

Abstract
Background
Adjuvant chemotherapy improves survival in premenopausal and postmenopausal women with early breast cancer. Taxanes are highly active chemotherapy agents used in metastatic breast cancer. Review authors examined their role in early breast cancer. This review is an update of a Cochrane Review first published in 2007.
Objectives
To assess the effects of taxane‐containing adjuvant chemotherapy regimens for treatment of women with operable early breast cancer.
Search methods
For this review update, we searched the Specialised Register of the Cochrane Breast Cancer Group, MEDLINE, Embase, CENTRAL (2018, Issue 6), the WHO International Clinical Trials Registry Platform (ICTRP), and ClinicalTrials.gov on 16 July 2018, using key words such as 'early breast cancer' and 'taxanes'. We screened reference lists of other related literature reviews and articles, contacted trial authors, and applied no language restrictions.
Selection criteria
Randomised trials comparing taxane‐containing regimens versus non‐taxane‐containing regimens in women with operable breast cancer were included. Studies of women receiving neoadjuvant chemotherapy were excluded.
Data collection and analysis
Two review authors independently extracted data and assessed risk of bias and quality of the evidence using the GRADE approach. Hazard ratios (HRs) were derived for time‐to‐event outcomes, and meta‐analysis was performed using a fixed‐effect model. The primary outcome measure was overall survival (OS); disease‐free survival (DFS) was a secondary outcome measure. Toxicity was represented as odds ratios (ORs), and quality of life (QoL) data were extracted when present.
Main results
This review included 29 studies (27 full‐text publications and 2 abstracts or online theses). The updated analysis included 41,911 randomised women; the original review included 21,191 women. Taxane‐containing regimens improved OS (HR 0.87, 95% confidence interval (CI) 0.83 to 0.92; high‐certainty evidence; 27 studies; 39,180 women; 6501 deaths) and DFS (HR, 0.88, 95% CI 0.85 to 0.92; high‐certainty evidence; 29 studies; 41,909 women; 10,271 reported events) compared to chemotherapy without a taxane. There was moderate to substantial heterogeneity across studies for OS and DFS (respectively).
When a taxane‐containing regimen was compared with the same regimen without a taxane, the beneficial effects of taxanes persisted for OS (HR 0.84, 95% CI 0.77 to 0.92; P < 0.001; 7 studies; 10,842 women) and for DFS (HR 0.84, 95% CI 0.78 to 0.90; P < 0.001; 7 studies; 10,842 women). When a taxane‐containing regimen was compared with the same regimen with another drug or drugs that were substituted for the taxane, a beneficial effect was observed for OS and DFS with the taxane‐containing regimen (OS: HR 0.80, 95% CI 0.74 to 0.86; P < 0.001; 13 studies; 16,196 women; DFS: HR 0.83, 95% CI 0.78 to 0.88; P < 0.001; 14 studies; 16,823 women). Preliminary subgroup analysis by lymph node status showed a survival benefit with taxane‐containing regimens in studies of women with lymph node‐positive disease only (HR 0.83, 95% CI 0.78 to 0.88; P < 0.001; 17 studies; 22,055 women) but less benefit in studies of women both with and without lymph node metastases or with no lymph node metastases. Taxane‐containing regimens also improved DFS in women with lymph node‐positive disease (HR 0.84, 95% CI 0.80 to 0.88; P < 0.001; 17 studies; 22,055 women), although the benefit was marginal in studies of women both with and without lymph node‐positive disease (HR 0.95, 95% CI 0.88 to 1.02; 9 studies; 12,998 women) and was not apparent in studies of women with lymph node‐negative disease (HR 0.99, 95% CI 0.86 to 1.14; 3 studies; 6856 women).
Taxanes probably result in a small increase in risk of febrile neutropenia (odds ratio (OR) 1.55, 95% CI 0.96 to 2.49; moderate‐certainty evidence; 24 studies; 33,763 women) and likely lead to a large increase in grade 3/4 neuropathy (OR 6.89, 95% CI 3.23 to 14.71; P < 0.001; moderate‐certainty evidence; 22 studies; 31,033 women). Taxanes probably cause little or no difference in cardiotoxicity compared to regimens without a taxane (OR 0.87, 95% CI 0.56 to 1.33; moderate‐certainty evidence; 23 studies; 32,894 women). Seven studies reported low‐quality evidence for QoL; overall, taxanes may make little or no difference in QoL compared to chemotherapy without a taxane during the follow‐up period; however, the duration of follow‐up differed across studies. Only one study, which was conducted in Europe, provided cost‐effectiveness data.
Authors' conclusions
This review of studies supports the use of taxane‐containing adjuvant chemotherapy regimens, with improvement in overall survival and disease‐free survival for women with operable early breast cancer. This benefit persisted when analyses strictly compared a taxane‐containing regimen versus the same regimen without a taxane or the same regimen with another drug that was substituted for the taxane. Preliminary evidence suggests that taxanes are more effective for women with lymph node‐positive disease than for those with lymph node‐negative disease. Considerable heterogeneity across studies probably reflects the varying efficacy of the chemotherapy backbones of the comparator regimens used in these studies. This review update reports results that are remarkably consistent with those of the original review, and it is highly unlikely that this review will be updated, as new trials are assessing treatments based on more detailed breast cancer biology.
Plain language summary
Taxane‐containing chemotherapy for women after surgery for early breast cancer
What is the aim of this review?
The aim of this Cochrane Review was to find out if adding taxane drugs to standard chemotherapy improves survival and is safe for women with early breast cancer. Cochrane Review authors collected and analysed all relevant studies to answer these questions and found 29 studies.
Key messages
Adding a taxane drug to standard chemotherapy improved survival (women lived longer) and reduced the chance of cancer returning in women with operable early breast cancer, but the use of taxanes probably led to increased risk of some side effects such as febrile neutropenia (low white cell count with fever) and neuropathy (damage to the nerves).
What was studied in this review?
Early breast cancer is cancer that has not spread beyond the breast or nearby lymph nodes. It may be curable with surgery alone, but there is a risk that after surgery the breast cancer may return. Chemotherapy and radiotherapy are needed after surgery to achieve a cure.
A combination of chemotherapy drugs, rather than one drug by itself, is usually used to treat early breast cancer.
One class of chemotherapy drugs commonly used is taxanes. Taxanes act by stalling the cellular processes that are needed for cells to divide. This action causes cancer cells to stop dividing and slows the growth of cancer or kills the cells. Two main taxane drugs are available ‐ paclitaxel and docetaxel.
The practice of adding taxanes to standard chemotherapy has increased over the last 10 years as data from clinical trials have become available. There is a need to review these data to find out the benefits of these drugs, any side effects of the drugs, and how treatment is affecting a woman's overall well‐being (quality of life).
What are the main results of this review?
Review authors found 29 relevant studies involving 41,911 women. These studies compared chemotherapy that contained a taxane against chemotherapy that did not contain a taxane. Around half of the studies used paclitaxel, and the other half used docetaxel. The decision whether to use paclitaxel or docetaxel generally was based on the availability of these drugs in the hospital. Researchers gave these drugs by injection into a vein.
The women's health was monitored for at least 12 months from the start of the study. Some studies monitored women for 10 years.
Review authors found that adding a taxane drug to chemotherapy:
• improves survival and reduces the risk of cancer coming back compared to chemotherapy with no taxane;
• probably leads to an increased chance of some side effects compared to chemotherapy with no taxane. Side effects that are more likely to occur due to taxanes are febrile neutropenia (low white cell count with fever) and neuropathy (damage to the nerves);
• probably makes little or no difference in heart function compared to chemotherapy with no taxane; and
• may make little or no difference in quality of life for women compared to chemotherapy with no taxane. Seven of 29 studies provided information on the quality of life of women.
Very little information is available on the costs of adding a taxane to chemotherapy; only one study, which was conducted in Europe, reported cost‐effectiveness data.
How up‐to‐date is this review?
The review authors searched for studies that had been published up to July 2018.
Summary of findings
Summary of findings for the main comparison. Taxane‐containing chemotherapy vs any chemotherapy without taxane for early breast cancer.
| Taxane‐containing chemotherapy compared to any chemotherapy without taxane for early breast cancer | ||||||
| Patient or population: women with early breast cancer (operable, stages I to IIIA) Setting: outpatient Intervention: taxane‐containing chemotherapy Comparison: any chemotherapy without taxanes | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with any chemotherapy without taxanes | Risk with taxane‐containing chemotherapy | |||||
| Overall survival Follow‐up: range 5 years to 10 years (baseline risks for low‐ and high‐risk groups in the control arm were estimated at 5 years) | Low risk of death | HR 0.87 (0.83 to 0.92) | 39,180 (27 studies) | ⊕⊕⊕⊕ HIGH | Additional analyses (including specific taxane, scheduling, treatment duration, doses, node positive, and risk of bias) showed equivalent efficacy | |
| 80 per 1000* | 70 per 1000 (67 to 73) | |||||
| High risk of death | ||||||
| 200 per 1000* | 176 per 1000 (169 to 184) | |||||
| Disease‐free progression Follow‐up: range 4 years to 10 years (*baseline risks for low‐ and high‐risk groups in the control arm were estimated at 5 years) | Low risk of recurrence | HR 0.88 (0.85 to 0.92) | 41,909 (29 studies, 30 comparisons) | ⊕⊕⊕⊕ HIGHa | As above for overall survival | |
| 140 per 1000* | 124 per 1000 (120 to 130) | |||||
| High risk of recurrence | ||||||
| 320 per 1000* | 288 per 1000 (280 to 299) | |||||
| Quality of life Follow‐up: 1 to 62 months | Not estimable. In general, there did not seem to be differences in quality of life scores between groups at long‐term follow‐up | ‐ | (7 studies) | ⊕⊕⊝⊝ LOWb | Studies used the validated EORTC‐C30 questionnaire and measures were patient‐reported | |
| Febrile neutropenia Follow‐up: 3 to 7 years | Study population | OR 1.43 (0.89 to 2.31) | 34,154 (23 studies, 24 comparisons) | ⊕⊕⊕⊝ MODERATEc | There was a higher incidence of febrile neutropenia with docetaxel‐containing regimens | |
| 56 per 1000 | 78 per 1000 (50 to 120) | |||||
| Neuropathy (including grade 3/4 sensory or motor neuropathy, or both) Follow‐up: 3 to 7 years | Study population | OR 6.89 (3.23 to 14.71) | 31,033 (22 studies, 23 comparisons) | ⊕⊕⊕⊝ MODERATEc | A test for subgroup differences by taxane type was not significant | |
| 6 per 1000 | 37 per 1,000 (18 to 76) | |||||
| Cardiotoxicity (including grade 3/4 and congestive cardiac failure) Follow‐up: 3 to 10 years | Study population | OR 0.87 (0.56 to 1.33) | 32,894 (23 studies) | ⊕⊕⊕⊝ MODERATEd | Risk of cardiotoxicity was reduced with lower planned dose of anthracycline | |
| 9 per 1000 | 8 per 1000 (5 to 12) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; OR: odds ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aHeterogeneity was detected (I² = 59%) mainly due to variations in chemotherapy backbones. The quality of evidence was not downgraded as variations in chemotherapy are likely to occur in clinical practice. bThis outcome was downgraded because all measures were patient‐reported, taking place in open‐label studies, and therefore at high risk of bias. Although all studies used the validated EORTC‐C30 questionnaires, the time frames when women were given the questionnaires was variable, and lengths of follow‐up were different. In one study, only 23% of participants completed baseline and end of chemotherapy questionnaires. cThere was significant heterogeneity across studies (I² = 96% for febrile neutropenia; I² = 82% for neuropathy). dThe confidence interval crosses the line of no effect and does not rule out a small increase in toxicity from taxanes.
Background
Description of the condition
Breast cancer is a major cause of morbidity and mortality among women worldwide. In 2012, an estimated 1.67 million new cases and over 522,000 deaths occurred (Ferlay 2015). Depending on the stage of early breast cancer, five‐year relative survival rates can range from 90% (stage II) to almost 100% (stage 0 or stage I) (AIHW 2012).
The primary treatment for early breast cancer is local, and surgery with or without radiotherapy is recommended for women with operable early breast cancer (NCCN 2007). Adjuvant polychemotherapy following surgery improves survival among premenopausal and postmenopausal women with early breast cancer (EBCTCG 2005).
Description of the intervention
Two taxanes are commercially available: paclitaxel (Taxol®, Bristol‐Myers Squibb) and docetaxel (Taxotere®, Sanofi‐Aventis). Extensive research has led to the creation of new second‐generation taxanes and additional non‐taxane microtubule‐targeting chemotherapies. Currently two new taxanes have been approved by the FDA: nab paclitaxel (Abraxane®, Celgene), which was approved in 2005 for treatment of refractory, relapsed, or metastatic breast cancer; and cabazitaxel (Jevtana®, Sanofi), which was approved in 2010 for use in hormone‐refractory metastatic prostate cancer. Additionally, two non‐taxane microtubule‐targeting agents have received FDA approval for use in breast cancer: ixabepilone (Ixempra®, Bristol‐Myers Squibb) in 2007, and eribulin (Halaven®, Eisai Co., Ltd.) in 2010.
The most common side effects differ slightly between the two available taxanes. Both agents cause neutropenia (low neutrophil count, a subset of white blood cells) and thrombocytopenia (low platelet count), as well as fatigue, nausea and vomiting, hair loss, diarrhoea, mouth ulcers, and joint and muscle pain. Paclitaxel also causes hypersensitivity reactions (skin rash and reactions to infusion of the agent) and peripheral neuropathy. Docetaxel also causes skin and nail changes and fluid accumulation (oedema). Both drugs can cause febrile neutropenia (serious infection due to low neutrophil count), which occasionally can be life‐threatening or fatal. Supportive therapies can modify many of these side effects.
How the intervention might work
Taxanes are cytotoxic chemotherapy agents that affect cellular structures needed for cancer cells to divide ‐ the microtubules. In a normal cell cycle, cells form microtubules at the beginning of cell division, and the microtubules are broken down when the cell stops dividing. Taxanes stabilise the microtubules, preventing them from breaking down normally. This causes the cancer cells to stop dividing, potentially slowing the growth of cancer or killing the cells.
Why it is important to do this review
Taxanes are among the most active agents in metastatic breast cancer (Bishop 1999; Chan 1999; Ghersi 2005); they are widely used (Crown 2002). Their incorporation into adjuvant regimens for early breast cancer has increased in recent years as mature data from clinical trials have become available. For this review update, data on an additional 19,416 participants were available, with time‐to‐event data provided for most of the randomised participants. Other systematic reviews have examined this topic: Bria 2006 and Qin 2011; however, these reviews did not include risk of bias assessments for the included studies as per Cochrane's risk of bias tool and did not grade the overall quality of evidence for each main outcome. An update of the efficacy and safety of taxanes in the form of an updated Cochrane Review seems warranted given the availability of mature follow‐up data and new trial data.
Objectives
To assess the effects of taxane‐containing adjuvant chemotherapy regimens for treatment of women with operable early breast cancer.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials are included. Quasi‐randomised trials were excluded.
We included studies with full‐text publications and studies published in abstract form only. We excluded studies available only as protocols and those published without the outcome measures of interest in this review.
Types of participants
We included women of any age with histologically confirmed operable breast cancer (stages I to IIIA).
We excluded women who received neoadjuvant chemotherapy. We included studies that included both women who received adjuvant chemotherapy and women who received neoadjuvant chemotherapy if data for the two groups were reported separately.
Types of interventions
We defined an intervention as any chemotherapy regimen that contains a taxane. We defined a comparator as any chemotherapy regimen that does not contain a taxane.
Comparisons included the following.
Question 1. Taxane‐containing regimen versus the same regimen without a taxane.
Question 2. Any taxane‐containing regimen versus any regimen without a taxane.
Question 3. Any taxane‐containing regimen versus the same regimen with another drug or drugs that were substituted for the taxane.
Endocrine treatment and targeted therapy were allowed if the same treatment was given to all groups.
The taxane drugs used were paclitaxel and docetaxel.
Types of outcome measures
Primary outcomes
Overall survival (OS), defined as time from randomisation/study entry until death from any cause
Secondary outcomes
Disease‐free survival (DFS), defined as time from date of randomisation to first date of a local, regional, or distant relapse, diagnosis of a second primary cancer, or death from any cause
Toxicity, defined by World Health Organization (WHO)/National Cancer Institute of Canada (NCIC) toxicity criteria
Quality of life (QoL), assessed by validated or trial‐specific instruments such as the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire
Cost‐effectiveness
Search methods for identification of studies
Electronic searches
We searched the following databases on 16 July 2018.
Specialised Register of the Cochrane Breast Cancer Group. Details of the search strategy used by the Group for identification of studies and the procedure used to code references are outlined in the Group's module (www.mrw.interscience.wiley.com/cochrane/clabout/articles/BREASTCA/frame.html). Studies coded as 'early breast cancer' and 'chemotherapy' on the Specialised Register were extracted and combined with the keywords 'taxol', 'docetaxel', and 'paclitaxel'. A search was carried out for the following text words: 'taxane', 'taxol', 'taxotere', 'paclitaxel', 'paxene', 'nsc‐12973', 'docetaxel', 'anzatax', 'taxanes', 'taxoids', and 'taxoid'.
Cochrane Central Register of Controlled Trials (CENTRAL; 2018, Issue 6) (see Appendix 1).
MEDLINE (via OvidSP) (see Appendix 2).
Embase (via Embase.com) (see Appendix 3).
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) search portal for all prospectively registered and ongoing trials (see Appendix 4).
ClinicalTrials.gov register (clinicaltrials.gov) for additional unpublished and ongoing studies (see Appendix 5).
Searching other resources
We searched the reference lists of other related literature reviews and articles.
We performed handsearching for abstracts published from 1995 to 2006 for presentations at the American Society of Clinical Oncology Annual Scientific Meeting, and up until 2009 for the San Antonio Breast Cancer Symposium.
Data collection and analysis
Selection of studies
In the original review and review update, two review authors (original review: AN, TF; review update: MW, LB) applied the selection criteria to each trial publication (if full publication available) or abstract (full publication not available). A third review author was available to resolve any disagreements regarding eligibility (review update: NW).
We have recorded excluded studies in the Characteristics of excluded studies table.
We applied no language restrictions.
Data extraction and management
For the original review and review update, two review authors (original review: AN, TF; review update: MW, LB) independently extracted data from the included studies. If required, a third review author (NW) was available to resolve any discrepancies regarding extraction of quantitative data. We collected information on study design, participants (including hormone receptor status and nodal involvement), settings, interventions, primary and secondary outcomes, follow‐up, and sources of funding. For studies with more than one publication, we extracted data from these publications, and we considered the final or updated version of each study as the primary reference.
Assessment of risk of bias in included studies
For the review update, we used Cochrane's 'Risk of bias' assessment tool to assess potential sources of bias in the included studies (Higgins 2011). Two review authors (MW, LB) independently assessed the potential risk of bias for each study and resolved any differences in judgement through discussion. The domains assessed were random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other bias. We assigned ratings of 'high', 'low', or 'unclear' risk of bias to each domain for each included study in keeping with the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Among phase III oncology studies, open‐label studies are common due to the difficulty involved in concealing different chemotherapy schedules and toxicities. The blinding of outcome assessment domain was therefore grouped with outcome measures most unlikely or most likely to be influenced by lack of blinding. Outcomes were segregated into (1) overall survival, (2) disease‐free survival and toxicity, and (3) quality of life.
Measures of treatment effect
The primary outcome for this review was overall survival, and the secondary outcome was disease‐free survival, with both considered as time‐to‐event outcomes. Hazard ratios (HRs) and variances were extracted from trial publications, when available. If not reported, statistics were extracted from publications via the methods described by Parmar et al using other summary statistics, or from data from published Kaplan‐Meier curves in the original review (Parmar 1998). Numbers at risk were adjusted based on estimated minimum and maximum follow‐up times. When these were not reported, minimum follow‐up was estimated from parameters given, including date of final accrual, date of study closure, date of submission, and estimated time to complete treatment. Maximum follow‐up time was similarly estimated from date of first accrual, date of analysis, date of submission, and last event on the time‐to‐event curve. For the review update, if required, we calculated summary statistics indirectly using the methods outlined by Tierney (Tierney 2007; indirect methods were recorded in the Notes section in the Characteristics of included studies tables). All efficacy analyses used an intention‐to‐treat population when this was reported. A pooled HR was calculated using observed (O) minus expected (E) event numbers, and variance from each trial was derived as above in a fixed‐effect model (Yusuf 1991).
The treatment effect was also analysed by subgroups for hormone receptor status. In this case, HRs and confidence intervals (CIs) reported in hormone receptor‐positive women and hormone receptor‐negative women were analysed, when available.
For two studies (ADEBAR; Taxit 216), missing data were estimated using the formula HR = [(taxane events)/(taxane participants)]/[(control events)/(control participants)]. For ADEBAR, this formula was used to estimate the number of participants per treatment group, and for Taxit 216, the formula was used to estimate the number of events per treatment group for overall survival and for disease‐free survival.
Toxicity data were extracted from each trial by one or two review authors (original review: RV; review update: MW, LB); when possible, this was done for the treated population rather than the intention‐to‐treat population. As definitions of toxic events varied between trials, events were extracted and summarised to best reflect clinically important outcomes. Pooled odds ratios (ORs) and 95% confidence intervals (CIs) were calculated for each toxicity via a random‐effects model, when that toxicity was reported in four or more trials. For this review, toxicity was abstracted only from the primary publication used to report efficacy or from a publication solely on toxicity, even when other published abstracts had reported separately on toxicity.
Quality of life (QoL) data were collected using the EORTC Common Toxicity Criteria (CTC) questionnaire, and four of the six trials reported data in a full publication. No attempt was made to statistically synthesise QoL data, which are summarised and reported qualitatively.
Pharmacoeconomic data were reported for only one trial, and a description was provided in the Results section.
Unit of analysis issues
Three trials were three‐arm studies (ECTO; NCIC‐CTG MA21a and NCIC‐CTG MA21b; UK TACT). For ECTO, data from two of the three arms were used for this review (the third being a neoadjuvant treatment arm). For NCIC CTG MA21, the control group was halved to allow a comparison with each of the two taxane arms (NCIC‐CTG MA21a; NCIC‐CTG MA21b). For UK TACT, the two control arms (epirubicin (e)‐cyclophosphamide, methotrexate, and fluorouracil (CMF) and fluorouracil/epirubicin/cyclophosphamide (FEC)) were combined.
Two trials were four‐arm studies (BIG 2‐98; CALGB 40101). For BIG 2‐98, the two control arms were combined, as were the two taxane arms. However, for the analysis related to sequential versus concurrent anthracycline/taxane, data comparing uncombined study arms were used (concurrent control vs concurrent taxane, and sequential control vs sequential taxane). For CALGB 40101, the two control arms (4‐ and 6‐cycle regimens) were combined, and the two taxane arms (four‐ and six‐cycle regimens) were combined.
Dealing with missing data
When data were missing, we contacted the original investigators (by written correspondence) to request missing data. For the review update, we contacted the following trialists for summary statistics, numbers of events for each treatment arm (for overall survival or disease‐free survival), and clarification on whether HRs were adjusted or unadjusted: ADEBAR; BIG 2‐98; CALGB 40101; E2197; FinHer; GEICAM 9906; GONO MIG‐5; HORG; Kader; NCIC‐CTG MA21a and NCIC‐CTG MA21b; RAPP‐01; Roy; Taxit 216; UK TACT. We received additional data from the trialists for seven studies: ADEBAR; CALGB 40101; E2197; FinHer; GEICAM 9906; GONO MIG‐5; HORG.
Assessment of heterogeneity
Heterogeneity was assessed by using the Chi² test and the I² statistic, as well as visual inspection of forest plots. The graphical representation of data was inspected; if confidence intervals for the results of individual studies had poor overlap, this generally indicated the presence of statistical heterogeneity.
We interpreted the I² statistic as per guidance provided in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): 0% to 40% might not be important; 30% to 60% represented moderate heterogeneity; 50% to 90% represented substantial heterogeneity; and 75% to 100% represented considerable heterogeneity.
Assessment of reporting biases
We followed the recommendations for testing for funnel plot asymmetry as described in Section 10.4.3.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Funnel plot asymmetry may be due to reporting bias; we addressed this possibility in the Results and Discussion sections of the review.
Data synthesis
For dichotomous outcome data (i.e. toxicity), we used a random‐effects (Mantel‐Haenszel method) model.
For time‐to‐event outcome data (i.e. overall survival and disease‐free‐survival), we used a fixed‐effect (exp[(O‐E)/Var] method) analysis. In the case of hormone receptor status subgroup analysis, we analysed the pooled HR using fixed‐effect (generic inverse variance method) analysis.
We performed all analyses using Review Manager software (RevMan).
Summary of findings
We used the GRADE approach to assess the quality of evidence for the following six main outcomes: mortality (overall survival), risk of recurrence (disease‐free survival), quality of life, febrile neutropenia, neuropathy (grade 3/4), and cardiotoxicity. We used GRADEproGDT software to develop the 'Summary of findings' table and followed GRADE guidance (GRADEproGDT; Schünemann 2011). Two review authors (MW, LB) graded the quality of the evidence for this review update.
To calculate absolute risk of the control group for time‐to‐event outcomes, we estimated the event rate at a specific time point (five years for OS and DFS) from the Kaplan‐Meier curves or reported event rates. We used a range for baseline event rates (i.e. low‐risk and high‐risk participants). We entered these estimated values into GRADEproGDT, and the corresponding absolute risks for the intervention group with low‐ and high‐risk subgroups at five years were automatically populated by GRADEproGDT.
Subgroup analysis and investigation of heterogeneity
We performed the following post hoc subgroup analyses for overall survival and disease‐free‐survival.
Type of taxane (paclitaxel or docetaxel).
Sequential or concurrent anthracycline and taxane.
Addition or substitution of a taxane.
Node positive only, node positive and negative, or node negative only.
Longer or the same duration of chemotherapy.
Fewer than four or four or more cycles of taxane.
Hormone receptor status.
We also conducted post‐hoc subgroup analyses by type of taxane for febrile neutropenia and neuropathy, because neutropenia and neuropathy are toxicities commonly seen when taxanes are used.
Sensitivity analysis
We performed the following sensitivity analyses.
Publication status: fully published trials versus trials published in abstract form only.
Differences in the definition of DFS: DFS versus relapse‐free survival (RFS); DFS versus time to recurrence (TTR).
Risk of bias assessments: low versus high/unclear risk of bias. Studies with more than five of the nine domain judgements with unclear/high risk of bias were assigned an overall assessment of unclear/high risk.
Results
Description of studies
Results of the search
For this review update, searching yielded 5118 records from the Specialised Register of the Cochrane Breast Cancer Group, MEDLINE, Embase, and CENTRAL, on 16 July 2018. Searching relevant review papers revealed an additional 16 records, and searching WHO ICTRP and ClinicalTrials.gov revealed one potentially eligible ongoing study. After removing duplicates, we screened the titles and abstracts of the 2802 remaining records and excluded 2735 of them based on information found in the abstract alone. We further assessed the full‐text articles or ongoing trial records for 67 records. We excluded nine records after full‐text review and provided reasons in the Characteristics of excluded studies table.
Of the 58 remaining records, 33 records related to 17 new studies (ADEBAR; Boccardo; CALGB 40101; DEVA; ELDA; GEICAM 2003‐02; GEICAM 9805; GOIM 9902; GONO MIG‐5; HORG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; RAPP‐01; Roy; Sakr; TITAN; UK TACT), 14 related to updated data for nine previously included studies (BCIRG 001; BIG 2‐98; E2197; ECTO; FinHer; GEICAM 9906; PACS 01; Taxit 216; US Oncology 9735), eight related to four studies classified as studies 'awaiting classification' due to insufficient reporting of the number of events per treatment arm and relevant effect estimates (EC‐DOC; Kader; EORTC 10041/BIG 3‐04 MINDACT; PACS 04), and three were classified as 'ongoing' studies (NNBC3; NCT01966471; NCT02549677).
The original Cochrane Review identified 23 potentially eligible studies: 12 included studies, three studies 'awaiting classification', and eight ongoing studies (refer to Ferguson 2007). When studies from the original review and those from the review update were combined, review authors had 37 potentially eligible studies involving 29 included studies (referring to 30 treatment comparisons), four studies awaiting inclusion, and four ongoing studies (see PRISMA flowchart: Figure 1). The PRISMA flowchart for the original review can be found in the previously published version of this review (Ferguson 2007).
1.
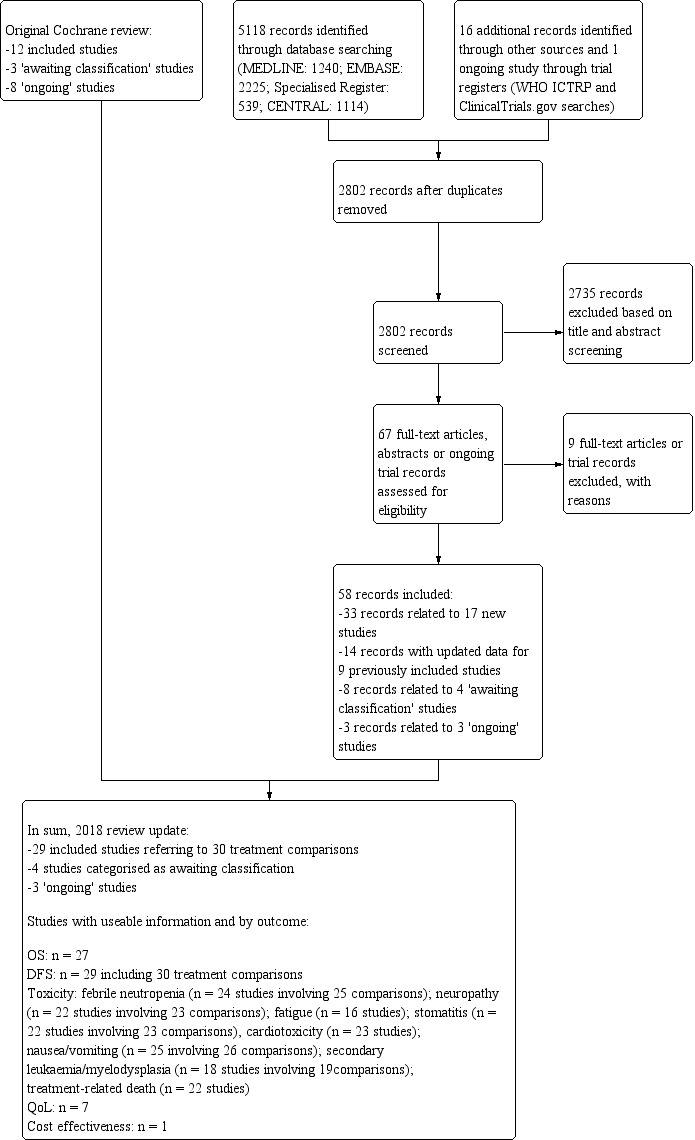
Review update: study flow diagram.
Of the 29 included studies, 27 studies published efficacy data in peer‐reviewed journals (ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 40101; CALGB 9344; DEVA; E2197; ECTO; ELDA; FinHer; GEICAM 2003‐02; GEICAM 9805; GEICAM 9906; GOIM 9902; GONO MIG‐5; HeCOG; HORG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; PACS 01; Roy; Sakr; TITAN; UK TACT; US Oncology 9735), one study had been reported only in abstract form (RAPP‐01), and for one study results were reported in an online thesis (Taxit 216). For seven studies, we received additional information or clarification of data from the trialists (ADEBAR; CALGB 40101; E2197; FinHer; GEICAM 9906; GONO MIG‐5; HORG).
Since publication of the original review, two studies formally categorised as 'awaiting assessment' ‐ ADEBAR and DEVA ‐ and six studies categorised as 'ongoing' studies ‐ GEICAM 9805; GOIM 9902; GONO MIG‐5; NCIC‐CTG MA21a and NCIC‐CTG MA21b; RAPP‐01; UK TACT ‐ have become included studies. We excluded one previously 'awaiting assessment' study (CALGB 9640: in the review update reclassified as SWOG S9623) due to confounders in the taxane (dose‐dense treatment) and non‐taxane (transplantation) treatment arms.
Included studies
See Characteristics of included studies.
For the updated review, we included 29 studies. Of these, seven studies addressed Question 1 (taxane‐containing regimen vs the same regimen without a taxane; BIG 2‐98; CALGB 9344; ECTO; GOIM 9902; HeCOG; NSABP B‐28; Taxit 216), nine addressed Question 2 (any taxane‐containing regimen vs any regimen without a taxane; ADEBAR; Boccardo; CALGB 40101; ELDA; GONO MIG‐5; HORG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; UK TACT), and 15 addressed Question 3 (any taxane‐containing regimen vs the same regimen with another drug or drugs substituted for the taxane; BCIRG 001; BIG 2‐98; DEVA; E2197; FinHer; GEICAM 2003‐02; GEICAM 9805; GEICAM 9906; PACS 01; RAPP‐01; Roy; Sakr; TITAN; UK TACT; US Oncology 9735). It is noted that BIG 2‐98 addressed Questions 1 and 3, and UK TACT addressed Questions 2 and 3.
The NCIC‐CTG MA21 study, which was a three‐arm study with two taxane‐containing arms, reported sufficient data that the study was split into NCIC‐CTG MA21a (comparing the taxane regimen of epirubicin/cyclophosphamide followed by paclitaxel (EC‐T) vs fluorouracil/epirubicin/cyclophosphamide (FEC)) and NCIC‐CTG MA21b (comparing the taxane regimen of doxorubicin/cyclophosphamide followed by paclitaxel (AC‐T) vs FEC).
Of the eight studies classified as 'awaiting classification' or 'ongoing', four addressed Question 2 (EC‐DOC; EORTC 10041/BIG 3‐04 MINDACT; NCI‐H99‐0038; NCT01966471) and four addressed Question 3 (Kader; NCT02549677; NNBC3; PACS 04).
Characteristics of patients
Axillary lymph node involvement
The included studies recruited patient populations with varying risk profiles. Seventeen trials entered participants who were positive for axillary node metastases (i.e. > 85% of included participants were lymph node positive; ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 9344; DEVA; FinHer; GEICAM 9906; GOIM 9902; GONO MIG‐5; HeCOG; HORG; NSABP B‐28; PACS 01; Roy; Sakr; Taxit 216). Nine studies included participants both with (node positive) and without (node negative) pathologically involved axillary lymph nodes (E2197; ECTO; ELDA; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; RAPP‐01; TITAN; UK TACT; US Oncology 9735). Three studies entered participants who were node negative (100%: GEICAM 2003‐02; GEICAM 9805; 94%: CALGB 40101). In GEICAM 2003‐02 and GEICAM 9805, participants also had a high‐risk factor for recurrence according to 1998 St. Gallen criteria. Baseline characteristics of participants included in the eligible studies are presented in the Characteristics of included studies table.
Menopausal status
Both premenopausal and postmenopausal women were included in all studies except for three studies (DEVA; ELDA; ICE II‐GBG 52), which included only postmenopausal patients. HeCOG excluded postmenopausal women with hormone receptor‐positive tumours and fewer than four positive axillary nodes.
Hormone receptor status
In nearly all studies, more than 58% of participants had tumours testing positive for oestrogen and/or progesterone receptors, except for TITAN, which included participants with triple‐negative breast cancer.
Interventions used in the trials
Twenty‐nine studies involved 41,911 women who were randomised to treatment groups: 21,791 to a taxane‐containing arm, and 20,120 to a non‐taxane‐containing arm.
Chemotherapy
Thirteen of the 29 included studies used paclitaxel (Boccardo; CALGB 40101; CALGB 9344; ECTO; GEICAM 2003‐02; GEICAM 9906; GONO MIG‐5; HeCOG; ICE II‐GBG 52; NCIC‐CTG MA21; NSABP B‐28; Roy; TITAN). The remaining 16 included studies used docetaxel (ADEBAR; BCIRG 001; BIG 2‐98; DEVA; E2197; ELDA; FinHer; GEICAM 9805; GOIM 9902; HORG; PACS 01; RAPP‐01; Sakr; Taxit 216; US Oncology 9735; UK TACT).
All studies except four used an anthracycline in both taxane‐containing and non‐taxane‐containing arms (CALGB 40101; ELDA; ICE II‐GBG 52; US Oncology 9735). US Oncology 9735 compared docetaxel and cyclophosphamide to doxorubicin and cyclophosphamide, CALGB 40101 compared paclitaxel alone to the doxorubicin plus cyclophosphamide regimen; ELDA compared docetaxel alone to cyclophosphamide, methotrexate, and fluorouracil, and ICE II‐GBG 52 compared paclitaxel and capecitabine against epirubicin, cyclophosphamide or cyclophosphamide, methotrexate, and fluorouracil.
The taxane and anthracycline were administered either sequentially ‐ ADEBAR; BIG 2‐98; Boccardo; CALGB 9344; DEVA; FinHer; GEICAM 2003‐02; GEICAM 9906; GOIM 9902; HeCOG; HORG; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; PACS 01; Roy; Sakr; Taxit 216; TITAN; UK TACT ‐ or concurrently ‐ BCIRG 001; BIG 2‐98; E2197; ECTO; GEICAM 9805; GONO MIG‐5; RAPP‐01. BIG 2‐98 randomised participants to four arms ‐ two control and two containing taxanes ‐ to simultaneously examine the effects of concurrent versus sequential administration of taxane and anthracycline. Updated published data from BIG 2‐98 provided the summary statistics for each group.
The total planned dose of anthracycline was the same in both arms in 10 studies (BCIRG 001; CALGB 9344; E2197; FinHer; GEICAM 9805; GOIM 9902; GONO MIG‐5; NCIC‐CTG MA21a; NSABP B‐28; Taxit 216); it was lower for the taxane‐containing arm in 14 studies (ADEBAR; BIG 2‐98; Boccardo; DEVA; ECTO; GEICAM 2003‐02; GEICAM 9906; HeCOG; HORG; PACS 01; RAPP‐01; Roy; Sakr; TITAN; UK TACT). UK TACT compared the taxane arm against two different control regimens of FEC or E‐CMF; however the total planned dose of anthracycline was lower in the taxane‐containing arm than in either of the control arms. NCIC‐CTG MA21b used doxorubicin in the taxane arm and epirubicin in the control arm, so doses of anthracycline used were not comparable. HeCOG administered dose‐dense chemotherapy in both taxane‐containing and non‐taxane‐containing arms. Dose density was unlikely to be a confounding factor, and the trial was included.
Five trials permitted granulocyte colony‐stimulating factor (G‐CSF) as primary prophylaxis. CALGB 9344 used primary prophylaxis with G‐CSF and ciprofloxacin with doxorubicin dosed at 90 mg/m², and this was not different between taxane and non‐taxane arms. HeCOG used G‐CSF during each cycle (days 3 to 10) with the same dose of G‐CSF given in the two treatment arms. TITAN stated that CSF could be used as per the American Society of Clinical Oncology (ASCO) guidelines, and its use was similar in the two treatment arms. GEICAM 9805 used primary prophylactic antibiotics for all participants receiving the taxane‐containing regimen and gave primary prophylactic G‐CSF after a protocol amendment. In NCIC‐CTG MA21 (i.e. NCIC‐CTG MA21a), G‐CSF was used as primary prophylaxis in the EC‐T taxane arm but not in the control arm. Use of G‐CSF as primary prophylaxis was identified as a potential confounder when it was used in only one treatment arm. In two studies (DEVA; FinHer), G‐CSF was recommended in the case of febrile neutropenia.
This review did not include neoadjuvant chemotherapy studies. ECTO enrolled patients before surgery and included a neoadjuvant treatment arm. The review included only the two adjuvant chemotherapy arms of this study, one if which contained a taxane drug in the treatment regimen and one that did not.
When the taxane‐containing chemotherapy regimens of the included studies was considered, the duration of chemotherapy varied across trials. The addition of extra cycles of chemotherapy to the taxane‐containing regimen has been considered as a potential confounding factor in favour of the taxane‐containing arms of these studies. Fifteen studies, involving 16 treatment comparisons, included chemotherapy treatment arms of equal duration (ADEBAR; BCIRG 001; CALGB 40101; E2197; ECTO; ELDA; FinHer; GEICAM 9805; NCIC‐CTG MA21a and NCIC‐CTG MA21b; PACS 01; RAPP‐01; Roy; Sakr; TITAN; US Oncology 9735), and eight studies used a taxane‐containing arm that delivered a longer duration or more cycles of chemotherapy than were used in the control arm (CALGB 9344; GEICAM 2003‐02; GEICAM 9906; GOIM 9902; HeCOG; HORG; NSABP B‐28; Taxit 216). Three studies involved a taxane‐containing arm of shorter chemotherapy duration than the non‐taxane‐containing arm (DEVA; Boccardo; GONO MIG‐5). In BIG 2‐98, a four‐arm study, the two taxane‐containing arms were of different duration: the sequential taxane arm was longer than the sequential control arm, and the concurrent taxane arm was the same duration as the concurrent control arm. UK TACT, a three‐arm study, provided two different control regimens whereby one control arm delivered a longer duration of chemotherapy than the second control arm. The control arms were not analysed separately and therefore were not included in analyses related to duration of chemotherapy.
Endocrine therapy
Tamoxifen 20 mg daily for five years for women with oestrogen receptor (ER)‐ and/or progesterone receptor (PR)‐positive tumours was used in most studies, with the following exceptions. HeCOG treated all hormone receptor‐positive premenopausal women with a gonadotropin‐releasing hormone (GnRH) analogue for one year in addition to tamoxifen; ECTO used tamoxifen for all participants before June 2000, but a subsequent protocol amendment mandated tamoxifen only for hormone receptor‐positive women; PACS 01 initially required tamoxifen treatment post chemotherapy only for postmenopausal women with hormone receptor‐positive or ‐negative tumours and amended the protocol in 1998 to require tamoxifen for premenopausal women with hormone receptor‐positive tumours; TITAN included women with triple‐negative breast cancer; in NSABP B‐28, all women older than 50 years of age at the time of surgery received tamoxifen regardless of hormone receptor status, and only those with positive hormone receptor status received tamoxifen if they were younger than 50 years of age. NSABP B‐28 also commenced tamoxifen concurrently with chemotherapy. It was not clear from published information whether any other included studies also commenced tamoxifen concurrently with chemotherapy. In eight studies, tamoxifen was initially given to women with ER‐ and/or PR‐positive tumours, but from 2003 to 2007, several protocol amendments allowed postmenopausal women to switch from tamoxifen to aromatase inhibitors (DEVA; E2197; FinHer; GEICAM 2003‐02; GEICAM 9906; GOIM 9902; NCIC‐CTG MA21a and NCIC‐CTG MA21b; UK TACT). GEICAM 2003‐02 allowed postmenopausal women with hormone receptor‐positive tumours to receive aromatase inhibitors as initial adjuvant therapy or after tamoxifen. All studies used uniform policies for hormonal treatment in both control and experimental arms.
Radiotherapy
Radiotherapy to the breast, when reported, was required when breast‐conserving surgery was performed. This information was not specified in the DEVA and GONO MIG‐5 study reports. Additional radiotherapy was 'as per institution' in BCIRG 001, BIG 2‐98, Boccardo, CALGB 40101, E2197, FinHer, GEICAM 9805, HORG, ICE II‐GBG 52, NCIC‐CTG MA21a and NCIC‐CTG MA21b, Sakr, TITAN, UK TACT, and US Oncology 9735. In the TITAN study, in some cases MammoSite brachytherapy radiation was permitted if immediately after surgery and before treatment. In ADEBAR, all participants received adjuvant radiotherapy given following completion of chemotherapy or intermittently after completion of 50% of chemotherapy. In Roy, participants received locoregional external beam radiotherapy following their modified radical mastectomy. Axillary or chest wall radiotherapy was given to participants with four or more positive axillary nodes or a primary tumour (T) > 5 cm in GOIM 9902, HeCOG, and GEICAM 9906. ECTO also gave chest wall radiotherapy to women with a primary T4 tumour. Radiation to the chest wall, supraclavicular area, and internal mammary chain was recommended following mastectomy in PACS 01. In CALGB 9344, axillary or chest wall radiotherapy was not permitted.
Targeted therapy
One study used secondary randomisation to examine the additional question of the addition of trastuzumab to adjuvant chemotherapy for women with human epidermal growth factor receptor 2 (HER2)‐positive tumours following adjuvant chemotherapy (FinHer). UK TACT allowed women with HER2‐positive tumours to enter clinical trials for trastuzumab, and CALGB 40101 and NCIC‐CTG MA21 (NCIC‐CTG MA21a and NCIC‐CTG MA21b) recommended trastuzumab for HER2‐positive tumours after 2005. ELDA included women with HER2‐positive tumours (19% of the population) who received adjuvant trastuzumab for one year after chemotherapy based on results from the HERA trial.
Outcomes assessed in trials
The median follow‐up for participants ranged from 24 months in Roy to 163 months (estimated follow‐up) in GONO MIG‐5.
Four studies reported overall survival as a primary outcome (Boccardo; GONO MIG‐5; NSABP B‐28; US Oncology 9735), and 26 studies reported DFS (or RFS, distant disease‐free survival (DDFS) if reported) as the primary outcome. Both NSABP B‐28 and US Oncology 9735 had two primary outcomes (overall survival and DFS).
Not all trials reported data on all outcomes for this review. Two studies did not report complete data on overall survival and were not included for this part of the meta‐analysis (NCIC‐CTG MA21a and NCIC‐CTG MA21b; RAPP‐01). For NCIC‐CTG Ma21, the number of events had not been reached to conduct a formal statistical comparison for survival analysis, and for RAPP‐01, overall survival data were not reported in abstract form nor in the full trial publication. All trials reported on DFS; however the terminology and definitions used differed slightly between trials (Table 14). Definitions were the same for both experimental and control arms of each trial, and, for the purposes of this review, all breast cancer recurrence events under the different definitions were combined.
1. Definition of "disease‐free survival".
| Study | Description | Definition |
| ADEBAR | DFS | Revised to iDFS (in 2016) in line with Standardized Definitions for Efficacy End Points (STEEP), where DFS referred to all invasive ipsilateral, regional, contralateral, and distant disease recurrences, second primary tumours, and death from any cause as events, and all non‐invasive in situ cancer events were excluded |
| BCIRG 001 | DFS | Time from randomisation to date of clinical relapse (histological or radiological evidence), a second cancer (except skin cancer other than melanoma or carcinoma in situ of breast or cervix), or death |
| BIG 2‐98 | DFS | Time from randomisation until first date of local, regional, or distant relapse; diagnosis of a second primary cancer, including contralateral invasive breast cancer; or death from any cause |
| Boccardo | RFS | Time from date of random assignment to date of locoregional and/or distant release |
| CALGB 40101 | RFS | Time from study entry until local recurrence, distant relapse, or death without relapse, whichever occurred first |
| CALGB 9344 | DFS | Time from randomisation to date of first locoregional recurrence, first distant metastasis, or death from any cause |
| DEVA | DFS | Time from date of randomisation to locoregional recurrence, distant recurrence, new primary breast cancer, or death |
| ECTO | Freedom from progression | Not reported in trial publication |
| ELDA | DFS | Interval between randomisation and locoregional or distant relapse or contralateral invasive breast cancer or second primary invasive non‐breast cancer or ipsilateral or contralateral in situ ductal carcinoma or death without cancer, whichever occurred first |
| E2197 | DFS | Time from date of random assignment to date of invasive breast cancer recurrence, invasive contralateral breast cancer, or death from any cause |
| FinHer | Recurrence‐free survival | Time from randomisation to date of detection of local or distant relapse (histological or radiological evidence) or contralateral invasive breast cancer or death without recurrence |
| GEICAM 2003‐02 | DFS | Time from random assignment to date of local, regional, or metastatic relapse; date of a second primary malignancy; or death form any cause, whichever occurred first |
| GEICAM 9805 | DFS | Time from date of randomisation to date of local, regional, or metastatic relapse; diagnosis of second primary cancer or death from any cause |
| GEICAM 9906 | DFS | According to the definition of invasive disease‐free survival (IDFS) in the STEEP system |
| GOIM 9902 | DFS | Time from randomisation to first relapse (local, regional, distant), contralateral breast cancer, or death from any cause |
| GONO MIG‐5 | EFS | Time from date of randomisation to date of local recurrence, distant metastases, contralateral breast cancer, second primary malignancy, or death from any cause, whichever occurred first |
| HeCOG | DFS | Time from randomisation until local recurrence, distant relapse, or death (without relapse) |
| HORG | DFS | Time from randomisation to date of breast cancer recurrence (local, regional, or distant), invasive contralateral breast cancer, non‐breast second primary cancer, or death from any cause |
| ICE II‐GBG 52 | iDFS and DDFS | Any local invasive or distant recurrence of breast cancer, any contralateral breast cancer, any second malignancy, and any death irrespective of its cause for iDFS |
| NCIC‐CTG MA21a | RFS | Time from random assignment to time of recurrence of the primary breast cancer (local, nodal, metastatic). Patients with contralateral breast cancer, second malignancy, non‐disease‐related death were censored |
| NCIC‐CTG MA21b | RFS | Time from random assignment to time of recurrence of the primary breast cancer (local, nodal, metastatic). Patients with contralateral breast cancer, second malignancy, non‐disease‐related death were censored |
| NSABP B‐28 | DFS | Time from randomisation until local, regional, or distant treatment failure, contralateral breast cancer, non‐breast second primary cancer, or death |
| PACS 01 | DFS | Time from randomisation until first relapse (local, regional, or distant), contralateral breast cancer, or death from any cause |
| RAPP‐01 | TTR | Time to locoregional relapse, contralateral breast cancer, or distant metastasis, whichever occurs first |
| Roy | DFS | Time from study entry to first local recurrence or distant metastasis, or death as a result of any cause |
| Sakr | DFS | Time from randomisation until first relapse (locoregional or distant), contralateral breast cancer, or death from any cause |
| UK TACT | DFS | Time from randomisation to first invasive relapse, new primary breast cancer (ipsilateral or contralateral), or death form any cause |
| Taxit 216 | DFS | Time from date of randomisation to local or distant recurrence or contralateral breast cancer or second primary malignancy or death from any case, whichever occurred first |
| TITAN | DFS | Time between randomisation and date of first documented disease recurrence or death from any cause |
| US Oncology 9735 | DFS | Time from first dose of chemotherapy until date of any recurrence of breast cancer, a new second breast cancer or any other type of cancer, death due to any cause without relapse or recurrence of breast cancer, or date of last patient contact |
DFS: disease‐free survival EFS: event‐free survival RFS: relapse‐free survival TTR: time to recurrence
All 29 studies reported some toxicity data; however data from one study could not be extracted and included in this review (Roy). This study presented toxicity data per week of treatment.
Seven studies presented quality of life measures (ADEBAR; BCIRG 001; DEVA; ELDA; GEICAM 9805; HeCOG; UK TACT). GONO MIG‐5, CALGB 40101, and NCIC‐CTG MA21 also listed quality of life as a secondary outcome; however these data have yet to be reported.
Excluded studies
Nine studies were excluded from this review update. One record reported results for GEICAM 9906 but was found to be a trial commentary (Dang), and another record reported on the prognostic and predictive significance of subtyping in the BCIRG 001 study (Hugh). Two studies involved neoadjuvant chemotherapy: in Albert, adjuvant data were unable to be separated from neoadjuvant data, and in NSABP B‐27, all participants received neoadjuvant chemotherapy before receiving adjuvant taxane. In four studies it was observed that all treatment arms received taxane therapy (NCT02838225; Sparano 2015; SWOG S0221; Wildiers). In SWOG S9623, dose‐dense chemotherapy in the taxane‐containing arm and high‐dose escalated chemotherapy with autologous haematopoietic progenitor cell transplantation in the non‐taxane arm were viewed as confounders, and the study was excluded. See Characteristics of excluded studies.
Risk of bias in included studies
Refer to Figure 2 for a summary of the risk of bias judgements for the included studies for each risk of bias domain.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The 29 studies, which related to 30 treatment comparisons, were described as randomised. The method of random sequence generation was described adequately (i.e. with low risk of bias) in 28 studies referring to 29 treatment comparisons (ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 40101; CALGB 9344; DEVA; E2197; ECTO; ELDA; FinHer; GEICAM 2003‐02; GEICAM 9805; GEICAM 9906; GOIM 9902; HeCOG; HORG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; PACS 01; RAPP‐01; Roy; Sakr; Taxit 216; TITAN; UK TACT). These studies reportedly used stratified randomisation, permuted block design, or minimisation. It was not possible to accurately assess the method of random sequence generation in one study owing to lack of information presented in the published trial report (US Oncology 9735). This study was classified as having unclear risk of bias.
Twenty‐three of the 29 studies were at low risk of bias for allocation concealment. These studies described central randomisation systems (computer or telephone/fax) (BIG 2‐98; Boccardo; CALGB 9344; DEVA; ECTO; ELDA; FinHer; GEICAM 2003‐02; GEICAM 9805; GOIM 9902; HeCOG; HORG; ICE II‐GBG 52; NCIC‐CTG MA21 (NCIC‐CTG MA21a and NCIC‐CTG MA21b); NSABP B‐28; PACS 01; RAPP‐01; Taxit 216;TITAN; UK TACT). Six studies did not describe methods of allocation concealment or did not provide sufficient detail in the trial publication or abstract and were judged as having unclear risk of bias (BCIRG 001; E2197; GEICAM 9906; Roy; Sakr; US Oncology 9735).
Blinding
Twenty‐one studies were described as 'open label' or 'non‐blinded' (ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 40101; CALGB 9344; DEVA; ECTO; ELDA; FinHer; GEICAM 2003‐02; GEICAM 9805; GEICAM 9906; GONO MIG‐5; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; PACS 01; Roy; Taxit 216; TITAN; UK TACT), and eight studies provided no information in the abstract or trial publication to allow a firm conclusion on whether they were 'open‐label' studies (E2197; GOIM 9902; HeCOG; HORG; NSABP B‐28; RAPP‐01; Sakr; US Oncology 9735). Performance bias due to lack of blinding of participants and personnel could not be ruled out, and these 29 studies were judged as having unclear risk of bias for this domain.
Detection bias was assessed by grouping outcomes with similar risks of bias: (1) overall survival, (2) disease‐free survival and toxicity, and (3) quality of life. For overall survival, lack of blinding was perceived as unlikely to have an impact on this outcome assessment. Therefore all studies were perceived to be at low risk of bias. For outcome measures that were more likely to be influenced by lack of blinding, that is, disease‐free survival and toxicity, we assessed whether outcome assessments were confirmed through imaging and biochemical tests and reviewed by independent panels/adjudication committees in each study. All 29 included studies were at unclear risk of bias because these outcomes were measured through scans and blood tests with no independent clinical review group. Quality of life measures were likely to be affected by lack of blinding to treatment. Seven of the ten studies that had planned to collect QoL data actually reported these data (data provided: ADEBAR; BCIRG 001; DEVA; ELDA; GEICAM 9805; HeCOG; UK TACT; data not reported: CALGB 40101; GONO MIG‐5; NCIC‐CTG MA21a and NCIC‐CTG MA21b). Quality of life questionnaires were completed by participants; the seven studies that reported these data were therefore considered to be at high risk.
Incomplete outcome data
Twenty‐three studies described intention‐to‐treat analysis and minimal patient loss to follow‐up that was accounted for; therefore we judged them to be at low risk of bias: BCIRG 001, BIG 2‐98, Boccardo, CALGB 40101, DEVA, E2197, ECTO, ELDA, FinHer, GEICAM 2003‐02, GEICAM 9805, GEICAM 9906, GONO MIG‐5, HeCOG, ICE II‐GBG 52, NCIC‐CTG MA21a and NCIC‐CTG MA21b, NSABP B‐28, PACS 01, Roy, Taxit 216, TITAN, UK TACT, and US Oncology 9735. Four studies were judged as having unclear risk of bias due to insufficient or no information provided for their analysis plan (ADEBAR; CALGB 9344; GOIM 9902; RAPP‐01), and two studies were considered at high risk of bias for this domain due to no intention‐to‐treat analyses and missing data with no reasons provided (HORG; Sakr).
Selective reporting
Twenty‐three studies, relating to 24 treatment comparisons, reported results for outcomes listed in the methods section of the trial publication (Boccardo; DEVA; GOIM 9902; HORG; Roy; Sakr; Taxit 216) or provided a trial registration record with listed outcomes found in the methods and results sections of the trial publication (ADEBAR; BCIRG 001; BIG 2‐98; CALGB 40101; CALGB 9344; E2197; ECTO; ELDA; GEICAM 9805; GEICAM 9906; HeCOG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; TITAN; UK TACT; US Oncology 9735). In the remaining six studies, there was partial reporting of results (i.e. for QoL) or some changes were noted in the primary or secondary outcomes (CALGB 40101; FinHer; GEICAM 2003‐02; GONO MIG‐5; PACS 01); these studies were ranked at unclear risk of bias for this domain. RAPP‐01 was judged as having high risk of bias for this domain, as data related to overall survival were not reported, although OS was listed as a secondary outcome in the trial publication.
Other potential sources of bias
Treatment groups were well balanced in most studies (low risk of bias: ADEBAR; BCIRG 001; BIG 2‐98; CALGB 40101; CALGB 9344; DEVA; E2197; ECTO; ELDA; GEICAM 2003‐02; GEICAM 9805; GOIM 9902; GONO MIG‐5; HORG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; RAPP‐01; Sakr; Taxit 216; TITAN; UK TACT; US Oncology 9735). When minor baseline imbalances (such as differences in tumour size and in hormone receptor status across treatment groups) were reported, these were considered by the review authors to be unlikely to bias trial outcomes; these trials therefore were ranked as having unclear risk (Boccardo; FinHer; GEICAM 9906; HeCOG; PACS 01; Roy).
Effects of interventions
See: Table 1
Refer to Table 1.
Overall survival
High‐quality evidence was obtained from 27 studies for analysis of overall survival (OS). The NCIC‐CTG MA21 ‐ NCIC‐CTG MA21a; NCIC‐CTG MA21b ‐ and RAPP‐01 studies did not report sufficient OS data to allow indirect calculations of the hazard ratio (HR) and confidence interval (CI). A total of 39,180 women were included in the overall survival analysis, with 6501 deaths reported. Taxane‐containing regimens improved survival when compared to non‐taxane‐containing controls (HR 0.87, 95% CI 0.83 to 0.92; P < 0.001; high‐certainty evidence; Analysis 1.1; Figure 3). Heterogeneity across trials was moderate (heterogeneity I² = 35%; P = 0.04).
1.1. Analysis.
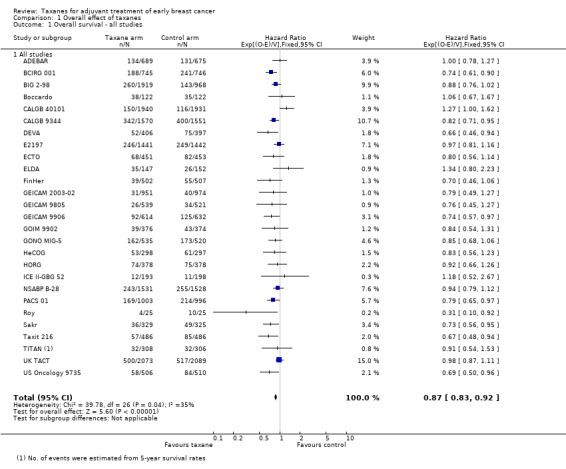
Comparison 1 Overall effect of taxanes, Outcome 1 Overall survival ‐ all studies.
3.
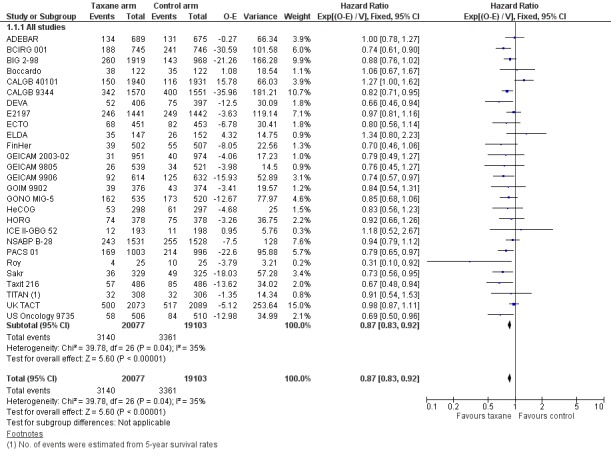
Forest plot of comparison: 1 Overall effect of taxanes, outcome: 1.1 Overall survival ‐ all studies.
Disease‐free survival
High‐certainty evidence was obtained from all 29 studies, involving 30 comparisons, for analysis of disease‐free survival (DFS). In this review, DFS analysis included freedom from progression (as measured in ECTO), time to recurrence (as measured in RAPP‐01), event‐free survival (as per GONO MIG‐5), and recurrence‐free survival (as measured in Boccardo, CALGB 40101, FinHer, and NCIC‐CTG MA21a and NCIC‐CTG MA21b). Although FinHer reported relapse‐free survival (RFS) and GONO MIG‐5 reported event‐free survival (EFS), these definitions were considered similar to those reported as DFS in other studies. Two studies ‐ ADEBAR; BCIRG 001 ‐ specifically excluded ductal carcinoma in situ (DCIS) and one study included DCIS in its DFS definition (ELDA). See Table 14 for the definition of DFS used in each study.
A total of 41,909 women from 29 studies were included in the DFS analysis, with 10,271 reported events. Taxane‐containing treatment improved disease‐free survival compared to control (HR 0.88, 95% CI 0.85 to 0.92; P < 0.001; high‐certainty evidence; Analysis 1.2; Figure 4). Heterogeneity across studies was substantial (heterogeneity I² = 59%; P < 0.001).
1.2. Analysis.
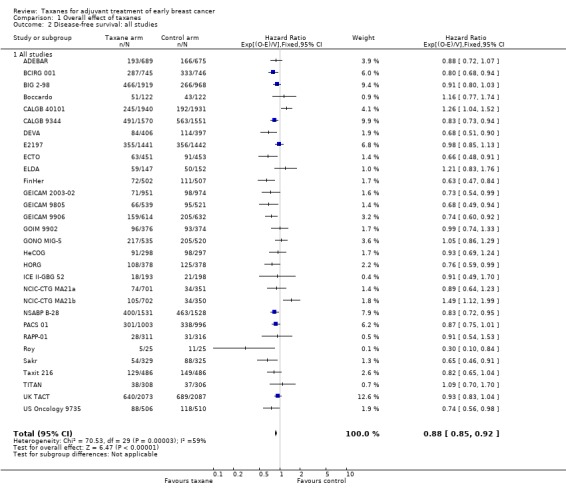
Comparison 1 Overall effect of taxanes, Outcome 2 Disease‐free survival: all studies.
4.
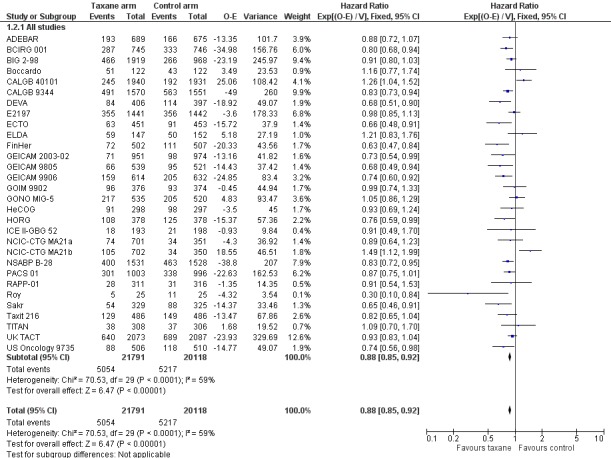
Forest plot of comparison: 1 Overall effect of taxanes, outcome: 1.2 Disease‐free survival: all studies.
We noted differences among the definitions used by each study, with death and contralateral breast cancer at times not counted among DFS events; in the first instance, we judged that this would have a minor impact on DFS analysis. To test this, we conducted a post‐hoc sensitivity analysis while excluding Boccardo, CALGB 40101, NCIC‐CTG MA21a and NCIC‐CTG MA21b and RAPP‐01. The result did not change significantly (HR 0.86, 95% CI 0.82 to 0.89; P < 0.001; Analysis 1.3), and heterogeneity was moderate (I² = 40%; P = 0.02). The definition of freedom from progression in ECTO would typically be consistent with DFS as defined in the other included studies; therefore we did not conduct a sensitivity analysis.
1.3. Analysis.
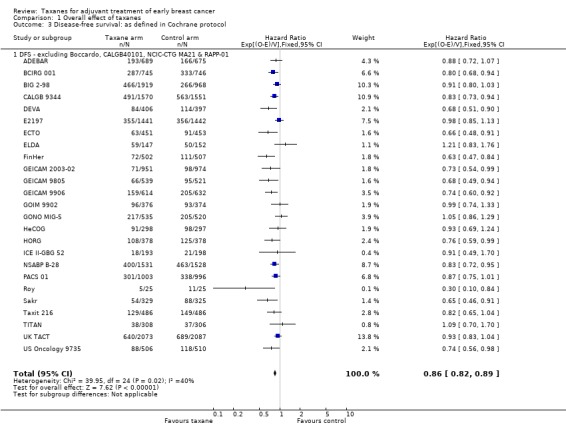
Comparison 1 Overall effect of taxanes, Outcome 3 Disease‐free survival: as defined in Cochrane protocol.
Subgroup analysis
Several post‐hoc subgroup analyses were performed in a side‐by‐side observational manner and are reported below. This exercise has limitations, although it was undertaken to address clinically relevant questions. Tests for interaction have not been performed as no differences between subgroups have been postulated.
Type of taxane
The two taxane drugs were analysed in separate groups in this post‐hoc analysis to assess whether there was a difference in efficacy between them.
Sixteen studies used docetaxel: 15 studies provided sufficient data for OS analyses (ADEBAR; BCIRG 001; BIG 2‐98; DEVA; E2197; ELDA; FinHer; GEICAM 9805; GOIM 9902; HORG; PACS 01; Sakr; Taxit 216; UK TACT; US Oncology 9735), and all 16 studies provided DFS data for analyses (ADEBAR; BCIRG 001; BIG 2‐98; DEVA; E2197; ELDA; FinHer; GEICAM 9805; GOIM 9902; HORG; PACS 01; RAPP‐01; Sakr; Taxit 216; UK TACT; US Oncology 9735). The remaining 13 studies used paclitaxel; OS data were available for 12 studies (Boccardo; CALGB 40101; CALGB 9344; ECTO; GEICAM 2003‐02; GEICAM 9906; GONO MIG‐5; HeCOG; ICE II‐GBG 52; NSABP B‐28; Roy; TITAN), and DFS data were available for 13 studies with 14 treatment comparisons (Boccardo; CALGB 40101; CALGB 9344; ECTO; GEICAM 2003‐02; GEICAM 9906; GONO MIG‐5; HeCOG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; Roy; TITAN). Both types of taxane showed a significant HR favouring taxane treatment over controls for both OS and DFS.
Overall survival
The docetaxel group included 22,105 women, and the paclitaxel group included 17,075 women. The HR for the docetaxel group was 0.86 (95% CI 0.81 to 0.92; P < 0.001; Analysis 2.1) with moderate heterogeneity (I² = 37%; P = 0.07), which was comparable to the paclitaxel group at HR 0.89 (95% CI 0.82 to 0.96; P = 0.003; Analysis 2.1) with moderate heterogeneity (I² = 37%; P = 0.10).
2.1. Analysis.
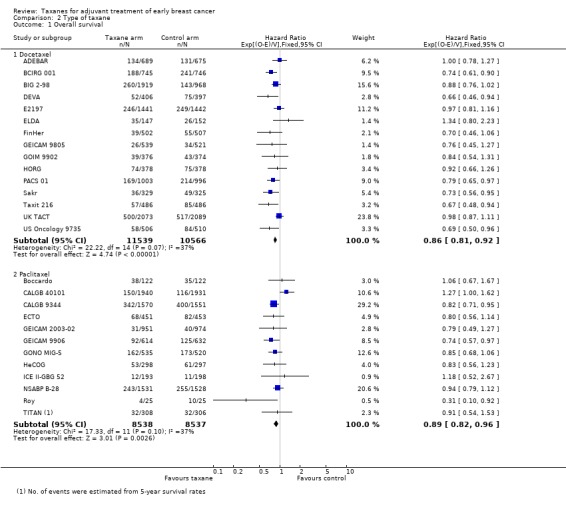
Comparison 2 Type of taxane, Outcome 1 Overall survival.
Disease‐free survival
The docetaxel group included 22,730 women, and the paclitaxel group had 19,179 women. The HR for the docetaxel group was 0.87 (95% CI 0.82 to 0.91; P < 0.001; Analysis 2.2) with moderate heterogeneity (I² = 39%; P = 0.06), and the HR for the paclitaxel group was 0.91 (95% CI 0.85 to 0.96; P = 0.002; Analysis 2.2) with substantial heterogeneity (I² = 71%; P < 0.001).
2.2. Analysis.
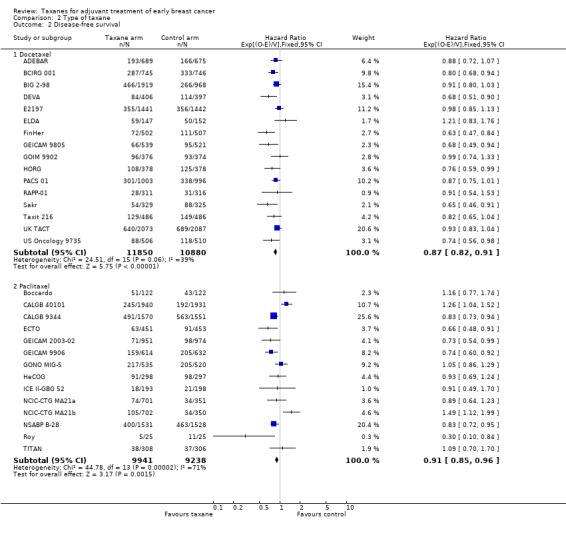
Comparison 2 Type of taxane, Outcome 2 Disease‐free survival.
Weekly versus three‐weekly taxane
A post‐hoc analysis was conducted to examine the difference in efficacy between weekly versus three‐weekly administered taxanes. All studies that administered docetaxel did so using a three‐weekly regimen, except ELDA, which administered weekly docetaxel, so efficacy data for docetaxel were not further examined. Two studies that administered paclitaxel were not included in this post‐hoc analysis as they could not be classified as weekly or three‐weekly regimens: CALGB 40101 administered paclitaxel either fortnightly or in three‐weekly cycles, and HeCOG used fortnightly paclitaxel for all women in the taxane group.
Overall survival
Four studies (4176 women) provided a regimen of weekly paclitaxel in the taxane arm (GEICAM 2003‐02; GEICAM 9906; ICE II‐GBG 52; TITAN). Analysis of these studies yielded an HR of 0.80, favouring the taxane arm (95% CI 0.65 to 0.98; P = 0.7; Analysis 3.1), with no heterogeneity (P = 0.70). Six studies (8433 women) administered paclitaxel every three weeks (Boccardo; CALGB 9344; ECTO; GONO MIG‐5; NSABP B‐28; Roy), with HR of 0.86 favouring taxane (95% CI 0.78 to 0.95; P = 0.002; Analysis 3.1), and with no significant heterogeneity (I² = 15%; P = 0.32).
3.1. Analysis.
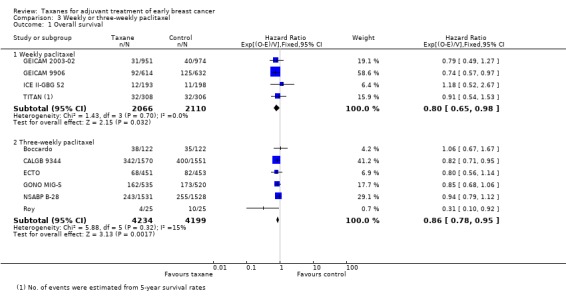
Comparison 3 Weekly or three‐weekly paclitaxel, Outcome 1 Overall survival.
Disease‐free survival
Four studies (4176 women) that administered weekly paclitaxel ‐ GEICAM 2003‐02; GEICAM 9906; ICE II‐GBG 52; TITAN ‐ gave a combined HR of 0.79 favouring the taxane (95% CI 0.67 to 0.92; P = 0.003; Analysis 3.2), with no heterogeneity (I² = 0%; P = 0.42). Six studies (8433) provided a regimen of paclitaxel administered three‐weekly (Boccardo; CALGB 9344; ECTO; GONO MIG‐5; NSABP B‐28; Roy). Analysis of these studies revealed an HR of 0.85 (95% CI 0.79 to 0.92; P < 0.001; Analysis 3.2), with significant heterogeneity (I² = 62%; P < 0.02).
3.2. Analysis.
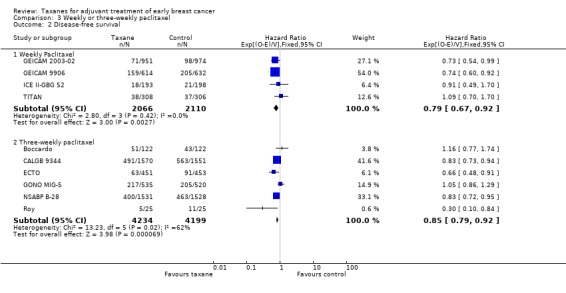
Comparison 3 Weekly or three‐weekly paclitaxel, Outcome 2 Disease‐free survival.
Sequential or concurrent taxane and anthracycline
Taxane treatment was given sequentially or concurrently with anthracycline, and it is unclear whether this scheduling impacts efficacy. Analysis was performed to examine this question. Toxicity differences between the two schedules are discussed separately.
Overall survival
Eighteen studies (24,764 women) administered the taxane and anthracycline sequentially in the experimental arm (ADEBAR; BIG 2‐98; Boccardo; CALGB 9344; DEVA; FinHer; GEICAM 2003‐02; GEICAM 9906; GOIM 9902; HeCOG; HORG; NSABP B‐28; PACS 01; Roy; Sakr; Taxit 216; TITAN; UK TACT). Analysis of these studies demonstrated an HR of 0.86 favouring the taxane‐containing group (95% CI 0.81 to 0.91; P < 0.001; Analysis 4.1), with no significant heterogeneity (I² = 17%; P = 0.25). Six studies (8839 women) administered the taxane and anthracycline concurrently in the experimental arm (BCIRG 001; BIG 2‐98; E2197; ECTO; GEICAM 9805; GONO MIG‐5). Analysis of these studies revealed an HR of 0.86, favouring the taxane‐containing group (95% CI 0.78 to 0.94; P = 0.002; Analysis 4.1), with no heterogeneity (P = 0.37).
4.1. Analysis.
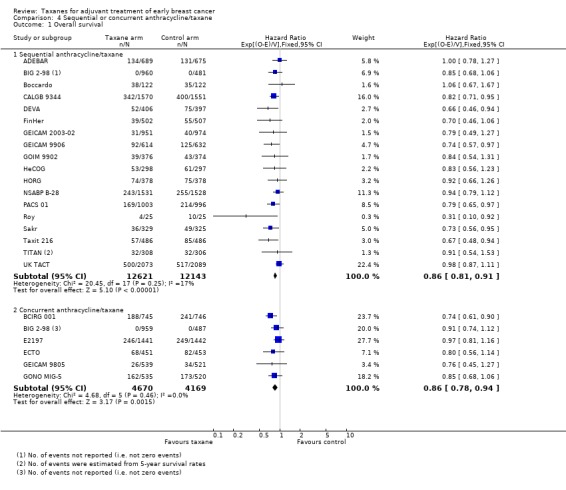
Comparison 4 Sequential or concurrent anthracycline/taxane, Outcome 1 Overall survival.
Disease‐free survival
Nineteen studies with 20 treatment comparisons (26,866 women) administered the taxane and anthracycline sequentially in the experimental arm (ADEBAR; BIG 2‐98; Boccardo; CALGB 9344; DEVA; FinHer; GEICAM 2003‐02; GEICAM 9906; GOIM 9902; HeCOG; HORG; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; PACS 01; Roy; Sakr; Taxit 216; TITAN; UK TACT). The estimated HR from analysis of these studies was 0.86, favouring the taxane‐containing group (95% CI 0.82 to 0.90; P < 0.001; Analysis 4.2), with moderate heterogeneity (I² = 51%; P = 0.005). Seven studies (9466 women) administered the taxane and anthracycline concurrently in the experimental arm (BCIRG 001; BIG 2‐98; E2197; ECTO; GEICAM 9805; GONO MIG‐5; RAPP‐01). The estimated HR was 0.89, favouring the taxane‐containing group (95% CI 0.83 to 0.97; P < 0.001; Analysis 4.2), with moderate heterogeneity (I² = 51%; P = 0.05). BIG 2‐98 reported both sequential and concurrent administration arms; thus separately reported arms were included in this analysis.
4.2. Analysis.
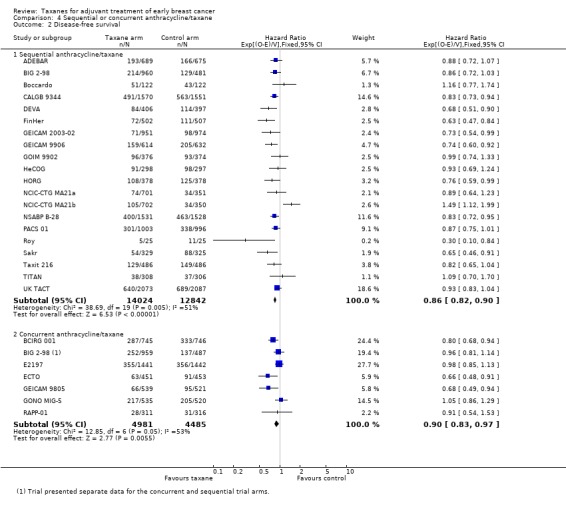
Comparison 4 Sequential or concurrent anthracycline/taxane, Outcome 2 Disease‐free survival.
Addition of taxane or substitution of taxane
This analysis separated groups into those where a taxane was added to control chemotherapy studies (Question 1) and those where a taxane was substituted for part of the control chemotherapy (Question 3). This analysis was performed, as it has been postulated that benefit from taxane treatment could be due in part to the addition of an extra non‐cross‐resistant drug rather than to superior efficacy of the taxane itself.
Overall survival
Data were available for seven studies (10,842 women) designed such that the experimental arm received a taxane administered in addition to control chemotherapy (BIG 2‐98; CALGB 9344; ECTO; GOIM 9902; HeCOG; NSABP B‐28; Taxit 216). Analysis of these studies yielded an HR of 0.84, favouring the taxane‐containing group (95% CI 0.77 to 0.92; P < 0.001; Analysis 5.1), with no heterogeneity (P = 0.60). Data were available for 13 (16,196 women) of 15 eligible studies, and these studies were designed with the experimental arm given a taxane substituted for one or more of the drugs from the control (BCIRG 001; BIG 2‐98; DEVA; E2197; FinHer; GEICAM 2003‐02; GEICAM 9805; GEICAM 9906; PACS 01; Roy; Sakr; TITAN; US Oncology 9735). This group also had an HR of 0.80 in favour of the taxane‐containing treatments (95% CI 0.74 to 0.86; P < 0.001; Analysis 5.1), with no significant heterogeneity (I² = 6%; P = 0.38).
5.1. Analysis.
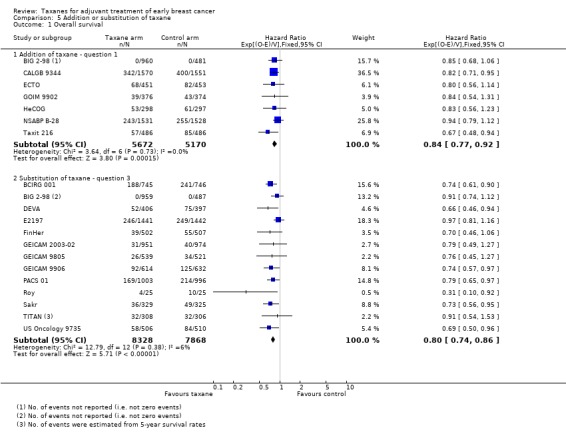
Comparison 5 Addition or substitution of taxane, Outcome 1 Overall survival.
Disease‐free survival
Seven studies (10,842 women) were designed such that the experimental arm received a taxane administered in addition to control chemotherapy (BIG 2‐98; CALGB 9344; ECTO; GOIM 9902; HeCOG; NSABP B‐28; Taxit 216). For these studies, an HR of 0.84 favoured the taxane‐containing group (95% CI 0.78 to 0.90; P < 0.001), with no heterogeneity (P = 0.66). Fourteen studies included 16,823 women and were designed with the experimental arm receiving a taxane substituted for one or more of the drugs from the control (BCIRG 001; BIG 2‐98; DEVA; E2197; FinHer; GEICAM 2003‐02; GEICAM 9805; GEICAM 9906; PACS 01; RAPP‐01; Roy; Sakr; TITAN; US Oncology 9735). This group also had an HR of 0.83, in favour of taxane‐containing treatments (95% CI 0.78 to 0.88; P < 0.001), with moderate heterogeneity (I² = 47%; P = 0.03).
Duration of chemotherapy
Studies have been examined post‐hoc to examine whether a longer duration of chemotherapy in the taxane arm may explain the observed improvement in outcomes. Groups were divided according to duration of total planned treatment rather than the total number of planned cycles to account for variation in cycle length between studies. BIG 2‐98 included two taxane‐containing arms ‐ one with longer duration and the other with the same duration as the control arm. As these arms were reported separately for OS and DFS, they are included in the analyses below.
Overall survival
Nine studies (13,865 women) included a taxane‐containing experimental arm that was of longer duration than the control arm (BIG 2‐98; CALGB 9344; GEICAM 2003‐02; GEICAM 9906; GOIM 9902; HeCOG; HORG; NSABP B‐28; Taxit 216). Analysis of these studies revealed an HR of 0.84, favouring the taxane‐containing group (95% CI 0.77 to 0.91; P < 0.001; Analysis 6.1), with no heterogeneity (P = 0.69). Fourteen studies (18,660 women) were designed with a taxane‐containing experimental arm of the same duration as the control arm (ADEBAR; BCIRG 001; BIG 2‐98; CALGB 40101; E2197; ECTO; ELDA; FinHer; GEICAM 9805; PACS 01; Roy; Sakr; TITAN; US Oncology 9735). This group also had an HR of 0.87 in favour of taxane‐containing treatments (95% CI 0.81 to 0.94; P < 0.001; Analysis 6.1), with moderate heterogeneity (I² = 52%; P = 0.01).
6.1. Analysis.
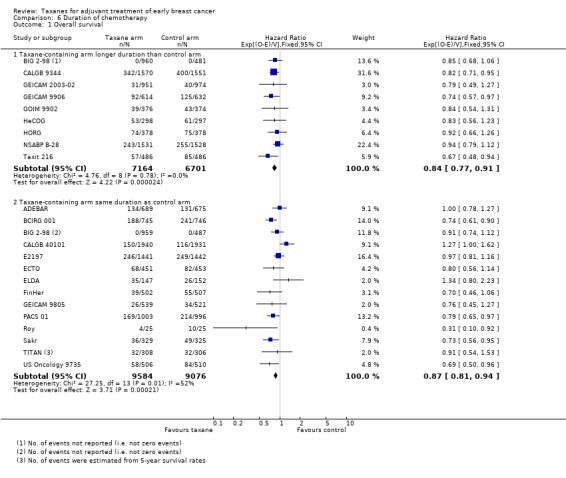
Comparison 6 Duration of chemotherapy, Outcome 1 Overall survival.
Disease‐free survival
Nine studies (13,865 women) were designed to include a taxane‐containing experimental arm of longer duration than the control arm (BIG 2‐98; CALGB 9344; GEICAM 2003‐02; GEICAM 9906; GOIM 9902; HeCOG; HORG; NSABP B‐28; Taxit 216). The HR for DFS was 0.83, favouring the taxane‐containing group (95% CI 0.77 to 0.88; P < 0.001; Analysis 6.2), with no heterogeneity (P = 0.85). Sixteen studies (17 treatment comparisons) involving 21,391 women were designed with the taxane‐containing arm having the same duration as the control arm (ADEBAR; BCIRG 001; BIG 2‐98; CALGB 40101; E2197; ECTO; ELDA; FinHer; GEICAM 9805; NCIC‐CTG MA21a and NCIC‐CTG MA21b; PACS 01; RAPP‐01; Roy; Sakr; TITAN; US Oncology 9735). An HR of 0.90 in favour of the taxane‐containing arms was found for this group of studies (95% CI 0.85 to 0.96; P < 0.001; Analysis 6.2), with substantial heterogeneity (I² = 70%; P < 0.001).
6.2. Analysis.
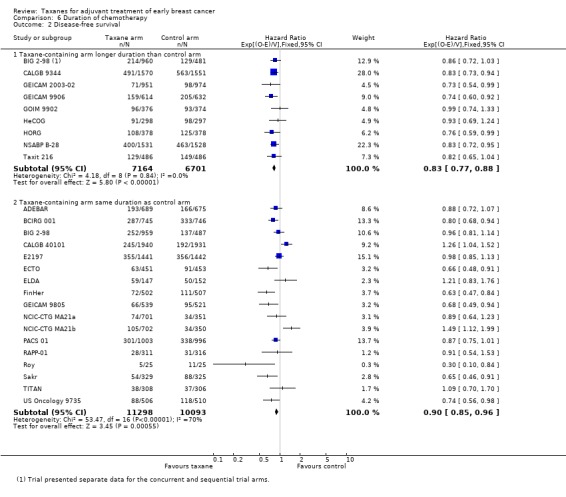
Comparison 6 Duration of chemotherapy, Outcome 2 Disease‐free survival.
Number of cycles of taxane‐containing chemotherapy
This post‐hoc analysis examined studies that administered three cycles of the taxane drug in comparison with studies that used four or more cycles of taxane in the experimental arm. This was done to determine if the number of cycles of taxane impacted efficacy. This analysis had limitations due to heterogeneity between studies with the use of different control regimens and varying doses and scheduling of the taxane drug. BIG 2‐98 included two taxane arms ‐ one administered three cycles of taxane, and the other four. These arms were separately reported for DFS and therefore were included separately in this analysis. Although TITAN administered paclitaxel weekly for 12 weeks, this is often considered as three weeks of paclitaxel (weekly) multiplied by four, and thus is classified as four cycles of taxane. GEICAM 2003‐02 and GEICAM 9906 used eight weekly doses of paclitaxel and could not be classified in either group. These results were not included in the analysis.
Overall survival
Seven studies (6551 women) used three cycles of taxane treatment in the experimental arm (BIG 2‐98; DEVA; FinHer; HeCOG; PACS 01; Roy; Sakr), reporting an HR of 0.77 favouring the taxane‐containing group (95% CI 0.69 to 0.86; P < 0.001; Analysis 7.1), with no heterogeneity (P = 0.60). Nineteen studies (29,458 women) ‐ ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 40101; CALGB 9344; E2197; ECTO; ELDA; GEICAM 9805; GOIM 9902; GONO MIG‐5; HORG; ICE II‐GBG 52; NSABP B‐28; Taxit 216; TITAN; UK TACT; US Oncology 9735 ‐ used four or more cycles of taxane and found an HR of 0.91 in favour of taxane‐containing treatments (95% CI 0.86 to 0.96; P < 0.001; Analysis 7.1), with moderate heterogeneity (I² = 32%; P = 0.09).
7.1. Analysis.
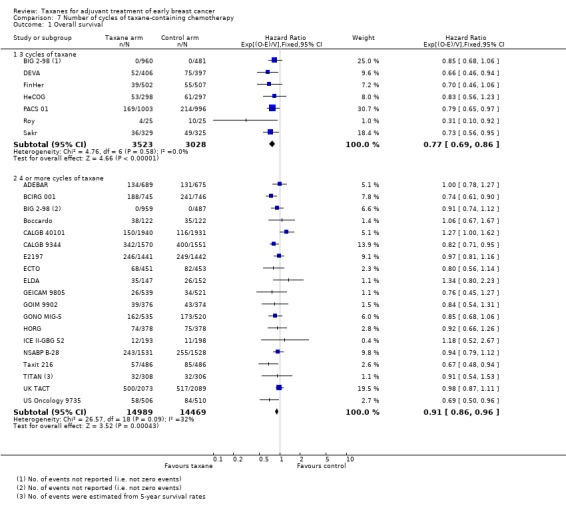
Comparison 7 Number of cycles of taxane‐containing chemotherapy, Outcome 1 Overall survival.
Disease‐free survival
Seven studies (6551 women) used three cycles of taxane treatment in the taxane arm (BIG 2‐98; DEVA; FinHer; HeCOG; PACS 01; Roy; Sakr). Analysis of these studies revealed an HR of 0.80, favouring the taxane‐containing group (95% CI 0.73 to 0.88; P < 0.001; Analysis 7.2), with moderate heterogeneity (I² = 48%; P = 0.07). Twenty‐one studies (32,187 women) with 22 treatment comparisons ‐ ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 40101; CALGB 9344; E2197; ECTO; ELDA; GEICAM 9805; GOIM 9902; GONO MIG‐5; HORG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; RAPP‐01; Taxit 216; TITAN; UK TACT; US Oncology 9735 ‐ used four or more cycles of taxane and also found an HR for DFS of 0.91 in favour of taxane‐containing treatments (95% CI 0.87 to 0.95; P < 0.001; Analysis 7.2), with moderate heterogeneity (I² = 57%; P < 0.001).
7.2. Analysis.
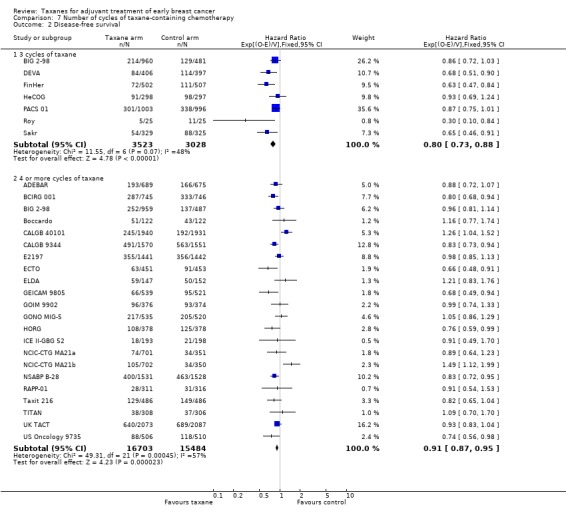
Comparison 7 Number of cycles of taxane‐containing chemotherapy, Outcome 2 Disease‐free survival.
Lymph node status
Variations in inclusion criteria between studies may have had an impact on the risk of recurrence. Analysis was performed to examine whether there was any indication that the benefit of taxane‐containing treatment was greater in studies that included only lymph node‐positive women as compared to studies that included both lymph node‐negative and lymph node‐positive women. It was identified that studies allowing participation of women without lymph node involvement generally required other high‐risk features for inclusion.
Overall survival
Seventeen studies included women with positive axillary lymph node metastasis (22,055 women; 4152 deaths) (ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 9344; DEVA; FinHer; GEICAM 9906; GOIM 9902; GONO MIG‐5; HeCOG; HORG; NSABP B‐28; PACS 01; Roy; Sakr; Taxit 216). Analysis of these studies revealed an HR of 0.83, favouring the taxane‐containing group (95% CI 0.78 to 0.88; P < 0.001; Analysis 8.1), with nominal heterogeneity (I² = 3%; P = 0.41). Seven studies included participants both with and without lymph node metastases (10,269 women; 1952 deaths) (E2197; ECTO; ELDA; ICE II‐GBG 52; TITAN; UK TACT; US Oncology 9735). Analysis of these studies yielded an HR of 0.95, with taxane‐containing groups resulting reporting little to no difference in survival compared to non‐taxane‐containing groups (95% CI 0.87 to 1.04; P = 0.26; Analysis 8.1), with no significant heterogeneity (I² = 12%, P = 0.34). Three studies included women with no lymph node metastases (6856 women; 397 deaths) (CALGB 40101; GEICAM 2003‐02; GEICAM 9805); researchers found an HR of 1.08, indicating that the taxane‐containing treatment group showed little to no difference in survival compared to the control group (95% CI 0.89 to 1.32; P = 0.43; Analysis 8.1), with substantial heterogeneity (I² = 62%; P = 0.07).
8.1. Analysis.
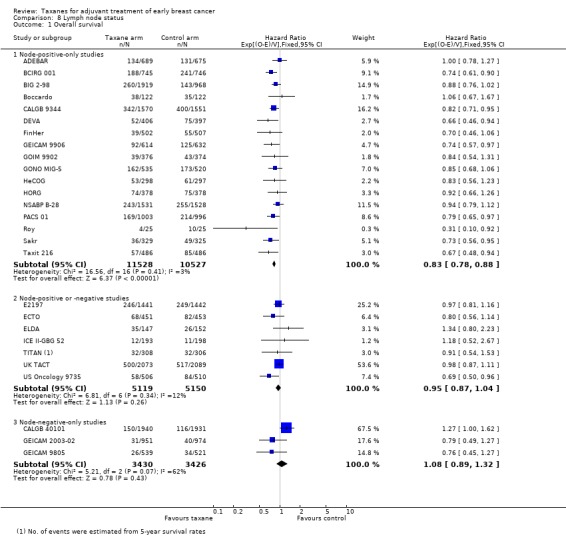
Comparison 8 Lymph node status, Outcome 1 Overall survival.
Disease‐free survival
Seventeen studies included participants with positive axillary lymph node metastasis (22,055 women; 6575 events) (ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 9344; DEVA; FinHer; GEICAM 9906; GOIM 9902; GONO MIG‐5; HeCOG; HORG; NSABP B‐28; PACS 01; Roy; Sakr; Taxit 216). The estimated HR was 0.84, favouring the taxane‐containing group (95% CI 0.80 to 0.88; P < 0.001; Analysis 8.2), with moderate heterogeneity (I² = 32%; P = 0.10). Nine studies with 10 treatment comparisons included women both with and without lymph node involvement (12,998 women; 2929 events) (E2197; ECTO; ELDA; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; RAPP‐01; TITAN; UK TACT; US Oncology 9735). The estimated HR was 0.95 and did not demonstrate a difference in risk of disease progression between taxane and non‐taxane groups (95% CI 0.88 to 1.02; P = 0.15; Analysis 8.2), with moderate heterogeneity (I² = 55%; P = 0.02). Three studies ‐ CALGB 40101; GEICAM 2003‐02; GEICAM 9805 ‐ included only women with negative lymph nodes (6856 women; 767 events); the pooled analysis did not demonstrate a difference in risk of disease progression between taxane‐containing and control chemotherapy regimens, with an estimated HR of 0.99 (95% CI 0.86 to 1.14; P = 0.85; Analysis 8.2), with substantial heterogeneity (I² = 87%; P < 0.001).
8.2. Analysis.
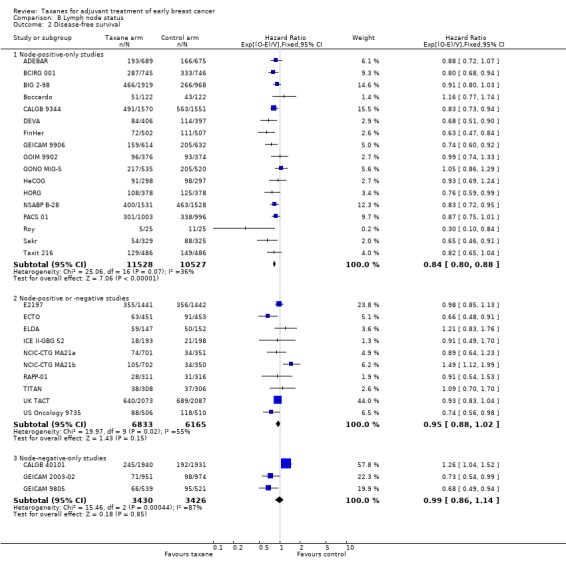
Comparison 8 Lymph node status, Outcome 2 Disease‐free survival.
Hormone receptor status
This post‐hoc analysis examined studies that reported treatment effects by subgroups for hormone receptor status. This analysis was performed to see whether there was any indication of benefit for taxane‐containing regimens in hormone receptor‐positive women as compared to women with hormone receptor‐negative tumours. Fifteen studies, 16 treatment comparisons, did not test or adequately report the effects of taxanes by hormone receptor subgroup for time‐to‐event analysis (ADEBAR; CALGB 40101; ECTO; ELDA; FinHer; GEICAM 2003‐02; GEICAM 9906; HeCOG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; RAPP‐01; Roy; Sakr; Taxit 216).
Overall survival
Five studies reported sufficient data for the subgroups of hormone receptor‐positive and ‐negative status (BCIRG 001; BIG 2‐98; GONO MIG‐5; PACS 01; TITAN). The subgroup of participants with hormone receptor‐positive tumours showed an HR of 0.79, in favour of taxane‐containing treatments (95% CI 0.70 to 0.89; P < 0.001; Analysis 9.1), with no heterogeneity (P = 0.61). The subgroup of participants with hormone receptor‐negative tumours showed that taxane‐containing regimens resulted in little to no difference in survival compared to non‐taxane‐containing regimens (HR 0.88, 95% CI 0.73 to 1.05; P = 0.15; Analysis 9.1), with no heterogeneity (P = 0.56).
9.1. Analysis.
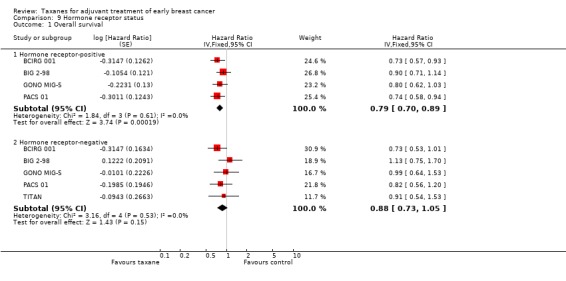
Comparison 9 Hormone receptor status, Outcome 1 Overall survival.
Disease‐free survival
Eleven studies published sufficient data on hormone receptor subgroups (BCIRG 001; BIG 2‐98; Boccardo; CALGB 9344; DEVA; E2197; GEICAM 9805; GOIM 9902; HORG; UK TACT; US Oncology 9735). For the subgroup of participants with hormone receptor‐positive (or oestrogen receptor‐positive only in some studies: DEVA; E2197; GOIM 9902; HORG; UK TACT) tumours, taxane‐containing regimens appeared to reduce the risk of disease recurrence compared to non‐taxane‐containing regimens, with an HR of 0.91 (95% CI 0.85 to 0.97; P = 0.005; 11 studies; 3367 participants; Analysis 9.2), although with substantial heterogeneity (I² = 56%; P = 0.01). The subgroup of participants with hormone receptor‐negative tumours was found to have a similar result in favour of the taxane‐containing regimen, with an HR of 0.80 (95% CI 0.73 to 0.88; P < 0.001; 12 studies; 1581 participants; Analysis 9.2), with no heterogeneity (P = 0.87).
9.2. Analysis.
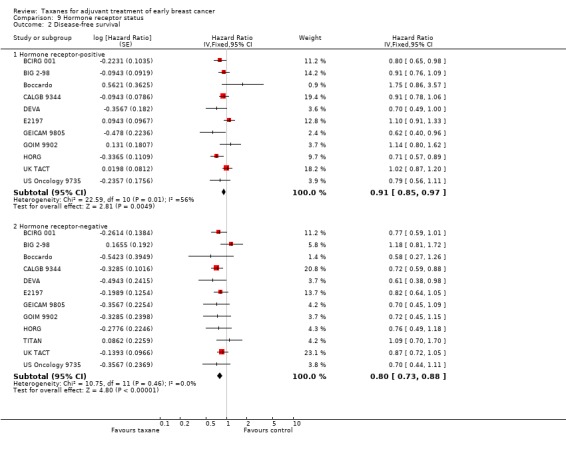
Comparison 9 Hormone receptor status, Outcome 2 Disease‐free survival.
Publication status
For a sensitivity analysis conducted to assess publication bias, studies with fully published efficacy papers were examined as a separate group from those published in non‐peer‐reviewed format only (i.e. abstracts or online theses).
Overall survival
Twenty‐five studies (38,208 women) had a published efficacy paper (ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 40101; CALGB 9344; DEVA; E2197; ECTO; ELDA; FinHer; GEICAM 2003‐02; GEICAM 9805; GEICAM 9906; GOIM 9902; GONO MIG‐5; HeCOG; HORG: NSABP B‐28; PACS 01; Roy; Sakr; TITAN; UK TACT; US Oncology 9735), and the HR of 0.88 favoured taxane‐containing treatment (95% CI 0.84 to 0.92; P < 0.001; Analysis 10.1), with moderate heterogeneity (I² = 33%; P = 0.05). One study (972 women) ‐ Taxit 216 ‐ had data available in an online thesis and indicated that the treatment effect persisted with an HR of 0.67, in favour of taxane‐containing treatment (95% CI 0.48 to 0.94; P = 0.05) (Analysis 10.1).
10.1. Analysis.
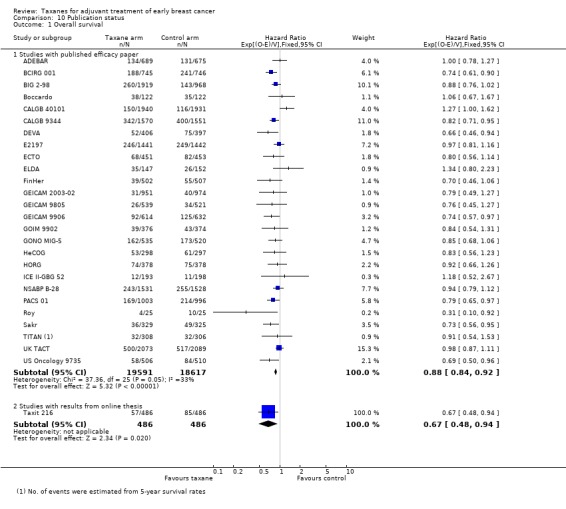
Comparison 10 Publication status, Outcome 1 Overall survival.
Disease‐free survival
Twenty‐seven studies with 28 treatment comparisons (40,310 women) included in this analysis had a published efficacy paper (ADEBAR; BCIRG 001; BIG 2‐98; Boccardo; CALGB 40101; CALGB 9344; DEVA; E2197; ECTO; ELDA; FinHer; GEICAM 2003‐02; GEICAM 9805; GEICAM 9906; GOIM 9902; GONO MIG‐5; HeCOG; HORG; ICE II‐GBG 52; NCIC‐CTG MA21a and NCIC‐CTG MA21b; NSABP B‐28; PACS 01; Roy; Sakr; TITAN; UK TACT; US Oncology 9735), estimating the HR as 0.88, favouring the taxane‐containing group (95% CI 0.85 to 0.92; P < 0.001; Analysis 10.2), with substantial heterogeneity (I² = 62%; P < 0.001). Two studies (1599 women) had data available in abstracts or online theses (RAPP‐01; Taxit 216), reporting an HR for DFS of 0.84 (95% CI 0.67 to 1.04; P = 0.10; Analysis 10.2), with no heterogeneity (P = 0.42).
10.2. Analysis.
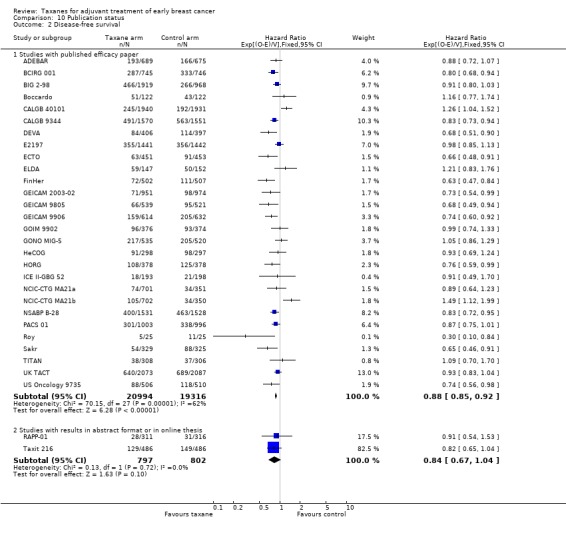
Comparison 10 Publication status, Outcome 2 Disease‐free survival.
A funnel plot did not support any publication bias for the studies reviewed (Figure 5).
5.
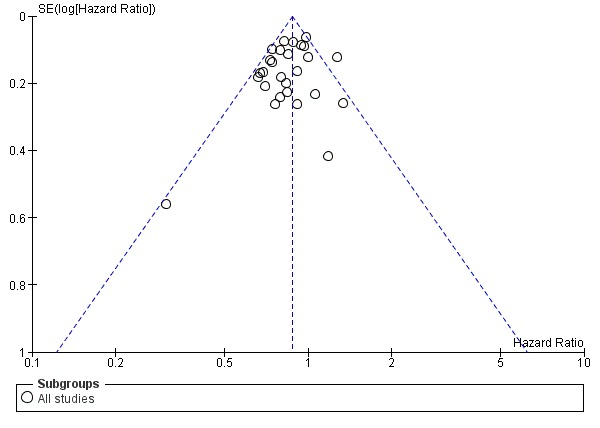
Funnel plot of comparison: 1 Overall effect of taxanes, outcome: 1.1 Overall survival ‐ all studies.
Toxicity
Toxic effects of taxane therapy have been well characterised and were expected to be observed in the taxane‐containing arms. We noted heterogeneity between studies with the use of different control chemotherapies and varying doses and scheduling of the taxane drug. This heterogeneity needs to be considered on a trial‐by‐trial basis to interpret the tolerability of each taxane‐containing regimen. Toxicity data were extracted and combined for analysis.
Of the 29 included studies, 28 provided extractable data on toxicity. No toxicity data could be extracted from Roy studies. Data were extracted and analysed for febrile neutropenia, grade 3 or 4 neuropathy (sensory and motor), grade 3 or 4 fatigue, grade 3 or 4 stomatitis, cardiotoxicity, grade 3 or 4 nausea and/or vomiting, and secondary leukaemia or myelodysplasia. These outcomes are illustrated in Analysis 11 (Analysis 11.1 to Analysis 11.10). Definitions of toxicity varied between studies, and the specific definitions from each study are reported in Table 15. When toxicity was reported as less than 1%, it was treated as 0% for the purposes of statistical comparison. It was not possible to determine the treated population (i.e. only those women receiving chemotherapy) in CALGB 9344. In this case, the randomised population was used for the denominator when the odds ratio was calculated.
11.1. Analysis.
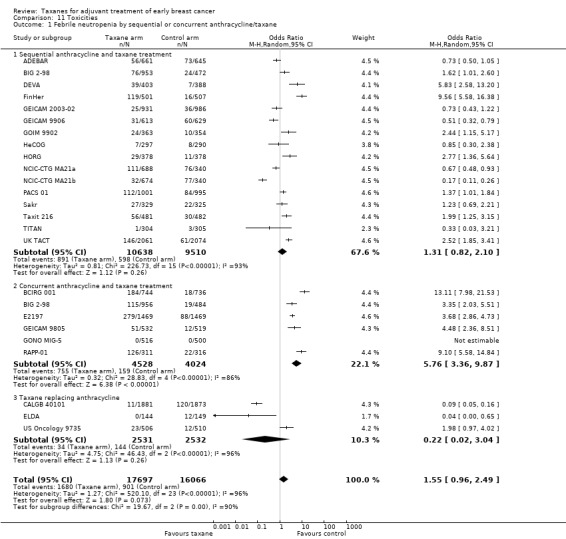
Comparison 11 Toxicities, Outcome 1 Febrile neutropenia by sequential or concurrent anthracycline/taxane.
11.10. Analysis.
Comparison 11 Toxicities, Outcome 10 Treatment‐related deaths.
2. Grouping of toxicity outcomes and description of individual study definitions.
| Trial | Cardiotoxicity | Febrile neutropenia | Neuropathy | Nausea/vomiting | Fatigue | Stomatitis |
| ADEBAR | Grade 3/4 cardiac symptoms: NCI CTC (version 2) | Reported as "febrile neutropenia": NCI CTC (version 2) | Grade 3/4 neurological symptoms: NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | NR | Grade 3/4 mucositis: NCI CTC (version 2) |
| BCIRG 001 | Congestive heart failure (cardiac function grade 3 or 4) | Reported as "febrile neutropenia": Protocol definition ‐ fever of grade 2 or more concomitant with grade 4 neutropenia requiring IV antibiotics, hospitalisation, or both | Grade 3/4 neurosensory effects: NCI CTC (version 1) | Grade 3/4 or severe vomiting: NCI CTC (version 2) | Grade 3/4 asthenia: NCI CTC (version 2) | Grade 3/4 stomatitis: NCI CTC (version 2) |
| BIG 2‐98 | Grade 3/4 cardiac function toxicity | Reported as "febrile neutropenia": protocol defined grade 4 neutropenia, fever with > 38 degrees Celsius, NCI CTC (version 1) | Grade 3/4 neurosensory effects: NCI CTC (version 1) | Grade 3/4 or severe vomiting: NCI CTC (version 1) | Grade 3 or higher asthenia: NCI CTC (version 1) | Grade 3 or higher stomatitis: NCI CTC (version 1) |
| Boccardo | Grade 3/4 cardiotoxicity: WHO toxicity criteria | NR | Grade 3/4 peripheral neuropathy: WHO toxicity criteria | Grade 3/4 nausea and/or vomiting: WHO toxicity criteria | NR | Grade 3/4 stomatitis: WHO toxicity criteria |
| CALGB 40101 | Grade 3 or higher cardiotoxicity (left ventricular systolic dysfunction, restrictive cardiomyopathy, general cardiac, cardiac deaths attributed to protocol treatment ‐ myocardial infarction and left ventricular dysfunction) | Reported as grade 3/4 febrile neutropenia: NCI CTC (version 2) | Grade 3/4 neuropathy (reported both sensory and motor): NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | Grade 3/4 fatigue: NCI CTC (version 2) | NR |
| CALGB 9344 | Congestive heart failure during treatment or post treatment follow‐up | NR | NR | NR | NR | NR |
| DEVA | Diagnosed congestive cardiac failure | Grade 3/4 febrile neutropenia: NCI CTC (version 2) | Grade 3/4 peripheral neuropathy: NCI CTC (version 2) | Grade 3/4 nausea and/or vomiting: NCI CTC (version 2) | NR | Grade 3/4 stomatitis: NCI CTC (version 2) |
| E2197 | Grade 3/4 congestive heart failure | Reported as "febrile neutropenia": toxicity criteria not specified | Grade 3/4 neuropathy (reported both sensory and motor) | Grade 3 or higher vomiting: toxicity criteria not specified | Grade 3 or higher fatigue: toxicity criteria not specified | Grade 3 or higher stomatitis: toxicity criteria not specified |
| ECTO | Symptomatic cardiac failure | NR | Grade 3 neuropathy reported only: NCI CTC (no version provided) | NR | NR | NR |
| ELDA | Grade 3/4 heart rhythm toxicity | Reported as "febrile neutropenia": NCT CTC (version 2) | Grade 3/4 neuropathy: NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | Grade 3/4 fatigue: NCI CTC (version 2) | Grade 3/4 mucositis: NCI CTC (version 2) |
| FinHer | NR | Reported as "neutropenic fever": NCI CTC (version 2) | Grade 3/4 neuropathy (reported both sensory and motor): NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | Grade 3/4 fatigue: NCI CTC (version 2) | Grade 3/4 stomatitis: NCI CTC (version 2) |
| GEICAM 2003‐02 | Grade 3/4 cardiac dysfunction, Grade 4 cardiac ischaemia, Grade 3 arrhythmia | Reported as "febrile neutropenia": NCI CTC (version 2) | Grade 3/4 sensory neuropathy: NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | Grade 3/4 fatigue: NCI CTC (version 2) | Grade 3/4 mucositis: NCI CTC (version 2) |
| GEICAM 9805 | Grade 3/4 arrhythmia, Grade 3/4 congestive heart failure | Reported as "febrile neutropenia": protocol defined fever Grade 2 or higher with Grade 4 neutropenia requiring intravenous antibiotics, hospitalisation, or both | Grade 3/4 peripheral neuropathy (reported both sensory and motor): NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | Grade 3/4 fatigue: NCI CTC (version 2) | Grade 3/4 stomatitis: NCI CTC (version 2) |
| GEICAM 9906 | NR | Reported as "febrile neutropenia" grade 3/4: NCI CTC (version 1) | Grade 3 peripheral sensory neuropathy: NCI CTC (version 1) | Grade 3/4 nausea and vomiting: NCI CTC (version 1) | Grade 3/4 fatigue: NCI CTC (version 1) | Grade 3/4 mucositis: NCI CTC (version 1) |
| GOIM 9902 | Reversible cardiotoxicity | Reported as "neutropenic fever" grade 3/4: NCI CTC (version 3) | Grade 3/4 neurotoxicity: NCI CTC (version 3) | Grade 3/4 nausea and/or vomiting: NCI CTC (version 3) | NR | Grade 3/4 mucositis: NCI CTC (version 3) |
| GONO MIG‐5 | Grade 3/4 cardiotoxicity: WHO criteria | Reported as "febrile neutropenia" grade 3/4: WHO criteria | Grade 3/4 neurological toxicity: WHO criteria | Grade 3/4 nausea and vomiting: WHO criteria | NR | Grade 3/4 stomatitis: WHO criteria |
| HeCOG | NR | Reported as "febrile neutropenia": WHO toxicity criteria | Grade 3/4 peripheral neuropathy: WHO toxicity criteria | Grade 3/4 nausea and/or vomiting: WHO toxicity criteria | Grade 3/4 fatigue: WHO toxicity criteria | NR |
| HORG | Grade 3/4 cardiotoxicity | Reported as "febrile neutropenia" grade 3/4: NCI CTC (version 2) | Grade 3/4 neurotoxicity: NCI CTC (version 2) | Grade 3/4 nausea: NCI CTC (version 2) | Grade 3/4 asthenia: NCI CTC (version 2) | Grade 3/4 stomatitis: NCI CTC (version 2) |
| ICE II‐GBG 52 | Grade 3 to 5 congestive heart failure and cardiac ischaemia | Reported as "febrile neutropenia" grade 1 to 5: NCI CTC (version 3) | Grade 3 to 5 sensory neuropathy: NCI CTC version 3 | Grade 3 to 5 vomiting: NCI CTC (version 3) | Grade 3 to 5 fatigue: NCI CTC (version 3) | Grade 3 to 5 mucositis, stomatitis, oesophagitis: NCI CTC (version 3) |
| NCIC‐CTG MA21a | Grade 3/4 cardiotoxicity | Reported as "febrile neutropenia" grade 3/4: NCI CTC (version 2) | Grade 3/4 neuropathy (reported both sensory and motor): NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | NR | Grade 3/4 stomatitis: NCI CTC (version 2) |
| NCIC‐CTG MA21b | Grade 3/4 cardiotoxicity | Reported as "febrile neutropenia" grade 3/4: NCI CTC (version 2) | Grade 3/4 neuropathy (reported both sensory and motor): NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | NR | Grade 3/4 stomatitis: NCI CTC (version 2) |
| NSABP B‐28 | Grade 3 or higher cardiac dysfunction | NR | NR | NR | NR | NR |
| PACS 01 | Any serious adverse cardiac event | Reported as "febrile neutropenia": WHO toxicity criteria | NR | Grade 3/4 nausea and/or vomiting: WHO toxicity criteria | NR | Grade 3/4 stomatitis: WHO toxicity criteria |
| RAPP‐01 | NR | Reported as "febrile neutropenia" defined as any Grade 3/4 neutropenia plus fever (> 38 degrees) requiring antibiotics: NCI CTC (version 2) | NR | Grade 3/4 nausea and/or vomiting: NCI CTC (version 2) | NR | Grade 3/4 mucositis: NCI CTC (version 2) |
| Sakr | Grade 3/4 cardiac event | Reported as "febrile neutropenia" Grade 3/4: WHO toxicity criteria | NR | Grade 3/4 nausea‐vomiting: WHO toxicity criteria | NR | Grade 3/4 stomatitis: WHO toxicity criteria |
| UK TACT | NR | Reported as "febrile neutropenia" Grade 3/4: NCI CTC (version 2) | Grade 3/4 neuropathy: NCI CTC (version 2) | Grade 3/4 nausea and/or vomiting: NCI CTC (version 2) | Grade 3/4 lethargy: NCI CTC (version 2) | Grade 3/4 stomatitis (mucositis): NCI CTC (version 2) |
| Taxit 216 | Cardiac function toxicity | Reported as "febrile neutropenia": NCI CTC (version 2) | Grade 3/4 neuropathy (reported both sensory and motor): NCI CTC (version 2) | Grade 3/4 vomiting: NCI CTC (version 2) | Grade 3/4 asthenia: NCI CTC (version 2) | Grade 3/4 stomatitis: NCI CTC (version 2) |
| TITAN | Cardiac events | Reported as "febrile neutropenia": NCI CTC (version 3) | Grade 3/4 peripheral neuropathy: NCI CTC (version 3) | Grade 3/4 nausea (vomiting not reported): NCI CTC (version 3) | Grade 3/4 fatigue: NCI CTC (version 3) | NR |
| US Oncology 9735 | Death due to cardiac event | Reported as "febrile neutropenia" Grade 3/4 : NCI CTC (version 1) | NR | Grade 3/4 vomiting: NCI CTC (version 1) | Grade 3/4 asthenia: NCI CTC (version 1) | Grade 3/4 stomatitis: NCI CTC (version 1) |
NCI CTC: National Cancer Institute Common Toxicity Criteria NR: not reported WHO: World Health Organziation
Febrile neutropenia
Twenty‐four studies with 25 treatment comparisons provided data on febrile neutropenia. Other myelosuppression toxicity data were reported in multiple studies (grade 3 to 4 neutropenia, grade 3 to 4 infection, and infection requiring antibiotics); however, febrile neutropenia was the most consistently reported outcome. Pooled analysis of these studies found that taxane‐containing regimens probably resulted in a small increase in risk of febrile neutropenia compared to non‐taxane‐containing regimens (OR 1.55, 95% CI 0.96 to 2.49; P = 0.07; 33,763 participants; 24 studies (25 treatment comparisons); moderate‐certainty evidence; Analysis 11.1; Figure 6). The risk was highest for studies that administered the taxane concurrently with an anthracycline (OR 5.76, 95% CI 3.36 to 9.87; P < 0.001; 8552 women; 6 studies; Analysis 11.1), with substantial heterogeneity (I² = 86%; P < 0.001) rather than sequential taxane and anthracycline treatment (OR 1.31, 95% CI 0.82 to 2.10; P = 0.26; 20,148 women; 15 studies (16 treatment comparisons); Analysis 11.1), also with substantial heterogeneity (I² = 93%; P < 0.001). There was little to no difference in the risk of febrile neutropenia among studies that compared a taxane replacement for an anthracycline versus control (OR 0.22, 95% CI 0.02 to 3.04; P = 0.26; 5063 women; 3 studies; Analysis 11.1), and heterogeneity was substantial (I² = 96%; P < 0.001). The test for differences between subgroups was significant (P < 0.001).
6.
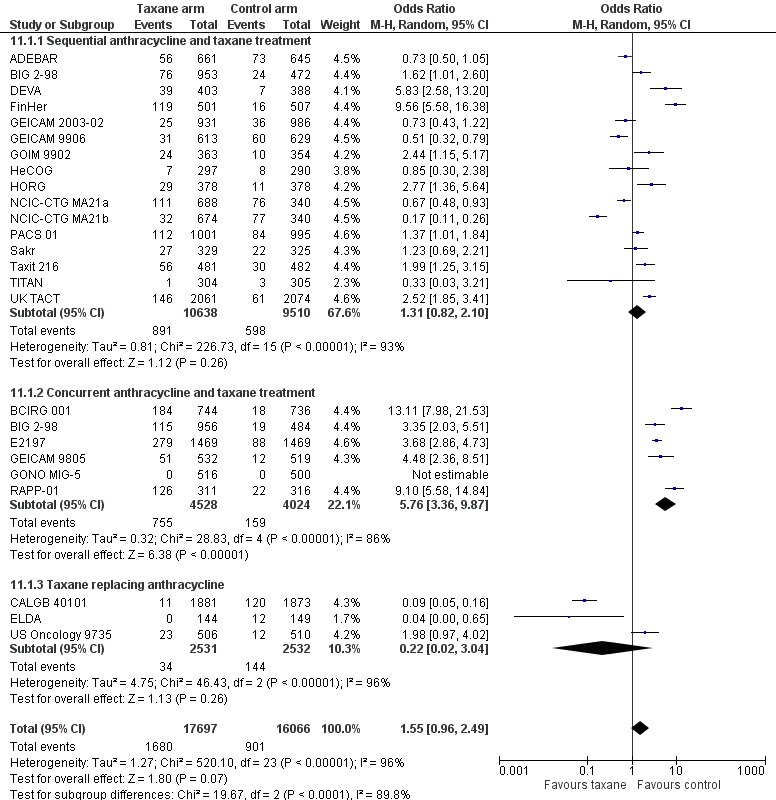
Forest plot of comparison: 11 Toxicities, outcome: 11.1 Febrile neutropenia by sequential or concurrent anthracycline/taxane.
A subgroup analysis indicated that increased risk of febrile neutropenia for taxane‐containing regimens (compared to non‐taxane regimens) appeared to be driven by docetaxel rather than paclitaxel (Analysis 11.2). Docetaxel‐containing regimens were likely to increase the risk of febrile neutropenia compared to non‐taxane regimens (OR 2.83, 95% CI 1.88 to 4.27; P < 0.001; 22,596 women; 16 studies; significant heterogeneity: I² = 92%; P < 0.001; Analysis 11.2.1), and paclitaxel‐containing regimens were likely to reduce the risk of febrile neutropenia compared to non‐taxane regimens (OR 0.36, 95% CI 0.19 to 0.67; P < 0.001; 11,558 women; 8 studies (9 treatment comparisons); substantial heterogeneity (I² = 88%; P < 0.001). The test for differences between subgroups was significant (P < 0.001).
11.2. Analysis.
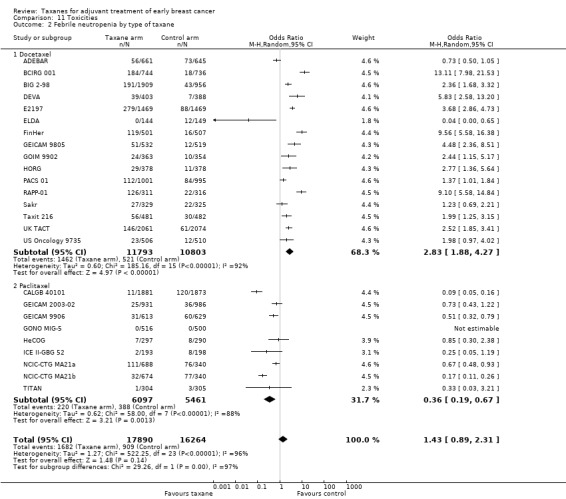
Comparison 11 Toxicities, Outcome 2 Febrile neutropenia by type of taxane.
Grade 3/4 neuropathy
Twenty‐two studies with 23 treatment comparisons reported grade 3 or 4 neuropathy. Of these, four studies provided data on neurosensory neuropathy only, six studies reported peripheral neuropathy only, seven studies reported sensory and motor neuropathy separately, and six reported neurotoxicity (no further specifics provided). Taxane‐containing regimens likely resulted in a large increase in neuropathy compared to controls (OR 6.89, 95% CI 3.23 to 14.71; P < 0.001; 31,033 women; moderate‐certainty evidence; Analysis 11.3), with substantial heterogeneity (I² = 82%; P < 0.001).
11.3. Analysis.
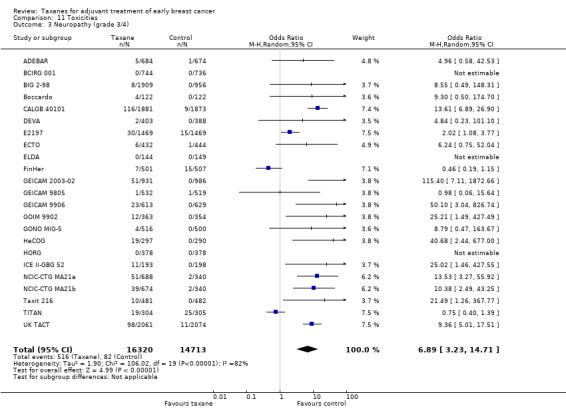
Comparison 11 Toxicities, Outcome 3 Neuropathy (grade 3/4).
A subgroup analysis suggested that the increase in risk of grade 3 or 4 neuropathy for taxane‐containing regimens compared to non‐taxane regimens tended to be worse for women receiving paclitaxel than docetaxel (Analysis 11.4); however the confidence intervals were very wide. For women receiving paclitaxel, the OR was 11.93 (95% CI 3.59 to 39.70; 10 studies (11 treatment comparisons); 12,678 women; substantial heterogeneity; I² = 85%; P < 0.001; Analysis 11.4.2), and for docetaxel, the OR was 3.74 (95% CI 1.33 to 10.53; 11 studies, 18,355 women; substantial heterogeneity; I² = 79%; P < 0.001; Analysis 11.4.1). The test for difference between subgroups was not statistically significant (P = 0.15).
11.4. Analysis.
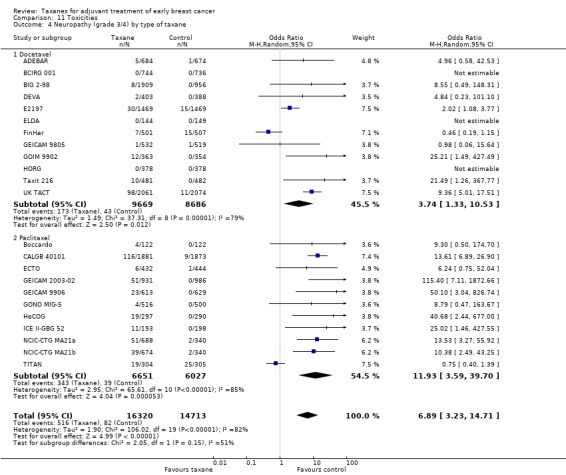
Comparison 11 Toxicities, Outcome 4 Neuropathy (grade 3/4) by type of taxane.
Grade 3/4 fatigue
Sixteen studies were pooled for analysis of grade 3 or 4 fatigue. Taxane‐containing regimens likely increased the risk of fatigue compared to non‐taxane‐containing regimens (OR 1.81, 95% CI 1.31 to 2.49; P < 0.001; 25,003 women; Analysis 11.5), with substantial heterogeneity (I2 = 83%; P < 0.001).
11.5. Analysis.
Comparison 11 Toxicities, Outcome 5 Fatigue (grade 3/4).
Grade 3/4 stomatitis
Twenty‐two studies with 23 treatment comparisons assessed grade 3 or 4 stomatitis. Pooled analysis of these studies revealed that taxane‐containing regimens likely resulted in little to no difference in stomatitis compared to controls (OR 1.29, 95% CI 0.93 to 1.78; P = 0.12; 22,648 women; Analysis 11.6), with substantial heterogeneity (I² = 81%; P < 0.001).
11.6. Analysis.
Comparison 11 Toxicities, Outcome 6 Stomatitis (grade 3/4).
A subgroup analysis indicated that the docetaxel‐containing regimens were likely to increase the risk of stomatitis compared to control regimens (OR 1.73, 95% 1.28 to 2.35; 16 studies; 22648 participants), with substantial heterogeneity (I² = 73%; P < 0.001), and paclitaxel‐containing regimens were likely to result in little to no difference in grade 3/4 stomatitis compared to control regimens (OR 0.64, 95% CI 0.31 to 1.32; 7 studies; 6852 participants), with substantial heterogeneity (I² = 81%; P < 0.001). The test for difference between subgroups was significant (P = 0.01).
Cardiotoxicity
Twenty‐three studies provided extractable data on cardiotoxicity. Pooled analysis of these studies indicated that administering taxane‐containing regimens probably resulted in little to no difference in cardiotoxicity compared to non‐taxane‐containing regimens (OR 0.87, 95% CI 0.56 to 1.33; 32,894 participants; 23 studies; moderate‐quality evidence; Analysis 11.7). When the same planned dose of anthracycline was used in the taxane‐containing arm and in the control arm, there was no difference in the risk of cardiotoxicity between groups (HR 1.27, 95% CI 0.88 to 1.84; 14,967 women; 9 studies; Analysis 11.7), with little heterogeneity (I² = 24%; P = 0.24). Risk of cardiotoxicity was reduced in studies that provided a lower planned dose of anthracycline in the taxane arm than in the control arm (OR 0.39, 95% CI 0.18 to 0.86; 12,473 women; 10 studies; Analysis 11.7), with little heterogeneity (I² = 24%; P = 0.22). Four studies were designed with the taxane replacing the anthracycline and showed little to no difference in cardiotoxicity between treatment groups (OR 1.01, 95% CI 0.26 to 3.91; 5454 women; 4 studies; Analysis 11.7), with moderate heterogeneity (I² = 58%; P = 0.07). The test for differences between subgroups was significant (P = 0.03).
11.7. Analysis.
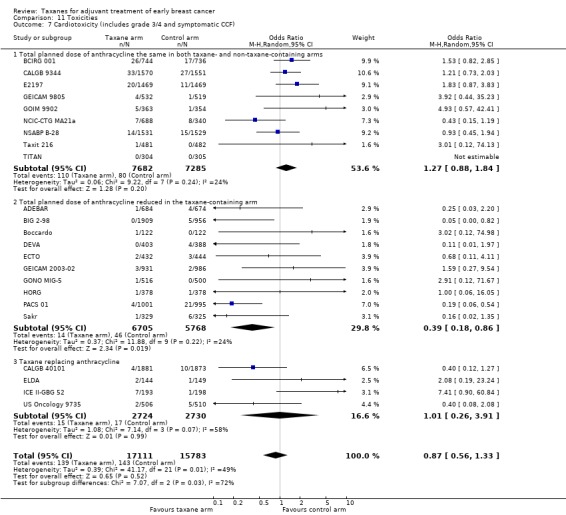
Comparison 11 Toxicities, Outcome 7 Cardiotoxicity (includes grade 3/4 and symptomatic CCF).
Grade 3/4 nausea and/or vomiting
Twenty‐five studies with 26 treatment comparisons were pooled for analysis of grade 3 or 4 nausea and/or vomiting. If studies did not report nausea/vomiting, we included data related to vomiting only if reported (refer to Table 15). Administration of taxane‐containing regimens likely resulted in little to no difference in nausea/vomiting compared to non‐taxane‐containing regimens (OR 0.83, 95% CI 0.67 to 1.04; 34,450 women; moderate‐certainty evidence; Analysis 11.8), with substantial heterogeneity (I² = 77%; P < 0.001).
11.8. Analysis.
Comparison 11 Toxicities, Outcome 8 Nausea and/or vomiting (grade 3/4).
Secondary leukaemia or myelodysplasia
A total of 86 cases of secondary leukaemia or myelodysplasia were reported from 18 studies, with 19 treatment comparisons: 39 cases from taxane‐containing regimens and 47 from control regimens. There was little to no difference in the risk of developing secondary leukaemia or myelodysplasia between groups (OR 0.85, 95% CI 0.54 to 1.33; 33,225 women; Analysis 11.9), with no heterogeneity (P = 0.62).
11.9. Analysis.
Comparison 11 Toxicities, Outcome 9 Secondary leukaemia/myelodysplasia.
Two studies reported on the development of other second malignancies. In PACS 01, 14 women in the taxane‐containing arm and 20 women in the control arm developed a second cancer, and in HeCOG, this occurred in five women from the taxane‐containing arm and in four women from the control arm.
Treatment‐related death
Treatment‐related death was uncommon for both taxane‐containing and non‐taxane‐containing groups. Treatment‐related deaths were defined in the trials as "toxic death", "treatment‐related death", and "death occurring during treatment or within 30 days post‐treatment". In all, 68 treatment‐related deaths were recorded from 22 studies during chemotherapy: 35 from taxane‐containing regimens and 33 from control regimens. Administering taxane‐containing regimens resulted in little to no difference in treatment‐related deaths compared to non‐taxane‐containing regimens (OR 1.24, 95% CI 0.63 to 2.47; 34,882 women; 22 studies; Analysis 11.10), with little heterogeneity (I² = 26%, P = 0.17). One trial reported deaths without describing the causes (three women; CALGB 9344). Several studies reported additional deaths but excluded these as they were not considered related to treatment.
For the purposes of this review, we have focused on the aforementioned toxicities commonly reported across most trials. Other reported toxicities ranged from grade 3/4 myalgia or arthralgia, anaemia, allergy, or oedema, to neurotoxicity, but reporting on the frequency of all of these toxicities was beyond the remit of this review.
Quality of life
Seven studies reported low‐certainty evidence for quality of life (QoL) (ADEBAR; BCIRG 001; DEVA; ELDA; GEICAM 9805; HeCOG; UK TACT); details of these findings are summarised in Table 16. NCIC‐CTG MA21 ‐ NCIC‐CTG MA21a and NCIC‐CTG MA21b ‐ stated that QoL data would be reported in a separate article. BCIRG 001 demonstrated a transient reduction in both treatment arms; the reduction in QoL score was greater in the taxane‐containing regimen, but by first follow‐up, both treatment arms had returned to baseline. Similarly, in ADEBAR, both groups had decreased scores on the EORTC questionnaire used to assess quality of life in cancer patients (EORTC‐C30), and changes in scores on the breast cancer‐specific EORTC QoL questionnaire (EORTC BR23) over time were similar in the two groups. DEVA and HeCOG did not report any differences in QoL scores between treatment arms either at the beginning or at the end of chemotherapy. ELDA reported no differences in global QoL scores, functioning scales, and other items; however, there was worsening of systemic therapy side effects in the docetaxel group compared to the CMF group at the end of one or more cycles (ELDA). GEICAM 9805 reported decreased QoL in the taxane‐containing group compared to the control group, but this was resolved by week 44, and there were no significant differences between groups during the follow‐up period. UK TACT noted a reduction in QoL global, physical, emotional functioning, social functioning, and fatigue scores compared to the control group; more nausea and vomiting was reported in the control group than in the taxane‐containing group.
3. Quality of life.
| Trial ID | Instruments used | Summary of findings |
| ADEBAR | Patients completed EORTC QLQ‐C30 and the breast cancer‐specific QLQ‐BR23 at baseline, before each course of chemotherapy, and at 4 weeks, 6 weeks, and 6 months after completion of chemotherapy | Both treatment groups had decreased EORTC‐C30 scores for physical, role, emotional, social, and cognitive functioning from baseline to 28 days after chemotherapy. Global health scores also decreased in this time frame for both groups. Changes in symptom scores were generally similar between treatment groups with the exception of nausea/vomiting and pain scores. Nausea/vomiting was worse for the FEC120 group than for the EC‐Doc group (+9.42 above baseline vs +1.88), and pain scores were worse in the EC‐Doc group (+9.18 for EC‐Doc vs ‐1.61 for FEC120). Changes in items on the EORTC BR23 over time were quantitatively and qualitatively similar in the 2 treatment groups |
| BCIRG 001 | Patients completed EORTC QLQ‐C30 (version 2.0) and the breast cancer‐specific QLQ‐BR23 (version 1.0) questionnaires at baseline, before cycles 3 and 5, at 3 to 4 weeks after completion, and at 6, 12, and 24 months | Both FAC and TAC arms led to a transient and statistically significant reduction in QoL scores, which returned to baseline levels at first follow‐up visit. QoL measures were similar between groups and were similar to baseline measures at 6 months and at the end of 2 years |
| CALGB 40101 | QoL results designed to study short‐ and long‐term toxicities on quality of life by treatment agent and duration. No further details provided | QoL to be reported in a companion study |
| DEVA | Patients completed EORTC QLQ‐C30 and the breast cancer‐specific QLQ‐BR23 in the clinic at baseline and 9 months, 2 years, and 5 years after random assignment | No significant difference in overall QoL or across any of the scales between treatments at 9 months, 2 years, or 5 years |
| ELDA | Patients completed EORTC QLQ‐C30 and the breast cancer‐specific QLQ BR23 at baseline, and at the end of the first, second, and third cycles of chemotherapy | No difference reported for QoL global functioning scales and other items. However there was worsening of systemic therapy side effects, future perspective, nausea and vomiting, diarrhoea, appetite loss, and upset by hair loss and body experience with docetaxel compared to CMF at the end of 1 or more cycles |
| GEICAM 9805 | Patients completed EORTC QLQ‐C30 and the breast cancer‐specific QLQ‐BR23 at baseline after each chemotherapy cycle and at 6, 12, and 24 months after completion of chemotherapy | During chemotherapy, QoL decreased and was worse with TAC than with FAC, but this effect resolved by week 44. There were no statistically significant differences in QoL between TAC+G‐CSF and FAC after cycle 6 or during further follow‐up |
| GONO MIG‐5 | Trial registry record indicates that QoL will be compared across treatment arms. No further details provided | QoL information not yet reported |
| HeCOG | Patients completed EORTC QLQ‐C30 questionnaire at baseline and on completion of chemotherapy | Only 23% of participants (72 participants in E‐T‐CMF and 67 participants in E‐CMF) completed baseline and end of chemotherapy questionnaires. There was no difference between the 2 treatment arms at the beginning or at the end of chemotherapy. Significant increase in nausea and vomiting for both groups. Social functioning significantly decreased in the taxane‐containing arm, and emotional functioning and pain significantly improved in the control arm |
| NCIC‐CTGMA21 (NCIC‐CTG MA21a; NCIC‐CTG MA21b) | Patients completed EORTC QLQ‐C30 and the breast cancer‐specific QLQ‐BR within 7 days before random assignment and afterwards | QoL results will be reported in a separate article |
| UK TACT | Selected centres invited participants to quality of life substudy. These patients completed EORTC QLQ‐C30, QLQ‐BR23, and Hospital Anxiety and Depression Scale (HADS) before randomisation, before fifth and after eighth cycles, and at 9, 12, 18, and 24 months post chemotherapy | FEC‐D was associated with worse global QoL (P = 0.001), physical (P < 0.0001), role (P = 0.002), emotional functioning (P = 0.008), social functioning (P = 0.003), pain (P = 0.001), and fatigue (P = 0.006) compared to control. In contrast, there was more nausea and vomiting in the control group (P = 0.01) than in the FEC‐D group |
CMF: cyclophosphamide, methotrexate and fluorouracil E‐CMF: epirubicin‐ cyclophosphamide, methotrexate and fluorouracil EC‐Doc: epirubicin, cyclophosphamide‐docetaxel EORTC QLQ‐C30: European Organisation for Research and Treatment of Cancer (EORTC) core Quality of Life Questionnaire C30 (QLQ‐C30) QLQ‐BR23: breast cancer‐specific Quality of Life Questionnaire BR23 E‐T‐CMF: epirubicin‐paclitaxel‐cyclophosphamide, methotrexate, fluorouracil FAC: fluorouracil, doxorubicin, cyclophosphamide FEC‐D: fluorouracil, epirubicin, cyclophosphamide ‐ docetaxel TAC: docetaxel, doxorubicin, cyclophosphamide
Cost‐effectiveness
One study presented data on cost‐effectiveness, finding that the extra cost of anthracycline plus docetaxel treatment compared to anthracycline alone was offset by the lower rate of recurrence in the anthracycline plus docetaxel group (PACS 01). The cost per quality‐adjusted life‐year (QALY) gained by adding the taxane was reported to be €2372 (upper CI €55,515). This value was reported to be a cost‐effective alternative.
Sensitivity analysis
Low versus high or unclear risk of bias
Post‐hoc subgroup analyses were conducted to investigate treatment effects in studies with low risk of bias compared to studies with unclear/high risk of bias. Of the 29 studies, 23 studies (24 treatment comparisons) were considered to be at low risk overall. Six studies were grouped as having unclear or high risk of bias overall (FinHer; GEICAM 9906; PACS 01; Roy; Sakr; US Oncology 9735).
Overall survival
Analysis of low risk of bias studies demonstrated an HR of 0.90, favouring the taxane‐containing group (95% CI 0.86 to 0.95; P < 0.001; Analysis 12.1), with moderate heterogeneity (I² = 28%; P = 0.12). For the six studies categorised as having unclear or high risk of bias, the HR favoured taxane‐containing regimens (HR 0.74, 95 CI 0.65 to 0.83; P < 0.001; Analysis 12.1), with no heterogeneity (P = 0.68).
12.1. Analysis.
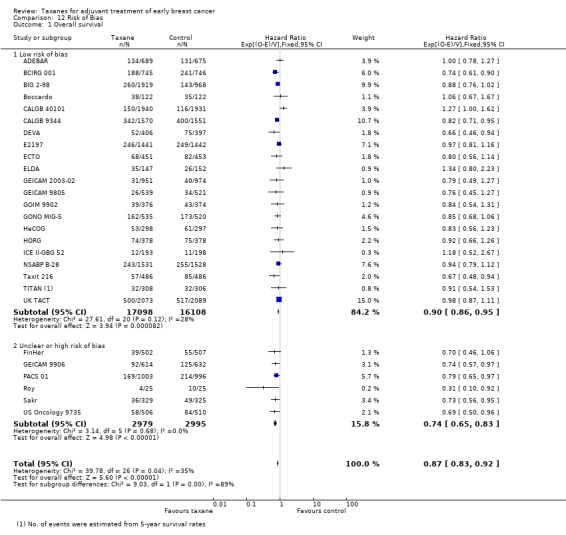
Comparison 12 Risk of Bias, Outcome 1 Overall survival.
Disease‐free survival
Twenty‐three low risk of bias studies (24 treatment comparisons) had data available for this outcome and showed an HR of 0.90 in favour of taxane‐containing regimens (95% CI 0.87 to 0.94; P < 0.001; 35,935 women; Analysis 12.2). Heterogeneity was moderate (I² = 57%; P < 0.001). For the six studies judged as having unclear or high risk of bias, an HR of 0.76 favoured taxane‐containing regimens (95% CI 0.69 to 0.84; P < 0.001; 5974 women), with moderate heterogeneity (I² = 42%; P = 0.12).
12.2. Analysis.
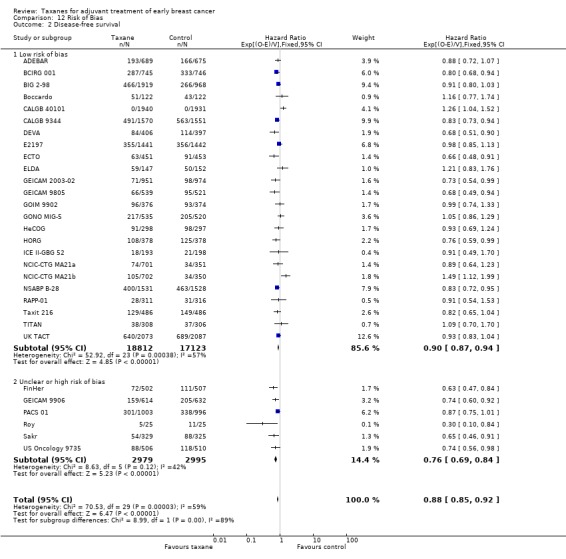
Comparison 12 Risk of Bias, Outcome 2 Disease‐free survival.
Discussion
Summary of main results
This review update provides high‐quality evidence supporting the conclusion that use of a taxane drug as part of the adjuvant chemotherapy regimen following surgery for early‐stage breast cancer in women with moderate to high risk of recurrence leads to improvement in overall survival (OS) and disease‐free survival (DFS) (see Table 1). Among these women, the hazard ratio for OS was 0.87, and for DFS 0.88, favouring use of a taxane as part of the adjuvant chemotherapy regimen when compared to adjuvant regimens that did not contain a taxane.
This review included 29 studies (totaling 41,911 randomised women, 6501 deaths, and 10,271 DFS events), which is an adequate number to establish conclusive results. With the addition of 17 new studies and updated follow‐up data from previously included studies, little evidence suggests that the hazard ratio will change over time. We identified three ongoing studies, and there may be further unpublished studies that have not been identified for this review update. It is possible that publication bias in favour of significant findings may have led to an overestimation of the overall treatment effect; however, the funnel plot does not support this.
Overall, taxane‐containing regimens were well tolerated but increased the risk of some adverse events. Moderate‐quality evidence shows that there was a difference in toxicity between taxane‐containing regimens and non‐taxane‐containing regimens. This is not unexpected based on established toxicity profiles for the taxane drugs. A small increase in rates of febrile neutropenia, neuropathy, and fatigue was evident for the taxane‐containing regimens. Researchers have noted no significant differences in the risk of developing cardiotoxicity, stomatitis, nausea and/or vomiting, secondary haematological or other second cancers, nor treatment‐related death. Less cardiotoxicity was seen with taxane‐containing regimens that employed less anthracycline than was seen with the control intervention. Seven studies reported quality of life (QoL) results. Overall, low‐quality evidence suggests no differences in QoL between groups during follow‐up.
The subgroup analysis looked at the relative efficacy for each type of taxane. For docetaxel and paclitaxel, the hazard ratio for OS was 0.86 (range 0.81 to 0.92) and 0.89 (range 0.82 to 0.96), respectively; and for DFS the hazard ratio was 0.87 (range 0.82 to 0.91) and 0.91 (range 0.85 to 0.96), respectively. No conclusions can be drawn about the relative efficacy of the two agents without a direct comparison. Such direct comparisons are underway, and some results have been published.
Overall completeness and applicability of evidence
This review provides preliminary evidence for restricting adjuvant taxane chemotherapy to lymph node‐positive women and shows that efficacy appeared equivalent in trials that included both node‐positive and node‐negative groups and only node‐negative groups. The absolute recurrence risk for an individual patient must be considered when one is making clinical decisions on the use of a taxane‐containing adjuvant chemotherapy regimen. Limited evidence suggests that taxane chemotherapy restricted to women with hormone receptor‐positive breast cancer could provide additional benefit in terms of survival, but for disease‐free survival, efficacy in trials of hormone receptor‐positive and hormone receptor‐negative groups is unclear. Further questions that are beyond the scope of this review include efficacy in women with more than four involved axillary lymph nodes and the role of human epidermal growth factor receptor 2 (HER2) in breast cancer treatment.
Quality of the evidence
This updated review contains 29 included studies involving over 41,000 women. High‐quality evidence supports the use of taxane‐containing regimens in the adjuvant setting with an increase in survival time and in time free of disease recurrence. Some clinical heterogeneity between studies was evident, with variation in choice of control chemotherapy, doses, and treatment scheduling. Post‐hoc analyses in a side‐by‐side comparison of pooled data were performed for several subgroups. Benefit appears to be no less when the taxane is substituted for part of the control regimen as opposed to adding it to the control regimen. This is also the case when taxane regimens of the same duration are compared with regimens of longer duration than the control regimen, when three cycles of taxane treatment rather than four or more are compared, and when sequential anthracycline and taxane combination is given rather than concurrent administration.
Potential biases in the review process
For the review update, trialists were contacted if data were not fully reported in the full‐text article or if no information aside from a conference proceeding abstract was available. We tested whether publication status (i.e. full text vs non‐peer‐reviewed information) had an impact on the effect estimate. It was reassuring that benefit from taxanes in the adjuvant setting persisted when data for overall survival were analysed; however, this benefit was not sustained for disease‐free survival, with data from abstracts, unpublished manuscripts, or online theses (related to four studies) showing no added benefit from taxanes compared to control interventions.
Agreements and disagreements with other studies or reviews
Results of this meta‐analysis are in accordance with the findings of previous meta‐analyses (Bria 2006; Qin 2011). The Bria 2006 meta‐analysis included nine studies with 15,598 and 15,074 women analysed for DFS and OS, respectively. Bria 2006 reported the risk ratios for DFS and OS as 0.86 (95% CI 0.81 to 0.90) and 0.87 (95% CI 0.81 to 0.93), in favour of the taxane‐containing treatment group (Bria 2006). The meta‐analysis by Bria et al included the MD Anderson CC trial, which was excluded from this Cochrane Review as published efficacy data did not distinguish results for adjuvant patients from those for neoadjuvant patients. Nineteen additional trials were included in the Cochrane Review, which provides even stronger supporting evidence for the efficacy of taxane‐containing regimens. Similarly, Qin 2011 reported a reduction in the number of deaths and in risk of disease recurrence for women receiving adjuvant taxane‐containing chemotherapy compared to those given chemotherapy without taxanes, and reported similar toxicity profiles as we have provided in our updated Cochrane Review.
Authors' conclusions
Implications for practice.
High‐certainty evidence supports the conclusion that use of a taxane drug as part of the adjuvant chemotherapy regimen following surgery for early‐stage breast cancer improves both overall survival and disease‐free survival in women with moderate to high risk of recurrence. Despite considerable heterogeneity across studies, taxane‐containing regimens provided benefit for survival and disease‐free survival compared to non‐taxane‐containing chemotherapy. A taxane‐containing regimen should be considered for women in this situation following assessment of individual risk of recurrence and comorbidities. Additional toxicity is associated with use of a taxane‐containing regimen. Toxicity implications should be discussed with individual women who are considering taxane‐based adjuvant chemotherapy regimens. Review authors found a paucity of studies examining effects of taxane‐containing chemotherapy on patient‐reported quality of life.
Implications for research.
A next generation of studies is required to further define the precise role of taxanes as adjuvant chemotherapy for early breast cancer. Future randomised trials comparing docetaxel with paclitaxel; addressing questions of dose density, scheduling, and duration; and looking at how best to combine taxane and anthracycline‐based treatment, and how to combine taxanes with trastuzumab in women with HER2‐positive disease will help to answer these questions. In many of the studies included in this review version, tumour profiling was not conducted at the time this review commenced. As this review update reported results that were remarkably consistent with those presented in the original review, even with the addition of over 20,000 women, it is highly unlikely that future studies will change the key findings; therefore we do not plan to update this review in the future. Instead, a new review topic would be warranted to assess taxane treatment based on detailed knowledge of the breast cancer subtype and to collect data related to toxicities and quality of life over the long term.
What's new
| Date | Event | Description |
|---|---|---|
| 11 September 2019 | Review declared as stable | It is highly unlikely that future studies will change the key findings of this review, therefore we do not plan to update this review topic. Instead, a new review topic would be warranted to assess taxane treatment based on detailed knowledge of the breast cancer subtype and to collect data related to toxicities and quality of life over the long term. |
History
Protocol first published: Issue 4, 2003 Review first published: Issue 4, 2007
| Date | Event | Description |
|---|---|---|
| 16 July 2018 | New citation required but conclusions have not changed | 17 new studies included, adding 20,720 participants |
| 16 July 2018 | New search has been performed | Search for new studies performed on 16 July 2018 |
| 3 September 2010 | Amended | Changes made to the 'Summary of findings' table |
| 13 May 2008 | Amended | Review converted to new review format |
| 22 August 2007 | New citation required and conclusions have changed | Substantive amendments made to first review publication |
| 15 May 2006 | Amended | Protocol first published |
Acknowledgements
We acknowledge the work of Professor Martin Stockler on the original protocol for this review. We are also grateful for statistical advice received from Kirsty Mann and Val Gebski (NHMRC Clinical Trials Centre, The Univeristy of Sydney) and from Sam Egger (Cancer Council New South Wales). We also acknowledge the assistance of Fergus Tai and Anne Parkhill in performing searches for this review update.
Appendices
Appendix 1. CENTRAL
#1 MeSH descriptor: [Breast Neoplasms] explode all trees
#2 (early breast cancer* or early breast neoplas* or early breast carcinoma* or early breast tumour* or early breast tumor*):ti,ab,kw
#3 (locally advanced breast cancer* or locally advanced breast neoplas* or locally advanced breast carcinoma* or locally advanced breast tumour* or locally advanced breast tumor*):ti,ab,kw
#4 #1 or #2 or #3
#5 (taxane containing regimen* or taxane containing chemotherapy regimen* or non‐taxane containing regimen* or non‐taxane containing chemotherapy regimen* or taxoid* or paclitaxel or docetaxel or texane* or taxol* or taxotere* or paxene* or anzatax or nsc‐125973 or 4alpha or 7‐epi‐taxol):ti,ab,kw
#6 MeSH descriptor: [Paclitaxel] explode all trees
#7 MeSH descriptor: [Taxoids] explode all trees
#8 #5 or #6 or #7
#9 #4 and #8
Appendix 2. MEDLINE
1. randomised controlled trial.pt.
2. randomized controlled trial.pt.
3. controlled clinical trial.pt.
4. randomized.ab.
5. randomised.ab
6. placebo.ab.
7. randomly.ab.
8. trial.ab.
9. groups.ab.
10.1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9
11.exp Breast Neoplasms/
12.(early adj6 breast adj6 cancer$).mp.
13.(early adj6 breast adj6 neoplasm$).mp.
14.(early adj6 breast adj6 carcinoma$).mp.
15.(early adj6 breast adj6 tumour$).mp.
16.(early adj6 breast adj6 tumor$).mp.
17.11 or 12 or 13 or 14 or 15 or 16
18.taxane containing regimens.mp
19.taxane containing chemotherapy regimens.mp
20.(taxane$ adj6 contain$ adj6 regimen$).mp
21.(taxane$ adj6 contain$ adj6 chemotherap$ adj6 regimen$).mp
22.non‐taxane containing regimens.mp
23.non‐taxane containing chemotherapy regimens.mp
24.(non adj3 taxane$ adj6 contain$ adj6 regimen$).mp
25.(non adj3 taxane$ adj6 contain$ adj6 chemotherap$ adj6 regimen$).mp
26.exp Taxoids/
27.exp Paclitaxel/
28.docetaxel.mp
29.taxane$.mp
30.taxol$.mp
31.taxotere.mp
32.paxene.mp
33.nsc‐125973.mp
34.anzatax.mp
35.4alpha.mp
36.7‐epi‐taxol.mp
37.18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36
38.10 and 17 and 37
39. Animals/
40. Humans/
41. 39 not 40
42. 38 not 41
Appendix 3. Embase
#1
random* OR factorial* OR crossover* OR cross NEXT/1 over* OR placebo* OR (doubl* AND blind*) OR (singl* AND blind*) OR assign* OR allocat* OR volunteer*OR 'crossover procedure'/exp OR 'double blind procedure'/exp OR 'randomized controlled trial'/exp OR 'single blind procedure'/exp
#2
'early breast cancer'
#3
'early breast neoplasm'
#4
'early breast carcinoma'
#5
'early breast tumour'
#6
'early breast tumor'
#7
(early OR 'early stage') NEAR/5 ('breast cancer' OR 'breast carcinoma' OR 'breast neoplasm' OR 'breast tumour' OR 'breast tumor')
#8
'locally advanced breast cancer'
#9
'locally advanced breast neoplasm'
#10
'locally advanced breast carcinoma'
#11
'locally advanced breast tumour'
#12
'locally advanced breast tumor'
#13
'locally advanced' NEAR/5 ('breast cancer' OR 'breast carcinoma' OR 'breast neoplasm' OR 'breast tumour' OR 'breast tumor')
#14
#2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13
#15
'taxane containing regimens'
#16
'taxane containing regimen'
#17
'taxane containing chemotherapy regimens'
#18
'taxane containing chemotherapy regimen'
#19
taxoid*
#20
'paclitaxel'/exp OR paclitaxel
#21
'docetaxel'/exp OR docetaxel
#22
taxane*
#23
'taxol'/exp OR taxol
#24
'taxoid'/exp OR taxoid
#25
'taxotere'/exp OR taxotere
#26
'paxene'/exp OR paxene
#27
taxotere*
#28
paxene*
#29
'nsc 125973'/exp OR 'nsc 125973'
#30
'anzatax'/exp OR anzatax
#31
4alpha
#32
'7 epi taxol'
#33
'non‐taxane containing regimen'
#34
'non‐taxane containing regimens'
#35
'non‐taxane containing chemotherapy regimen'
#36
'non‐taxane containing chemotherapy regimens'
#37
#15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR#35 OR #36
#38
#1 AND #14 AND #37
#39
#38 NOT ([animals]/lim NOT [humans]/lim)
#40
#39 AND [embase]/lim
Appendix 4. WHO ICTRP
Basic Searches:
1. early breast cancer AND taxane containing regimen
2. early breast cancer AND taxane containing regimens
3. early breast cancer AND non taxane containing regimen
4. early breast cancer AND non taxane containing regimens
5. early stage breast cancer AND taxane
6. early stage breast cancer AND non taxane
Advanced Searches:
1. Title: Taxanes for adjuvant treatment of early breast cancer
Recruitment Status: All
2. Condition: early breast cancer*
Intervention: chemotherapy AND taxane containing regimen*
Recruitment Status: All
3. Condition: early breast cancer*
Intervention: chemotherapy AND non taxane containing regimen*
Recruitment Status: All
4. Title: chemotherapy AND taxane containing regimen*
Condition: early breast cancer*
Recruitment Status: All
5. Title: chemotherapy AND non taxane containing regimen*
Condition: early breast cancer*
Recruitment Status: All
6. Condition: early breast cancer*
Intervention: taxane OR taxol OR taxotere OR paclitaxel OR paxene OR nsc‐125973 OR docetaxel OR anzatax OR taxanes OR taxane containing regimen OR taxane containing regimens OR non taxane containing regimen OR non taxane containing regimens
Recruitment Status: All
7. Condition: early stage breast cancer*
Intervention: taxane OR taxol OR taxotere OR paclitaxel OR paxene OR nsc‐125973 OR docetaxel OR anzatax OR taxanes OR taxane containing regimen OR taxane containing regimens OR non taxane containing regimen OR non taxane containing regimens
Recruitment Status: All
Appendix 5. ClinicalTrials.gov
Basic Searches:
1. taxanes for adjuvant treatment of early breast cancer
2. early breast cancer AND taxane containing regimen
3. early breast cancer AND non taxane containing regimen
4. early breast cancer AND (chemotherapy AND taxane containing regimen)
5. early breast cancer AND (chemotherapy AND non taxane containing regimen)
6. early stage breast cancer AND taxane containing regimen
7. early stage breast cancer AND non taxane containing regimen
8. early stage breast cancer AND (chemotherapy AND taxane containing regimen)
9. early stage breast cancer AND (chemotherapy AND non taxane containing regimen)
10. operable breast cancer AND taxane containing regimen
11. operable breast cancer AND non taxane containing regimen
12. operable breast cancer AND (chemotherapy AND taxane containing regimen)
13. operable breast cancer AND (chemotherapy AND non taxane containing regimen)
Advanced Searches:
1. Search Terms: Taxanes for adjuvant treatment of early breast cancer
Recruitment: All studies
Study Results: All studies
Study Type: All studies
2. Conditions: early breast cancer
Interventions: taxane OR taxol OR taxotere OR paclitaxel OR paxene OR nsc‐125973 OR docetaxel OR anzatax OR taxanes OR taxane containing regimen OR taxane containing regimens OR non taxane containing regimen OR non taxane containing regimens
Recruitment: All studies
Study Results: All studies
Study Type: All studies
3. Conditions: early stage breast cancer
Interventions: taxane OR taxol OR taxotere OR paclitaxel OR paxene OR nsc‐125973 OR docetaxel OR anzatax OR taxanes OR taxane containing regimen OR taxane containing regimens OR non taxane containing regimen OR non taxane containing regimens
Recruitment: All studies
Study Results: All studies
Study Type: All studies
4. Conditions: operable breast cancer
Interventions: taxane OR taxol OR taxotere OR paclitaxel OR paxene OR nsc‐125973 OR docetaxel OR anzatax OR taxanes OR taxane containing regimen OR taxane containing regimens OR non taxane containing regimen OR non taxane containing regimens
Recruitment: All studies
Study Results: All studies
Study Type: All studies
Data and analyses
Comparison 1. Overall effect of taxanes.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival ‐ all studies | 27 | 39180 | Hazard Ratio (95% CI) | 0.87 [0.83, 0.92] |
| 1.1 All studies | 27 | 39180 | Hazard Ratio (95% CI) | 0.87 [0.83, 0.92] |
| 2 Disease‐free survival: all studies | 30 | 41909 | Hazard Ratio (95% CI) | 0.88 [0.85, 0.92] |
| 2.1 All studies | 30 | 41909 | Hazard Ratio (95% CI) | 0.88 [0.85, 0.92] |
| 3 Disease‐free survival: as defined in Cochrane protocol | 25 | 35063 | Hazard Ratio (95% CI) | 0.86 [0.82, 0.89] |
| 3.1 DFS ‐ excluding Boccardo, CALGB40101, NCIC‐CTG MA21 & RAPP‐01 | 25 | 35063 | Hazard Ratio (95% CI) | 0.86 [0.82, 0.89] |
Comparison 2. Type of taxane.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 27 | Hazard Ratio (95% CI) | Subtotals only | |
| 1.1 Docetaxel | 15 | 22105 | Hazard Ratio (95% CI) | 0.86 [0.81, 0.92] |
| 1.2 Paclitaxel | 12 | 17075 | Hazard Ratio (95% CI) | 0.89 [0.82, 0.96] |
| 2 Disease‐free survival | 30 | Hazard Ratio (95% CI) | Subtotals only | |
| 2.1 Docetaxel | 16 | 22730 | Hazard Ratio (95% CI) | 0.87 [0.82, 0.91] |
| 2.2 Paclitaxel | 14 | 19179 | Hazard Ratio (95% CI) | 0.91 [0.85, 0.96] |
Comparison 3. Weekly or three‐weekly paclitaxel.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 10 | Hazard Ratio (95% CI) | Subtotals only | |
| 1.1 Weekly paclitaxel | 4 | 4176 | Hazard Ratio (95% CI) | 0.80 [0.65, 0.98] |
| 1.2 Three‐weekly paclitaxel | 6 | 8433 | Hazard Ratio (95% CI) | 0.86 [0.78, 0.95] |
| 2 Disease‐free survival | 10 | Hazard Ratio (95% CI) | Subtotals only | |
| 2.1 Weekly Paclitaxel | 4 | 4176 | Hazard Ratio (95% CI) | 0.79 [0.67, 0.92] |
| 2.2 Three‐weekly paclitaxel | 6 | 8433 | Hazard Ratio (95% CI) | 0.85 [0.79, 0.92] |
Comparison 4. Sequential or concurrent anthracycline/taxane.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 23 | Hazard Ratio (95% CI) | Subtotals only | |
| 1.1 Sequential anthracycline/taxane | 18 | 24764 | Hazard Ratio (95% CI) | 0.86 [0.81, 0.91] |
| 1.2 Concurrent anthracycline/taxane | 6 | 8839 | Hazard Ratio (95% CI) | 0.86 [0.78, 0.94] |
| 2 Disease‐free survival | 26 | Hazard Ratio (95% CI) | Subtotals only | |
| 2.1 Sequential anthracycline/taxane | 20 | 26866 | Hazard Ratio (95% CI) | 0.86 [0.82, 0.90] |
| 2.2 Concurrent anthracycline/taxane | 7 | 9466 | Hazard Ratio (95% CI) | 0.90 [0.83, 0.97] |
Comparison 5. Addition or substitution of taxane.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | Hazard Ratio (95% CI) | Subtotals only | |
| 1.1 Addition of taxane ‐ question 1 | 7 | 10842 | Hazard Ratio (95% CI) | 0.84 [0.77, 0.92] |
| 1.2 Substitution of taxane ‐ question 3 | 13 | 16196 | Hazard Ratio (95% CI) | 0.80 [0.74, 0.86] |
| 2 Disease‐free survival | 20 | Hazard Ratio (95% CI) | Subtotals only | |
| 2.1 Addition of taxane ‐ question 1 | 7 | 10842 | Hazard Ratio (95% CI) | 0.84 [0.78, 0.90] |
| 2.2 Substitution of taxane ‐ question 3 | 14 | 16823 | Hazard Ratio (95% CI) | 0.83 [0.78, 0.88] |
5.2. Analysis.
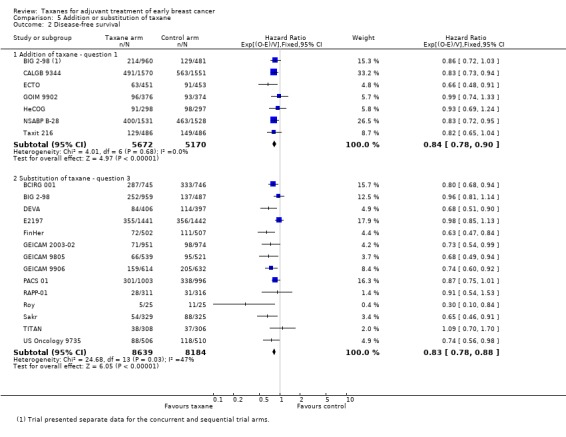
Comparison 5 Addition or substitution of taxane, Outcome 2 Disease‐free survival.
Comparison 6. Duration of chemotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 22 | Hazard Ratio (95% CI) | Subtotals only | |
| 1.1 Taxane‐containing arm longer duration than control arm | 9 | 13865 | Hazard Ratio (95% CI) | 0.84 [0.77, 0.91] |
| 1.2 Taxane‐containing arm same duration as control arm | 14 | 18660 | Hazard Ratio (95% CI) | 0.87 [0.81, 0.94] |
| 2 Disease‐free survival | 25 | Hazard Ratio (95% CI) | Subtotals only | |
| 2.1 Taxane‐containing arm longer duration than control arm | 9 | 13865 | Hazard Ratio (95% CI) | 0.83 [0.77, 0.88] |
| 2.2 Taxane‐containing arm same duration as control arm | 17 | 21391 | Hazard Ratio (95% CI) | 0.90 [0.85, 0.96] |
Comparison 7. Number of cycles of taxane‐containing chemotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 25 | Hazard Ratio (95% CI) | Subtotals only | |
| 1.1 3 cycles of taxane | 7 | 6551 | Hazard Ratio (95% CI) | 0.77 [0.69, 0.86] |
| 1.2 4 or more cycles of taxane | 19 | 29458 | Hazard Ratio (95% CI) | 0.91 [0.86, 0.96] |
| 2 Disease‐free survival | 28 | Hazard Ratio (95% CI) | Subtotals only | |
| 2.1 3 cycles of taxane | 7 | 6551 | Hazard Ratio (95% CI) | 0.80 [0.73, 0.88] |
| 2.2 4 or more cycles of taxane | 22 | 32187 | Hazard Ratio (95% CI) | 0.91 [0.87, 0.95] |
Comparison 8. Lymph node status.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 27 | Hazard Ratio (95% CI) | Subtotals only | |
| 1.1 Node‐positive‐only studies | 17 | 22055 | Hazard Ratio (95% CI) | 0.83 [0.78, 0.88] |
| 1.2 Node‐positive or ‐negative studies | 7 | 10269 | Hazard Ratio (95% CI) | 0.95 [0.87, 1.04] |
| 1.3 Node‐negative‐only studies | 3 | 6856 | Hazard Ratio (95% CI) | 1.08 [0.89, 1.32] |
| 2 Disease‐free survival | 30 | Hazard Ratio (95% CI) | Subtotals only | |
| 2.1 Node‐positive‐only studies | 17 | 22055 | Hazard Ratio (95% CI) | 0.84 [0.80, 0.88] |
| 2.2 Node‐positive or ‐negative studies | 10 | 12998 | Hazard Ratio (95% CI) | 0.95 [0.88, 1.02] |
| 2.3 Node‐negative‐only studies | 3 | 6856 | Hazard Ratio (95% CI) | 0.99 [0.86, 1.14] |
Comparison 9. Hormone receptor status.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 5 | Hazard Ratio (Fixed, 95% CI) | Subtotals only | |
| 1.1 Hormone receptor‐positive | 4 | Hazard Ratio (Fixed, 95% CI) | 0.79 [0.70, 0.89] | |
| 1.2 Hormone receptor‐negative | 5 | Hazard Ratio (Fixed, 95% CI) | 0.88 [0.73, 1.05] | |
| 2 Disease‐free survival | 12 | Hazard Ratio (Fixed, 95% CI) | Subtotals only | |
| 2.1 Hormone receptor‐positive | 11 | Hazard Ratio (Fixed, 95% CI) | 0.91 [0.85, 0.97] | |
| 2.2 Hormone receptor‐negative | 12 | Hazard Ratio (Fixed, 95% CI) | 0.80 [0.73, 0.88] |
Comparison 10. Publication status.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 27 | Hazard Ratio (95% CI) | Subtotals only | |
| 1.1 Studies with published efficacy paper | 26 | 38208 | Hazard Ratio (95% CI) | 0.88 [0.84, 0.92] |
| 1.2 Studies with results from online thesis | 1 | 972 | Hazard Ratio (95% CI) | 0.67 [0.48, 0.94] |
| 2 Disease‐free survival | 30 | Hazard Ratio (95% CI) | Subtotals only | |
| 2.1 Studies with published efficacy paper | 28 | 40310 | Hazard Ratio (95% CI) | 0.88 [0.85, 0.92] |
| 2.2 Studies with results in abstract format or in online thesis | 2 | 1599 | Hazard Ratio (95% CI) | 0.84 [0.67, 1.04] |
Comparison 11. Toxicities.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Febrile neutropenia by sequential or concurrent anthracycline/taxane | 24 | 33763 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [0.96, 2.49] |
| 1.1 Sequential anthracycline and taxane treatment | 16 | 20148 | Odds Ratio (M‐H, Random, 95% CI) | 1.31 [0.82, 2.10] |
| 1.2 Concurrent anthracycline and taxane treatment | 6 | 8552 | Odds Ratio (M‐H, Random, 95% CI) | 5.76 [3.36, 9.87] |
| 1.3 Taxane replacing anthracycline | 3 | 5063 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.02, 3.04] |
| 2 Febrile neutropenia by type of taxane | 25 | 34154 | Odds Ratio (M‐H, Random, 95% CI) | 1.43 [0.89, 2.31] |
| 2.1 Docetaxel | 16 | 22596 | Odds Ratio (M‐H, Random, 95% CI) | 2.83 [1.88, 4.27] |
| 2.2 Paclitaxel | 9 | 11558 | Odds Ratio (M‐H, Random, 95% CI) | 0.36 [0.19, 0.67] |
| 3 Neuropathy (grade 3/4) | 23 | 31033 | Odds Ratio (M‐H, Random, 95% CI) | 6.89 [3.23, 14.71] |
| 4 Neuropathy (grade 3/4) by type of taxane | 23 | 31033 | Odds Ratio (M‐H, Random, 95% CI) | 6.89 [3.23, 14.71] |
| 4.1 Docetaxel | 12 | 18355 | Odds Ratio (M‐H, Random, 95% CI) | 3.74 [1.33, 10.53] |
| 4.2 Paclitaxel | 11 | 12678 | Odds Ratio (M‐H, Random, 95% CI) | 11.93 [3.59, 39.70] |
| 5 Fatigue (grade 3/4) | 16 | 25003 | Odds Ratio (M‐H, Random, 95% CI) | 1.81 [1.31, 2.49] |
| 5.1 Docetaxel | 10 | 16503 | Odds Ratio (M‐H, Random, 95% CI) | 2.14 [1.65, 2.78] |
| 5.2 Paclitaxel | 6 | 8500 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.58, 2.84] |
| 6 Stomatitis (grade 3/4) | 23 | 29500 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.93, 1.78] |
| 6.1 Docetaxel | 16 | 22648 | Odds Ratio (M‐H, Random, 95% CI) | 1.73 [1.28, 2.35] |
| 6.2 Paclitaxel | 7 | 6852 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.31, 1.32] |
| 7 Cardiotoxicity (includes grade 3/4 and symptomatic CCF) | 23 | 32894 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.56, 1.33] |
| 7.1 Total planned dose of anthracycline the same in both taxane‐ and non‐taxane‐containing arms | 9 | 14967 | Odds Ratio (M‐H, Random, 95% CI) | 1.27 [0.88, 1.84] |
| 7.2 Total planned dose of anthracycline reduced in the taxane‐containing arm | 10 | 12473 | Odds Ratio (M‐H, Random, 95% CI) | 0.39 [0.18, 0.86] |
| 7.3 Taxane replacing anthracycline | 4 | 5454 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.26, 3.91] |
| 8 Nausea and/or vomiting (grade 3/4) | 26 | 34450 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.67, 1.04] |
| 8.1 Docetaxel | 16 | 22648 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.62, 1.03] |
| 8.2 Paclitaxel | 10 | 11802 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.57, 1.39] |
| 9 Secondary leukaemia/myelodysplasia | 19 | 33225 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.54, 1.33] |
| 9.1 Docetaxel | 13 | 24911 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.60, 1.59] |
| 9.2 Paclitaxel | 6 | 8314 | Odds Ratio (M‐H, Random, 95% CI) | 0.45 [0.15, 1.32] |
| 10 Treatment‐related deaths | 22 | 34882 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.63, 2.47] |
| 10.1 Docetaxel | 15 | 26021 | Odds Ratio (M‐H, Random, 95% CI) | 1.54 [0.76, 3.15] |
| 10.2 Paclitaxel | 7 | 8861 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.14, 3.85] |
Comparison 12. Risk of Bias.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 27 | 39180 | Hazard Ratio (95% CI) | 0.87 [0.83, 0.92] |
| 1.1 Low risk of bias | 21 | 33206 | Hazard Ratio (95% CI) | 0.90 [0.86, 0.95] |
| 1.2 Unclear or high risk of bias | 6 | 5974 | Hazard Ratio (95% CI) | 0.74 [0.65, 0.83] |
| 2 Disease‐free survival | 30 | 41909 | Hazard Ratio (95% CI) | 0.88 [0.85, 0.92] |
| 2.1 Low risk of bias | 24 | 35935 | Hazard Ratio (95% CI) | 0.90 [0.87, 0.94] |
| 2.2 Unclear or high risk of bias | 6 | 5974 | Hazard Ratio (95% CI) | 0.76 [0.69, 0.84] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
ADEBAR.
| Methods | Randomised controlled trial Multi‐centre (Germany), open‐label Stratified randomisation, according to metastatic axillary lymph node involvement, hormone receptor status, and timing of adjuvant radiotherapy Accrual September 2001 to May 2005 Baseline patient and tumour characteristics appear well balanced | |
| Participants | Female, premenopausal and postmenopausal Aged 18 to 70 years, median age 55 years (25 to 71) Operable breast cancer with clear surgical margins Axillary node positive: 100% (pN2‐3m ≥ 4 metastatic lymph nodes) Exclusion of metastatic disease or inflammatory breast cancer HR positive: 75% in each treatment arm ECOG < 2 |
|
| Interventions | ARM 1 (EC‐Doc):
EC × 4 21‐day cycles (epirubicin 90 mg/m², cyclophosphamide 600 mg/m²) followed by Doc × 4 21‐day cycles (docetaxel 100 mg/m²) ARM 2 (FEC): FEC × 6 28‐day cycles (fluorouracil 500 mg/m² and epirubicin, 60 mg/m² IV on days 1 and 8, cyclophosphamide 750 mg/m² PO on days 1 to 14) Tamoxifen for 5 years for all patients who are ER and/or PR positive. Tamoxifen could be substituted with exemestane, letrozole, or anastrozole in postmenopausal patients with contraindications or who have tolerability issues with tamoxifen. Patients < 40 years of age with restart of menstrual bleeding within 6 months of completion of cytostatic treatment or with premenopausal hormone levels received goserelin 3.6 mg subcutaneously every 4 weeks for 2 years All patients received adjuvant radiotherapy either following completion of chemotherapy or intermittently after completion of 50% of chemotherapy Granulocyte colony‐stimulating factor could be used as secondary prophylaxis in cases of febrile neutropenia |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 60.6 months in EC‐Doc and 59.5 months in FEC120
Clinical Trial Identifier: NCT00047099 (see clinicaltrials.gov/ct2/show/record/NCT00047099) Trial supported by Sanofi‐Aventis, Astra‐Zeneca, Amgen, Wilex, and Novartis Trial was stopped prematurely in 3.7% of participants in the EC‐Doc arm and in 8.0% in the FEC120 arm due to toxicity (P = 0.0009) For this review update, the hazard ratio for OS was derived using Method 3 (Tierney 2007). Outcome data and numbers of participants included in the analysis for DFS were provided by trial authors and the trial publication (in 2016), and the HR was derived using Method 7 (Tierney 2007) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation based on prognostic variables, including metastatic axillary lymph node involvement, hormone receptor status, and timing of radiotherapy |
| Allocation concealment (selection bias) | Low risk | Trial co‐ordinated by a central office Comment: allocation concealment probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Toxicity evaluated using NCI CTC and ECG before each cycle of chemotherapy and 28 days after chemotherapy. ECG also performed 6 months after chemotherapy and whenever indicated. DFS assessment not reported Comment: no apparent involvement of an independent adjudication committee reassessing outcomes |
| Blinding of outcome assessment ‐ QoL (detection bias) | High risk | Measured using QLQ‐C30 and QLQ‐BR23. Quality of life assessed at baseline, before each course of chemotherapy, and at 4 weeks, at 6 weeks, and then at 6 months after completion of chemotherapy |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 92% (689/748) of patients in the taxane arm and 91% (675/745) in the comparator arm included in the efficacy analysis. Reasons provided and appeared to be similar across groups |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes in Clinical Trials.gov record reported across various publications, including QoL data in the 2014 unpublished manuscript (clinicaltrials.gov/ct2/show/record/NCT00047099). Outcomes specified in methods section and results section of trial publications consistent |
| Other bias | Low risk | No other sources of bias identified Quote: "Patient characteristics after randomization were well‐balanced between the two treatment arms" |
BCIRG 001.
| Methods | Randomised controlled trial Multi‐centre, international (20 countries participated) Computer‐generated randomisation lists balanced with a block size of 4, stratified according to institution, and number of involved nodes Accrual June 1997 to June 1999 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal Median age 49 years (23 to 70) Unilateral, operable breast cancer with clear surgical margins Axillary node positive: 100% HR positive: 76% in each treatment arm Exclusion of T4, N2/3, and M1 disease | |
| Interventions | ARM 1:
TAC × 6 21‐day cycles (doxorubicin 50 mg/m², cyclophosphamide 500 mg/m², docetaxel 75 mg/m²)
ARM 2:
FAC × 6 21‐day cycles (doxorubicin 50 mg/m², fluorouracil 500 mg/m², cyclophosphamide 500 mg/m²) Primary prophylaxis with G‐CSF not permitted. Tamoxifen 20 mg/d for 5 years given to all patients with ER‐ and/or PR‐positive tumours. Radiotherapy given as mandatory following breast‐conserving surgery |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Intention‐to‐treat analysis
Median follow‐up: 124 months
Clinical Trial Identifier: NCT00688740 (see clinicaltrials.gov/ct2/show/NCT00688740) Funded by Sanofi. Interim efficacy analysis done by a statistician as part of an independent DMC; final efficacy analysis completed by Sanofi’s statistician |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation Quote: "computer‐generated randomisation lists were used for each stratum (centre and number of nodes) and were balanced with a block size of four" |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Random assignment was done with an interactive voice response system and treatment allocation was immediately communicated to the investigator" Comment: methods not described in sufficient detail |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unblinded Quote: "patients and treating physicians could not be masked to allocation because of the nature of the interventions" |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Quote: "investigators were not masked since the outcomes (relapse, death) were objective" |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Chest radiography and mammography performed every year of follow‐up. Blood counts, general biochemical and clinical assessments each cycle and every 6 months for 5 years, then annually Comment: no independent assessment committee overseeing assessment of outcomes |
| Blinding of outcome assessment ‐ QoL (detection bias) | High risk | Assessed using European Organisation for Research and Treatment of Cancer QoL Questionnaire (QLQ‐C30, version 2.0) and the Breast‐cancer‐specific QLQ‐BR21 (version 1.0). Patients asked to complete both at baseline, before cycles 3 and 5, and at 1, 6, 12, and 24 months after last cycle |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Intention‐to‐treat efficacy and safety analyses were done as originally and prospectively defined in the study protocol" "fewer than 6% of participants... lost to 10‐year follow up" 43/745 in the TAC group and 39/746 in the FAC group |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes in Clinical Trials.gov record ‐ clinicaltrials.gov/ct2/show/NCT00688740 ‐ and in methods section of the trial publication the same. All outcomes reported in 5‐year follow‐up data. All outcomes (excluding quality of life) reported in 10‐year follow‐up data |
| Other bias | Low risk | Quote: "specific demographic, clinical, and molecular phenotypic characteristics of patients were well‐balanced between the group[s]" |
BIG 2‐98.
| Methods | Randomised controlled trial Open‐label, multi‐centre Randomisation method not specified, stratified to participating centre, number of nodes 1 to 3 or 4+ and age < 50 or ≥ 50 Randomisation 1:1:2:2 Accrual June 1998 to June 2001 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal Median age 49 years (21 to 70) Histologically proven, following surgery for operable, node‐positive breast cancer (T1 to T3) Axillary node positive: 100% HR positive: 76% T4 tumours and distant metastases excluded | |
| Interventions | ARM 1a (A‐CMF):
A × 4 21‐day cycles (doxorubicin 75 mg/m²), then CMF × 3 28‐day cycles (cyclophosphamide 100 mg/m² days 1 to 14 orally, methotrexate 40 mg/m² days 1 and 8, 5‐FU days 1 and 8) ARM 1b (AC‐CMF): AC × 4 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²), then CMF × 3 28‐day cycles (as in arm 1a) ARM 2 (A‐T‐CMF): A × 3 21‐day cycles (doxorubicin 75 mg/m²), then T × 3 21‐day cycles (docetaxel 100 mg/m²), then CMF × 3 (as in arm 1a) ARM 3 (AT‐CMF): AT × 4 21‐day cycles (doxorubicin 50 mg/m², docetaxel 75 mg/m²), then CMF × 3 (as in arm 1a) Tamoxifen 20 mg/d for 5 years for ER‐ and/or PR‐positive patients Protocol amended in 2004 to allow AI in postmenopausal women and ovarian suppression in premenopausal women Radiotherapy when indicated following chemotherapy No primary G‐CSF permitted |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 121 months (max 153 months) Clinical Trial Identifier: NCT00174655 (see clinicaltrials.gov/ct2/show/NCT00174655) Funded by Sanofi‐Aventis; trial conducted by BIG. Analyses done entirely independent of Sanofi‐Aventis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned" Treatment allocation done through a "minimization procedure with stratification for centre…" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "treatment allocation was done centrally by use of a minimisation procedure" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Quote: "clinical, haematological and biochemical assessments required before each cycle, including assessing of toxic effects according to the NCI CTC version" Follow‐up visits every 3 months for first 2 years, every 6 months for years 3 to 5, then once a year Comment: no independent adjudication committee involved in these assessments |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Results analysed by intention‐to‐treat Quote: "overall, 2.8% of patients were lost to follow‐up, with equal percentages from control and docetaxel treatment groups" |
| Selective reporting (reporting bias) | Low risk | All outcomes reported as specified in the ClinicalTrials.gov record (see clinicaltrials.gov/ct2/show/NCT00174655) |
| Other bias | Low risk | No other sources of bias identified Quote: "baseline characteristics of enrolled patients were well balanced" |
Boccardo.
| Methods | Randomised controlled trial Multi‐centre, international, open‐label Central randomisation by random numbers tables Accrual April 1997 to January 2004 Baseline patient and tumour characteristics well balanced excluding tumour size | |
| Participants | Female, premenopausal and postmenopausal
Aged 18 to 70 years. Median age not reported
Operable, unilateral breast cancer, completely resected with clear surgical margins
Axillary node positive: 100% (3 or more lymph nodes)
Exclusion of metastatic disease HR positive: ER positive: 89% to 90% in each treatment arm |
|
| Interventions | ARM 1 (E‐CMF)
E × 4 21‐day cycles (epirubicin 100 mg/m²) followed by CMF × 4 28‐day cycles (cyclophosphamide 600 mg/m², methotrexate 40 mg/m², fluorouracil 600 mg/m²) ARM 2 (Paclitaxel‐EV) Paclitaxel × 4 21‐day cycles (paclitaxel 175 mg/m²) followed by EV × 4 21‐day cycles (epirubicin 75 mg/m², vinorelbine 25 mg/m²) Tamoxifen 20 mg/d for 5 years given to all patients who were ER and/or PR positive Radiotherapy given as mandatory following breast‐conserving surgery, and used after mastectomy according to local guidelines |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 102 months No trial record identified Funding: National Research Council and University of Research Italian Minister In the review update, O‐E and V for OS and DFS derived using Method 3 (Tierney 2007) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization... was based on random number tables" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "randomization was carried out by telephone from a central office of the coordinating center" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Clinical examinations every 3 months for 2 years, every 6 months for years 3 to 5, and annually thereafter. Chest X‐ray, liver ultrasound, and/or CT abdomen and a bone scan repeated annually during first 5 years of follow‐up. CBC and biochemistry repeated before each chemotherapy cycle. Toxicity scored using WHO criteria Comment: no involvement of an independent adjudication committee for outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All 244 patients randomised accounted for in results (122 per treatment arm) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in the methods section included in the results section of the trial publication. No trial registry record or protocol found |
| Other bias | Unclear risk | Quote: "treatment arms were well balanced with respect to major pretreatment variables, excluding tumour size (more patients in the E‐CMF arm were affected by tumours < 2 cm in size, P = 0.01)" |
CALGB 40101.
| Methods | Randomised controlled trial Multi‐centre, international Randomisation stratified according to menopausal status, hormone receptor status, and HER2 status Accrual May 2002 to July 2010 Baseline patient and tumour characteristics well balanced 2 × 2 factorial design | |
| Participants | Female, premenopausal and postmenopausal
18 years of age and older
Operable breast cancer with clear surgical margins
90% of participants node negative; 10% with 1 to 3 positive axillary lymph nodes
Exclusion of metastatic disease ER positive: 64% to 65% in each treatment arm |
|
| Interventions | Arm 1:
AC × 4 21/14‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) Arm 2: AC × 6 21/14‐day cycles (as in arm 1) Arm 3: T × 4 21/14‐day cycles (paclitaxel 80 mg/m² when given weekly (for 12 or 18 weeks – 3 weeks equalling 1 cycle), or paclitaxel 175 mg/m² every 2 weeks) Arm 4: T × 6 21/14‐day cycles (as in arm 3) Tamoxifen recommended for patients with hormone receptor‐positive tumours Radiotherapy given as mandatory following breast‐conserving surgery, and at the discretion of physicians post mastectomy After 2005, trastuzumab recommended to women with HER2‐positive tumours |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: approximately 73 months Clinical Trial Identifier: NCT00041119 (see clinicaltrials.gov/ct2/show/record/NCT00041119) Trial supported by grants from the National Cancer Institute (USA) For the review update, 2014 full‐text publications reported HRs for RFS and OS | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization used a permuted block design with fixed block size of 12 allocated patients with equal probability to one of the four possible treatment arms. Randomization was stratified by menopausal status, hormone receptor status, and after October 2005, HER2 status" |
| Allocation concealment (selection bias) | Low risk | Centralised system (CALGB online Patient Registration system where randomisation accepted only through CALGB main member/selected institutions using the online patient registration system) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label study |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Patients followed up every 6 months for the first 2 years and annually thereafter for 15 years. Adverse events reported using NCI common toxicity criteria Comment: no apparent involvement of an independent adjudication committee for outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "efficacy analyses used an intention‐to‐treat approach" and "at the time of reporting, 45 patients (1%) were lost to follow‐up, and 57 patients (2%) had withdrawn consent to receive follow‐up" |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes in Clinical Trials.gov record reported (clinicaltrials.gov/ct2/show/record/NCT00041119). Trial publication describes a companion trial on QoL and induction of menopause that has yet to be reported |
| Other bias | Low risk | No other sources of bias identified |
CALGB 9344.
| Methods | Randomised controlled trial Open‐label, multi‐centre (516 sites), USA Central randomisation, stratified for number of positive axillary nodes Accrual May 1994 to April 1999 No significant imbalance between groups 3 × 2 factorial design | |
| Participants | Female, premenopausal and postmenopausal Median age not provided Operable breast cancer with clear surgical margins Axillary node positive: 100% HR positive: ER positive 59%; ER or PR positive 66% | |
| Interventions | ARM 1 (AC‐T):
AC × 4 21‐day cycles (doxorubicin (60, 75, or 90 mg/m²), cyclophosphamide 600 mg/m²) followed by T × 4 21‐day cycles (paclitaxel 175 mg/m² over 3 hours)
ARM 2 (AC):
AC × 4 21‐day cycles (doxorubicin (60, 75, or 90 mg/m², cyclophosphamide 600 mg/m²) followed by NO paclitaxel Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive Radiotherapy following chemotherapy required for all patients after breast‐conserving surgery. Primary prophylaxis with G‐CSF and ciprofloxacin given routinely with doxorubicin 90 mg/m², but only as secondary prophylaxis for dosing of 60 or 75 mg/m² doxorubicin |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 69 months; minimum follow‐up: 12 months
98.5% of participants eligible/available for analysis Trial protocol available (see cancer.gov/about‐cancer/treatment/clinical‐trials/search) National Cancer Institute (USA) sponsored the trial. Bristol‐Myers Squibb provided a grant to CALGB for statistical support and for data updates |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned at the statistical centre with equal probability to one of six treatment combinations using a stratified random permuted block design" |
| Allocation concealment (selection bias) | Low risk | Quote: "randomly assigned at the Statistical Centre" Comment: central allocation probably took place |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Mammogram and chest X‐ray obtained at entry and yearly thereafter. CBC obtained twice weekly. Evaluation every 3 months during year 1, twice annually for next 2 years, then annually thereafter Comment: no independent adjudication committee involved in outcome assessments |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "although 3170 women were randomised, 49 patients never received any protocol therapy, usually because the patient withdrew consent. Because no information is available on the treatment the cancelled patients received, their disease‐free survival, or their overall survival, all analyses in this article are based on the remaining 3,121 patients" |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in the trial publication as outlined in the trial registry record (see cancer.gov/about‐cancer/treatment/clinical‐trials/search) |
| Other bias | Low risk | Quote: "there was no significant imbalances in the randomizations" |
DEVA.
| Methods | Randomised controlled trial with partial 2 × 2 factorial design Multi‐centre (36 centres in 5 European countries) Randomisation with computer‐generated permuted blocks. Stratified according to institution and intention‐to‐treat with tamoxifen Accrual August 1997 to December 2005 Baseline patient and tumour characteristics well balanced Randomised 1:1 | |
| Participants | Female, postmenopausal
Complete tumour excision with clear surgical margins
Axillary node positive: 100%
Exclusion of metastatic disease HR positive: 77% to 78% in each treatment arm |
|
| Interventions | ARM 1 (EPI)
EPI × 6 28‐day cycles (epirubicin 50 mg/m² days 1 and 8)
ARM 2 (EPI‐Doc)
EPI × 3 28‐day cycles (epirubicin 50 mg/m² days 1 and 8) followed by Doc × 3 21‐day cycles (docetaxel 100 mg/m² on day 1) Tamoxifen 20 mg/d for 5 years given to women with ER‐ and/or PR‐positive tumours; some centres randomising to administer concurrently or sequential to chemotherapy Use of prophylactic G‐CSFs and antibiotics recommended in the case of febrile neutropenia |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Intention‐to‐treat analysis
Median follow‐up 64.7 months
Clinical Trial Identifier: ISRCTN89772270 (see isrctn.com/ISRCTN89772270) Supported by unrestricted educational trials from Pfizer and Sanofi‐Aventis, and docetaxel provided by Sanofi‐Aventis For the review update, O‐E and V for OS and DFS derived using Method 3 (Tierney 2007) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer generated permuted blocks were used" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "independent random assignment was by telephone/fax to the International Collaborative Cancer Group Data centre, London, England" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Toxicity assessed according to NCI CTC version 2. Assessed after each chemotherapy cycle, with follow‐up every 3 months for first year, every 4 months for second year, every 6 months for years 3 and 4, and annually thereafter until minimum 10 years. No other information on outcome assessment included in the report Comment: no apparent involvement of an independent adjudication committee; therefore this domain assessed as having 'unclear' risk |
| Blinding of outcome assessment ‐ QoL (detection bias) | High risk | Measured by QLQ‐C30 and QLQ‐BR23. Assessed at baseline and at 9 months, 2 years, and 5 years after random assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "only one patient withdrew consent for additional treatment and follow‐up (in the EPI‐DOC arm) and approximately 3% were classified as lost to follow‐up; all patients were included in the intention‐to‐treat analyses" |
| Selective reporting (reporting bias) | Low risk | Outcomes specified in the methods section and reported in the results section consistent. Primary and secondary outcomes not provided at time of registration on ISRCTN |
| Other bias | Low risk | Quote: "baseline clinicopathologic characteristics of patients were evenly balanced between treatment groups" |
E2197.
| Methods | Randomised controlled trial, conducted in USA Randomisation method not specified; stratified according to nodal, hormone receptor, and menopausal status Accrual July 1998 to January 2000 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal following surgery for operable breast cancer Median age 51 years Following complete surgical excision of the primary tumour 66% lymph node negative with T > 1 cm, 34% node positive (1 to 3 N+) HR positive: approximately 68% either ER or PR positive Excluded locally advanced bilateral or metastatic cancer | |
| Interventions | ARM 1 (AT):
AT × 4 21‐day cycles (doxorubicin 60 mg/m², docetaxel 60 mg/m²)
ARM 2 (AC):
AC × 4 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive. In June 2005, protocol changed to allow women to switch from tamoxifen to aromatase inhibitors Radiotherapy given after chemotherapy to all patients following breast‐conserving surgery and to select high‐risk patients following mastectomy at physician’s discretion |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Intention‐to‐treat analysis
Median follow‐up: 11.5 years
97.8% of randomised patients eligible and analysable Clinical Trial Identifier: NCT00003519 (see clinicaltrials.gov/ct2/show/NCT00003519) Funded by Department of Health and Human Services and National Institutes of Health (USA). Study co‐ordinated by ECOG For the review update, data on numbers of events and participants per treatment arm for OS and DFS provided by trial authors |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned to arm A or B… Treatments were assigned using permuted blocks within strata" |
| Allocation concealment (selection bias) | Unclear risk | Quote: "treatments were assigned using permuted blocks within strata with dynamic balancing within main institutions and their affiliate networks" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided in trial publication |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Physical examinations every 3 months for 2 years, then every 6 months for the next 3 years. Mammography and blood testing performed annually Quote: "patients were seen before each course of chemotherapy for physical and hematologic evaluations" Comment: no independent assessment committee overseeing outcome assessments |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Of 2952 patients randomised, 70 considered ineligible with reasons provided. 35 participants excluded from both treatment groups for similar reasons Quote: "when all patients, eligible and ineligible, were analysed. Results for this analysis were similar to the results for patients classified as eligible" |
| Selective reporting (reporting bias) | Low risk | All outcomes reported as specified in ClinicalTrials.gov (clinicaltrials.gov/ct2/show/NCT00003519). Methods and results sections of the trial publication also consistent |
| Other bias | Low risk | Quote: "patient characteristics were well balanced between treatment groups" |
ECTO.
| Methods | Randomised controlled trial Open‐label, multi‐centre (31 European centres), international Central randomisation stratified by centre, tumour size, tumour grade, and hormone receptor status Randomised at 1:1:1 Accrual November 1996 to May 2002 | |
| Participants | Female, premenopausal and postmenopausal Aged 18 to 70 years; median age range not reported in 2009 article Untreated, unilateral operable breast cancer (T2 to 3, N0 to 1, M0) Node‐positive (47%) or ‐negative (53%) breast cancer HR positive: ER/PR positive: around 68% in each treatment arm 1 and 2 Excluded locally advanced metastatic or bilateral cancer | |
| Interventions | ARM 1 (Surgery‐A‐CMF):
A × 4 21‐day cycles (doxorubicin 75 mg/m²) followed by CMF × 4 28‐day cycles (cyclophosphamide 600 mg/m², methotrexate 40 mg/m², fluorouracil 600 mg/m²) ARM 2 (Surgery‐AT‐CMF): AT × 4 21‐day cycles (doxorubicin 60 mg/m², paclitaxel 200 mg/m²) followed by CMF × 4 28‐day cycles (cyclophosphamide 600 mg/m², methotrexate 40 mg/m², fluorouracil 600 mg/m²) ARM 3 (AT‐CMF‐Surgery): Neoadjuvant AT‐CMF dosed as per arm 2 followed by surgery All patients undergoing either mastectomy or breast‐conserving surgery with clear margins. All patients who received breast‐conserving surgery undergoing postoperative irradiation. All patients with pT4 disease given chest wall irradiation following mastectomy Tamoxifen 20 mg/d for 5 years to all patients before June 2000, then protocol amended and limited to ER‐ and/or PR‐positive group |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Only arms 1 and 2 used in this review Median follow‐up: 43 months Trial identifier not retrieved Supported by an “unrestricted grant from Bristol‐Myers Squibb”. Data collection, analysis, and interpretation independent of sponsors and completed by ECTO Group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "treatment was allocated centrally using a minimization algorithm" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "treatment was allocated centrally" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Assessed by physical exam before each cycle of chemotherapy and 2, 6, 12, 18, and 24 months after completion of treatment, then yearly thereafter. Mammography performed yearly after completion of radiotherapy. Cardiac function assessed by physical examination, ECG, and measurement of LVEF at baseline, at completion of chemotherapy, and every 6 months for 2 years followed by yearly Comment: no involvement of an independent assessment committee |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all randomly assigned patients were included in the intention‐to‐treat analyses" |
| Selective reporting (reporting bias) | Low risk | All outcomes in the trial publication reported as specified in ClinicalTrials.gov (http://clinicaltrials.gov/ct2/show/NCT00003013?term=european+cooperative+trial+in+operable+breast+cancer&rank=1). All prespecified outcomes in the methods section reported in the results section of the trial publication |
| Other bias | Low risk | Quote: "baseline characteristics were well balanced between the three treatment arms" |
ELDA.
| Methods | Randomised controlled trial Multi‐centre, international Central randomisation, using a minimisation procedure with centre, pT and pN category, planned number of chemotherapy cycles, age as strata Open‐label study Accrual July 2003 to April 2011 Baseline tumour characteristics well balanced although slight imbalance in distribution of comorbidities (e.g. no comorbidity in 7% and 13%, and previous cerebrovascular disease in 1% and 6% in docetaxel and CMF groups, respectively) | |
| Participants | Female, postmenopausal Aged 65 to 79 years (median 70) Operable breast cancer with clear surgical margins Axillary node positive or high risk node negative ECOG performance status ≤ 2 HR positive: ER positive/PgR positive: 74% to 76% in each treatment group Exclusion of metastatic disease | |
| Interventions | ARM 1 (CMF)
CMF × 4 to 6 28‐day cycles (cyclophosphamide 600 mg/m², methotrexate 40 mg/m², fluorouracil 600 mg/m² on days 1 and 8) ARM 2 (Doc) Doc × 4 to 6 28‐day cycles (docetaxel 35 mg/m² on days 1, 8, and 15) 6 cycles planned for tumours < 10% positive for both ER and PgR, 4 cycles for those with ER or PR ≥ 10% Tamoxifen or aromatase inhibitors according to standard schedules given after chemotherapy to patients with tumour positive for ER/PR in at least 1% of cells Patients with HER2‐positive tumour given adjuvant trastuzumab for 1 year after chemotherapy. Radiotherapy performed when indicated after the end of chemotherapy and within 6 months after surgery |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up 70 months (95% confidence interval 66 to 73 months)
Clinical Trial Identifier: NCT00331097 (see clinicaltrials.gov/ct2/show/NCT00331097) Sponsored by the Clinical Trials Unit of the National Cancer Institute of Naples |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “randomization was carried out centrally at the Clinical Trials Unit of the NCI Naples, with a computer‐based minimization procedure…” |
| Allocation concealment (selection bias) | Low risk | Central randomisation Quote: “…clinicians contacted the Clinical Trials Unit by telephone or by fax” |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label study |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Assessment including clinical visits with biochemistry and haematological tests every 3 months for 3 years, then every 6 months for 2 years, then annually for 5 years. Chest X‐ray, ultrasonography, and mammography included. Treatment toxicity graded according to National Cancer Institute Common Toxicity Criteria Comment: no involvement of an independent adjudication committee for outcome assessment |
| Blinding of outcome assessment ‐ QoL (detection bias) | High risk | Patients completing EORTC QLQ‐C30a and BR23 at baseline, and at end of first, second, and third cycles of chemotherapy |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 302 patients randomised (docetaxel: 150; CMF: 152) and 299 patients included in modified intention‐to‐treatment analysis (99.1%) |
| Selective reporting (reporting bias) | Low risk | Methods consistent with trial registry record (clinicaltrials.gov/ct2/show/NCT00033683). Outcomes reported in the methods and results sections of the trial publication consistent. Primary and secondary outcomes not listed at the time of registration |
| Other bias | Low risk | No other sources identified |
FinHer.
| Methods | Randomised controlled trial Multi‐centre (17 Finnish centres), open‐label Central randomisation stratified for HER2 status and institution Accrual October 2000 to September 2003 Baseline patient and tumour characteristics well balanced | |
| Participants | Female premenopausal and postmenopausal (age < 66 years) within 12 weeks following surgery for operable unilateral invasive breast cancer Performance score 0 or 1 Median age 51 years (range 25.5 to 65.8) 89% lymph node positive; 11% lymph node negative with T > 20 mm and PR negative 72.2% of cancers were ER positive | |
| Interventions | ARM 1 (T‐FEC):
T × 3 21‐day cycles (docetaxel 100 mg/m² (changed to 80 mg/m² in Feb 2002 by independent study monitoring committee)), then FEC × 3 21‐day cycles (fluorouracil 600 mg/m², epirubicin 60 mg/m², cyclophosphamide 600 mg/m²)
ARM 2 (V‐FEC):
V × 3 21‐day cycles (vinorelbine 25 mg/m² days 1, 8, and 15), then FEC × 3 21‐day cycles (fluorouracil 600 mg/m², epirubicin 60 mg/m², cyclophosphamide 600 mg/m²) HER2‐positive patients randomised to receive trastuzumab or not (weekly dose for 9 weeks commencing with first cycle of docetaxel or vinorelbine, first dose 4 mg/kg, then 2 mg/kg) Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive until protocol amendment in 2005, which allowed postmenopausal women to switch to aromatase inhibitors to complete 5 years of hormonal therapy Radiotherapy as per institution’s guidelines G‐CSF not recommended unless 1 or more episodes of febrile neutropenia or severe infection |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Intention‐to‐treat analysis
Median follow‐up 62 months
No patients lost to follow‐up Clinical Trial Identifier: ISRCTN76560285 (see isrctn.com/ISRCTN76560285) Supported by Sanofi‐Aventis, Pierre Fabre, Phamacia, Roche, and state of Finland For the review update, we received clarification on the unadjusted hazard ratio for RFS from trial authors |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "permuted blocks were used to randomly assign all participants" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "participants were randomly assigned (central and with computer‐assisted blinding)" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Quote: "patients were scheduled for follow‐up for a minimum of five years. Mammography was performed at one‐to‐two‐year intervals, but otherwise follow‐up was carried out according to institution's guidelines. Patients were assessed for adverse effects of therapy on day 21 of each cycle, and 12 and 36 months after completing chemotherapy" Comment: no independent adjudication committee providing oversight |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote from Joensuu 2006: "No patient was lost to follow up. Two women who did not receive the study treatments because of abnormal results on liver‐function tests were excluded from the safety analyses, and one patient ... With overt distant metastases at randomisation was excluded from the survival analyses" Efficacy analyses based on intention‐to‐treat principle |
| Selective reporting (reporting bias) | Unclear risk | All outcomes reported in the trial registry reported in 1 or more trial publications. Primary endpoint changed from recurrence‐free survival in 2006 to distant disease‐free survival (DDFS) in the 2009 publication, whereby DDFS did not include contralateral breast cancers. Trialists stated in 2009: "DDFS was preferred to time to any recurrence as the primary endpoint because it allowed a longer follow‐up time and collection of more endpoints before final analysis and distant recurrences are more closely associated with mortality than local ones" |
| Other bias | Unclear risk | Quote: "the baseline characteristics of the patients in the treatment groups were balanced, except that larger breast tumours (> 20 mm in diameter) were more common in the docetaxel group than in the vinorelbine group" |
GEICAM 2003‐02.
| Methods | Randomised controlled trial Open‐label, multi‐centre trial involving 67 Spanish institutions Centralised randomisation in blocks of 4. Stratified according to institution, menopausal status, node status diagnostic method, hormone receptor status Accrual September 2003 to October 2008 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal
Age 18 to 70 years
Median age 50 years
Operable breast cancer with clear surgical margins
High risk, lymph node negative (St Gallen criteria)
Exclusion of metastatic disease
Exclusion of HER2‐positive patients after 2005. Overall 9.4% HER2 positive HR positive: 73% in each treatment arm |
|
| Interventions | ARM 1 (FAC)
FAC × 6 21‐day cycles (5‐fluorouracil 500 mg/m², doxorubicin 50 mg/m², cyclophosphamide 500 mg/m²) ARM 2 (FAC‐wP) FAC × 4 21‐day cycles (doxorubicin 50 mg/m², fluorouracil 500 mg/m², cyclophosphamide 500 mg/m²) followed by wP × 8 weekly cycles (paclitaxel 100 mg/m²) Premenopausal women with hormone receptor‐positive tumours given tamoxifen 20 mg/d for 5 years post chemotherapy. Postmenopausal women with hormone receptor‐positive tumours allowed to receive aromatase inhibitors as initial adjuvant therapy or after tamoxifen Radiotherapy given as mandatory following breast‐conserving surgery |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 63.3 months Clinical Trial Identifier: NCT00129389 (see clinicaltrials.gov/ct2/show/record/NCT00129389?term=GEICAM) Supported partially by Bristol‐Myers Squibb; company not involved in trial design, data collection, data analysis, manuscript writing, or decisions related to publication of results For the review update, O‐E and V for OS and DFS derived using Method 3 (Tierney 2007) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation Quote: "patients were randomly assigned" and "patients were stratified... In blocks of four" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "random assignment was centralized at GEICAM headquarters" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Follow‐up visits every 3 months for first 2 years, every 6 months for years 3 to 5, annually for years 6 to 10. For first 5 years, haematology and biochemistry performed every 6 months, and chest radiography and mammograms performed annually Quote: "toxicities were assessed after each chemotherapy cycle and graded according to the NCI CTC version 2.0. ECG and LVEF were repeated as clinically indicated" Comment: no independent adjudication committee involved in assessing outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "we performed the primary analysis on the intention‐to‐treat population, whereas safety analyses were performed on all patients who received at least one dose of chemotherapy according to the treatment received" |
| Selective reporting (reporting bias) | Unclear risk | Primary and secondary outcomes reported as consistent with trial registry 2009 update (clinicaltrials.gov/ct2/show/record/NCT00129389?term=GEICAM). Methods section and results section in the trial publication consistent. The original secondary outcome measure of QoL listed in the 2005 trial registry record not reported |
| Other bias | Low risk | Quote: "baseline patient and tumor characteristics were well balanced between arms" |
GEICAM 9805.
| Methods | Randomised controlled trial
Multi‐centre international (49 centres in Spain, 2 in Poland, 4 in Germany), open‐label
Central, block randomisation. Stratified according to institution and menopausal status
Accrual from July 1999 to March 2003 Baseline patient and tumour characteristics well balanced |
|
| Participants | Female, premenopausal and postmenopausal
18 to 70 years of age
Median age 50 years (23 to 74)
Operable breast cancer, resected with clear surgical margins
High risk for recurrence (according to 1998 St Gallen criteria), axillary lymph node negative
Exclusion of T4 tumours or metastatic disease HR positive: 64% to 67% in each treatment arm |
|
| Interventions | ARM 1 (TAC)
TAC × 6 21‐day cycles (docetaxel 75 mg/m², doxorubicin 50 mg/m², cyclophosphamide 500 mg/m²) ARM 2 (FAC) FAC × 6 21‐day cycles (fluorouracil 500 mg/m², doxorubicin 50 mg/m², cyclophosphamide 500 mg/m²) Primary prophylactic antibiotics for all TAC patients. Primary prophylactic G‐CSF for TAC after protocol amended July 2000 Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive Radiotherapy given as mandatory following breast‐conserving surgery, and given according to individual institutional guidelines for post‐mastectomy patients |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 77 months
Clinical Trial Identifier: NCT00121992 (see clinicaltrials.gov/ct2/show/NCT00121992) Trial sponsored by GEICAM and Sanofi‐Aventis For the review update, O‐E and V for OS and DFS derived using Method 3 (Tierney 2007) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation Quote: "patients underwent randomization... According to a center‐specific randomization block" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "randomization was centralized and stratified for the participating institution and for menopausal status" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Chest radiography and mammography performed yearly during first 5 years of follow‐up. Toxic effects assessed before each chemotherapy cycle and graded according to the NCICTC version 2.0. CBC mandatory on days 7 to 10 and 20 to 21. No independent adjudication committee involved in outcome assessment |
| Blinding of outcome assessment ‐ QoL (detection bias) | High risk | EORTC QLQ‐C30 and QLQ‐BR23 used. Self‐administered to patients at baseline and at size prospective time points corresponding to chemotherapy cycles |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the primary analysis was performed for the intention‐to‐treat population" < 1% of the participant population not included in the safety analyses, with reasons provided |
| Selective reporting (reporting bias) | Low risk | Primary and secondary outcomes reported as consistent with study protocol (clinicaltrials.gov/ct2/show/NCT00121992). Outcomes specified in the methods and results sections consistent |
| Other bias | Low risk | Quote: "baseline characteristics were well balanced between the treatment groups" |
GEICAM 9906.
| Methods | Randomised controlled trial Multi‐centre, Spain, open‐label Randomisation by a computer programme Stratified for institution, menopausal status, affected lymph nodes (1 to 3 or > 3) Accrual November 1999 to June 2002 Baseline patient and tumour characteristics well balanced, except for HR‐positive tumours; FEC‐paclitaxel with higher percentage of HR‐positive tumours compared to FEC alone (P = 0.024) | |
| Participants | Female, premenopausal and postmenopausal following primary curative surgery for operable node‐positive breast cancer
Age 18 to 70 years. Median age: 50 years Axillary node positive: 100% HR positive: > 63% in each treatment arm Exclusion of advanced disease (T4, N2/3, M1) |
|
| Interventions | ARM A (FEC):
FEC × 6 21‐day cycles (fluorouracil 600 mg/m², epirubicin 90 mg/m², cyclophosphamide 600 mg/m²) ARM B (FEC‐T): FEC × 4 21‐day cycles followed by T × 8 weekly cycles (paclitaxel 100 mg/m²) Tamoxifen 20 mg/d for 5 years for all ER‐ and/or PR‐positive patients An amendment in 2005 allowed administration of aromatase inhibitors to menopausal women Radiotherapy mandatory after breast‐conserving surgery and recommended for patients with > 4 axillary lymph nodes and tumours > 5 cm |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 66 months
Intention‐to‐treat analysis
Clinical Trial Identifier: NCT00129922 (see clinicaltrials.gov/ct2/show/NCT00129922) Supported by unrestricted grants for the conduct of this trial by Bristol‐Myers Squibb and Pharmacia For the review update, we received clarification on the unadjusted hazard ratio for OS from trial authors |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation used Quote: "eligible patients were stratified according to....and randomly assigned to the control or experimental arms by means of a computer program" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Quotes: "haematologic and biochemical tests, chest X‐ray and mammography for first 5 years of follow up" and "blood counts, biochemical and clinical assessments performed each cycle of chemotherapy and continued after completion of therapy every 3 months for 2 years, then every 6 months in years 3‐5 followed by annually thereafter" Comment: no independent adjudication committee involved in assessing these outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the primary analysis was conducted according to the intention‐to‐treat principle" |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes in Clinical Trials.gov record (clinicaltrials.gov/ct2/show/NCT00129922) reported with the addition of distant relapse‐free survival in the 2008 publication. Methods and results sections of the trial publication also consistent |
| Other bias | Unclear risk | Quote: "two treatment arms were well balanced in terms of demographic and tumour characteristics, except for hormone receptor status" |
GOIM 9902.
| Methods | Randomised controlled trial Multi‐centre, 20 Italian centres Centralised, computer‐generated randomisation. Stratified according to institution, number of metastatic lymph nodes, age, and hormone receptor status Accrual April 1999 to October 2005 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal
Age 18 to 70 years
Median age 50 years (range 43 to 60)
Operable breast cancer with clear surgical margins
Axillary node positive: 100%
Exclusion of metastatic disease
Performance status 0 to 1 HR positive: 77% in each treatment arm |
|
| Interventions | ARM 1 (EC)
EC × 4 21‐day cycles (epirubicin 120 mg/m², cyclophosphamide 600 mg/m²) ARM 2 (D‐EC) D × 4 21‐day cycles (docetaxel 100 mg/m²) followed by EC × 4 21‐day cycles (epirubicin 120 mg/m², cyclophosphamide 600 mg/m²) Primary prophylaxis with G‐CSF not permitted Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive. From January 2003, postmenopausal women given anastrozole for 5 years Radiotherapy given following breast‐conserving surgery and in cases of > 4 positive lymph nodes |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 64 months Trial registration record not retrieved GOIM 9902 funded by Sanofi‐Aventis and the Gruppo Oncologica Italia Meridionale For the review update, O‐E and V for OS and DFS derived using Method 3 (Tierney 2007) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomisation procedures were computer generated... And patients assigned according to the minimization technique" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "randomisation procedures were computer generated, centralized at the Regina Elena National Cancer Institute" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided in the trial publication |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Imaging studies (chest X‐ray, liver ultrasound) carried out every 6 months for 5 years and yearly thereafter. Mammography and bone scan performed every year. Toxicity evaluated each cycle, graded according to NCI CTC (version 3.0) criteria. LVEF evaluated with MGAS or echocardiography at baseline, after for EC cycles, and during follow‐up Comment: no independent adjudication committee involved in outcome assessments |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "data on treatment and follow‐up were completely lacking from 14 (arm A) and 8 (arm B) patients" ITT analysis carried out on remaining patients for whom treatment and follow‐up data were available |
| Selective reporting (reporting bias) | Low risk | Primary and secondary outcomes reported in the results section consistent with study methods section |
| Other bias | Low risk | Quote: "in general, the two treatment arms were well balanced in terms of demographics and tumour characteristics" |
GONO MIG‐5.
| Methods | Randomised controlled trial Multi‐centre (30 sites), Italy Randomisation method not specified Accrual November 1996 to January 2001 | |
| Participants | Female, premenopausal and postmenopausal
Aged under 70 years
Operable, unilateral breast cancer, stage IIa, IIb, or III, completely resected with clear surgical margins
Axillary node positive: 100% with at least 1 and fewer than 10 involved nodes
Exclusion of metastatic disease
ECOG performance status 0 HR positive: 76% in CEF and 81% in ET No prior chemotherapy. Surgery performed not more than 5 weeks before randomisation |
|
| Interventions | Arm 1 (CEF)
CEF × 6 21‐day cycles (cyclophosphamide 600 mg/m², epirubicin 60 mg/m², 5‐fluorouracil 600 mg/m²) Arm 2 (ET) ET × 4 21‐day cycles (epirubicin 90 mg/m², paclitaxel 175 mg/m²) Tamoxifen 20 mg/d for 5 years to women with ER‐ and/or PR‐positive tumours Postoperative radiotherapy given to patients who received breast‐conserving surgery. For those who had a mastectomy, radiotherapy done according to local guidelines |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 12.8 years Clinical Trial Identifier: NCT00005581 (see clinicaltrials.gov/ct2/show/record/NCT00005581) and NCT02450058 (see clinicaltrials.gov/ct2/show/NCT02450058) Funded by National Institute for Cancer Research Italy For the review update, numbers of events for OS and EFS, as well as unadjusted hazard ratio and confidence interval for each analysis, provided by trial authors and confirmed in a follow‐up trial publication in 2016. O‐E and V for OS and EFS derived using Method 3 (Tierney 2007) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "eligible patients were randomly allocated 1:1 to one of the two study arms by telephone or fax at the central operation office …Patients were assigned to a treatment arm according to stratified random lists that were balanced in blocks of various sizes in random sequence” |
| Allocation concealment (selection bias) | Low risk | Quote: “…randomly allocated 1:1 to one of the two study arms by telephone or fax at the central operational office of the Trials Center of National Cancer Research Institute” Comment: central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label study |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Assessment including clinical visits every 3 months for first 3 years, then every 6 months during the fourth and fifth years, followed by annual visits. Mammography and blood count performed yearly. Toxicity graded according to WHO toxicity criteria Comment: no involvement of an independent adjudication committee for outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Analyses conducted according to intention‐to‐treat principle. At median follow‐up of 12.8 years, 3.2% of patients lost to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | Trial registry record specifies OS, DFS, and QoL; abstract and trial publication report on DFS, OS, and toxicity but not quality of life (clinicaltrials.gov/ct2/show/record/NCT00005581) |
| Other bias | Low risk | No other sources of bias identified |
HeCOG.
| Methods | Randomised controlled trial Multi‐centre, Greece Stratified randomisation balanced by centre for number of positive nodes, hormonal receptor status, and menopausal status Accrual June 1997 to November 2000 Baseline patient and tumour characteristics well balanced except for tumour grade (significantly higher tumour grade in group A) Treatment delays and dose reductions similar in each group | |
| Participants | Female, premenopausal and postmenopausal following surgery for operable breast cancer, pathological stage T1 to 3 N1 M0 or T3 N0 M0 Axillary node positive: 98% Postmenopausal women with 1 to 3 positive nodes and hormone receptor positive excluded Median age: 50 years (22 to 78) HR positive: 75% to 76% in each group |
|
| Interventions | ARM A (E‐T‐CMF):
E × 3 14‐day cycles (epirubicin 110 mg/m²) then T × 3 14‐day cycles (paclitaxel 250 mg/m² over 3 hours) followed by intensified CMF × 3 14‐day cycles (cyclophosphamide 840 mg/m², methotrexate 57 mg/m², fluorouracil 840 mg/m²). G‐CSF (5 mcg/kg) days 3 to 10 of each cycle ARM B (E‐CMF): E × 4 14‐day cycles, then intensified CMF × 4 14‐day cycles. Doses and G‐CSF given as in arm A Tamoxifen 20 mg/d for 5 years for all ER‐ and/or PR‐positive patients All premenopausal patients given ovarian suppression for 1 year (IM triptorelin) Radiotherapy for all patients with breast‐conserving therapy and/or T > 5 cm |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Intention‐to‐treat analysis Median follow up 61.7 months (Group A) and 62 months (Group B) Clinical Trial Identifier: ACTRN12611000506998 (see anzctr.org.au/Trial/Registration/TrialReview.aspx?id=336915) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "stratified randomisation balanced by centre" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "...randomisation... was performed at the HeCOG Data Office in Athens" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided in trial publication or trial registry record |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Chest X‐ray, abdominal US, and bone scan done every 6 months for 3 years, then annually. Blood count and biochemistry repeated before each cycle. CBC, biochemistry, and physical exams repeated every 3 months for 2 years, then every 6 months thereafter Comment: no independent adjudication committee involved in assessing these outcomes |
| Blinding of outcome assessment ‐ QoL (detection bias) | High risk | Completed by participants at baseline and at completion of chemotherapy using QLQ‐C30 questionnaire |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | At 5‐year follow‐up, 4/298 in the E‐T‐CMF group and 7/297 in the E‐CMF group lost to follow‐up. Analyses conducted according to intention‐to‐treat principle |
| Selective reporting (reporting bias) | Low risk | All outcomes reported as specified in ANZCTR (anzctr.org.au/Trial/Registration/TrialReview.aspx?id=336915) and in trial publications reported consistently |
| Other bias | Unclear risk | Quote: "no significant differences in major characteristics between the two treatment groups with the exception of tumour grade" E‐T‐CMF group had 36% II, 57% III vs E‐CMF group with 52% II and 45% III |
HORG.
| Methods | Randomised controlled trial Multi‐centre, 9 centres in Greece and Cyprus Central randomisation, stratified according to number of positive lymph nodes and menopausal status Accrual June 1995 to October 2004 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal
Aged 18 to 75 years
Median age 56 years (26 to 73)
Operable early stage (II to IIIa) breast cancer with clear surgical margins
Axillary node positive: 100%
ECOG performance status: 0 to 2
Exclusion of metastatic disease HR positive: 68% to 74% in each treatment arm |
|
| Interventions | ARM 1 (FEC)
FEC × 6 21‐day cycles (5‐fluorouracil 700 mg/m², epirubicin 75 mg/m², cyclophosphamide 700 mg/m²) ARM 2 (D‐EC) D × 4 21‐day cycles (docetaxel 100 mg/m²) followed by EC × 4 21‐day cycles (epirubicin 75 mg/m², cyclophosphamide 700 mg/m²) Tamoxifen 20 mg/d for 5 years given to all patients with ER‐ and/or PR‐positive tumours Postoperative radiotherapy given after chemotherapy to all patients after breast‐conserving surgery and to high‐risk patients following mastectomy No prior chemotherapy, no endocrine or radiation therapy allowed Prophylactic G‐CSF not permitted |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 62.5 months Conducted by the Hellenic Cooperative Oncology Group (HeCOG) No record of trial registration found For the review update, number of events per treatment arm for DFS and unadjusted hazard ratio and confidence intervals for OS and DFS provided by trial authors. O‐E and V for OS and DFS derived using Method 3 (Tierney 2007) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation for number of lymph nodes and menopausal status |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "treatment allocation was done centrally" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | History, physical exam, and routine bloods performed every 3 months for first 2 years, every 6 months for following 3 years, and yearly thereafter. Imaging (mammography, chest X‐ray, liver ultrasound) performed 1 year post surgery and yearly for 5‐year follow‐up. Physical examination, full blood count, and biochemistry performed before each course of chemotherapy. Toxicity graded according to NCI CTC version 2.0. LVEF measured by radio‐isotopic or echocardiographic methods at baseline, after completion of chemotherapy, and at 1‐year follow‐up Comment: no independent adjudication committee involved in outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "there were 16 (4.2%) patients in the D/EC arm and 27 (7.1%) in the FEC who were lost to follow‐up (P=0.084); all these patients were censored in DFS and OS analyses" Efficacy analyses based only on participants who received treatment |
| Selective reporting (reporting bias) | Low risk | Outcomes specified in the methods and results sections in the trial publication consistent. No record of trial registration found |
| Other bias | Low risk | Quote: "baseline patient characteristics were well balanced between the two treatment arms" |
ICE II‐GBG 52.
| Methods | Randomised controlled trial Multi‐centre (63 sites), Germany (part of German Breast Group), open‐label Randomisation performed centrally and stratified by participating centre, risk assessment method (pT3/4, pN2/3, or clinicopathological, or high urokinase plasminogen activator (uPA) or high plasminogen activator inhibitor 1 (PAI‐1)), age, oestrogen receptor/progesterone receptor/HER2 status Accrual April 2009 to April 2013 Baseline characteristics balanced between treatment groups | |
| Participants | Female or male patients aged ≥ 65 years with Charlson co‐morbidity index ≤ 2 Median age 72 (range 65 to 84) Two‐thirds of patients had clinopathologically medium‐ to high‐risk pT1/2 pN0/1 breast cancer 65.5% hormone receptor‐positive/HER2‐negative disease; 16.9% HER2‐positive disease; 17.6% triple‐negative breast cancer Exclusion of metastatic disease Prior chemotherapy for any malignancy and concurrent or previous systemic investigational or established anti‐tumour treatment not permitted | |
| Interventions | ARM 1 (EC or CMF)
EC × 4 21‐day cycles (epirubicin 90 mg/m², cyclophosphamide 600 mg/m²) or CMF × 6 28‐day cycles (cyclophosphamide 500 mg/m², methotrexate 40 mg/m², 5‐fluorouracil 600 mg/m² on days 1 and 8) based on investigators’ decision ARM 2 (nPX) nPX × 6 21‐day cycles (nab‐paclitaxel 100 mg/m² on days 1, 8, and 15 every 3 weeks with a week of rest every 6 weeks) plus capecitabine (1000 mg/m²) twice daily on days 1 to 14 every 3 weeks Sequential radiotherapy, anti‐HER2 therapy, and endocrine treatment recommended as per national guidelines Primary prophylaxis with granulocyte colony‐stimulating factor not recommended |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
Toxicity, assessed using Common Terminology Criteria for Adverse Events (version 3.0) |
|
| Notes | Median follow‐up: 22.8 months Clinical Trial Identifier: NCT01204437 (see clinicaltrials.gov/ct2/show/NCT01204437) Funded: supported received from Celgene and Roche; sponsored/collaborators were the German Breast Group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “…randomization at a 1:1 ratio was performed centrally and stratified according to participating center, risk assessment method, age, and estrogen receptor/progesterone receptor/HER2 status…” |
| Allocation concealment (selection bias) | Low risk | Randomisation performed centrally |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label study |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Assessment including clinical visits and breast imaging techniques every 3 months for 2 years, then every 6 months from year 2 to 5 for the diagnosis of local, locoregional, ipsilateral, or contralateral recurrence; distant metastasis, or death. Toxicity graded according to National Cancer Institute Common Toxicity Criteria Comment: no involvement of an independent adjudication committee for outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All efficacy analysis intention‐to‐treat; safety analysis including patients who received treatment |
| Selective reporting (reporting bias) | Low risk | Trial registry record outcomes reported in the trial publication |
| Other bias | Low risk | No other sources identified |
NCIC‐CTG MA21a.
| Methods | Randomised controlled trial, 3‐arm trial Multi‐centre, international Central randomisation by minimisation procedure Stratified according to number of positive nodes, type of surgery, ER status Accrual December 2000 to May 2005 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal
Age 60 years or younger. Median age not provided
Operable breast cancer, resected with clear surgical margins
Approximately 72% axillary node positive and 28% high risk node negative
Exclusion of metastatic disease HR positive: ER positive: 59% to 60% in each treatment arm |
|
| Interventions | ARM 1 (CEF):
CEF × 6 28‐day cycles (cyclophosphamide 75 mg/m² orally days 1 to 14, epirubicin 60 mg/m² days 1 and 8, fluorouracil 500 mg/m² days 1 and 8) ARM 2 (EC‐T): EC with G‐CSF × 6 14‐day cycles (epirubicin 120 mg/m², cyclophosphamide 830 mg/m²) followed by paclitaxel × 4 21‐day cycles (paclitaxel 175 mg/m²) Tamoxifen 20 mg/d for 5 years given to all patients with ER‐positive tumours. After October 2004, aromatase inhibitors allowed After June 2005, trastuzumab for 1 year allowed for patients with HER2‐positive cancer Radiotherapy given to all women following breast‐conserving surgery, and given following mastectomy according to institutional practice |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 30.4 months
Clinical Trial Identifier: NCT00014222 (see clinicaltrials.gov/ct2/show/NCT00014222) Supported in part by the Canadian Cancer Society, National Institutes of Health, and the following companies; Pfizer, Bristol‐Myers Squibb For the review update, O‐E and V for DFS derived using Method 3 (Tierney 2007) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were assigned using a minimization procedure to one of three regimens" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "patients were assigned... by the NCIC CTG central office" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Patients undergoing history, physical exam, CBC count, platelet count, and liver function tests at each follow‐up every 3 months for first year, every 4 months in second year, every 6 months to the end of 5 years, and yearly thereafter. Mammography performed yearly. Toxicity evaluations by NCI CTC Version 2.0, performed on day 1 of each cycle of chemotherapy Comment: no independent adjudication committee involved for outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all 2,104 patients randomly assigned to the study are included in the efficacy analysis" ITT principle used |
| Selective reporting (reporting bias) | Low risk | Method consistent with trial registry record (clinicaltrials.gov/ct2/show/NCT00014222). To date, only RFS, OS, and toxicity outcomes presented, DFS and QoL data yet to be published |
| Other bias | Low risk | Quote: "baseline characteristics were similar among treatments" |
NCIC‐CTG MA21b.
| Methods | See details in NCIC‐CTG MA21a | |
| Participants | See details in NCIC‐CTG MA21a | |
| Interventions | Three‐arm trial. For MA21b: ARM 1 (CEF): CEF × 6 28‐day cycles (cyclophosphamide 75 mg/m² orally days 1 to 14, epirubicin 60 mg/m² days 1 and 8, fluorouracil 500 mg/m² days 1 and 8) ARM 3 (AC‐T): AC × 4 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) followed by T × 4 21‐day cycles (paclitaxel 175 mg/m²) |
|
| Outcomes | See details in NCIC‐CTG MA21a | |
| Notes | See details in NCIC‐CTG MA21a | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | See NCIC‐CTG MA21a |
| Allocation concealment (selection bias) | Low risk | See NCIC‐CTG MA21a |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | See NCIC‐CTG MA21a |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | See NCIC‐CTG MA21a |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | See NCIC‐CTG MA21a |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | See NCIC‐CTG MA21a |
| Selective reporting (reporting bias) | Low risk | See NCIC‐CTG MA21a |
| Other bias | Low risk | See NCIC‐CTG MA21a |
NSABP B‐28.
| Methods | Randomised controlled trial Multi‐centre Central randomisation, stratified for number of positive nodes, type of surgery, and tamoxifen use Accrual August 1995 to May 1998 Baseline patient and tumour characteristics well balanced between treatment arms | |
| Participants | Female, premenopausal and postmenopausal Operable breast cancer with free surgical margins Axillary node positive: 100% HR positive: 66% in each treatment arm Exclusion of metastatic disease | |
| Interventions | ARM 1 (AC):
AC × 4 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) ARM 2 (AC‐T): AC × 4 21‐day cycles (as per control arm) followed by T × 4 21‐day cycles (paclitaxel 225 mg/m²) Tamoxifen 20 mg/d for 5 years given to all patients over 50 years and to those younger than 50 years with ER and/or PR positive Whole‐breast irradiation given to patients treated with breast‐conserving surgery |
|
| Outcomes | Primary endpoints:
Secondary endpoints:
Post‐hoc analyses presented in 2005 publication but not part of the trial protocol (as described in the publication):
|
|
| Notes | Intention‐to‐treat analysis
Median follow‐up: 64.6 months
79% continuing follow‐up at 5 years
Trial identifier not retrieved Supported by Public Health Service grants from NCI and NIH (USA) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patient assignment to the two treatment arms was balanced… using a biased‐coin minimisation algorithm" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "random assignment was performed centrally" at the NSABP Biostatistical Centre in Pittsburgh, PA |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Gynaecological exam (where applicable), chest X‐ray, and bilateral/unilateral mammogram yearly for first 5 years. Physical exam, gynaecological exam, and mammogram annually after 5‐year follow‐up. History, physical exam, and haematological studies and chemistries on day 1 before each cycle and every 6 months for first 5 years Comment: no independent adjudication committee involved in assessing these outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat analysis. 79% of participants continuing follow‐up at 64.6 months. Only 1 patient contributing no follow‐up in the non‐taxane treatment arm |
| Selective reporting (reporting bias) | Low risk | Outcomes listed in methods and results sections of the trial publications consistent with trial listing at the NCI (cancer.gov/about‐cancer/treatment/clinical‐trials/search/) |
| Other bias | Low risk | Quote: "patient and tumour characteristics were distributed evenly between the two groups" |
PACS 01.
| Methods | Randomised controlled trial Multi‐centre (85 centres across France and Belgium) Central randomisation and balanced per block Stratification by age, number of positive nodes, and centre Accrual June 1997 to March 2000 Baseline characteristics well balanced between treatment arms, except for combined HR status and ER status | |
| Participants | Female, premenopausal and postmenopausal, ages 18 to 64
Median age: 50 years (25 to 67)
Following surgery for operable node‐positive unilateral breast cancer (all had axillary dissection) Axillary node positive: 100% WHO performance status: < 2 HR positive: 81% in FEC‐D and 77% in FEC arm Exclusion of metastatic disease |
|
| Interventions | ARM A (FEC):
FEC × 6 21‐day cycles (fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m² day 1)
ARM B (FEC‐D):
FEC × 3 21‐day cycles (as in arm A), then docetaxel (D) × 3 21‐day cycles (docetaxel 100 mg/m² day 1) Tamoxifen 20 mg/d for 5 years for all ER‐ and/or PR‐positive patients and at investigators' discretion for ER/PR‐negative women Radiotherapy mandatory for all women following breast‐conserving surgery. Chest wall, supraclavicular fossa (SCF), and internal mammary chain radiotherapy recommended following mastectomy. Irradiation to axilla prohibited |
|
| Outcomes | Primary endpoints:
Secondary endpoints:
|
|
| Notes | Intention‐to‐treat analysis
Median follow‐up: 92.8 months
Trial identifier not retrieved Supported by Ligue Nationale Contre le Cancer, Sanofi‐Aventis, and Amgen |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation. |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "randomisation procedures were centralized" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Physical examination performed every 4 months from 2 years then 6 monthly for the next 3 years. Imaging studies (mammography, chest X‐ray, liver ultrasound, and bone scan) performed 1 year post surgery then annually for 5 years Quote: "ECG and absolute blood count were performed on day 21... Toxicity was graded according to WHO criteria" Comment: no independent adjudication committee involved in assessing these outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Results analysed by intention‐to‐treat principle. No apparent loss to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | Data on prespecified outcomes reported as outlined in the NCI trial registry record (see cancer.gov/about‐cancer/treatment/clinical‐trials/search/) except for QoL |
| Other bias | Unclear risk | Quote: "baseline characteristics were well balanced between treatment arms, except for combined hormone‐receptor status (HR) and estrogen‐receptor status" |
RAPP‐01.
| Methods | Randomised controlled trial Multi‐centre (11 French centres) Central randomisation using computerised random number generator. Stratified according to centre, node status, and proliferation Accrual June 1999 to January 2003 Baseline patient and tumour characteristics well balanced Closed prematurely for toxicity in 2003 | |
| Participants | Female, premenopausal and postmenopausal
Age 18 to 70 years
Median age: 52 years (range 26 to 70)
Unilateral, operable breast cancer with clear surgical margins
57% limited axillary node positive (≤ 3); 43% high risk node negative
Exclusion of metastatic disease HR positive: 80% to 81% in each treatment arm |
|
| Interventions | ARM 1 (AT):
AT × 4 21‐day cycles (doxorubicin 50 mg/m², docetaxel 75 mg/m²) ARM 2 (AC): AC × 4 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive Radiotherapy given as mandatory following breast‐conserving surgery Chemotherapy delivered without primary G‐CSF prophylaxis |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 64 months Trial registry record not identified Rene Huguenin Cancer Centre sponsored the trial supported in part by Aventis‐Oncology France and Ligue Regionale Contre le Cancer due Department des Yvelines For the review update, O‐E and V for DFS derived using Method 3 (Tierney 2007) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation Quote: "using a computerized random‐number generator" |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "central randomization was performed by fax or telephone in the Biostatistics Department of Rene Huguenin Cancer Center (Saint‐Cloud, France)" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information provided |
| Selective reporting (reporting bias) | High risk | Not all outcomes reported as outlined in the methods section of the trial publication. Primary outcome listed as DFS in 2005 full‐text article but reported as TTR in the 2009 abstract, and data related to overall survival not reported (although listed as a secondary outcome measure) |
| Other bias | Low risk | Quote: "the patients' characteristics were well balanced between the two treatment groups" |
Roy.
| Methods | Randomised controlled trial Open‐label, single institution Computer‐based randomisation procedure, 1:1 Accrual July 2007 to January 2010 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal
Median age: 47 years (18 to 66)
Operable, unilateral breast cancer, stage II
Post modified radical mastectomy with clear surgical margins Axillary node positive: 100% Exclusion of metastatic disease Karnofsky performance status: > 70 HR positive: 60% AC‐T and 58% AC |
|
| Interventions | ARM 1 (AC)
AC × 6 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) ARM 2 (AC‐T) AC × 3 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) followed by T × 3 21‐day cycles (paclitaxel 175 mg/m²) Tamoxifen 20 mg/d for 5 years to women with ER‐ and/or PR‐positive tumours, or tumours with unknown hormone receptor status Surgical treatment of mastectomy All patients given locoregional external beam radiotherapy post chemotherapy |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 24 months Trial registry record not found in Clinical Trials Registry ‐ India Funding source: not reported in trial publication For the review update, O‐E and V for OS and DFS derived using Method 3 (Tierney 2007) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the patient[s] were randomly assigned into two treatment arms by a computer‐based randomization procedure" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Mammography, CXR, and in patients receiving tamoxifen, pelvic and rectal examination with Pap smear performed annually. Examination and evaluation before the start of each chemotherapy cycle; CBC, liver and kidney function tests. Follow‐up performed 3 weeks after chemotherapy, then 2 monthly for the first year, followed by 3 monthly until end of the study. LVEF evaluated at baseline and at 18 months Comment: no involvement of an independent adjudication committee for outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "there was no loss of patients in any of the arms due to 'lost to follow‐up'" |
| Selective reporting (reporting bias) | Low risk | Outcomes specified in the methods and results sections of the trial publication consistent. No trial registry or protocol found on Clinical Trials Registry ‐ India |
| Other bias | Unclear risk | Baseline patient and tumour characteristics appearing well balanced, excluding tumour status with 28% T2 and 44% T3 in AC‐T treatment group vs 44% T2 and 24% T3 in AC treatment group |
Sakr.
| Methods | Randomised controlled trial Randomisation method not specified Accrual January 2006 to January 2010 Baseline patient and tumour characteristics well balanced | |
| Participants | Female, premenopausal and postmenopausal
Aged 18 to 65. Median age: 45 years (24 to 69)
Operable, unilateral breast cancer, completely resected with clear surgical margins
Axillary lymph node positive or high risk (T3/4) node negative Axillary node positive: 100% Exclusion of metastatic disease ECOG performance status: 0 to 1 HR positive: 81% FEC‐D and 77% FEC |
|
| Interventions | ARM 1 (FEC)
FEC × 6 21‐day cycles (fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m²) ARM 2 (FEC‐D) FEC × 3 21‐day cycles (fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m²) followed by D × 3 21‐day cycles (docetaxel 100 mg/m²) Tamoxifen 20 mg/d for 5 years to women with ER‐ and/or PR‐positive tumours Radiotherapy mandatory following breast‐conserving surgery, and used after mastectomy according to local guidelines |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 61 months No trial record identified Funding source: not reported in the trial publication For the review update, O‐E and V for OS derived using Method 3; Method 7 used for DFS (Tierney 2007) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the randomisation scheme was a permuted block design with an equal probability of assignment to either treatment arms" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Physical exam and metastatic work‐up (mammogram, chest X‐ray, abdominal ultrasound) performed 3 weeks after completion of chemotherapy, then every 3 months for first year, then yearly thereafter. Toxicity graded according to WHO criteria. Assessed with ECG, absolute blood count, and tolerability before each cycle Comment: no involvement of an independent adjudication committee for outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "data of three patients who did not receive treatment (2 FEC, 1 FEC‐D) were deleted list wise from the study" Not apparent that ITT analyses were conducted |
| Selective reporting (reporting bias) | Low risk | Outcomes specified in the methods and results sections of the trial publication consistent. No trial registry or protocol found |
| Other bias | Low risk | Quote: "baseline characteristics... were well balanced among the both treatment groups" |
Taxit 216.
| Methods | Randomised controlled trial Multi‐centre, open‐label Computer programme allocation via dynamic balancing algorithm Balancing factors: centre, number of lymph nodes involved, ER status, menopausal status Accrual from July 1998 to July 2002 | |
| Participants | Female, premenopausal and postmenopausal
Median age: 51 years (range 23 to 74)
Following surgery for operable breast cancer
Axillary node positive: 100% HR status reported; 66% ER positive |
|
| Interventions | ARM 1 (E‐CMF):
E × 4 21‐day cycles (epirubicin 120 mg/m² day 1) followed by CMF × 4 28‐day cycles (cyclophosphamide 600 mg/m², methotrexate 40 mg/m², and fluorouracil 600 mg/m² all IV days 1 and 8)
ARM 2 (E‐T‐CMF):
E × 4 21‐day cycles (dose as per arm 1) followed by T × 4 21‐day cycles (docetaxel 100 mg/m² on day 1) followed by CMF × 4 28‐day cycles (dose as in arm 1) Radiation therapy mandatory after breast‐conserving surgery and commencing after completion of chemotherapy Tamoxifen 20 mg/d recommended after chemotherapy for ER‐positive premenopausal women and for all postmenopausal women irrespective of ER status |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 62 months Trial identifier not retrieved | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "treatment allocation was performed by a computer program using a dynamic balancing algorithm" |
| Allocation concealment (selection bias) | Low risk | Quote: "randomization was done centrally by fax at the coordinating center (University of Naples Federico II, Italy)" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Physical exam and blood chemistry every 3 weeks and haematology weekly during chemotherapy. Follow‐up every 3 months for 2 years, every 6 months for years 3 to 5, and annually for years 6 to 10 Comment: no independent adjudication committee involved in assessing these outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy analyses done as per intention‐to‐treat principle. All participants apparently included in efficacy and safety analysis (i.e. no withdrawals) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes in the methods section reported in the results section of the thesis |
| Other bias | Low risk | Baseline characteristics well balanced |
TITAN.
| Methods | Randomised controlled trial Multi‐centre (66 sites), USA, open‐label Randomisation method (using web system) stratified according to number of involved axillary notes (0, 1 to 3, 4, more) Accrual December 2008 to January 2011 Baseline characteristics well balanced between treatment groups | |
| Participants | Females > 18 years, 70% postmenopausal
Median age: 54 years
Unilateral or synchronous bilateral, operable breast cancer with clear surgical margins, < pT4
Triple negative (ER/PR negative < 10% IHC, HER2 negative)
Axillary node positive: 33% (pN0 to pN3a eligible)
Exclusion of metastatic disease Prior anthracycline exposure not permitted ECOG: 0 to 2 |
|
| Interventions | ARM 1 (AC‐Ixa)
AC × 4 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) followed by Ixa × 4 21‐day cycles (ixabepilone 40 mg/m²) ARM 2 (AC‐T) AC × 4 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m²) followed by T × 12 weekly (paclitaxel 80 mg/m²) Colony‐stimulating growth factor allowed as per American Society of Clinical Oncology (ASCO) guidelines or at discretion of the treating physician MammoSite Brachytherapy radiation permitted if immediately following surgery and before study treatment. Radiotherpy following BCS or postmastectomy radiotherapy as per institutional guidelines except to those who had MammoSite Brachytherapy |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 48 months Clinical Trial Identifier: NCT00789581 (see clinicaltrials.gov/ct2/show/NCT00789581) Funded: supported in part by a grant from Bristol‐Myers Squibb; sponsored also by SCRI Development Innovations, LLC | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “…randomized …using an Interactive Web Response System and stratified according to the number of involved axillary lymph nodes (0, 1‐3, 4, or more)…” |
| Allocation concealment (selection bias) | Low risk | Randomisation via a web‐based system Comment: central randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label study |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Assessment including clinical visits every 3 months for 2 years, then every 6 months for 3 years; breast imaging performed annually. Toxicity graded according to National Cancer Institute Common Toxicity Criteria Comment: no involvement of an independent adjudication committee for outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All efficacy analysis intention‐to‐treat; safety analysis including patients who received treatment |
| Selective reporting (reporting bias) | Low risk | Trial registry record outlining disease‐free survival and overall survival as outcomes (clinicaltrials.gov/ct2/show/NCT00789581); trial publication adding toxicity data, which are considered important outcome data Outcomes reported in the methods and results sections of the trial publication consistent |
| Other bias | Low risk | No other sources identified |
UK TACT.
| Methods | Randomised controlled trial Multi‐centre (103 centres in the UK and 1 in Belgium), open‐label Computer‐generated permuted block randomisation 1:1 taxane regimen or control regimen Accrual February 2001 to July 2003 | |
| Participants | Female, premenopausal and postmenopausal
Aged over 18 years. Median age not reported
Operable, unilateral breast cancer, completely resected with clear surgical margins
80% axillary node positive; 20% high risk node negative
Exclusion of metastatic disease
WHO performance status: 0 to 1 HR positive: ER positive: 69% in each treatment group |
|
| Interventions | ARM 1 (FEC‐D)
FEC × 4 21‐day cycles (fluorouracil 600 mg/m², epirubicin 60 mg/m², cyclophosphamide 600 mg/m²) followed by D × 4 21‐day cycles (docetaxel 100 mg/m²) ARM 2a (Control) Regimen a (FEC) FEC × 8 21‐day cycles (fluorouracil 600 mg/m², epirubicin 60 mg/m², cyclophosphamide 600 mg/m²) ARM 2b (Control) Regimen b (E‐CMF) E × 4 21‐day cycles (epirubicin 100 mg/m²) followed by CMF × 4 28‐day cycles (cyclophosphamide 600 mg/m², methotrexate 40 mg/m², fluorouracil 600 mg/m² all IV on days 1 and 8) Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive. From 2005, aromatase inhibitors could be used as an alternative to tamoxifen Radiotherapy given as mandatory following breast‐conserving surgery, and used after mastectomy according to local guidelines Patients with HER2‐positive tumours allowed to enter clinical trials for trastuzumab |
|
| Outcomes | Primary endpoint:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 97.5 months Clinical Trial Identifier: NCT00033683 (see clinicaltrials.gov/ct2/show/NCT00033683) Funded by Cancer Research UK, Sanofi‐Aventis, Pfizer, and Roche For the review update, O‐E and V for OS and DFS derived using Method 3 (Tierney 2007) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly assigned by computer‐generated permuted block randomization" |
| Allocation concealment (selection bias) | Low risk | Quote: "independent randomisation was by telephone to the ICR‐CTSU or one of four regional clinical trial units" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Clinical follow‐up as per local policy with relevant details forwarded to the clinical trials unit. Adverse events assessed after every cycle of chemotherapy and every 3 months for 2 years of follow‐up. Graded according to NCI CTC criteria Comment: no involvement of an independent adjudication committee for outcome assessment |
| Blinding of outcome assessment ‐ QoL (detection bias) | High risk | Patients completing EORTC QLQ‐C30, BR23, HADS questionnaires before randomisation, before fifth and after eighth cycles of chemotherapy, and at 9, 12, 18, and 24 months of follow‐up |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "complete follow‐up data available for 3191 (94%) of 3410 alive patients" Analyses using ITT principle |
| Selective reporting (reporting bias) | Low risk | Methods consistent with the trial registry record (clinicaltrials.gov/ct2/show/NCT00033683). Outcomes reported in the methods and results sections of the trial publication consistent. Primary and secondary outcomes not listed at the time of registration |
| Other bias | Low risk | Baseline characteristics apparently balanced |
US Oncology 9735.
| Methods | Randomised controlled trial Randomisation method not specified Patients stratified by age and nodal status Accrual July 1997 to December January 2000 Baseline characteristics well balanced between treatment arms | |
| Participants | Female, premenopausal and postmenopausal Patients aged 18 to 75 years. Median age: 52 years (28 to 78) Following surgery for operable breast cancer, stage I to III Approximately 48% node negative and 52% node positive included Locally advanced tumours excluded HR positive: 71% of patients | |
| Interventions | ARM 1 (AC):
AC × 4 21‐day cycles (doxorubicin 60 mg/m², cyclophosphamide 600 mg/m² day 1)
ARM 2 (TC):
TC × 4 21‐day cycles (docetaxel 75 mg/m², cyclophosphamide 600 mg/m² day 1) Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive Radiotherapy as indicated |
|
| Outcomes | Primary endpoints:
Secondary endpoints:
|
|
| Notes | Median follow‐up: 84 months
Trial identifier not retrieved Supported by Sanofi‐Aventis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned" No further details provided in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment ‐ OS (detection bias) | Low risk | Assessment of overall survival unlikely to be influenced by no or incomplete blinding |
| Blinding of outcome assessment ‐ DFS & Toxicity (detection bias) | Unclear risk | Quote: "follow‐up was done at 6‐month intervals for 5 years and annually thereafter to 7 years. Lab work, annual chest X‐rays, mammograms (if indicated), and assessments of health status occurred at these visits" Quote: "toxicity was assessed at each patient visit and for 30 days after the last dose" Graded according to NCI CTC Comment: no independent adjudication committee involved in assessing these outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients reported as randomised (1016) accounted for in the results presented: 506 in the TC group and 510 in the AC group. Efficacy analyses conducted as intention‐to‐treat and safety analyses including patients who had received at least 1 dose of study drug |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes in the methods section reported in the results section of the trial publication |
| Other bias | Low risk | Quote: "patient characteristics were well balanced between treatment arms" |
AC: doxorubicin, cyclophosphamide. CBC: complete blood count. CMF: cyclophosphamide, methotrexate, fluorouracil. CR: complete response. CT: computed tomography. DCIS: ductal carcinoma in situ. DDFS: distant disease‐free survival. DFS: disease‐free survival. DMC: data monitoring committee. EC: epirubicin, cyclophosphamide. ER: oestrogen receptor. EV: epirubicin, vinorelbine. FAC: fluorouracil, doxorubicin, cyclophosphamide. FEC: fluorouracil, epirubicin, cyclophosphamide. G‐CSF: granulocyte colony‐stimulating factor. HER2: human epidermal growth factor 2. HR: hormone receptor. ITT: intention‐to‐treat. LVEF: left ventricular ejection fraction. NCI‐CTC: National Cancer Institute Common Toxicity Criteria. OS: overall survival. PR/PgR: progesterone receptor. QoL: quality of life. SD: stable response. TAC: docetaxel, doxorubicin, cyclophosphamide. V: vinorelbine.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Albert | Efficacy results for adjuvant group and neoadjuvant group could not be extracted |
| Dang | Paper presented some results of GEICAM 9906 trial, but it was a trial commentary |
| Di Leo | All arms contain taxane |
| Dunphy | This is not a randomised study |
| Hugh | Prognostic data were provided only for the BCIRG001 trial |
| Kummel | Dose‐dense treatment in the taxane‐containing arm confounds results |
| MD Anderson CC | Efficacy results for adjuvant group and neoadjuvant group could not be extracted |
| NCT02838225 | Both arms contain taxane |
| NSABP B‐27 | Patients received neoadjuvant chemotherapy before adjuvant taxane |
| Sparano 2015 | All 4 treatment arms contain taxane |
| SWOG S0221 | All arms contain taxane |
| SWOG S9623 | (1) Dose‐dense treatment in the taxane‐containing arm only, and (2) high‐dose dose‐escalated treatment with autologous haematopoietic progenitor cell transplantation in non‐taxane arm only confound the results |
| Wildiers | All arms contain taxane |
Characteristics of studies awaiting assessment [ordered by study ID]
EC‐DOC.
| Methods | Randomised controlled trial Multi‐centre, international Randomisation method not specified Accrual March 2000 to August 2005 Baseline patient and tumour characteristics well balanced |
| Participants | Female, premenopausal and postmenopausal Mean age: 51.5 years Operable breast cancer with clear surgical margins 1 to 3 positive lymph nodes Exclusion of metastatic disease HR positive: 78% |
| Interventions | ARM 1 (EC‐Doc):
EC × 4 21‐day cycles (epirubicin 90 mg/m², cyclophosphamide 600 mg/m²) followed by docetaxel × 4 21‐day cycles (docetaxel 100 mg/m²) ARM 2 (CEF/CMF): FEC × 6 21‐day cycles (fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m²) or CMF × 6 28‐day cycles (cyclophosphamide 600 mg/m², methotrexate 40 mg/m², fluorouracil 600 mg/m² on days 1 and 8) |
| Outcomes | Primary endpoint:
Secondary endpoints:
|
| Notes | Median follow‐up: 64 months Numbers of events and time‐to‐event data not reported in the abstract (64‐month follow‐up) |
EORTC 10041/BIG 3‐04 MINDACT.
| Methods | Randomised controlled trial Multi‐centre, international Randomisation method not specified |
| Participants | Women, premenopausal and postmenopausal Operable breast cancer Node‐negative or fewer than 3 positive lymph nodes Exclusion of metastatic disease For inclusion in chemotherapy randomisation, 1 of the following criteria must be met: (a) high risk of recurrence according to both clinical‐pathological criteria and 70‐gene signature, (b) high risk of recurrence according to clinical‐pathological criteria and low risk of recurrence according to 70‐gene signature and randomised to use the clinical‐pathological criteria for chemotherapy decision, or (c) low risk of recurrence according to clinical‐pathological criteria and high risk of recurrence according to 70‐gene signature and randomised to use the 70‐gene signature for chemotherapy decision |
| Interventions | ARM 1 (anthracycline‐based): patients can receive 1 of the following regimens:
FEC 100: on day 1 × 6 21‐day cycles
Canadian CEF: cyclophosphamide on days 1 to 14 and epirubicin and fluorouracil on days 1 and 8, × 6 28‐day cycles
CAF: cyclophosphamide, doxorubicin, and fluorouracil on day 1 × 6 28‐day cycles FAC: cyclophosphamide, doxorubicin, and fluorouracil on days 1 and 8 × 6 21‐day cycles E‐CMF: epirubicin on day 1, × 4 21‐day cycles ARM 2 (docetaxel and capecitabine): Docetaxel on day 1 and oral capecitabine twice daily on days 1 to 14 × 6 21‐day cycles Endocrine therapy (for all postmenopausal and some premenopausal patients who have endocrine‐responsive tumours) |
| Outcomes | Primary endpoints:
Secondary endpoints:
|
| Notes | Awaiting full‐text publication following conference proceedings abstract |
Kader.
| Methods | Randomised controlled trial Single institutional, Egypt Computer randomisation, 1:1 Accrual June 2007 to July 2008 Baseline patient and tumour characteristics well balanced between groups |
| Participants | Female, premenopausal and postmenopausal
Aged over 18 years. Median age: 50 years (28 to 69)
Operable, unilateral breast cancer, completely resected with clear surgical margins
Axillary node positive: 86.6% (FEC) to 90% (FEC‐D) Exclusion of metastatic disease |
| Interventions | ARM 1 (FEC‐D)
FEC × 3 (fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m²) followed by D × 3 (docetaxel 100 mg/m²) ARM 2 (FEC) FEC × 6 (fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m²) Tamoxifen 20 mg/d for 5 years to premenopausal or postmenopausal women, or aromatase inhibitors to postmenopausal women given for ER‐ and/or PR‐positive tumours. Radiotherapy given as mandatory following breast‐conserving surgery, and used after mastectomy according to local guidelines |
| Outcomes | Primary endpoint:
Secondary endpoint:
|
| Notes | The number of events for DFS not reported |
PACS 04.
| Methods | Randomised controlled trial Multi‐centre (82 centres in France and Belgium), open‐label Randomisation method not specified. Stratified according to institution and number of involved nodes Accrual February 2001 to August 2004 Baseline patient and tumour characteristics well balanced |
| Participants | Female, premenopausal and postmenopausal Age: 18 to 64 years. Median age: 50 Localised, unilateral, operable breast cancer, resected with clear surgical margins Axillary node positive (N1 to 3) Exclusion of metastatic disease |
| Interventions | Part 1 Arm 1 (FEC) FEC × 6 21‐day cycles (fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m²) Arm 2 (ED) ED × 6 21‐day cycles (epirubicin 75 mg/m², docetaxel 75 mg/m²) Part 2: for patients with HER2/neu‐positive tumours only, secondarily randomised to receive trastuzumab for 1 year or observation Tamoxifen 20 mg/d for 5 years given to all patients with ER and/or PR positive. Radiotherapy given as mandatory following breast‐conserving surgery |
| Outcomes | Primary endpoint:
Secondary endpoints:
|
| Notes | Median follow‐up: 59.3 months Clinical Trial Identifier: NCT0054587 (see clinicaltrials.gov/ct2/show/NCT00054587) Number of events for DFS not reported; additional data for OS to be provided with longer follow‐up period |
CMF: cyclophosphamide, methotrexate, fluorouracil. D: docetaxel. DFS: disease‐free survival. EC: epirubicin, cyclophosphamide. ED: epirubicin, docetaxel. ER: oestrogen receptor. FAC: fluorouracil, doxorubicin, cyclophosphamide. FEC: fluorouracil, epirubicin, cyclophosphamide. HR: hormone receptor. N: node. OS: overall survival. PR: progesterone receptor.
Characteristics of ongoing studies [ordered by study ID]
NCI‐H99‐0038.
| Trial name or title | Randomised Phase II Study of Adriamycin, Cytoxan/Taxol (ACT) vs. Cytoxan, Thiotepa, Carboplatin (STAMP V) in Patients With High‐Risk Primary Breast Cancer |
| Methods | Randomised controlled trial |
| Participants | Women, age 60 or younger; menopausal status not specified High‐risk breast cancer (stage 2 with at least 10 positive nodes, or stage 3a or 3b) |
| Interventions | All patients receiving conventional dose adjuvant chemotherapy (FAC × 4 cycles), then randomised to 1 of 2 high‐dose chemotherapy treatment arms
ARM 1 (ACT) (clinical trials.gov record states that it is closed to accrual as of 4/6/2006): doxorubicin, cyclophosphamide, and paclitaxel with peripheral blood stem cell rescue and G‐CSF support
ARM 2 (STAMP V): cyclophosphamide, carboplatin, and thiotepa with peripheral blood stem cell rescue and G‐CSF support as in arm 1 Tamoxifen (twice per day) for all hormone receptor‐positive patients for 5 years |
| Outcomes | Primary outcomes:
Secondary outcomes:
|
| Starting date | May 1999 Estimated completion date: November 2014 |
| Contact information | George Somlo, Chair, Cancer Center and Beckman Research Institute, City of Hope, Duarte, California, USA |
| Notes | Clinical Trial Identifier: NCT00004092; see clinicaltrials.gov/show/NCT00004092 |
NCT01966471.
| Trial name or title | A Randomized, Multicenter, Open‐Label, Phase III Trial Comparing Trastuzumab plus Pertuzumab plus a Taxane Following Anthracyclines vs Trastuzumab Emtansine plus Pertuzumab Following Anthracyclines as Adjuvant Therapy in Patients With Operable HER2‐Positive Primary Breast Cancer |
| Methods | Randomised controlled trial, parallel assignment Multi‐centre Open‐label |
| Participants | Women aged 18 years and older Operable primary invasive breast carcinoma HER2 positive Node‐positive or node‐negative disease |
| Interventions | ARM 1: Trastuzumab (8‐mg/kg loading dose, then 21‐day cycles of 6 mg/kg) + pertuzumab (840‐mg loading dose, then 21 day cycles of 420 mg) + taxane (21‐day cycles of 80 mg/m² paclitaxel or docetaxel) + standard anthracycline‐based chemotherapy ARM 2: Trastuzumab emtansine (Kadcyla (21‐day cycles of 3.6 mg/kg) + pertuzumab (840‐mg loading dose, then 21‐day cycles of 420 mg) + standard anthracycline‐based chemotherapy) Three to four cycles of standard anthracycline‐based chemotherapy |
| Outcomes | Primary outcome:
Secondary outcomes:
|
| Starting date | January 2014 Estimated completion date: January 2024 |
| Contact information | Hoffmann‐La Roche |
| Notes | Clinical Trial Identifier: NCT01966471 (see clinicaltrials.gov/ct2/show/NCT01966471) |
NCT02549677.
| Trial name or title | Epirubicin vs Docetaxel plus Cyclophosphamide in Lymph Node‐Negative, ER‐Positive, HER2‐Negative Breast Cancer (ELEGANT) |
| Methods | Randomised controlled trial |
| Participants | Women, aged > 18 and < 70 years
Pathologically verified no lymph node involvement, ER‐positive, HER2‐negative breast cancer Life expectancy > 12 months, ECOG 0 to 1 |
| Interventions | ARM 1: epirubicin (90 mg/m²) and cyclophosphamide (600 mg/m²) on day 1, every 3 weeks ARM 2: docetaxel (75 mg/m²) and cyclophosphamide (600 mg/m²) on day 1, every 3 weeks |
| Outcomes | Primary outcome:
Secondary outcomes:
|
| Starting date | October 2015 Estimated completion date: September 2019 |
| Contact information | Jiayi Wu or Yan Fang (fangyan4743@163.com), Ruijin Hospital, Shanghai, China |
| Notes | Clinical Trial Identifier: NCT02549677; see clinicaltrials.gov/ct2/show/NCT02549677 |
NNBC3.
| Trial name or title | Randomized Multicentre Study Comparing 6× FEC with 3× FEC‐3× Doc in High‐Risk Node Negative Patients With Operable Breast Cancer: Comparison of Efficacy and Evaluation of Clinico‐pathological and Biochemical Markers as Risk Selection Criteria |
| Methods | Randomised controlled trial, parallel assignment
Multi‐centre (Germany) Open‐label |
| Participants | Women aged 18 to 70 years; menopausal status not specified Histologically proven primary breast cancer (pT1b‐pT2, pN0, M0) Complete surgical excision with clear margins Lymph node negative |
| Interventions | Experimental: Arm A taxane‐containing FEC × 3 21‐day cycles (5‐fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m²) followed by Doc × 3 21‐day cycles (docetaxel 100 mg/m²) Active comparator: Arm B standard anthracycline FEC × 6 21‐day cycles (5‐fluorouracil 500 mg/m², epirubicin 100 mg/m², cyclophosphamide 500 mg/m²) |
| Outcomes | Primary outcome:
Secondary outcomes:
|
| Starting date | January 2002 Estimated completion date: February 2019 |
| Contact information | Eva J Kantelhardt Klinik und Poliklinik fur Gynakologie, Martin‐Luther Universitat, Halle Saale, Germany Email: eva.kantelhardt@medizin.uni‐halle.de |
| Notes | Clinical Trial Identifier: NCT01222052 (see clinicaltrials.gov/show/NCT01222052) |
ACT: adriamycin, cytoxan/taxol. DFS: disease‐free survival. DRFI: distant recurrence‐free interval. ER: oestrogen receptor FAC: fluorouracil, doxorubicin, cyclophosphamide. FEC: fluorouracil, epirubicin, cyclophosphamide. G‐CSF: granulocyte‐colony stimulating factor HER2: human epidermal growth factor 2. IDFS: invasive disease‐free survival.
Differences between protocol and review
Differences between the original review and the review update include the following.
For the inclusion criteria, we included studies where co‐interventions included the same targeted therapy in both treatment arms.
We revised the MEDLINE search strategy to include a new search syntax for effectively filtering search results to include human studies only. We also revised the Embase search strategy to update the search syntax used, including removal of irrelevant search terms and using more effective filters for limiting search results to randomised controlled trials, controlled clinical trials, and human studies only. We also revised search strategies for the WHO ICTRP and ClinicalTrials.gov, removing all irrelevant search terms. As part of Cochrane's conduct standards, we included in the Appendix a new search string for CENTRAL.
Given the different definitions used for DFS, we added a sensitivity analysis in the review update to assess whether results remained consistent irrespective of slight differences in DFS definitions.
We performed additional post‐hoc subgroup analyses that were clinically relevant (i.e. related to hormone receptor status) as well as sensitivity analyses based on risk of bias assessments.
We presented toxicity data in Analysis 11; therefore we removed the toxicity table (previously labelled as Table 16) in the original review from the review update. Outcomes selected for the main analysis were the same as those chosen for the original review, except for neuropathy. In the original version of the review, neuropathy was reported in only three studies; however 19 treatment comparisons reported grade 3 or 4 neuropathy in this updated version; therefore we conducted a pooled analysis. We conducted post‐hoc subgroup analyses by taxane type for the outcomes of neutropenia and neuropathy.
Contributions of authors
For the updated review: Screening studies and retrieving papers: LB, MW, NW. Conducting risk of bias assessments: LB, MW, NW. Extracting data: LB, MW, NW. Entering data into Review Manager: MW, LB. Analysing and interpreting data: LB, MW, NW. Providing clinical oversight: NW, AN. For the original review: Conceiving the review: Dr Anna Nowak. Designing the review: Dr Anna Nowak. Co‐ordinating the review: Dr Anna Nowak. Collecting data for the review: Dr Anna Nowak, Dr Tom Ferguson. Designing search strategies: Dr Anna Nowak. Undertaking searches: Dr Anna Nowak, Dr Tom Ferguson. Screening search results: Dr Anna Nowak, Dr Tom Ferguson. Organising retrieval of papers: Dr Anna Nowak, Dr Tom Ferguson. Screening retrieved papers against inclusion criteria: Dr Anna Nowak, Dr Tom Ferguson. Appraising quality of papers: Dr Anna Nowak, Dr Tom Ferguson. Extracting data from papers: Dr Anna Nowak, Dr Tom Ferguson, Ms Rosmary Vagg. Obtaining and screening data from unpublished studies: Dr Anna Nowak, Dr Tom Ferguson. Managing data for the review: Dr Anna Nowak, Dr Tom Ferguson, Ms Rosmary Vagg. Developing a protocol: Dr Anna Nowak, Dr Nicholas Wilcken, Dr Davina Ghersi. Entering data into RevMan: Dr Anna Nowak, Dr Tom Ferguson. Analysing data: Dr Anna Nowak, Dr Tom Ferguson. Interpreting data: Dr Anna Nowak, Dr Tom Ferguson, Dr Nicholas Wilcken. Providing a methodological perspective: Dr Anna Nowak, Dr Nicholas Wilcken, Dr Davina Ghersi. Providing a clinical perspective: Dr Anna Nowak, Dr Tom Ferguson, Dr Nicholas Wilken. Providing a policy perspective: Dr Anna Nowak, Dr Nicholas Wilken, Dr Davina Ghersi. Writing the review: Dr Anna Nowak, Dr Tom Ferguson. Providing general advice on the review: Dr Anna Nowak, Dr Nicholas Wilken, Dr Davina Ghersi. Securing funding for the review: Dr Anna Nowak. Performing previous work that was the foundation of the current study: Dr Anna Nowak, Dr Nicholas Wilken, Dr Davina Ghersi.
Sources of support
Internal sources
NHMRC Clinical Trials Centre, Australia.
University of Western Australia, Australia.
Sir Charles Gairdner Hospital, Australia.
Royal Perth Hospital, Australia.
External sources
National Breast Cancer Centre, Australia.
Declarations of interest
MW: none known. LB: none known. TF: none known. DG: none known. AN: no relevant conflicts of interest relevant to the topic under review, breast cancer. All declarations relate directly to consulting work and clinical trials in relation to malignant mesothelioma or brain cancer. NW: intermittently served on advisory boards for pharmaceutical companies and been paid honoraria for educational lectures sponsored by pharmaceutical companies though not related to the topic under review.
These authors contributed equally to this work
These authors contributed equally to this work
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
ADEBAR {published data only}
- Gauger K, Bismarck FV, Heinrigs M, Janni W, Steinfeld, Augustin D, et al. Phase III study evaluating the role of docetaxel in the adjuvant setting of breast cancer patients with ≥ 4 involved lymph nodes: ADEBAR‐Study. Journal of Clinical Oncology 2005;23(Suppl 16):908. [Google Scholar]
- Janni W, Harbeck N, Rack B, Augustin D, Jueckstock J, Wischnik A, et al. Randomised phase III trial of FEC120 vs EC‐docetaxel in patients with high‐risk node‐positive primary breast cancer: final survival analysis of the ADEBAR study. British Journal of Cancer 2016;114(8):863‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janni W, Harbeck N, Sommer H, Rack B, Augustin D, Jueckstock J, et al. Randomized phase III trial of fluorouracil/epirubicin/cyclophosphamide (FEC) versus epirubicin/cyclophosphamide/docetaxel in patients with node positive primary breast cancer: final analysis of the ADEBAR study. Author provided draft manuscript. Received on the 10th of March 2014.
- Janni W, Harbeck N, Sommer H, Rack B, Augustin D, Simon W, et al. Sequential treatment with epirubicin/cyclophosphamide followed by docetaxel is equieffective, but less toxic than FEC120, in the adjuvant treatment of breast cancer patients with extensive lymph node involvement: the German ADEBAR phase III study. Cancer Research 2009;69:604. [Google Scholar]
- Janni W, Harbeck N, Sommer H, Rack B, Augustin D, Simon W, et al. Sequential treatment with epirubicin/cyclophosphamide followed by docetaxel versus FEC120 in the adjuvant treatment of node‐positive breast cancer patients: final survival analysis of the German ADEBAR phase III study. Journal of Clinical Oncology 2012;30:1081. [Google Scholar]
- Janni WJ, Harbeck N, Sommer H, Rack B, Salmen J, Augustin D, et al. Sequential treatment with epirubicin/cyclophosphamide, followed by docetaxel vs. FEC120, in the adjuvant treatment of breast cancer patients with extensive lymph node involvement: final survival analysis of the German ADEBAR phase III study. Cancer Research 2011;71:PD07‐01. [Google Scholar]
- NCT00047099. Combination chemotherapy in treating women with breast cancer. clinicaltrials.gov/ct2/show/NCT00047099 (published 27 January 2003).
- Schonherr A, Aivazova‐Fuchs V, Annecke K, Juckstock J, Hepp P, Andergassen U, et al. Toxicity analysis in the ADEBAR trial: sequential anthracycline‐taxane therapy compared with FEC120 for the adjuvant treatment of high‐risk breast cancer. Breast Care 2012;7(4):289‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwentner L, Harbeck N, Singer S, Eichler M, Rack B, Forstbauer H, et al. Short term quality of life with epirubicin‐fluorouracil‐cyclophosphamide (FEC) and sequential/cyclophosphamide‐docetaxel (EC‐DOC) chemotherapy in patients with primary breast cancer ‐ results from the prospective multi‐center randomized ADEBAR trial. The Breast 2016;27:69‐77. [DOI] [PubMed] [Google Scholar]
BCIRG 001 {published data only}
- Mackey JR, Martin M, Pienkowski T, Rolski J, Guastalla JP, Sami A, et al. Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node‐positive breast cancer: 10‐year follow‐up of the phase 3 randomised BCIRG 001 trial. Lancet Oncology 2013;14(1):72‐80. [DOI] [PubMed] [Google Scholar]
- Martin M, Pienkowski T, Mackey J, Pawlicki M, Guastalla JP, Weaver C, et al. Adjuvant docetaxel for node‐positive breast cancer. New England Journal of Medicine 2005;352(22):2302‐13. [DOI] [PubMed] [Google Scholar]
BIG 2‐98 {published data only}
- Crown JP, Francis P, Leo A. Docetaxel given concurrently with or sequentially to anthracycline‐based adjuvant therapy for patients with node‐positive breast cancer, in comparison with non‐taxane combination chemotherapy: first results of the BIG 2‐98 trial at 5 years median follow‐up. Journal of Clinical Oncology 2006;24(Suppl 18):LBA519. [Google Scholar]
- Francis P, Crown J, Leo A, Buyse M, Balil A, Andersson M, et al. Adjuvant chemotherapy with sequential or concurrent anthracycline and docetaxel: Breast International Group 02‐98 randomized trial. Journal of the National Cancer Institute 2008;100(2):121‐33. [DOI] [PubMed] [Google Scholar]
- Sonnenblick A, Francis PA, Azim Jr HA, Azambuja E, Nordenskjöld B, Gutiérez J, et al. Final 10‐year results of the Breast International Group 2‐98 phase III trial and the role of Ki67 in predicting benefit of adjuvant docetaxel in patients with oestrogen receptor positive breast cancer. European Journal of Cancer 2015;51(12):1481‐9. [DOI] [PubMed] [Google Scholar]
Boccardo {published data only}
- Boccardo F, Amadori D, Guglielmini P, Sismondi P, Farris A, Agostara B, et al. Epirubicin followed by cyclophosphamide, methotrexate and 5‐fluorouracil versus paclitaxel followed by epirubicin and vinorelbine in patients with high‐risk operable breast cancer. Oncology (United States) 2010;78(3‐4):274‐81. [DOI] [PubMed] [Google Scholar]
CALGB 40101 {published data only}
- Shulman LN, Berry DA, Cirrincione CT, Becker H, Perez EA, O'Regan R, et al. Comparison of doxorubicin and cyclophosphamide (AC) versus single‐agent paclitaxel (T) as adjuvant therapy for breast cancer in women with 0‐3 positive axillary nodes: CALGB 40101. Journal of Clinical Oncology 2013;31:1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman LN, Berry DA, Cirrincione CT, Becker HP, Perez EA, O'Regan R, et al. Comparison of doxorubicin and cyclophosphamide versus single‐agent paclitaxel as adjuvant therapy for breast cancer in women with 0 to 3 positive axillary notes: GALGB40101 (Alliance). Journal of Clinical Oncology 2014;32(22):2311‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman LN, Cirrincione CT, Berry DA, Becker HP, Perez EA, O'Regan R, et al. Six cycles of doxorubicin and cyclophosphamide or paclitaxel are not superior to four cycles as adjuvant chemotherapy for breast cancer in women with zero to three positive axillary nodes: Cancer and Leukemia Group B 40101. Journal of Clinical Oncology 2012;30(33):4071‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
CALGB 9344 {published data only}
- Henderson IC, Berry DA, Demetri GD, Cirrincione CT, Goldstein LJ, Martino S, et al. Improved outcomes from adding sequential paclitaxel but not from escalating doxorubicin dose in an adjuvant chemotherapy regimen for patients with node‐positive primary breast cancer. Journal of Clinical Oncology March 2003;21(6):976‐83. [DOI] [PubMed] [Google Scholar]
DEVA {published data only}
- Coombes RC, Bliss JM, Espie M, Erdkamp F, Wals J, Tres A, et al. Randomized, phase III trial of sequential epirubicin and docetaxel versus epirubicin alone in postmenopausal patients with node‐positive breast cancer. Journal of Clinical Oncology 2011;29(24):3247‐54. [DOI] [PubMed] [Google Scholar]
- Coombes RC, Bliss JM, Espie M, Erdkamp F, Wals JJ, Tres A, et al. DEVA: randomized trial of sequential epirubicin and docetaxel versus epirubicin alone in node‐positive postmenopausal early breast cancer (EBC) patients. Journal of Clinical Oncology 2010;28:15. [DOI] [PubMed] [Google Scholar]
- International Cancer Collaborative Group. A multicentre randomised trial of sequential epirubicin and docetaxel versus epirubicin in node positive postmenopausal breast cancer patients. Protocol only 1997.
E2197 {published data only}
- Goldstein L, O'Neill A, Sparano J, et al. E2197. Phase III AT vs. AC in the adjuvant treatment of node‐positive and high‐risk node‐negative breast cancer. Journal of Clinical Oncology 2005;23(16 Suppl 7):Abstract 512. [Google Scholar]
- Goldstein LJ, O'Neill A, Sparano JA, Perez EA, Shulman LN, Martino S, et al. Concurrent doxorubicin plus docetaxel is not more effective than concurrent doxorubicin plus cyclophosphamide in operable breast cancer with 0 to 3 positive axillary nodes: North American Breast Cancer Intergroup Trial E 2197. Journal of Clinical Oncology 2008;26(25):4092‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparano J, O'Neil A, Gray R, Perez E, Shulman L, Martino S, et al. 10‐year update of E2197: phase III doxorubicin/docetaxel (AT) versus doxorubicin/cyclophosphamide (AC) adjuvant treatment of LN+ and high‐risk LN‐ breast cancer and the comparison of the prognostic utility of the 21‐gene recurrence score (RS) with clinicopathologic features. Journal of Clinical Oncology 2012;30:1021. [Google Scholar]
ECTO {published data only}
- Gianni L, Baselga J, Eiermann W, Guillem Porta V, Semiglazov V, Lluch A, et al. European Cooperative Trial in Operable Breast Cancer (ECTO). Improved freedom from progression from adding paclitaxel to doxorubicin followed by cyclophosphamide methotrexate and fluorouracil. Journal of Clinical Oncology 2005;23(Suppl 7):513. [DOI] [PubMed] [Google Scholar]
- Gianni L, Baselga J, Eiermann W, Guillem Porta V, Semiglazov V, Lluch A, et al. Feasibility and tolerability of sequential doxorubicin/paclitaxel followed by cyclophosphamide, methotrexate, and fluorouracil and its effects on tumor response as preoperative therapy. Clinical Cancer Research 2005;11(24):8715‐21. [DOI] [PubMed] [Google Scholar]
- Gianni L, Baselga J, Eiermann W, Porta VG, Semiglazov V, Lluch A, et al. Phase III trial evaluating the addition of paclitaxel to doxorubicin followed by cyclophosphamide, methotrexate, and fluorouracil, as adjuvant or primary systemic therapy: European Cooperative Trial in Operable Breast Cancer. Journal of Clinical Oncology 2009;27(15):2474‐81. [DOI] [PubMed] [Google Scholar]
ELDA {published data only}
- Nuzzo F, Morabito A, Maio E, Rella F, Gravina A, Labonia V, et al. Weekly docetaxel versus CMF as adjuvant chemotherapy for elderly breast cancer patients: safety data from the multicentre phase 3 randomised ELDA trial. Critical Reviews in Oncology/Hematology 2008;66(2):171‐80. [DOI] [PubMed] [Google Scholar]
- Perrone F, Nuzzo F, Rella F, Gravina A, Iodice G, Labonia V, et al. Weekly docetaxel versus CMF as adjuvant chemotherapy for older women with early breast cancer: final results of the randomized phase III ELDA trial. Annals of Oncology 2015;26(4):675‐82. [DOI] [PubMed] [Google Scholar]
FinHer {published data only}
- Joensuu H, Bono P, Kataja V, Alanko T, Kokko R, Asola R, et al. Fluorouracil, epirubicin, and cyclophosphamide with either docetaxel or vinorelbine, with or without trastuzumab, as adjuvant treatments of breast cancer: final results of the FinHer Trial. Journal of Clinical Oncology 2009;27(34):5685‐92. [DOI] [PubMed] [Google Scholar]
- Joensuu H, Kellokumpu‐Lehtinen PL, Bono P, Alanko T, Kataja V, Asola R, et al. Adjuvant docetaxel or vinorelbine with or without Trastuzumab for breast cancer. New England Journal of Medicine 2006;354(8):809‐20. [DOI] [PubMed] [Google Scholar]
GEICAM 2003‐02 {published data only}
- Martin M. Fluorouracil plus doxorubicin and cyclophosphamide (FAC) versus FAC plus weekly paclitaxel as adjuvant treatment of node negative high risk breast cancer patients. Physician Data Query (PDQ) 2005.
- Martin M, Ruiz A, Ruiz‐Borrego M, Barnadas A, Gonzales S, Calavo L, et al. Fluorouracil, doxorubicin, and cyclophosphamide (FAC) versus FAC followed by weekly paclitaxel as adjuvant therapy for high‐risk, node‐negative breast cancer: results form the GEICAM/2003‐02 study. Journal of Clinical Oncology 2013;31(20):2593‐9. [DOI] [PubMed] [Google Scholar]
GEICAM 9805 {published data only}
- Martin M, Lluch A, Segui MA, Anton A, Fernandez‐Chacon C, Ruiz A, et al. Toxicity and health‐related quality of life in node negative breast cancer patients receiving adjuvant treatment with TAC or FAC: impact of adding prophylactic growth factors to TAC. GEICAM Study 9805. Journal of Clinical Oncology 2005;23:604. [Google Scholar]
- Martin M, Lluch A, Segui MA, Anton A, Ruiz A, Ramos A. Prophylactic growth factor support with adjuvant docetaxel, doxorubicin, and cyclophosphamide (TAC) for node‐negative breast cancer: an interim safety analysis of the GEICAM 9805 study. Journal of Clinical Oncology 2004;22:620. [Google Scholar]
- Martin M, Segui MA, Anton A, Ruiz A, Ramos M, Adrover E, et al. Adjuvant docetaxel for high‐risk, node‐negative breast cancer. New England Journal of Medicine 2010;363(23):2200‐10. [DOI] [PubMed] [Google Scholar]
- Martín M, Lluch A, Seguí MA, Ruiz A, Ramos M, Adrover E, et al. Toxicity and health‐related quality of life in breast cancer patients receiving adjuvant docetaxel, doxorubicin, cyclophosphamide (TAC) or 5‐fluorouracil, doxorubicin and cyclophosphamide (FAC): impact of adding primary prophylactic granulocyte‐colony stimulating factor to the TAC regimen. Annals of Oncology 2006;17(8):1205‐12. [DOI] [PubMed] [Google Scholar]
GEICAM 9906 {published data only}
- Martin M, Rodriguez‐Lescure A, Ruiz A, Alba E, Calvo L, Ruiz‐Borrego M, et al. Molecular predictors of efficacy of adjuvant weekly paclitaxel in early breast cancer. Breast Cancer Research and Treatment 2010;123(1):149‐57. [DOI] [PubMed] [Google Scholar]
- Martin M, Rodriguez‐Lescure A, Ruiz A, Alba E, Calvo L, Ruiz‐Borrego M, et al. Randomized phase 3 trial of fluorouracil, epirubicin, and cyclophosphamide alone or followed by paclitaxel for early breast cancer. Journal of the National Cancer Institute 2008;100(11):805‐14. [DOI] [PubMed] [Google Scholar]
- Martín M, Rodríguez‐Lescure A, Ruiz A, Alba E, Calvo L, Ruiz‐Borrego M, et al. Multicentre, randomized phase 3 study of adjuvant chemotherapy for node positive breast cancer comparing 6 cycles of FEC(90) versus 4 cycles of FEC(90) followed by 8 weekly paclitaxel administrations: interim efficacy analysis of GEICAM 9906 Trial. Breast Cancer Research and Treatment 2005;94(Suppl 1):39. [Google Scholar]
- Rodriguez‐Lescure A, Martin M, Ruiz A, Alba E, Calvo M, Ruiz‐Borrego M. Multicenter, randomized phase 3 study of adjuvant chemotherapy for axillary positive breast cancer (APBC) comparing 6 cycles of FEC vs 4 cycles of FEC followed by 8 weekly paclitaxel administrations: safety analysis of GEICAM 9906 trial. Journal of Clinical Oncology 2004;22(Suppl 14):596. [Google Scholar]
GOIM 9902 {published data only}
- Participating Institutions of GOIM 9902 Trial, Italian Cooperative Group, Rome, Italy. Epirubicin and cyclophosphamide (EC) vs docetaxel followed by EC in adjuvant treatment of node positive breast cancer. A multicenter randomized phase 3 study. Journal of Clinical Oncology 2001;20:1836. [Google Scholar]
- Vici P, Brandi M, Giotta F, Foggi P, Schittulli F, lauro L, et al. A multicenter phase III prospective randomized trial of high‐dose epirubicin in combination with cyclophosphamide (EC) versus docetaxel followed by EC in node‐positive breast cancer. GOIM (Gruppo Oncologico Italia Meridionale) 9902 study. Annals of Oncology 2012;23(5):1121‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
GONO MIG‐5 {published data only}
- Mastro L, Costantini M, Durando A, Michelotti A, Danese S, Aitini E, et al. Cyclophosphamide, epirubicin, and 5‐fluorouracil versus epirubicin plus paclitaxel in node‐positive early breast cancer patients: a randomized, phase III study of Gruppo Oncologico Nord Ovest‐Mammella Intergruppo Group. Journal of Clinical Oncology 2008;26(Suppl 10):516. [Google Scholar]
- Mastro L, Levaggi A, Michelotti A, Cavazzini G, Adami F, Scotto T, et al. 5‐fluorouracil, epirubicin and cyclophosphamide versus epirubicin and paclitaxel in node‐positive early breast cancer: a phase III randomized GONO‐MIG5 trial. Breast Cancer Research and Treatment 2016;155(1):117‐26. [DOI] [PubMed] [Google Scholar]
- NCT02450058. Adjuvant FEC versus EP in breast cancer (MIG5). clinicaltrials.gov/ct2/show/NCT02450058 (first received 21 May 2015).
- Participating Institutions to GONO‐MIG 5 study. Absence of clinically relevant cardiotoxicity in early breast cancer patients treated with the association of epirubicin plus paclitaxel: results from the Italian MIG 5 Study. Journal of Clinical Oncology 2000;19:363. [Google Scholar]
HeCOG {published data only}
- Fountzilas G, Skarlos D, Dafni U, Gogas H, Briasoulis E, Pectasides D, et al. Postoperative dose‐dense sequential chemotherapy with epirubicin, followed by CMF with or without paclitaxel, in patients with high risk operable breast cancer: a randomized phase 3 study conducted by the Hellenic Cooperative Oncology Group. Annals of Oncology 2005;16(11):1762‐71. [DOI] [PubMed] [Google Scholar]
HORG {published data only}
- Polyzos A, Malamos N, Boukovinas I, Adamou A, Ziras N, Kalbakis K, et al. FEC versus sequential docetaxel followed by epirubicin/cyclophosphamide as adjuvant chemotherapy in women with axillary node‐positive early breast cancer: a randomized study of the Hellenic Oncology Research Group (HORG). Breast Cancer Research and Treatment 2010;119(1):95‐104. [DOI] [PubMed] [Google Scholar]
ICE II‐GBG 52 {published data only}
- NCT01204437. Adjuvant chemotherapy for elderly non frail patients with an increased risk for relapse of a primary carcinoma of the breast (ICE‐II). clinicaltrials.gov/ct2/show/NCT01204437 (first received 17 September 2010).
- Minckwitz G, Conrad B, Reimer T, Decker T, Eidtmann H, Eiermann W, et al. A randomized phase 2 study comparing EC or CMF versus nab‐paclitaxel plus capecitabine as adjuvant chemotherapy for nonfrail elderly patients with moderate to high‐risk early breast cancer (ICE II‐GBG 52). Cancer 2015;121(20):3639‐48. [DOI] [PubMed] [Google Scholar]
NCIC‐CTG MA21a {published data only}
- Burnell M, Levine M, Chapman JA, et al. A randomized trial of CEF versus dose dense EC followed by paclitaxel versus AC followed by paclitaxel in women with node positive or high risk node negative breast cancer, NCIC CTG MA.21: results of an interim analysis. Breast Cancer Research and Treatment, San Antonio Breast Cancer Symposium. 2006:53.
- Burnell M, Levine MN, Chapman JAW, Bramwell V, Gelmon K, Walley B, et al. Cyclophosphamide, epirubicin, and fluorouracil versus dose‐dense epirubicin and cyclophosphamide followed by paclitaxel versus doxorubicin and cyclophosphamide followed by paclitaxel in node‐positive or high‐risk node‐negative breast cancer. Journal of Clinical Oncology 2010;28(1):77‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnell MJ, Shepherd L, Gelmon K, Bramwell V, Walley B, Vandenberg E, et al. A randomized trial of CEF versus dose dense EC followed by paclitaxel versus AC followed by paclitaxel in women with node positive or high risk node negative breast cancer, NCIC CTG MA.21: results of the final relapse free survival analysis. Cancer Research 2012;72(Suppl 24):P1‐13‐01. [Google Scholar]
- Participating Organisations. Phase 3 randomized study of adjuvant cyclophosphamide, epirubucin, and fluorouracil versus cyclophosphamide, epirubicin, filgrastim (G‐CSF), and epoetin alfa followed by paclitaxel versus cyclophosphamide and doxorubicin followed by paclitaxel in premenopausal or early postmenopausal women with previously resected node positive or high‐risk node negative stage 1‐3A breast cancer. Protocol only 2001.
NCIC‐CTG MA21b {published data only}
- Burnell M, Levine M, Chapman JA, et al. A randomized trial of CEF versus dose dense EC followed by paclitaxel versus AC followed by paclitaxel in women with node positive or high risk node negative breast cancer, NCIC CTG MA.21: results of an interim analysis. Breast Cancer Research and Treatment, San Antonio Breast Cancer Symposium. 2006:53.
- Burnell M, Levine MN, Chapman JAW, Bramwell V, Gelmon K, Walley B, et al. Cyclophosphamide, epirubicin, and fluorouracil versus dose‐dense epirubicin and cyclophosphamide followed by paclitaxel versus doxorubicin and cyclophosphamide followed by paclitaxel in node‐positive or high‐risk node‐negative breast cancer. Journal of Clinical Oncology 2010;28(1):77‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnell MJ, Shepherd L, Gelmon K, Bramwell V, Walley B, Vandenberg E, et al. A randomized trial of CEF versus dose dense EC followed by paclitaxel versus AC followed by paclitaxel in women with node positive or high risk node negative breast cancer, NCIC CTG MA.21: results of the final relapse free survival analysis. Cancer Research 2012;72(Suppl 24):P1‐13‐01. [Google Scholar]
- Participating Organisations. Phase 3 randomized study of adjuvant cyclophosphamide, epirubucin, and fluorouracil versus cyclophosphamide, epirubicin, filgrastim (G‐CSF), and epoetin alfa followed by paclitaxel versus cyclophosphamide and doxorubicin followed by paclitaxel in premenopausal or early postmenopausal women with previously resected node positive or high‐risk node negative stage 1‐3A breast cancer. Protocol only issue 2001.
NSABP B‐28 {published data only}
- Mamounas EP, Bryant J, Lembersky B, Fehrenbacher L, Sedlacek SM, Fisher B, et al. Paclitaxel after doxorubicin plus cyclophosphamide as adjuvant chemotherapy for node‐positive breast cancer: results from NSABP B‐28. Journal of Clinical Oncology 2005;23(16):3686‐96. [DOI] [PubMed] [Google Scholar]
PACS 01 {published data only}
- Coudert B, Asselain B, Campone M, Spielmann M, Machiels JP, Penault‐Llorca F, et al. Extended benefit from sequential administration of docetaxel after standard fluorouracil, epirubicin, and cyclophosphamide regimen for node‐positive breast cancer: the 8‐year follow‐up results of the UNICANCER‐PACS01 trial. Oncologist 2012;17(7):900‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino P, Siani C, Roché H, Protière C, Fumoleau P, Spielmann M, et al. Cost‐effectiveness of adjuvant docetaxel for node‐positive breast cancer patients: results of the PACS 01 economic study. Annals of Oncology 2010;21(7):1448‐54. [DOI] [PubMed] [Google Scholar]
- Roche H, Fumoleau P, Spielmann M, et al. Five years analysis of the PACS 01 Trial: 6 cycles of FEC100 vs 3 cycles of FEC100 followed by 3 cycles of docetaxel for the adjuvant treatment of node positive breast cancer. San Antonio Breast Cancer Symposium. 2004.
- Roché H, Fumoleau P, Spielmann M, Canon JL, Delozier T, Serin D, et al. Sequential adjuvant epirubicin‐based and docetaxel chemotherapy for node‐positive breast cancer patients: the FNCLCC PACS 01 Trial. Journal of Clinical Oncology 2006;24(36):5664‐71. [DOI] [PubMed] [Google Scholar]
RAPP‐01 {published data only}
- Brain E, Debled M, Eymard J, Bachelot T, Extra J, Serin D, et al. Final results of the RAPP‐01 phase III trial comparing doxorubicin and docetaxel with doxorubicin and cyclophosphamide in the adjuvant treatment of high‐risk node negative and limited node positive (≤ 3) breast cancer patients. Cancer Research 2009;69(2 Suppl 1):4101. [Google Scholar]
- Brain EG, Bachelot T, Serin D, Kirscher S, Graic Y, Eymard J, et al. Life‐threatening sepsis associated with adjuvant doxorubicin plus docetaxel for intermediate‐risk breast cancer. Journal of the American Medical Asssociation 2005;293(19):2367‐71. [DOI] [PubMed] [Google Scholar]
- Brain EGC, Bachelot T, Serin D, Graic Y, Eymard JC, Extra JM, et al. Phase III trial comparing doxorubicin docetaxel (AT) with doxorubicin cyclophosphamide (AC) in the adjuvant treatment of high‐risk node negative (pN0) and limited node positive (pN+</=3) breast cancer (BC) patients (pts): first analysis of toxicity. Journal of Clinical Oncology 2004;22(Suppl 14):617. [Google Scholar]
Roy {published data only}
- Roy C, Choudhury KB, Pal M, Saha A, Bag S, Banerjee C. Adjuvant chemotherapy with six cycles of AC regimen versus three cycles of AC regimen followed by three cycles of paclitaxel in node‐positive breast cancer. Indian Journal of Cancer 2012;49(3):266‐71. [DOI] [PubMed] [Google Scholar]
Sakr {published data only}
- Sakr H, Hamed RH, Anter AH, Yossef T. Sequential docetaxel as adjuvant chemotherapy for node‐positive or/and T3 or T4 breast cancer: clinical outcome (Mansoura University). Medical Oncology 2013;30(1):457. [DOI] [PubMed] [Google Scholar]
Taxit 216 {published data only}
- Bianco AR, Matteis A, Manzione L, Boni C, Palazzo S, Palma M, et al. Sequential epirubicin‐docetaxel‐CMF as adjuvant therapy of early breast cancer: results of the Taxit216 multicenter phase III trial. Journal of Clinical Oncology 2006;24:LBA520. [Google Scholar]
- Cognetti F, Laurentiis M, Matteis A, Manzione L, Boni C, Palazzo S, et al. Sequential epirubicin‐docetaxel‐CMF as adjuvant therapy for node‐positive early stage breast cancer: updated results of the taxit216 randomized trial. Annals of Oncology 2008;19(Suppl 8):viii77–viii88: 1820. [Google Scholar]
- Forestieri V. Docetaxel in adjuvant therapy of breast cancer: results of the TAXIT 216 multicenter phase III trial. Docetaxel in Adjuvant Therapy of Breast Cancer: Results of the TAXIT 216 Multicenter Phase III Trial. Naples, Italy: University of Naples Federico II, 2008. [Google Scholar]
TITAN {published data only}
- NCT00789581. A randomized trial of Ixempra versus Taxol in adjuvant therapy of triple negative breast cancer (TITAN). clinicaltrials.gov/ct2/show/NCT00789581 (first received 13 November 2008).
- Yardley DA, Arrowsmith ER, Daniel BR, Eakle J, Brufsky A, Drosick DR, et al. TITAN: phase III study of doxorubicin/cyclophosphamide followed by ixabepilone or paclitaxel in early‐stage triple‐negative breast cancer. Breast Cancer Research and Treatment 2017;164(3):649‐58. [DOI] [PubMed] [Google Scholar]
- Yardley DA, Hainsworth JD, Harwin WN, Goble SA, Daniel BR, Ackerman MA, et al. TITAN: ixabepilone versus weekly paclitaxel following doxorubicin/cyclophosphamide (AC) adjuvant chemotherapy in triple‐negative breast cancer (TNBC): preliminary toxicity of a Sarah Cannon Research Institute phase III trial. Journal of Clinical Oncology. 2011; Vol. 29:1103.
UK TACT {published data only}
- Bliss JM, Ellis P, Kilburn L, Bartlett J, Bloomfield D, Cameron D, et al. Mature analysis of UK Taxotere as Adjuvant Chemotherapy (TACT) trial (CRUK 01/001); effects of treatment and characterisation of patterns of breast cancer relapse. Cancer Research 2012;72:P1‐13‐03. [Google Scholar]
- Ellis P, Barrett‐Lee P, Johnson L, Cameron D, Wardley A, O'Reilly S, et al. Sequential docetaxel as adjuvant chemotherapy for early breast cancer (TACT): an open‐label, phase III, randomised controlled trial. The Lancet 2009;373(9676):1681‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopwood P, Ellis P, Barrett‐Lee P, Bliss J, Hall E, Johnson L, et al. Impact of quality of life (QL) during chemotherapy (CT) of FEC‐T compared to FEC or E‐CMF: results from the UK NCRI Taxotere as Adjuvant Chemotherapy trial (TACT). Journal of Clinical Oncology 2005;23(Suppl 16):661. [Google Scholar]
- Participating Organizations. Phase 3 randomized adjuvant study of fluorouracil, epirubicin, and cyclophosphamide (FEC) or epirubicin followed by cyclophosphamide, methotrexate, and fluorouracil versus FEC followed by sequential docetaxel in women with resected stage 1 or 2 breast cancer. Protocol only 2002.
US Oncology 9735 {published data only}
- Jones S, Holmes FA, O'Shaughnessy J, Blum JL, Vukelja SJ, Mclntyre KJ, et al. Docetaxel with cyclophosphamide is associated with an overall survival benefit compared with doxorubicin and cyclophosphamide: 7‐year follow‐up of US oncology research trial 9735. Journal of Clinical Oncology 2009;27(8):1177‐83. [DOI] [PubMed] [Google Scholar]
- Jones SE, Savin M, Holmes FA, et al. Preliminary results of a prospective randomized trial of adjuvant chemotherapy for patients with stage 1‐3 operable, invasive breast cancer comparing 4 cycles of AC to 4 courses of TC. Journal of Clinical Oncology 2001;20:128. [Google Scholar]
- Jones SE, Savin MA, Holmes FA, O'Shaughnessy JA, Blum JL, Vukelja S, et al. Phase III trial comparing doxorubicin plus cyclophosphamide with docetaxel plus cyclophosphamide as adjuvant therapy for operable breast cancer. Journal of Clinical Oncology 2006;24(34):5381‐7. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Albert {published data only}
- Albert JM, Buzdar AU, Guzman R, Allen PK, Strom EA, Perkins GH, et al. Prospective randomized trial of 5‐fluorouracil, doxorubicin, and cyclophosphamide (FAC) versus paclitaxel and FAC (TFAC) in patients with operable breast cancer: impact of taxane chemotherapy on locoregional control. Breast Cancer Research and Treatment 2011;128(2):421‐7. [DOI] [PubMed] [Google Scholar]
Dang {published data only}
- Dang C. Randomized phase 3 trial of fluorouracil, epirubicin, and cyclophosphamide alone or followed by paclitaxel for early breast cancer. Current Breast Cancer Reports 2009;1(1):1‐2. [DOI] [PubMed] [Google Scholar]
Di Leo {published data only}
- Leo A, Crown J, Nogaret JM, Duffy K, Bartholomeus S, Dolci S, et al. Feasibility of docetaxel‐containing regimens in the adjuvant treatment of breast cancer. Annals of Oncology 2000;11(2):169‐75. [DOI] [PubMed] [Google Scholar]
Dunphy {published data only}
- Dunphy F, Rodriguez J, Petruska P, Velasquez W, McIntyre W, Spitzer G. High dose therapy for high risk (stage 3) breast cancer. Phase II trials of two treatment regimens cytoxan‐etoposide‐cisplatin and cytoxan‐etoposide‐cisplatin‐taxol‐carboplatin. Breast Cancer Research and Treatment 1997;46(1):308. [Google Scholar]
Hugh {published data only}
- Hugh J, Hanson J, Cheang MCU, Nielsen TO, Perou CM, Dumontet C, et al. Breast cancer subtypes and response to docetaxel in node‐positive breast cancer: use of an immunohistochemical definition in the BCIRG 001 trial. Journal of Clinical Oncology 2009;27(8):1168‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kummel {published data only}
- Kummel S, Krocker J, Kohls A, Breitbach GP, Morack G, Budner M. Randomised trial: survival benefit and safety of adjuvant dose‐dense chemotherapy for node‐positive breast cancer. British Journal of Cancer 2006;94(9):1237‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
MD Anderson CC {published data only}
- Buzdar AU, Singletary SE, Valero V, Booser DJ, Ibrahim NK, Rahman Z, et al. Evaluation of paclitaxel in adjuvant chemotherapy for patients with operable breast cancer: preliminary data of a prospective randomized trial. Clinical Cancer Research 2002;8(5):1073‐9. [PubMed] [Google Scholar]
NCT02838225 {published data only}
- NCT02838225. DA versus DAC as postoperative adjuvant treatment for early‐stage breast cancer. clinicaltrials.gov/ct2/show/NCT02838225 (first received 20 July 2016).
NSABP B‐27 {published data only}
- Bear HD, Anderson S, Smith RE, Geyer CE Jr, Mamounas EP, Fisher B, et al. Sequential preoperative or postoperative docetaxel added to preoperative doxorubicin plus cyclophosphamide for operable breast cancer: National Surgical Adjuvant Breast and Bowel Project Protocol B‐27. Journal of Clinical Oncology 2006;24(13):2019‐27. [DOI] [PubMed] [Google Scholar]
Sparano 2015 {published data only}
- Sparano JA, Zhao F, Martino S, Ligibel JA, Perez EA, Saphner T, et al. Long‐term follow‐up of the E1199 phase III trial evaluating the role of taxane and schedule in operable breast cancer. Journal of Clinical Oncology 2015;33(21):2353‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
SWOG S0221 {published data only}
- Budd GT, Barlow WE, Moore HCF, Hobday TJ, Stewart JA, Isaacs C, et al. First analysis of SWOG S0221: a phase III trial comparing chemotherapy schedules in high‐risk early breast cancer. Journal of Clinical Oncology 2011;29:1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
SWOG S9623 {published data only}
- Moore H, Green S, Gralow J, Bearman S, Lew D, Barlow W, et al. Intensive dose‐dense compared with high‐dose adjuvant chemotherapy for high‐risk operable breast cancer: Southwest Oncology Group/Intergroup Study 9623. Journal of Clinical Oncology 2007;25(13):1677‐82. [DOI] [PubMed] [Google Scholar]
- Mortimer JE. A comparison of intensive sequential chemotherapy using doxorubicin plus paclitaxel plus cyclophosphamide with high dose chemotherapy and autologous hematopoietic progenitor cell support for primary breast cancer in women with 4‐9 involved axillary lymph nodes, phase 3, Intergroup. Protocol only 2001. [CALGB 9960]
Wildiers {published data only}
- Wildiers H. A randomized phase II trial exploring feasibility of densification and optimal sequencing of postoperative adjuvant fluorouracil, epirubicin plus cyclophosphamide (FEC) and docetaxel chemotherapy in patients with high risk primary operable breast cancer. Physician Data Query (PDQ) 2006.
References to studies awaiting assessment
EC‐DOC {published data only}
- Gluz O, Erber R, Kates R, Kreipe H, Liedtke C, Pelz E, et al. Predictive value of HER2, topoisomerase‐II (Topo‐II) and tissue inhibitor of metalloproteinases (TIMP‐1) for efficacy of taxane‐based chemotherapy in intermediate risk breast cancer ‐ results of the EC‐Doc Trial. Cancer Research 2011;71:P1‐06‐03. [DOI] [PubMed] [Google Scholar]
- Nitz U, Huober J, Lisboa B, Harbeck N, Fischer H, Moebus V, et al. Interim results of Intergroup EC‐Doc Trial: a randomized multicentre phase III trial comparing adjuvant CEF/CMF to EC‐Docetaxel in patients with 1‐3 positive lymph nodes. Journal of Clinical Oncology 2008;26(Suppl 15):515. [Google Scholar]
- Nitz U, Huober J, Lisboa B, Harbeck N, Fischer H, Moebus V, et al. Superiority of sequential docetaxel over standard FE100C in patients with intermediate risk breast cancer: survival results of the randomized intergroup phase III trial EC‐Doc. Cancer Research 2008;69:78. [Google Scholar]
EORTC 10041/BIG 3‐04 MINDACT {published data only}
- Cardoso F, Piccart‐Gebhart MJ, Rutgers EJ, Litiere S, Van't VL, Viale G, et al. Standard anthracycline‐based vs docetaxel‐capecitabine in early breast cancer: results from the chemotherapy randomization (R‐C) of EORTC 10041/BIG3‐04 MINDACT phase III trial. Journal of Clinical Oncology; 2017 Annual meeting of the American Society of Clinical Oncology. 2017; Vol. 35:15 Suppl 1.
Kader {published data only}
- Kader YA, El‐Nahas T, Sakr A. Adjuvant chemotherapy for luminal A breast cancer: a prospective study comparing two popular chemotherapy regimens. OncoTargets and Therapy 2013;6:1073‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
PACS 04 {published data only}
- Participating Organizations. Phase 3 randomized study of adjuvant docetaxel and epirubicin versus adjuvant cyclophosphamide, epirubucin, and fluorouracil with or without trastuzumab in women with nonmetastatic adenocarcinoma of the breast with lymph node invasion. Protocol only 2003.
- Roché H, Allouache D, Romieu G, Bourgeois H, Canon J, Serin D, et al. Five‐year analysis of the FNCLCC‐PACS04 Trial: FEC100 vs ED75 for the adjuvant treatment of node positive breast cancer. Cancer Research 2009;69:602. [Google Scholar]
- Spielmann M, Roche H, Delozier T, Canon JL, Romieu G, Bourgeois H, et al. Trastuzumab for patients with axillary‐node‐positive breast cancer: results of the FNCLCC‐PACS 04 trial. Journal of Clinical Oncology 2009;27(36):6129‐34. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCI‐H99‐0038 {published data only}
- Participating Organizations. Phase 2 randomized study of doxorubicin, cyclophosphamide, and paclitaxel vs cyclophosphamide, thiotepa, and carboplatin in patients with high‐risk primary breast cancer. Protocol only 2001.
NCT01966471 {published data only}
- NCT01966471. A study of Kadcyla (trastuzumab emtansine) plus Perjeta (pertuzumab) following anthracyclines in comparison with Herceptin (trastuzumab) plus Perjeta and a taxane following anthracyclines as adjuvant therapy in patients with operable HER2‐positive primary breast cancer. clinicaltrials.gov/show/NCT01966471 (first received 21 October 2013).
NCT02549677 {published data only}
- NCT02549677. Epirubicin versus docetaxel plus cyclophosphamide in lymph node negative, ER‐positive, Her2‐negative breast cancer (ELEGANT). clinicaltrials.gov/ct2/show/NCT02549677 (first received 15 September 2015).
NNBC3 {published data only}
- Kantelhardt E, Vetter M, Schmidt M, Veyret C, Augustin D, Hanf V, et al. Prospective evaluation of prognostic factors uPA/PAI‐1 in node‐negative breast cancer: phase III NNBC3‐Europe trial (AGO, GBG, EORTC‐PBG) comparing 6 x FEC versus 3 x FEC/3 x docetaxel. BioMed Central 2011;11:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
AIHW 2012
- Breast cancer in Australia: an overview. Australian Institute of Health and Welfare & Cancer Australia. Case series no. 71. Canberra: Cat no CAN 67, October 2012.
Bishop 1999
- Bishop JF, Dewar J, Toner GC, Smith J, Tattersall MH, Olver IN, et al. Initial paclitaxel improves outcome compared with CMFP combination chemotherapy as front‐line therapy in untreated metastatic breast cancer. Journal of Clinical Oncology 1999;17(8):2355‐64. [DOI] [PubMed] [Google Scholar]
Bria 2006
- Bria E, Nistico C, Cuppone F, Carlini P, Ciccarese M, Milella M, et al. Benefit of taxanes as adjuvant chemotherapy for early breast cancer: pooled analysis of 15,500 patients. Cancer 2006;106(11):2337‐44. [DOI] [PubMed] [Google Scholar]
Chan 1999
- Chan S, Friedrichs K, Noel D, Pinter T, Belle S, Vorobiof D, et al. Prospective randomized trial of docetaxel versus doxorubicin in patients with metastatic breast cancer. The 303 Study Group. Journal of Clinical Oncology 1999;17(8):2341‐54. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Crown 2002
- Crown J, Dieras V, Kaufmann M, Minckwitz G, Kaye S, Leonard R, et al. Chemotherapy for metastatic breast cancer ‐ report of a European expert panel. Lancet Oncology 2002;3(12):719‐27. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
EBCTCG 2005
- Early Breast Cancer Trialists' Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15‐year survival: an overview of the randomised trials.. Lancet 2005;365:1687‐1717. [DOI] [PubMed] [Google Scholar]
Ferlay 2015
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International Journal of Cancer 2015;136:E359‐86. [DOI] [PubMed] [Google Scholar]
Ghersi 2005
- Ghersi D, Wilcken N, Simes J, Donoghue E. Taxane containing regimens for metastatic breast cancer. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD003366] [DOI] [PubMed] [Google Scholar]
GRADEproGDT
- GRADEproGDT: GRADEpro Guideline Development Tool [software]. McMaster University, 2015 (developed by Evidence Prime, Inc). Available from www.gradepro.org.
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Joensuu 2006
- Joensuu H, Kellokumpu‐Lehtinen P, Bono P, Alanko T, Kataja V, Asola R, et al. Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. New England Journal of Medicine 2006;354:809‐20. [DOI] [PubMed] [Google Scholar]
NCCN 2007
- National Comprehensive Cancer Network Practice Guidelines in Oncology: Breast Cancer; Version 2, 2007. www.nccn.org/professionals/physician_gls/PDF/breast.pdf (accessed 30 April 2007).
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Qin 2011
- Qin YY, Li H, Guo KJ, Ye XF, Wei X, Zhou YH, et al. Adjuvant chemotherapy, with or without taxanes, in early or operable breast cancer: a meta‐analysis of 19 randomized trials with 30698 patients. PLoS ONE 2011;6(11):e26946. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glaziou P, et al. Chapter 12. Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Tierney 2007
- Tierney J, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Methodology 2007;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yusuf 1991
- Yusuf S, Wittes J, Probstfield J, Tyroler HA. Analysis and interpretation of treatment effects in subgroups of patients in randomized clinical trials. JAMA 1991;266(1):93‐8. [PubMed] [Google Scholar]
References to other published versions of this review
Ferguson 2007
- Ferguson T, Wilcken N, Vagg R, Ghersi D, Nowak AK. Taxanes for adjuvant treatment of early breast cancer. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD004421.pub2] [DOI] [PubMed] [Google Scholar]