

Abstract
Background
Acquired severe aplastic anemia is a rare and potentially fatal disease, which is characterized by hypocellular bone marrow and pancytopenia. The major signs and symptoms are severe infections, bleeding, and exhaustion. First‐line allogeneic hematopoietic stem cell transplantation (HSCT) of a human leukocyte antigen (HLA)‐matched sibling donor (MSD) is a treatment for newly diagnosed patients with severe aplastic anemia. First‐line treatment with ciclosporin and/or antithymocyte or antilymphocyte globulin (as first‐line immunosuppressive therapy) is an alternative to MSD‐HSCT and is indicated for patients where no MSD is found.
Objectives
To evaluate the effectiveness and adverse events of first‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors compared to first‐line immunosuppressive therapy including ciclosporin and/or antithymocyte or antilymphocyte globulin in patients with acquired severe aplastic anemia.
Search methods
We searched the electronic databases MEDLINE (Ovid), EMBASE (Ovid), and The Cochrane Library CENTRAL (Wiley) for published articles from 1946 to 22 April 2013. Further searches included trial registries, reference lists of recent reviews, and author contacts.
Selection criteria
The following prospective study designs were eligible for inclusion: randomized controlled trials (RCTs) and non‐randomized controlled trials if the allocation of patients to treatment groups was consistent with 'Mendelian randomization'. We included participants with newly diagnosed severe aplastic anemia who received MSD‐HSCT or immunosuppressive therapy without prior HSCT or immunosuppressive therapy, and with a minimum of five participants per treatment group. We did not apply limits on publication year or languages.
Data collection and analysis
Two review authors abstracted the data on study and patient characteristics and assessed the risk of bias independently. We resolved differences by discussion or by appeal to a third review author. The primary outcome was overall mortality. Secondary outcomes were treatment‐related mortality, graft failure, no response to first‐line immunosuppressive therapy, graft‐versus‐host‐disease (GVHD), relapse after initial successful treatment, secondary clonal and malignant disease, health‐related quality of life, and performance score.
Main results
We identified three trials that met the inclusion criteria. None of these trials was a RCT. 302 participants are included in this review. The three included studies were prospectively conducted and had features consistent with the principle of 'Mendelian randomization' as defined in the present review. All studies had a high risk of bias due to the study design. All studies were conducted more than 10 years ago and may not be applicable to the standard of care of today. Primary and secondary outcome data showed no statistically significant difference between treatment groups. We present results for first‐line allogeneic hematopoietic stem cell transplantation of an HLA‐matched sibling donor, which we denote as the MSD‐HSCT group, versus first‐line treatment with ciclosporin and/or antithymocyte or antilymphocyte globulin, which we denote as the immunosuppressive therapy group in the following section.
The pooled hazard ratio for overall mortality for the MSD‐HSCT group versus the immunosuppressive therapy group was 0.95 (95% confidence interval 0.43 to 2.12, P = 0.90, low quality evidence). Therefore, overall mortality was not statistically significantly different between the groups. Treatment‐related mortality ranged from 20% to 42% for the MSD‐HSCT group and was not reported for the immunosuppressive therapy group (very low quality evidence). The authors reported graft failure from 3% to 16% for the MSD‐HSCT group and GVHD from 26% to 51% (both endpoints not applicable for the immunosuppressive therapy group, very low quality evidence). The authors did not report any data on response and relapse for the MSD‐HSCT group. For the immunosuppressive therapy group, the studies reported no response from 15% (not time point stated) to 64% (three months) and relapse in one of eight responders after immunosuppressive therapy at 5.5 years (very low quality evidence). The authors reported secondary clonal disease or malignancies for the MSD‐HSCT group versus the immunosuppressive therapy group in 1 of 34 versus 0 of 22 patients in one study and in 0 of 28 versus 4 of 86 patients in the other study (low quality evidence). None of the included studies addressed health‐related quality of life. The percentage of the evaluated patients with a Karnofsky performance status score in the range of 71% to 100% was 92% in the MSD‐HSCT group and 46% in the immunosuppressive therapy group.
Authors' conclusions
There are insufficient and biased data that do not allow any conclusions to be made about the comparative effectiveness of first‐line allogeneic hematopoietic stem cell transplantation of an HLA‐matched sibling donor and first‐line treatment with ciclosporin and/or antithymocyte or antilymphocyte globulin (as first‐line immunosuppressive therapy). We are unable to make firm recommendations regarding the choice of intervention for treatment of acquired severe aplastic anemia.
Keywords: Humans; Siblings; Anemia, Aplastic; Anemia, Aplastic/immunology; Anemia, Aplastic/mortality; Anemia, Aplastic/therapy; Antilymphocyte Serum; Antilymphocyte Serum/therapeutic use; Cyclosporine; Cyclosporine/therapeutic use; Graft Rejection; Graft Rejection/immunology; Graft vs Host Disease; Graft vs Host Disease/immunology; HLA Antigens; HLA Antigens/immunology; Hematopoietic Stem Cell Transplantation; Hematopoietic Stem Cell Transplantation/adverse effects; Hematopoietic Stem Cell Transplantation/methods; Hematopoietic Stem Cell Transplantation/mortality; Histocompatibility; Histocompatibility/immunology; Immunosuppressive Agents; Immunosuppressive Agents/therapeutic use; Mendelian Randomization Analysis; T‐Lymphocytes; T‐Lymphocytes/immunology; Transplantation, Homologous
Plain language summary
Stem cell transplantation of sibling donors compared with specific immunosuppressive therapy for acquired severe aplastic anemia
Acquired severe aplastic anemia is rare. Stem cells from the bone marrow usually replace naturally dying blood cells in the peripheral blood. Severe aplastic anemia is probably caused by an irregular, attacking immune response against these blood producing stem cells within the body. If supplies are not maintained, functional blood cells are lacking and infections, bleeding, and exhaustion will occur. Patients may experience paleness, weakness, fatigue, and shortness of breath. Disease progression is associated with severe infections, which are a major cause of death.
The transplantation of stem cells from a human leukocyte antigen (HLA)‐matched sibling donor without prior therapy (first‐line therapy) is a treatment option for newly diagnosed patients with severe aplastic anemia. An HLA‐matched sibling donor, a brother or a sister, serves as a donor of stem cells that carry identical (matched) genetic characteristics to the HLA genes. The harvested cells are transfused intravenously and produce new blood cells. Problems may arise when the cells do not settle down sufficiently to produce blood cells (graft failure) or if the donor immune cells recognize body cells of the recipient as foreign and attack them (graft‐versus‐host disease). Both problems may lead to early death.
The application of the drugs ciclosporin and/or antithymocyte or antilymphocyte globulin as immunosuppressive therapy without prior therapy (i.e. as first‐line therapy) is an alternative to transplantation and can be used for patients where no HLA‐matched sibling donor is found. Immunosuppressive therapy means the drugs suppress reactions of the immune system. The aim is to reduce abnormal immune reactions. Problems may arise when patients do not respond well or show no response at all.
We identified three studies meeting our quality criteria for inclusion in the review. All had methodological limitations meaning that we could not draw firm conclusions. With respect to the primary outcomes, they showed ambiguous results for overall mortality when comparing treatment arms: one study favored transplantation and two studies favored treatment with ciclosporin and/or antithymocyte or antilymphocyte globulin. Treatment‐related mortality, that is death caused by complications of the treatment, was considerable for patients in the transplantation arm. Treatment failure, that is no response to treatment, was substantial for patients in the ciclosporin and/or antithymocyte or antilymphocyte globulin arm. Graft failure was reported for 3% to 16% and graft‐versus‐host‐disease for 26% to 51% of the transplanted patients. Because the data are scarce and biased it is not possible to determine which treatment is better ‐ transplantation for HLA‐matched sibling donors or ciclosporin and/or antithymocyte or antilymphocyte globulin for patients that do not have such a sibling.
Several reasons make it highly probable that there will not be any good evidence comparing these interventions in the future. One reason is that randomized controlled trials are unlikely to be conducted due to ethical constraints and strong patient and clinician preferences. Studies with 'Mendelian randomization' could provide some good evidence in theory, however, in practice they are difficult to conduct properly and have been disappointing. 'Mendelian randomization' means the view that nature itself has already 'randomized' the paternal and maternal part of a gene given that donor and recipient are siblings. Another reason is that the outcome of transplantation has improved considerably. This is true for matched donor transplantation including both related and unrelated donors. It means that ciclosporin and/or antithymocyte or antilymphocyte globulin may not be a first treatment choice if a matched sibling or even a matched unrelated donor is available.
Summary of findings
Summary of findings for the main comparison. First‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors compared with first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin for acquired severe aplastic anemia.
| First‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors compared with first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin for acquired severe aplastic anemia | ||||||
| Patient or population: patients with acquired SAA Settings: hospital Intervention: first‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors (MSD‐HSCT) Comparison: first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin (IST) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| First‐line ciclosporin and/or antithymocyte or antilymphocyte globulin (IST) | First‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors (MSD‐HSCT) | |||||
| Overall mortality Follow‐up: 2 years | Moderate1 | HR 0.95 (0.45 to 1.91) | 203 (3 studies) | ⊕⊕⊝⊝ low2,3 | ||
| 31 per 100 | 30 per 100 (15 to 51) | |||||
|
Treatment‐related mortality Follow‐up: not reported |
See comment | See comment | Not estimable | 54 (2 studies4) | ⊕⊝⊝⊝ very low4 | Case series; 15 of 54 of the MSD‐HSCT group affected |
| Health‐related quality of life | See comment | See comment | Not estimable | 0 (0) | See comment | No studies identified |
|
Graft failure Follow‐up: not reported |
See comment | See comment | Not estimable | 54 (2 studies4) | ⊕⊝⊝⊝ very low4 | Case series; 4 of 54 of the MSD‐HSCT group affected |
|
No response to IST Follow‐up: not reported |
See comment | See comment | Not estimable | 35 (2 studies4) | ⊕⊝⊝⊝ very low4 | Case series; 16 of 35 of the IST group affected |
|
Graft‐versus‐host disease Follow‐up: not reported |
See comment | See comment | Not estimable | 52 (2 studies4) | ⊕⊝⊝⊝ very low4 | Case series; 22 of 52 of the MSD‐HSCT group affected |
|
Secondary clonal disease or malignancies Follow‐up: not reported |
Moderate1 | Peto OR 0.54 (0.07 to 4) | 170 (2 studies) | ⊕⊕⊝⊝ low2,3 | ||
| 2 per 100 | 1 per 100 (0 to 10) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; IST: immunosuppressive therapy; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched (identical) sibling donor; OR: odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Median control group risk across studies. 2'Mendelian randomization'. 3Wide confidence interval. 4Case series.
Background
Description of the condition
Epidemiology
Acquired severe aplastic anemia is a rare (GARD 2012) and potentially fatal disease which is characterized by hypocellular bone marrow and pancytopenia; it mainly affects young adults. The incidence rate of aplastic anemia is estimated at 0.7 to 4.1 per million people per year (Kaufman 2006). The age‐standardized incidence rate (West Germany in 1987) is estimated at 2 per million children younger than 15 years of age per year (GCCR 2006).
Pathophysiology
The underlying pathophysiology is thought to be an aberrant immune response involving the T‐cell mediated destruction of hematopoietic stem cells. In most cases, the cause is unknown, although various triggers such as drugs, radiation, toxins, and viruses have been reported (Brodsky 2005; Killick 2000; Young 2006).
Signs and symptoms
The major signs and symptoms are severe infections, bleeding, and exhaustion. Infections are caused by the diminished number of granulocytes and lymphocytes to prevent bacteria and viruses. The risk of spontaneous, longer‐lasting and major bleeding increases the lower the level of platelets. Physical and mental exhaustion goes along with the reduced ability of the red blood cells to transport oxygen as the major energy source. Patients may experience paleness, weakness, fatigue, and shortness of breath. Disease progression is associated with severe infections, which are a major cause of death.
Diagnostic criteria
Diagnosis is established by full blood count and bone marrow biopsy and aspirate, all of which also serve to exclude other possible causes of aplasia, and/or peripheral pancytopenia (Marsh 2009). A blood cell count of less than 0.5 G/L polymorphonuclear neutrophils is crucial for the diagnosis of severe aplastic anemia. A very low polymorphonuclear neutrophils count of less than 0.2 G/L may be called very severe aplastic anemia (VSAA), a subdivision of severe aplastic anemia. The diagnostic criteria used today derive from reports from the 1970s (Camitta 1975). Severe aplastic anemia should be clearly differentiated from other diseases with similar appearing blood cell counts, such as Fanconi anemia, paroxysmal nocturnal hemoglobinuria, myelodysplastic syndromes, and secondary bone marrow failure by malignant bone marrow infiltration, for example leukemia (Marsh 2009).
Description of the intervention
Intervention: first‐line allogeneic hematopoietic stem cell transplantation from human leukocyte antigen (HLA)‐matched sibling donors (MSD‐HSCT)
Description of the procedure
The impaired hematopoiesis, including the patients' dysfunctional immune cells, is destroyed by myeloablative chemotherapy. New healthy hematopoietic stem cells then need to be transferred. Usually, they are collected from an HLA‐identical sibling donor. The harvested cells are transfused intravenously, flow through the body via the blood circulation system and end up in the bone marrow to nestle, grow, and produce a complete hematopoietic system including granulocytes, red blood cells, and platelets. Problems may arise when the cells do not settle down sufficiently to produce blood cells, which is called graft failure. The donor immune cells recognize epithelial cells of the recipient as foreign, which may lead to graft‐versus‐host disease. This may lead to serious organ toxicities. Therefore, the patient is exposed to immunosuppressive treatment to minimize a graft‐versus‐host reaction. The type of drugs, the standard dose, and the recommended duration of the conditioning regimen is listed in Table 2.
1. Conditioning for transplantation.
| Medicinal product | MSD‐HSCT1 | Bayever 1984 | Führer 2005 |
| Dose and duration of therapy | |||
| Cyclophosphamide | 50 mg/kg/day for 4 days (‐5, ‐4, ‐3, ‐2); BMT preparative regimen | Yes, as recommended | Yes, as recommended |
| Antithymocyte globulin | 30 mg/kg/day for 3 days (‐5, ‐4, ‐3); horse ATG; BMT preparative regimen | NR | Führer 1998 reports antilymphocyte globulin 0.75 ml/kg/day for 4 days Führer 2005 reports horse ATG 0.75 ml/kg/day for 4 days |
| Methylprednisolone | 2 mg/kg/day for 3 days (‐5, ‐4, ‐3); BMT preparative regimen; usually not used for children | Yes, 2 mg/kg/day prednisone in patients who developed acute GVHD of grade 2 or higher | NR |
| Methotrexate | 15 mg/m2/day for 1 day (+1) and 10 mg/m2/day for 3 days (+3, +6, +11); BMT post‐transplant immunosuppression | Yes, as recommended | NR Führer 1994 (protocol): methotrexate was planned |
| Ciclosporin | 5 mg/kg/day; BMT post‐transplant immunosuppression; starting on day ‐1 continued for 12 months with tapering beginning at 9 months; Marsh 2009 | NR | NR Führer 1994 (protocol): ciclosporin was planned |
| Irradiation2 | Radiation‐based regimens should be avoided | 3 Gy total body irradiation on day ‐1 | NR Führer 1994 (protocol): irradiation was not planned |
1Recommended dose and duration of therapy according to Schrezenmeier 2000 unless indicated.
2Schrezenmeier 2000: "While irradiation‐based programs have been effective in reducing rejection, they have accomplished their goal at the price of more transplant‐related complications. A combination of cyclophosphamide and antithymocyte globulin was found as effective as irradiation in preventing rejection with better long‐term outcome. In regards to conditioning regimens, radiation‐based regimens should be avoided because of the higher associated likelihood of inducing secondary cancer, the deleterious effects on fertility, and the potential detrimental effects on growth and development, a policy that would be particularly important for pediatric patients."
Abbreviation: ATG: antithymocyte globulin; BMT: bone marrow transplantation; GVHD: graft‐versus‐host disease; Gy: Gray; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; NR: not reported
Indication
According to the 2009 Guidelines for the diagnosis and management of aplastic anaemia of the British Committee for Standards in Haematology (Marsh 2009), first‐line allogeneic HSCT from the bone marrow of an HLA‐matched sibling donor (MSD) is regarded as the initial treatment of choice for newly diagnosed patients with severe aplastic anemia younger than 40 years. The harvesting of hematopoietic stem cells from the bone marrow is recommended rather than from mobilized peripheral blood stem cells (Schrezenmeier 2007). Radiation‐based conditioning regimens are not recommended because of an increased risk of inducing secondary cancer and other unwanted effects, such as infertility or impairment of growth and development of children (Schrezenmeier 2000).
Treatment failure and severe adverse events
Graft failure may lead to early death. The conditioning regimen leads to hematological and non‐hematological organ toxicities. Severe adverse events (SAE) such as acute graft‐versus‐host disease (GVHD), grade II to IV, and extensive chronic GVHD endanger the function of certain organs.
Comparator: first‐line treatment with ciclosporin and/or antithymocyte or antilymphocyte globulin (as first‐line immunosuppressive therapy)
Description of the procedure
First‐line treatment with ciclosporin and/or antithymocyte or antilymphocyte globulin (as first‐line immunosuppressive therapy) means the suppression of reactions of the immune system by the specific drugs: ciclosporin alone, antithymocyte globulin alone, antilymphocyte globulin alone, or ciclosporin combined with either antithymocyte or antilymphocyte globulin. First‐line means that patients have not received immunosuppressive therapy or any hematopoietic stem cell transplantation (HSCT) before except replacement of blood cells. Other substances that affect the immune reaction, such as corticosteroids, are not included. The aim is to reduce the abnormal immune reaction of the patient against their own hematopoiesis. When successful, the hematopoiesis of the patient can recover to the extent that they can lead a normal life. Immunosuppressive therapy may have a curative potential but probably a lower frequency than MSD‐HSCT. The long immune suppression may be associated with adverse events. The type of drugs, the standard dose, and the recommended duration of treatment is listed in Table 3.
2. Immunosuppressive treatment.
| Medicinal product | IST1 | Bayever 1984 | Führer 2005 |
| Dose and duration of therapy | |||
| Antithymocyte globulin | 40 mg/kg/day horse ATG for 4 days (1, 2, 3, 4) or 3.5 mg/kg/day; rabbit ATG for 5 days (1, 2, 3, 4, 5) Scheinberg 2011 |
20 mg/kg/day for 8 days (1, 2, 3, 4, 5, 6, 7, 8) | Führer 1998 reported antilymphocyte globulin 0.75 ml/kg/day for 8 days Führer 2005 reported horse ATG 0.75 ml/kg/day for 8 days |
| Methylprednisolone | 1 mg/kg/day for 14 days (1 to 14) and tapering off until day 28 | 40 mg/m2/day prednisone for 5 days (8, 9, 10, 11, 12) | Yes, as recommended |
| Ciclosporin | 5 mg/kg/day; starting on day 1 continued at least until day 112, further treatment depending upon response | NR | Yes, dose as recommended for at least 6 months |
1Recommended dose and duration of therapy according to Schrezenmeier 2000 unless indicated.
Abbreviation: ATG: antithymocyte globulin; IST: immunosuppressive therapy including ciclosporin and/or antithymocyte or antilymphocyte globulin; NR: not reported
Indication
Immunosuppressive therapy is indicated for younger patients where no MSD is found, which can be expected for 70% of patients with severe aplastic anemia (Brodsky 2005). Some of these patients respond to immunosuppressive therapy.
Treatment failure and adverse events
Some patients do not respond well or show no response at all. A major risk of immunosuppressive therapy is the development of clonal diseases (Guinan 2009). Frequent transfusions increase the risk of adverse events such as iron overload and early death.
Why it is important to do this review
The treatment of severe aplastic anemia mainly includes immunosuppressive therapy with antithymocyte/antilymphocyte globulin and/or cyclosporine, or MSD‐HSCT. Clinical treatment algorithms have been suggested to assist decisions that satisfy individual conditions, personal preferences, and prognostic factors. Current recommendations are mainly based onthe results of analyses of secondary data such as bone marrow registries. MSD‐HSCT is seen as the treatment of choice for younger patients with an HLA‐matched sibling donor. This type of donor may only be available for about one‐third of eligible patients. The upper age limit for transplantation is controversial because lack of co‐morbidities may be associated with a favorable outcome in older patients. MSD‐HSCT can cure severe aplastic anemia, however, it is associated with graft failure, graft‐versus‐host disease (GVHD), and organ toxicities. These severe adverse events can lead to treatment‐related mortality.
Immunosuppressive therapy is regarded as an alternative treatment for patients without a suitable donor, for patients who do not favor MSD‐HSCT, for those who have an increased risk due to impaired general health, comorbidities, or other health conditions incompatible with the stem cell transplantation procedure, and for older patients. Patients may not respond to immunosuppressive therapy at all, which can lead to early mortality. Extended immunosuppressive therapy may be associated with the development of clonal diseases later in life. Pre‐treatment with immunosuppressive therapy and transfusion of blood products may have an unfavorable effect on the outcome of MSD‐HSCT. First‐line MSD‐HSCT could prevent these additional risks. If a diagnosis of severe aplastic anemia is established at an early patient age, then it is crucial to know which treatment promises more benefit and less harm in the long run. Evidence based on randomized controlled trials is not available. Non‐randomized controlled studies can differ substantially in their quality and have additional risks of bias. Thus, there is a need to search and appraise the evidence in a rigorous way to inform practice.
Objectives
To evaluate the effectiveness and severe adverse events of first‐line hematopoietic stem cell transplantation of HLA‐matched sibling donors compared to first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin in patients with acquired severe aplastic anemia.
Methods
Criteria for considering studies for this review
Types of studies
Inclusion criteria
Since we expected RCTs to be rare or not available, we additionally included non‐randomized controlled trials and other prospective study designs that are consistent with the principle of 'Mendelian randomization' in allocating patients to treatment groups. We required a minimum of 80% of relevant patients per group and we set a minimum sample size of five participants per group. We set no limits on language, year of publication, or year of treatment.
Exclusion criteria
Studies with a retrospective design.
Rationale for including non‐randomized clinical trials consistent with the principle of 'Mendelian randomization'
There are ethical concerns around randomization of patients with severe aplastic anemia to transplantation versus non‐transplantation. In general, MSD‐HSCT is a life‐threatening treatment that can lead to early severe adverse events including death. Acquired severe aplastic anemia is a rare disease and, according to the results of a previous literature search, currently there are no published RCTs available. However, there are considerable numbers of studies with comparative data available.
Gray 1991 and Wheatley 2004 described the potential of 'Mendelian randomization' to minimize bias when comparing MSD‐HSCT with an alternative therapy. The base concept has been ascribed to Katan 1986. 'Mendelian randomization' by definition accepts only siblings as transplant donors and these sibling donors are required to have 'identical' or matched features of specific transplant‐relevant HLA sites when compared with the transplant recipient. Therefore, patients with an HLA‐matched sibling will be allocated to the MSD‐HSCT group. On the other hand, patients with siblings that are not HLA compatible will be allocated to the immunosuppressive therapy group. The term 'Mendelian randomization' refers to the fact that the genetic distribution of paternal and maternal alleles follows a random process and is determined before birth. This concept takes advantage of an instrumental variable for allocating the patients to treatment groups and, at the same time, this variable is neither associated with the treatment nor associated with the outcome.
If this is true, then this allocation by HLA features of siblings can reduce bias. Bias is introduced when a perceived high risk or a certain age group affects the decision of the person who allocates patients to the treatment groups. We suppose that studies with a prospective design and application of the principles of 'Mendelian randomization' have a higher quality than other non‐randomized studies. For this reason we included these studies after careful evaluation. Limitations of 'Mendelian randomization' are addressed in the Discussion.
Types of participants
Inclusion criteria
We included participants with newly diagnosed acquired severe aplastic anemia without prior HSCT or immunosuppressive therapy. This included idiosyncratic causes, such as idiopathic etiology, hepatitis, drugs and chemicals, and predictable causes, such as radiation or chemotherapy (Killick 2000). We have adopted the diagnostic criteria and classification of severity for severe aplastic anemia according to the guidelines of the British Committee for Standards in Haematology (Marsh 2009). Severe aplastic anemia is characterized by a count of less than 0.5 G/L polymorphonuclear neutrophils. Very severe aplastic anemia (VSAA), a subdivision of severe aplastic anemia, is characterized by a polymorphonuclear neutrophils count of less than 0.2 G/L. We did not set any age limits for participants.
Exclusion criteria
We excluded studies on participants with inherited aplastic anemia, such as Fanconi anemia, dyskeratosis congenita, or Shwachman syndrome (Killick 2000). We also excluded studies on participants with malignant aplastic anemia, such as childhood lymphoblastic leukemia.
Types of interventions
Intervention
We included allogeneic hematopoietic stem cell transplants harvested from any source of HLA‐matched sibling donors (MSD‐HSCT), serving as a first‐line therapy (Passweg 2010). No other HSCT or immunosuppressive therapy has been offered to the patients before.
Comparator
We included immunosuppressive therapy with either antithymocyte/antilymphocyte globulin or ciclosporin or a combination of the two (Passweg 2010). The combination has recently been reported as the standard immunosuppressive therapy by the British Committee for Standards in Haematology (BCSH) (Marsh 2009). Antithymocyte globulin can be prepared in horses or in rabbits. Both preparations are accepted, although horse has been recently recommended as first‐line immunosuppressive therapy over rabbit antithymocyte globulin, based on study results (Marsh 2011).
Types of outcome measures
Primary outcomes
Overall mortality
Secondary outcomes
Treatment‐related mortality (TRM reported for both arms)
Graft failure (reported only for MSD‐HSCT arm)
Graft‐versus‐host disease (reported only for MSD‐HSCT arm)
No response to immunosuppressive therapy (reported only for immunosuppressive therapy arm)
Relapse after initial successful treatment (reported for both treatment arms)
Secondary clonal disease or malignancies (reported for both treatment arms)
Health‐related quality of life scores measured by validated questionnaires (reported for both treatment arms)
Performance scores measured by validated questionnaires (reported for both treatment arms)
Search methods for identification of studies
Electronic searches
We modified the search strategies suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2011). We did not use the RCT filter in developing the search strategy as we suspected that the studies using 'Mendelian randomization' might be missed.
We conducted an electronic literature database search in MEDLINE (Ovid) including articles published between 1946 and 22 April 2013. The search strategy is shown in Appendix 1. We searched EMBASE (Ovid), including articles published between 1980 and 22 April 2013, by using the search strategy shown in Appendix 2. We searched The Cochrane Library CENTRAL (Wiley), including articles published from inception to 22 April 2013, by using the search strategy shown in Appendix 3.
We searched for ongoing trials by scanning the online registries ClinicalTrials.gov (ClinicalTrials.gov 2012) and World Health Organization International Clinical Trials Registry Platform (ICTRP 2012) using the term 'severe aplastic anemia' in the field condition and 'stem cell transplantation' in the field intervention.
We searched for oral and poster abstracts presented at the last five consecutive American Society of Hematology (ASH) annual meetings from 2007 to 2012 available for online searching. We requested an online search using the term 'transplantation AND immunosuppressive AND aplastic AND anemia' in the search field (ASH meeting abstracts 2012). We searched for abstracts presented at the last two consecutive European Group for Blood and Marrow Transplantation (EBMT) annual meetings from 2010 to 2012 available for online searching. We requested an online search using the term 'aplastic AND (anemia OR anaemia)' in the search field (EBMT meeting abstracts 2012). Conference proceedings of three societies were covered by EMBASE (Ovid) and searched on 22 April 2013: BMT Tandem Meetings of the American Society for Blood and Marrow Transplantation (ASBMT) and the Center for International Blood and Marrow Transplant Research (CIBMTR) published in Biology of Blood and Marrow Transplantation (BBMT) and Annual Meeting of the American Society of Hematology (ASH) published in Blood Conference.
Searching other resources
We located information about trials not registered in electronic databases by searching the reference lists of relevant articles and review articles. We contacted authors to replenish missing information.
Data collection and analysis
Selection of studies
We endorsed the PRISMA statement, adhered to its principles, and conformed to its checklist (Moher 2009). We retrieved all titles and abstracts by electronic searching and downloaded them to the reference management database EndNote Version X3 (Thomson Reuters Corp 2012). We removed duplicates and two review authors examined the remaining references independently (FP, CB). We excluded those studies that clearly did not meet the inclusion criteria and we obtained copies of the full texts of potentially relevant references. Two authors (FP, CB) assessed the eligibility of retrieved papers independently. We resolved disagreement by discussion and it was not necessary to consult a third review author. We considered studies written in languages other than English and asked peers familiar with the particular language and with the principles of study evaluation to translate major methodological issues. We also used the online assistance of the Google Translate 2012 program. We documented reasons for the exclusion of studies.
Prospective studies
We judged studies to be prospective if an explicit statement was reported or there were clues suggesting a prospective design (e.g. prior approval of treatment, informed consent). We judged studies to be retrospective if an explicit statement was reported or it was implied by description that data were reviewed from an existing source. We regarded each of the following items as an indication of a retrospective design: registry reports and reviewing of medical records.
Studies with 'Mendelian randomization'
We judged studies as consistent with the principle of 'Mendelian randomization' if all transplant donors were clearly siblings and if the allocation of patients to treatment groups was not based on age. We regarded studies as not consistent with the principle of 'Mendelian randomization' if age was not balanced between groups, indicating that age played a role in group assignment. Examples: distribution of age categories was statistically not comparable (P value less than 0.05).
Data extraction and management
Two review authors (FP, UG) independently abstracted data on study characteristics, patients and interventions, duration of follow‐up, outcomes, and deviations from protocol. In addition, two review authors (FP, UG) independently assessed the risk of bias. We resolved differences between review authors by discussion or by appeal to a third review author (CB). All three included studies were full‐text publications. We found selective outcome reporting in the study by Führer 1998 because survival was reported only for subgroups by disease severity. It appeared that the authors reported interim results of a then ongoing study and we asked whether data from the concluded study or updated data were available. We contacted the first author after confirming the new address including the e‐mail address. The author did not answer the request.
We extracted the following data:
General information on author, title, source, publication date.
Study characteristics: trial design, setting, inclusion/exclusion criteria, comparability of patients' characteristics between groups, treatment allocation, blinding, subgroup analysis, length of follow‐up.
Participant characteristics: age, gender, number of participants recruited/allocated/affected/analyzed, additional diagnoses, participants lost to follow‐up.
Interventions: type of transplantation and type of immunosuppressive treatment.
Outcomes: overall survival, treatment‐related mortality, graft failure, no response to immunosuppressive therapy, graft‐versus‐host disease, relapse after initial successful treatment, secondary clonal disease or malignancies, health‐related quality of life scores, and performance scores.
Assessment of risk of bias in included studies
Two review authors (FP, UG) independently assessed the risk of bias in the included studies using six criteria. We have used four criteria from The Cochrane Collaboration's tool for assessing risk of bias (Higgins 2011a):
blinding of outcome assessment (detection bias);
incomplete outcome data such as missing data (attrition bias);
selective reporting such as not reporting pre‐specified outcomes (reporting bias); and
other sources of bias such as bias related to the specific study design and competing interest.
We extended the Cochrane tool for assessing risk of bias with two additional criteria that are specific to the inclusion criteria for the present review and critical for confidence in results:
comparable baseline characteristics; and
concurrent control.
As all studies were supposed to apply 'Mendelian randomization', we removed the criteria random sequence generation (selection bias), allocation concealment (selection bias), and blinding of participants and personnel (performance bias) because these criteria are determined by the study design. The process of allocation of alleles is a random process of nature and is therefore concealed (Lewis 2010). Blinding of participants and personnel does not seem possible with respect to MSD‐HSCT and immunosuppressive therapy. We would not have applied the removal of these items in the case of RCTs.
We applied The Cochrane Collaboration's criteria for judging risk of bias (Higgins 2011b). In general, a 'low risk' of bias judgement indicates that the bias is unlikely to seriously alter the results, for example, participants and investigators enrolling participants could not foresee assignment. A 'high risk' of bias judgement indicates that the bias seriously weakens confidence in the results, for example, participants or investigators enrolling participants could possibly foresee assignments. An 'unclear' risk of bias judgement indicates that the bias raises some doubt about the results, for example, the method of concealment is not described or not described in sufficient detail to allow a definite judgement.
Measures of treatment effect
The primary effect measure was the hazard ratio (HR) for time‐to‐event data. If the hazard ratio was not directly given in the publication, we estimated hazard ratios according to methods proposed by Parmar 1998 and Tierney 2007. We planned to calculate odds ratios (ORs) with 95% confidence intervals (CIs) for dichotomous outcomes. In case of rare events, we planned to use the Peto odds ratio instead. We planned to analyze continuous data and to present them as mean differences, if all results were measured on the same scale (e.g. length of hospital stay). If this was not the case (e.g. pain or quality of life), we planned to use standardized mean differences.
Dealing with missing data
We conformed to The Cochrane Collaboration's principal methods for dealing with missing data and analyzed only the available data (Higgins 2011c). If data were missing or only imputed data were reported we contacted trial authors to request data on the outcomes among participants who were assessed. We contacted the first author of the study by Führer 1998 to ask for missing survival data. The author did not answer the request.
Assessment of heterogeneity
We assessed heterogeneity between studies by visual inspection of forest plots, by estimation of the percentage of heterogeneity between trials which cannot be ascribed to sampling variation (I2 statistic) (Higgins 2003), by a formal statistical test of the significance of the heterogeneity (Cochran's Q) (Deeks 2011) and, if possible, by subgroup analyses (see Subgroup analysis and investigation of heterogeneity). We planned to investigate and report possible reasons if there was evidence of substantial heterogeneity. We used the random‐effects model with inverse variance weighting for statistical pooling (DerSimonian 1986).
Assessment of reporting biases
We conformed to The Cochrane Collaboration's criteria and planned to evaluate reporting biases such as publication bias, time lag bias, multiple (duplicate) publication bias, location bias, citation bias, language bias, and outcome reporting bias (Sterne 2011). We did not assess reporting bias because of the low number of identified studies.
Data synthesis
One review author (FP) entered the data into Review Manager 2011. Another review author (UG) checked the entered data. Methods of synthesizing the studies depended on the quality, design, and heterogeneity of the studies identified. We synthesized data on mortality (MSD‐HSCT versus immunosuppressive therapy) by using the hazard ratio as the effect measure with a random‐effects model. We also synthesized data on secondary clonal disease or malignancies (MSD‐HSCT versus immunosuppressive therapy) by using the Peto odds ratio as the effect measure with a fixed‐effect model. We did not identify other data suitable for quantitative pooling.
We used the software GRADEpro 3.2 (GRADEpro 2008) to create Table 1 as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011). We presented overall mortality, secondary clonal disease and malignancies, and Karnofsky Performance Index 70% or less because data for both treatment arms were available for these three outcomes. For other outcomes, data were available for only one treatment or were not available at all.
Subgroup analysis and investigation of heterogeneity
We planned subgroup analyses on age and time period of treatment. However, we found no appropriate data to conduct these analyses.
Sensitivity analysis
We planned sensitivity analyses to compare the results of studies with low versus high risk of bias. As all included studies had a high risk of bias, sensitivity analyses were obsolete.
Results
Description of studies
Results of the search
Of 4356 retrieved publications, 94 were regarded as reporting potentially relevant comparative data (Figure 1). We identified three non‐randomized, prospective, parallel, controlled clinical trials reported in seven articles including four follow‐up articles (Bayever 1984; Führer 1998; Gratwohl 1981). The three trials were consistent with the principles of 'Mendelian randomization' that met the inclusion criteria. We did not identify any RCTs.
1.
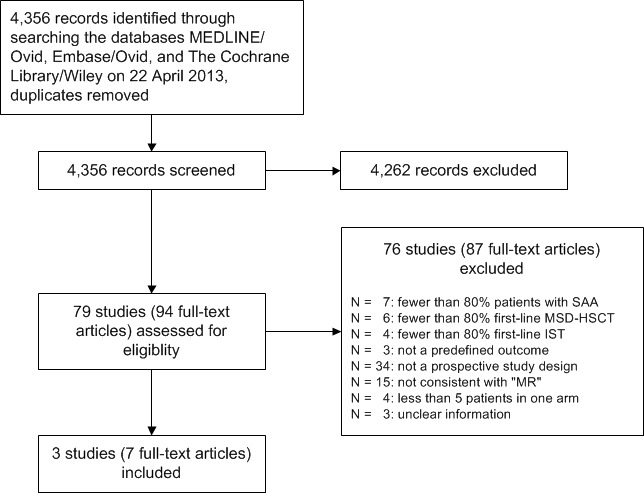
MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched (identical) sibling donor; IST: immunosuppressive therapy; "MR": 'Mendelian randomization'; SAA: severe aplastic anemia
Included studies
The characteristics of the three included studies are also described in the Characteristics of included studies table.
Design
We included three included studies that were judged to be non‐randomized, prospective, parallel, controlled clinical trials. The study by Bayever 1984 was conducted from 1977 to 1982, the study by Führer 1998 from 1993 to 2001, and the study by Gratwohl 1981 from 1976 to 1980. Bayever 1984 did not explicitly describe a prospective design. Nevertheless, we found clues in the text of the article sufficient to assume a prospective design. Quotes from the article: "[...]concurrently underwent therapy[...]; [...]experimental use[...]; [...]therapy was approved by[...]committee[...]; [...]informed consent[...]prior to the initiation of therapy[...]". Führer 1998 described a prospective, multi‐center study and published a study protocol in 1994. Gratwohl 1981 described a prospective single‐center study.
All three studies had features of allocation consistent with the principle of 'Mendelian randomization' as defined in the present review. Bayever 1984, Führer 1998, and Gratwohl 1981 clearly described that the allocation of patients to treatment groups depended on the availability of an HLA‐matched sibling donor. Quote by Bayever 1984: "All 35 patients who had an HLA‐identical donor underwent bone marrow transplantation. The 22 patients without an HLA‐identical donor received ATG therapy." Comment by Bayever 1984: "This high proportion of patients who underwent transplantation reflects the local referral pattern rather than the proportion of patients with an HLA‐identical donor in the general population." Quote by Führer 1998: "By biologic selection depending on the availability of an MSD, patients were assigned to either the HSCT or the IST group." Translated quote by Gratwohl 1981: "Group A: 10 patients with an HLA‐identical sibling received a bone marrow transplantation[...]Group C: 13 patients did not have a donor and received anti‐lymphoglobulin only."
Sample sizes
The study by Bayever 1984 included 57 patients, 35 in the MSD‐HSCT group and 22 in the immunosuppressive therapy group. The study by Führer 1998 included 213 patients, 67 in the MSD‐HSCT group and 146 in the immunosuppressive therapy group. The study by Gratwohl 1981 included 58 patients, 19 in the MSD‐HSCT group, 13 in the immunosuppressive therapy group, and 26 in a third group with characteristics not included in the present review.
Setting
The study by Bayever 1984 was conducted as a single‐center study in the United States, the study by Führer 1998 as a multi‐center study in Germany and Austria, and the study by Gratwohl 1981 was conducted as a single‐center study in Switzerland.
Participants
The age of the participants in the study by Bayever 1984 was 1 to 24 years and age was comparable between treatment groups with a median age of 17 years in the MSD‐HSCT group and 15 years in the immunosuppressive therapy group. The proportion of males was 67% and gender was also comparable with 66% males in the MSD‐HSCT group and 68% males in the immunosuppressive therapy group. In the immunosuppressive therapy group, data for one patient were not eligible because the patient had paroxsymal nocturnal hemoglobinuria.
The age of the participants in the study by Führer 1998 was 0 to 16 years with a median of 8.9 years. The proportion of males was 59%. The authors did not provide data per treatment group and assessment of comparability with respect to age and gender is limited. We found two relevant articles for the study, one article published in 1998 and another in 2005. In the 2005 article, results were reported for the two separate subgroups, VSAA only and severe aplastic anemia without VSAA.
The age of the participants in the study by Gratwohl 1981 was 4 to 37 years and age was comparable between treatment groups with a median age of 18 years in the MSD‐HSCT group and 23 years in the immunosuppressive therapy group. Gender was also comparable with 53% males in the MSD‐HSCT group and 54% males in the immunosuppressive therapy group.
Interventions
In the study by Bayever 1984, 35 of 57 patients received stem cells from HLA‐matched sibling donors (source: bone marrow) and 22 of 57 patients did not have an HLA‐matched sibling donor and thus received antithymocyte globulin only (source: horse). The authors stated that the proportion of patients in the MSD‐HSCT group was unusually high and did not reflect the accordant proportion in the general population.
In the study by Führer 1998, 62 of 213 patients received stem cells from HLA‐matched sibling donors (source: bone marrow), five patients dropped out of the transplantation group, and 146 of 213 patients did not have an HLA‐matched sibling donor and thus received ciclosporin combined with antithymocyte globulin (source: horse). The proportion of transplanted patients can be expected as about two‐thirds of patients will not find an HLA‐matched sibling donor. Nevertheless, the authors did not discuss this issue.
In the study by Gratwohl 1981, 19 of 58 patients received stem cells from HLA‐matched sibling donors (source: bone marrow) and 13 of 58 patients did not have an HLA‐matched sibling donor and thus received antilymphocyte globulin only (source: not reported). In the immunosuppressive therapy group, data for two patients were not eligible because the patients had HLA‐matched (identical) siblings. The siblings were not available and thus the patients were allocated to the immunosuppressive therapy group. After the two patients did not respond to immunosuppressive therapy, both patients received a second‐line HSCT and died after the therapy.
Dose and duration of conditioning for transplantation (Table 2) as well as immunosuppressive therapy (Table 3) was different from the recommendations of Schrezenmeier 2000.
Primary outcome
All included studies reported overall survival (Bayever 1984; Führer 1998; Gratwohl 1981).
Secondary outcomes
Bayever 1984 reported treatment‐related mortality, graft failure, no response to immunosuppressive therapy, graft‐versus‐host disease, relapse after initial successful therapy, and secondary clonal disease and malignancies for both treatment groups. Führer 1998 reported relapse after initial successful therapy and secondary clonal disease and malignancies separate for each treatment group. The authors did not report treatment‐related mortality, graft failure, no response to immunosuppressive therapy, graft‐versus‐host disease, and relapse after initial successful therapy separately for each treatment group. Gratwohl 1981 reported no response to immunosuppressive therapy and reported causes of death including graft failure, graft‐versus‐host disease, infection, and bleeding.
Excluded studies
We excluded 76 comparative studies that were reported in 87 articles, including 11 follow‐up articles (Figure 1). Excluded studies are described in the Characteristics of excluded studies. We identified nine articles in a language other than English (one in Chinese, one in Croatian, one in Czech, two in German, two in Russian, one in Serbian, and one in Swedish) for full‐text screening and were able to contact peers to translate the text and/or used the online assistance of the Google Translate 2012 program.
Risk of bias in included studies
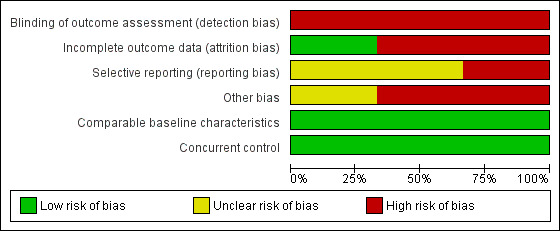
All three included studies had a high risk of bias due to the study design (Characteristics of included studies; Figure 2; Figure 3). As outlined in Assessment of risk of bias in included studies, we chose four items from the 'Risk of bias' tool: blinding of outcome assessment (detection bias), incomplete outcome data (attrition bias), selective reporting (reporting bias), and other bias, and added two items: comparable baseline characteristics and concurrent control.
2.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
3.
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Blinding of outcome assessment (detection bias)
All three studies had a high risk of bias for the criterion blinding of outcome assessment because it was not done in any of the three included studies.
Incomplete outcome data
Presumably, patients did not drop out in the study by Bayever 1984 and we judged the risk of bias as low.
In the study by Führer 1998, 7.5% (5 of 67) patients assigned to the MSD‐HSCT group dropped out because the parents refused the treatment. Thus we judged the risk of bias as high.
In the study by Gratwohl 1981, the data for 58 of 61 consecutively recruited patients that fulfilled the eligibility criteria have been analyzed. Therefore three patients dropped out and we judged the risk of bias as high.
Selective reporting
In the study by Bayever 1984, relapse was reported for the immunosuppressive therapy group but not for the MSD‐HSCT group and we judged the risk of bias as unclear.
In the study by Führer 1998, preliminary results were published in 1998 covering the study period from 1993 to 1997. Overall survival and secondary clonal disease or malignancies were reported separately for the MSD‐HSCT group versus the immunosuppressive therapy group. In the update report published in 2005, covering the study period from 1993 to 2001, overall survival, secondary clonal disease or malignancies, and also relapse were not reported separately for the two distinct treatment groups. Rather, the results were presented for two subgroups according to disease severity. The authors dichotomized the patients into a group that consisted of patients with severe aplastic anemia but not with very severe aplastic anemia (VSAA) and in another group that consisted of patients with VSAA only. The combined results of both subgroups were not reported. Subsequently, we cannot compare the subgroup results with the results of other studies that generally report the results of all severe aplastic anemia patients per treatment group. We have contacted Monika Führer, first author of the 1998 and the 2005 paper, to enquire whether data for all patients per group are available and could be calculated, and whether another update was presumably conducted, however we did not receive a reply. Treatment‐related mortality, treatment failure, and GVHD were not reported in either of the two publications of the study by Führer 1998. We judged the risk of bias as high.
In the study by Gratwohl 1981, besides overall survival, no other outcome was reported systematically, such as treatment‐related mortality, graft failure, no response to immunosuppressive therapy, GVHD, relapse, secondary clonal disease or malignancies, or performance status. Gratwohl 1981 did report the number of patients with graft failure (N = 3) and GVHD (N = 5) if these adverse events were regarded as the cause of death. Gratwohl 1981 did report the number of patients with no response to immunosuppressive therapy (N = 2) only for two patients that actually had a MSD and were offered a second‐line MSD‐HSCT. We judged the risk of bias as unclear.
Other potential sources of bias
Competing interests
Bayever 1984 reported that the study was supported in part by non‐for‐profit institutions such as the National Institutes of Health and others and we judged the risk of bias as high.
Führer 1998 reported that the 'SAA 94' study was supported by 'AMGEN'(Amgen Inc. headquarters in Thousand Oaks, California, USA, a manufacturer of filgrastim, a human granulocyte colony‐stimulating factor (G‐CSF)). The authors stated that G‐CSF was given in addition to immunosuppressive therapy but did not mention the manufacturer in the methods chapter. The type of support, such as providing G‐CSF or financial support, was not specified. Führer 1998 reported that the 'SAA 94' study was also supported by 'JMTIX'. This term does not link to any commercial company when searched for using Google. We judged the risk of bias as high.
Gratwohl 1981 reported that 15% (2 of 13) patients assigned to the immunosuppressive therapy group had an HLA‐identical sibling donor and were actually eligible to be allocated to the MSD‐HSCT group. There was no other bias such as competing interest or cross‐over. We judged the risk of bias as unclear.
Comparable baseline characteristics
In all three included studies, there were no differences in baseline characteristics between the two treatment groups, in particular with reference to prognostic factors such as age and gender. We therefore judged risk of bias as low in all the studies.
Concurrent control
In all three included studies, data for the control group were collected during the same time period as data for the test group. We therefore judged risk of bias as low in all the studies.
Effects of interventions
See: Table 1
See: Table 1 for the comparison of first‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors versus first‐line immunosuppressive treatment including ciclosporin and/or antithymocyte or antilymphocyte globulin for acquired severe aplastic anemia.
Primary outcome
Overall survival/mortality
Overall survival was not statistically significantly different between matched sibling donor hematopoietic stem cell transplantation (MSD‐HSCT) and immunosuppressive therapy: in the study by Bayever 1984 (at two years 72% (95% confidence interval (CI) 64 to 80) versus 45% (95% CI 29 to 61), P = 0.18), in the study by Führer 1998 (at two years 84% (95% CI not reported) versus 87% (95% CI not reported), P = 0.43), and in the study by Gratwohl 1981 (at five years 47% (95% CI not reported) versus 60% (95% CI not reported), P = 0.56) (Table 4). The pooled hazard ratio estimate for overall mortality was 0.95 with a 95% confidence interval of 0.43 to 2.12 (P value = 0.90) (Analysis 1.1; Figure 4). According to the meta‐analysis based on data from all three included studies, overall mortality was not statistically significantly different between MSD‐HSCT and immunosuppressive therapy. The overall mortality across all three included studies was 28.0% (23 of 82 patients) in the MSD‐HSCT arm and was 17.4% (21 of 121 patients) in the immunosuppressive therapy arm (Table 5). Data on overall mortality are also presented in Table 1.
3. Overall survival.
| Study | MSD‐HSCT | IST | Time1 | P value | ||
| N | OS (95% CI) | N | OS (95% CI) | Year | ||
| Bayever 1984 | 35 | 72% (64 to 80) | 22 | 45% (29 to 61) | 2 | 0.18 |
| Führer 1998 | 28 | 84% (NR) | 86 | 87% (NR) | 4 | 0.43 |
| Gratwohl 1981 | 19 | 47% (NR) | 13 | 69%2 (NR) | 5 | 0.563 |
1Time point of Kaplan‐Meier estimate.
2Gratwohl 1981: Two of 13 patients were eligible for MSD‐HSCT but donors were not available in the first place; the two patients died after they received a second‐line HSCT from the then again available MSD that was offered after the patients showed no response to IST.
3 The P value was not reported and we calculated the P value using Fisher's exact test.
Abbreviations: CI: confidence interval; IST: immunosuppressive therapy including ciclosporin and/or antithymocyte or antilymphocyte globulin; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; N: number of analyzed patients; NR: not reported; OS: overall survival
1.1. Analysis.
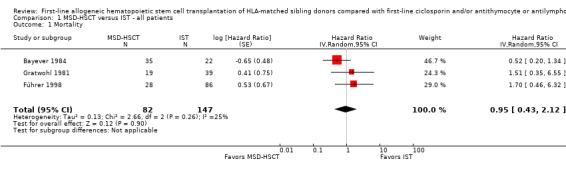
Comparison 1 MSD‐HSCT versus IST ‐ all patients, Outcome 1 Mortality.
4.
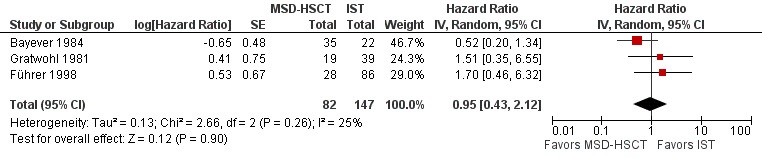
Mortality (HSCT vs. IST); effect: hazard ratio; random‐effects model. Standard error calculated from data presented in the Kaplan‐Meier graph of the article or deduced from it by the method of Parmar 1998 and the applicable tool by Tierney 2007. Data from the study Führer 1998 are taken from the article Führer 1998 (114 participants) because results in the article by Führer 2005 (213 participants) were not sufficient to allow calculation of the respective standard error. We included the data for 203 participants in the meta‐analysis. We were not able to include the data for the additional 99 participants of the update.
Abbreviations: CI: confidence interval; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation of bone marrow of HLA‐matched sibling donors; log: logarithm; IST: first‐line immunosuppressive therapy; IV: inverse variance; SE: standard error
4. Mortality.
| Study | MSD‐HSCT | IST | ||||
| N | Died | % | N | Died | % | |
| Bayever 1984 | 35 | 9 | 25.7 | 22 | 9 | 40.9 |
| Führer 1998 | 28 | 4 | 14.3 | 86 | 8 | 9.3 |
| Gratwohl 1981 | 19 | 10 | 52.6 | 13 | 4 | 30.8 |
| Total | 82 | 23 | 28.0 | 121 | 21 | 17.4 |
Note: This table is meant to support the information about overall mortality. The difference between the two treatment groups MSD‐HSCT and IST is calculated by the hazard ratio shown in Figure 4. Therefore, we did not calculate P values.
Abbreviations: IST: immunosuppressive therapy including ciclosporin and/or antithymocyte or antilymphocyte globulin; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; N: number of analyzed patients
Secondary outcomes
Treatment‐related mortality
Treatment‐related mortality was reported for the MSD‐HSCT group at 20% (7 of 35 patients) by Bayever 1984 and at 42% (8 of 19 patients) by Gratwohl 1981 (Table 6). Time point, follow‐up, and P value were not reported. This outcome was not addressed in the study by Führer 1998.
5. Treatment‐related mortality after MSD‐HSCT.
| Study | MSD‐HSCT |
| % (N affected of N evaluable) | |
| Bayever 1984 | 20 (7 of 35) |
| Führer 1998 | NR |
| Gratwohl 19811 | 42 (8 of 19) |
1Gratwohl 1981: Treatment‐related mortality not stated in article but implied by the authors of the present review as 3 patients died of graft failure and 5 patients died of GVHD in the MSD‐HSCT group; in general graft failure and GVHD are complications of HSCT.
Abbreviations: MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; N: number; NR: not reported
Graft failure
Bayever 1984 reported serious problems with engraftment in 3% (1 of 35) and Gratwohl 1981 in 16% (3 of 19) transplanted patients (Table 7). This single‐arm outcome was not addressed in the study by Führer 1998.
6. Graft failure after MSD‐HSCT.
| Study | Any graft failure |
| % (N affected of N evaluable) | |
| Bayever 1984 | 3 (1 of 35) |
| Führer 1998 | NR |
| Gratwohl 1981 | 16 (3 of 19) |
Abbreviations: MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; N: number; NR: not reported
Graft‐versus‐host‐disease (GVHD)
GVHD was reported for a considerable number of transplanted patients with successful engraftment in the study by Bayever 1984: 51% (17 of 33) patients (Table 8). Acute and chronic GVHD was not presented separately. Gratwohl 1981 reported 26% (5 of 19) patients with GVHD. This single‐arm outcome was not addressed in the study by Führer 1998.
7. GVHD after MSD‐HSCT.
| Study | GVHD |
| % (N affected of N evaluable) | |
| Bayever 1984 | 51 (17 of 33)1 |
| Führer 1998 | NR |
| Gratwohl 1981 | 26 (5 of 19) |
1Bayever 1984: 51 patients with GVHD, acute or chronic. Of 35 patients receiving MSD‐HSCT, 2 were not able to develop GVHD because 1 patient died of sepsis 1 day after transplant and 1 patient experienced a graft rejection. Of 33 patients, 17 patients developed GVHD according to the results section of the article, without specifying the acute or chronic type. In the discussion section, the authors report moderate to severe GVHD in 10 patients. This could be the number of patients with acute GVHD. However, it is not clearly described how many patients had acute versus chronic GVHD.
Abbreviations: GVHD: graft‐versus‐host disease; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; N: number; NR: not reported
No response to immunosuppressive therapy
Bayever 1984 reported a considerable proportion of patients that did not responded to immunosuppressive therapy after three months follow‐up: 64% (14 of 22) patients (Table 9). Gratwohl 1981 reported a much lower proportion of 15% (2 of 13) patients. This single‐arm outcome was not addressed in the study by Führer 1998.
8. No response to IST.
| Study | No response |
| % (N affected of N evaluable) | |
| Bayever 1984 | 64 (14 of 22) |
| Führer 1998 | NR |
| Gratwohl 1981 | 15 (2 of 13) |
Abbreviations: IST: immunosuppressive therapy including ciclosporin and/or antithymocyte or antilymphocyte globulin; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; N: number; NR: not reported
Relapse
The three included studies did not report relapse in the MSD‐HSCT groups. Bayever 1984 reported that 12.5% (1 of 8) responders after immunosuppressive therapy had recurrent severe aplastic anemia and were alive at 5.5 years (Table 10). This outcome was not addressed across treatment groups in the study by Führer 1998. Rather, in a follow‐up in 2005, Führer 1998 reported a relapse‐free survival at five years of 80% (95% CI 70 to 91) in the 'VSAA only' subgroup, and of 67% (95% CI 51 to 83) in the 'SAA without VSAA' subgroup. This outcome was not addressed in the study by Gratwohl 1981.
9. Relapse.
| Study | MSD‐HSCT | IST |
| % (N affected of N evaluable) | ||
| Bayever 1984 | NR | 12.5 (1 of 8) at 5 years |
| Führer 1998 | NR | NR |
| Gratwohl 1981 | NR | NR |
Abbreviations: IST: immunosuppressive therapy including ciclosporin and/or antithymocyte or antilymphocyte globulin; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; N: number; NR: not reported
Secondary clonal disease or malignancies
Secondary clonal disease or malignancies were reported in the HSCT group versus immunosuppressive therapy group in 1 of 34 versus 0 of 22 patients in the study by Bayever 1984 and in 0 of 28 versus 4 of 86 patients in the study by Führer 1998 (Table 11). This outcome was not addressed in the study by Gratwohl 1981.
10. Secondary clonal disease or malignancies.
| Study | MSD‐HSCT | IST | P value1 | Type of secondary clonal disease or malignancy |
| % (N affected of N evaluable) | ||||
| Bayever 1984 | 3 (1 of 34) | 0 (0 of 22) | 1.00 | MSD‐HSCT: 1 x T‐cell lymphoma |
| Führer 1998 | 0 (0 of 28) | 5 (4 of 86) | 0.57 | IST: 4 x acute myelogenous leukemia |
| Gratwohl 1981 | NR | NR | NR | NR |
1We calculated the P value using Fisher's exact test.
Abbreviations: IST: immunosuppressive therapy including ciclosporin and/or antithymocyte or antilymphocyte globulin; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from bone marrow of HLA‐matched sibling donor; N: number; NR: not reported
To combine the results we conducted a meta‐analysis of secondary clonal disease or malignancies in the two studies that reported events for this outcome. We applied the Peto odds ratios for dichotomous data with a fixed‐effect model because the number of events was low. The pooled Peto odds ratio estimate was 0.54 with a 95% confidence interval of 0.07 to 4.00(P value = 0.55) (Analysis 1.2; Figure 5). According to the meta‐analysis including data from two of three included studies, the frequency of secondary clonal disease or malignancies was not statistically significantly different between MSD‐HSCT and immunosuppressive therapy.
1.2. Analysis.
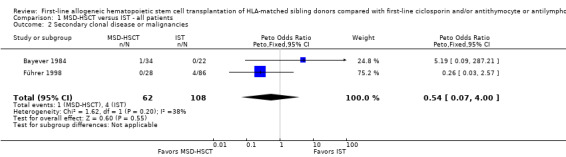
Comparison 1 MSD‐HSCT versus IST ‐ all patients, Outcome 2 Secondary clonal disease or malignancies.
5.
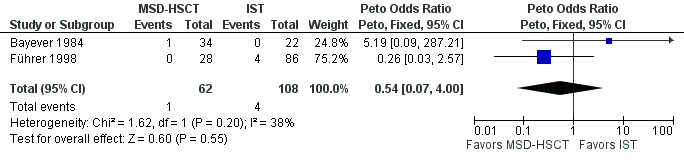
Forest plot of comparison: 1 MSD‐HSCT vs. IST ‐ all patients, outcome: 1.2 Secondary clonal disease or malignancies.
Abbreviations: CI: confidence interval; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation of bone marrow of HLA‐matched sibling donors; IST: first‐line immunosuppressive therapy
Health‐related quality of life
Health‐related quality of life scales were not used in any of the included studies.
Performance scores
One study (Bayever 1984) reported results for Karnofsky Performance Status (Karnofsky 1949) at the end of follow‐up. According to the National Palliative Care Research Center (NPCRC 2012), a performance status of higher than 70% means that the patient is able to carry on normal activity and to work with no special care needed. This good performance status was present for almost all transplanted and evaluable patients (92%) but for less than half of the patients in the immunosuppressive therapy group (46%) (Table 12). Therefore, transplanted patients had a significantly better physical functional performance status than patients in the immunosuppressive therapy group. This difference is dependent on the time point of assessment. Performance status was not addressed in the studies by Führer 1998 and Gratwohl 1981.
11. Karnofsky performance status.
| Study | Score 71% to 100% | Score 41% to 70% | Score 0% to 40% | P value1 | |||
| MSD‐HSCT | IST | MSD‐HSCT | IST | MSD‐HSCT | IST | ||
| % (N affected of N evaluable) | |||||||
| Bayever 1984 | 92 (24 of 26) | 46 (6 of 13) | 4 (1 of 26) | 54 (7 of 13) | 4 (1 of 26) | 0 (0 of 13) | < 0.001 |
| Führer 1998 | NR | NR | NR | NR | NR | NR | NR |
| Gratwohl 1981 | NR | NR | NR | NR | NR | NR | NR |
1We calculated the P value using Fisher's exact test.
Karnofsky performance status scale definitions according to NPCRC 2012: 71% to 100%: able to carry on normal activity and to work; no special care needed (100%: normal, no complaints); 41% to 70%: unable to work; able to live at home and care for most personal needs; varying amount of assistance needed; 0% to 40%: unable to care for self; requires equivalent of institutional or hospital care; disease may be progressing rapidly (0%: dead).
Abbreviations: IST: immunosuppressive therapy including ciclosporin and/or antithymocyte or antilymphocyte globulin; MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched sibling donor; N: number; NR: not reported
Discussion
Summary of main results
We identified three prospective, non‐randomized controlled trials (Bayever 1984; Führer 1998; Gratwohl 1981) including 302 participants; 121 received matched sibling donor hematopoietic stem cell transplantation (MSD‐HSCT) and 181 received immunosuppressive therapy. Based on these trials we found insufficient evidence to clarify whether first‐line allogeneic MSD‐HSCT leads to better overall survival than first‐line immunosuppressive therapy.
Bayever 1984 and Gratwohl 1981 reported considerable treatment‐related mortality: from 20% to 42% in the MSD‐HSCT with no event reported for the immunosuppressive therapy group. Both studies were conducted roughly 30 years ago and are certainly not applicable to the standard of care of today. The graft failure rate was variable and caused the death of 16% of transplanted patients in the study by Gratwohl 1981; graft failure was a problem in 3% in the study by Bayever 1984. No response to immunosuppressive therapy was observed in more than half of patients in the study by Bayever 1984 and was also still considerable in the study by Gratwohl 1981. Graft‐versus‐host‐disease (GVHD) affected a quarter to a half of patients, without distinction between acute and chronic GVHD. Relapse was reported only in one study, affecting up to one in eight patients. Secondary clonal disease or malignancies were detected in the immunosuppressive therapy group in the study by Führer 1998 and in the MSD‐HSCT group by Bayever 1984. This result is certainly not representative but shows that malignancies may occur after and could possibly be caused by both treatments.
Overall completeness and applicability of evidence
Patients were treated in the time period from 1976 to 1982 (Bayever 1984; Gratwohl 1981) and from 1993 to 2001 (Führer 1998). The applicability of these data to current clinical practice is considerably restricted as medical knowledge and terms of health care, especially in HSCT, have progressed and changed significantly over that time period.
Quality of the evidence
The low number of studies included in the present review limits the inferences we can make from the extracted data. One of the two included studies does not report data for all participants but rather restricts the presentation to subgroups according to severity of severe aplastic anemia. In general, studies handle very severe aplastic anemia (VSAA) data as part of severe aplastic anemia data, in agreement with the classification of severe aplastic anemia. Absence of randomized controlled trials (RCTs) may be associated with ethical problems in HSCT, especially regarding children (Chybicka 2008).
One of the strengths of this review is the broadness of the search strategy such that study retrieval bias is very unlikely. Nevertheless, there remains a slight possibility that an unknown number of studies were not registered and not published. Duplicate publication bias is very unlikely because we searched for follow‐up papers of a single study, to ensure that we included the updated version, and we excluded secondary analyses of registers or databases, which may use data that have been published previously by individual contributing study centers. We identified outcome reporting bias with respect to treatment‐related mortality.
Potential biases in the review process
The low number of studies included in the present review is caused by our approach in excluding studies with obvious excess risk of bias in addition to a non‐randomized design such as retrospective evaluations and registry analyses. Another main reason for excluding studies was the unbalanced distribution of patients' characteristics across treatment groups. Control for confounders was not addressed in any of the potentially relevant studies.
Gray 1991 and Wheatley 2004 addressed the problem of bias in studies comparing allogeneic hematopoietic stem cell transplantation with another treatment using a non‐randomized study design. They suggested 'Mendelian randomization' as a principle to avoid bias. Use of 'Mendelian randomization' is no guarantee, however, that bias is minimized. This may be because tissue typing data may not be accurate. Patients may have only one sibling either in the donor or in the no donor group. Large families have a greater chance of finding a donor. Therefore, designing a non‐randomized controlled trial by applying 'Mendelian randomization' requires careful thought to effectively reduce bias and control for potential confounders. There is a time lag in patients with siblings because tissue typing and readiness for assignment to treatment group may possibly take several months (Wheatley 2004). On the other hand, patients with no siblings can be assigned immediately and are at earlier risk for adverse events. Nitsch 2006 describes the limits to causal inference based on 'Mendelian randomization'.
Agreements and disagreements with other studies or reviews
Primary outcome
The results of the studies included in the present systematic review match the recent estimates reported by others. We found that Bayever 1984 reported an overall survival of recipients of human leukocyte antigen (HLA)‐matched transplantation from sibling donors (MSD‐HSCT group) of 72% at two years. The patients had a median age of 17 years and were treated in the time period from 1977 to 1982. Führer 1998 reported higher overall survival for the MSD‐HSCT group of 84% at two years. In an update in 2005, Führer 1998 reported even higher overall survival for the MSD‐HSCT group of 96% (95% confidence interval (CI) 89 to 100) at five years for the subgroup of patients with severe aplastic anemia but without VSAA. In the other subgroup that included VSAA only overall survival was 89% (95% CI 80 to 99) at five years. Führer 1998 suggested that "[...] in children with SAA a more severe disease stage at diagnosis indicates a favorable outcome with immunosuppressive therapy." However, this finding was not confirmed in a comprehensive evidence‐based guideline by Marsh 2009: "Allogeneic BMT from a human leucocyte antigen (HLA)‐identical sibling donor is the initial treatment of choice for newly diagnosed patients if they have severe or very severe aplastic anaemia, are <40 years old and have an HLA‐compatible sibling donor." Marsh 2009 estimated that this type of transplantation provides a 75% to 90% chance of long‐term cure. Guinan 2009 reported in a review of studies with younger patients that overall survival ranges between 75% and 95% at three to five years. In a recent review, Eapen 2012 sets a cut‐off at the age of 20 years and describes overall survival of about 80% in younger and 50% to 70% in older patients. The (now unusual) low overall survival of 47% at two years, remaining stable at 47% up to five years, in the study by Gratwohl 1981 is probably associated with the age of the study (starting in 1976): it is certainly not applicable to current expectations regarding overall survival.
It is assumed that the risks of transplant‐related mortality increase with age. Gupta 2010 reported the increased risk with age in 1307 patients with severe aplastic anemia after MSD‐HSCT. Therefore, age plays a major role in clinical decisions about assigning patients to treatment. Marsh 2009 proposed an algorithm for the treatment of severe aplastic anemia that directs patients younger than 40 years of age to the MSD‐HSCT group and patients older than 40 years of age to the immunosuppressive therapy group for first‐line treatment. The upper age limit for recommending transplantation is a matter of debate because younger patients could have increased risk if co‐morbidities are present and older patients could have a favorable prognosis if in a state of physical well‐being and if transplanted as early as possible after diagnosis. Gupta 2010 also reported that mortality risks were associated with a poor performance score and a long interval between diagnosis and transplantation of more than three months. Sangiolo 2010 reported on 23 consecutive patients ranging in age from 40 to 68 years and showed an overall survival of 65% after a median follow‐up of 9.1 years. The authors concluded that their data favors extending MSD‐HSCT to patients older than 40 years of age who are without significant co‐morbidities. The Third Consensus Conference on the treatment of aplastic anemia agreed on 21 February 2010 that bone marrow should be used as the source of stem cells and that the upper age limit should be 50 years (Kojima 2011). In a recent publication (ahead of print) Bacigalupo 2012 reported a survival advantage for bone marrow as compared to peripheral blood as a stem cell source in all age groups.
Concerning immunosuppressive therapy, overall survival was 45% in the study by Bayever 1984 and ranged from 81% to 93% in the study by Führer 1998. Marsh 2009 reported five‐year survival from 75% to 85% and Guinan 2009 reported 10‐year survival from 80% to 83%, as summarized in other studies. Deyell 2011 estimated five‐year overall survival of 96% in 45 children with a median age of 7.3 years and therefore gives reason to believe that children can have a very good prognosis if treated with immunosuppressive therapy. The Third Consensus Conference on the treatment of aplastic anemia agreed in 2010 that the combination of antithymocyte globulin and ciclosporin remains the gold standard for immunosuppressive therapy (Kojima 2011).
Bacigalupo 2008 reported that the outcome has improved since 1996 for HSCT but not for immunosuppressive therapy. Peinemann 2011 identified three studies that reported a statistically significant improvement of overall survival in the HSCT group but not in the immunosuppressive therapy group (Bacigalupo 2000; Locasciulli 1990; Locasciulli 2007) and one study that found an improvement in both treatment groups (Pitcher 1999). Several factors may have contributed to recent improvements in HSCT, such as detailed HLA‐matching, less irradiation‐based conditioning, less acute GVHD with a prophylaxis combination of methotrexate plus ciclosporin instead of methotrexate alone (Bacigalupo 2000), refinement of the type and dosage of conditioning drugs, and general advancement of medical and nursing clinical science.
It is assumed that frequent prior transfusion of blood products and prior immunosuppressive therapy is associated with a worse outcome after HSCT. Therefore, first‐line HSCT is recommended in eligible patients with selected characteristics. The data in the included studies were too scarce to allow any reference to this important topic. Immunosuppressive therapy is an alternative treatment option for patients who are not eligible for sibling donor HSCT and bone marrow transplantation (BMT) is the recommended source of stem cells (Marsh 2009). This recommendation was derived from the results of registry analyses that showed worse survival in patients that received peripheral blood stem cells (Schrezenmeier 2007).
Peinemann 2011 conducted a systematic review without applying restrictions such as excluding studies with a retrospective design and included as many comparative studies as possible so as not to miss any valuable outcome information. Data from patients with varying characteristics, such as young and advanced age, may have compromised the applicability of the results. They found high risk of bias among 26 identified non‐randomized comparative studies. Of these, 19 studies were included in a meta‐analysis of overall mortality but considerable heterogeneity did not justify a pooled estimate. Rather the forest plot was meant only to visualize the effect and the corresponding broad confidence intervals.
Secondary outcomes
Treatment‐related mortality was high in the MSD‐HSCT group at 20% to 42% of transplanted patients. Graft failure and GVHD are adverse events that are unique complications of allogeneic transplantation. Thus, treatment‐related mortality events are easy to define and to detect after transplantation. For the immunosuppressive therapy group, treatment‐related mortality was not observed. Treatment‐related mortality events are very difficult to determine. An infection, for example, can be caused by the disease or can be caused by the treatment. It is almost impossible to clarify this situation.
Bayever 1984 reported graft failure in 3% of patients and Gratwohl 1981 in 16% of patients, which lies at either end of the range of 4% to 14% summarized by Marsh 2009. Bayever 1984 reported that 64% of patients did not respond to immunosuppressive therapy at three months and that 12.5% of responders relapsed at five years. While the 'no response' rate of 64% appears rather high, the 'no response' rate of 15% reported by Gratwohl 1981 is probably rather low compared with others. Führer 1998 reported a higher proportion of responders (69% and 44%) compared to Bayever 1984 and response and relapse was significantly favorable in the VSAA only subgroup. Marsh 2009 described the combination of antithymocyte globulin and ciclosporin as being associated with response rates of between 60% and 80%. Guinan 2009 described a response rate from 27% to 79% in pediatric studies at six months and a relapse rate among responders between 16% to 33% at 10 years. According to Deyell 2011, the five‐year cumulative incidence of relapse was 12.9% in 45 children with a median age of 7.3 years; according to Kamio 2011 a response was observed in 59.8% (264 of 441) children and the 10‐year cumulative incidence of relapse was 11.9% at 10 years. The recent findings about response and relapse match well with the very early findings. This leads to the assumption that the serious problems of not responding to immunosuppressive therapy or relapsing after an initial successful response may not have improved sufficiently over the last three decades. However, a reporting bias for these late events cannot be ruled out. Bayever 1984 reported acute GVHD (grade II to IV) in 29% of patients, which may appear high compared to the range of 12% to 30% (but grade III to IV) summarized by Marsh 2009. The 21% risk of extensive chronic GVHD reported by Bayever 1984 matches the range from 30% to 40% summarized by Marsh 2009 (any chronic GVHD). The condition regimen and graft manipulation has substantially changed over time, so that comparisons over three decades appear biased. Gratwohl 1981 reported 26% GVHD but did not distinguish between acute and chronic GVHD, which makes it difficult to compare the reported rates.
The risk for later secondary clonal disease and malignancies, including hemolytic paroxysmal nocturnal hemoglobinuria and myelodysplastic syndrome, was 3% in the HSCT group reported by Bayever 1984 and 5% in the immunosuppressive therapy group reported by Führer 1998. According to Deyell 2011, the five‐year cumulative incidence of clonal evolution was 3.2% in 45 children with a median age of 7.3 years. We believe that this risk is probably highly underestimated because of an insufficient length of follow‐up. Indeed, Frickhofen 2003 found much higher proportions among 30 responders (14 complete, 16 partial). Five patients developed hemolytic paroxysmal nocturnal hemoglobinuria, four myelodysplastic syndrome or leukemia, and four solid tumors after a median follow‐up of 11.3 years. The actual probability at 11 years was 25% for clonal or malignant disease, 10% for hemolytic paroxysmal nocturnal hemoglobinuria, 8% for myelodysplastic syndrome or leukemia, and 11% for solid tumors. It should be mentioned that 30/17 patients of a total of 47 patients were treated with/without ciclosporin. Guinan 2009 stressed that recurrence of severe aplastic anemia and clonal evolution are hurdles to overcome in improving overall survival. Frickhofen 2003 showed in a RCT that ciclosporin is an essential part of immunosuppressive therapy along with antithymocyte globulin. Recently, Scheinberg 2011 showed in a RCT that horse is superior to rabbit antithymocyte globulin, as indicated by hematologic response and survival. However, horse antithymocyte globulin is currently not available on the pharmaceutical market (Marsh 2011).
Bayever 1984 reported a good Karnofsky performance status of 92% in the HSCT group versus 46% in the immunosuppressive therapy group. This result may point to a better performance after HSCT than after immunosuppressive therapy for patients that have survived. We did not find any other study, review, or guideline addressing Karnofsky performance status.
Recent publications and potential treatment outlook
While updating the literature search, we identified recent publications that are loosely associated with the topic of the present Cochrane review. Burroughs 2012 observed three groups of HLA‐matched related HSCTs with respect to conditioning and GVHD prophylaxis. In group 1 the regimen consisted of cyclophosphamide and methotrexate, in group 2 cyclophosphamide was given alone, and in group 3 cyclophosphamide and methotrexate was extended with antithymocyte globulin and ciclosporin. With a median follow‐up of 25 years, the five‐year survival was 66%, 95%, and 100% for groups 1, 2, and 3, respectively (P < 0.0001). The authors concluded: "Advances in preparative and GVHD prophylaxis regimens, and supportive care during the past 40 years have led to improved outcomes for children with SAA. These results confirm the use of allogeneic marrow transplantation for children with SAA who have HLA‐matched related donors". Battiwalla 2012 found that secondary graft failure at two years after HLA‐matched sibling bone marrow transplantation for severe aplastic anemia was lower in patients who carried HLA DR15+ compared to HLA DR15‐ but did not find a significant impact on survival. Kim 2012 found that overall survival did not differ significantly between the younger patient group (< 40 years old) and the older patient group (>= 40 years old) in patients with severe aplastic anemia who received transplants from HLA‐matched related donors. The authors reported that patients between the ages of 41 and 50 years should undergo allogeneic HSCT as early as possible to optimize survival. Buchbinder 2013 described autologous cord blood HSCT as an effective and safe alternative if no related donor is available. Risitano 2013 recommended that alemtuzumab‐based immunosuppression may be a treatment option as a second‐line therapy for severe aplastic anemia patients who are not indicated for allogeneic HSCT and who did not respond to initial immunosuppressive therapy. Scheinberg 2012a and Scheinberg 2012b provided an overview and update of various treatment options for severe aplastic anemia including immunosuppressive therapy and transplantation.
Authors' conclusions
Implications for practice.
The included data are too scarce and too biased to allow any conclusion on the comparative effectiveness of first‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors and first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin. We are unable to make firm recommendations regarding choice of intervention. The rates of adverse events, such as treatment‐related mortality, graft failure, no response to first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin, and graft‐versus‐host disease, are unusually high, which may be explained by the age of the studies (starting in 1976). The results are not applicable to current modern standard care.
Patients should be made aware of the early treatment‐related mortality and the burden of acute graft‐versus‐host disease after first‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors. Patients treated with first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin should also be made aware that the disease may recur after initial successful treatment, and that life‐threatening late clonal and malignant disease after first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin may occur in a higher percentage compared to first‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors. Unrelated matched donor transplantation after first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin in patients without a first‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors was not the scope of the present review.
Implications for research.
Randomized controlled trials that compare first‐line allogeneic hematopoietic stem cell transplantation of HLA‐matched sibling donors with first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin are unlikely to be conducted due to ethical constraints and strong patient and clinician preferences for one or the other intervention based on patient characteristics and disease status. In theory, prospective studies that use 'Mendelian randomization' offer a useful alternative to randomized controlled trials to reduce the risk of bias. In practice, it appears unlikely that a well‐designed comparative study based on 'Mendelian randomization' will be accomplished in the future. Some experts in this field have admitted that expectations have been disappointed. One reason might be the rarity of severe aplastic anemia, although collaboration across cancer centers could facilitate the recruitment of sufficient numbers of participants. Another reason is that the outcome of transplantation has improved considerably. This is true not only for matched related donor transplantation but especially for matched unrelated donors. It means that ciclosporin and/or antithymocyte or antilymphocyte globulin may not be a first treatment choice if a matched sibling or a matched related or even a matched unrelated donor is available.
History
Protocol first published: Issue 1, 2007 Review first published: Issue 7, 2013
| Date | Event | Description |
|---|---|---|
| 9 November 2006 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We thank the staff of the Cochrane Haematological Malignancies Group (CHMG), especially Nicole Skoetz for advice on the review. We thank Tatjana Kvitkina for language translation.
Appendices
Appendix 1. MEDLINE Ovid search strategy
1 exp ANEMIA, APLASTIC/ 2 (aplast$ anem$ or aplast$ anaem$).tw,kf,ot. 3 or/1‐2 4 exp STEM CELL TRANSPLANTATION/ 5 exp TRANSPLANTATION, HOMOLOGOUS/ 6 transplant$.tw,kf,ot. 7 graft$.tw,kf,ot. 8 (allograft$ or allo‐graft$).tw,kf,ot. 9 (homograft$ or homo‐graft$).tw,kf,ot. 10 or/4‐9 11 RANDOMIZED CONTROLLED TRIALS.sh. 12 RANDOMIZED CONTROLLED TRIAL.pt. 13 random$.tw,kf,ot. 14 CONTROLLED CLINICAL TRIAL.pt. 15 RANDOM ALLOCATION.sh. 16 DOUBLE BLIND METHOD.sh. 17 SINGLE BLIND METHOD.sh. 18 (ANIMALS NOT HUMANS).sh. 19 exp CLINICAL TRIALS/ 20 CLINICAL TRIAL.pt. 21 (clin$ adj25 trial$).tw,kf,ot. 22 ((singl$ or doubl$ or trebl$ or tripl$) adj25 (blind$ or mask$)).tw,kf,ot. 23 PLACEBOS.sh. 24 placebo$.tw,kf,ot. 25 RESEARCH DESIGN.sh. 26 COMPARATIVE STUDY.sh. 27 exp EVALUATION STUDIES/ 28 FOLLOW‐UP STUDIES.sh. 29 PROSPECTIVE STUDIES.sh. 30 (control$ or prospectiv$ or volunteer$).tw,kf,ot. 31 (metaanaly$ or (meta and analy$) or (( review or search$) and (medical database$ or medline or pubmed or embase or cochrane or systemat$))).tw,kf,ot. 32 META‐ANALYSIS.sh. 33 META‐ANALYSIS.pt. 34 exp REGISTRIES/ 35 (registr$ or register$ or ibmtr$ or ebmt$).tw,kf,ot. 36 ((group or regist$) and (blood or stem cell or marrow) and tranplant$ and (europ$ or international)).tw,kf,ot. 37 or/11‐36 38 (ANIMALS NOT HUMANS).sh. 39 37 not 38 40 and/3,10,39
Appendix 2. EMBASE Ovid search strategy
1 APLASTIC ANEMIA.sh. 2 (aplast$ anem$ or aplast$ anaem$).tw,hw,ot. 3 or/1‐2 4 exp STEM CELL TRANSPLANTATION/ 5 exp BONE MARROW TRANSPLANTATION/ 6 transplant$.tw,hw,ot. 7 graft$.tw,hw,ot. 8 (allograft$ or allo‐graft$).tw,hw,ot. 9 (homograft$ or homo‐graft$).tw,hw,ot. 10 or/4‐9 11 RANDOMIZED CONTROLLED TRIAL.sh. 12 RANDOMIZATION.sh. 13 random$.tw,hw,ot. 14 exp CLINICAL TRIAL/ 15 (clin$ adj25 trial$).tw,hw,ot. 16 DOUBLE BLIND PROCEDURE.sh. 17 SINGLE BLIND PROCEDURE.sh. 18 ((singl$ or doubl$ or trebl$ or tripl$)adj25 (blind$ or mask$)).tw,hw,ot. 19 PLACEBO.sh. 20 placebo$.tw,hw,ot. 21 FOLLOW UP.sh. 22 COMPARATIVE STUDY.sh. 23 PROSPECTIVE STUDY.sh. 24 (control$ or prospectiv$ or volunteer$).tw,hw,ot. 25 META ANALYSIS.sh. 26 (metaanaly$ or (meta and analy$) or ((review or search$) and (medical database$ or medline or pubmed or embase or cochrane or sytemat$))).tw,hw,ot. 27 REGISTER.sh. 28 (registr$ or register$ or ibmtr$ or ebmt$).tw,hw,ot. 29 ((group or regist$) and (blood or stem cell or marrow) and transplant$ and (europ$ or international)).tw,hw,ot. 30 or/11‐29 31 (ANIMAL not HUMAN).sh. 32 30 not 31 33 and/3,10,32
Appendix 3. The Cochrane Library CENTRAL search strategy
#1 MeSH descriptor Anemia, Aplastic explode all trees in MeSH products #2 aplast* anem* OR aplast* anaem* in All Fields in all products #3 (#1 OR #2) #4 MeSH descriptor Stem Cell Transplantation explode all trees in MeSH products #5 MeSH descriptor Transplantation, Homologous explode all trees in MeSH products #6 transplant* in All Fields in all products #7 graft* in All Fields in all products #8 allograft* OR allo‐graft* in All Fields in all products #9 homograft* OR homo‐graft* in All Fields in all products #10 (#4 OR #5 OR #6 OR #7 OR #8 OR #9) #11 (#3 AND #10)
Data and analyses
Comparison 1. MSD‐HSCT versus IST ‐ all patients.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 3 | 229 | Hazard Ratio (Random, 95% CI) | 0.95 [0.43, 2.12] |
| 2 Secondary clonal disease or malignancies | 2 | 170 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.54 [0.07, 4.00] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bayever 1984.
| Methods | Duration
Randomization
Treatment
Median follow‐up time
|
|
| Participants | Setting
Eligibility criteria
Number of patients
Age
Gender
Further inclusion criteria to define SAA
|
|
| Interventions | MSD‐HSCT arm
IST arm
|
|
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessor was not reported for any outcome |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The data for all 57 included patients have been analyzed and it is conceivable that the outcome data were complete (Table 3 of the article) |
| Selective reporting (reporting bias) | Unclear risk | Relapse was reported for the IST group but not for the MSD‐HSCT group. "All eight patients with severe aplastic anemia who responded after ATG therapy are alive. One of the responders has relapsed but still survives(..)" |
| Other bias | High risk | The authors reported the study results at an early time point before all planned data had been gathered. "We present this interim report(...)". No other bias such as competing interest or cross‐over. |
| Comparable baseline characteristics | Low risk | There were no differences of baseline characteristics between the 2 treatment groups, in particular with reference to age (Table 1 of the article) |
| Concurrent control | Low risk | Data for the control group were collected during the same time period as the data for the test group. "Fifty‐seven consecutive patients younger than 25 years with severe aplastic anemia underwent treatment at UCLA between November 1977 and October 1982." |
Führer 1998.
| Methods | Duration
Randomization
Treatment
Median follow‐up time
|
|
| Participants | Setting
Eligibility criteria
Number of patients
Age
Gender
Further inclusion criteria to define SAA
|
|
| Interventions | Interval from diagnosis to treatment
MSD‐HSCT arm
IST arm
|
|
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessor was not reported for any outcome |
| Incomplete outcome data (attrition bias) All outcomes | High risk | In the 2005 update, 7.5% (5 of 67) patients assigned to the MSD‐HSCT group dropped out because the parents refused the treatment. "Sixty‐two patients received BMT after a conditioning treatment with ATG (...) and cyclophosphamide (...). In the remaining 5 patients, 3 with SAA and 2 with vSAA, parents refused BMT." |
| Selective reporting (reporting bias) | High risk | In the 2005 update, overall survival, secondary clonal disease or malignancies, and also relapse were not reported separately for the 2 distinct treatment groups. Rather, the results were presented for 2 subgroups according to disease severity. This was different from the earlier report of the same study published in 1998 covering the study period from 1993 to 1997. See Fig. 1 and Tab. 1 of the article. "In 137 (64%) of 213 patients very SAA was diagnosed. Within the BMT group, patients with very SAA (N = 40) and with SAA (N = 27) reached comparable survival rates (5‐year survival rate: very SAA 89% (95% CI 80% to 99%); SAA 96% (95% CI 89% to 100%)." |
| Other bias | High risk | Financial support was provided by 2 pharmaceutical companies. "The SAA 94 study was supported by AMGEN and JMTIX". |
| Comparable baseline characteristics | Low risk | There were no differences in baseline characteristics between the 2 treatment groups, in particular with reference to age. See Table 1 and Table 2 of the 1998 article. |
| Concurrent control | Low risk | Data for the control group were collected during the same time period as the data for the test group. "Two hundred and thirteen patients newly diagnosed with SAA younger than the age of 17 years in 53 centers in Germany and Austria were included in the study between November 1993 and December 2001." |
Gratwohl 1981.
| Methods | Duration
Randomization
Treatment
Median follow‐up time
|
|
| Participants | Setting
Eligibility criteria
Number of patients
Age
Gender
Further inclusion criteria to define SAA
|
|
| Interventions | MSD‐HSCT arm
IST arm
Haploidentical family donor arm
|
|
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessor was not reported for any outcome |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The data for 58 of 61 consecutively recruited patients that fulfilled the eligibility criteria have been analyzed. Therefore 3 patients have dropped out. "Von den 61 überwiesenen Patienten sind 3 trotz intensivem Zellersatz und Antibiotika gestorben, bevor die spezifische Behandlung fUr die SAA begonnen werden konnte." |
| Selective reporting (reporting bias) | Unclear risk | Besides overall survival, no other outcome was reported systematically |
| Other bias | Unclear risk | 15% (2 of 13) patients assigned to the IST group had an HLA‐identical sibling donor and were actually eligible to be allocated to the MSD‐HSCT group. "13 Patienten hatten keinen Spender und erhielten ALG allein. Von diesen 13 Patienten in Gruppe C hatten 2 einen HLA‐identischen Spender". No other bias such as competing interest or cross‐over. The study was supported by non‐for‐profit institutions such as a Swiss National Research Foundation, a Swiss Cancer Association, and the Swiss Department for Health. |
| Comparable baseline characteristics | Low risk | There were no differences of baseline characteristics between the 2 treatment groups, in particular with reference to age. "Diese drei Gruppen sind vergleichbar bezüglich Alter, Ätiologie, vorhergehender Behandlung und Schwere der SAA bei Einweisung (Tab. 2)." |
| Concurrent control | Low risk | Data for the MSD‐HSCT arm were collected during the same time period as the data for the IST arm. "Dieser Bericht beschreibt alle Patienten, die dem Kantonsspital Basel zwischen Januur 1976 und Dezember 1980 wegen SAA überwiesen wurden." |
Definitions of HLA‐identical or matched bone marrow vary with time and between different studies. In this table, expressions are quoted as given in the publications. Death from secondary malignancy was not considered treatment‐related mortality in the present review.
ALG: antilymphocyte globulin
ATG: antithymocyte globulin
BMT: bone marrow transplantation
G‐CSF: granulocyte colony‐stimulating factor
GVHD: graft‐versus‐host‐disease
HSCT: hematopoietic stem cell transplantation
IST: immunosuppressive therapy
MSD: matched sibling donor
MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched (identical) sibling donor
PMN: polymorphonuclear neutrophils
SAA: severe aplastic anemia
VSAA: very severe aplastic anemia
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ACS/NIH 1976 | Study design: not prospective: registry report |
| Ahn 2003 | Allocation: not consistent with 'Mendelian randomization': age unequally distributed HSCT versus IST: 83 versus 61 patients in age category 14 to 40 years, 73 versus 3 patients in age category >= 41 years, P < 0.001 |
| Arranz 1994 | Allocation: not consistent with 'Mendelian randomization': assignment by age, cut‐off 40 years |
| Bacigalupo 1988 | Study design: not prospective: registry report |
| Bacigalupo 1996 | Study design: not prospective: registry report |
| Bacigalupo 2000a | Study design: not prospective: registry report |
| Bacigalupo 2000b | Study design: not prospective: registry report |
| Camitta 1976 | Control: fewer than 80% of patients with first‐line IST |
| Camitta 1978 | Control: fewer than 80% of patients with first‐line IST |
| Camitta 1979 | Control: fewer than 80% of patients with first‐line IST |
| Cesaro 2010 | Study design: not prospective: registry report |
| Champlin 1984 | Allocation: not consistent with 'Mendelian randomization': assignment by age, cut‐off 45 years |
| Chen 1992 | Fewer than 5 patients in one arm: 3 patients in MSD‐HSCT group |
| Crump 1992 | Intervention: fewer than 80% of patients with first‐line MSD‐HSCT: 100% second‐line HSCT |
| de Planque 1990 | Intervention: fewer than 80% of patients with first‐line MSD‐HSCT: 42% second‐line HSCT |
| Deyell 2009 | Study design: not prospective |
| Doney 1981 | Intervention: fewer than 80% of patients with first‐line HSCT: haploidentical marrow |
| Doney 1997 | Allocation: not consistent with 'Mendelian randomization': assignment by age, cut‐off 55 years |
| Fouladi 2000 | Study design: not prospective, no clues to prospective design |
| Ganapiev 2010 | Study design: not prospective, no clues to prospective design [Russian] |
| Garanito 2009 | Study design: not prospective |
| George 2010 | Study design: not prospective |
| Ghavamzadeh 2004 | Allocation: not consistent with 'Mendelian randomization': assignment by age, cut‐off 45 years |
| Gillio 1997 | Study design: not prospective, reporting of retrospective design |
| Gluckman 1979 | Study design: not prospective, no clues to prospective design; a randomized allocation was reported for the last year of investigation, however, the number of patients randomized and the years of transplantation were not reported |
| Gluckman 1981 | Study design: not prospective: registry report |
| Gluckman 1987 | Study design: not prospective: registry report |
| Golubovskaya 2010 | Unclear information: conference abstract without required information, authors did not clearly report whether allocation was based on availability of MSD and not on patient age |
| Halperin 1989 | Study design: not prospective, no clues to prospective design |
| Hirabayashi 1995 | Study design: not prospective: registry report |
| Horowitz 1989 | Study design: not prospective: registry report |
| Horowitz 1997 | Study design: not prospective: registry report |
| Howard 2004 | Outcome: not a predefined outcome |
| Höcker 1987 | Allocation: not consistent with 'Mendelian randomization': by age [German] |
| Ilhan 2001 | Patients: fewer than 80% of patients with SAA |
| Kahn 2002 | Allocation: not consistent with 'Mendelian randomization': assignment by age, cut‐off 40 years |
| Kim 1994 | Allocation: not consistent with 'Mendelian randomization' |
| Kim 2003 | Allocation: not consistent with 'Mendelian randomization': assignment by age, cut‐off 50 years |
| Kodera 1999 | Study design: not prospective: registry report |
| Kojima 1988 | Study design: not prospective, no clues to prospective design |
| Kojima 2000 | Study design: not prospective, reporting of retrospective design |
| Kosaka 2004 | Intervention: fewer than 80% of patients with first‐line MSD‐HSCT: 100% second‐line |
| Kulagin 2006 | Fewer than 5 patients in one arm |
| Lawlor 1997 | Study design: not prospective, reporting of retrospective design |
| Ljungman 1991 | Study design: not prospective, no clues to prospective design [Swedish] |
| Locasciulli 1990 | Study design: not prospective: registry report |
| Locasciulli 2007 | Study design: not prospective: registry report |
| McCann 1994 | Study design: not prospective: registry report |
| Milosevic 1998 | Intervention: fewer than 80% of patients with first‐line MSD‐HSCT [Serbian] |
| Mollee 2001 | Allocation: not consistent with 'Mendelian randomization': by age |
| Montante 2009 | Fewer than 5 patients in one arm |
| Mrsic 2009 | Allocation: not consistent with 'Mendelian randomization' [Croatian] |
| Nagler 2001 | Patients: fewer than 80% of patients with SAA |
| Paquette 1995 | Allocation: not consistent with 'Mendelian randomization': age unequally distributed HSCT versus IST, 36% versus 15% in age category 16 to 19 years, 42% versus 40% in age category 19 to 29 years, 22% versus 45% in age category >= 30 years, P = 0.02 |
| Pitcher 1999 | Patients: not at least 80% SAA: 23% of patients with non‐severe aplastic anemia |
| Podesta 1998 | Outcome: not a predefined outcome |
| Pokorski 1998 | Study design: not prospective: registry report |
| Shaw 1999 | Patients: fewer than 80% of patients with SAA |
| Socié 1993 | Patients: fewer than 80% of patients with SAA |
| Sovinz 2010 | Unclear information: conference abstract without required information, authors did not clearly report whether allocation was based on availability of MSD and not on patient age |
| Stalder 2009 | Study design: not prospective |
| Starý 1998 | Study design: not prospective [Czech] |
| Tichelli 1994 | Allocation: not consistent with 'Mendelian randomization': by age |
| Tschiedel 2010 | Fewer than 5 patients in one arm [German] |
| Tsukimoto 1989 | Outcome: not a predefined outcome |
| Tukic 2009 | Unclear information: conference abstract without required information, authors did not clearly report whether allocation was based on availability of MSD and not on patient age |
| Tzeng 1989 | Study design: not prospective, no clues to prospective design |
| UCLA 1976 | Control: fewer than 80% of patients with first‐line IST: androgens only |
| Uss 1999 | Study design: not prospective [Russian] |
| Viollier 2005 | Allocation: not consistent with 'Mendelian randomization': assignment by age, cut‐off 40 years |
| Wagner 1996 | Patients: fewer than 80% of patients with SAA |
| Webb 1991 | Study design: not prospective, reporting of retrospective design |
| Werner 1989 | Study design: not prospective, reporting of retrospective design |
| Windass 1987 | Patients: fewer than 80% of patients with SAA |
| Yan 2011 | Intervention: fewer than 80% of patients with first‐line MSD‐HSCT [Chinese] |
| Yoshida 2011 | Allocation: not consistent with 'Mendelian randomization': donors were family members and not siblings |
HSCT: hematopoietic stem cell transplantation IST: immunosuppressive therapy MSD: matched sibling donor MSD‐HSCT: first‐line allogeneic hematopoietic stem cell transplantation from HLA‐matched (identical) sibling donor SAA: severe aplastic anemia
Differences between protocol and review
We changed the objective to evaluation of transplants from HLA‐matched (identical) siblings only and not from HLA‐matched related family members. We wanted to create a homogeneous group of patients with an identical close relationship. We also wanted to react to common practice which recommends first‐line allogeneic hematopoietic stem cell transplantation preferably to HLA‐matched (identical) siblings.
We included studies with a prospective design only and we did not include retrospective studies to reduce the risk of bias introduced by retrospective designs.
We amended the outcomes. We deleted "treatment‐free survival" as a primary outcome measure because it is not an established outcome in transplantation science. We deleted "transfusion requirements" as a secondary outcome measure because it is a risk factor before treatment. We changed "complete and partial response" to "no response to first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin" and subsumed it into the outcome "treatment failure" together with "graft failure" in transplanted patients.
We confined the immunosuppressive treatment to first‐line ciclosporin and/or antithymocyte or antilymphocyte globulin.
Contributions of authors
FP: concept, developing search strategy, selecting and appraising studies, extracting and analyzing data, interpretation of results, primary manuscript preparation.
CB: selecting studies, interpretation of results, manuscript review.
UG: appraising studies, extracting and analyzing data, interpretation of results, manuscript review.
Sources of support
Internal sources
-
University of Cologne, Germany.
Access to full‐text publications
External sources
No sources of support supplied
Declarations of interest
The authors declare that they have no conflict of interest.
New
References
References to studies included in this review
Bayever 1984 {published data only}
- Bayever E, Champlin R, Ho W, Lenarsky C, Storch S, Ladisch S, et al. Comparison between bone marrow transplantation and antithymocyte globulin in treatment of young patients with severe aplastic anemia. Journal of Pediatrics 1984;105:920‐5. [DOI] [PubMed] [Google Scholar]
Führer 1998 {published data only}
- Führer M, Bender‐Götze C, Ebell W, Friedrich W, Kohne E. Treatment of aplastic anemia‐‐aims and development of the SAA 94 pilot protocol [Die Behandlung der Aplastischen Anamie‐‐Zielsetzung und Aufbau des Pilotprotokolls SAA 94]. Klinische Pädiatrie 1994;206:289‐95. [DOI] [PubMed] [Google Scholar]
- Führer M, Burdach S, Ebell W, Gadner H, Haas R, Harbott J, et al. Relapse and clonal disease in children with aplastic anemia (AA) after immunosuppressive therapy (IST): The SAA 94 experience. Klinische Pädiatrie 1998;210:173‐9. [DOI] [PubMed] [Google Scholar]
- Führer M, Rampf U, Baumann I, Faldum A, Niemeyer C, Janka‐Schaub G, et al. Immunosuppressive therapy for aplastic anemia in children: a more severe disease predicts better survival. Blood 2005;106:2102‐4. [DOI] [PubMed] [Google Scholar]
- Führer M, Rampf U, Burdach S. Immunosuppressive therapy (IST) and bone marrow transplantation (BMT) for aplastic anemia (AA) in children. Blood 1998;92 Suppl 1:156a, Abstract 631. [Google Scholar]
- Führer M, Rampf U, Niemeyer CM, Faldum A, Baumann I, Friedrich W, et al. Bone marrow transplantation and immunosuppressive therapy in children with aplastic anemia: data from a prospective multinational trial in Germany, Austria and Switzerland. Blood 2004;104:Abstract 1439. [Google Scholar]
Gratwohl 1981 {published data only}
- Gratwohl A, Osterwalder B, Nissen C, Leibundgut U, Signer E, Luthy A, et al. Treatment of severe aplastic anemia [Behandlung der schweren aplastischen Anämie]. Schweizerische Medizinische Wochenschrift 1981;111:1520‐2. [PubMed] [Google Scholar]
References to studies excluded from this review
ACS/NIH 1976 {published data only}
- ACS/NIH. Bone marrow transplantation from donors with aplastic anemia. A report from the ACS/NIH bone marrow transplant registry. JAMA 1976;236(10):1131‐5. [DOI] [PubMed] [Google Scholar]
Ahn 2003 {published data only}
- Ahn MJ, Choi JH, Lee YY, Choi IY, Kim IS, Yoon SS, et al. Outcome of adult severe or very severe aplastic anemia treated with immunosuppressive therapy compared with bone marrow transplantation: multicenter trial. International Journal of Hematology 2003;78:133‐8. [DOI] [PubMed] [Google Scholar]
Arranz 1994 {published data only}
- Arranz R, Otero MJ, Ramos R, Steegmann JL, Lamana ML, Tomas JF, et al. Clinical results in 50 multiply transfused patients with severe aplastic anemia treated with bone marrow transplantation or immunosuppressive therapy. Bone Marrow Transplantation 1994;13:383‐7. [PubMed] [Google Scholar]
- Arranz R, Steegmann JL, Fernandez‐Garesse D, Figuera A, Vazquez L, Fernandez‐Ranada JM. Treatment of severe aplastic anemia with bone marrow transplants or with antithymocyte globulin. Evaluation of 27 patients [Tratamiento de la anemia aplasica severa con trasplante de medula osea o con globulina antitimocitica. Evaluacion de 27 pacientes]. Revista Clínica Espanola 1989;184:121‐4. [PubMed] [Google Scholar]
Bacigalupo 1988 {published data only}
- Bacigalupo A, Hows J, Gluckman E, Nissen C, Marsh J, Lint MT, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. British Journal of Haematology 1988;70:177‐82. [DOI] [PubMed] [Google Scholar]
Bacigalupo 1996 {published data only}
- Bacigalupo A. Aetiology of severe aplastic anaemia and outcome after allogeneic bone marrow transplantation or immunosuppression therapy. Working Party on Severe Aplastic Anaemia of the European Blood and Marrow Transplantation Group. European Journal of Haematology 1996;60:16‐9. [PubMed] [Google Scholar]
Bacigalupo 2000a {published data only}
- Bacigalupo A, Oneto R, Bruno B, Socie G, Passweg J, Locasciulli A, et al. Current results of bone marrow transplantation in patients with acquired severe aplastic anemia. Report of the European Group for Blood and Marrow transplantation. On behalf of the Working Party on Severe Aplastic Anemia of the European Group for Blood and Marrow Transplantation. Acta Haematologica 2000;103:19‐25. [DOI] [PubMed] [Google Scholar]
Bacigalupo 2000b {published data only}
- Bacigalupo A, Brand R, Oneto R, Bruno B, Socie G, Passweg J, et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy‐‐The European Group for Blood and Marrow Transplantation experience. Seminars in Hematology 2000;37:69‐80. [DOI] [PubMed] [Google Scholar]
Camitta 1976 {published data only}
- Camitta BM, Thomas ED, Nathan DG, Santos G, Gordon‐Smith EC, Gale RP, et al. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood 1976;48:63‐70. [PubMed] [Google Scholar]
Camitta 1978 {published data only}
- Camitta BM, Thomas ED. Severe aplastic anaemia: a prospective study of the effect of androgens or transplantation on haematological recovery and survival. Clinical Haematology 1978;7:587‐95. [PubMed] [Google Scholar]
Camitta 1979 {published data only}
- Camitta BM, Thomas ED, Nathan DG, Gale RP, Kopecky KJ, Rappeport JM, et al. A prospective study of androgens and bone marrow transplantation for treatment of severe aplastic anemia. Blood 1979;53:504‐14. [PubMed] [Google Scholar]
Cesaro 2010 {published data only}
- Cesaro S, Marsh J, Tridello G, Rovo A, Maury S, Montante B, et al. Retrospective survey on the prevalence and outcome of prior autoimmune diseases in patients with aplastic anemia reported to the registry of the European group for blood and marrow transplantation (Conference abstract). Acta Haematologica 2010;124(1):19‐20. [DOI] [PubMed] [Google Scholar]
Champlin 1984 {published data only}
- Champlin R, Ho W, Bayever E, Winston DJ, Lenarsky C, Feig SA, et al. Treatment of aplastic anemia: results with bone marrow transplantation, antithymocyte globulin, and a monoclonal anti‐T cell antibody. Progress in Clinical Biological Research 1984;148:227‐38. [PubMed] [Google Scholar]
Chen 1992 {published data only}
- Chen RL, Lin KH, Chen BW, Su S, Lin DT, Chuu WM, et al. Long‐term observation of pediatric aplastic anemia. Journal of the Formosan Medical Association 1992;91:390‐5. [PubMed] [Google Scholar]
Crump 1992 {published data only}
- Crump M, Larratt LM, Maki E, Curtis JE, Minden MD, Meharchand JM, et al. Treatment of adults with severe aplastic anemia: primary therapy with antithymocyte globulin (ATG) and rescue of ATG failures with bone marrow transplantation. American Journal of Medicine 1992;92:596‐602. [DOI] [PubMed] [Google Scholar]
de Planque 1990 {published data only}
- Planque MM, Richel DJ, Fibbe WE, Ottolander GJ, Guiot HF, Zwaan FE. Acquired severe aplastic anaemia in adults‐‐a single centre study with 13 years follow‐up. The Netherlands Journal of Medicine 1990;37:103‐10. [PubMed] [Google Scholar]
Deyell 2009 {published data only}
- Deyell RJ, Shereck EB, Schultz KR. Immunosuppressive therapy without granulocyte‐colony stimulating factor in pediatric acquired aplastic anemia (Conference abstract). Pediatric Blood and Cancer 2009 2009;52(6):705. [Google Scholar]
Doney 1981 {published data only}
- Doney KC, Weiden PL, Buckner CD, Storb R, Thomas ED. Treatment of severe aplastic anemia using antithymocyte globulin with or without an infusion of HLA haploidentical marrow. Experimental Hematology 1981;9(8):829‐34. [PubMed] [Google Scholar]
Doney 1997 {published data only}
- Doney K, Leisenring W, Storb R, Appelbaum FR. Primary treatment of acquired aplastic anemia: outcomes with bone marrow transplantation and immunosuppressive therapy. Seattle Bone Marrow Transplant Team. Annals of Internal Medicine 1997;126(2):107‐15. [DOI] [PubMed] [Google Scholar]
Fouladi 2000 {published data only}
- Fouladi M, Herman R, Rolland‐Grinton M, Jones‐Wallace D, Blanchette V, Calderwood S, et al. Improved survival in severe acquired aplastic anemia of childhood. Bone Marrow Transplantation 2000;26:1149‐56. [DOI] [PubMed] [Google Scholar]
Ganapiev 2010 {published data only}
- Ganapiev AA, Golubovskaia IK, Zalialov IR, Estrina MA, Afanas'ev BV. The use of allogeneic bone marrow transplantation and immunosuppressive therapy in the treatment of patients with acquired aplastic anemia [Russian]. Terapevticheskii Arkhiv 2010;82(7):48‐52. [PubMed] [Google Scholar]
Garanito 2009 {published data only}
- Garanito MP, Filho VO, Dos Santos MV, Velloso E, Dulley FL, Carneiro JDA. Long‐term clinical outcome of children with acquired aplastic anemia in Brazil (Conference abstract). Blood 2009;114(22):4216. [Google Scholar]
George 2010 {published data only}
- George B, Mathews V, Viswabandya A, Lakshmi KM, Srivastava A, Chandy M. Allogeneic hematopoietic stem cell transplantation is superior to immunosuppressive therapy in Indian children with aplastic anemia single‐center analysis of 100 patients. Pediatric Hematology and Oncology 2010;27(2):122‐31. [DOI] [PubMed] [Google Scholar]
Ghavamzadeh 2004 {published data only}
- Ghavamzadeh A, Iravani M, Vafaiezadeh F, Jahani M, Mousavi A. Bone marrow transplantation versus immunosuppressive therapy in severe aplastic anemia, 1990 ‐ 2001. Archives of Iranian Medicine 2004;7:272‐8. [Google Scholar]
Gillio 1997 {published data only}
- Gillio AP, Boulad F, Small TN, Kernan NA, Reyes B, Childs BH, et al. Comparison of long‐term outcome of children with severe aplastic anemia treated with immunosuppression versus bone marrow transplantation. Biology of Blood and Marrow Transplantation 1997;3:18‐24. [PubMed] [Google Scholar]
Gluckman 1979 {published data only}
- Gluckman E, Devergie A, Faille A, Bussel A, Benbunan M, Bernard J. Antilymphocyte globulin treatment in severe aplastic anemia‐‐comparison with bone marrow transplantation. Report of 60 cases. Haematology and Blood Transfusion 1979;24:171‐9. [DOI] [PubMed] [Google Scholar]
Gluckman 1981 {published data only}
- Gluckman E, Barrett AJ, Arcese W, Devergie A, Degoulet P. Bone marrow transplantation in severe aplastic anaemia: a survey of the European Group for Bone Marrow Transplantation (E.G.B.M.T.). British Journal of Haematology 1981;49:165‐73. [DOI] [PubMed] [Google Scholar]
Gluckman 1987 {published data only}
- Gluckman E. Current status of bone marrow transplantation for severe aplastic anemia: a preliminary report from the International Bone Marrow Transplant Registry. Transplantation Proceedings 1987;19:2597‐9. [PubMed] [Google Scholar]
Golubovskaya 2010 {published data only}
- Golubovskaya I, Zalyalov Y, Ganapiev A, Afanasyev B. Treatment of acquired aplastic anaemia by bone marrow transplantation and antithymocyte globulin: A 5‐year single‐centre experience (Conference abstract). Bone Marrow Transplantation 2010;45:S171. [Google Scholar]
Halperin 1989 {published data only}
- Halperin DS, Grisaru D, Freedman MH, Saunders EF. Severe acquired aplastic anemia in children: 11‐year experience with bone marrow transplantation and immunosuppressive therapy. American Journal of Pediatric Hematology/Oncology 1989;11:304‐9. [PubMed] [Google Scholar]
Hirabayashi 1995 {published data only}
- Hirabayashi N, Kodera Y, Matsuyama T, Tanimoto M, Horibe K, Naoe T, et al. Bone marrow transplantation in 614 patients: Twenty year experience of the Nagoya Bone Marrow Transplantation Group (NBMTG). Transplantation Proceedings 1995;27:1380‐2. [PubMed] [Google Scholar]
Horowitz 1989 {published data only}
- Horowitz MM, Bortin MM, Rimm AA. Report from the International Bone Marrow Transplant Registry. Bone Marrow Transplantation 1989;4:221‐8. [PubMed] [Google Scholar]
Horowitz 1997 {published data only}
- Horowitz MM, Rowlings PA. An update from the International Bone Marrow Transplant Registry and the Autologous Blood and Marrow Transplant Registry on current activity in hematopoietic stem cell transplantation. Current Opinion in Hematology 1997;4:395‐400. [DOI] [PubMed] [Google Scholar]
Howard 2004 {published data only}
- Howard SC, Naidu PE, Hu XJ, Jeng MR, Rodriguez‐Galindo C, Rieman MD, et al. Natural history of moderate aplastic anemia in children. Pediatric Blood & Cancer 2004;43:545‐51. [DOI] [PubMed] [Google Scholar]
Höcker 1987 {published data only}
- Höcker P, Hinterberger W, Gadner H, Hajek‐Rosenmayr A, Fischer‐Hinterberger M, Kos M, et al. Therapy of severe aplastic anemia (SAA) by bone marrow transplantation (BMT) or immunosuppression? [Therapie der schweren aplastischen Anämie (SSA) durch Knochenmarktransplantation (KMT) oder Immunsuppression?]. Beiträge zu Infusiontherapie und Klinischer Ernährung 1987;18:329‐31. [PubMed] [Google Scholar]
Ilhan 2001 {published data only}
- Ilhan O, Bakanay SM, Arat M, Ozcan M, Celebi H, Gurman G, et al. Treatment of aplastic anemia: Experience of Ibn‐i Sina Hospital. Turkish Journal of Haematology 2001;18:33‐40. [Google Scholar]
Kahn 2002 {published data only}
- Kahn Q, Ellis RJ, Skikne BS, Mayo MS, Allgood JW, Bodensteiner DM, et al. A retrospective analysis of long‐term survival in severe aplastic anemia patients treated with allogeneic bone marrow transplantation or immunosuppressive therapy with antithymocyte globulin and cyclosporin A at a single institution. Military Medicine 2002;167:541‐5. [PubMed] [Google Scholar]
Kim 1994 {published data only}
- Kim CC, Jin JY, Kim DJ. Treatment of aplastic anemia: Allogeneic bone marrow transplantation versus immunomodulation therapy with antilymphocyte globulin. Chinese Medical Journal 1994;107:735‐6. [Google Scholar]
Kim 2003 {published data only}
- Kim I, Yoon SS, Park S, Kim BK, Kim NK. The treatment of severe aplastic anemia: outcomes of bone marrow transplantation and immunosuppressive therapy in a single institution of Korea. Journal of Korean Medical Science 2003;18:365‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kodera 1999 {published data only}
- Kodera Y, Morishima Y, Kato S, Akiyama Y, Sao H, Matsuyama T, et al. Analysis of 500 bone marrow transplants from unrelated donors (UR‐BMT) facilitated by the Japan Marrow Donor Program: confirmation of UR‐BMT as a standard therapy for patients with leukemia and aplastic anemia. Bone Marrow Transplantation 1999;24:995‐1003. [DOI] [PubMed] [Google Scholar]
Kojima 1988 {published data only}
- Kojima S, Fukuda M, Horibe K, Matsuyama K, Takeyama K, Matsushita T, et al. Comparison between bone marrow transplantation and immuno‐suppressive therapy in treatment of patients younger than 20 years with severe aplastic anemia. Acta Haematologica Japonica 1988;51(1):28‐35. [PubMed] [Google Scholar]
Kojima 2000 {published data only}
- Kojima S, Horibe K, Inaba J, Yoshimi A, Takahashi Y, Kudo K, et al. Long‐term outcome of acquired aplastic anaemia in children: comparison between immunosuppressive therapy and bone marrow transplantation. British Journal of Haematology 2000;111:321‐8. [DOI] [PubMed] [Google Scholar]
Kosaka 2004 {published data only}
- Kosaka Y, Kobayashi R, Ayukawa H, Kato K, Kaneko T, Kigasawa H, et al. Prospective multicenter trial comparing repeated immunosuppressive therapy (IST) with stem cell transplantation (SCT) from an alternative donor as a second‐line treatment for children with acquired aplastic anemia (AA). Blood (ASH Annual Meeting Abstracts) 2004;104:Abstract 2815. [Google Scholar]
Kulagin 2006 {published data only}
- Kulagin AD, Lisukov IA, Kriuchkova IV, Sizikova SA, Gilevich AV, Denisova VV, et al. Diagnosis and treatment of acquired aplastic anaemia. Terapevticheskii Arkhiv 2006;78(11):48‐54. [PubMed] [Google Scholar]
Lawlor 1997 {published data only}
- Abella E, Ravindranth Y. Immunosuppressive therapy vs. bone marrow transplant for severe aplastic anemia. Journal of Pediatric Hematology/Oncology 1997;19:533‐4. [DOI] [PubMed] [Google Scholar]
- Lawlor ER, Anderson RA, Davis JH, Fryer CJ, Pritchard SL, Rogers PC, et al. Immunosuppressive therapy: a potential alternative to bone marrow transplantation as initial therapy for acquired severe aplastic anemia in childhood?. Journal of Pediatric Hematology/Oncology 1997;19:115‐23. [DOI] [PubMed] [Google Scholar]
Ljungman 1991 {published data only}
- Ljungman P, Aschan J, Bolme P, Lindqvist R, Lonnqvist B, Ringden O, et al. Bone marrow transplantation or antithymocyte globulin in aplastic anemia? Both therapeutic alternatives result in good quality of life [Benmärgstransplantation eller antitymocytglobulin vid aplastisk anemi? Bada behandlingsalternativen ger god livskvalitet]. Läkartidningen 1991;88:2308‐11. [PubMed] [Google Scholar]
Locasciulli 1990 {published data only}
- Locasciulli A, van't Veer L, Bacigalupo A, Hows J, Lint MT, Gluckman E, et al. Treatment with marrow transplantation or immunosuppression of childhood acquired severe aplastic anemia: a report from the EBMT SAA Working Party. Bone Marrow Transplantation 1990;6:211‐7. [PubMed] [Google Scholar]
Locasciulli 2007 {published data only}
- Locasciulli A, Oneto R, Bacigalupo A, Socié G, Korthof E, Bekassy A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation. Haematologica 2007;92:11‐8. [DOI] [PubMed] [Google Scholar]
McCann 1994 {published data only}
- McCann SR, Bacigalupo A, Gluckman E, Hinterberger W, Hows J, Ljungman P, et al. Graft rejection and second bone marrow transplants for acquired aplastic anaemia: a report from the Aplastic Anaemia Working Party of the European Bone Marrow Transplant Group. Bone Marrow Transplantation 1994;13:233‐7. [PubMed] [Google Scholar]
Milosevic 1998 {published data only}
- Milosevic R, Antonijevic N, Jankovic G, Babic D, Colovic M. Aplastic anemia‐‐clinical characteristics and survival analysis [Aplasticna anemija‐‐klinicke odlike i analiza prezivnjavanja]. Srpski Arhiv za Celokupno Lekarstvo 1998;126:234‐8. [PubMed] [Google Scholar]
Mollee 2001 {published data only}
- Mollee P, Woodward N, Durrant S, Lockwood L, Gillett EA, Morton J, et al. Single institution outcomes of treatment of severe aplastic anaemia. Internal Medicine Journal 2001;31:337‐42. [DOI] [PubMed] [Google Scholar]
Montante 2009 {published data only}
- Montante B, Pinazzi MB, Severino A, Proia A, Majolino I, Locasciulli A. Cyclosporin monitoring at 2 hours from oral intake leads to less toxicity and better compliance in patients undergoing hematopoietic stem cells transplantation and immunosuppressive treatment for severe aplastic anemia (Conference abstract). Haematologica 2009;94:219‐20. [Google Scholar]
Mrsic 2009 {published data only}
- Mrsic M, Serventi Seiwerth R, Labar B, Bogdanic V, Nemet D, Durakovic N, et al. Twenty years of severe aplastic anemia treatment at Department of Hematology, University Department of Medicine, Zagreb University Hospital Center, Zagreb, Croatia [Dvadeset godina lijecenja teske aplasticne anemije u Zavodu za Hematologiju Klinike za Unutrasnje Bolesti Klinickog Bolnickog Centra Zagreb]. Acta Medica Croatica 2009;63(3):209‐14. [PubMed] [Google Scholar]
Nagler 2001 {published data only}
- Nagler A, Aker M, Or R, Naparstek E, Varadi G, Brautbar C, et al. Low‐intensity conditioning is sufficient to ensure engraftment in matched unrelated bone marrow transplantation. Experimental Hematology 2001;29(3):362‐70. [DOI] [PubMed] [Google Scholar]
Paquette 1995 {published data only}
- Paquette RL, Tebyani N, Frane M, Ireland P, Ho WG, Champlin RE, et al. Long‐term outcome of aplastic anemia in adults treated with antithymocyte globulin: comparison with bone marrow transplantation. Blood 1995;85:283‐90. [PubMed] [Google Scholar]
Pitcher 1999 {published data only}
- Pitcher LA, Hann IM, Evans JP, Veys P, Chessells JM, Webb DK. Improved prognosis for acquired aplastic anaemia. Archives of Disease in Childhood 1999;80(2):158‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Podesta 1998 {published data only}
- Podesta M, Piaggio G, Frassoni F, Pitto A, Zikos P, Sessarego M, et al. The assessment of the hematopoietic reservoir after immunosuppressive therapy or bone marrow transplantation in severe aplastic anemia. Blood 1998;91:1959‐65. [PubMed] [Google Scholar]
Pokorski 1998 {published data only}
- Pokorski RJ. 5‐year survival after bone marrow transplantation for aplastic anemia. Journal of Insurance Medicine 1998;30:237‐9. [PubMed] [Google Scholar]
Shaw 1999 {published data only}
- Shaw PH, Haut PR, Olszewski M, Kletzel M. Hematopoietic stem‐cell transplantation using unrelated cord‐blood versus matched sibling marrow in pediatric bone marrow failure syndrome: one center's experience. Pediatric Transplantation 1999;3:315‐21. [DOI] [PubMed] [Google Scholar]
Socié 1993 {published data only}
- Socie G, Henry‐Amar M, Bacigalupo A, Hows J, Tichelli A, Ljungman P, et al. Malignant tumors occurring after treatment of aplastic anemia. European Bone Marrow Transplantation‐Severe Aplastic Anaemia Working Party. New England Journal of Medicine 1993;329:1152‐7. [DOI] [PubMed] [Google Scholar]
Sovinz 2010 {published data only}
- Sovinz P, Lackner H, Schwinger W, Benesch M, Nebl A, Urban C. Management of severe aplastic anaemia in childhood: A single‐centre experience over 30 years (Conference abstract). Bone Marrow Transplantation. 2010; Vol. 45:S272.
Stalder 2009 {published data only}
- Stalder MP, Rovo A, Halter J, Heim D, Silzle T, Passweg J, et al. Aplastic anemia and concomitant autoimmune diseases. Annals of Hematology 2009;88(7):659‐65. [DOI] [PubMed] [Google Scholar]
Starý 1998 {published data only}
- Stary J, Sedlacek P, Kobylka P, Komrska V, Syruckova Z, Smisek P, et al. Significant progress in the treatment of acquired aplastic anemia in children in 1990s [Vyznamny pokrok v lecbe ziskane aplasticke anemie u deti v 90. Letech]. Ceskoslovenska Pediatrie 1998;53:260‐6. [Google Scholar]
Tichelli 1994 {published data only}
- Speck B, Gratwohl A, Nissen C. Severe aplastic anemia: A prospective study on the value of different therapeutic approaches in 37 successive patients. Blut 1980;41:160‐3. [Google Scholar]
- Speck B, Gratwohl A, Nissen C, Leibundgut U, Ruggero D, Osterwalder B, et al. Treatment of severe aplastic anaemia with antilymphocyte globulin or bone‐marrow transplantation. British Medical Journal 1981;282:860‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck B, Gratwohl A, Nissen C, Osterwalder B, Signer E, Jeannet M. Bone marrow graft versus ALG in patients with aplastic anaemia. Biomedicine & Pharmacotherapy 1983;37:139‐43. [PubMed] [Google Scholar]
- Speck B, Gratwohl A, Nissen C, Osterwalder B, Signer E, Jeannet M. Treatment of severe aplastic anemia: a prospective study of antilymphocyte globulin versus bone marrow transplantation. Progress in Clinical Biological Research 1984;148:249‐58. [PubMed] [Google Scholar]
- Speck B, Gratwohl A, Nissen C, Osterwalder B, Wursch A, Tichelli A, et al. A comparison between ALG and bone marrow transplantation in treatment of severe aplastic anemia. Thymus 1987;10:147‐58. [DOI] [PubMed] [Google Scholar]
- Speck B, Gratwohl A, Nissen C, Osterwalder B, Wursch A, Tichelli A, et al. Treatment of severe aplastic anemia. Experimental Hematology 1986;14:126‐32. [PubMed] [Google Scholar]
- Speck R, Tichelli A, Gratwohl A, Nissen C. Treatment of severe aplastic anemia: A longterm followup of 175 patients on antilymphocyte globulin or bone marrow transplantation. Chinese Medical Journal 1994;107:739. [Google Scholar]
- Tichelli A, Gratwohl A, Nissen C, Speck B. Late clonal complications in severe aplastic anemia. Leukemia & Lymphoma 1994;12:167‐75. [DOI] [PubMed] [Google Scholar]
- Tichelli A, Gratwohl A, Nissen C, Wursch A, Signer E, Speck B. Late complications in patients with aplastic anemia [Spätkomplikationen bei Patienten mit aplastischer Anämie]. Schweizerische Medizinische Wochenschrift 1988;118:1528‐32. [PubMed] [Google Scholar]
- Tichelli A, Gratwohl A, Wursch A, Nissen C, Speck B. Late haematological complications in severe aplastic anaemia. British Journal of Haematology 1988;69(3):413‐8. [DOI] [PubMed] [Google Scholar]
Tschiedel 2010 {published data only}
- Tschiedel E, Gierenz N, Wieland R, Wulff B, Ballauff A. Severe aplastic anaemia in six children after non‐A‐E hepatitis without hepatic failure [Schwere aplastische Anamie bei 6 Kindern nach Non‐A‐E‐Hepatitis ohne Leberversagen]. Zeitschrift fur Gastroenterologie 2010;48(8):825‐8. [DOI] [PubMed] [Google Scholar]
Tsukimoto 1989 {published data only}
- Tsukimoto I, Tsuchida M, Ohara A, Akabane T, Nakahata T, Akatsuka J, et al. Long‐term prognosis and residual abnormalities of idiopathic acquired aplastic anemia in children. Nippon Ketsueki Gakkai Zasshi 1989;52:1370‐8. [PubMed] [Google Scholar]
Tukic 2009 {published data only}
- Tukic L, Stamatovic D, Tarabar O, Elez M, Tatomirovic Z, Radic‐Tasic O, et al. Long‐term outcome of acquired aplastic anemia: Comparation between immunosuppressive therapy and bone marrow transplantation (Conference abstract). Haematologica. 2009; Vol. 94:241.
Tzeng 1989 {published data only}
- Tzeng CH, Chen PM, Chuang MW, Liu JH, Hsieh RK, Liu CJ, et al. Treatment of severe aplastic anemia: comparison of bone marrow transplantation to immunotherapy. Chung Hua I Hsueh Tsa Chih 1989;43:21‐8. [PubMed] [Google Scholar]
UCLA 1976 {published data only}
- UCLA Bone Marrow Transplant Team. Bone‐marrow transplantation in severe aplastic anaemia. Lancet 1976;308(7992):921‐3. [DOI] [PubMed] [Google Scholar]
Uss 1999 {published data only}
- Uss AL, Zmachinskii VA, Milanovich NF, Skriagin AE, Batan ZE, Dziuba EV, et al. Experience in the use of allogeneic bone marrow transplantation in severe forms of aplastic anemia at the Byelorussian hematological center [Opyt primeneniia transplantatsii allogennogo kostnogo mozga pri tiazheloi forme aplasticheskoi anemii v Belorusskom gematologicheskom tsentre]. Terapevticheskii Arkhiv 1999;71:69‐72. [PubMed] [Google Scholar]
Viollier 2005 {published data only}
- Viollier R, Passweg J, Gregor M, Favre G, Kühne T, Nissen C, et al. Quality‐adjusted survival analysis shows differences in outcome after immunosuppression or bone marrow transplantation in aplastic anemia. Annals of Hematology 2005;84:47‐55. [DOI] [PubMed] [Google Scholar]
Wagner 1996 {published data only}
- Wagner JE, Rosenthal J, Sweetman R, Shu XO, Davies SM, Ramsay NK, et al. Successful transplantation of HLA‐matched and HLA‐mismatched umbilical cord blood from unrelated donors: analysis of engraftment and acute graft‐versus‐host disease. Blood 1996;88:795‐802. [PubMed] [Google Scholar]
Webb 1991 {published data only}
- Webb DK, Hann IM, Chessells JM. Acquired aplastic anaemia: still a serious disease. Archives of Disease in Childhood 1991;66:858‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Werner 1989 {published data only}
- Werner EJ, Stout RD, Valdez LP, Harris RE. Immunosuppressive therapy versus bone marrow transplantation for children with aplastic anemia. Pediatrics 1989;83:61‐5. [PubMed] [Google Scholar]
Windass 1987 {published data only}
- Windass B, Vowels MR, Hughes DO, White L. Aplastic anaemia in childhood: prognosis and approach to therapy. Medical Journal of Australia 1987;146:15‐9. [DOI] [PubMed] [Google Scholar]
Yan 2011 {published data only}
- Yan HM, Liu J, Xue M, Wang ZD, Zhu L, Ding L, et al. Comparison of hematopoietic stem cell transplantation and non‐transplantation for treatment of severe aplastic anemia [Chinese]. Journal of Clinical Rehabilitative Tissue Engineering Research 2011;15(1):63‐7. [Google Scholar]
Yoshida 2011 {published data only}
- Yoshida N, Kikuchi A, Kobayashi R, Yabe H, Kosaka Y, Muramatsu H, et al. Outcomes in children with severe aplastic anemia receiving bone marrow transplantation from an HLA‐matched family donor or intensive immunosuppressive therapy as first‐line treatment. Blood (ASH Annual Meeting Abstracts). Washington, DC: American Society of Hematology, 2011; Vol. 118:Abstract 3421.
Additional references
ASH meeting abstracts 2012
- American Society of Hematology. ASH meeting abstracts. Blood. Washington, DC: American Society of Hematology, 2012 (http://bloodjournal.hematologylibrary.org/site/misc/ASH_Meeting_Abstracts_Info.xhtml).
Bacigalupo 2000
- Bacigalupo A, Brand R, Oneto R, Bruno B, Socie G, Passweg J, et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy‐‐The European Group for Blood and Marrow Transplantation experience. Seminars in Hematology 2000;37:69‐80. [DOI] [PubMed] [Google Scholar]
Bacigalupo 2008
- Bacigalupo A. Treatment strategies for patients with severe aplastic anemia. Bone Marrow Transplantation 2008;42 Suppl 1:S42‐4. [DOI] [PubMed] [Google Scholar]
Bacigalupo 2012
- Bacigalupo A, Socie G, Schrezenmeier H, Tichelli A, Locasciulli A, Führer M, et al. Bone marrow versus peripheral blood sibling transplants in acquired aplastic anemia: survival advantage for marrow in all age groups. Haematologica 2012;97(8):1142‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Battiwalla 2012
- Battiwalla M, Wang T, Carreras J, Deeg HJ, Ayas M, Bajwa RPS, et al. HLA‐matched sibling transplantation for severe aplastic anemia: Impact of HLA DR15 antigen status on engraftment, graft‐versus‐host disease, and overall survival. Biology of Blood and Marrow Transplantation 2012;18(9):1401‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brodsky 2005
- Brodsky RA, Jones RJ. Aplastic anaemia. Lancet 2005;365(9471):1647‐56. [DOI] [PubMed] [Google Scholar]
Buchbinder 2013
- Buchbinder D, Hsieh L, Puthenveetil G, Soni A, Stites J, Huynh V, et al. Successful autologous cord blood transplantation in a child with acquired severe aplastic anemia. Pediatric Transplantation 2013;17(3):E104‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Burroughs 2012
- Burroughs LM, Woolfrey AE, Storer BE, Deeg HJ, Flowers MED, Martin PJ, et al. Success of allogeneic marrow transplantation for children with severe aplastic anaemia. British Journal of Haematology 2012;158(1):120‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Camitta 1975
- Camitta BM, Rappeport JM, Parkman R, Nathan DC. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood 1975;45:355‐63. [PubMed] [Google Scholar]
Chybicka 2008
- Chybicka A. Quality of life and ethical and legal dilemmas in children during and after hematopoietic SCT procedure. Bone Marrow Transplantation 2008;42 Suppl 2:S87‐9. [DOI] [PubMed] [Google Scholar]
ClinicalTrials.gov 2012
- NIH National Institutes of Health. ClinicalTrials.gov. Department of Health and Human Services (http://clinicaltrials.gov/) 2012.
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG. Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org 2011.
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Deyell 2011
- Deyell RJ, Shereck EB, Milner RA, Schultz KR. Immunosuppressive therapy without hematopoietic growth factor exposure in pediatric acquired aplastic anemia. Pediatric Hematology and Oncology 2011;28(6):469‐78. [DOI] [PubMed] [Google Scholar]
Eapen 2012
- Eapen M. Allogeneic transplantation for aplastic anemia. Hematology 2012;17 Suppl 1:15‐7. [DOI] [PubMed] [Google Scholar]
EBMT meeting abstracts 2012
- European Group of Blood and Marrow Transplantation. EBMT meeting abstracts. EBMT conference proceedings. London: European Group of Blood and Marrow Transplantation, 2012 (http://www.ebmt.org/Contents/Annual‐Meeting/PastEBMTAnnualMeetings/Pages/Past‐EBMT‐Annual‐Meetings.aspx).
Frickhofen 2003
- Frickhofen N, Heimpel H, Kaltwasser JP, Schrezenmeier H. German Aplastic Anemia Study Group. Antithymocyte globulin with or without cyclosporin A: 11‐year follow‐up of a randomized trial comparing treatments of aplastic anemia. Blood 2003;101:1236‐42. [DOI] [PubMed] [Google Scholar]
GARD 2012
- Genetic and Rare Diseases Information Center (GARD). Aplastic anemia. The Office of Rare Diseases Research (ORDR) and the National Human Genome Research Institute (NHGRI) of the National Institutes of Health (NIH) (http://rarediseases.info.nih.gov/GARD/Condition/5836/Aplastic_anemia.aspx) 2012.
GCCR 2006
- Annual Report 2006. Routine Analyses: diagnoses and cases, time trends, regional differences, survival probabilities, and mortality. Summary data for selected diagnoses not defined in ICCC‐3 of patients under 15 in Germany (2001‐2005). German Childhood Cancer Registry (GCCR) (http://www.kinderkrebsregister.de/external/publications/annual‐reports/jb2006gb/index.html?L=1) 2006.
Google Translate 2012 [Computer program]
- Google, Inc. Google Translate (http://translate.google.com/). Mountain View: Google, Inc, 2012.
GRADEpro 2008 [Computer program]
- Brozek J, Oxman A, Schünemann H. GRADEpro (GRADE profiler) (http://ims.cochrane.org/gradepro). Version Version 3.2 for Windows. Copenhagen: Information Management System (IMS), Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Gray 1991
- Gray R, Wheatley K. How to avoid bias when comparing bone marrow transplantation with chemotherapy. Bone Marrow Transplantation 1991;7(Suppl 3):9‐12. [PubMed] [Google Scholar]
Guinan 2009
- Guinan EC. Acquired aplastic anemia in childhood. Hematology/Oncology Clinics of North America 2009;23(2):171‐91. [DOI] [PubMed] [Google Scholar]
Gupta 2010
- Gupta V, Eapen M, Brazauskas R, Carreras J, Aljurf M, Gale RP, et al. Impact of age on outcomes after bone marrow transplantation for acquired aplastic anemia using HLA‐matched sibling donors. Haematologica 2010;95(12):2119‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC. Table 8.5.a The Cochrane Collaboration's tool for assessing risk of bias. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC. Table 8.5.d Criteria for judging risk of bias in the 'Risk of bias' assessment tool. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011c
- Higgins JPT, Deeks JJ, Altman DG. 16.1 Missing data. Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
ICTRP 2012
- ICTRP International Clinical Trials Registry Platform. ICTRP Search Platform. WHO World Health Organization (http://www.who.int/ictrp/en/) 2012.
Kamio 2011
- Kamio T, Ito E, Ohara A, Kosaka Y, Tsuchida M, Yagasaki H, et al. Relapse of aplastic anemia in children after immunosuppressive therapy: a report from the Japan Childhood Aplastic Anemia Study Group. Haematologica 2011;96(6):814‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karnofsky 1949
- Karnofsky DA, Burchenal JH. The clinical evaluation of chemotherapeutic agents in cancer. In: MacLeod CM editor(s). Evaluation of Chemotherapeutic Agents. New York City: Columbia University Press, 1949:191. [Google Scholar]
Katan 1986
- Katan MB. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet 1986;327(8479):507‐8. [DOI] [PubMed] [Google Scholar]
Kaufman 2006
- Kaufman DW, Kelly JP, Issaragrisil S, Laporte JR, Anderson T, Levy M, et al. Relative incidence of agranulocytosis and aplastic anemia. American Journal of Hematology 2006;81(1):65‐7. [DOI] [PubMed] [Google Scholar]
Killick 2000
- Killick SB, Marsh JCW. Aplastic anaemia: management. Blood Reviews 2000;14:157‐71. [DOI] [PubMed] [Google Scholar]
Kim 2012
- Kim H, Lee KH, Yoon SS, Sohn SK, Joo YD, Kim SH, et al. Allogeneic hematopoietic stem cell transplant for adults over 40 years old with acquired aplastic anemia. Biology of Blood and Marrow Transplantation 2012;18(10):1500‐8. [DOI] [PubMed] [Google Scholar]
Kojima 2011
- Kojima S, Nakao S, Young N, Bacigalupo A, Gerard G, Hirano N, et al. The Third Consensus Conference on the treatment of aplastic anemia. International Journal of Hematology 2011;93(6):832‐7. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Lewis 2010
- Lewis SJ. Mendelian randomization as applied to coronary heart disease, including recent advances incorporating new technology. Circulation Cardiovascular Genetics 2010;3(1):109‐17. [DOI] [PubMed] [Google Scholar]
Marsh 2009
- Marsh JCW, Ball SE, Cavenagh J, Darbyshire P, Dokal I, Gordon‐Smith EC, et al. Guidelines for the diagnosis and management of acquired aplastic anaemia. British Journal of Haematology 2009;147:43‐70. [DOI] [PubMed] [Google Scholar]
Marsh 2011
- Marsh JCW, Cavenagh J, Darbyshire P, Dokal I, Gordon‐Smith EC, Keidan J, et al. Guidance from the BCSH Aplastic Anaemia Writing Committee relating to recent prospective studies of rabbit antithymocyte globulin (ATG) used for a first course of immunosuppressive therapy (IST) in aplastic anaemia. British Committee for Standards in Haematology (BCSH), London, UK (http://www.bcshguidelines.com/documents/Use_of_rabbit_ATG_July_2011.pdf) 2011.
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Medicine 2009;6:e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nitsch 2006
- Nitsch D, Molokhia M, Smeeth L, DeStavola BL, Whittaker JC, Leon DA. Limits to causal inference based on Mendelian randomization: a comparison with randomized controlled trials. American Journal of Epidemiology 2006;163(5):397‐403. [DOI] [PubMed] [Google Scholar]
NPCRC 2012
- Karnofsky performance status scale definitions rating (%) criteria. National Palliative Care Research Center (NPCRC) (http://www.npcrc.org/usr_doc/adhoc/functionalstatus/Karnofsky%20Performance%20Scale.pdf) 2012.
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Passweg 2010
- Passweg JR, Marsh JCW. Aplastic Anemia: First‐line treatment by immunosuppression and sibling marrow transplantation. Hematology American Society of Hematology Education Program 2010;2010:36‐42. [DOI] [PubMed] [Google Scholar]
Peinemann 2011
- Peinemann F, Grouven U, Kroger N, Bartel C, Pittler MH, Lange S. First‐line matched related donor hematopoietic stem cell transplantation compared to immunosuppressive therapy in acquired severe aplastic anemia. PLoS One 2011;6(4):e18572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Risitano 2013
- Risitano AM, Schrezenmeier H. Alternative immunosuppression in patients failing immunosuppression with ATG who are not transplant candidates: Campath (Alemtuzumab). Bone Marrow Transplantation 2013;48(2):186‐90. [DOI] [PubMed] [Google Scholar]
Sangiolo 2010
- Sangiolo D, Storb R, Deeg HJ, Flowers MED, Martin PJ, Sandmaier BM, et al. Outcome of allogeneic hematopoietic cell transplantation from HLA‐identical siblings for severe aplastic anemia in patients over 40 years of age. Biology of Blood and Marrow Transplantation 2010;16(10):1411‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scheinberg 2011
- Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Biancotto A, Wu CO, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. New England Journal of Medicine 2011;365(5):430‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scheinberg 2012a
- Scheinberg P. Aplastic anemia: therapeutic updates in immunosuppression and transplantation. Hematology / the Education Program of the American Society of Hematology 2012;2012:292‐300. [DOI] [PubMed] [Google Scholar]
Scheinberg 2012b
- Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood 2012;120(6):1185‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schrezenmeier 2000
- Schrezenmeier H, Bacigalupo A, Aglietta M, Frickhofen N, Führer M, Gluckman E, et al. Guidelines for treatment of aplastic anemia. Consensus document of a group of international experts. In: Schrezenmeier H, Bacigalupo A editor(s). Aplastic Anemia. Pathophysiology and Treatment. Cambridge: Cambridge University Press, 2000:308‐15. [Google Scholar]
Schrezenmeier 2007
- Schrezenmeier H, Passweg JR, Marsh JCW, Bacigalupo A, Bredeson CN, Bullorsky E, et al. Worse outcome and more chronic GVHD with peripheral blood progenitor cells than bone marrow in HLA‐matched sibling donor transplants for young patients with severe acquired aplastic anemia. Blood 2007;110(4):1397‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Sterne 2011
- Sterne JAC, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Thomson Reuters Corp 2012 [Computer program]
- Thomson Reuters. EndNote. Version X3. New York City: Thomson Reuters, 2012.
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wheatley 2004
- Wheatley K, Gray R. Commentary: Mendelian randomization: an update on its use to evaluate allogeneic stem cell transplantation in leukaemia. International Journal of Epidemiology 2004;33:15‐7. [DOI] [PubMed] [Google Scholar]
Young 2006
- Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood 2006;108(8):2509‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]