan software so they were dichotomised where possible (see below).
4.1 Dichotomous data Where the original authors of the studies gave outcomes such as 'clinically improved' or 'not clinically improved' based on their clinical judgement, predetermined criteria or any scale this was recorded in RevMan. If data were from a rater not clearly stated to be independent then it was included if it did not change the results, otherwise it was presented separately with a label 'prone to bias'. Where possible, efforts were made to convert relevant categorical or continuous outcome measures to dichotomous data by identifying cut off points on rating scales and dividing subjects accordingly into groups. This was with the cut off points 'moderate or severe impairment' for end of study data or 'no better or worse' for change data wherever possible.
4.1.1 Summary statistic: for dichotomous outcomes we calculated the relative risk (RR) and its 95% confidence interval (CI) using a fixed effects model. If heterogeneity was found (see section 5) we used a random effects model. We also calculated the number needed to treat/harm statistic (NNT/H) when outcomes were statistically significant.
4.2 Continuous data 4.2.1 Normal data Continuous scale derived data if often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data the following standards were applied to continuous endpoint data: (a) Standard deviations and means were reported in the paper or were obtainable from the authors; (b) The standard deviation (SD), when multiplied by 2 was less than the mean (as otherwise the mean was unlikely to be an appropriate measure of the centre of the distribution) (Altman 1996). Data that did not meet the first or second standard were not analysed in RevMan software, but were entered into other data tables and reported as skewed data in the results section. Endpoint scores on scales often have a finite start and endpoint and this rule can be applied to them. If a scale starts from a positive value (such as PANSS, which can have values from 30‐210) the calculation described above in (b) should be modified to take the scale starting point into account. In these cases skew is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score.
4.2.2 Endpoint versus change data: endpoint scale‐derived data are finite, ranging from one score to another. Change data (endpoint minus baseline) are more problematic and in the absence of individual patient data it is impossible to know if data are skewed, though this is likely. After consulting the ALLSTAT electronic statistics mailing list, we presented change data in MetaView in order to summarise available information. In doing this, it was assumed either that data were not skewed or that the analyses could cope with the unknown degree of skew. Where possible we presented endpoint data, and if both endpoint and change data were available for the same outcomes, then we reported only the former.
4.2.3 Summary statistic: for continuous outcomes we estimated a weighted mean difference (WMD) fixed effect model between groups. Again, if heterogeneity was found (see section 5) we used a random effects model.
4.3 Intention to treat data We excluded data from studies where more than 40% of participants in any group were lost to follow up (this does not include the outcome of 'leaving the study early'). In studies with less than 40% dropout rate, we considered people leaving the study early to have had the negative outcome, except for the event of death.
4.4 Scale derived data Unpublished scales are known to be subject to bias in trials of treatments for schizophrenia (Marshall 2000). Therefore we only included continuous data from rating scales if the measuring instrument had been described in a peer‐reviewed journal.
In many included studies in this review it was unclear that scale based data were rated independently of treatment (see Included studies tables) so we presented the data with a label 'prone to bias'.
4.5 Cluster trials Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997, Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster randomised study, but will adjust them for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
5. Test for heterogeneity Firstly, we considered all the included studies within any comparison to judge clinical heterogeneity. Then we visually inspected graphs to investigate the possibility of statistical heterogeneity. This was supplemented, primarily, by employing the I‐squared statistic. This provides an estimate of the percentage of inconsistency thought to be due to chance. Where the I‐squared estimate was equal to, or greater than 75%, this was interpreted as evidence of high levels of heterogeneity (Higgins 2003). In such cases, we sought to remove outlying trial(s) and perform and report sensitivity analyses both with and without these outlying trials. Where no obvious outlying trial(s) could be identified we analysed and reported the result using a random effects model, which takes into account that the effects being estimated are not identical.
6. Assessing the presence of publication bias Data from all included trials were entered into a funnel graph (trial effect versus trial size or 'precision') in an attempt to investigate the likelihood of overt publication bias. Where only 3‐4 studies reported an outcome or there was little variety in sample size (or precision estimate) between studies ‐ funnel plot analysis was not appropriate. There is currently no consensus about the validity of formal statistical tests to investigate funnel plot asymmetry, one test, proposed by Egger 1997 has been subject to criticism (Stuck 1998). Further versions of this review will include such tests when their validity has been proven.
7. Sensitivity analyses 7.1 Outcomes for intention‐to‐treat analysis were compared with completer analysis. Where there were differences these were either reported or presented graphically. 7.2 Results for the elderly with schizophrenia were to be analysed separately and compared with the results for younger trial participants (cut‐off of age 65 where possible) however this was not possible (see below).
8. General Where possible, we entered data into RevMan so the area to the left of the line of no effect indicated a favourable outcome for thioridazine.
Results
Description of studies
1. Excluded studies We excluded 87 studies, details of which are in the 'Excluded studies' table. Most were excluded due to being non‐randomised studies. We excluded more than 20 studies because of irrelevant interventions. The remainder had to be excluded because we could find no usable data. For example, in several there were no outcomes reported, or in the crossover studies there were no data from the first pre‐crossover stage. We were unable to include the most recent study we found, Mahmoud 2004, as no reported data were available from the thioridazine arm when tit was compared with risperidone.
2. Awaiting assessment One Japanese study (Tanimukai 1973) is awaiting translation.
3. Ongoing studies We are not aware of any ongoing studies.
4. Included Studies During the 2006 update we found four 'new' studies to include (Carranza 1974, Ju 1997, Schiele 1961, Zhang 1999), and three further reports of trials already included in the review, one of which provided additional data (Liu 1994). A total of 42 studies are included.
4.1 Length of trials Study durations ranged from 28 days to 40 months. Most studies (n=30) were short‐term evaluations (up to 12 weeks), although ten were of intermediate duration (13 weeks to one year) and two were longer‐term trials (Grinspoon 1967 24 months, Rasmussen1976 40 months).
4.2 Participants A total of 3498 people have participated in the 42 trials, most of whom had a diagnosis of schizophrenia. Judah 1958 reported participants had 'schizophrenia in 80% of the treated group and 73% of the control group'. Kramer 1978 included one person with schizoaffective disorder. Somerville 1960 randomised 56 people with schizophrenia or "paraphrenic psychosis" and four with bipolar disorder. These studies were included because the great majority of randomised patients had schizophrenia. Only 16 studies used predefined diagnostic criteria, Diagnostic Statistical Manual (DSM), International Classification of Diseases (ICD), NIMH criteria, Feighner's criteria, and Chinese Classification of Mental Diseases (CCMD). The remainder appeared to have made a clinical diagnosis. Many studies (n=25) involved people with chronic illnesses; four of these involved people with chronic illness but who were experiencing an acute exacerbation. The rest included acutely ill people and first episode patients; five specified a high level of symptomatology on the Brief Psychiatric Rating Scale (BPRS). Ages ranged from seven to above 81 years. Only one study specifically focussed on older patients (Phanjoo 1990).
4.3 Setting Most studies were conducted in hospital settings. Only three trials were undertaken in an outpatient environment (Clark 1975, Nishikawa 1985, Rada 1972). Most trial centres were in North America or Europe, but five were from China (Chen 1995, Gui‐Yun 1988, Ju 1997, Liu 1994, Zhang 1999).
4.4 Study size The number of people in the included studies ranged from 10 to 512. Most studies had 60 or fewer participants.
4.5 Interventions The mean dose of thioridazine, based on 13 studies which reported it, was about 468 mg/day (SD 208 mg/day) and the range, taken from 39 studies, was 25 to 1600 mg/day.
Fourteen studies had a separate placebo arm; two used an 'active placebo' which was phenobarbital with atropine to reproduce the adverse effects of the antipsychotic drugs. Montgomery 1992 involved people allocated to placebo taking thioridazine for one week post‐randomisation. We included this study to increase generalisability of data (including it did not change the overall findings). Twenty‐seven studies compared thioridazine with oral typical neuroleptics such as fluphenazine or chlorpromazine. Three studies (Keks 1994, McCreadie 1988, Phanjoo 1990) compared thioridazine with the atypical remoxipride (which was withdrawn in 1994 following reports of aplastic anaemia), and another three, Carranza 1974, Liu 1994 and Ju 1997, compared thioridazine to the atypicals, sulpiride, and clozapine.
4.6 Outcomes 4.6.1 Missing outcomes No study reported on negative symptoms as an outcome, neither were there usable cognitive outcomes. No included study attempted to quantify levels of satisfaction or quality of life, or any direct economic evaluation of thioridazine.
4.6.2 Scales Most outcomes were reported as dichotomous (yes‐no/binary outcomes), and are presented as such. Scale derived data was obtained from five scales, details of these are given below. We have reported reasons for exclusion of data from other scales in the 'Included studies' table. Scales that provided usable data are reported below.
4.6.2.1 Global State 4.6.2.1.1 Clinical Global Impression ‐ CGI (Guy 1976) The CGI is a three‐item scale commonly used in studies on schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement. The items are: severity of illness; global improvement and efficacy index. A seven‐point scoring system is usually used with low scores indicating decreased severity and/or greater recovery. Clark 1971, Clark 1975, Liu 1994 reported usable data from this scale.
4.6.2.1.2 Global Assessment Scale ‐ GAS (Endicott 1976) Used to evaluate the overall functioning of a person during a specified time period in terms of psychological well‐being or sickness. The scale ranges from 1 (hypothetically sickest person) to 100 (hypothetically healthiest person) and is divided into 10 equal intervals. High score indicates good outcome. Montgomery 1992 reported usable data from this scale.
4.6.2.2 Mental state 4.6.2.2.1 Brief Psychiatric Rating Scale ‐ BPRS (Overall 1962) The BPRS is an 18‐item scale measuring positive symptoms, general psychopathology and affective symptoms. The original scale has sixteen items, but a revised eighteen‐item scale is commonly used. Scores can range from 0‐126. Each item is rated on a seven‐point scale varying from 'not present' to 'extremely severe', with high scores indicating more severe symptoms. Chen 1995 and Liu 1994 reported usable data from this scale.
4.6.2.3 Behaviour 4.6.2.3.1 Nurses Observational Scale of Inpatients Evaluation ‐ NOSIE (Honingfeld 1965). An 80‐item scale with items rated on a five‐point scale from zero (not present) to four (always present). Ratings are based on behaviour over the previous three days. The seven headings are social competence, social interest, personal neatness, cooperation, irritability, manifest psychosis and psychotic depression. The total score ranges from 0‐320 with high scores indicating a poor outcome. Mena 1966 reported usable data from this scale.
4.6.2.4 Adverse events 4.6.2.4.1 Treatment Emergent Symptoms Scale ‐ TESS (Guy 1976) This checklist assesses a variety of characteristics for each adverse event, including severity, relationship to the drug, temporal characteristics (timing after a dose, duration and pattern during the day), contributing factors, course, and action taken to counteract the effect. Symptoms can be listed a priori or can be recorded as observed by the investigator. The TESS records the presence or absence of a list of side effects. Chen 1995, Gui‐Yun 1988 and Ju 1997 reported usable data from this check list.
Risk of bias in included studies
1. Randomisation Only five studies described the method used to generate random allocation (Bergling 1975, Gardos 1978, Granacher 1982, Gui‐Yun 1988, Ju 1997). They all used tables of random numbers, except one, which used a coin toss. Six studies reported that allocation was undertaken independently (Bergling 1975, Somerville 1960, Stabenau 1964, Wolpert 1968, Herrera 1990, Miyakawa 1973). NIMH 1964 described a form of allocation concealment (sealed envelopes). For other studies readers were given little assurance that bias was minimised during the allocation procedure. Clark 1971 and Keks 1994 used block randomisation. Seventeen studies reported that the numbers allocated to each treatment group were identical, without reporting the use of block randomisation.
2. Blindness Thirty trials were double blind, seven trials did not report whether blinding was attempted, although some report using identical capsules. Three trials were single blind, and one trial was not blinded. Two studies (Mena 1966, Cohler 1966) tested the quality of blinding using a questionnaire.
3. Leaving the study early Thirty eighty of the 42 included studies report data for leaving the study early, and 16 of these described the reasons for this attrition. In the thioridazine versus placebo comparison 24% (n=492) of all participants left the study; in the thioridazine versus typical comparison 16% (n=1587), and; in the thioridazine versus atypical antipsychotics comparison, 26% (n=344).
4. Data reporting Only 12 studies reported that those rating outcome were independent of the treatment (see Included studies table). Largely, a person unlikely to be disinterested in the final result, rated scale outcomes. Most are therefore presented in this review with a warning 'prone to bias'. In any case, continuous scale data were often poorly reported. Frequently they lacked explicit statements regarding the denominator or variance, were only presented as significance tests or within graphs, or simply reported insufficient or no data at all. Liu 1994 reported that all participants in the thioridazine group experienced dry mouth and 60% experienced tachycardia and 45% dizziness, but we were unable to present this data as the frequency of these adverse effects were not reported in the control group. Twelve studies reported scale‐based categorical data that appeared to use the 'last outcome carried forward' (LOCF) approach for those who left the study. Sometimes this was stated in the text, but in other instances it was only apparent from the tables (see Included studies table). We have presented these data in this review. Where they substantially affected the results we reported these instances in the text. Clark 1975 and Nishikawa 1985 reported relapse as a reason for leaving the study early but did not make criteria for this explicit. We could not be sure that these studies reported all relapses in the study population. These sparse outcome data were presented both as 'Relapse, clinically diagnosed' and 'leaving the study'.
Effects of interventions
1. The search The original search (2000) yielded 809 citations, and after removal of duplicate records, 152 were obtained as full publications. A further 15 were acquired after hand searching references of the other papers, but none of the latter could be included. To date, contacting the relevant drug company (Novartis) has not led to further usable data being obtained as limited records of early unpublished research were found (see Acknowledgements). The 2002 update search identified 61 abstracts, 54 were obtained as full publications, and three further studies were included in the review. We found 93 citations during the 2006 update search, and were able to include four additional studies (Carranza 1974, Ju 1997, Schiele 1961, Zhang 1999), and three further reports of studies already included in the review (Gallant 1972, Liu 1994, Rada 1972). This review includes 42 randomised trials with a total of 3498 participants.
2. THIORIDAZINE versus PLACEBO Six hundred and sixty eight participants were randomised within 14 studies.
2.1 Global state 2.1.1 No change or worse Change in global state during short‐term assessment (three months or less) favoured thioridazine compared with placebo (n=100, 3 RCTs, RR 0.66 CI 0.4 to 1.0, NNT 5 CI 3 to 81). At six months, data continued to favour thioridazine (n=105, 3 RCTs, RR 0.32 CI 0.2 to 0.5) with NNT of 2 (CI 2 to 3).
2.1.2 Clinical Global Impression Clinical Global Impression data dichotomised to 'moderately or severely ill' were not significantly different at 28 days or by six months. Clark 1975 used 'last observation carried forward' for about 30% of CGI endpoint data at six months, and we found results favoured thioridazine (n=23, WMD ‐0.99 CI ‐1.8 to ‐0.2) compared with placebo.
2.1.3 Global Assessment Scale Global Assessment Scale data from one four‐week study (Montgomery 1992) favoured thioridazine (n=50, WMD 14.26 CI 3.4 to 25.1).
2.2 Mental state 2.2.1 Relapse The number of participants experiencing a (short‐term) relapse were significantly fewer in the thioridazine group compared with placebo (n=261, RR 0.09 CI 0.03 to 0.3) but data are heterogeneous (I2 =88%). Six‐month data found no difference (Clark 1975, n=25, RR 0.33 CI 0.1 to 1.0).
2.2 Not improved or worse For the outcome 'not improved or worse' no differences were found at six weeks (Somerville 1960) or seven months (Wolpert 1968). We found dichotomised data 'moderately or severely ill' (Clark 1971) were equivocal (n=43, RR 0.78 CI 0.4 to 1.5) at four‐week assessment. Brief Psychiatric Rating Scale data (Montgomery 1992) contained wide confidence intervals (skewed data) and are not reported.
2.2.3 Depression We found no significant differences in rates of depression at short‐term (n=88, 2 RCTs, RR 0.95 CI 0.2 to 4.2) and medium‐term (n=82, 2 RCTs, RR 2.68 CI 0.8 to 9.6) assessment.
2.3 Leaving the study early We found attrition rates 'any reason' (three months or less) significantly favoured thioridazine (n=510, 9 RCTs, RR 0.42 CI 0.3 to 0.6, NNT 6 CI 5 to 9). Fourteen percent in the thioridazine group left early compared with 32% of people allocated to placebo. The four trials reporting data between three and 12 months (medium‐term) did not clearly favour thioridazine or placebo (n=115, RR 0.67 CI 0.3 to 1.4). Where reasons for leaving the study were reported, more significant differences emerged if negative outcomes were assumed for all those who left the study due to 'relapse or worsening/no improvement' (n=396, 6 RCTs, RR 0.10 CI 0.1 to 0.2, NNT 4 CI 4 to 5). However, where adverse effects were blamed as the reason for leaving, we found no indication that thioridazine promoted this. The same applies to leaving due to refusal of treatment.
2.4 Adverse events 2.4.1 Anticholinergic Very comprehensive lists of adverse effects were reported by several studies. Few differences between thioridazine and placebo were apparent. Limited data from trials suggests that thioridazine is not strongly anticholinergic (blurred vision at six months, n=65, RR 0.76 CI 0.2 to 3.4). The thioridazine group experienced significantly more occurrences of dry mouth in the short‐term (n=324. 3 RCTs, RR 6.75 CI 3.1 to 14.9, NNH 6 CI 3 to 15), but longer‐term data were equivocal (n=82, 2 RCTs, RR 1.62 CI 0.5 to 4.9). We found nasal congestion at short‐term assessment favoured the placebo group (n=279, 2 RCTs, RR 3.42 CI 1.4 to 8.3, NNH 11 CI 4 to 61), but again longer‐term data from one study (Clark 1975) were equivocal (n=30, RR 0.5 CI 0.1 to 4.9).
2.4.2 Arousal Significant data relating to arousal, specifically drowsiness, suggest that thioridazine is sedating both up to three months (n=324, 3 RCTs, RR 5.37 CI 3.2 to 9.1, NNH 4 CI 2 to 7), and from three months to one year (n=162, 4 RCTs RR 2.41 CI 1.3 to 4.5, NNH 6 CI 3 to 27). We found no significant differences for insomnia or excitement from small studies.
2.4.3 Cardiovascular When cardiovascular adverse effects were recorded we found one outcome (faintness, dizziness and weakness) did favour the placebo group (Clark 1971, n=43, RR 4.30 CI 1.1 to 17.6, NNH 4 CI 2 to 211). However, another small study (n=25) reporting the same outcome did not reveal any significant differences (Clark 1975, RR 0.67, CI 0.2 to 2.7). Other measures, chest pain, hypotension, and tachycardia were not found to be significantly more prevalent in the thioridazine group.
2.4.4 Central nervous system In the earlier version of this review we found data from the NIMH 1964 study were heavily influenced by the assumption of poor outcome for people who had left early, and suggested that placebo promoted headache, fainting and even seizures. We removed this ITT data set from the NIMH 1964 study (adverse events) and analysed without assuming that those lost to follow‐up had had a negative outcome, as those leaving the NIMH 1964 study left due to either treatment failure or administration problems. We found all data for confusion, headache, memory defects, seizures, and syncope were not significantly different between thioridazine and placebo.
2.4.5 Endocrine Breast swelling and lactation were monitored over six weeks and we found no significant data to suggest that thioridazine promotes this compared with placebo (NIMH 1964). In the Clark 1975 study we again did not find any significant data to suggest that participants given thioridazine for six months had higher occurrences of lactation than the placebo group.
2.4.6 Movement disorders Thioridazine may cause more movement disorders than placebo but most data are equivocal (akathisia, akinesia, dystonia, oculogyric crisis, parkinsonism, rigidity). Only tremor (n=279, 2 RCTs, RR 3.03 CI 1.2 to 7.4, NNH 13 CI 4 to 102), and use of antiparkinsonian drugs (NIMH 1964, n=236, RR 2.53 CI 1.2 to 5.6, NNH 11 CI 4 to 79) were significantly higher in the thioridazine group at short‐term assessment. However, medium‐term follow up (three months to one year) for tremor and use of antiparkinsonian drugs did not reveal any significant difference between thioridazine and placebo.
2.4.7 Gastrointestinal We found most outcomes were equivocal. Short‐term data suggests that thioridazine is constipating (n=279, 2 RCTs, RR 2.47 CI 1.3 to 4.8, NNH 10 CI 4 to 45), although medium‐term data (n=82, 2 RCTs, RR 1.80 CI 0.3 to 11.1) did not reveal any significant difference between thioridazine and placebo. Diarrhoea (short and medium‐term) data were equivocal. We found reports of nausea from the NIMH 1964 study to be significantly higher in the thioridazine group (NIMH 1964, n=236, RR 12.01 CI 3.8 to 38.2, NNH 4 CI 2 to 15). However, reports of nausea from (Clark 1975) were not significantly different. Reports of vomiting came from only one study (NIMH 1964) with significantly more participants experiencing vomiting in the thioridazine group (n=236, RR 25.88 CI 1.5 to 434.1, NNH 5 CI 2 to 186) compared with placebo. We found weight loss (n=25, RR 0.17 CI 0.02 to 1.3) and weight gain (n=25, RR 2.00 CI 0.2 to 16.6) were equivocal at six months assessment (Clark 1975).
2.4.8 Genitourinary We found non‐specific reports of urinary disturbances, from two studies (Clark 1971, NIMH 1964) were significantly higher in the thioridazine group (n=279, RR 3.82, CI 1.1 to 13.0, NNH 18 CI 5 to 407) compared with placebo at short‐term assessment.
2.4.9 Haematology We found no significant differences (n=65, 2 RCTs, RR 0.80 CI 0.4 to 1.7) between thioridazine and placebo for the outcome of 'abnormal laboratory results' (short‐term assessment).
2.4.10 Other adverse effects We found most outcomes were not significantly different (infections, liver abnormalities, oedema, pyrexia, salivation, sweating, photosensitivity, rash) between thioridazine and placebo. Reports of weakness were significantly higher in the thioridazine group (medium‐term assessment) (n=97, 2 RCTs, RR 4.88 CI 1.1 to 21.4, NNH 7 CI 2 to 241).
3. THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC A total of 2598 participants were randomised within 27 studies.
3.1 Death We found no significant differences in death with Gui‐Yun 1988 reporting one death from physical illness in the thioridazine group by three months.
3.2 Global state 3.2.1 No change or worse (LOCF) No significant differences were found (short‐term) in the thioridazine group compared with typical antipsychotics for the number of participants reported as 'not improved or worse' (n=743, 11 RCTs, RR 0.98 CI 0.8 to 1.2). Medium‐term data from three trials (Clark 1975, Schiele 1961, Stabenau 1964) were also equivocal (n=142, RR 0.99, CI 0.6 to 1.6). Excluding studies that used LOCF did not change this.
3.2.2 Clinical Global Impression (Moderately or severely ill ‐ LOCF) We found no significant difference during short and medium‐term assessments with CGI scale data dichotomised to 'moderately or severely ill. Clinical Global Impression average endpoint scores by six months (Clark 1975) were also equivocal (n=26, RR ‐0.21 CI ‐0.9 to 0.5). 3.3 Mental state 3.3.1 Relapse We found no significant differences in relapse rates between thioridazine and typical antipsychotics at short (n=368, 2 RCTs, RR 0.55 CI 0.2 to 1.7) and medium‐term (n=76, 2 RCTs RR 1.07 CI 0.7 to 1.6) assessments.
3.3.2 No change or worse (BPRS) Short‐term assessments (by three months) were not significantly different (n=208, 5 RCTs, RR 1.26 CI 1.0 to 1.7) between thioridazine and typical antipsychotic. Wolpert 1968 reported data at seven months and again we found no significant differences when BPRS derived data were dichotomised to 'no change or worse'.
3.3.3 Average endpoint BPRS We found BPRS endpoint scores favoured thioridazine over chlorpromazine at six weeks (n=121, WMD ‐2.04 CI ‐3.9 to ‐0.2) (Chen 1995).
3.3.4 Moderately or severely ill (LOCF) We found no significant difference by three months assessment between thioridazine and typical antipsychotics (n=85, 2 RCTs, RR 1.35 CI 0.8 to 2.4).
3.3.5 Depressed No significant differences were found in rates of depression between thioridazine and the other typical antipsychotics group at short (n=95, 2 RCTs, RR 0.91 CI 0.3 to 3.0) and medium‐term (n=94 2 RCTs, RR 1.11 CI 0.5 to 2.7) assessment.
3.4 Behaviour (NOSIE) Just Mena 1966 reported usable data derived from a nurse‐rated scale. We found no significant difference for the outcome 'no better or worse' at five weeks (n=40, RR 2.33 CI 0.7 to 7.8).
3.5 Leaving the study early The number of people who left the study early during short‐term assessment (up to three months) did not reveal any statistically significant differences between thioridazine and typical antipsychotics (n=1587, 19 RCTs, RR 1.07 CI 0.9 to 1.3). Sixteen percent of participants from each group left the study early. Five medium‐term studies (n=612) also suggested no significant difference. Attrition rates from Rasmussen 1976 (n=30) were also equivocal at three and a half years (RR 1.50 CI 0.3 to 7.7). Where reasons were cited for study attrition, due to absence or refusal to continue, no significant differences were found. The strongest data relate to attrition due to adverse effects. These favoured typical antipsychotic drugs over thioridazine (n=871, RR 2.24 CI 1.2 to 4.2, NNT 26 CI 10 to 164). Medium‐term data from two studies were equivocal. Leaving due to refusal of medication/poor compliance, or relapse/no change or worsening of heath did not reveal any significant difference.
3.6 Adverse events 3.6.1 Anticholinergic There were no clear differences between thioridazine and other typical antipsychotics for the majority of anticholinergic adverse effects. Incidences of dry mouth were significantly higher in the thioridazine group (n=829, 5 RCTs, RR 1.47 CI 1.2 to 1.9, NNH 12 CI 7 to 34). However, medium‐term data (three months to one year) were equivocal (n=146, 3 RCTs RR 1.11 CI 0.6 to 2.1). Blurred vision, nasal congestion, and urinary retention were not significantly different between groups.
3.6.2 Arousal About half of those allocated thioridazine felt drowsy or sedated but these data are no different from typical antipsychotics (n=891, 8 RCTs, RR 1.10 CI 0.9 to 1.3). All other measures of arousal, excitement, and insomnia were not significantly different.
3.6.3 Cardiovascular We found data from Gui‐Yun 1988 favoured chlorpromazine for the outcome 'any cardiovascular adverse event' (n=74, RR 3.17 CI 1.4 to 7.0, NNH 3 CI 2 to 5) by three months. Results from two studies (Chen 1995, Gallant 1972) measuring changes in electrocardiogram (ECG) were significantly higher in the thioridazine group (n=254, RR 2.38, CI 1.6 to 3.6, NNH 4 CI 3 to 10). All other cardiovascular outcomes (chest pain, faintness/dizziness/weakness, hypotension, and tachycardia) did not reveal any significant differences.
3.6.4 Central nervous system We found data from four studies favoured other typical antipsychotics for the outcome 'syncope' (n=519, 4 RCTs, RR 3.21 CI 1.3 to 7.8, NNH 22 CI 7 to 156). However, we found data reported at four months (Schiele 1961) were not significantly different (syncope, n=60, RR 1.00 CI 0.1 to 10.4). One participant in the thioridazine group developed pigmented retinopathy (Chen 1995, n=234, RR 2.80 CI 0.1 to 68.1). We found no difference in ocular deposits between chlorpromazine and thioridazine (Rasmussen 1976, n=30). All other outcomes, ataxia, confusion, concentration difficulties concentration difficulties, headache, memory defects, and seizure did not reveal any significant differences.
3.6.5 Endocrine We found no significant differences between thioridazine and other typical antipsychotics for the adverse effects of breast swelling, and lactation.
3.6.6 Movement disorders Extrapyramidal adverse events that required use of antiparkinsonian drugs were significantly lower in the thioridazine group (n=1082, 7 RCTs, RR 0.45 CI 0.4 to 0.6), but data are heterogeneous (I2 statistic 82%). Medium‐term data by Schiele 1961 and Stabenau 1964 did not reveal any significant difference for the same outcome. We found reports of parkinsonism were significantly higher in the other typical antipsychotic group (n=340, RR 0.29 CI 0.1 to 0.7, NNH 9 CI 8 to 22) during three months of assessment. Short‐term reports of rigidity were equivocal (n=509, 4 RCTs, RR 0.60 CI 0.4 to 1.0), but medium‐term data suggests rigidity occurs more frequently in the other typical antipsychotics (n=154, 3 RCTs, RR 0.44, C 0.2 to 0.9, NNH 6 CI 4 to 23). Akathisia data (short and medium‐term) were not significantly different. All other assessments (akinesia, dyskinesia, dystonia, oculogyric crisis, and tremor) did not reveal any significant differences.
3.6.7 Gastrointestinal We found data from NIMH 1964 favoured the typical antipsychotics for the outcome 'nausea' (n=338, RR 2.35 CI 1.5 to 3.7, NNH 7 CI 4 to 18) by six weeks. However, another study (Clark 1975) revealed no significant differences in rates of nausea by 6 months (n=30, RR 0.50 CI 0.1 to 2.3). Reports of vomiting from three studies (Galbrecht 1968, NIMH 1964, Weston 1973) favoured the typical antipsychotic group (short‐term) with significantly more participants in the thioridazine experiencing vomiting (n=734, RR 1.82 CI 1.1 to 3.0, NNH 20 CI 9 to 150). Only two studies reported on weight gain (Rada 1972, n=30, RR 1.00 CI 0.6 to 1.7 by 3 months, and Clark 1975, n=30, RR 0.6 CI 0.2 to 2.1 by 6 months) but no significant differences were apparent. All other outcomes constipation, diarrhoea and weight loss were not significantly different between thioridazine and typical antipsychotics. 3.6.8 Genitourinary Difficulty with urination did not reveal any significant difference between thioridazine and typical antipsychotics (n=799, 4 RCTs, RR 1.62 CI 0.9 to 2.8) by three months assessment.
3.6.9 Laboratory tests We found no significant differences in abnormal laboratory results between treatment groups for blood cell tests or liver and renal functioning.
3.6.10 Other adverse events Reports of photosensitivity were significantly higher in the typical antipsychotics (n=181, 3 RCTs, RR 0.60 CI 0.4 to 0.9, NNH 7 CI 5 to 32) during short‐term assessment, but data from Stabenau 1964 at ten months were not significantly different (n=52, RR 1.71 CI 0.7 to 4.3). We found reports of allergic reactions, infections, odema, pyrexia, salivation, sweating, rash, and weakness did not reveal any significant differences between thioridazine and other typical antipsychotics.
4. THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC A total of 344 participants were randomised within six studies.
4.1 Death Keks 1994 reported one death (by suicide) in the remoxipride group by six weeks.
4.2 Global state 4.2.1 Not improved or worse We found three studies (Ju 1997, Keks 1994, Phanjoo 1990) reporting global state 'not improved or worse', and found no significant differences between thioridazine and the atypical antipsychotics, remoxipride and sulpiride (n=203, RR 1.00 CI 0.8 to 1.3) during short‐term assessment.
4.2.2 Clinical Global Impression We found CGI endpoint data at 6 weeks were not significantly different between thioridazine and clozapine (Liu 1994, n=33 RR ‐0.21 CI ‐0.7 to 0.3).
4.3 Mental state 4.3.1 No significant change (BPRS) Phanjoo 1990 reported on 'no important change' on the BPRS scale by six weeks, with 50% of participants dropping out of the study. We found no significant differences between groups (n=18, RR 0.50 CI 0.2 to 1.4).
4.3.2 Average endpoint change scores We found no significant difference in BPRS endpoint scores (Liu 1994, n=33, WMD ‐1.89 CI ‐7.6 to 3.8) at 6‐week assessment between thioridazine and clozapine. Liu 1994 assessed participants using the SAPS and SANS scale but data were found to contain wide confidence intervals (skewed data) and are not reported here. Keks 1994, McCreadie 1988 both reported BPRS data but again these contained wide confidence intervals and are not reported.
4.3.3 Use on benzodiazepines Only McCreadie 1988 reported on this outcome and we found the thioridazine group needed significantly fewer benzodiazepines compared with the remoxipride group (n=61, RR 0.47 CI 0.3 to 0.8, NNT 3 CI 2 to 8).
4.4 Leaving the study early We found the number of participants leaving the studies early were not significantly different between thioridazine and atypical antipsychotics (n=344, 6 RCTs, RR 0.86 CI 0.6 to 1.2) during short‐term assessment. We also found no significant differences for leaving the studies due to adverse events, refusal of medication/poor compliance, or relapse/worsening between groups.
4.5 Adverse effects 4.5.1 Anticholinergic We found no significant differences between thioridazine and atypicals for the outcomes of hypotension (n=162, 2 RCTs RR 1.58 CI 0.8 to 3.0) and dry mouth (Phanjoo 1990, n=18, RR 2.0 CI 0.2 to 18.3) at short‐term assessment.
4.5.2 Arousal We found data reported by Phanjoo 1990 for the outcomes 'drowsiness/sedation' were equivocal and data by (Ju 1997, Phanjoo 1990) for 'insomnia' revealed no significant differences between thioridazine and atypical antipsychotics.
4.5.3 Cardiovascular For the outcome 'faintness, dizziness, weakness' no significant differences were apparent (Phanjoo 1990, n=18, RR 2.00, CI 0.2 to 18.3).
4.5.4 Central nervous system We found data reported by Phanjoo 1990 for the outcomes 'concentration difficulties' and 'headache' were not significantly different between thioridazine and remoxipride.
4.5.5 Movement disorders All data by Phanjoo 1990 (n=18) were equivocal for the outcomes 'rigidity' and 'tremor'. Extrapyramidal symptoms reported in two studies (Liu 1994, Ju 1997) were not significantly different between thioridazine and atypicals (n=81, RR 1.22 CI 0.5 to 2.8).
4.5.6 Gastrointestinal We found no significant differences for the adverse events, constipation, diarrhoea or nausea from the small study (n=18) by Phanjoo 1990.
4.5.7 Hepatic abnormalities Ju 1997 reported the only data for this outcome and we found no significant difference between thioridazine and sulpiride (n=41, RR 0.48, CI 0.1 to 4.9).
5. Publication bias Funnel plots were planned to investigate the possibility of publication bias (see Methods). No overt asymmetry was detected but many of the outcomes had a small number of trials which limits the value of the plot. Such plots are not powerful investigative tools and are further weakened when there is little variation in study size (Egger 1997).
6. Sensitivity analysis 6.1 Intention to treat Sensitivity analysis in which those who left the study were not assumed to have a bad outcome did not substantially change most of the main outcomes. An exception was placebo‐controlled data on the breakdown of reasons for leaving the study. The a priori protocol for this review stated that if there was a difference in the results between completer analysis and 'intention to treat' analysis, then the latter would be preferred. The other exception was placebo‐controlled adverse event data for extrapyramidal, gastrointestinal, endocrine and miscellaneous other adverse events which favoured thioridazine over placebo. This was due to the differential drop‐out rate in the NIMH 1964 study. None of the NIMH 1964 placebo group were removed from the study due to complications of treatment (i.e. adverse events), but participants were removed mainly due to treatment failure (relapse), or administrative reasons. Therefore, outcome data from the NIMH 1964 study were presented in the results section using ITT only for relapse. Adverse events data were reported without using ITT assumptions. We feel this provides a more accurate appraisal of the data.
6.2 The elderly No sensitivity analyses were possible for the elderly, as had been planned, as only one small study (Phanjoo 1990) focused on an older age group.
6.3 Last observation carried forward A planned sensitivity analysis where data using 'last outcome carried forward' (LOCF) was removed (see Methodological quality of included studies) gave essentially the same outcomes as the main analyses with a few exceptions. These have already been noted and did not appear clinically or statistically significant. The LOCF data were therefore included in the main analysis and the overall stability of the findings on sensitivity testing appears high.
Discussion
1. Generalisability of findings Overall, we felt generalisability to be good as the 42 included studies involved people with schizophrenia who would be recognisable in everyday practice. In most of the studies the diagnosis was clinical and only a few used operational criteria. Both those with acute and chronic illness participated. Studies were, however, undertaken mostly in hospital settings. The daily doses of thioridazine largely reflected present practice, although seven studies did employ higher levels. Non‐Western cultures were represented by only five studies and therefore applicability to those in the developing world may be limited. People with both schizophrenia and substance misuse were frequently excluded which may reduce applicability of findings, as co‐existence of the two problems is common (Turner 1990).
Only one small study (Phanjoo 1990) included people in the older age group. For 18 people aged 67‐70 years, it compared thioridazine with remoxipride; the latter was withdrawn in 1994 following reports of aplastic anaemia. In addition, Judah 1958 reported a median age of 63 years for trial participants but did not give a range. Other trials did include older patients but did not present their data separately and, although attempts were made to contact authors, it was not possible to obtain individual patient data. Many studies excluded elderly people. Thioridazine has been considered a drug of choice in the elderly (see Background) and is thought to have been widely used in this group (King 1995); however, this clinical preference does not appear to be based on good quality, trial‐based evidence for elderly patients with schizophrenia.
2. THIORIDAZINE versus PLACEBO
2.1 Global state People given placebo were significantly more likely to have the negative outcome 'no change or worse' than the thioridazine group, with short‐term NNT of about five. This treatment effect became more apparent during medium‐term assessment with NNT lessening to about two. Clinical Global Impression scores 'moderate or severely ill' from two small studies did not reveal any significant differences; larger sample sizes are needed before we can have confidence in this result. Medium‐term (six months) endpoint CGI data significantly favoured thioridazine using the last observation carried forward method, but with assumptions being made for many of the (n=23) participants more robust data is needed to inform clinicians of its true efficacy. Global Assessment Scale data (Montgomery 1992) also significantly favoured thioridazine, again from limited numbers. Nevertheless, thioridazine does appear to confer an advantage over placebo when rated as 'no change or worse'.
2.2 Mental state For the comparison of thioridazine versus placebo, few data exist to support its effect on mental state. It is a sign of changing times that this drug could, for so long, be a widely used antipsychotic and favoured for elderly people, on the back of such limited trial data. The only statistically significant outcome was relapse. To prevent one person relapsing about four people need treating.
2.2 Leaving the study early Indirect data on global effect, or acceptability of the treatments may be seen in the attrition data. Significantly fewer people allocated to thioridazine left studies early (NNT 6). The reasons for this, hopefully but not necessarily, were that they were better, or at least encouraged by improvement. When specific reasons are cited for leaving early the data are not very helpful, although fewer people given thioridazine leave due to relapse. All studies suffered considerably fewer losses than more 'sophisticated' studies currently prevalent (Thornley 1998). This could be for a variety of reasons including selection of participants, drug regimens used, outcomes measured and general conduct of the studies. Whatever the reason, it would seem prudent for trialists to investigate these old studies in order to improve the wasteful loss of data in current randomised trials.
2.3 Adverse events Despite comprehensive lists of adverse effects, few differences between thioridazine and placebo were apparent. Trial data support the clinical impression that thioridazine is not strongly anticholinergic. Thioridazine appears to be sedating during short‐term (NNH 4) and medium‐term (NNH 6) assessment. No clear differences emerged for cardiovascular adverse effects with all but one outcome being non‐significant; the four‐week assessed outcome of significance, 'faintness, dizziness, weakness' became equivocal at the longer six months evaluation. All outcomes categorised as central nervous system adverse events were equivocal. We found no significant data to suggest that thioridazine causes breast swelling or lactation. Data relating to movement disorders do not fall into a clear pattern. Only tremor (NNH 13) and use of antiparkinsonian drugs (NNH 11) were higher in the thioridazine group, but the same outcomes were equivocal when assessed over a longer period of time. Similarly, data for gastrointestinal adverse effects did not reveal a clear pattern to indicate that thioridazine causes such problems, although constipation (NNH 10), and vomiting (NNH 5) may be increased by thioridazine, but without more robust data uncertainties remain. Genitourinary disturbances may also be an adverse effect of thioridazine (NNH 18), but we are unable to specify the type of disturbance, so such data are of limited use. We found no significant data on haematological abnormalities from two small studies. Other adverse events data were inconclusive. We found no data reporting on retinal changes. This is not entirely surprising, as trials, especially small, short‐term trials, are poor at detecting rare, important adverse effects. Also, most trials were not using the high doses associated with retinopathy (Rennie 1993).
3. THIORIDAZINE versus TYPICAL ANTIPSYCHOTICS
3.1 Death There was just one death for a total of about 800 person‐years (crude mortality rate: one death per 423 person‐years). Gui‐Yun 1988 reported one death in the thioridazine group from an unspecified physical illness. The lifetime incidence of suicide for people suffering from schizophrenia is 10‐13% (Caldwell 1992). The use of high doses of antipsychotic drugs has been associated with sudden death (Jusic 1994). Seven studies did employ higher doses than in modern practice but no sudden or cardiac deaths were reported in the included studies. This was with about 566 person/years exposure to thioridazine. This might have been because of careful screening for physical illness, because the level of monitoring is high in clinical trials and because polypharmacy is prevented by the study protocol. Alternatively, this meta‐analysis may not have had sufficient power to detect what might be a rare event. Thioridazine has also been reported to be associated with sudden death at normal doses (Mehtonen 1991).
3.2 Global state Analyses of various measures of global state consistently failed to find clear differences between thioridazine and other typical antipsychotics such as the 'benchmark' drug chlorpromazine (Thornley 2003). This was also the case when studies using LOCF were excluded.
3.3 Mental state Generally thioridazine had a similar efficacy to other typical antipsychotic drugs for various measures of mental state, 'relapse', 'no change or worse', 'moderately or severely ill', 'depression'. Only BPRS endpoint scores measured at 6 weeks were significant in favour of thioridazine when compared with chlorpromazine, but with all other outcomes being equivocal, more data are needed to have confidence in this single finding.
3.4 Behaviour Only one study (Mena 1966) measures changes in behaviour using the NOSIE scale, but data were equivocal between thioridazine and mesoridazine.
3.5 Leaving the study early Sixteen percent left the thioridazine groups, and also 16% in the control group by three months, which is low for clinical trials of people with schizophrenia, but thioridazine did not confer any advantage over other typical antipsychotics. Medium and longer‐term evaluation also revealed no significant differences. Specific reasons for leaving the study early did not reveal any significant differences, except for 'due to adverse events' which favoured typical antipsychotics (NNT 26), however, two studies providing data up to one year indicated no significant differences.
3.6 Adverse events We found no difference, on intention to treat analysis, between the overall tolerability of thioridazine and other drugs as measured indirectly by leaving the study due to adverse events.
A great number of adverse effects were listed in the included studies but few showed clear differences between thioridazine and other typical antipsychotics. Thioridazine caused dry mouth (NNH 12) by three months assessment (n=829), but this outcome did not remain statistically significant in the three trials (n=148) collecting data for one year. We found no convincing data to suggest that thioridazine is any more or less anticholinergic that other typical antipsychotics. Although about half of all participants given thioridazine felt either drowsy or sedated, the control group also experienced similar levels of this adverse event. Cardiovascular adverse events were mostly non‐significant, but the outcome 'any cardiovascular adverse event' was higher in the thioridazine group (NNH 3), but this non‐descriptive event is not informative to clinicians. Electrocardiogram changes were more frequent in the thioridazine (NNH 4) group, confirming its recognised potential to affect heart rhythm (Psychotropics 2006), although we do not know whether these changes were torsades de pointe or other changes. Hypotension affected about 40% of those given thioridazine compared with about 30% of the other typical antipsychotics, and was not significantly different (n=106). Larger scale studies are required to determine if thioridazine causes hypotension more frequently and severely than other typical antipsychotics. Fainting occurred more often in the thioridazine group (NNH 22, n=519) during short‐term assessment, whereas data from Schiele 1961 were equivocal at four months, but is based on a sample of just 60 participants.
One case of pigmented retinopathy on thioridazine was reported (Chen 1995). This was with about 566 person/years exposure to thioridazine. This implies that it is a rare adverse event or that it may have been underreported. Pigmented retinopathy is associated with doses above 800 mg which were only permitted in seven of the reviewed studies. Also, many studies were short‐term and pigmented retinopathy is associated with prolonged use (see Background). Endocrine adverse effects were infrequent and non‐significant between thioridazine and other typical antipsychotics. Thioridazine is less likely to cause extrapyramidal adverse effects, but data are heterogeneous. This is largely a function of McCreadie 1988, but the reasons why this study introduces heterogeneity are unclear. If data from this study are excluded the effect is even more in favour of thioridazine. However, medium‐term extrapyramidal adverse events (up to one year) (Schiele 1961, Stabenau 1964) were not significantly different. Parkinsonism (NNH 9) and rigidity (NNH 6) affected the control group more than the thioridazine group, but larger trials are needed to add weight to limited data. Other measures (akinesia, dyskinesia, dystonia, oculogyric crisis, and tremor) did not reveal any great differences. Gastrointestinal adverse events were mostly equivocal. Nausea did occur more in the thioridazine group (NNH 7) at short‐term, but one small six month study did not substantiate this initial finding. Vomiting was also higher in the thioridazine group but more trial data are needed to have confidence that a real difference exists between thioridazine and other typicals. Reports of weight loss and gain were equivocal, but detecting differences from such small studies (n=30) was unlikely. Genitourinary and laboratory tests did not reveal any significant differences. Most other adverse events were equivocal, except for photosensitivity which affected the other typical group more (NNH 7), but this finding was not sustained during one year follow up (Stabenau 1964, n=52).
4. THIORIDAZINE versus ATYPICAL ANTIPSYCHOTICS
4.1 Death One control group death was due to suicide (Keks 1994) giving a lifetime rate of about 14% as would be expected for this group.
4.2 Global state Both global state assessment 'not improved or worse' and continuous CGI scale data did not reveal any significant differences between thioridazine and atypical antipsychotics at short‐terms assessments. Larger studies of longer duration are needed to determine if the atypical antipsychotics are more beneficial than thioridazine.
4.3 Mental state Data were limited and mostly equivocal for the assessment of mental state. Only 'use of benzodiazepines' resulted in a significant difference favouring thioridazine (NNT 3) in comparison with the withdrawn drug remoxipride.
4.4 Leaving the study early Study attrition for any reason and specific reasons were all non‐significant, and as a proxy measure for treatment acceptability thioridazine was no more or less acceptable than atypical antipsychotics.
4.5 Adverse events Adverse events data all came from small scale studies and most came from (Phanjoo 1990) (n=18). All adverse event categories, anticholinergic, arousal, cardiovascular, central nervous system, movement disorders, gastrointestinal adverse events, and hepatic abnormalities were equivocal. We were unlikely to find statistically significant data from such small data sets. Without adequate sample sizes detecting differences between thioridazine and typical antipsychotics is unlikely unless large treatment effects are present.
Authors' conclusions
1. For clinicians Although there are shortcomings and gaps in the data, there appears to be enough overall consistency over different outcomes and time scales to confirm that thioridazine is an antipsychotic of similar efficacy to other commonly used neuroleptics, such as chlorpromazine, for people with schizophrenia. The adverse event profile of thioridazine seemed similar to that of typical drugs overall, but it may have a lower overall level of extrapyramidal adverse events. Considering that this adverse effect is the only clinical feature that makes the atypicals (except clozapine) really different from the typicals, it is not surprising that thioridazine has been suggested as having an 'atypical' profile. Electrocardiogram changes were significantly higher in the thioridazine group, which fits with the guidelines of monitoring patients for cardiovascular abnormalities. Thioridazine has been widely used in the elderly (King 1995), but this clinical preference does not appear to be based on good quality, trial based evidence for elderly people with schizophrenia. This is of concern to clinicians as there are many reasons why the elderly might be vulnerable to thioridazine's adverse events. In view of the lack of evidence, possible benefits versus harm of prescribing thioridazine must be carefully looked at for these patients, and alternative treatments considered. Clinicians in the UK will not be faced with these clinical decisions after the voluntary withdrawal of thioridazine from the market by Novartis, and the MHRA decision to stop licensing the drug in the UK. Thioridazine may still be obtained under generic label, and clinicians in some countries, where these restrictions do not apply will need to consider the advantages and disadvantages of prescribing thioridazine carefully.
2. For people with schizophrenia Thioridazine is probably as effective as other commonly used antipsychotic treatments for schizophrenia and it may have a lower level of extrapyramidal adverse events. It may therefore be a matter of personal preference as to which treatment is best, although since the recent withdrawal of thioridazine in the UK, this consideration is unlikely to apply to most people with schizophrenia, especially those in Europe and North America. In the elderly, there is no strong evidence that it is an effective treatment or that it is preferable to other antipsychotics, and may present an unacceptable risk considering the concerns of cardiotoxicity. However, with the withdrawal of thioridazine people in the older age groups may wish to ask what other treatments are available, which are better supported by evidence.
3. For managers, funders, decision makers Following the recent withdrawal of thioridazine in 2005, managers may consider whether other antipsychotics are able to provide a low risk of extrapyramidal adverse events for elderly patients.
1. General The trials reviewed predated the CONSORT statement (Begg 1996). Had this been anticipated much more data would have been available to inform practice. Allocation concealment gives the assurance that selection bias is kept to the minimum and should be properly described. Only seven studies in this review reported independent allocation or allocation concealment. For the other studies readers were given little assurance that bias was minimised during the allocation procedure. Well reported and tested blinding could have encouraged confidence in the control of performance and detection bias. Twenty of the reviewed trials described precautions to make the investigation blind but only two studies (Mena 1966, Cohler 1966) tested the quality of blinding using a questionnaire. It is also important to know how many, and from which groups, people were withdrawn in order to evaluate exclusion bias. Raters should be independent of treatment. This was the case in only ten of the reviewed studies (see Included studies table). Continuous data were poorly reported in the reviewed studies. It would have been helpful if authors had presented data in a way which reflects associations between intervention and outcome, for example, relative risk, odds‐ratio, risk or mean differences, as well as raw numbers. Binary outcomes should be calculated in preference to continuous results, as they are easier to interpret. Trials should report service utilisation data as well as satisfaction with care and economic outcomes.
2. Specific Thioridazine's efficacy seems comparable to that of other typical antipsychotics but it has not been tested adequately against placebo ‐ probably because licensing requirements were less stringent at the time thioridazine was developed. There were only limited data for the comparison with atypical neuroleptic so the claim that thioridazine is an 'atypical' is largely untested by a direct comparison. Should thioridazine be used as the control group for trials of atypical drugs, it is entirely feasible that fewer differences would be seen, especially for extrapyramidal effects, than is currently the case with the preferred comparator, haloperidol (Thornley 1998). There is a lack of trial based, good quality evidence to guide the use of thioridazine in the elderly. However, further research is now unlikely, and probably not justified.
Acknowledgements
Clive Adams (Cochrane Schizophrenia Group, UK) gave invaluable help, advice and encouragement.
Grateful thanks to Dr Luk Wai Ho (Lecturer in Psychiatry, Addenbrooke's NHS trust, Cambridge, UK) who helped with data extraction and assessment of three Chinese studies.
Dr Min Jiang (Department of Physiology, University of Turku, Finland), for helping to data extract and assess three Chinese studies.
Dr Kristian Wahlbeck (lecturer, editor, University of Helsinki, Cochrane Schizophrenia Group, Finland) very kindly provided data and assessment of one Chinese study.
Jane Dilworth (Novartis Pharma) kindly replied to inquiries about unpublished data.
Elaine Sultana (spouse) helped with correspondence and was always tolerant and supportive during the hours of computer work.
Thanks to Jun Xia (Cochrane Schizophrenia Group, UK) for data extracting Chinese language papers and Tessa Grant for her editorial support.
Data and analyses
Comparison 1.
THIORIDAZINE versus PLACEBO
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. No change or worse (LOCF) | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 by 3 months | 3 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.44, 0.98] |
| 1.2 > 3 months ‐ 1 year | 3 | 105 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.21, 0.48] |
| 2 Global state: 2. Moderate or severely ill (CGI >=4, LOCF) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 by 28 days | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.41, 1.49] |
| 2.2 by 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.11, 1.03] |
| 3 Global state: 3. Average endpoint change score by 6 months (CGI, high=poor, LOCF) | 1 | 23 | Mean Difference (IV, Fixed, 95% CI) | ‐0.99 [‐1.79, ‐0.19] |
| 4 Global state: 4. Average endpoint change score by 4 weeks (GAS, low=poor, LOCF) | 1 | 50 | Mean Difference (IV, Fixed, 95% CI) | 14.26 [3.38, 25.14] |
| 5 Mental state: 1. Relapse | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 by 3 months | 2 | 261 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.09 [0.03, 0.27] |
| 5.2 by 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.11, 1.03] |
| 6 Mental state: 2. No improved or worse (LOCF) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 by 6 weeks | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.51, 1.30] |
| 6.2 by 7 months | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Mental state: 3. Moderately or severely ill by 4 weeks (LOCF) | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.41, 1.49] |
| 8 Mental state: 4. Average endpoint chage score by 4 weeks (BPRS, high=poor, skewed data) | Other data | No numeric data | ||
| 9 Mental state: 5. Depression | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 depression ‐ 3 months | 2 | 88 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.22, 4.21] |
| 9.2 depression ‐ >3 months to 1 year | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.68 [0.75, 9.63] |
| 10 Leaving the study early: 1a. Any reason | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 by 3 months | 9 | 510 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.30, 0.60] |
| 10.2 by 3 months to 1 year | 4 | 115 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.32, 1.40] |
| 11 Leaving the study early: 1b. Due to adverse events ‐ by 3 months | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 any adverse events | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.30, 7.11] |
| 11.2 dystonia ‐ severe | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.3 hypotension | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.38 [0.14, 82.01] |
| 11.4 jaundice | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.5 parkinsonism ‐ severe | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.02, 9.11] |
| 11.6 seizure | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.7 skin reaction, facial oedema ‐ severe | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12 Leaving the study early: 1c. Due to refusal of treatment | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 by 3 months | 2 | 79 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.30, 4.52] |
| 13 Leaving the study early: 1d. Due to relapse / worsening or no improvement | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 by 3 months | 6 | 396 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.05, 0.24] |
| 13.2 by 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.06, 1.12] |
| 14 Adverse events: 1. Anticholinergic | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 blurred vision ‐ 3 months | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [0.39, 9.35] |
| 14.2 blurred vision ‐ 6 months | 2 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.17, 3.39] |
| 14.3 dry mouth ‐ 3 months | 3 | 324 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.75 [3.05, 14.94] |
| 14.4 dry mouth ‐ >3 months to 1 year | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.62 [0.54, 4.88] |
| 14.5 nasal congestion ‐ upto 6 weeks | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.42 [1.42, 8.25] |
| 14.6 nasal congestion ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.05, 4.94] |
| 15 Adverse events: 2. Arousal | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 drowsiness ‐ 3 months | 3 | 324 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.37 [3.18, 9.08] |
| 15.2 drowsiness ‐ >3 months to 1 year | 4 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.41 [1.28, 4.52] |
| 15.3 excitement ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.03, 3.20] |
| 15.4 excitement ‐ >3 months to one year | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.76 [0.89, 51.46] |
| 15.5 insomnia ‐ 3 months | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.43, 5.84] |
| 15.6 insomnia ‐ >3 months to 1 year | 3 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.47, 1.55] |
| 16 Adverse events: 3. Cardiovascular | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 chest pain ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.14, 12.82] |
| 16.2 faintness, dizziness, weakness ‐ 4 weeks | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.30 [1.05, 17.61] |
| 16.3 faintness, dizziness, weakness ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.17, 2.67] |
| 16.4 hypotension ‐ 4 weeks | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [0.39, 9.35] |
| 16.5 tachycardia ‐ 3 months | 2 | 88 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.18, 1.32] |
| 17 Adverse events: 4. Central nervous system ‐ other | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 confusion ‐ >3 months to one year | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.90 [0.32, 26.21] |
| 17.2 headache ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.44, 2.90] |
| 17.3 headache ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.25, 3.15] |
| 17.4 memory defects ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.14, 12.82] |
| 17.5 seizure ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.07, 17.79] |
| 17.6 syncope ‐ 3 months | 3 | 324 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.08 [0.90, 10.48] |
| 17.7 syncope ‐ 4 months | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 14.90] |
| 18 Adverse events: 5. Endocrine | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 breast swelling ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 18.2 lactation ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 18.3 lactation ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.14, 12.82] |
| 19 Adverse events: 6. Movement disorders | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 akathisia +/‐ restlessness ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.96, 2.12] |
| 19.2 akathisia ‐ >3 months to 1 year | 3 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.30, 2.35] |
| 19.3 akinesia ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.20, 3.09] |
| 19.4 dystonia ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.38 [0.14, 82.01] |
| 19.5 dystonia ‐ >3 months to 1 year | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.35, 6.01] |
| 19.6 oculogyric crisis ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 19.7 parkinsonism ‐ 3 months | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 19.8 parkinsonism ‐ >3 months to 1 year | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.4 [0.59, 9.84] |
| 19.9 rigidity ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.55, 2.94] |
| 19.10 rigidity ‐ >3 months to 1 year | 3 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.88 [0.66, 5.32] |
| 19.11 tremor ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.03 [1.24, 7.39] |
| 19.12 tremor ‐ >3 months to 1 year | 3 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.49, 3.23] |
| 19.13 use of antiparkinsonian drugs ‐ 3 months | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.53 [1.15, 5.60] |
| 19.14 use of antiparkinsonian drugs ‐>3 months to 1 year | 2 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.36 [0.49, 3.82] |
| 20 Adverse events: 7. Gastrointestinal | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 20.1 constipation ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [1.27, 4.83] |
| 20.2 constipation ‐ >3 months to 1 year | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [0.29, 11.06] |
| 20.3 diarrhoea ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.38 [0.14, 82.01] |
| 20.4 diarrhoea ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.28] |
| 20.5 nausea ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 12.01 [3.78, 38.15] |
| 20.6 nausea ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.07, 1.49] |
| 20.7 vomiting ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 25.88 [1.54, 434.08] |
| 20.8 weight loss ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.28] |
| 20.9 weight gain ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.24, 16.61] |
| 21 Adverse events: 8. Genitourinary | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 21.1 urinary disturbance ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.82 [1.12, 12.97] |
| 22 Adverse events: 9. Haematology ‐ abnormal laboratory results ‐ >3 months to 1 year | 2 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.37, 1.71] |
| 23 Adverse events: 10. Other | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 23.1 infections ‐ 6 weeks | 1 | 236 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.25 [0.42, 12.06] |
| 23.2 liver function abnormality (laboratory test) ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.0 [0.56, 28.40] |
| 23.3 oedema ‐ facial ‐ 6 weeks | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 23.4 oedema ‐ peripheral ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [0.41, 7.83] |
| 23.5 pyrexia ‐ 6 weeks | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.02, 0.99] |
| 23.6 salivation increased ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.64 [0.66, 10.58] |
| 23.7 salivation increased ‐ >3 months to 1 year | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 23.8 sweating ‐ >3 months to 1 year | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.9 [0.12, 68.33] |
| 23.9 weakness ‐ >3 months to 1 year | 2 | 97 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.88 [1.11, 21.35] |
| 23.10 photosensitivity ‐ 3 months | 2 | 88 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.27, 7.73] |
| 23.11 rash ‐ 3 months | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.26, 3.90] |
| 23.12 rash ‐ 6 months | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.09, 2.20] |
Analysis 1.1.
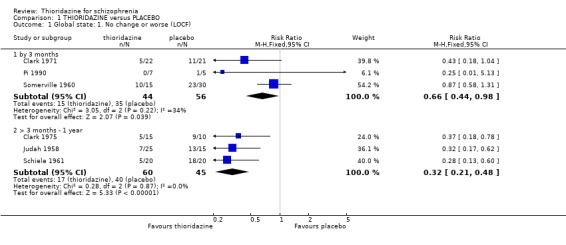
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 1 Global state: 1. No change or worse (LOCF).
Analysis 1.2.
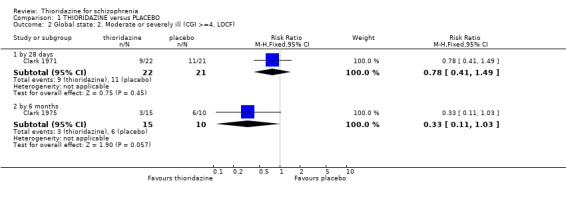
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 2 Global state: 2. Moderate or severely ill (CGI >=4, LOCF).
Analysis 1.3.
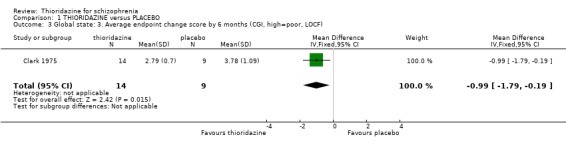
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 3 Global state: 3. Average endpoint change score by 6 months (CGI, high=poor, LOCF).
Analysis 1.4.
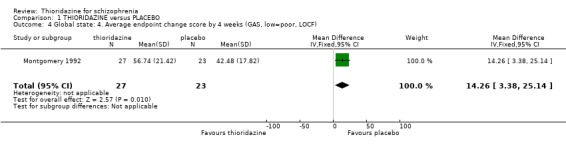
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 4 Global state: 4. Average endpoint change score by 4 weeks (GAS, low=poor, LOCF).
Analysis 1.5.
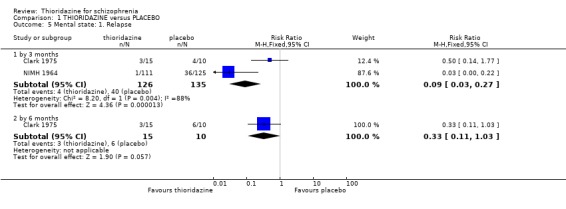
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 5 Mental state: 1. Relapse.
Analysis 1.6.
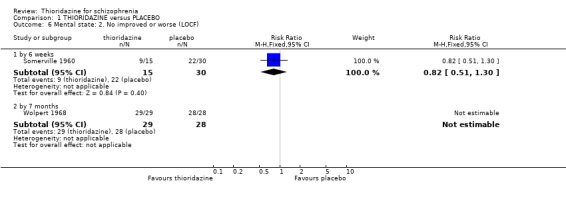
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 6 Mental state: 2. No improved or worse (LOCF).
Analysis 1.7.
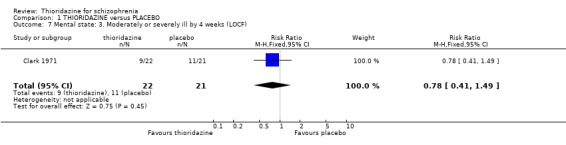
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 7 Mental state: 3. Moderately or severely ill by 4 weeks (LOCF).
Analysis 1.8.
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 8 Mental state: 4. Average endpoint chage score by 4 weeks (BPRS, high=poor, skewed data).
Mental state: 4. Average endpoint chage score by 4 weeks (BPRS, high=poor, skewed data)
| Study | Intervention | Mean | SD | N |
|---|---|---|---|---|
| Montgomery 1992 | Thioridazine | 16.77 | 11.33 | 27 |
| Montgomery 1992 | Placebo | 25.39 | 15.18 | 23 |
Analysis 1.9.
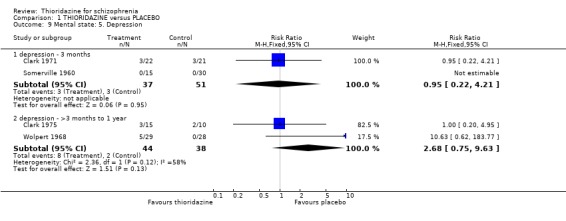
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 9 Mental state: 5. Depression.
Analysis 1.10.
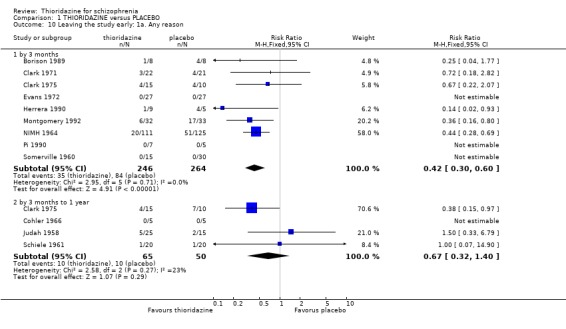
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 10 Leaving the study early: 1a. Any reason.
Analysis 1.11.
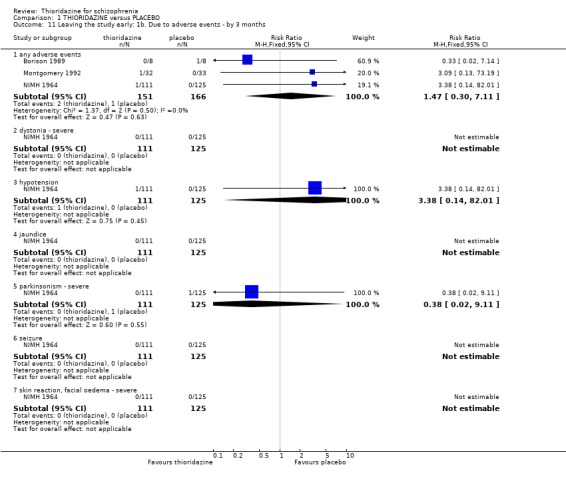
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 11 Leaving the study early: 1b. Due to adverse events ‐ by 3 months.
Analysis 1.12.
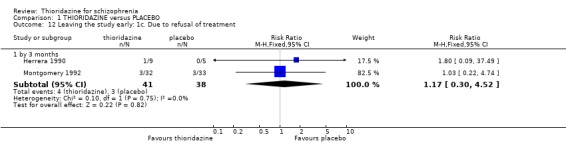
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 12 Leaving the study early: 1c. Due to refusal of treatment.
Analysis 1.13.
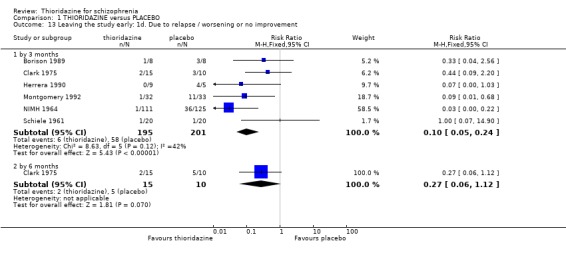
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 13 Leaving the study early: 1d. Due to relapse / worsening or no improvement.
Analysis 1.14.

Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 14 Adverse events: 1. Anticholinergic.
Analysis 1.15.
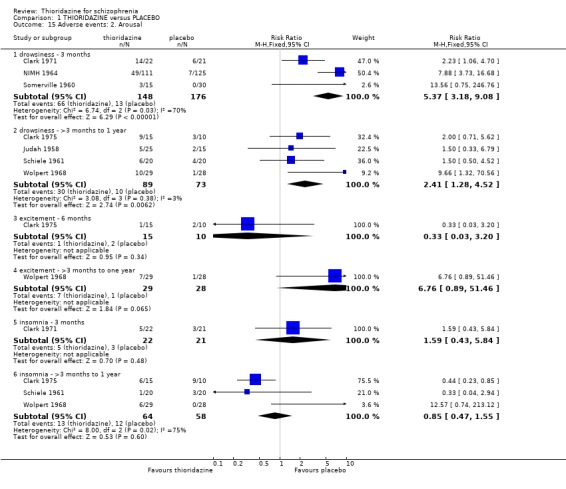
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 15 Adverse events: 2. Arousal.
Analysis 1.16.
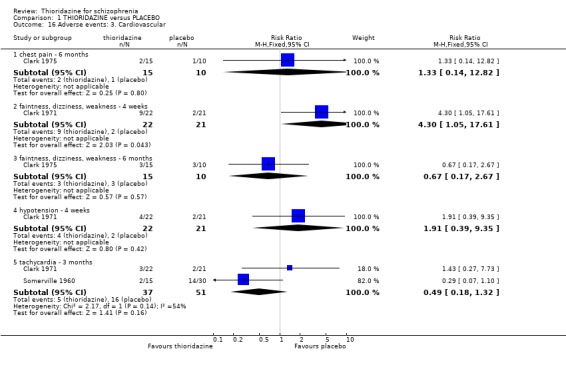
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 16 Adverse events: 3. Cardiovascular.
Analysis 1.17.
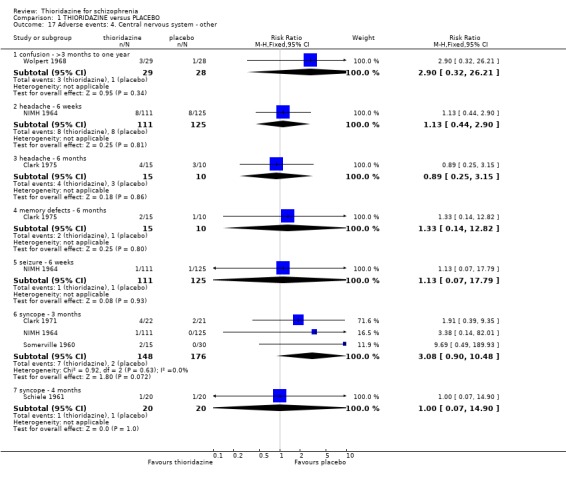
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 17 Adverse events: 4. Central nervous system ‐ other.
Analysis 1.18.
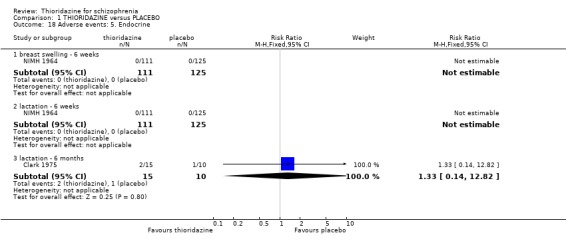
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 18 Adverse events: 5. Endocrine.
Analysis 1.19.
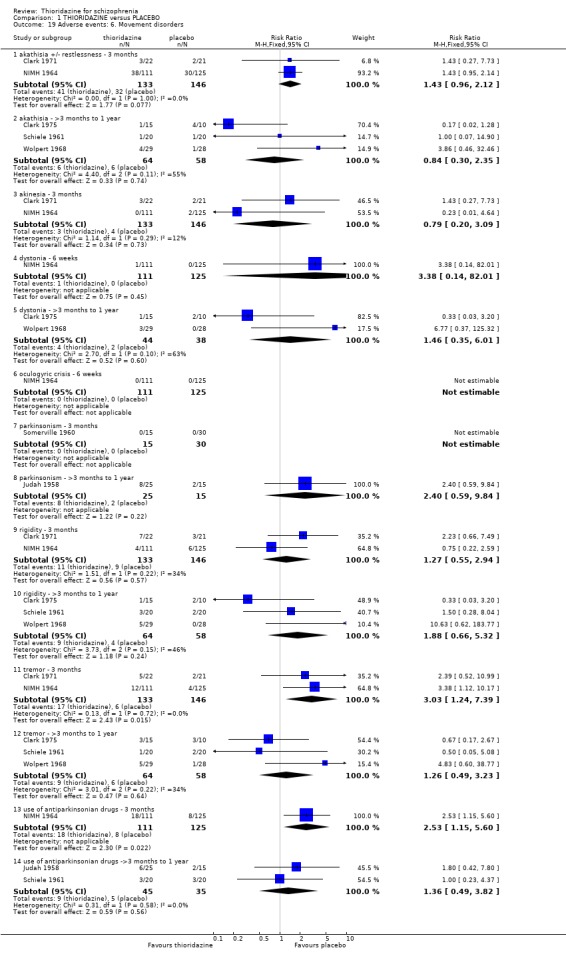
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 19 Adverse events: 6. Movement disorders.
Analysis 1.20.
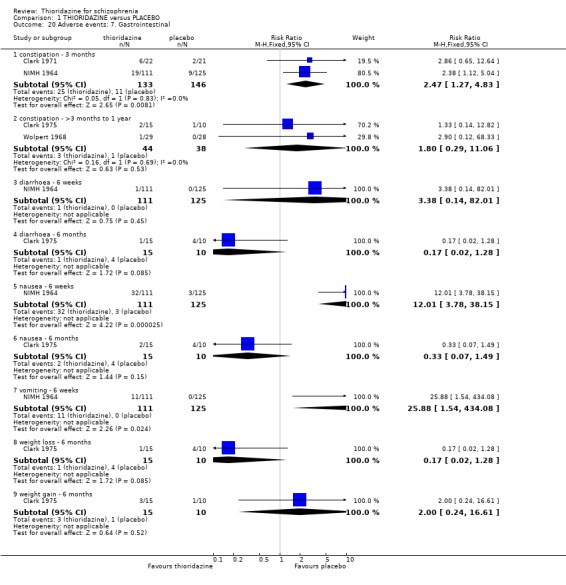
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 20 Adverse events: 7. Gastrointestinal.
Analysis 1.21.
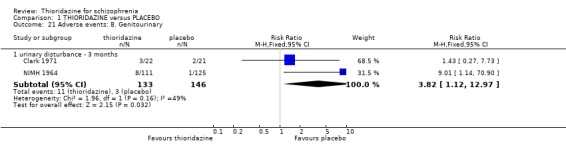
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 21 Adverse events: 8. Genitourinary.
Analysis 1.22.
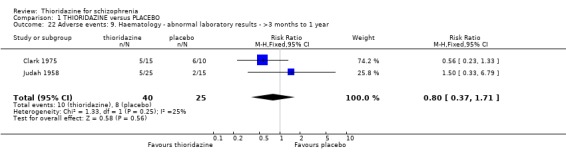
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 22 Adverse events: 9. Haematology ‐ abnormal laboratory results ‐ >3 months to 1 year.
Analysis 1.23.
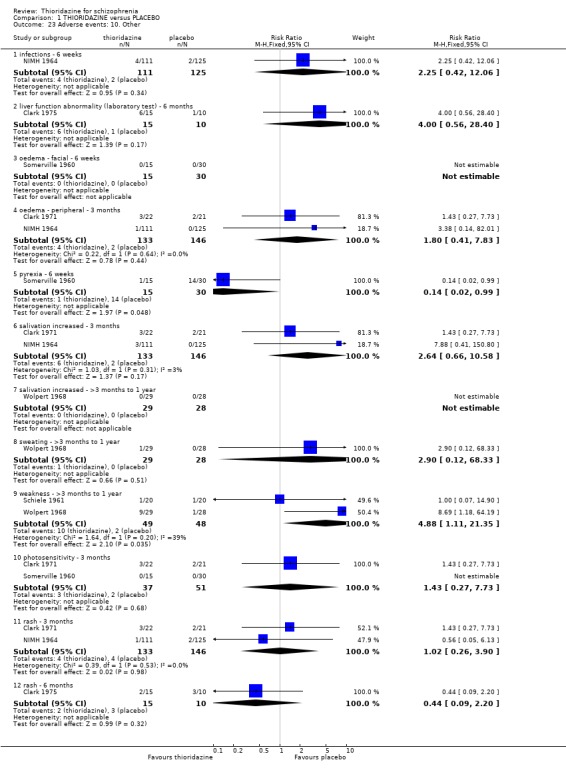
Comparison 1 THIORIDAZINE versus PLACEBO, Outcome 23 Adverse events: 10. Other.
Comparison 2.
THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 by 3 months (physical illness) | 1 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.34] |
| 2 Global state: 1. No change or worse (LOCF) | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 by 3 months | 11 | 743 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.83, 1.16] |
| 2.2 by 3 months to 1 year | 3 | 142 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.62, 1.59] |
| 3 Global state: 2. Moderately or severely ill (CGI >=4 (LOCF) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 by 28 days | 1 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.69, 2.66] |
| 3.2 by 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.14, 1.35] |
| 4 Global state: 3. Average endpoint change score by 6 months (CGI, high=poor, LOCF) | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | ‐0.21 [‐0.90, 0.48] |
| 5 Mental state: 4. Relapse | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 by 3 months | 2 | 368 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.18, 1.70] |
| 5.2 by 3 months to 1 year | 2 | 76 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.73, 1.58] |
| 6 Mental state: 5. No change or worse (LOCF) | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 by 3 months | 5 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.97, 1.65] |
| 6.2 by 7 months | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Mental state: 8. Average endpoint score at 6 weeks (BPRS, high=poor, LOCF) | 1 | 234 | Mean Difference (IV, Fixed, 95% CI) | ‐2.04 [‐3.92, ‐0.16] |
| 8 Mental state: 7. Moderately or severely ill by 3 months (LOCF) | 2 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.75, 2.44] |
| 9 Mental state: 10. Average endpoint score by 8 weeks (SAPS total, high score=poor, skewed data) | Other data | No numeric data | ||
| 10 Mental state: 11. Depression (clinical diagnosis) | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 depression ‐ 3 months | 2 | 95 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.28, 3.03] |
| 10.2 depression ‐ >3 months to 1 year | 2 | 94 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.46, 2.68] |
| 11 Behaviour: 1. Not improved or worse by 5 weeks (NOSIE, LOCF) | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.33 [0.70, 7.76] |
| 12 Leaving the study early: 1a. Any reason | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 by 3 months | 19 | 1587 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.85, 1.34] |
| 12.2 by 3 months to 1 year | 5 | 612 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.85, 1.15] |
| 12.3 by 1 to 4 years | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.29, 7.73] |
| 13 Leaving the study early: 1b. Due to absence without leave or refusing to continue | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 by 3 months | 1 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.08, 1.87] |
| 13.2 by 6 months | 1 | 424 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.07, 1.11] |
| 14 Leaving the study early: 1c. Due to adverse events | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 by 3 months | 8 | 871 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.24 [1.19, 4.22] |
| 14.2 by > 3 months to 1 year | 2 | 470 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.52, 2.54] |
| 15 Leaving the study early: 1d. Due to refusal of medication/poor compliance | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 by > 3 months to 1 year | 2 | 470 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.15, 1.25] |
| 16 Leaving the study early: 1e. Due to relapse, worsening or no improvement | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 by 3 months | 5 | 507 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.31, 1.35] |
| 16.2 by > 3 months to 1 year | 4 | 560 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.54, 1.24] |
| 17 Adverse events: 1. Anticholinergic | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 blurred vision ‐ 3 months | 4 | 482 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.59, 1.61] |
| 17.2 blurred vision ‐ 6 months | 2 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.18, 2.49] |
| 17.3 dry mouth ‐ 3 months | 5 | 829 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [1.16, 1.87] |
| 17.4 dry mouth ‐ >3 months to 1 year | 3 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.60, 2.06] |
| 17.5 nasal congestion ‐ 3 months | 2 | 403 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.82, 2.47] |
| 17.6 nasal congestion ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.04, 2.85] |
| 17.7 urinary retention ‐ 12 weeks | 1 | 86 | Risk Ratio (M‐H, Fixed, 95% CI) | 12.42 [0.72, 213.88] |
| 18 Adverse events: 2. Arousal | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 drowsiness / sedation ‐ 3 months | 8 | 891 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.93, 1.30] |
| 18.2 drowsiness / sedation ‐ >3 months to 1 year | 3 | 154 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.91, 2.36] |
| 18.4 excitement ‐ >3 months to one year | 4 | 211 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.68, 1.45] |
| 18.5 insomnia ‐ 3 months | 1 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.63 [0.56, 4.75] |
| 18.6 insomnia ‐ >3 months to 1 year | 3 | 154 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.51, 1.64] |
| 19 Adverse events: 3. Cardiovascular | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 any cardiovascular adverse event ‐ 3 months | 1 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.17 [1.43, 7.02] |
| 19.2 chest pain ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.13, 3.44] |
| 19.3 ECG changes ‐ 3 months | 2 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.38 [1.58, 3.59] |
| 19.4 faintness, dizziness, weakness ‐ 3 months | 4 | 482 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.96, 2.10] |
| 19.5 faintness, dizziness, weakness ‐ > 3 months to 1 year | 2 | 94 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.46, 1.61] |
| 19.6 hypotension ‐ 3 months | 3 | 106 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.69, 1.95] |
| 19.7 hypotension ‐ orthostatic ‐ >3 months to 1 year | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.71 [0.68, 4.32] |
| 19.8 tachycardia ‐ 3 months | 2 | 95 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.18, 1.01] |
| 20 Adverse events: 4. Central nervous system ‐ other | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 20.1 ataxia ‐ 3 months | 1 | 21 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.23, 7.74] |
| 20.2 confusion ‐ 3 months | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.02, 1.05] |
| 20.3 confusion ‐ >3 months to one year | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.13, 1.55] |
| 20.4 headache ‐ 3 months | 2 | 424 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.50, 1.85] |
| 20.5 headache ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.17, 1.13] |
| 20.6 memory defects ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.13, 3.44] |
| 20.7 seizure ‐ 3 months | 2 | 648 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.41, 3.22] |
| 20.8 syncope ‐ 3 months | 4 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.21 [1.32, 7.84] |
| 20.9 syncope ‐ 4 months | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.10, 10.38] |
| 20.10 retinopathy ‐ pigmented ‐ 6 weeks | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.80 [0.12, 68.12] |
| 20.11 lens deposits | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 20.12 corneal deposits | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.67, 1.76] |
| 20.13 conjunctival deposits | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.38 [0.28, 101.96] |
| 21 Adverse events: 5. Endocrine | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 21.1 breast swelling ‐ 6 weeks | 1 | 338 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.02, 5.58] |
| 21.2 lactation ‐ 6 weeks | 1 | 338 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.01, 2.76] |
| 21.3 lactation ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.13, 3.44] |
| 22 Adverse events: 6. Movement disorders | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 22.1 akathisia +/‐ restlessness ‐ 3 months | 5 | 819 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.70, 1.13] |
| 22.2 akathisia ‐ >3 months to 1 year | 3 | 154 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.18, 1.00] |
| 22.3 akinesia ‐ 3 months | 3 | 489 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.23, 1.06] |
| 22.4 dyskinesia ‐ acute | 1 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.26, 1.98] |
| 22.5 dystonia ‐ 3 months | 4 | 754 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.45, 1.85] |
| 22.6 dystonia ‐ >3 months to 1 year | 2 | 94 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.26, 2.98] |
| 22.7 extrapyramidal / use of antiparkinsonian drugs ‐ 3 months | 7 | 1082 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.36, 0.55] |
| 22.8 extrapyramidal / use of antiparkinsonian drugs ‐ > 3 months to 1 year | 2 | 112 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.40, 1.30] |
| 22.9 oculogyric crisis ‐ 3 months | 2 | 424 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.25 [0.67, 15.67] |
| 22.10 parkinsonism ‐ 3 months | 2 | 340 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.12, 0.70] |
| 22.11 rigidity ‐ 3 months | 4 | 509 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.36, 1.00] |
| 22.12 rigidity ‐ >3 months to 1 year | 3 | 154 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.22, 0.86] |
| 22.13 tremor ‐ 3 months | 4 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.92, 1.67] |
| 22.14 tremor ‐ >3 months to 1 year | 3 | 154 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.29, 1.19] |
| 23 Adverse events: 7. Gastrointestinal | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 23.1 constipation ‐ 3 months | 3 | 489 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.64, 1.38] |
| 23.2 constipation ‐ >3 months to 1 year | 2 | 94 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.17, 2.48] |
| 23.3 diarrhoea ‐ 3 months | 1 | 338 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.07, 6.48] |
| 23.4 diarrhoea ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.03, 1.98] |
| 23.5 nausea ‐ 6 weeks | 1 | 338 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.35 [1.48, 3.73] |
| 23.6 nausea ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.11, 2.33] |
| 23.8 vomiting ‐ 3 months | 3 | 734 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.82 [1.11, 2.99] |
| 23.9 weight loss ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.03, 1.51] |
| 23.10 weight gain ‐ 3 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.60, 1.66] |
| 23.11 weight gain ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.17, 2.07] |
| 24 Adverse events: 8. Genitourinary | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 24.1 urinary disturbance ‐ 3 months | 4 | 799 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.62 [0.93, 2.84] |
| 25 Adverse events: 9. Laboratory tests ‐ abnormal results | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 25.1 blood cells ‐ decrease in haematocrit, haemoglobin ‐ 3 months | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 7.32] |
| 25.2 blood cells ‐ leucopenia, WCC<5000 ‐ >3 months to 1 year | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.71 [0.68, 4.32] |
| 25.3 liver function tests abnormal ‐ 3 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.27, 2.41] |
| 25.4 liver function tests abnormal ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.53, 4.26] |
| 25.5 liver function tests ‐ cephaline phosphatase >2 ‐ 3 months to 1 year | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.71 [0.68, 4.32] |
| 25.6 liver function tests ‐ SGOT, SGPT elevated ‐ 3 months | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.08 [0.00, 1.21] |
| 25.7 renal function ‐ decreased calcium ‐ 3 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.16, 6.20] |
| 25.8 renal function ‐ elevated phosphate ‐ 3 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.27, 2.41] |
| 25.9 renal function ‐ abnormal urea / nitrogen ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.04, 2.85] |
| 26 Adverse events: 10. Other | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 26.1 allergic reactions ‐ 3 months | 1 | 86 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.73 [0.72, 45.59] |
| 26.2 infections ‐ 6 weeks | 1 | 338 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.05 [0.52, 8.03] |
| 26.3 oedema ‐ facial ‐ 6 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.90] |
| 26.4 oedema ‐ peripheral ‐ 3 months | 2 | 403 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.32, 3.07] |
| 26.5 pyrexia ‐ 6 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.03, 1.98] |
| 26.6 salivation increased ‐ 3 months | 3 | 489 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.46, 1.87] |
| 26.7 salivation increased ‐ >3 months to 1 year | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.01, 2.38] |
| 26.8 sweating ‐ >3 months to 1 year | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.06, 6.32] |
| 26.9 photosensitivity ‐ 3 months | 3 | 181 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.40, 0.92] |
| 26.10 photosensitivity ‐ > 3 months to 1 year | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.71 [0.68, 4.32] |
| 26.11 rash ‐ 3 months | 4 | 734 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.51, 1.96] |
| 26.12 rash ‐ 6 months | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.11, 2.33] |
| 26.13 weakness ‐ 4 months | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.06, 4.18] |
Analysis 2.1.
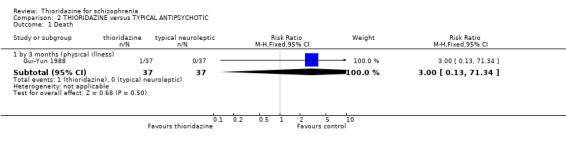
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 1 Death.
Analysis 2.2.
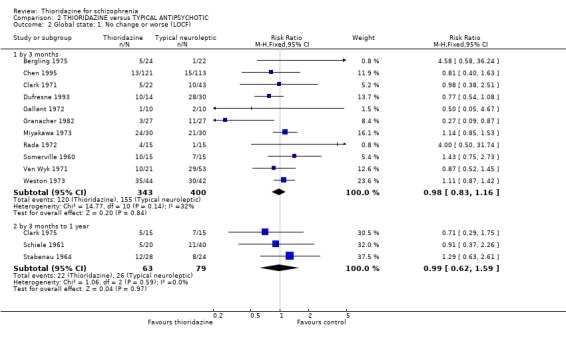
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 2 Global state: 1. No change or worse (LOCF).
Analysis 2.3.
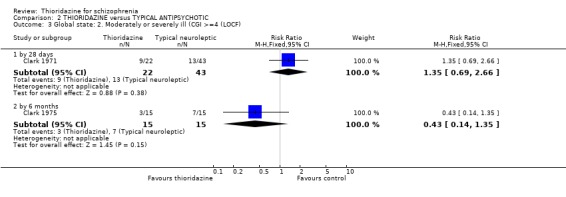
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 3 Global state: 2. Moderately or severely ill (CGI >=4 (LOCF).
Analysis 2.4.

Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 4 Global state: 3. Average endpoint change score by 6 months (CGI, high=poor, LOCF).
Analysis 2.5.
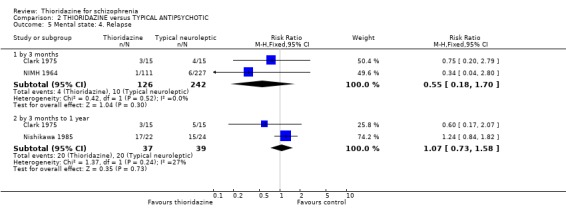
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 5 Mental state: 4. Relapse.
Analysis 2.6.
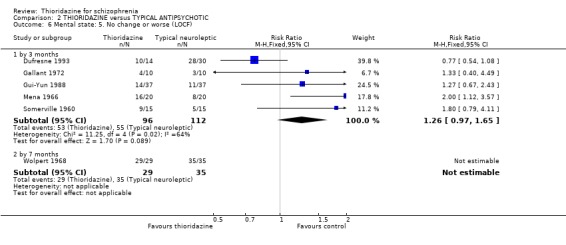
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 6 Mental state: 5. No change or worse (LOCF).
Analysis 2.7.
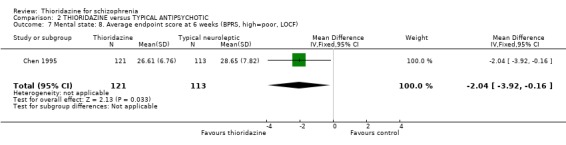
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 7 Mental state: 8. Average endpoint score at 6 weeks (BPRS, high=poor, LOCF).
Analysis 2.8.
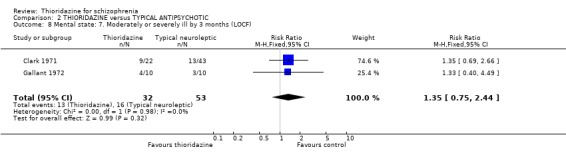
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 8 Mental state: 7. Moderately or severely ill by 3 months (LOCF).
Analysis 2.9.
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 9 Mental state: 10. Average endpoint score by 8 weeks (SAPS total, high score=poor, skewed data).
Mental state: 10. Average endpoint score by 8 weeks (SAPS total, high score=poor, skewed data)
| Study | Intervention | Mean | SD | N |
|---|---|---|---|---|
| Zhang 1999 | Thioridazine | 16. 72 | 3. 11 | 40 |
| Zhang 1999 | Clozapine | 18. 25 | 10. 21 | 30 |
Analysis 2.10.
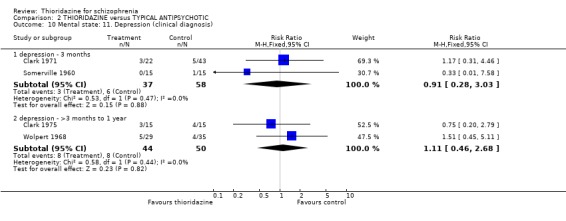
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 10 Mental state: 11. Depression (clinical diagnosis).
Analysis 2.11.
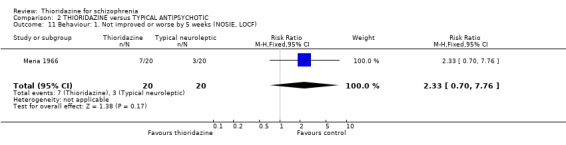
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 11 Behaviour: 1. Not improved or worse by 5 weeks (NOSIE, LOCF).
Analysis 2.12.
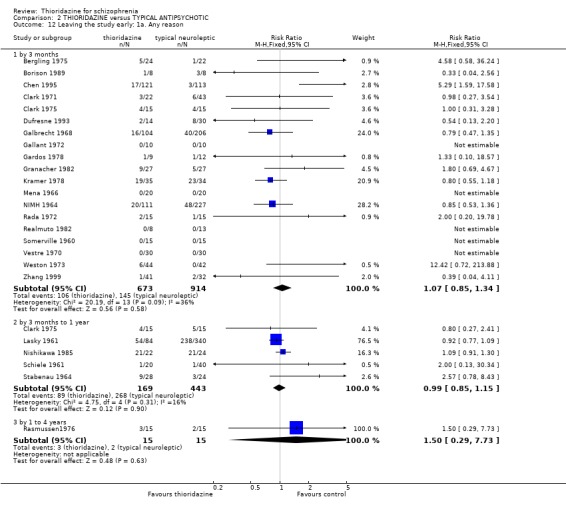
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 12 Leaving the study early: 1a. Any reason.
Analysis 2.13.
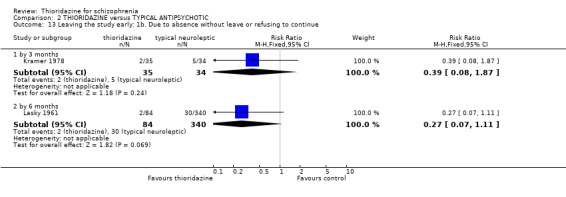
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 13 Leaving the study early: 1b. Due to absence without leave or refusing to continue.
Analysis 2.14.
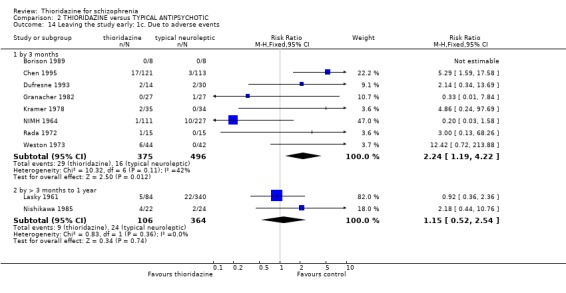
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 14 Leaving the study early: 1c. Due to adverse events.
Analysis 2.15.
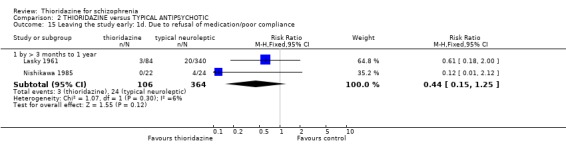
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 15 Leaving the study early: 1d. Due to refusal of medication/poor compliance.
Analysis 2.16.
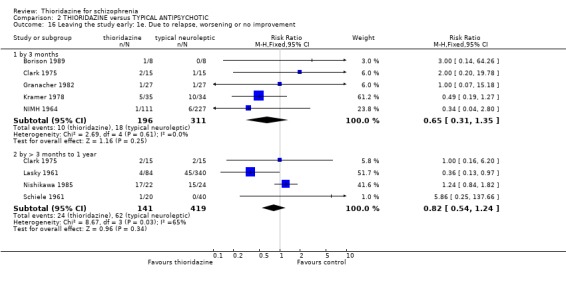
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 16 Leaving the study early: 1e. Due to relapse, worsening or no improvement.
Analysis 2.17.
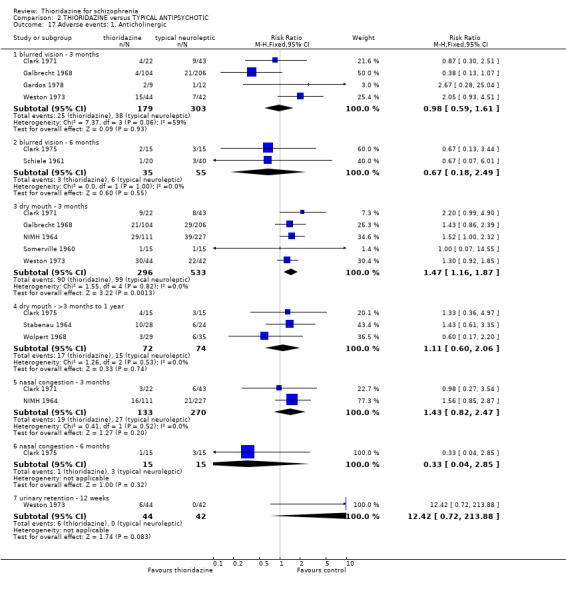
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 17 Adverse events: 1. Anticholinergic.
Analysis 2.18.
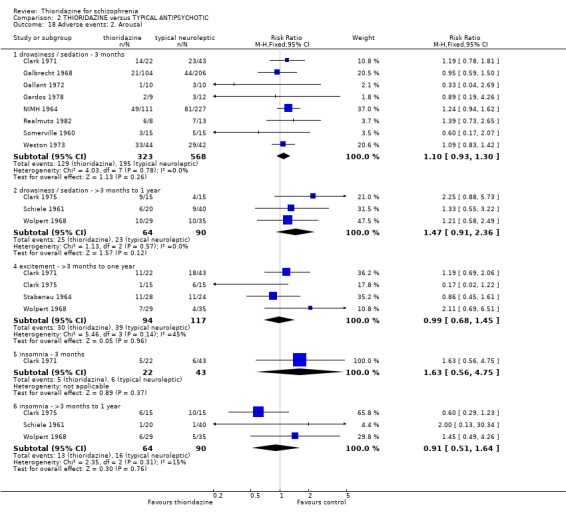
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 18 Adverse events: 2. Arousal.
Analysis 2.19.
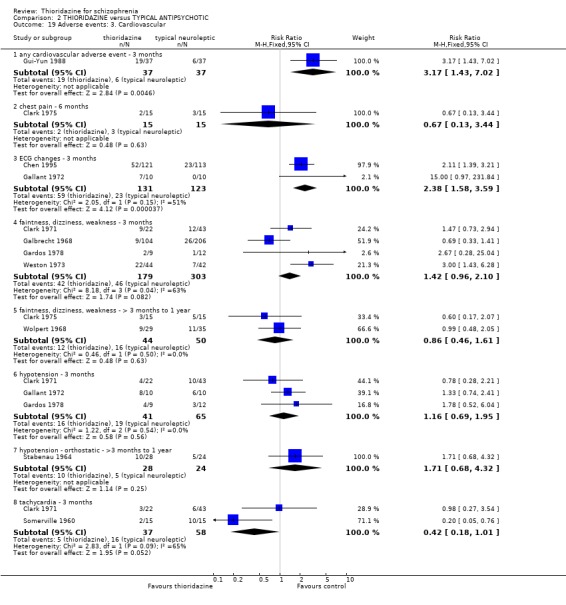
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 19 Adverse events: 3. Cardiovascular.
Analysis 2.20.
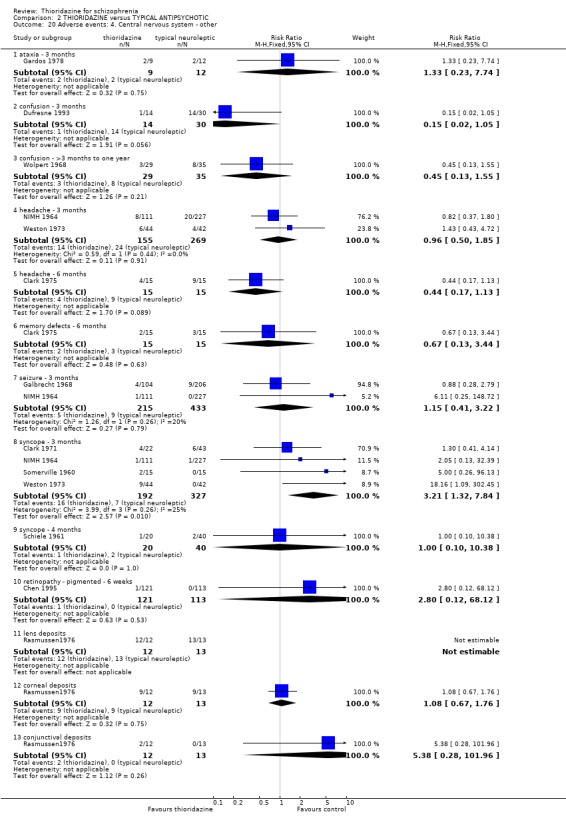
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 20 Adverse events: 4. Central nervous system ‐ other.
Analysis 2.21.
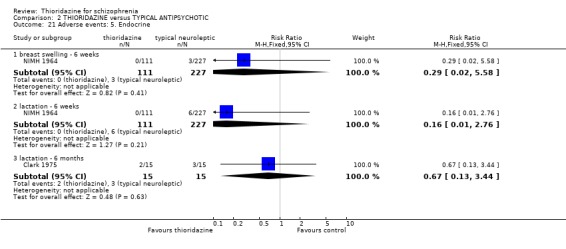
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 21 Adverse events: 5. Endocrine.
Analysis 2.22.
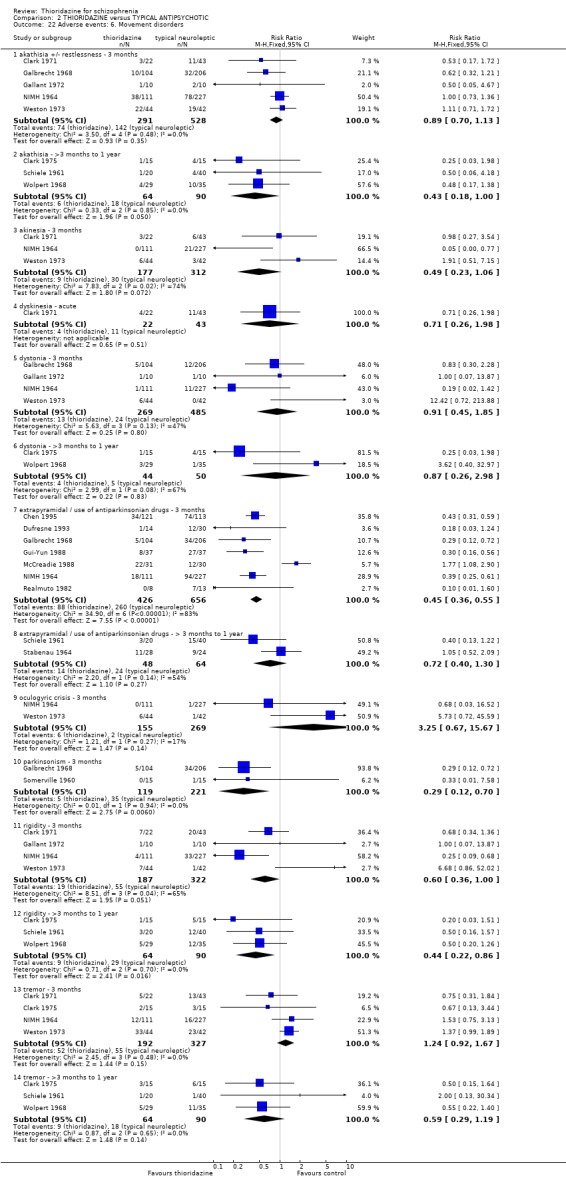
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 22 Adverse events: 6. Movement disorders.
Analysis 2.23.
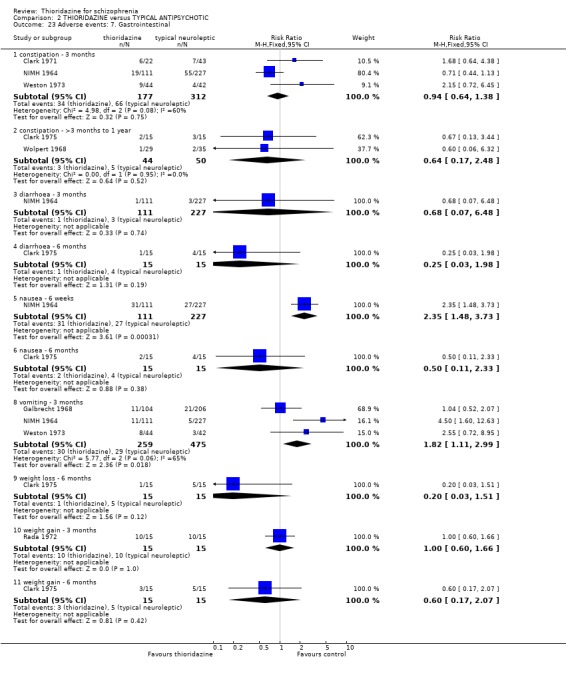
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 23 Adverse events: 7. Gastrointestinal.
Analysis 2.24.
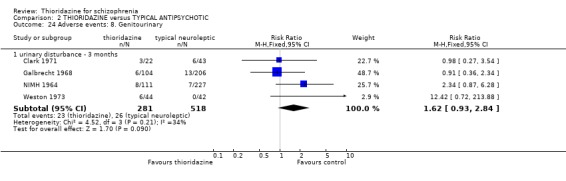
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 24 Adverse events: 8. Genitourinary.
Analysis 2.25.
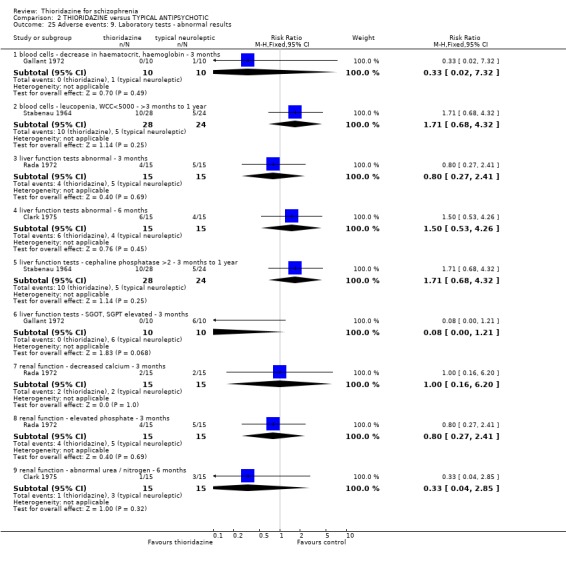
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 25 Adverse events: 9. Laboratory tests ‐ abnormal results.
Analysis 2.26.
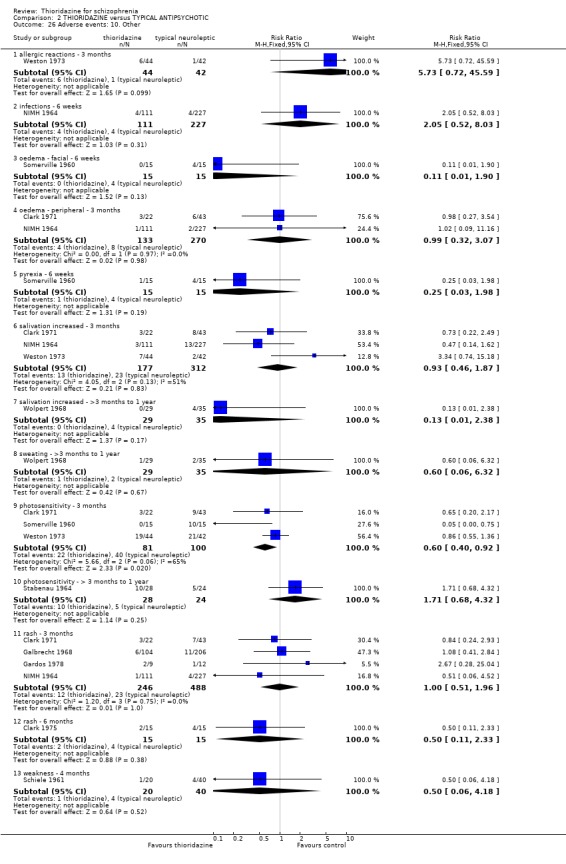
Comparison 2 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC, Outcome 26 Adverse events: 10. Other.
Comparison 3.
THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 by 6 weeks (suicide) | 1 | 144 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.27] |
| 2 Global state: 1. Not improved or worse (short term) | 3 | 203 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.81, 1.25] |
| 3 Global state: 2. Average endpoint change score by 6 weeks (CGI, high=poor, LOCF) | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | ‐0.21 [‐0.70, 0.28] |
| 4 Mental state: 1. No important change (50% drop) by 6 weeks (BPRS, LOCF) | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.18, 1.40] |
| 5 Mental state: 2. Average endpoint change score at 6 weeks (BPRS, high=poor, LOCF) | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | ‐1.89 [‐7.60, 3.82] |
| 6 Mental state: 3. Average endpoint change score (SAPS, skewed data) | Other data | No numeric data | ||
| 7 Mental state: 4. Average endpoint change score (SANS, skewed data) | Other data | No numeric data | ||
| 8 Mental state: 5. Average endpoint score at 6 weeks (BPRS, high=poor, skewed) | Other data | No numeric data | ||
| 9 Mental state: 6. Use of benzodiazepines | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.27, 0.82] |
| 10 Leaving the study early: 1a. Any reason ‐ by 3 months | 6 | 344 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.62, 1.22] |
| 11 Leaving the study early: 1b. Due to adverse events | 2 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.16 [0.73, 6.36] |
| 12 Leaving the study early: 1c. Due to refusal of medication/poor compliance | 2 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.33, 1.74] |
| 13 Leaving the study early: 1d. Due to relapse, worsening or no improvement | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 by 3 months | 2 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.19, 1.03] |
| 14 Adverse effects: 1. Anticholinergic | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 hypotension ‐ 3 months | 2 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.84, 2.95] |
| 14.2 dry mouth ‐ 3 months | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.22, 18.33] |
| 15 Adverse events: 2. Arousal | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 drowsiness / sedation ‐ 3 months | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.25, 1.28] |
| 15.2 insomnia ‐ 3 months | 2 | 59 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.58, 4.16] |
| 16 Adverse events: 3. Cardiovascular | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 faintness, dizziness, weakness ‐ 3 months | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.22, 18.33] |
| 17 Adverse events: 4. Central nervous system ‐ other | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 concentration difficulties ‐ 6 weeks | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 7.24] |
| 17.2 headache ‐ 3 months | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.05, 4.58] |
| 18 Adverse effects: 5. Movement disorders | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 rigidity ‐ 3 months | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.14, 65.16] |
| 18.2 tremor ‐ 3 months | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.14, 65.16] |
| 18.3 extrapyramidal symptoms ‐ 3 months | 2 | 81 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.54, 2.76] |
| 19 Adverse events: 6. Gastrointestinal | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 constipation ‐ 3 months | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 7.24] |
| 19.2 diarrhoea ‐ 3 months | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 7.24] |
| 19.3 nausea ‐ 6 weeks | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.22, 18.33] |
| 20 Adverse effects: 7. Hepatic abnormality ‐ 12 weeks | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.05, 4.85] |
Analysis 3.1.
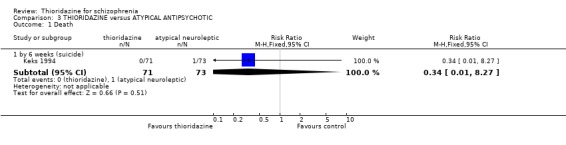
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 1 Death.
Analysis 3.2.
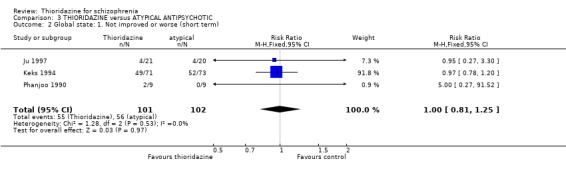
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 2 Global state: 1. Not improved or worse (short term).
Analysis 3.3.
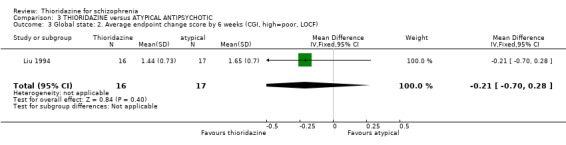
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 3 Global state: 2. Average endpoint change score by 6 weeks (CGI, high=poor, LOCF).
Analysis 3.4.
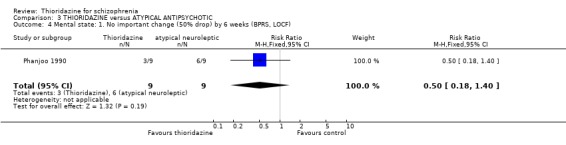
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 4 Mental state: 1. No important change (50% drop) by 6 weeks (BPRS, LOCF).
Analysis 3.5.
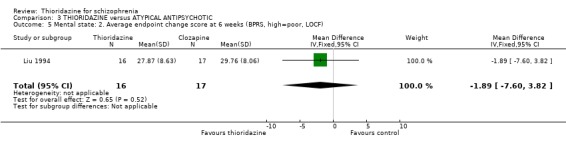
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 5 Mental state: 2. Average endpoint change score at 6 weeks (BPRS, high=poor, LOCF).
Analysis 3.6.
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 6 Mental state: 3. Average endpoint change score (SAPS, skewed data).
Mental state: 3. Average endpoint change score (SAPS, skewed data)
| Study | Intervention | mean | SD | N |
|---|---|---|---|---|
| Liu 1994 | Thioridazine | 2.00 | 3.42 | 20 |
| Liu 1994 | Clozapine | 4.41 | 5.37 | 20 |
Analysis 3.7.
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 7 Mental state: 4. Average endpoint change score (SANS, skewed data).
Mental state: 4. Average endpoint change score (SANS, skewed data)
| Study | Intervention | mean | SD | N |
|---|---|---|---|---|
| Liu 1994 | Thioridazine | 8.06 | 16.70 | 20 |
| Liu 1994 | Clozapine | 13.41 | 13.05 | 20 |
Analysis 3.8.
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 8 Mental state: 5. Average endpoint score at 6 weeks (BPRS, high=poor, skewed).
Mental state: 5. Average endpoint score at 6 weeks (BPRS, high=poor, skewed)
| Study | Intervention | Mean | SD | N |
|---|---|---|---|---|
| Keks 1994 | Thioridazine | 21.3 | 11.1 | 71 |
| Keks 1994 | Remoxipride | 21.3 | 10.9 | 73 |
| McCreadie 1988 | Thioridazine | 10.1 | 8.1 | 28 |
| McCreadie 1988 | Remoxipride | 14.3 | 8.1 | 26 |
Analysis 3.9.
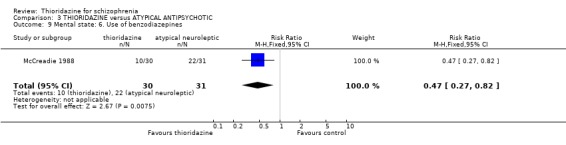
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 9 Mental state: 6. Use of benzodiazepines.
Analysis 3.10.
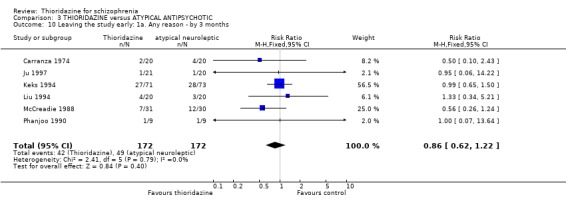
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 10 Leaving the study early: 1a. Any reason ‐ by 3 months.
Analysis 3.11.
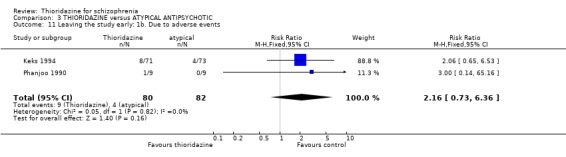
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 11 Leaving the study early: 1b. Due to adverse events.
Analysis 3.12.

Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 12 Leaving the study early: 1c. Due to refusal of medication/poor compliance.
Analysis 3.13.
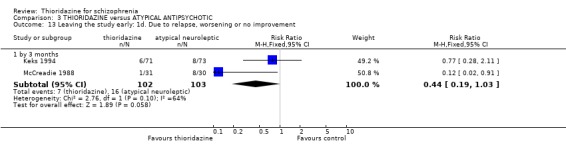
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 13 Leaving the study early: 1d. Due to relapse, worsening or no improvement.
Analysis 3.14.
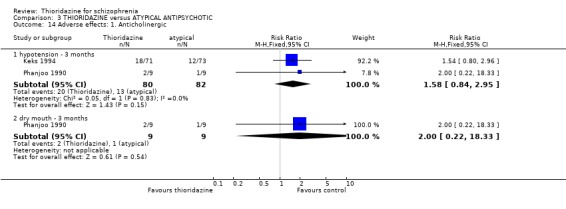
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 14 Adverse effects: 1. Anticholinergic.
Analysis 3.15.
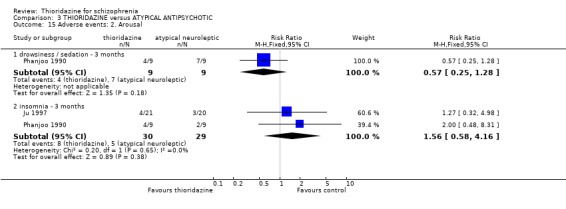
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 15 Adverse events: 2. Arousal.
Analysis 3.16.
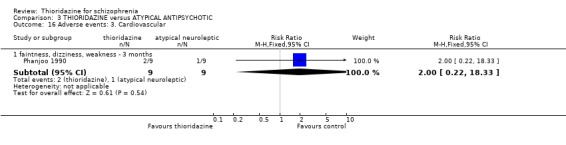
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 16 Adverse events: 3. Cardiovascular.
Analysis 3.17.
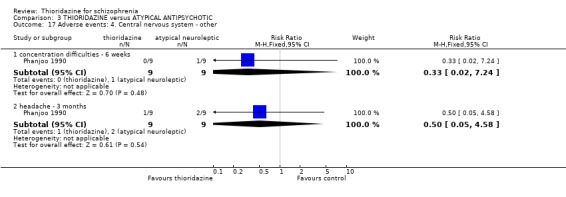
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 17 Adverse events: 4. Central nervous system ‐ other.
Analysis 3.18.
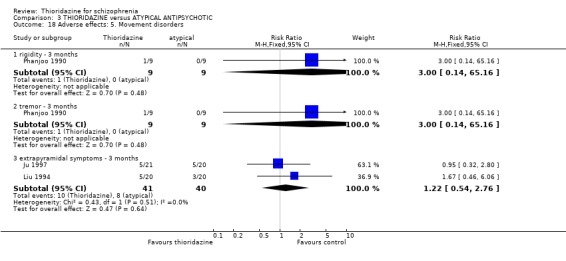
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 18 Adverse effects: 5. Movement disorders.
Analysis 3.19.
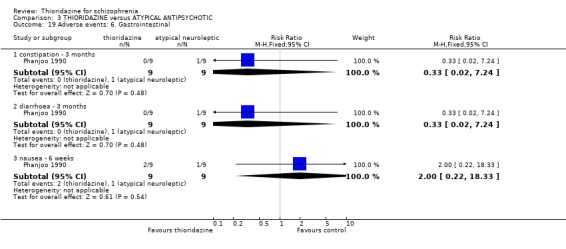
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 19 Adverse events: 6. Gastrointestinal.
Analysis 3.20.
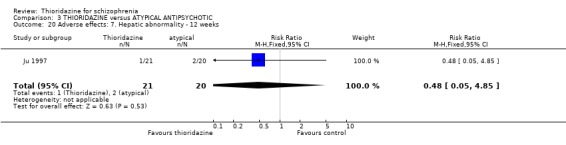
Comparison 3 THIORIDAZINE versus ATYPICAL ANTIPSYCHOTIC, Outcome 20 Adverse effects: 7. Hepatic abnormality ‐ 12 weeks.
Comparison 4.
THIORIDAZINE versus PLACEBO ‐ Intention to treat analysis for leaving the study
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1a. Due to adverse events ‐ by 6 weeks | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 any adverse event | 3 | 317 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.28, 0.61] |
| 1.2 dystonia, severe | 1 | 236 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.28, 0.69] |
| 1.3 hypotension | 1 | 236 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.28, 0.69] |
| 1.4 jaundice | 1 | 236 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.28, 0.69] |
| 1.5 parkinsonism, severe | 1 | 236 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.28, 0.69] |
| 1.6 seizure | 1 | 236 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.28, 0.69] |
| 1.7 skin reaction, facial oedema | 1 | 236 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.28, 0.69] |
| 2 Leaving the study early: 1b. Due to refusal of treatment ‐ by 1 month | 2 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.15, 0.66] |
| 3 Leaving the study early: 1c. Due to relapse ‐ by 6 months | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.15, 0.97] |
| 4 Leaving the study early: 1d. Due to worsening or no improvement ‐ by 3 months | 4 | 331 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.27, 0.58] |
Analysis 4.1.
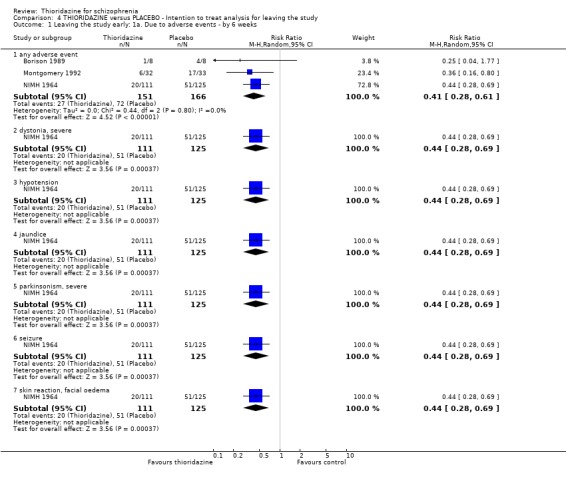
Comparison 4 THIORIDAZINE versus PLACEBO ‐ Intention to treat analysis for leaving the study, Outcome 1 Leaving the study early: 1a. Due to adverse events ‐ by 6 weeks.
Analysis 4.2.
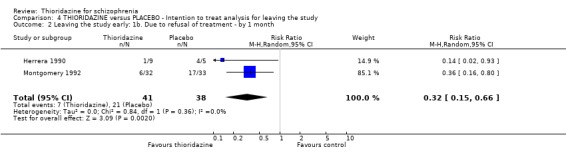
Comparison 4 THIORIDAZINE versus PLACEBO ‐ Intention to treat analysis for leaving the study, Outcome 2 Leaving the study early: 1b. Due to refusal of treatment ‐ by 1 month.
Analysis 4.3.

Comparison 4 THIORIDAZINE versus PLACEBO ‐ Intention to treat analysis for leaving the study, Outcome 3 Leaving the study early: 1c. Due to relapse ‐ by 6 months.
Analysis 4.4.
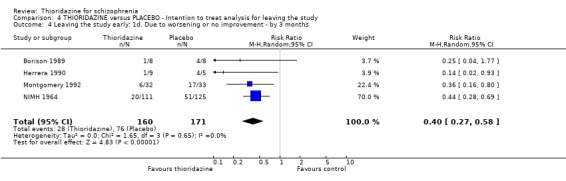
Comparison 4 THIORIDAZINE versus PLACEBO ‐ Intention to treat analysis for leaving the study, Outcome 4 Leaving the study early: 1d. Due to worsening or no improvement ‐ by 3 months.
Comparison 5.
THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC ‐ Intention to treat analysis for leaving the study
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1a. Due to any adverse event ‐ by 3 months | 10 | 860 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.70, 1.11] |
| 2 Leaving the study early: 1b. Due to no improvement or worsening | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 by 3 months | 6 | 682 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.70, 1.09] |
| 2.2 by 6 months | 1 | 424 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.77, 1.09] |
Analysis 5.1.
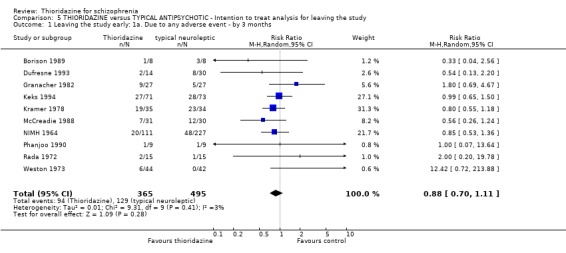
Comparison 5 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC ‐ Intention to treat analysis for leaving the study, Outcome 1 Leaving the study early: 1a. Due to any adverse event ‐ by 3 months.
Analysis 5.2.
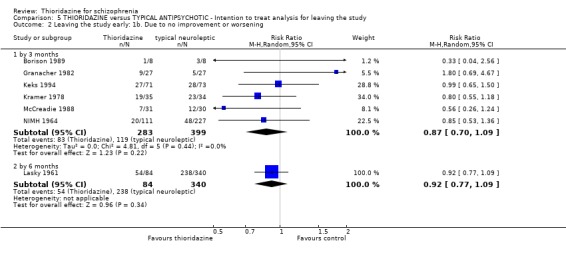
Comparison 5 THIORIDAZINE versus TYPICAL ANTIPSYCHOTIC ‐ Intention to treat analysis for leaving the study, Outcome 2 Leaving the study early: 1b. Due to no improvement or worsening.
What's new
| Date | Event | Description |
|---|---|---|
| 13 April 2011 | Amended | Contact details updated. |
History
Protocol first published: Issue 1, 2000 Review first published: Issue 3, 2000
| Date | Event | Description |
|---|---|---|
| 5 August 2009 | Amended | Contact details updated. |
| 31 October 2008 | Amended | Converted to new review format. |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Allocation: randomised (no further description). Blindness: double, cross‐over (medication matched for appearance, sealed key to identity with pharmacist). Duration: 24 weeks* (preceded by 12 weeks placebo wash out). Setting: hospital. Raters: authors rated independently of each other and two nurses also rated the participants. | |
| Participants | Diagnosis: schizophrenia. N=50. Age: mean 43 years, range 25‐70. Sex: 26M, 21F, 3 not given. History: chronically ill, two years continuous hospitalisation. Exclusions: epilepsy, mental subnormality, organic cerebral disease and physical illness. | |
| Interventions | 1. Thioridazine: dose 300 mg/day in first five days then according to response. N=25. 2. Pericyazine: dose 30 mg/day in first five days then according to response. N=25.** |
|
| Outcomes | Leaving the study early. Death. Unable to use ‐ Mental state: Wing rating scale (no usable data). |
|
| Notes | *Only data from the first 12 week arm was presented in this review. **Two withdrawals from the pericyazine group were during the placebo wash out phase, these were included. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (random numbers table with code only known to pharmacologist and dispensing chemist). Blindness: double (medication in identical capsules). Duration: 8 weeks. Setting: hospital. Raters: 2 doctors (not stated to be independent of treatment). | |
| Participants | Diagnosis: schizophrenia, 'paranoic syndromes'. N=46. Age: median 22 years, range 7‐43. Sex: 36M, 6F*. History: chronic, inpatient (illness duration >6 years, hospital stay >3 years). Exclusions: depressive symptoms, physical illness, substance abuse, aggression in hospital. | |
| Interventions | 1. Thioridazine: dose not given. N=24. 2. Thiothixene: dose not given. N=22. Dose adjusted in first 3 weeks then fixed. Trihexyphenidyl 5 mg/tds for extrapyramidal symptoms, nitrazepam 5 mg for sleep disturbance. |
|
| Outcomes | Leaving the study early. Global state: improved/not improved.** Unable to use ‐ Mental state: modified scale of Martens (only significance tests). |
|
| Notes | * sex of 4 participants not given. ** uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
| Methods | Allocation: randomised (no further description). Blindness: double (no further description). Duration: 6 weeks (preceded by 7 day wash out). Setting: hospital. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: schizophrenia (DSM III). N=32. Age: 18‐60 years. Sex: not given. History: inpatient at time of study. Exclusions: unstable physical health, BPRS score <34. | |
| Interventions | 1. Thioridazine: dose range 150‐750 mg/day. N=8. 2. Haloperidol: dose range 15‐75 mg/day. N=8. 3. Tiospirone: dose range 45‐225 mg/day. N=8.* 4. Placebo: 3 times a day. N=8. Dose adjusted according to response. Chloral hydrate 500 mg for severe agitation. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Mental state: BPRS (data unusable ‐ presented in graph only). |
|
| Notes | * Tiospirone data excluded (unsure whether valid comparator). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double. Duration: 8 weeks. Setting: hospital. Raters: not reported. | |
| Participants | Diagnosis: schizophrenia (paranoid). N=40. Age: 15‐40 years. Sex: 10M, 30F. History: not reported. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose range 300‐900 mg (no further details). N=20. 2. Sulpiride: dose range 1200 mg (no further details). N=20. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Global state: CGI (no usable data). Mental state: BPRS (no usable data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: not stated. Duration: 6 weeks (preceded by 1 week wash out). Setting: hospital. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: schizophrenia (116 paranoid, 107 undifferentiated, 7 hebephrenic, 4 simple). N=234. Age: mean 31 years, range 16‐75. Sex: 125M, 109F. History: inpatient at time of study. Exclusions: major physical illness, organic brain disorder, BPRS score <35. | |
| Interventions | 1. Thioridazine: dose range 300‐800 mg/day. N=121. 2. Chlorpromazine: dose range 300‐800 mg/day. N=113. Treatment started at unspecified low dose, built up to therapeutic range in first week. |
|
| Outcomes | Leaving the study early. Mental state: BPRS*. Global state: Chinese Psychiatric Association Scale*. Adverse events: TESS*. | |
| Notes | * uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised in blocks of 4 (no further description). Blindness: double (medication in identical capsules). Duration: 28 days. Setting: hospital. Raters: research psychiatrists, nurses, psychologists, independent of treatment. | |
| Participants | Diagnosis: schizophrenia (NIMH criteria). N=86. Age: mean 33 years range 18‐22. Sex: 22M, 53 F* . History: no hospitalisations for at least 6 months prior to inclusion; current acute exacerbation of chronic illness, moderately or severely ill on admission, 2 or more previous admissions. Exclusions: people <18 years of age or over 45 years; childhood autism, childhood schizophrenia, chronic or acute brain syndrome, I.Q. < 70, alcoholism, epilepsy, drug addiction; diabetes, hepatitis, chronic physical illness requiring continuous medication. | |
| Interventions | 1. Thioridazine: dose increased on a sliding scale to 1000 mg/day. N=22. 2. Placebo: N=21. 3. Fluphenazine oral: dose increased on a sliding scale to 10 mg/day. N=20. 4. Chlorpromazine: dose increased on a sliding scale to 1000 mg/day. N=23. Dose adjusted for intolerance; nighttime sedation allowed. |
|
| Outcomes | Leaving the study early. Global state: improved/not improved.** Adverse events. Unable to use ‐ Mental state: BPRS (no SD). Behaviour: NOSIE (no SD). |
|
| Notes | * sex of 1 participant not given. ** uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: random (no further description). Blindness: double (medication in identical capsules). Duration: 6 months. Setting: outpatient. Raters: research nurse and project psychiatrist. | |
| Participants | Diagnosis: schizophrenia. N=40. Age: range 24‐60 years. Sex: all female. History: chronic, outpatient taking medication for at least 3 months at time of study. Exclusions: poor physical health. | |
| Interventions | 1. Thioridazine: maximum dose 750 mg/day. N=15. 2. Placebo. N=10. 3. Pimozide: maximum dose 20 mg/day. N=15. |
|
| Outcomes | Leaving the study early. Global state: CGI improved/not improved, CGI score.* Relapse*. Adverse events*. Unable to use ‐ Global state: CGI data from psychiatrist (authors used CGI data from research nurse as thought this more likely to be independent). Mental state: BPRS (no SD). Social functioning scale: Social Adjustment Scale (used modified version of Katz‐Lyerly scale). |
|
| Notes | * uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: random (no further description). Blindness: double (active placebo with similar side effects to medication given, blindness then tested by guessing questionnaire). Duration: 8 months. Setting: hospital. Raters: 2 nurses independently made each rating. | |
| Participants | Diagnosis: schizophrenia. N=10. Age: not given. Sex: not given. History: chronic, inpatient at time of study. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose not given. N=5. 2. Phenobarbital and atropine sulphate: dose not given. N=5. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Behaviour: Behavioural Disturbance Index (data unusable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules). Duration: 6 weeks (preceded by at least 1 week wash out). Setting: hospital. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: schizophrenia (DSM III). N=44. Age: mean 34 years, range 18‐63. Sex: not given. History: chronic, inpatient with several hospitalisations. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: maximum dose 800 mg/day. N=14. 2. Haloperidol: maximum dose 40 mg/day. N=16. 3. Molindone: maximum dose 200 mg/day. N=14. Dose adjusted according to response. Chloral hydrate for insomnia, agitation, amantadine for extrapyramidal symptoms. |
|
| Outcomes | Leaving the study early. Global state: CGI improved/not improved. Mental state: BPRS improved/not improved. Adverse events. Unable to use ‐ Mental state: HAMD, BPRS (data unusable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication indistinguishable in taste and appearance). Duration: 28 days (preceded by 1 week wash out). Setting: hospital. Raters: ward nurse (not stated to be independent of treatment). | |
| Participants | Diagnosis: schizophrenia (NIMH criteria). N=54. Age: mean 27 years. Sex: all male. History: acute, recently hospitalised, no previous hospitalisation within 12 months of study. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose 400 mg/day. N=27. 2. Placebo: N=27. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Behaviour: NOSIE (data unusable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules). Duration: 8 weeks. Setting: hospital. Raters: psychologist, psychiatrist independently rated mental state, treating physician recorded adverse events. | |
| Participants | Diagnosis: schizophrenia (˜155 paranoid), (˜93 chronic undifferentiated), (˜31 catatonic), (˜31 other subtypes). N=310. Age: < 55 years. Sex: all male. History: acute, newly hospitalised. Exclusions: inability to take oral medication, substance abuse, prefrontal lobotomy, any physical illness. | |
| Interventions | 1. Thioridazine: dose mean 700 mg/day, range 200‐1600 mg/day. N=104. 2. Chlorpromazine: dose mean 750 mg/day, range 200‐1600 mg/day. N=102. 3. Fluphenazine: dose mean 8.4 mg/day, range 2.5‐20 mg/day. N=104. Dose fixed first 2 weeks, then adjusted according to response. Antiparkinsonian medication allowed. |
|
| Outcomes | Leaving the study early. Adverse events*. Unable to use ‐ Mental state: IMPS (no data). |
|
| Notes | * uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (no further description). Duration: 12 weeks (preceded by 21 days medication free period). Setting: hospital. Raters: 2 psychiatrists, 1 research nurse (all independent of treatment). | |
| Participants | Diagnosis: schizophrenia. N=20. Age: mean 44 years, range 31‐53. Sex: 10M, 10F. History: chronic, inpatient at time of study, medial length of hospitalisation ˜ 16 years). Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose increased from 200 mg/day to 800 mg/day on dose schedule. N=10. 2. Piperacetazine: dose 50 mg/day increased to 150 mg/day on dose schedule. N=10. |
|
| Outcomes | Leaving the study early. Mental state: BPRS improved/not improved. Global state: improved/not improved. Adverse events. Unable to use ‐ Behaviour: NOSIE (data unusable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (random code). Blindness: single (with 2 raters), one rater not blind, cross‐over. Duration: 24 weeks*. Setting: hospital. Raters: clinical changes were evaluated by raters blind to drug assignment; rating on clinical improvement, severity of hallucinations, side effects, AIMS and Dyskinesia Rating Scale were completed by a non‐blinded ward psychiatrist. | |
| Participants | Diagnosis: schizophrenia (Feighner's criteria). N=21. Age: mean 39 years, range 27‐50. Sex: 14M, 5F**. History: chronic, treatment resistant (illness duration 11‐34 years). Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose final mean 479 mg/day, range 100‐800 mg/day. N=9. 2. Mesoridazine: dose final mean 284 mg/day, range 50‐400 mg/day. N=12. Initial dose according to previous dosing level then adjusted according to response. |
|
| Outcomes | Leaving the study early. Adverse events. Unable to use ‐ Global state: improved/not improved (data unusable). Mental state: BPRS (no SD). Behaviour: NOSIE (no SD). |
|
| Notes | * data taken from first 12 week arm only? ** two participants not accounted for. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (table of random numbers). Blindness: double (medication in identical capsules). Duration: 12 weeks (preceded by 2 weeks psychotropic drug free period). Setting: hospital. Raters: physician and nurse (not stated to be independent of treatment). | |
| Participants | Diagnosis: psychosis. N=54*. Age: mean 41 years, range 21‐64. Sex: male and female. History: inpatient at time of study, have marked to extremely severe psychosis with at least 3 moderately severe psychotic BPRS symptoms. Exclusions: other physical illness, those receiving high dose treatment and/or treatment resistant, substance misuse. | |
| Interventions | 1. Thioridazine: dose range 100‐800 mg/day. N=27. 2. Thiothixene: dose range 10‐60 mg/day. N=27. Dose titrated upwards according to clinical response; antiparkinsonian medication allowed. |
|
| Outcomes | Leaving the study early. Global state: improved/not improved.** Unable to use ‐ Mental state: BPRS (data unusable). Behaviour: NOSIE (data unusable). |
|
| Notes | * 5 participants excluded for protocol violations (allocation not given). ** uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules and an 'active placebo'). Duration: 2 years (preceded by 13 week wash out period). Setting: research unit. Raters: 9 nurses (not stated to be independent of treatment). | |
| Participants | Diagnosis: schizophrenia. N=10. Age: range 24‐34 years. Sex: all male. History: chronic, ill for at least 3 years. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose range 300‐1000 mg/day. N=5. 2. 'Active' placebo: phenobarbital dose range 60‐200 mg/day, atropine dose range 0.36‐1.2 mg up to 4 months. Inert placebo after 4 months. N=5. Dose built up to 300 mg/day thioridazine or 60 mg/day phenobarbital, 0.36 mg atropine over 4 weeks then blindly varied by administrator within dose range. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Adverse events: data unusable. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (by tossing coin). Blindness: double (medication in identical capsules). Duration: 3 months. Setting: hospital. Raters: fully trained in BPRS (not stated to be independent of treatment). | |
| Participants | Diagnosis: schizophrenia ‐ simple 8, hebephrenic 42, paranoid 21, unspecified 3 (Chinese Psychiatric Association criteria 1984). N=74. Sex: all female. Age: mean 35 years, range 17‐61. History: inpatient at time of study, both acute, chronic illness (duration mean ˜ 10 years, range 8 days ‐ 31 years), BPRS <35. Exclusions: other physical illness, allergy to medications, EPS or ECG abnormalities. | |
| Interventions | 1. Thioridazine: dose maximum 800 mg/day. N=37. 2. Chlorpromazine: dose maximum 800 mg/day. N=37. |
|
| Outcomes | Mental state: BPRS improved/not improved*. Adverse events: TESS*. Unable to use ‐ Leaving the study early: (no data from treatment group). |
|
| Notes | *uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules). Duration: 4 weeks (preceded by 1 week placebo wash out period) Setting: hospital. Raters: research staff. | |
| Participants | Diagnosis: schizophrenia. N=14. Age: mean 27 years, range 19‐44. Sex: all male. History: acutely ill at time of study with duration under 1 year and no more than one previous admission. Exclusions: other physical illness, substance misuse, likely to need concomitant medication. | |
| Interventions | 1. Thioridazine slow release: dose range 400‐1000 mg/day. N=9. 2. Placebo: N=5. Dose titrated slowly upwards according to clinical response; chloral hydrate for insomnia and antiparkinsonian allowed. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Mental state: BPRS (data unusable, presented in graphs only). Behaviour: NOSIE (data unusable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (using random numbers). Blindness: not reported. Duration: 12 weeks. Setting: hospital. Raters: not reported. | |
| Participants | Diagnosis: schizophrenia (CCMD‐2). N=41. Age: 18‐65 years. Sex: male. History: chronic illness 7 to 46 years. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose initially 100‐200 mg/day, gradually increased to 300‐600 mg/day within the first week. N=21. 2. Sulpiride: dose initially 200‐300 mg/day, gradually increased to 600‐1000 mg/day. N=20. |
|
| Outcomes | Leaving the study early. Global state: not improved . Adverse effects: TESS, insomnia, hepatic abnormality. Unable to use ‐ Mental state: BPRS, SANS, SAPS (no usable data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised* (no further description). Blindness: partially double.** Duration: 15 weeks (at 9 weeks medication stopped for 1 week). Setting: hospital. Raters: ward psychiatrist, ward nurse, nursing assistant (not independent of treatment). | |
| Participants | Diagnosis: schizophrenia in 80% of treated group, 73% of control group. N=40. Age: median 63 years. Sex: not given. History: chronic inpatient at time of study (median hospital stay ˜ 27 years, median symptom duration ˜ 30 years). Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose 9 weeks on 500 mg/day, 5 weeks on 700 mg/day. N=25. 2. Placebo: maintained throughout. N=15. |
|
| Outcomes | Leaving the study early. Global state: improved/not improved. Unable to use ‐ Mental state: Lorr ‐ Multidimensional Scale (no data). |
|
| Notes | *matched for psychiatric morbidity on MSRPP (Lorr‐Multidimensional Scale for Rating Psychiatric Patients). ** 'not a completely double blind procedure' ‐ thioridazine stopped for 1 week on 500 mg while placebo continued. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (in blocks of 4, balanced within centres, no further description). Blindness: double (no further description). Duration: 6 weeks (preceded by ˜ 1 week wash out period). Setting: hospital. Raters: received training in BPRS, CGI (not stated to be independent of treatment). | |
| Participants | Diagnosis: schizophrenia or schizophreniform disorder (DSM IIIR). N=144. Age: mean 31 years. Sex: 110M, 34F. History: inpatient at time of study, moderately ill (BPRS score at least 18). Exclusions: depot within 4 weeks of study, other physical illness, substance misuse. | |
| Interventions | 1. Thioridazine: dose range 150‐600 mg. N=71. 2. Remoxipride: dose range 150‐600 mg. N=73. Dose adjusted according to response; oral benzodiazepine for disturbed behaviour; chloral hydrate or short acting benzodiazepine as hypnotic. |
|
| Outcomes | Leaving the study early. Global state: CGI improved/not improved. Adverse events.* Unable to use ‐ Mental state: BPRS (skewed data). |
|
| Notes | * uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: not reported, but medication in identical capsules. Duration: 4 weeks (preceded by 2 week wash out). Setting: hospital. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: schizophrenia (DSM II). N=69. Age: mean 32 years, range 18‐57. Sex: 21M, 35F*. History: inpatient at time of study. Exclusions: other serious physical illness. | |
| Interventions | 1. Thioridazine: dose mean ˜ 425 mg/day, range 100‐800 mg/day. N=35. 2. Loxapine: dose mean ˜ 79 mg, range 20‐200 mg/day. N=34. Dose titrated upwards until therapeutic effect achieved or side effects prevented further increases. Antiparkinsonian allowed. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Global state: CGI (data unusable). Mental state: BPRS (data unusable). Behaviour: NOSIE (data unusable). Adverse events: >40% loss. |
|
| Notes | *13 not given. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules). Duration: 24 weeks. Setting: hospital. Raters: psychologist, psychiatrist, nurse, nursing assistant independently rated mental state, treating physician rated side effects and reason for leaving study. | |
| Participants | Diagnosis: schizophrenia (half paranoid, one third undifferentiated). N=512. Age: <55 years. Sex: all male. History: acutely ill inpatients at time of study, able to take oral medication. Exclusions: systemic or CNS illness, prefrontal lobotomy. | |
| Interventions | 1. Thioridazine: dose mean 845 mg/day, range 200‐1600 mg. N=84. 2. Chlorpromazine: dose mean 746 mg/day, range 200‐1600 mg/day. N=86. 3. Fluphenazine: dose mean 10 mg/day, range 2.5‐20 mg/day. N=84. 4. Chlorprothixene: dose mean 224 mg/day, range 50‐400 mg/day. N=87. 5. Trifluoperazine: dose mean 208 mg/day, range 50‐400 mg/day. N=83. 6. Reserpine: dose mean 6 mg/day, range 1.5‐12 mg/day. N=88.* |
|
| Outcomes | Leaving the study early. Unable to use ‐ Mental state: Multidimensional Psychiatric Scale, Psychotic Reaction Profile ( >40% loss). Adverse events: ( >40% loss). |
|
| Notes | * Reserpine data not used in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (no further description). Duration: 6 weeks. Setting: hospital. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: schizophrenia. N=40. Age: mean 26 years. Sex: 24M, 16F. History: inpatient at time of study, both new and chronic patients, illness duration ˜ 6 months to 2 years. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose range 50‐150 mg/tds. N=20. 2. Clozapine: dose range 25‐75 mg/tds. N=20. |
|
| Outcomes | Leaving the study early. Mental state: BPRS, SAPS, SANS. Global state: CGI score. Unable to use ‐ Adverse events: TESS (data unusable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (no further description). Duration: 6 weeks (preceded by 1 week placebo wash out period). Setting: hospital and outpatient. Raters: psychiatrists met regularly to ensure inter rater agreement (not stated to be independent of treatment). | |
| Participants | Diagnosis: schizophrenia, schizophreniform disorder (DSM III). N=61. Age: mean 38 years, range 19‐67. Sex: 27M, 34F. History: acute, moderately ill (BPRS >/= 15). Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose range 50‐750 mg/day. N=31. 2. Remoxipride: dose range 25‐375 mg/day. N=30. Dose increased in steps according to response; short acting benzodiazepine as hypnotic, benzodiazepine for gross mental or behavioral disturbance, anticholinergic for extrapyramidal symptoms allowed. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Global state: CGI (data unusable). Mental state: BPRS (skewed data). Adverse events: data unusable. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules, blindness checked by survey). Duration: 5 weeks. Setting: hospital. Raters: 2 nurse's aides, 1 nurse, 2 psychiatric residents. | |
| Participants | Diagnosis: schizophrenia. N=40. Age: mean 42 years. Sex: all male. History: chronic (mean duration of illness ˜ 15 years). Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose mean 580 mg/day, range 300‐1200 mg/day. N=20. 2. Mesoridazine: dose mean ˜ 259 mg/day, range 75‐600 mg/day. N=20. Dose adjusted according to response; no antiparkinsonian allowed; chloral hydrate or paraldehyde for agitation or irritability. |
|
| Outcomes | Leaving the study early. Mental state: BPRS improved/not improved. Behaviour: NOSIE improved/not improved. Unable to use ‐ Adverse events: no data. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (by a neutral controller). Blindness: double, cross‐over (medication in identical appearing, sugar coated tablets). Duration: 24 weeks* (preceded by one week placebo wash out for some patients). Setting: hospital. Raters: nursing staff and doctors. | |
| Participants | Diagnosis: schizophrenia. N=60. Age: range 19‐60 years. Sex: 40M, 20F. History: chronic illness. Exclusions: patients with cardiac, hepatic, renal and hematopoietic disorders. | |
| Interventions | 1. Thioridazine: dose 150 mg/day in first week, then flexible to maximum 600 mg/day. N=30. 2. Mesoridazine: dose 150 mg/day in first week, then flexible to a maximum 600 mg/day. N=30. |
|
| Outcomes | Leaving the study early. Global state: improved/not improved. Unable to use ‐ Global state: Global Severity Rating, Symptom improvement rating, Psychiatric Evaluation Scale (data unusable). Behaviour: Behaviour Rating Scale (data unusable). Adverse events: data unusable. |
|
| Notes | *only data from the first 12 week arm of the cross‐over are presented in this metanalysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
| Methods | Allocation: randomised (no further description). Blindness: double (double dummy technique used). Duration: 4 weeks. Setting: hospital. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: schizophrenia (DSM III). N=96*. Age: range 20‐60 years. Sex: 53M, 43F. History: chronic with acute relapse. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose 200 mg/day for 1 week then 400 mg/day for 3 weeks. N=32. 2. Placebo: dose thioridazine 200 mg/day for 1 week then placebo for 3 weeks. N=33. 3. Des‐enkephalin‐gamma‐endorphin: dose 10 mg/day IM. N=30**. Antiparkinsonian allowed. Lorazepam as 'rescue medication'. |
|
| Outcomes | Leaving the study early. Global state: GAS score. Unable to use ‐ Mental state: BPRS, Montgomery Schizophrenia Scale (data skewed). Adverse events: no data. |
|
| Notes | * one person left the study before receiving active treatment. The authors did not report their allocation. **data for des‐enkephalin‐gamma‐endorphin was not used in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules or vials for injection). Duration: 6 weeks. Setting: multicentre. Raters: doctors and nurses (not stated to be independent of treatment). | |
| Participants | Diagnosis: schizophrenia (Feighner criteria). N=463. Age: mean 28 years, range 16‐45. Sex: male and female. History: acute inpatient recently admitted (half of the participants were first admissions). Exclusions: any significant hospitalisation 12 months prior to study enrolment. | |
| Interventions | 1. Thioridazine: dose (i) oral mean 700 mg/day, range 200‐1600 mg/day, (ii) parenteral in 26%, mean 75 mg/day, range 50‐400 mg/day. N=111. 2. Placebo: dose (i) oral mean 8.5 doses, range 2‐16 doses, (ii) parenteral in 22%, mean 6 ampules, range 2‐16 injections. N=125. 3. Chlorpromazine: dose (i) oral mean 655 mg/day, range 200‐1600 mg/day, (ii) parenteral in 22%, mean 100 mg/day, range 50‐400 mg/day. N=112. 4. Fluphenazine: dose (i) oral mean ˜ 6 mg, range 2‐16 mg, (ii) parenteral in 20%, mean 6 mg, range 1‐8 mg. N=115. |
|
| Outcomes | Leaving the study early. Adverse events. Unable to use ‐ Global state: improved/not improved (no individual group data). Mental state: Inpatient Multidimensional Psychiatric Scale (no individual group data). Behaviour: Burdock Ward Behaviour Rating Scale(data unusable). |
|
| Notes | Due to the large numbers of people dropping out from the placebo group, we made a post hoc decision not to use a intention to treat analysis for adverse effects from this study, as the ITT data would have created a large over‐estimate of effect in the placebo group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication identical in taste and appearance). Setting: outpatient. Duration: 1 year. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: schizophrenia (DSM III). N=106*. Age: mean 40 years. Sex: 78M, 28F. History: in remission or residual phase. Exclusions: not reported. | |
| Interventions | 1. Thioridazine 25 mg/day. N=12. 2. Thioridazine 75 mg/day. N=10. 3. Pimozide 2 mg/day. N=13. 4. Pimozide 2 mg/day + thioridazine 25 mg/day. N=11. 5. Pimozide 2 mg/day + thioridazine 75 mg/day. N=11. 6. Pimozide 6 mg/day. N=11. 7. Pimozide 6 mg/day + thioridazine 25 mg/day. N= 12.* 8. Pimozide 6 mg/day + thioridazine 75 mg/day. N= 13.* Nitrazepam 10 mg for insomnia, trihexyphenidyl 2 mg for EPS combined with each drug. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Mental state: >40% loss. |
|
| Notes | * data extracted only for pimozide and thioridazine. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (no further description). Duration: 6 weeks (preceded by 2 &1/2 day placebo washout)*. Setting: hospital. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: 5 schizophrenia paranoid type, 5 paranoia, 5 acute paranoia, 3 atypical paranoia. N=18. Age: range 67‐80 years. Sex: 1M, 17F. History: chronic (median duration of illness 3 years) Exclusions: major affective disorder, severe dementia, substance dependence, history of serious adverse drug reaction. | |
| Interventions | 1. Thioridazine: dose initially 25 mg/bid with fortnightly 50 mg increases, mean last week 133 mg/day, maximum 200 mg/day. N=9. 2. Remoxipride: dose initially 25 mg/bid with fortnightly 50 mg/day increases, mean last week 200 mg/day, maximum 200 mg/day. N=9. Dose adjusted according to response; chloral hydrate or benzodiazepine as hypnotic, procyclidine for extrapyramidal symptoms. |
|
| Outcomes | Leaving the study early. Global state: CGI improved/not improved.* Mental state: BPRS improved/not improved.* Adverse events.** | |
| Notes | * responders to remoxipride continued on open basis for 12 months. * *uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules). Duration: 4 weeks (preceded by 3‐7 day wash out). Setting: hospital. Raters: not stated to be independent of treatment. | |
| Participants | Diagnosis: schizophrenia (DSM III). N=12. Age: mean 21 years, range 19‐44. Sex: both sexes (no further details). History: chronic with acute exacerbation of positive symptoms, responsive neuroleptics past, at least one or more previous admissions. Exclusion: other significant physical illness. | |
| Interventions | 1. Thioridazine: dose range 100‐800 mg/day. N=7. 2. Placebo: N=5. Dose titrated to at least 200 mg/day over 3‐7 days depending on response. |
|
| Outcomes | Global state: CGI improved/not improved. Unable to use ‐ Mental state: BPRS (no SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: not stated. Duration: 8 weeks (preceded by 2 week placebo wash out). Setting: outpatient. Rater: psychiatrist, "rater blind study". | |
| Participants | Diagnosis: schizophrenia, undifferentiated or paranoid. N=30. Age: range 21‐60 years. Sex: all female. History: currently experiencing at least 2 positive or negative symptoms. Exclusions: other significant physical illness. | |
| Interventions | 1. Thioridazine: maximum dose 800 mg/day. N=15. 2. Piperacetazine: maximum dose 160 mg/day. N=15. Dose adjusted for adverse events. |
|
| Outcomes | Leaving the study early. Global state: CGI improved/not improved. Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: not stated. Duration: three years, six months. Setting: not stated. Rater: not stated. | |
| Participants | Diagnosis: schizophrenia. N=30. Age: mean 57 years. Sex: all female. History: treated at least 3 years with chlorpromazine. Exclusions: not stated. | |
| Interventions | 1. Thioridazine: dose mean 375 mg/day, range 200‐600 mg/day. N=15. 2. Chlorpromazine: dose mean 325 mg, range 100‐800 mg/day. N=15. |
|
| Outcomes | Leaving the study early. Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: single (no further description). Duration: 4‐6 weeks. Setting: hospital. Raters: 2 psychiatrists, one independent. | |
| Participants | Diagnosis: schizophrenia (DSM III). N=21. Age: mean 16 years, range 11‐19. Sex: 13M, 8F. History: not reported. Exclusions: seizure disorder, substance misuse, drug induced psychosis, IQ>70, other significant physical illness, need for other psychotropics. | |
| Interventions | 1. Thioridazine: dose mean 0.26 mg/kg. N=8. 2. Thiothixene: dose mean 3.07 mg/kg. N=13. Dose adjusted according to response. |
|
| Outcomes | Leaving the study early. Adverse events. Unable to use ‐ Global state: CGI (data unusable). Mental state: BPRS (data unusable). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blinding: double (identical capsules used, hospital pharmacist held code). Duration: 16 weeks. Setting: hospital. Raters: not reported. | |
| Participants | Diagnosis: schizophrenia. N=80. Sex: male. Age: mean 40.6 years. History: all hospitalised for 10 years. | |
| Interventions | 1. Thioridazine: dose 200‐1000 mg/day. N=20. 2. Trifluoperazine: dose 10‐50 mg/day. N=20. 3. Chlorpromazine: dose 200‐1000 mg/day. N=20. 4. Placebo. N=20. Dose adjusted according to response. Additional medication, phenobarbital for sedation and benztropine for extrapyramidal symptoms. |
|
| Outcomes | Leaving the study early. Global state: improved/not improved (clinical judgement). Adverse effects. Additional medications: antiparkinsonian drugs. Unable to use ‐ Behaviour: The manifest behaviour scale (no SD). Mental state: MMPI (>50% not accounted for). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (by doctor unconnected to ward but no further description). Blindness: not stated but medication in identical tablets. Duration: 6 weeks (preceded by 1 month medication free wash out). Setting: hospital. Raters: psychiatrist, ward medical officer, nursing staff (not independent of treatment). | |
| Participants | Diagnosis: 56 schizophrenia or "paraphrenic psychosis", 4 bipolar. N=60. Age: mean 22 years, range 20‐60. Sex: female. History: long stay, 'poor prognosis'. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose increased on a sliding scale to 800 mg/day. N=15. 2. Placebo: N=30. 3 . Chlorpromazine: dose increased on a sliding scale to 800 mg/day. N=15. |
|
| Outcomes | Leaving the study early. Global state: improved/not improved. Mental state: FFS improved/not improved. Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (by hospital pharmacist but no further description). Blindness: double (medication in identical capsules). Duration: 10 & 1/2 months. Setting: hospital. Raters: patient's physician or nurse (not independent of treatment). | |
| Participants | Diagnosis: schizophrenia.* N=52. Age: range 18‐50 years. Sex: 21M, 19F** History: acute, high proportion of first admissions, felt by house officer to need phenothiazine due to aggressive behaviour, severe anxiety, hyperactivity, thought disorder, delusions, hallucinations. Exclusions: neurotic, organic, personality disorder, need other treatment like ECT. | |
| Interventions | 1. Thioridazine: dose initially 100‐300 mg/day, then 50‐100 mg/day increments; mean ˜400 mg/day, range 300‐500 mg/day. N=28. 2. Chlorpromazine: dose initially 100‐300 mg/day, then 50‐100 mg/day increments; mean ˜400 mg/day, range 300‐500 mg/day. N=24. |
|
| Outcomes | Leaving the study early. Global state: CGI improved/not improved.*** Adverse events. Unable to use ‐ Service utilisation: duration of admission (data unusable). Mental state: Wittenborn Psychiatric Rating Scale, Mental State Check List, Mood Adjective Check List (data unusable). |
|
| Notes | * for the majority of participants. ** 12 not given. *** uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: not blind. Duration: 12 weeks. Setting: not reported. Raters: not reported. | |
| Participants | Diagnosis: schizophrenia with (n=27) also reported as having 'toxic psychosis' .* N=74. Sex: male. Age: not given. History: physically healthy bantu males suffering from acute psychosis. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose 600 mg/day**. N=21. 2. Clothiapine: dose 120 mg/day. N=28. 3. Chlorpromazine: dose 600 mg/day***. N=25. |
|
| Outcomes | Global state: improved/not improved. | |
| Notes | *patients with 'toxic psychosis' not included in this review. ** 10 patients received 900 mg on the first day for agitation, restlessness. ***100 mg given IM initially to 4 patients |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description), crossover study. Blindness: raters reported as blind (medication in identical capsules). Duration: 2 week placebo wash out, 7 weeks on one drug, 2 week placebo wash out, 7 weeks on second drug. Setting: hospital. Raters: 4 psychiatrists not independent of treatment, 2 nurses independent of treatment, nurses did not know design. | |
| Participants | Diagnosis: schizophrenia. N=60. Age: mean 43 years, range 23‐65. Sex: all male. History: length of previous hospitalisations ranged from 1.75 years to 33 years, mean 15 years. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose range 200‐800 mg/day. N=30. 2. Trifluoperazine: dose range 5‐40 mg/day. N=30. Dose adjusted according to response. For extrapyramidal symptoms dose reduced then Cogentin if not alleviated. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Mental state: BPRS, Psychotic Reaction Profile (data unusable). Adverse events: data unusable. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further description). Blindness: double (medication in identical capsules). Duration: 12 weeks (preceded by 4 week placebo wash out). Setting: hospital. Raters: 2 sessional psychiatrists, 4 research nurses (independent of treatment); supervising psychiatrist (not independent of treatment) rated status, progress & adverse events. | |
| Participants | Diagnosis: schizophrenia. N=86. Age: mean 49 years. Sex: 45M, 41F. History: chronic illness, duration at least 2 years, majority of time inpatient, diagnosis same throughout contact, at least 2 positive or negative symptoms. Exclusions: other significant organic or physical illness, substance misuse, leucotomy. | |
| Interventions | 1. Thioridazine: dose mean 333 mg/day, range 300‐600 mg/day. N=44. 2. Haloperidol: dose mean ˜ 5 mg/day, range 4.5‐7.5 mg/day. N=42. Dose adjusted according to response. Orphenadrine 50 mg/tds for extrapyramidal symptoms allowed.** |
|
| Outcomes | Leaving the study early. Global state: improved/not improved.* Adverse events.* Unable to use ‐ Mental state: Inpatient Multidimensional Scale (data unusable). Behaviour: Psychiatric Reaction Profile (data unusable). |
|
| Notes | * uses LOCF. **Use of other tranquillizers, ECT or drugs other than analgesics led to withdrawal. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (by independent staff member, no further description). Blindness: double (medication in identical capsules). Duration: 7 months. Setting: hospital. Raters: independent. | |
| Participants | Diagnosis: schizophrenia (2 'childhood onset', 6 'organic' factors in diagnosis'). N=92. Age: mean 54 years. Sex: all male. History: chronic, mean length of admission ˜20 years. Exclusions: abnormal lab results, other physical abnormalities. | |
| Interventions | 1. Thioridazine: dose mean last month 200 mg/day, maximum 1200 mg/day. N=29. 2. Placebo: N=28. 3. Thiothixene: dose mean last 3 months 10 mg/day, maximum 60 mg/day. N=35. Dose increased on fixed schedule then flexible schedule in second month; trihexyphenidyl 2.5‐20 mg for extrapyramidal symptoms. |
|
| Outcomes | Mental state: BPRS improved/not improved.* Adverse events.* Unable to use ‐ Mental state: MMPI (data unusable). Behaviour: NOSIE (data unusable). Leaving the study early: participants were changed from initial random allocation. |
|
| Notes | * uses LOCF. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Allocation: randomised (no further details). Blindness: not reported. Duration: 8 weeks. Setting: hospital. Raters: not reported | |
| Participants | Diagnosis: schizophrenia (CCMD‐II & ICD‐10). N=73. Age: 18‐50 years. Sex: male and female. History: illness 1‐22 years. Exclusions: not reported. | |
| Interventions | 1. Thioridazine: dose mean 521 mg/day. N=41. 2. Chlorpromazine: dose mean 480 mg/day. N=32. |
|
| Outcomes | Leaving the study early. Mental state: SAPS. Unable to use ‐ Mental state: BPRS (no usable data). Adverse events: TESS (no usable data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Diagnostic classifications CCMD ‐ Chinese Classification of Mental Diseases DSM ‐ Diagnostic and Statistical Manual ICD ‐ International Classification of Diseases
Rating scales FFS ‐ Fergus Falls Scale
Behaviour ‐ NOSIE ‐ Nurses Observational Scale of Inpatients Evaluation
Global state ‐ CGI ‐ Clinical Global Impression GAS ‐ Global Assessment Scale
Mental state ‐ BPRS ‐ Brief Psychiatric Rating Scale HAMD ‐Hamilton Rating Scale for Depression IMPS ‐ Inpatient Multidimensional Psychiatric Scale MMPI ‐ Minnesota Multiphasic Personality Inventory SAPS ‐ Scale for the Assessment of Positive Symptoms SANS ‐ Scale for the Assessment of Negative Symptoms
Adverse effects ‐ TESS ‐ Treatment Emergent Symptom Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Acker 1965 | Allocation: not randomised, case series. |
| Altman 1973 | Allocation: randomised, crossover design. Participants: people with schizophrenia. Interventions: chlorpromazine versus thioridazine. Outcome: no pre‐crossover data. |
| Askar 1970 | Allocation: not randomised, not double blind. |
| Bandelow 1992 | Allocation: randomised. Participants: people with schizophrenia. Interventions: continuing neuroleptic maintenance versus early intervention (neuroleptic reinstatement for early symptoms) versus crisis intervention (reinstatement only on relapse); various antipsychotics given ‐ perazine, fluphenazine, levomepromazine, thioridazine, clozapine, haloperidol, haloperidol decanoate, perphenazine enanthate, fluphenazine decanoate, fluphenthixol decanoate, fluspirilene. Outcomes: no data by individual drug. |
| Barker 1969 | Allocation: randomised, crossover design. Participants: people with schizophrenia. Interventions: thioridazine versus pericyazine. Outcome: no pre‐crossover data. |
| Bigelow 1980 | Allocation: not randomised, case series. |
| Bishop 1965 | Allocation: not randomised, review. |
| Blum 1969 | Allocation: not randomised, allocation described as ' by consecutive assignment'. |
| Branchey 1978 | Allocation: unsure if randomised, study described as 'double blind crossover' but no further details. Participants: people with schizophrenia. Interventions: thioridazine versus fluphenazine. Outcomes: no usable pre‐crossover data. |
| Caffey 1963 | Allocation: randomised. Participants: people with schizophrenia. Interventions: dose comparison (thioridazine, chlorpromazine at same dose versus thioridazine, chlorpromazine at reduced dose and intermittent schedule). |
| Carpenter 1990 | Allocation: 'randomised block'. Participants: people with schizophrenia. Interventions: continuous versus targeted medication; generally participants continued on the same medication prior to study enrolment (fluphenazine decanoate, haloperidol, loxapine, thioridazine, molindone, thiothixene, fluphenazine hydrochloride, mesoridazine, trifluoperazine, chlorpromazine, perphenazine). Outcomes: no data by individual group. |
| Claghorn 1972 | Allocation: randomised. Participants: not schizophrenia, people with childhood psychiatric disorder. |
| Claveria 1975 | Allocation: not randomised, case series. |
| Cottereau 1979 | Allocation: not randomised, case series. |
| Cowley 1979 | Allocation: randomised. Participants: not schizophrenia, people with psychosis associated with organic brain syndrome. |
| Crowley 1981 | Allocation: randomised, crossover design. Participants: people with schizophrenia. Interventions: thioridazine versus thiothixene. Outcome: no pre‐crossover data. |
| Deutsch 1971 | Allocation: unsure if randomised, allocation not described ‐ not double blind. |
| Dillenkofer 1974 | Allocation: not randomised. |
| Downing 1963 | Allocation: randomised. Participants: people with schizophrenia. Interventions: thioridazine, chlorpromazine, fluphenazine, placebo. Outcome: no usable data. |
| Dubin 1985 | Allocation: randomised. Participants: <50% people with schizophrenia. Intervention: haloperidol versus thiothixene versus thioridazine. Outcome: no usable data. |
| Eccleston 1985 | Allocation: randomised. Participants: people with schizophrenia. Intervention: thioridazine versus propranolol. |
| Eitan 1992 | Allocation: unsure if randomised, study described as a 'double blind crossover' but no further details. Participants: people with schizophrenia. Intervention: thioridazine versus chlorpromazine, trifluoperazine or haloperidol. Outcome: no usable data. |
| Essock 1996 | Allocation: fully random in 84 participants, 'biased coin' randomisation with 2/3 likelihood of clozapine assignment for 143 participants. Participants: people with schizophrenia. Interventions: clozapine versus continuing treatment with typical neuroleptic; typical neuroleptic could be changed. Outcomes: no data by individual typical neuroleptic. |
| Fragoso Mendes 1965 | Allocation: not randomised, case series. |
| Friedman 1961 | Allocation: randomised, 'Latin square design'. Participants: people with schizophrenia. Interventions: thioridazine versus prochlorperazine or perphenazine. Outcome: no data from first allocation. |
| Gallant 1963 | Allocation: unsure if randomised, allocation not described. |
| Gallant 1966 | Allocation: unsure if randomised, allocation not described. |
| Gardos 1974 | Allocation: randomised. Participants: people with schizophrenia. Interventions: thiothixine versus chlorpromazine or continuing doctor's choice medication. |
| Geogotas 1981 | Allocation: randomised. Participants: people with schizophrenia. Interventions: thioridazine versus trebenzomine (unlicensed compound, uncertain efficacy). |
| Gerlach 1977 | Allocation: not randomised, case series. |
| Gillis 1977 | Allocation: not randomised, case series. |
| Goldberg 1965 | Allocation: randomised. Participants: people with schizophrenia. Interventions: thioridazine versus chlorpromazine, fluphenazine or placebo. Outcome: no usable data. |
| Goldberg 1967 | Allocation: not randomised, review. |
| Goldstein 1969 | Allocation: unsure if randomised, study described as 'double blind' but no further details. Participants: people with schizophrenia. Interventions: thioridazine and placebo, unclear if groups were parallel. Outcomes: no usable data. |
| Gonier 1970 | Allocation: quasi‐randomisation. |
| Gottschalk 1975 | Allocation: unsure if randomised, study described as 'double blind' but no further details. Participants: people with schizophrenia. Interventions: thioridazine plasma levels study versus placebo. Outcomes: no usable data. |
| Guo 1988 | Allocation: unsure if randomised, allocation not described. |
| Hanlon 1965 | Allocation: randomised. Participants: acutely disturbed new admissions, 84% psychotic. Interventions: thioridazine versus chlorpromazine, trifluoperazine, prochlorperazine, perphenazine, thiopropazate or fluphenazine. Outcomes: no usable data. |
| Hanlon 1975 | Allocation: randomised. Participants: acutely ill psychiatric hospital people. Intervention: thioridazine‐chlordiazepoxide, thioridazine‐imipramine, thioridazine‐placebo or any direct choice treatment by physician. Outcome: no usable data. |
| Hardeman 1970 | Allocation: not randomised. |
| Harris 1992 | Allocation: randomised. Participant: "psychiatric patients". Interventions: thioridazine versus haloperidol. Outcomes: no usable data. |
| Holden 1968 | Allocation: randomised. Participants: people with schizophrenia. Interventions: thioridazine combined with chlordiazepoxide. |
| Holden 1969 | Allocation: randomised. Participants: people with schizophrenia. Interventions: chlordiazepoxide + thioridazine versus chlordiazepoxide + thioridazine (using different dosages). |
| Hollister 1974 | Allocation: not randomised. |
| Judd 1973 | Allocation: not randomised. |
| Jus 1974 | Allocation: randomised. Participants: people with schizophrenia. Intervention: penfluridol versus thioridazine, penfluridol + chlorpromazine or thioridazine + chlorpromazine. Outcome: no usable data. |
| Klerman 1970 | Allocation: randomised. Participants: people with schizophrenia. Interventions: thioridazine versus placebo, chlorpromazine or fluphenazine. Outcomes: no usable data. |
| Kulkarni 1996 | Allocation: not randomised, case series. |
| Lambert 1982 | Allocation: not randomised, case series. |
| Lapolla 1969 | Allocation: randomisation unclear, double blind. Participants: people with schizophrenia. Interventions: thioridazine versus acetophenazine. Outcomes: no usable data. |
| Linn 1979 | Allocation: randomised. Participants: people with schizophrenia. Intervention: day hospital and neuroleptics versus outpatient care and neuroleptics; various neuroleptics given, chlorpromazine, haloperidol, thioridazine and other neuroleptics which were not specified. |
| Lonowski 1978 | Allocation: randomised. Participants: people with schizophrenia. Intervention: dose comparison, continuing neuroleptic at same dose (thioridazine, chlorpromazine, haloperidol) versus 50% dose reduction. |
| Mahmoud 2004 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone versus conventional antipsychotics (no individual data for thioridazine). |
| McCarthy 1986 | Allocation: not randomised, case series. |
| McClelland 1974 | Allocation: randomised. Participants: people with schizophrenia. Intervention: continuing versus withdrawing antiparkinsonian medication. |
| Mellinger 1965 | Allocation: not randomised, case series. |
| Melnyk 1966 | Allocation: randomised. Participants: people with schizophrenia. Interventions: continuing chlorpromazine or thioridazine versus substituting placebo. Outcomes: no usable data. |
| Meltzer 1969 | Allocation: not randomised, case series. |
| Menolascino 1985 | Allocation: not randomised, review. |
| Montero 1971 | Allocation: unsure if randomised, study described as double blind but no further details. Participants: people with schizophrenia. Interventions: thioridazine versus chlorpromazine + trifluoperazine. Outcomes: no data given. |
| Nelson 1975 | Allocation: randomised. Participants: people with schizophrenia. Intervention: self medication versus traditional drug administration during hospitalisation using either thioridazine or chlorpromazine. |
| Nordstrom 1996 | Allocation: not randomised, case series. |
| Overall 1964 | Allocation: unsure if randomised, allocation not described and double blinding not reported. |
| Payne 1974 | Allocation: randomised. Participants: people with schizophrenia. Interventions: thioridazine versus placebo. Outcomes: no data given. |
| Peselow 1989 | Allocation: unsure if randomised, allocation not described, double blind. Participants: people with schizophrenia and neuroleptic induced tardive dyskinesia. Intervention: not thioridazine (placebo versus GMI ganglioside). |
| Prien 1973 | Allocation: randomised. Participants: people with schizophrenia. Intervention: dose comparison ‐ continuos versus intermittent treatment of pre‐enrolment antipsychotic (thioridazine, chlorpromazine, trifluoperazine and perphenazine). |
| Rainaut 1975 | Allocation: unsure if randomised, study described as double blind but no further details. Participants: people with schizophrenia (n=36) and people reported as having psychosis (n=13) without schizophrenia. Interventions: thioridazine versus loxapine. Outcomes: no usable data. |
| Rasmussen 1976 | Allocation: not randomised, case series. |
| Reiter 1971 | Allocation: unsure if randomised, study described as double blind but no further details. Participants: people with schizophrenia. Intervention:chlorprothixene, mesoridazine and TPS‐23. Outcomes: no usable data. |
| Remr 1974 | Allocation: randomised, double blind crossover. Participants: people with schizophrenia. Interventions: thioridazine versus oxypertine. Outcomes: no usable data. |
| Remr 1975 | Allocation: unsure if randomised, study described as double blind but no further details. Participants: people with schizophrenia. Interventions: thioridazine versus oxypertine or placebo. Outcomes: no usable data. |
| Sandison 1960 | Allocation: not randomised, review. |
| Schrodt 1982 | Allocation: not randomised, case series. |
| Shavartsburd 1984 | Allocation: randomised (depending on prestudy medication, people already on haloperidol or thioridazine were kept on their prestudy medication). Participants: people with schizophrenia. Intervention: thioridazine versus haloperidol. Outcome: no usable data. |
| Smith 1974 | Allocation: randomised. Participants: not schizophrenia, people with chronic brain syndrome and senile psychosis. |
| Smith 1987 | Allocation: not randomised, case series. |
| Smythies 1974 | Allocation: unsure if randomised, study described as 'double‐blind crossover' but no further details. Participants: people with schizophrenia. Interventions: thioridazine versus pimozide. Outcome: no usable data. |
| Stucke 1969 | Allocation: not randomised, case series. |
| Tetreault 1969 | Allocation: randomised. Participants: people with schizophrenia. Interventions: TPS‐23 (derivative of thioridazine) versus chlorpromazine versus placebo. |
| Ucer 1969 | Allocation: unsure if randomised, no description of allocation given. Participants: not schizophrenia, emotionally disturbed children with mental retardation. |
| Vaisanen 1981 | Allocation: randomised. Participants: not schizophrenia, people with mental retardation and behavioural disturbance. |
| Van Wyck 1971 | Allocation: not randomised, case series. |
| Versiani 1968 | Allocation: randomised. Participants: people with psychosis associated with organic brain syndrome or mental retardation. |
| Vital‐Herne 1976 | Allocation: randomised. Participants: people with schizophrenia. Intervention: thioridazine versus mesoridazine. Outcome: no usable data. |
| Walinder 1976 | Allocation: not randomised, case series. |
| Wittenborn 1975 | Allocation: randomised. Participants: mainly people without schizophrenia. |
| Youssef 1991 | Allocation: unsure if randomised, allocation not described. Participants: people with psychosis. Intervention: thioridazine versus haloperidol, pimozide, flupenthixol decanoate, clopenthixol, haloperidol decanoate or fluphenazine decanoate. Outcomes: no usable data. |
Contributions of authors
Joe Reilly ‐ data extraction, support with producing report.
John Rathbone ‐ selected studies, extracted data, summated data, produced report.
Mark Fenton ‐ checked selection of abstracts and studies, support with producing report.
Alec Sultana ‐ prepared protocol, selected studies, extracted data, summated data, produced report.
Sources of support
Internal sources
Tees & North Yorkshire Health Services Trust, UK.
External sources
No sources of support supplied
Declarations of interest
None.
Edited (no change to conclusions)
References
References to studies included in this review
- Barker JC, Miller M. A double blind comparative trial of pericyazine and thioridazine in chronic schizophrenia. British Journal of Psychiatry 1969;115(519):169‐72. [DOI] [PubMed] [Google Scholar]
- Bergling R, Mjorndal T, Oreland L, Rapp W, Wold S. Plasma levels and clinical effects of thioridazine and thiothixene. Journal of Clinical Pharmacology 1975;15(2‐3):178‐86. [DOI] [PubMed] [Google Scholar]
- Borison RL, Sinha D, Haverstock S, McLarnon MC, Diamond BI. Efficacy and safety of tiospirone vs. haloperidol and thioridazine in a double‐blind, placebo‐controlled trial. Psychopharmacology Bulletin 1989;5(2):190‐3. [PubMed] [Google Scholar]
- Carranza Acevedo J, Angel Toro L, Gomez‐Mont F. Double blind evaluation of sulpiride and thioridazine in paranoid schizophrenia. 9th Congres de Neuropsychopharmacologie. Nouveaux Developements du Sulpiride; 1974 Jul 10, Paris. 1974. [MEDLINE: ] [Google Scholar]
- Chen J, Zhao J, Chen Y. A randomized control study of thioridazine and chlorpromazine on treatment of schizophrenia. Chinese Journal of Neurology and Psychiatry 1995;28:329‐32. [Google Scholar]
- Clark ML, Huber WK, Charalampous KD, Serafetinides EA, Trousdale W, Colmore JP. Drug treatment in newly admitted schizophrenic patients. Archives of General Psychiatry 1971;25(5):404‐9. [DOI] [PubMed] [Google Scholar]
- Clark ML, Huber WK, Hill D, Wood F, Costiloe JP. Pimozide in chronic Schizophrenic outpatients. Diseases of the Nervous System 1975;36:137‐41. [PubMed] [Google Scholar]
- Cohler J, Grinspoon L, Shader R, Chatterjee S. Behavioral correlates of the guessing game. Side effects and double‐blind studies. Archives of General Psychiatry 1966;15(3):279‐87. [DOI] [PubMed] [Google Scholar]
- Dufresne RL, Valentino D, Kass DJ. Thioridazine improves affective symptoms in schizophrenic patients. Psychopharmacology Bulletin 1993;29(2):249‐55. [PubMed] [Google Scholar]
- Evans JR, Rodnick EH, Goldstein MJ, Judd LL. Premorbid adjustment, phenothiazine treatment, and remission in acute schizophrenics. Archives of General Psychiatry 1972;27(4):486‐90. [DOI] [PubMed] [Google Scholar]
- Galbrecht CR, Klett CJ. Predicting response to phenothiazines: the right drug for the right patient. Journal of Nervous and Mental Disease 1968;147:173‐83. [DOI] [PubMed] [Google Scholar]
- Gallant. Piperacetazine vs. thioridazine‐1. Early Clinical Drug Evaluation Unit Reports1971. [MEDLINE: ] ; Gallant DM, Bishop MP. Quide vs. Mellaril in chronic schizophrenic patients. Current Therapeutic Research January 1972;14(1):10‐6. [PubMed] [Google Scholar]
- Gardos G, Tecce J, Harteman E, Bowers P, Cole J. Treatment with mesoridazine and thioridazine in chronic schizophrenia. I. Assessment of clinical and electrophysiologic responses in refractory hallucinating schizophrenics. Comprehensive Psychiatry 1978;19(6):517‐25. [DOI] [PubMed] [Google Scholar]
- Granacher RP, Douglas R. A comparison of thioridazine (mellaril) and thiothixene (navane) in the treatment of hospitalized psychotic patients. Current Therapeutic Research 1982;31:692‐705. [Google Scholar]
- Grinspoon L, Ewalt JR, Shader R. Long‐term treatment of chronic schizophrenia. A preliminary report. International Journal of Psychiatry 1967;4(2):116‐28. [PubMed] [Google Scholar]
- Gui‐yun G, Guoging Z. A double blind controlled study for the efficacy and the side effects of thioridazine and chlorpromazine in schizophrenia. Chinese Journal of Neurology and Psychiatry 1988;21(5):300‐2. [PubMed] [Google Scholar]
- Herrera John, Costa J, Sramek John, Heh C. The efficacy of sustained release thioridazine in the treatment of schizophrenic inpatients. Current Therapeutic Research 1990;48:1006‐11. [Google Scholar]
- Ju FenYing, Zhu DongSheng, Liu Yan Lu Wei hong, Wang XiuHua, Jiao YuMei, Wang WenZhong, Zhu WeiMing. The randomised control trial of thioridazine and sulpiride treating schizophrenia with negative symptoms. Sichuan Mental Health 1997;10(3):171‐72. [Google Scholar]
- Judah L, Murphree O, Seager L. Psychiatric response of geriatric‐psychiatric patients to Mellaril (TP‐21 Sandoz). American Journal of Psychiatry 1959;115:1118‐9. [DOI] [PubMed] [Google Scholar]
- Keks N, McGrath J, Lambert T, Catts S, Vaddadi K, Burrows G, Varghese F, George T, Hustig H, Burnett P, Kerr K, Zorbas A, Hill C, Steadman T, Johnson G, Lerbert B, Copolov D, Mackenzie M, Dillenbeck C. The Australian multicentre double‐blind comparative study of remoxipride and thioridazine in schizophrenia. Acta Psychiatrica Scandinavica 1994;90(5):358‐65. [DOI] [PubMed] [Google Scholar]; Lambert T, Keks N, McGrath J, Catts S, Hustig H, Vaddadi K, Burrows G, Varghese F, George T, Kerr K, Johnson G, Burnett P, Zorbas A, Hill. Remoxipride versus thioridazine in the treatment of first episodes of schizophrenia in drug‐naive patients ‐ A case for specific, low potency D2 antagonists. Human Psychopharmacology 1995;10(6):455‐60. [EMBASE 1996026897] [Google Scholar]
- Kramer M, Roth T, Salis PJ. Relative efficacy and safety of loxapine succinate (Loxitane(R)) and thioridazine hydrochloride (Mellaril(R)) in the treatment of acute schizophrenia. Current Therapeutic Research 1978;23(5, Pt 2):619‐31. [Google Scholar]
- Lasky JJ, Klett CJ, Caffey EM, Bennett JL, Rosenblum MP, Hollister LE. Drug treatment of schizophrenic patients. A comparative evaluation of chlorpromazine, chloprothixene, fluphenazine, reserpine, thioridazine and triflupromazine. Diseases of the Nervous System 1962;23:698‐706. [Google Scholar]
- Liu BingLang, Chen YY, Yang DS. Effects of thioridazine on schizophrenics and clinical utility of plasma levels. Chinese Journal of Neurology and Psychiatry 1994;27:364‐7. [Google Scholar]; Liu BingLun, Cheng YangGuang, Yang DeSen, YangLing, Zuo ChengYe. Thioridazine in the treatment of schizophrenia: a double‐blind controlled clinical trial compared with clozapine. Shanghai Archives of Psychiatry 1995;7(4):253‐55. [Google Scholar]
- McCreadie RG, Todd N, Livingston M, Eccleston D, Watt JA, Herrington RN, Tait D, Crocket G, Mitchell MJ, Huitfeldt B. A double‐blind comparative study of remoxipride and thioridazine in the acute phase of schizophrenia. Acta Psychiatrica Scandinavica Supplementum 1990;358:136‐7. [DOI] [PubMed] [Google Scholar]; McCreadie RG, Todd N, Livingston M, Eccleston D, Watt JA, Tait D, Crocket G, Mitchell MJ, Huitfeldt B. A double blind comparative study of remoxipride and thioridazine in the acute phase of schizophrenia. Acta Psychiatrica Scandinavica 1988;78(1):49‐56. [DOI] [PubMed] [Google Scholar]
- Mena A, Grayson H, Cohen S. A study of thioridazine and its side‐chain derivative, mesoridazine, in chronic male hospitalized psychiatric patients. Journal of New Drugs 1966;6(6):345‐8. [DOI] [PubMed] [Google Scholar]
- Miyakawa K, Mori H, Arai M, Yamauchi N, Hasue I, Mori H, Oana Y, Kamiya T, Shinozaki M, Satoh Y. Double blind controlled study of mesoridazine and thioridazine on schizophrenic inpatients. Clinical Evaluation 1973;1(2,3):181‐212. [Google Scholar]
- Montgomery SA, Green M, Rimon R, Heikkila L, Forsstrom R, Hirsch SR, Hallstrom C, Hippius H, Naber R, Khan MC, Sitsen JMA. Inadequate treatment response to des‐enkephalin‐gamma‐endorphin compared with thioridazine and placebo in schizophrenia. Acta Psychiatrica Scandinavica 1992;86(2):97‐103. [DOI] [PubMed] [Google Scholar]
- Cole JO, Goldberg SC, Klerman GL. Phenothiazine treatment in acute schizophrenia. Archives of general psychiatry 1964;10:246‐61. [PubMed] [Google Scholar]
- Nishikawa T, Tsuda A, Tanaka M, Koga I, Uchida Y. Prophylactic effects of neuroleptics in symptom‐free schizophrenics: roles of dopaminergic and noradrenergic blockers. Biological Psychiatry 1985;20:1161‐6. [DOI] [PubMed] [Google Scholar]
- Phanjoo‐AL, Link‐C. Remoxipride versus thioridazine in elderly psychotic patients. Acta Psychiatrica Scandinavica Supplementum 1990;358:181‐5. [DOI] [PubMed] [Google Scholar]
- Pi E, Sramek J, Johnson T, Herrera J, Heh C, Costa J, Cutler N, Anath J. Subjective neuroleptic response and treatment outcome under open and double blind conditions ‐ a preliminary report. Progressive Neuro‐Psychopharmacology and Biological Psychiatry 1990;14:971‐5. [DOI] [PubMed] [Google Scholar]
- Rada. Piperacetazine vs. thioridazine. Early Clinical Drug Evaluation Unit Reports1972. [MEDLINE: ] [DOI] [PubMed]; Rada RT, Donlon PT. Piperacetazine vs. thioridazine for the control of schizophrenia in outpatients. Psychosomatics 1972;13(6):373‐6. [DOI] [PubMed] [Google Scholar]
- Rasmussen K, Kirk I, Fairby A. Deposits in the lens and cornea of the eye during long term chlorpromazine medication. Acta Psyciatrica Scandinavica 1976;53:1‐6. [DOI] [PubMed] [Google Scholar]
- Realmuto GM, Erickson WD, Yellin AM, Hopwood JH, Greenberg LM. Clinical comparison of thiothixene and thioridazine in schizophrenic adolescents. American Journal of Psychiatry 1984;141(3):440‐2. [DOI] [PubMed] [Google Scholar]
- Schiele BC, Vestre ND, Stein KE. A comparison of thioridazine, trifluoperazine, chlorpromazine, and placebo: a double‐blind controlled study on the treatment of chronic hospitalized, schizophrenic patients. Journal of Clinical and Experimental Psychopathology 1961;22(3):151‐62. [MEDLINE: ] [PubMed] [Google Scholar]
- Somerville DM, Cohen PH, Graves GD. Phenothiazine side‐effects. Comparison of two major tranquillizers. British Journal of Psychiatry 1960;106:1417‐24. [Google Scholar]
- Stabenau JR, Grinols DR. A double‐blind comparison of thioridazine and chlorpromazine. Psychiatric Quarterly 1964;38(1):42‐63. [DOI] [PubMed] [Google Scholar]
- Wyk AJ, Marais GF. Chlorpromazine, clotiapine and thioridazine ‐ a comparative clinical trial on Bantu psychotic patients. South African Medical Journal 1971;45(34):945‐7. [PubMed] [Google Scholar]
- Vestre ND, Schiele BC. Differential drug effects of two phenothiazines (a controlled comparison of thioridazine and trifluoperazine in chronic schizophrenics). Diseases of the Nervous System 1970;31(12):821‐5. [PubMed] [Google Scholar]
- Weston MJ, Bentley R, Unwin A, Morris M, Harper MA. A comparative trial of haloperidol and thioridazine: management of chronic schizophrenia. Australia and New Zealand Journal of Psychiatry 1973;7(1):52‐7. [DOI] [PubMed] [Google Scholar]
- Wolpert A, Sheppard C, Merlis S. Thiothixene, thioridazine, and placebo in male chronic schizophrenic patients. Clinical Pharmacology and Therapeutics 1968;9(4):456‐64. [DOI] [PubMed] [Google Scholar]
- Zhang NaiXuan, Wang TianHua, Zhong TianYong. Efficacy comparison of Thioridazine and chlorpromazine treating type I schizophrenia. Journal of Guangdon Medical College 1999;17(2):122‐26. [Google Scholar]
References to studies excluded from this review
- Acker CW. An exploration of the autonomic effects of phenothiazines. Psychopharmacologia 1965;7(2 (Feb 15)):150‐8. [DOI] [PubMed] [Google Scholar]
- Altman H, Mehta D, Evenson RC, Sletten IW. Behavioral effects of drug therapy on psychogeriatric inpatients. I. Chlorpromazine and thioridazine. Journal of the American Geriatric Society 1973;21(6):241‐8. [DOI] [PubMed] [Google Scholar]
- Askar A, Rakhawy Y, El‐Grindy T. Thioridazine in treatment of psychiatric patients. Acta Psychiatrica Scandinavica Supplementum 1970;211:91. [MEDLINE: ] [Google Scholar]
- Bandelow B, Muller P, Frick U, Gaebel W, Linden M, Muller Spahn F, Pietzcker A, Tegeler J. Depressive syndromes in schizophrenic patients under neuroleptic therapy. European Archives of Psychiatry and Clinical Neuroscience 1992;241(5):291‐5. [DOI] [PubMed] [Google Scholar]
- Barker JC, Miller M. A double blind comparative trial of pericyazine and thioridazine in chronic schizophrenia. British Journal of Psychiatry 1969;115(519):169‐72. [DOI] [PubMed] [Google Scholar]
- Bigelow LB, Weinberger DR, Wyatt RJ. Synergism of combined lithium‐neuroleptic therapy: a double blind, placebo controlled case study. American Journal of Psychiatry 1981;138(1):81‐3. [DOI] [PubMed] [Google Scholar]
- Bishop MP, Gallant DM, Sykes TF. Extrapyramidal side effects and therapeutic response. Archives of General Psychiatry 1965;13:155‐62. [DOI] [PubMed] [Google Scholar]
- Blum RA, Livingston PB, Shader RI. Changes in cognition, attention and language in acute schizophrenia. Diseases of the Nervous System 1969;30(1):31‐6. [PubMed] [Google Scholar]
- Branchey MH, Lee JH, Amin R, Simpson GM. High‐ and low‐potency neuroleptics in elderly psychiatric patients. JAMA 1978 May 5;239:1860‐2. [PubMed] [Google Scholar]
- Caffey EM, Forrest IS, Frank TV, Klett CJ. Phenothiazine excretion in chronic schizophrenics. American Journal of Psychiatry 1963;120:578‐82. [DOI] [PubMed] [Google Scholar]
- Carpenter W, Hanlon T, Heinrichs D, Summerfelt A, Kirkpatrick B, Levine J, Buchanan R. Continuous versus targeted medication in schizophrenic outpatients: outcome results. American Journal of Psychiatry 1990;147:9. [DOI] [PubMed] [Google Scholar]
- Claghorn JL. A double‐blind comparison of haloperidol (Haldol) and thioridazine (Mellaril) in outpatient children. Current Therapeutic Research 1972;14(12):785‐9. [PubMed] [Google Scholar]
- Claveria LE, Teychenne PF, Calne‐DB, Haskayne L, Petrie A, Pallis CA, Lodge Patch IC. Tardive dyskinesia treated with pimozide. Journal of Neurological Science 1975 Apr;24(4):393‐401. [DOI] [PubMed] [Google Scholar]
- Cottereau M, Porier M, Loo H, Deniker P. no English title available [Le succinate de loxapine: un noveau neuroleptique]. L' Encephale 1979;5:251‐67. [PubMed] [Google Scholar]
- Cowley LM, Glen RS. Double‐blind study of thioridazine and haloperidol in geriatric patients with a psychosis associated with organic brain syndrome. Journal of Clinical Psychiatry 1979;40(10):411‐9. [PubMed] [Google Scholar]
- Crowley TJ, Hydinger Macdonald M. Motility, Parkinsonism, and prolactin with thiothixene and thioridazine. Archives of General Psychiatry 1981 June;38:668‐75. [DOI] [PubMed] [Google Scholar]
- Deutsch M, Ananth JV, Ban TA. A clinical study with propericiazine in chronic psychotic patients. Current Therapeutic Research Clinical and Experimental 1971;13(6):353‐8. [PubMed] [Google Scholar]
- Dillenkoffer RL, George RB, Bishop‐MP, Gallant‐DM. Electrocardiographic evaluation of thiothixene: a double‐blind comparison with thioridazine. Advances in Biochemistry and Psychopharmacology 1974;9:487‐95. [PubMed] [Google Scholar]
- Downing RW, Ebert JN, Shubrooks SJ. Effect of phenothiazines on the thinking of acute schizophrenics. Perceptual and Motor Skills 1963;17(2):511‐20. [DOI] [PubMed] [Google Scholar]
- Dubin WR, Waxman HM, Weiss KJ, Ramchandani D, Tavani Petrone C. Rapid tranquillization: the efficacy of oral concentrate. Journal of Clinical Psychiatry 1985;46(11):475‐8. [PubMed] [Google Scholar]
- Eccleston D, Fairbairn AF, Hassanyeh F, McClelland HA, Stephens DA. The effect of propranolol and thioridazine on positive and negative symptoms of schizophrenia. British Journal of Psychiatry 1985;147:623‐30. [DOI] [PubMed] [Google Scholar]
- Eitan N, Levin Y, Ben Artzi E, Levy A, Neumann M. Effects of antipsychotic drugs on memory functions of schizophrenic patients. Acta Psychiatrica Scandinavica 1992;85(1):74‐6. [DOI] [PubMed] [Google Scholar]
- Essock SM, Hargreaves WA, Covell NH, Goethe J. Clozapine's effectiveness for patients in state hospitals: results from a randomized trial. Psychopharmacology Bulletin 1996;32(4):683‐97. [PubMed] [Google Scholar]
- Fragoso Mendes, JM, Nunes Ferreira CN. Clinical trial with TPN 12 (thioridazine metabolite) in the treatment of schizophrenia [Ensaio clinico com o TPN 12 (metabolito da tioridazina) no tratamento da esquizofrenia]. Actas Luso Esp Neurol Psiquiatr 1965;3:139‐60. [PubMed] [Google Scholar]
- Friedman DX, Jong J. Treshold for drug induced akathisia. American Journal of Psychiatry 1964;vol unknown:930. [DOI] [PubMed] [Google Scholar]
- Gallant DM, Bishop MP, Timmons E, Steele CA. A controlled evaluation of trifluperidol: a new potent psychopharmacologic agent. Current Therapeutic Research 1963;5(9):463‐71. [PubMed] [Google Scholar]
- Gallant DM, Bishop MP, Timmons E, Gould AR. Thiothixene (P‐4657B): a controlled evaluation in chronic schizophrenic patients. Current Therapeutic Research 1966 Apr;8:153‐8. [PubMed] [Google Scholar]
- Gardos G. Are antipsychotic drugs interchangeable?. Journal of Nervous and Mental Diseases 1974;159(5):343‐8. [DOI] [PubMed] [Google Scholar]
- Georgotas A, Gerbino L, Jordan B, McCarthy M, Gershon S, Kleinberg D, Lautin A, Stanley M, Rotrosen J. A double blind comparison of trebenzomine and thioridazine in the treatment of schizophrenia. Psychopharmacology 1981 May;73:292‐4. [DOI] [PubMed] [Google Scholar]
- Gerlach J, Rasmussen PT, Hansen L, Kristjansen P. Antiparkinsonian agents and long‐term neuroleptic treatment. Effect of G 31.406, orphenadrine, and placebo on parkinsonism, schizophrenic symptoms, depression and anxiety. Acta Psychiatrica Scandinavica 1977;55(4):251‐60. [DOI] [PubMed] [Google Scholar]
- Gillis JS. The effects of selected antipsychotic drugs of human judgement. Current Therapeutic Research Clinical and Experimental 1977;21:224‐32. [PubMed] [Google Scholar]
- Goldberg SC, Klerman GL, Cole JO. Changes in schizophrenia psychopathology and ward behaviour as a function of phenothiazine treatment. British Journal of Psychiatry 1965 Feb;111:120‐33. [DOI] [PubMed] [Google Scholar]
- Goldberg SC, Mattsson N, Cole JO, Klerman GL. Prediction of improvement in schizophrenia under four phenothiazines. Archives of General Psychiatry 1967;16:107‐17. [DOI] [PubMed] [Google Scholar]
- Goldstein MJ, Judd LL, Rodnick EH, LaPolla A. Psychophysiological and behavioral effects of phenothiazine administration in acute schizophrenics as a function of premorbid status. Journal of Psychiatric Research 1969;6(4):271‐87. [DOI] [PubMed] [Google Scholar]
- Gonier T, Schiele BC, Vestre‐ND. A comparison of haloperidol and thioridazine HC1 in chronic treatment‐resistant schizophrenics. Behavioral Neuropsychiatry 1970 Jun‐Jul;2(3):47‐9. [PubMed] [Google Scholar]
- Gottschalk L A, Biener R, Noble EP, Birch H, Wilbert DE, Heiser JF. Thioridazine plasma levels and clinical response. Comprehensive Psychiatry 1975;4:323‐37. [DOI] [PubMed] [Google Scholar]
- Guo GY. A double‐blind controlled study of the efficacy and side effects of thioridazine and chlorpromazine in schizophrenia. Chung Hua Shen Ching Ching Shen Ko Tsa Chih Chinese Journal of Neurology and Psychiatry 1988 October;21:300‐2, 319‐20. [PubMed] [Google Scholar]
- Hanlon TE, Michaux MH, Ota KY, Shaffer JW, Kurland AA. The comparative effectiveness of eight phenothiazines. Psychopharmacologia 1965 Feb 15;7(2):89‐106. [DOI] [PubMed] [Google Scholar]
- Hanlon TE, Blatchley RJ, Kurland AA. Effects of control techniques on therapeutic outcome in a controlled clinical trial. International Pharmacopsychiatry 1975;10(3):169‐76. [DOI] [PubMed] [Google Scholar]
- Hardeman WJ, Bakker SJ. Double blind comparative study of the effects of thiothixene and thioridazine in twenty chronic schizophrenic patients. Psychiatria Neurologia Neurochirurgia 1970 Jan‐Feb;73(1):1‐7. [PubMed] [Google Scholar]
- Harris MJ, Panton D, Caligiuri MP, Krull AJ, Tran Johnson TK, Jeste DV. High incidence of tardive dyskinesia in older outpatients on low doses of neuroleptics. Psychopharmacology Bulletin 1992;28(1):87‐92. [MEDLINE: ] [PubMed] [Google Scholar]
- Holden JM, Itil TM, Keskiner A, Fink M. Thioridazine and chlordiazepoxide, alone and combined, in the treatment of chronic schizophrenia. Comprehensive Psychiatry 1968;9(6):633‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Holden JM, Itil TM. Laboratory changes with chlordiazepoxide and thioridazine, alone and combined. Canadian Psychiatric Association Journal 1969;14(3):299‐301. [DOI] [PubMed] [Google Scholar]
- Hollister LE, Overall JE, Kimbell I Jr, Pokorny A. Specific indications for different classes of phenothiazines. Archives of General Psychiatry 1974;30(1):94‐9. [DOI] [PubMed] [Google Scholar]
- Judd LL, Goldstein MJ, Rodnick EH, Jackson NL Jr. Phenothiazine effects in good premorbid schizophrenics divided into paranoid‐nonparanoid status. Archives of General Psychiatry 1973;29:207‐21. [DOI] [PubMed] [Google Scholar]
- Jus A, Pineau R, Jus K, Villeneuve A, Gautier J, Pires‐P, Drolet A, Cote M, Villeneuve R. Penfluridol: a long‐acting oral neuroleptic as therapeutic agent in chronic schizophrenia. Current Therapeutic Research Clinical and Experimental 1974;16(10):1041‐58. [PubMed] [Google Scholar]
- Klerman GL, Goldberg SG, Davis D. Relationship between the hospital milieu and the response to phenothiazines in the treatment of schizophrenics. Acta Psychiatrica Belgica 1970 Nov;70:716‐29. [PubMed] [Google Scholar]
- Kulkarni J, Castella A, Smith D, Taffe J, Keks N, Copolov D. A clinical trial of the effects of estrogens in acutely psychotic women. Schizophrenia Research 1996;20:247‐52. [DOI] [PubMed] [Google Scholar]
- Lambert PA, Chabannes JP, Tell GP, Schechter PJ, Koch‐Weser J. no english title available [Essai therapeutique du vinyl GABA , un inhibiteur de la GABA‐transanimase, dans les dyskinesies tardives induites par les neuroleptiques]. L'Encephale 1982;VII:371‐6. [PubMed] [Google Scholar]
- Lapolla. Acetophenazine versus thioridazine in the elderly. Psychopharmacology Bulletin 1969;6(3):101‐3. [JN: Psychopharmacology Bulletin] [Google Scholar]
- Linn MW, Caffey EM Jr, Klett CJ, Hogarty GE, Lamb‐HR. Day treatment and psychotropic drugs in the aftercare of schizophrenic patients. A Veterans Administration cooperative study. Archives of General Psychiatry 1979 Sep;36(10):1055‐66. [DOI] [PubMed] [Google Scholar]
- Lonowski DJ, Sterling FE, Kennedy JC. Gradual reduction of neuroleptic drugs among chronic schizophrenics. A double‐blind controlled study. Acta Psychiatrica Scandinavica 1978 Feb;57(2):97‐102. [DOI] [PubMed] [Google Scholar]
- Mahmoud RA, Engelhart LM, Janagap CC, Oster G, Ollendorf D. Risperidone versus conventional antipsychotics for schizophrenia and schizoaffective disorder: Symptoms, quality of life and resource use under customary clinical care. Clinical Drug Investigation 2004;24(5):275‐86. [EMBASE 2004231261] [DOI] [PubMed] [Google Scholar]
- McCarthy ST, John SM, McCarthy GL, Wollner L. The influence of chlormethiazole in comparison to thioridazine on body temperature and postural hypotension in healthy adults and healthy elderly volunteers. Acta Psychiatrica Scandinavica Supplementum 1986;329:40‐4. [DOI] [PubMed] [Google Scholar]
- McClelland HA, Blessed G, Bhate S, Ali N, Clarke PA. The abrupt withdrawal of antiparkinsonian drugs in schizophrenic patients. British Journal of Psychiatry 1974;124(579):151‐9. [DOI] [PubMed] [Google Scholar]
- Mellinger TJ. Serum concentrations of thioridazine after different oral medication forms. American Journal of Psychiatry 1965;122:1119‐22. [DOI] [PubMed] [Google Scholar]
- Melnyk WT, Worthington AG, Laverty SG. Abrupt withdrawal of chlorpromazine and thioridazine from schizophrenic in‐patients. Canadian Psychiatric Association Journal 1966;11(5):410‐3. [Google Scholar]
- Meltzer H, Shader R, Grinspoon L. The behavioral effects of Nicotinamine Adenine Dinucleotide in chronic schizophrenia. Psychopharmacologia 1969;15:144‐52. [DOI] [PubMed] [Google Scholar]
- Menolascino FJ, Ruedrich SL, Golden CJ, Wilson TE. Diagnosis and pharmacotherapy of schizophrenia in the retarded. Psychopharmacology Bulletin 1985;21(2):316‐22. [PubMed] [Google Scholar]
- Montero E. Thioridazine compared with a combination of chlorpromazine and trifluoperazine hydrochloride in schizophrenic patients. 5th World Congress of Psychiatry, Ciudad de Mexico. November 28 ‐ December 4, 1971:878. [Google Scholar]
- Nelson AA Jr, Gold BH, Hutchinson RA, Benezra E. Drug default among schizophrenic patients. American Journal of Hospital Pharmacology 1975 Dec;32(12):1237‐42. [PubMed] [Google Scholar]
- Nordström A, Nyberg S, Farde L. Dopamine and serotonin receptor occupancy in patients treated with antipsychotics. 8th Congress of the Association of European Psychiatrists, London, UK. 1996. [Google Scholar]
- Overall JE, Hollister LE, Meyer F, Kimbell I, Shelton J. Imipramine and thioridazine in depressed and schizophrenic patients. Are there specific antidepressant drugs?. Journal of the American Medical Association 1964 Aug;189:605‐8. [DOI] [PubMed] [Google Scholar]
- Payne RW. Phenothiazine and Schizophrenic Thought Disorder. Unknown source 1974;10:69. [Google Scholar]
- Peselow ED, Irons S, Rotrosen J, Teresa Alonso M, Dorsey F. GMI as a potential treatment in tardive dyskinisia. Psychopharmacology Bulletin 1989;25(2):277‐80. [PubMed] [Google Scholar]
- Prien RF, Gillis RD, Caffey EM Jr. Intermittent pharmacotherapy in chronic schizophrenia. Hospital and Community Psychiatry 1973 May;24(5):317‐22. [DOI] [PubMed] [Google Scholar]
- Rainaut J. Statistical analysis of the activity of loxapine and thioridazine in a double‐blind study [Analyse statistique de l'etude comparative double insu de l'activite de la loxapine et de la thioridazine]. Encephale 1975;1(2):147‐60. [PubMed] [Google Scholar]
- Rasmussen K, Kirk L, Faurbye A. Deposits in the lens and cornea of the eye during long‐term chlorpromazine medication. Acta Psychiatrica Scandinavica 1976 Jan;53:1‐6. [DOI] [PubMed] [Google Scholar]
- Reiter L, Hofmann G. [Differenzierte Indikationsstellung bei akuten psychotischen Zustõnden und Exazerbationen anhand einer kontrollierten Vergleichsstudie mit TPS 23 (Mesoridazin)]. V World Congress of Psychiatry, Ciudad de Mexico 28 N. 1971. [Google Scholar]
- Remr J, Nekolova J, Heinzl Z. A comparison of the therapeutic effect of oxypertine and thioridazine in chronic schizophrenia (a controlled trial). Activitas Nervosa Superior 1974;4:261‐2. [PubMed] [Google Scholar]
- Remr J, Nekolova J, Heinzl Z. Comparison of the effect of oxypertine and thioridazine on sensorimotor activity in chronic schizophrenics: a controlled study. Activitas Nervosa Superior 1975;17(4):213‐4. [PubMed] [Google Scholar]
- Sandison RA, Whitelaw E, Currie JDC. Clinical trials with Melleril (TP21) in the treatment of schizophrenia. A two‐year study. British Journal of Psychiatry 1960 Apr;106:732‐41. [DOI] [PubMed] [Google Scholar]
- Schrodt Jr GR, Wright JH, Simpson R, Moore DP, Sheldon C. Treatment of tardive dyskinesia with propranolol. Journal of Clinical Psychiatry 1982;43(8I):328‐31. [PubMed] [Google Scholar]
- Shavartsburd A, Sajadi C, Morton V, Mirabi M, Gordon J, Smith RC. Blood levels of haloperidol and thioridazine during maintenance neuroleptic treatment of schizophrenic outpatients. Journal of Clinical Psychopharmacology 1984 Aug;4(4):194‐8. [PubMed] [Google Scholar]
- Smith GR, Taylor CW, Linkous P. Haloperidol versus thioridazine for the treatment of psychogeriatric patients: a double‐blind clinical trial. Psychosomatics 1974;15(3):134‐8. [Google Scholar]
- Smith RC, Baumgartner R, Ravichandran GK, Largen J, Calderon M, Burd A, Mauldin M. Cortical atrophy and white matter density in the brains of schizophrenics and clinical response to neuroleptics. Acta Psychiatrica Scandinavica 1987 Jan;75:11‐9. [DOI] [PubMed] [Google Scholar]
- Smythies JR, Beaton JM. A pilot study of pimozide in schizophrenia. Journal of Psychiatric Research 1974;11:71‐2. [DOI] [PubMed] [Google Scholar]
- Stucke W. Report on clinical trials of a new neuroleptic drug [Klinischer Erfahrungsbericht uber ein neues Neuroleptikum]. Medizinische Welt 1969 Feb;7:356‐9. [PubMed] [Google Scholar]
- Tetreault L. Comparative study of 2 drugs and a placebo in chronic schizophrenia [Etude comparative de deux medicaments et d'un placebo chez le schizophrene chronique]. Actualites Pharmacologiques 1969;22:1‐8. [PubMed] [Google Scholar]
- Ucer E, Kreger KC. A double‐blind study comparing haloperidol with thioridazine in emotionally disturbed, mentally retarded children. Current Therapeutic Research Clinical and Experimental 1969 May;11:278‐83. [PubMed] [Google Scholar]
- Vaisanen K, Viukari M, Rimon R, Raisanen P. Haloperidol, thioridazine and placebo in mentally subnormal patients ‐ serum levels and clinical effects. Acta Psychiatrica Scandinavica 1981 Mar;63:262‐71. [DOI] [PubMed] [Google Scholar]
- Wyk AJ, Marais GF. Chlorpromazine, clotiapine and thioridazine‐‐a comparative clinical trial on Bantu psychotic patients. South African Medical Journal 1971 Aug 28;45(34):945‐7. [PubMed] [Google Scholar]
- Versiani M, Silva JAR, Mundim FD. Loxapine versus thioridazine in the treatment of organic psychosis. Journal of Internal Medical Research 1980;8:22. [DOI] [PubMed] [Google Scholar]
- Vital‐Herne J, Gerbino L, Kay R, Katz IR, Opler LA. Mesoridazine and thioridazine: clinical effects and blood levels in refractory schizophrenics. Journal of Clinical Psychiatry 1986 Jul;47(7):375‐9. [PubMed] [Google Scholar]
- Walinder J, Skot A, Carlsson A, Roos BE. Potentiation by metyrosine of thioridazine effects in chronic schizophrenics. A long‐term trial using double‐blind crossover technique. Archives of General Psychiatry 1976 Apr;33(4):501‐5. [DOI] [PubMed] [Google Scholar]
- Wittenborn JR, Kiremitci N. A comparison of antidepressant medications in neurotic and psychotic patients. Archives of General Psychiatry 1975 Sep;32(9):1172‐6. [DOI] [PubMed] [Google Scholar]
- Youssef HA. Duration of neuroleptic treatment and relapse rate: a 5‐year follow‐up study with haloperidol decanoate. Clinical Neuropharmacology 1991;14:16‐21; discussion S22‐3. [PubMed] [Google Scholar]
References to studies awaiting assessment
- Tanimukai H, Inui M, Takahashi H, Kaneko Z. A double blind comparison of sulpiride with thioridazine on schizophrenia. Seishin Igaku (Clinical Psychiatry) 1973;15(2):197‐207. [MEDLINE: ] [Google Scholar]
Additional references
- Altman DG, Bland JM. Detecting skewness from summary information. British Medical Journal 1996;313:1200. [THI020600] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annane D, Girard F, Fabre JE, Raphael JC, Gajdos P. Near fatal case of self‐poisoning with thioridazine. Intensive Care Medicine 1996 Dec;22(12):1463‐4. [DOI] [PubMed] [Google Scholar]
- Assie MB, Sleight AJ, Koek W Eur J. Biphasic displacement of [3H]YM‐09151‐2 binding in the rat brain by thioridazine, risperidone and clozapine, but not by other antipsychotics. Pharmacology 1993;237(2‐3 (Jun 24)):183‐9. [DOI] [PubMed] [Google Scholar]
- Bain TA, Healy D, Shorter E (eds). The rise of psychopharmacology and the story of CINP. Budapest: Animula Publishing House, 1998. [Google Scholar]
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, Pitkin R, Rennie D, Schulz KF, Simel D, Stroup DF. Improving the quality of reporting of randomized controlled trials: the CONSORT statement. JAMA 1996;276(8):637‐9. [DOI] [PubMed] [Google Scholar]
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- British Medical Association, Royal Pharmaceutical Society of Great Britain. British National Formulary Number 35. The Pharmaceutical Press, 1998. [Google Scholar]
- Borison RL, Diamond BI. Regional selectivity of neuroleptic drugs: an argument for site specificity. Brain Research Bulletin 1983 Aug;11(2):215‐8. [DOI] [PubMed] [Google Scholar]
- Bowers MB. Thioridazine: central dopamine turnover and clinical effects of antipsychotic drugs. Journal of Clinical Pharmacolgy and Therapeutics 1975;17(1):73‐8. [DOI] [PubMed] [Google Scholar]
- Buckley NA, Whyte IM, Dawson AH. Cardiotoxicity more common in thioridazine overdose than with other neuroleptics. Journal of Toxicology ‐ Clinical Toxicology 1995;33(3):199‐204. [DOI] [PubMed] [Google Scholar]
- Caldwell CB, Gottesman II. Schizophrenia ‐ a high‐risk factor for suicide: clues to risk reduction. Suicide and Life‐Threatening Behavior 1992;22:479‐93. [PubMed] [Google Scholar]
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
- Drolet B, Vincent F, Rail J, Chahine M, Deschenes D, Nadeau S, Khalifa M, Hamelin BA, Turgeon J. Thioridazine lengthens repolarization of cardiac ventricular myocytes by blocking the delayed rectifier potassium current. Journal of Pharmacology and Experimental Therapeutics 1999 Mar;288(3):1261‐8. [PubMed] [Google Scholar]
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott J, Spitzer RL, Fleiss JL. The Global Assessment Scale. Archives of General Psychiatry 1976;33:766‐71. [DOI] [PubMed] [Google Scholar]
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
- Guy W. ECDEU Assessment manual for psychopharmacology. Rockville, MD: National Institute of Mental Health. DHEW publication No (ADM), 1976:124‐585. [Google Scholar]
- Heiman EM. Cardiac toxicity with thioridazine‐tricyclic antidepressant combination. Journal Of Nervous And Mental Disease 1977 Aug;165(2):139‐43. [DOI] [PubMed] [Google Scholar]
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]. The Cochrane Library. Chichester: John Wiley & Sons, Ltd, 2005, issue Issue 3.
- Hollister LE. Electrocardiographic screening in psychiatric patients. The Journal of Clinical Psychiatry 1995 Jan;56(1):26‐9. [PubMed] [Google Scholar]
- Honingfeld G, Klett CJ. The Nurses Observation Scale for Inpatient Evaluation (NOSIE): a new scale for measuring improvement in chronic schizophrenia. Journal of Clinical Psychology 1965;21:65‐71. [DOI] [PubMed] [Google Scholar]
- Jadad A, Moore A, Carrol D, Jenkinson C, Reynolds DJM, Gavanagh DJ, Quay HJ. Assessing the quality of reports of randomised controlled trials: is blinding necessary?. Controlled Clinical Trials 1996;17:1‐12. [DOI] [PubMed] [Google Scholar]
- Jusic N, Lader M. Post‐mortem antipsychotic drug concentrations and unexplained deaths. British Journal of Psychiatry 1994;165:787‐91. [DOI] [PubMed] [Google Scholar]
- King DJ. Neuroleptics and schizophrenia. Seminars in clinical psychopharmacology. Gaskell1995:259‐327.
- Lipscomb PA. Cardiovascular side effects of phenothiazines and tricyclic antidepressants. A review with precautionary measures. Postgraduate Medicine 1980;67(3):189‐92, 195‐6. [DOI] [PubMed] [Google Scholar]
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
- Mehtonen OP, Aranko K, Malkonen L. A study of sudden death associated with the use of antipsychotic or antidepressant drugs. Acta Psychiatrica Scandinavica 1991;84:58‐64. [DOI] [PubMed] [Google Scholar]
- Medicines and Healthcare products Regulatory Agency. http://www.mhra.gov.uk.
- Moreau A, Jones BD, Banno V. Chronic central anticholinergic toxicity in manic depressive illness mimicking dementia. Canadian Journal of Psychiatry 1986;31(4 (May)):339‐41. [DOI] [PubMed] [Google Scholar]
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
- Psychotropics. http://www.psychotropics.dk/.
- Rennie IG. Clinically important ocular reactions to systemic drug therapy. Drug Safety 1993;9(3):196‐211. [DOI] [PubMed] [Google Scholar]
- Roden DM. Torsade de pointes. Clinical Cardiology 1993;16(9):683‐6. [DOI] [PubMed] [Google Scholar]
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
- Sedvall G, Pauli S, Farde L, Karlsson P, Nyberg S, Nordstrom AL. Recent developments in PET scan imaging of neuroreceptors in schizophrenia. Israel Journal of Psychiatry and Related Sciences 1995;32(1):22‐9. [PubMed] [Google Scholar]
- Seeman P, Ulpian C. Neuroleptics have identical potencies in human brain limbic and putamen regions. European Journal of Pharmacology 1983;94(1‐2):145‐8. [DOI] [PubMed] [Google Scholar]
- Stuck AE, Rubenstein LZ, Wieland D, Vandenbroucke JP, Irwig L, Macaskill P, et al. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1998;316:469. [PMC free article] [PubMed] [Google Scholar]
- Thornley B, Adams C. Content and quality of 2000 controlled trials in schizophrenia over 50 years. British Medical Journal 1998;317:1181‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornley B, Rathbone J, Adams CE, Awad G. Chlorpromazine versus placebo for schizophrenia (Cochrane Review). Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD000284.pub2] [DOI] [PubMed] [Google Scholar]
- Trevitt J, Atherton A, Aberman J, Salamone JD. Effects of subchronic administration of clozapine, thioridazine and haloperidol on tests related to extrapyramidal motor function in the rat. Psychopharmacology (Berl) 1998;137(1):61‐6. [DOI] [PubMed] [Google Scholar]
- Turner WM, Tsuang MT. The impact of substance abuse on the course and outcome of schizophrenia. Schizophrenia Bulletin 1990;16:18. [DOI] [PubMed] [Google Scholar]
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
References to other published versions of this review
- A Sultana, J Reilly, M Fenton. Thioridazine for schizophrenia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD001944.pub2] [DOI] [PubMed] [Google Scholar]