

Abstract
Background
Charcot‐Marie‐Tooth disease (CMT) comprises a large variety of different forms of motor and sensory neuropathies. The most frequent are demyelinating forms (CMT1) and axonal forms (CMT2). The molecular basis of several CMT forms has been clarified during the last 15 years. Since muscle wasting and sensory disturbance are the main features of these syndromes, treatments aim to improve motor impairment and sensory disturbances. Specific treatment trials are rare.
Objectives
The objective was to review systematically all randomised and quasi‐randomised studies of any treatment for CMT.
Search methods
We searched the Cochrane Neuromuscular Disease Group Trials Register, MEDLINE (January 1966 to August 2007), EMBASE (January 1980 to August 2007), LILACS (January 1982 to August 2007) for randomised controlled trials of treatment for CMT.
Selection criteria
We included randomised and quasi‐randomised trials of any treatment for people with CMT. Where a study aimed to evaluate the treatment of general neuromuscular symptoms of people with peripheral neuropathy including CMT, we included the study if we were able to identify the effect of treatment in the CMT group. Observational studies and case reports on the treatment of people with CMT were not included.
Data collection and analysis
Two review authors (PY and TBB) extracted the data, assessed study quality and performed data extraction independently.
Main results
Only one trial with only eight participants met all the inclusion criteria and provided the primary outcome measure for this review. In this trial, four participants treated with neurotrophin‐3 had more improvement after six months on the Neuropathy Impairment Score, mean difference ‐9.50 (95% CI ‐13.77 to ‐5.23), than those four treated with placebo. Small trials of exercise training, creatine monohydrate, orthoses and purified bovine brain ganglioside injections (Cronassial) showed no significant benefit in people with genetically undefined CMT1 or CMT2.
Authors' conclusions
Small trials of exercise, creatine, purified brain gangliosides, and orthoses have been performed. None showed significant benefit. A very small trial of neurotrophin‐3 showed possible minor benefit which needs to be replicated in a larger trial. None of the two trials were large enough to detect moderate benefit or harm. Larger RCTs are needed for any form of pharmacological intervention as well as as for any form of physical intervention. Outcome measures should include a validated composite scale such as the Charcot‐Marie‐Tooth neuropathy scale.
Keywords: Humans, Charcot‐Marie‐Tooth Disease, Charcot‐Marie‐Tooth Disease/therapy, Creatine, Creatine/administration & dosage, Exercise Therapy, Gangliosides, Gangliosides/administration & dosage, Neurotrophin 3, Neurotrophin 3/therapeutic use, Orthotic Devices
Treatment for Charcot‐Marie‐Tooth disease (hereditary motor and sensory neuropathy)
Charcot‐Marie‐Tooth disease is a broad spectrum of different types of inherited peripheral neuropathy. The most common types affect motor and sensory nerves and cause muscle wasting and sensory loss. There have been few trials of treatment for Charcot‐Marie Tooth disease. One very small trial of the nerve growth promoting factor, neurotrophin‐3, showed possible benefit but needs to be replicated. Trials of exercise, orthosis, creatine and ganglioside injections have been done but did not show significant benefit. These were all too small to identify moderate benefit or harm. Trials of vitamin C for the commonest type of Charcot‐Marie Tooth disease are in progress.
Background
Introduction
Charcot‐Marie‐Tooth disease (CMT) comprises a huge group of inherited neuropathies which primarily affect either the myelin sheath or the axon of the peripheral nerve (Dyck 2005a). Primary loss of the myelin sheath, called demyelination, leads in most cases to secondary axonal degeneration resulting in muscular atrophy (Martini 2001). Thus the main motor clinical signs are atrophy and weakness starting in the feet and calves and later affecting the hands (Dyck 2005a). Sensory features are less prominent but may cause much discomfort and add to the disability. Skeletal deformities like scoliosis or foot deformities can be additional features (Dyck 2005a). Depending on the underlying molecular genetic defect the phenotypic features vary from mild weakness to severe paralysis and disability (Kuhlenbaumer 2002). Histopathological findings in peripheral nerve biopsies reveal demyelination or axonal degeneration depending on the specific type of CMT. Different patterns of demyelination range from pure demyelination with adequate remyelination to focal demyelinated stretches or pathological folding of the myelin sheath (Gabreels‐Festen 1993). Demyelination and axonal loss can be differentiated (Dyck 2005a). Demyelination results in severely slowed nerve conduction velocities (NCVs). Types of CMT with primary axonal loss and preserved myelin are characterised by normal or near normal NCVs. Weakness and sensory deficits are the relevant symptoms to be treated and are the focus of this review. Established strategies are lacking for the treatment of CMT and its symptoms (Young 2001). Some trials which aimed to improve muscle strength or sensory deficits have been undertaken. Trials have been performed with corticosteroids and antioxidants, and with physiotherapy for improving muscle function (Grandis 2005; Young 2001).
With respect to the genetic basis of CMT syndromes, several animal models have been generated over the last ten years (Martini 1997; Young 2001). In particular, animal models for the most frequent form of CMT, Charcot‐Marie‐Tooth type 1A (CMT1A) suggested new approaches to treatment. Vitamin C (Ascorbic acid) (Passage 2004) and the progesterone antagonist onapristone (Sereda 2003) each improved muscle function and reduced demyelination in animal models of CMT1A.
Genetics
A broad spectrum of different gene mutations have been identified as a cause of CMT (Grandis 2005; Pareyson 2004; Young 2001). We will give a short introduction of the most frequent forms of CMT, autosomal dominant demyelinating (CMT1), X‐chromosome linked Charcot‐Marie‐Tooth disease (CMTX) and autosomal dominant axonal Charcot‐Marie‐Tooth disease type 2 (CMT2). CMT1 is primarily demyelinating and the most common form of autosomal dominant inherited CMT (Nelis 1996). In the last two decades the molecular genetic basis of this syndrome has largely been elucidated (Young 2003).The most frequent genetic defect in CMT1 is an intrachromosomal duplication on chromosome 17p11.2 (Lupski 1991; Nelis 1996). The gene encoding the peripheral myelin protein 22 (PMP22) is located within the duplicated region (Timmerman 1992). This gene has been shown to be the culprit for the genetic subgroup CMT1A (Kuhlenbaumer 2002). CMT1B is defined by the presence of mutations in the gene encoding Myelin Protein Zero (MPZ) (Hayasaka 1993a; Hayasaka 1993b; Hayasaka 1993c). CMT1C is caused by mutation of a gene encoding the LITAF/SIMPLE‐protein (LITAF) and was identified recently (Street 2003). The gene encoding the transcription factor early growth arrest protein 2 (EGR2) is responsible for CMT1D (Young 2003).
CMTX is caused by mutations in the gene encoding the gap junction protein beta1 (GJB1) which is localised on the X‐chromosome. CMTX is characterised by the histopathological signs of axonal degeneration and demyelination (Hahn 2001; Nicholson 1993) which cause the typical electrophysiological features of reduced NCV and reduction of compound muscle action potential as well.
The pure axonal form of CMT is called CMT2 (Kuhlenbaumer 2002). Recently mutations in the gene encoding the mitochondrial fusion protein mitofusin 2 were shown to be a frequent cause of CMT2A (Zuchner 2004; Verhoeven 2006).
Hereditary Neuropathy with Liability to Pressure Palsies (HNPP) represents the only form of CMT which is characterised by recurrent focal nerve palsies without signs of a generalised progressive neuropathy. The underlying genetic defect is the opposite state of CMT1A, namely the loss of one allele of PMP22 due to the loss of the chromosomal fragment on chromosome 17p11.2.
Many more rare dominant and recessive inherited forms of CMT have now been classified (see the Inherited Peripheral Neuropathies Mutations Database http://www.molgen.ua.ac.be/CMTMutations).
Biology
The biological mechanisms which lead to the clinical phenotype are still unclear for many of the responsible genes implicated in specific forms of CMT (Street 2003; Young 2003). For some the biological function has been elucidated with the aid of animal models (Martini 1997; Martini 2000) and in vitro experiments (Suter 2003). In general mutations in CMT disease associated genes lead to disturbed function of either Schwann cells or axons (Martini 2001). The change in gene dosage in the case of the duplication or the deletion of PMP22 is clear (Lupski 1992) but other mutations in CMT associated genes cause the expression of a mutated protein (Suter 2003). Specific causative gene therapies should regulate the expression of a mutated protein either at the DNA or RNA level (Young 2001). In the diseases with different mutations within the same gene, one would have to establish gene therapy strategies for each individual mutation. For CMT1A, in which alteration of the gene dosage of PMP22 is the underlying mechanism, therapeutic approaches will need to aim at regulating the amount of expressed PMP22 protein. After an initial phase of correct myelination during development, overexpression or underexpression of PMP22 in the myelinating Schwann cell causes dysmyelination or demyelination, as was shown in a PMP22 deficient mouse model (Adlkofer 1995; Adlkofer 1997). Further PMP22 protein dosage alterations cause incorrect incorporation of myelin proteins into the myelin membranes (Suter 2003).
In the CMT1B and CMTX subgroups, mouse models showed that the demyelinating phenotype was associated with an immune response (Maurer 2002). Reducing this immune response reduced the degree of demyelination and in parallel the behavioural deficits in these animals. This observation raised the question whether modulation of the immune system could be a helpful therapeutic strategy for CMT1B and CMTX.
CMT disease is commonly due to genes encoding structural proteins. It may also be due to other genes such as those encoding transcription factors, or enzymes or genes associated with mitochondria (Grandis 2005). The biological function of many of these genes is still unknown. The variety of the involved genes emphasises the fact that myelination, myelin maintenance and axonal integrity are the product of complex interactions of many proteins besides structural ones (Martini 2001). These interactions, if understood in more detail, will become the focus of future therapeutic approaches.
Clinical features
Although molecular genetic methods for the identification of new genes in CMT have been developed quickly, CMT1A remains the most common (Nelis 1996). The most prominent symptom in all forms of CMT1 is progressive distal wasting of peroneal muscles resulting in distal weakness beginning in the second decade (Kuhlenbaumer 2002). Many people with CMT1 have foot deformities. Sensory symptoms include tingling, burning or aching of feet and legs. Autonomic nervous system involvement is rare and not prominent. Erectile dysfunction may occur due to progressive involvement of the relevant motor and sensory nerves (Dyck 2005a). Need for a wheelchair is rare. People carrying point mutations in the PMP22 gene may be more severely affected (Young 2001).
The pathobiological hallmarks of CMT1 are demyelination and axonal degeneration with consequent muscle atrophy (Krajewski 1999; Krajewski 2000). With the aid of motor nerve conduction velocity (MNCV) measurements demyelination can be identified by reduction of motor NCV. Reduction in NCV does not reflect the clinical severity of CMT1 (Krajewski 1999; Krajewski 2000). For the diagnosis of CMT1 a reduced MNCV of less than 38 m/sec in the ulnar nerve is required (Dyck 2005a). Axonal CMT2 cannot be differentiated by clinical means from CMT1 but the MNCV is normal or only slightly reduced in CMT2 although the compound motor action amplitudes (CMAP) are significantly decreased (Dyck 2005a; Harding 1980).
Objectives
The objective was to review systematically all randomised and quasi‐randomised studies of any treatment for CMT.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised and quasi‐randomised trials of any treatment for people with CMT. Where a study aimed to evaluate the treatment of general neuromuscular symptoms of people with peripheral neuropathy including CMT we included the study if we were able to identify the effect of treatment in the CMT group. Observational studies and case reports on the treatment of patients with CMT were not included.
Types of participants
Because of the heterogeneity of CMT we included people with all types of CMT regardless of whether clinical, electrophysiological, histopathological or molecular genetic criteria were used. Due to the fast progress in the elucidation of the genetic basis of CMT, we considered rare forms of CMT. Experimental data from animal models were not included.
Types of interventions
We included all drug and physical treatments. Drugs were likely to include corticosteroids, antioxidants such as Vitamin E, lipoic acid and coenzyme Q. With regard to future developments, studies using molecular approaches like RNA interference (RNAi) with the aid of small interfering RNAs (siRNAs), antisense or ribozymes were included when available. Physical treatments included exercise and different physiotherapy strategies. Treatment should extend for at least 12 successive weeks. Twelve weeks of treatment was chosen as a minimum because the very slow progressive course of CMT makes it seem unlikely to us that a shorter period of treatment would be an adequate test of its efficacy.
Types of outcome measures
Changes and numbers of events occurring, over time were re‐scaled to `change or number of events in a fixed period`, for example three months, to permit the pooling of studies with differing follow‐up periods.
Primary outcomes
Change in impairment after at least 12 weeks. We accepted any measure of impairment made with a validated scale, such as the MRC sum score, Neuropathy Impairment Score (NIS) (Dyck 2005b), Total Neuropathy Score (Cornblath 1999), or CMT Neuropathy score (Shy 2005). The last two are composite scores of different aspects of impairment.
Secondary outcomes
Secondary outcome measures included:
Change in disability after at least 12 weeks. We accepted any measure of disability made with a validated scale such as the Rankin Scale (van Swieten 1988) or the Inflammatory Neuropathy Cause and Treatment Overall Disability Status Scale (Merkies 2000).
Change of maximum compound muscle action potential amplitudes (CMAP) of the ulnar median or peroneal nerve one year after randomisation because it has been shown that disability is best correlated to CMAP measures (Krajewski 1999; Krajewski 2000).
Change in sensory impairment one year after randomisation measured with a validated scale.
Change in sensory nerve action potential amplitude (SNAP) of the sural or other sensory nerves.
Change of grip force as measured with the aid of dynamometer or vigorimeter after one year of randomisation.
Quality of life assessment using validated scores as SF‐36 health related quality of life scale (Jenkinson 1993) or EuroQol (Anderson 1996) one year following randomisation.
Adverse events, whether minor or severe, caused by any form of treatment. We distinguished between minor adverse events and severe adverse events (those which were fatal, life‐threatening, required or prolonged hospitalisation or caused discontinuation of the treatment).
Search methods for identification of studies
Electronic searches
We searched the Cochrane Neuromuscular Disease Group Trials Register, MEDLINE (January 1966 to August 2007), EMBASE (January 1980 to August 2007), LILACS (January 1982 to August 2007) for randomised controlled trials using 'Charcot‐Marie‐Tooth disease' and its synonyms 'hereditary motor and sensory neuropathy', 'inherited neuropathy', 'peroneal muscular atrophy' and 'distal spinal muscular atrophy' as the search terms. For MEDLINE, EMBASE and LILACS search terms see Appendix 1, Appendix 2 and Appendix 3 respectively.
Searching other resources
Authors from clinical trials and experts from the field were contacted to identify unpublished trials. We also checked the bibliographies of trials identified.
Data collection and analysis
Selection of studies
Two review authors checked the titles and abstracts of the articles identified by the search.
Data extraction and management
Data were extracted independently by at least two review authors onto a specially designed data extraction form. We contacted trial authors to obtain missing data or raw data if necessary.
Assessment of risk of bias in included studies
Two review authors (TBB and PY) decided which trials fitted the inclusion criteria and graded the methodological quality using the Cochrane approach: A: adequate, B: unclear, C: inadequate or not done. The methodological quality assessment took into account security of randomisation, allocation concealment, patient blinding, observer blinding, explicit inclusion criteria, extent to which the study took into account any imbalance in prognostic important variables present at time of randomisation, and completeness of follow‐up.
Data synthesis
If data were available for more than one trial with a specific intervention, we used the Cochrane Review Manager 4.2 (RevMan) software to pool data. We summarised continuous data with weighted mean differences (WMD) and 95% confidence intervals (CIs). In case we found outcomes measured with different scales, standardised mean differences (SMD) were used. Dichotomised data were analysed with relative risks with 95% CIs.
In the 'Discussion' we considered adverse events and cost‐effectiveness taking into account information from the non‐randomised literature.
Subgroup analysis and investigation of heterogeneity
If heterogeneity was found we explored and described possible reasons for differences between studies such as types of participants, intervention or quality.
We separately compared CMT1, CMT2 and where possible other CMT subgroups. Due to the fact that CMT1A was the most frequent form of CMT, we considered CMT1A separately whenever possible.
Sensitivity analysis
We performed sensitivity analyses by omitting trials which lacked one or more of the methodological attributes. If necessary we used the random‐effects model to obtain the pooled estimates.
Results
Description of studies
We finally identified 41 references including 38 different studies describing treatment of any form of CMT but only six of them were randomised studies. As in two other systematic reviews dealing with exercise for people with peripheral neuropathies (White 2004) and with strength training and aerobic exercise training for muscle disease (van der Kooi 2005), we identified three articles dealing with the same randomised controlled study group (Lindemann 1994; Lindeman 1995; Lindeman 1999). In the following we only refer to the article from Lindeman 1995. In total we only included six small studies whose details are given in the Table 'Characteristics of included studies'. The interventions were exercise (Lindeman 1995), oral creatine monohydrate (Smith 2006; Chetlin 2004), parenteral Neurotrophin‐3 (NT‐3) (Sahenk 2005), subcutaneous preparation of purified bovine gangliosides (Cronassial) (Bradley 1988) and foot orthoses (Burns 2006).
Risk of bias in included studies
The methodological quality scores of the included studies as assessed by the review authors are given in Additional Table 4 'Methodological quality'. The study analysing strength training in CMT participants (Lindeman 1995) was individually matched in relation to muscle strength and timed stair climbing. Detailed description of the method of randomisation and allocation concealment were not given in the paper but the first author of the study gave information on request. Randomisation and allocation were done in calculating comparable z‐scores for each participant after having calculated peak/body weight ratios (mean knee extension torques divided by body weight) and performance on a stair climbing test (seconds). A statistician made a computer program to calculate all possible combinations of 15 pairs, looking for the optimal mix. Matching was done by two independent people who drew a sealed lot per matched pair and allocated it by tossing a coin to the training or non‐training group. Explicit diagnostic criteria for specific CMT subforms were not given but differentiation of CMT1 and CMT2 was claimed in accordance to clinical and electrophysiological data. For the main article about the study testing the combination of resistance training and creatine monohydrate (Chetlin 2004), explicit methods of randomisation and expicit inclusion criteria were not given. However, another article about the same participants described randomisation (Smith 2006). One of the coauthors of this study (S. Alway) provided comprehensive information. Randomisation was done by using block randomisation to assign participants to their respective groups (creatine or placebo). Numerical block positions were designated 1 to 8, 9 to 16, and 17 to 20. Twenty paper squares, 10 for each of the two groups, were evenly apportioned and drawn from a container sight unseen. Once drawn, each paper square was discarded in order to assure an equal number of group designations for each block. CMT diagnosis was done on a clinical, electrophysiological and genetic basis without recording the exact genotypes included in the study.
Table 1.
Methodological quality
| Study | Randomisation | Concealed allocation | Follow up | Blinding observer | Blinding patient | Inclusion criteria |
| Bradley 1988 | B | B | A | A | C | A |
| Burns 2006 | A | A | A | D | A | A |
| Chetlin 2004 | A | A | A | B | A | A |
| Doherty 2001 | B | B | A | B | B | B |
| Lindeman 1995 | A | A | A | A | D | A |
| Sahenk | A | A | A | B | A | A |
| Smith 2006 | A | A | A | B | A | A |
In the study analysing the effect of creatine monohydrate without physical training, genetic testing was also done (Doherty 2001). The method of randomisation was not given. Unfortunately detailed information was not made available.
In 1988 genetic testing was not available for CMT syndromes and inclusion criteria for the study using Cronassial were based on clinical and electrophysiological data (Bradley 1988). In this study individuals suffering from other neuropathy causing diseases (e.g. diabetes) were excluded (Bradley 1988). From these data CMT participants were subdivided in CMT1 and CMT2. In the study analysing the impact of NT‐3 in CMT1A participants (Sahenk 2005), the method of randomisation was given in detail. The presence of the duplication on choromosome 17p11.2 was an explicit inclusion criterion.
None of the randomised studies defined a minimum level of impairment or disability at baseline. All studies explicitly recorded the number and reasons for drop outs in the different experimental groups. For statistical assessment drop outs were claimed to be considered.
Blinding of the observer as of the participant was reported in all studies in which medication was tested (Bradley 1988; Chetlin 2004; Doherty 2001; Sahenk 2005). Side effects at the injection sites which were reported in the Cronassial (Bradley 1988) studies and in the NT‐3 study (Sahenk 2005) were not reported to be causes for unblinding of the participants. Due to the nature of physical exercise, patient blinding was not possible in the study on strength training (Lindeman 1995).
None of the studies considered explicitly differences in baseline characteristics. Age of onset and duration of the disease were not given in any of the studies. Two studies used impairment scores to compare groups. The NIS was used in one (Sahenk 2005) and an undefined score in the other (Bradley 1988). The studies of physical training (Lindeman 1995), physical training in combination with creatine monohydrate (Chetlin 2004; Smith 2006) or impact of foot orthoses did not use validated scores to measure impairment. The trial analysing the effect of orthoses concerning pain used an unspecified quality of life questionaire (Burns 2006).
Recordings of the completeness of the trials were given in all studies. Drop outs were reported in three studies (Bradley 1988; Lindeman 1995; Chetlin 2004). In the trial by Lindeman 1995, the matched pairs design demanded removal of one matched control participant.
All studies described interventions and the related procedures in detail. Two trials using physical training as an interventional parameter gave detailed information about duration, intensity and setting of the therapy (Lindeman 1995; Chetlin 2004). Doses of medication, duration of intake and mode of application were also recorded in detail in the studies analysing pharmacological intervention in CMT (Bradley 1988; Doherty 2001; Sahenk 2005; Smith 2006). In the study using orthoses, detailed information on the treatment and placebo were given (Burns 2006)
Definition and description of outcome criteria were given in all studies we selected. Because only one study used an impairment score as a primary outcome measure (Sahenk 2005), the review authors decided to include all randomised studies in which defined outcome measures were reported. Furthermore in one trial the observation period was shorter than the minimum of 12 weeks (Doherty 2001) which we defined as a prerequisite for inclusion. Due to the clear definition of the outcome measure this trial was included. Quality of life was assessed in one study. However, defined and validated questionaires were not used for quality of life assessement (Burns 2006). In the trial in which muscle fibre size (Chetlin 2004) and biochemically proven amount of myosin heavy chain composition (Smith 2006) were analysed as an outcome measure, techniques to assemble the data were described in detail as in the study of Neurotrophin‐3 (Sahenk 2005) in CMT1A, too.
None of the selected studies had top scores on all items using the Cochrane approach. Without the unpublished data, even quality assessment of the randomisation items would have been unclear in three trials. Scoring for each of the included studies, done independently by two of the review authors (PY and TBB) are shown in Table 4. Their scores coincided for all included trials.
Effects of interventions
Due to the heterogeneity of the different studies and the great variability of presentation of the data, pooling of data was not possible. The results are given for each intervention separately in relation to the predefined outcome measures.
Neurotrophin‐3
(Sahenk 2005) One NT‐3 study was identified in which impairment was measured with the use of a composite scale. In this parallel group double blind study (Sahenk 2005) with eight participants, four receiving NT‐3 and four receiving placebo, the NIS scale was used at baseline and after 24 weeks of treatment. Subscoring was performed with respect to motor, sensory and reflex status of the participants, which were people with genetically proven CMT1A. Mean differences between the treatment group and the placebo group were ‐9.50 (95% Cl ‐13.77 to ‐5.23) (see Analysis 01.01) in the overall NIS. Subscoring for motor items showed a mean difference of ‐0.50 (95% CI ‐2.42 to 1.42) (see Analysis 01.02). Mean difference for sensory scores was ‐4.80 (95% CI ‐8.08 to ‐1.52) (see Analysis 01.03), and mean difference for reflex score was ‐4.50 (95% CI ‐7.65 to ‐1.35) (see Analysis 01.04). These data indicate a significant but slight improvement of sensory and reflex scores but not of motor function.
Exercise
(Lindeman 1995) Grip force as indicating change in muscle strength was not measured with the aid of dynamometer or vigorimeter in any of the studies. However muscle strength was measured in one study in people suffering from neuropathies (Lindeman 1995; Lindeman 1994b). This study showed a significant reduction in time needed for six metre comfortable walking at 24 weeks after starting the exercise with a WMD of 0.70 (95% CI 0.23 to 1.17) (see Analysis 02.04). This study also reported significant improvement of isokinetic knee extension torque in the exercise group at 24 weeks after starting with a WMD of 17.70 (95% CI 5.11 to 30.29) (see Analysis 02.01). Isokinetic knee flexion torque was not improved, WMD 6.10 (95% CI 0.29 to 11.91) (see Analysis 02.02). Maximal isometric voluntary contraction force was also not significantly improved in the exercise group, WMD 12.60 (95% CI ‐1.51 to 26.71) (see Analysis 02.03).
Foot orthoses
(Burns 2006) Pain was measured in one study analysing the benefit of foot orthoses in a subset of CMT participants. The use of foot orthoses did not significantly improve functional outcome in nine CMT participants compared with seven given sham insoles (Burns 2006), MD 6.00 (CI ‐15.25 to 27.25) (see Analysis 03.02). Pain was also not significantly improved in the orthoses group, WMD 12.90 (95% CI ‐21.09 to 46.89) (see Analysis 03.01). Quality of life was assessed in this study using the SF36 Health Survey with the items physical functioning WMD of 4.80 (95% CI ‐12.09 to 21.69) (see Analysis 03.03), general health WMD of 1.90 (95% CI ‐19.57 to 23.37) (see Analysis 03.04), vitality WMD of 10.20 (95% CI ‐2.69 to 23.09) (see Analysis 03.05) and social functioning WD of ‐1.50 (95% CI ‐19.18 to 16.18) (see Analysis 03.06).
Subcutaneous bovine brian gangliosides injections
(Bradley 1988) In this trial none of the predefined outcome measures were reported.
Oral Creatine monohydrate
In this trial none of the predefined outcome measures were reported.
Adverse events, whether minor or severe, caused by any form of treatment
We distinguished between minor and severe adverse events (those which were fatal, life‐threatening, required or prolonged hospitalisation or caused discontinuation of the treatment). The adverse events from included and excluded studies are given in Additional Table 5 'Adverse events for interventions with drugs or food supplements' and in Additional Table 6 'Adverse events for non‐drug, non surgical interventions'. Further information is also given in the 'Characteristics of excluded studies' Table.
Table 2.
Adverse events for interventions with drugs or food supplements
| Study | Intervention | Patients | Major adverse events | Minor adverse events |
| Bradley 1988 | Purified bovine brain gangliosides (Cronassial), five months 40 mg Cronassial versus five months 100mg Cronassial versus placebo | 30 CMT (clinical) | Allergic rash seen with 100mg in 2 patients, pain at injection site in one patient | 16.9% increased pain with medication versus 7.4% in the placebo group, 2.7% redness/itching versus 0.9% in placebo group, 6.8% skin rash versus 2.7% in the placebo group, tinnitus at the dose of 100mg in 2.7% versus 0.3% in the placebo group. |
| Smith 2006, Chetlin 2004 | Creatine monohydrate 5g/d combined with exercise | 39 CMT patients | None reported. | Mild gastrointestinal upset, leg cramps, fatigue, delayed‐onset soreness. |
| Sahenk 2005 | Neurotrophin‐3 starting with 50 µg/kg increasing up to 150 µg/kg | 4 CMT1A (genetically proven) | None reported. | Irritation at injection site, mild diarrhoea. |
| Prensky 1984 | Methylprednisolone 40 to 60 mg/m2 | 14 CMT clinical | Weight gain of 15 to 18 pounds in 3 patients, proximal myopathy and psychiatric illness in 1 patient (drop out), compression fracture of dorsal vertebrae T‐12 due to significant worsening of osteoporosis in 1 patient, severe leg cramps in 1 patient, pseudotumour when steroid was stopped in 1 patient, depression in 1 patient, hypertrophic nerve root in one patient. | None reported. |
| Russell 1995 | 3,4‐diaminopyridine (3,4‐DAP) 4x5mg/day up to 4x20mg/day | 27 CMT patients (clinical) | None reported. | 71% facial pain and predominantly perioral parasthesia with a maximum duration of 90 minutes, 29% muscle fatigue, 18% nauseas and abdominal discomfort, 15% insomnia, 12% transient urinary frequency, 6% increased tearing or nasal dicharge, 6% facial flushing, 6% transient hand tremor or muscle twitching, 6% headache, 6% transient parageusia. |
| Dyck 1982a, Dyck 1982b | Prednisone | Single cases | None reported. | None reported. |
| Folkers 1985, Folkers 1995 | Q10 | Single cases | None reported. | None reported. |
| Lisett 1998 | Growth hormone replacement | Single case | None reported. | None reported. |
| Jaradeh 1992 | Levodopa | 6 unspecified CMT cases. | None reported. | None reported. |
| Williams 1986 | Vitamin E combined with Cis‐linolein, gamma‐linoleic, oleic acid | 20 CMT patients (clinical). | None reported. | None reported. |
Table 3.
Adverse events for non‐drug, non surgical interventions
| Study | Intervention | Patients | Major adverse events | Minor adverse events |
| Lindeman 1998, Lindeman 1994b, Lindeman 1995, Lindeman 1999 | Strength training | 29 CMT patients | None reported. | 1 drop out due to knee problems, complaints owing to overuse. |
| Burns 2006 | Foot orthoses | 16 CMT | None reported specifically for CMT. | None specific for CMT group, additional foot pain in 12% of total number of (154) participants, ankle instability and foot irritation. |
| Bean 2001 | Ankle foot orthoses | 1 CMT patient | None reported. | None reported. |
| Aras 2005 | Exercise and electrical stimulation | 4 CMT patients | None reported. | None reported. |
| Dahl 2004 | Treatment in warm climate | 21 CMT patients | None reported. | None reported. |
| Lobzin 1986; Lobzin 1987; Lobzin 1988; Lobzin 1990; Lobzin 1992 | Proserin in electrophoresis with benzohexonium, and sine modelled electricity in electrophoresis with benzohexonium | 433 CMT patients | None reported. | None reported. |
Adverse events for surgical interventions
Interventions on the lower extremities found in the literature were triple arthrodesis (Mann 1992; Saltzman 1999; Wetmore 1989), posterior tibial tendon transfers (Miller 1982), various surgical interventions (Brown 1992), use of a hinged distraction apparatus (Oganesyan 1996), and periacetabular osteotomy (Trumble 1999). Interventions on the upper extremities comprised tendon transfers of upper limb (Wood 1995) and extensor pollicis longus‐opponensplasty (Riley 1980). Additionally there was an article about microvascular decompression for trigeminal neuralgia in three CMT patients (de Matas 2000). In summary adverse events were non‐specific and were not peculiar to the CMT phenotype.
Discussion
Included studies, number of participants in total
In total we identified six different trials describing the medical or physical treatment of CMT syndromes in a randomised controlled study design. Only one study fulfilled a subset of the inclusion criteria defined in the protocol. In this study in total eight people with genetically proven CMT1A were included (Sahenk 2005). It was the only study to be judged as adequate. None of the other included studies met the predefined inclusion criteria. However two more studies were analysed due to their study design fulfilling the criteria to be a randomised clinical trial with regard to the Cochrane approach (Lindeman 1995; Burns 2006). These two trials were small and did not focus on CMT but CMT turned out to be a subgroup in these studies. Three other trials did not report the outcome measures selected for this review (Smith 2006; Doherty 2001; Bradley 1988).
The NT‐3 study and the study testing the use of foot orthoses were the only studies in which people with genetically proven CMT1A were included. In all other studies, CMT1 and CMT2 were diagnosed by clinical and electrophysiological aspects. How far participants who were included in these studies were comparable is unclear since data on severity evaluated in a composite scale (Shy 2005) were not given in any of these studies.
In the studies in which the muscle fibre diameter was analysed following a defined orally administered dosage of creatine monohydrate in combination with defined physical exercise (Chetlin 2004), no change on outcome parameters was reported, but detailed data were not given. The amount of myosin heavy chain composition was not changed in the placebo group nor in the group receiving creatine monohydrate in combination with strength exercise (Doherty 2001). The administration of creatine alone in CMT1 and 2 did not show any effect on the amount of myosin heavy chain (Doherty 2001).
Only one study focused on people with CMT1 but the number of participants included was very small (Sahenk 2005). People with CMT represented subgroups in all other studies which were designed for people with different kinds of neuromuscular diseases.
Quality of study design
The studies which were identified as RCTs used appropriate randomisation methods.
Blinding was done in all studies except studies in which physical exercise was evaluated (Lindeman 1995) and blinding was not possible. Numbers of CMT participants were low in all studies and statistical power analysis was not given.
Types of interventions
None of the trials had significantly long follow‐up to detect differences in rate of progression of CMT between treatment groups (Reilly 2006). Furthermore two outcome measures, and especially the CMT‐neuropathy score, now favoured for CMT trials (Reilly 2006) were not used.
Limitation of the review
The small number of trials which fulfilled the selection criteria for this review reflects the difficulties of undertaking clinical trials in CMT. This review demonstrates the lack of adequate RCTs in CMT.
Since CMT is biologically and genetically heterogeneous and shows great variability in severity, we had wanted to analyse different subgroups but the lack of data made this impossible. In updates of this review we intend to consider CMT1 and CMT2 separately. The selection of outcome measures was specified for CMT taking in account the slow natural history of CMT1 and CMT2. Thus study quality was low in all selected studies with regard to this selection criterion.
The lack of availability of genetic testing in these old studies limited the definition of different CMT syndromes. In future CMT1 and CMT2 will be of the most clinical importance (Nelis 1996; Young 2003; Verhoeven 2006).
Since in a mouse model of CMT1A, vitamin C was shown to be effective in improving motor function as well as to enhance remeylination (Passage 2004) several vitamin C studies were initiated within the last two years (see Ongoing trials). Most of these trials are conducted in accordance to expert recommendations (Reilly 2006) and will offer the first set of data from treating CMT1A in multicentre trials (Pareyson 2006). Data from these trials will be included in review when available.
Authors' conclusions
For clinical practice none of the studies showed significant benefit from the interventions tested for any CMT syndrome. Possible benefit from NT‐3 was shown in a very small trial. However the use of NT‐3 may be promising but is at this point far from being applicable in clinical routine. None of the trials was large enough to identify or exclude even moderate benefit or harm.
There is a need for properly designed trials of interventions in CMT. Such trials should have adequate follow‐up, use appropriate outcome measures such as the validated CMT‐NS composite outcome measure, and be powered to detect a clinically important result. The results of trials such as vitamin C are awaited.
Acknowledgements
The authors wish to thank Professor Richard Hughes and Janice Fernandes for critical reading of the review. Kate Jewitt's continuous help not just to understand the differences between RefMan and RevMan but to use the last one as well is very much appreciated.
Appendices
Appendix 1. PubMed MEDLINE Search Strategy
(randomized controlled trial [pt] OR controlled clinical trial [pt] OR randomized controlled trials [mh] OR random allocation [mh] OR double‐blind method [mh] OR single‐blind method [mh] OR clinical trial [pt] OR clinical trials [mh] OR ("clinical trial" [tw]) OR ((singl* [tw] OR doubl* [tw] OR trebl* [tw] OR tripl* [tw]) AND (mask* [tw] OR blind* [tw])) OR ( placebos [mh] OR placebo* [tw] OR random* [tw] OR research design [mh:noexp] OR comparative study [mh] OR evaluation studies [mh] OR follow‐up studies [mh] OR prospective studies [mh] OR control* [tw] OR prospectiv* [tw] OR volunteer* [tw])) NOT (animals [mh] NOT human [mh]) AND ((charcot OR (heredit* AND neuropath*) OR (inherit* AND neuropath*) OR (peroneal and atroph*) OR (distal and atroph*)) OR (Charcot‐Marie‐Tooth Disease [mh] OR hereditary motor and sensory neuropathies [mh] OR muscular atrophy, spinal [mh]))
Appendix 2. Ovid EMBASE Search Strategy
1. Randomized Controlled Trial/ 2. Clinical Trial/ 3. Multicenter Study/ 4. Controlled Study/ 5. Crossover Procedure/ 6. Double Blind Procedure/ 7. Single Blind Procedure/ 8. exp RANDOMIZATION/ 9. Major Clinical Study/ 10. PLACEBO/ 11. Meta Analysis/ 12. phase 2 clinical trial/ or phase 3 clinical trial/ or phase 4 clinical trial/ 13. (clin$ adj25 trial$).tw. 14. ((singl$ or doubl$ or tripl$ or trebl$) adj25 (blind$ or mask$)).tw. 15. placebo$.tw. 16. random$.tw. 17. control$.tw. 18. (meta?analys$ or systematic review$).tw. 19. (cross?over or factorial or sham? or dummy).tw. 20. ABAB design$.tw. 21. or/1‐20 22. human/ 23. nonhuman/ 24. 22 or 23 25. 21 not 24 26. 21 and 22 27. 25 or 26 28. Hereditary Motor Sensory Neuropathy/ 29. Charcot Marie Tooth.mp. 30. (charcot or (heredit$ and neuropath$)).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 31. or/28‐30 32. 27 and 31
Appendix 3. LILACS Search Strategy
((Pt RANDOMIZED CONTROLLED TRIAL OR Pt CONTROLLED CLINICAL TRIAL OR Mh RANDOMIZED CONTROLLED TRIALS OR Mh RANDOM ALLOCATION OR Mh DOUBLE‐BLIND METHOD OR Mh SINGLE‐BLIND METHOD) AND NOT (Ct ANIMALS AND NOT (Ct HUMAN AND Ct ANIMALS)) OR (Pt CLINICAL TRIAL OR Ex E05.318.760.535$) OR (Tw clin$ AND (Tw trial$ OR Tw ensa$ OR Tw estud$ OR Tw experim$ OR Tw investiga$)) OR ((Tw singl$ OR Tw simple$ OR Tw doubl$ OR Tw doble$ OR Tw duplo$ OR Tw trebl$ OR Tw trip$) AND (Tw blind$ OR Tw cego$ OR Tw ciego$ OR Tw mask$ OR Tw mascar$)) OR Mh PLACEBOS OR Tw placebo$ OR (Tw random$ OR Tw randon$ OR Tw casual$ OR Tw acaso$ OR Tw azar OR Tw aleator$) OR Mh RESEARCH DESIGN) AND NOT (Ct ANIMALS AND NOT (Ct HUMAN AND Ct ANIMALS)) OR (Ct COMPARATIVE STUDY OR Ex E05.337$ OR Mh FOLLOW‐UP STUDIES OR Mh PROSPECTIVE STUDIES OR Tw control$ OR Tw prospectiv$ OR Tw volunt$ OR Tw volunteer$) AND NOT (Ct ANIMALS AND NOT (Ct HUMAN AND Ct ANIMALS))) AND NOT Mh ANIMALS [Words] AND (charcot OR (heredit$ AND neuropath$) OR (inherit$ AND neuropath$) OR (peroneal AND atroph$) OR (distal AND atroph$)) [Words]
Data and analyses
Comparison 1.
Neurotrophin‐3 versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Neuropathy impairment score | 1 | 8 | Mean Difference (IV, Fixed, 95% CI) | ‐9.5 [‐13.77, ‐5.23] |
| 2 Neuropathy impairment score motor | 1 | 8 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐2.42, 1.42] |
| 3 Neuropathy impairment score sensory | 1 | 8 | Mean Difference (IV, Fixed, 95% CI) | ‐4.8 [‐8.08, ‐1.52] |
| 4 Neuropathy impairment score reflex | 1 | 8 | Mean Difference (IV, Fixed, 95% CI) | ‐4.5 [‐7.65, ‐1.35] |
Analysis 1.1.
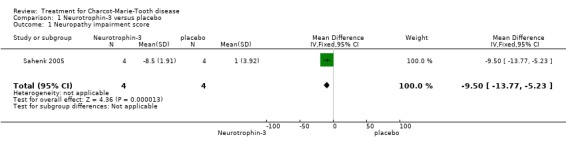
Comparison 1 Neurotrophin‐3 versus placebo, Outcome 1 Neuropathy impairment score.
Analysis 1.2.
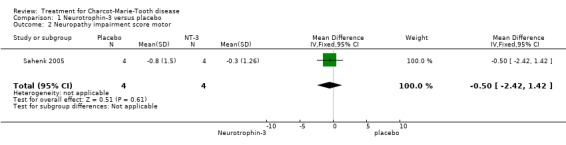
Comparison 1 Neurotrophin‐3 versus placebo, Outcome 2 Neuropathy impairment score motor.
Analysis 1.3.
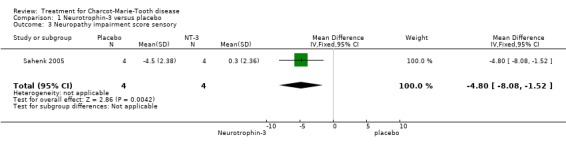
Comparison 1 Neurotrophin‐3 versus placebo, Outcome 3 Neuropathy impairment score sensory.
Analysis 1.4.
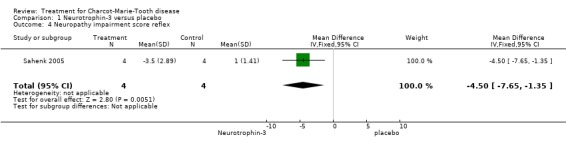
Comparison 1 Neurotrophin‐3 versus placebo, Outcome 4 Neuropathy impairment score reflex.
Comparison 2.
Strength training versus no training
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Isokinetic knee torque extension | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 17.7 [5.11, 30.29] |
| 2 Isokinetic knee torque flexion | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 6.1 [0.29, 11.91] |
| 3 Maximal voluntary contraction | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 12.60 [‐1.51, 26.71] |
| 4 Walking 6m (comfortable) | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 0.7 [0.23, 1.17] |
Analysis 2.1.
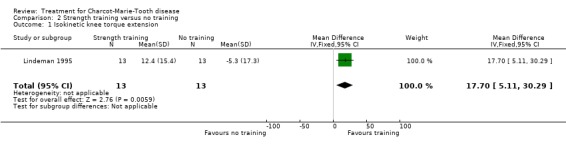
Comparison 2 Strength training versus no training, Outcome 1 Isokinetic knee torque extension.
Analysis 2.2.
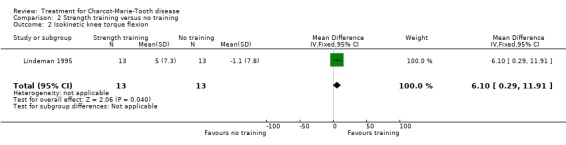
Comparison 2 Strength training versus no training, Outcome 2 Isokinetic knee torque flexion.
Analysis 2.3.
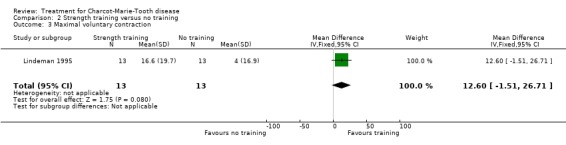
Comparison 2 Strength training versus no training, Outcome 3 Maximal voluntary contraction.
Analysis 2.4.
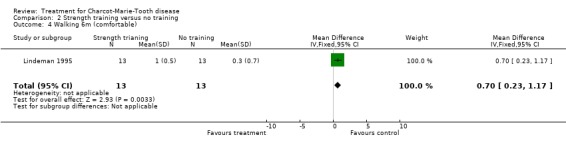
Comparison 2 Strength training versus no training, Outcome 4 Walking 6m (comfortable).
Comparison 3.
Custom foot orthoses versus sham insoles
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Foot pain | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 12.90 [‐21.09, 46.89] |
| 2 Function | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 6.0 [‐15.25, 27.25] |
| 3 Physical functioning | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 4.8 [‐12.09, 21.69] |
| 4 General health | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 1.90 [‐19.57, 23.37] |
| 5 Vitality | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 10.2 [‐2.69, 23.09] |
| 6 Social functioning | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | ‐1.5 [‐19.18, 16.18] |
Analysis 3.1.
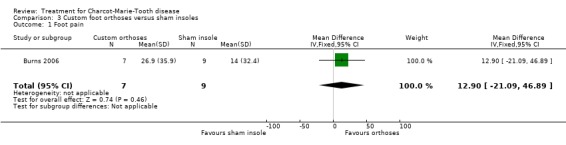
Comparison 3 Custom foot orthoses versus sham insoles, Outcome 1 Foot pain.
Analysis 3.2.
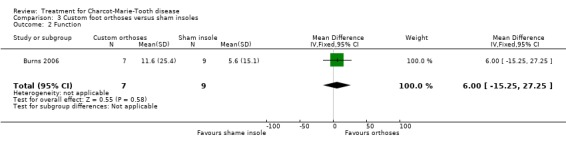
Comparison 3 Custom foot orthoses versus sham insoles, Outcome 2 Function.
Analysis 3.3.
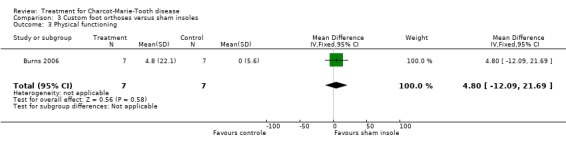
Comparison 3 Custom foot orthoses versus sham insoles, Outcome 3 Physical functioning.
Analysis 3.4.
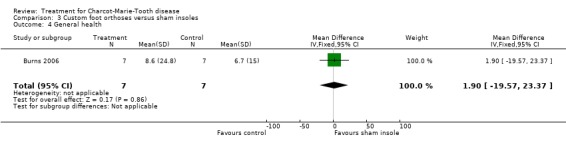
Comparison 3 Custom foot orthoses versus sham insoles, Outcome 4 General health.
Analysis 3.5.
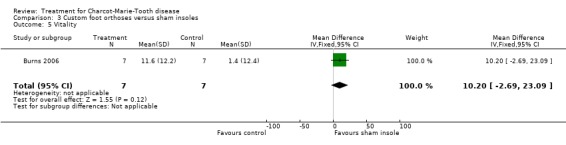
Comparison 3 Custom foot orthoses versus sham insoles, Outcome 5 Vitality.
Analysis 3.6.
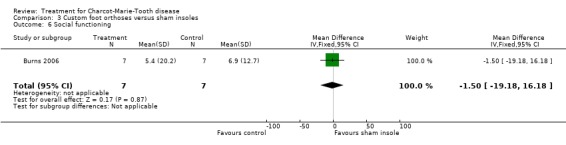
Comparison 3 Custom foot orthoses versus sham insoles, Outcome 6 Social functioning.
What's new
| Date | Event | Description |
|---|---|---|
| 27 April 2008 | Amended | Converted to new review format. |
History
Protocol first published: Issue 2, 2006 Review first published: Issue 1, 2008
| Date | Event | Description |
|---|---|---|
| 14 November 2007 | New citation required and conclusions have changed | Substantive amendment |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Randomised double‐blind trial. Method of randomisation not given. | |
| Participants | Two groups with 15 CMT participants each. CMT 1 and CMT2 included diagnosed clinically and electrophysiologically, no genetic testing. | |
| Interventions | Initially 2 months placebo in both arms followed by 40mg. Cronassial versus placebo for 5 months followed by Cronassial 40mg versus 100mg for another 5 months, no wash out phase in between, mode of administration i.m. 12 weeks. | |
| Outcomes | Sensory measures, strength measures, EMG, Nerve conduction velocities and sensory and motor action potentials. | |
| Notes | 1 drop out because of allergic rash under 100 mg Cronnassial, 1 drop out because of pain at injection site under 40 mg after 8 months. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Randomised controlled trial. Method of randomisation central computer based. | |
| Participants | In total inclusion of 7 genetically proven CMT1A participants. | |
| Interventions | Custom foot orthoses for 3 months. | |
| Outcomes | Foot pain, foot function and physical functioning, general health, vitality and social functioning as secondary outcomes. | |
| Notes | Subgroup analysis was given on request. AE were not recorded seperately for CMT group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
| Methods | Randomised double‐blind trial testing combination of resistance training and creatine. Method of randomisation was given on request. | |
| Participants | CMT1 and CMT2 patients based on clinical, electrophysiological and genetic diagnosis, 10 patients allocated to each group. | |
| Interventions | One group performing resistance training daily in combination with additional oral intake of 5mg creatine per day, one group receiving resistance training in combination with placebo 12 weeks. | |
| Outcomes | Muscle fibre typing and size, quantitative muscle assessment. | |
| Notes | 3 participants reported to have decreased their training due to delayed onset soreness. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
| Methods | Randomised double‐blind placebo controlled cross‐over trial. Method of randomisation unclear. | |
| Participants | A total of 39 CMT1 and CMT2 participants based on clincal, electrophysiological and genetic diagnosis. | |
| Interventions | 5mg creatine orally taken per day versus placebo, before cross over 5 week wash out. | |
| Outcomes | Maximal voluntary strength, fatigue, 30‐m timed walking, stair climbing. | |
| Notes | In the placebo group, side effects gastrointestinal upset and leg cramps in both groups, 1 drop out due to unrelated illness, 1 drop out due to muscle cramps. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
| Methods | Randomised controlled single blind trial. Method of randomisation was given on request. | |
| Participants | 29 participants in total either CMT1 or CMT2 based on clinical data. | |
| Interventions | One group strength training for 24 weeks, control group no strength training. | |
| Outcomes | Muscle strength voluntary contraction, isokinetic knee torques, timed functional activities. | |
| Notes | One drop out from exercise group due to knee problems. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
| Methods | Randomised controlled single blind trial. Method of randomisation was given on request (same study as Lindeman 1995) | |
| Participants | 29 participants in total either CMT1 or CMT2 based on clinical data. | |
| Interventions | One group strength training for 24 weeks, control group no strength training. | |
| Outcomes | Muscle strength voluntary contraction, isokinetic knee torques, timed functional activities. | |
| Notes | One drop out from exercise group due to knee problems. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
| Methods | Randomised controlled single blind trial. Method of randomisation was given on request (same study as Lindeman 1995) | |
| Participants | 29 participants in total either CMT1 or CMT2 based on clinical data. | |
| Interventions | Randomised controlled single blind trial. Method of randomisation was given on request (same study as Lindeman 1995). | |
| Outcomes | Muscle strength voluntary contraction, isokinetic knee torques, timed functional activities. | |
| Notes | One drop out from exercise group due to knee problems. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
| Methods | Randomised controlled double‐blind trial. | |
| Participants | 8 patients with CMT1A carrying the duplication on chromosome 17p11.2 harbouring the PMP22 gene. | |
| Interventions | One group 50 µg/ kg 3x/w recombinant human NT‐3 subcutaneously. Control group placebo injections for 6 months. | |
| Outcomes | Nerve biopsies, Neuropathy impairment scale, pegboard test, density of myelinated fibers and Schwann cells in sural nerve biopsies. | |
| Notes | No drop outs | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
| Methods | Describes the same study as Chetlin 2004 | |
| Participants | ||
| Interventions | ||
| Outcomes | Myosin heavy chain content. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aras 2005 | Not a randomised controlled trial. Electric stimulation in four children suffering from unspecified CMT. Outcome measure: 10 minute timed walking and fatigue. |
| Bean 2001 | Single subject study analysing effect of brace modification on aerobic exercise. |
| Brown 1992 | Not a randomised controlled trial. Retrospective evaluation of upper limb neuropathies in CMT. No intervention done. No report on adverse effects. |
| Chetlin 2004b | Trial describes same participants as in Chetlin 2004 without creatine monohydrate supplementation. |
| Congia 1991 | TRH low dose treatment for a total of two weeks in four unspecified CMT patients. Observational period too short. |
| Dahl 2004 | Not a randomised controlled trial. Comparison of the effect of physiotherapy in a group of patients with neuromuscular diseases including 21 CMT patients and two Dejerine‐Sottas patients in warm climate. |
| de Matas 2000 | Not a randomised controlled trial. Description of three CMT patients suffering from trigeminal neuralgia and the effect of microvascular decompression. |
| Dyck 1982a | Not a randomised controlled trial, case series. |
| Dyck 1982b | Not a randomised controlled trial, case series. Same trial as Dyck 1982 |
| Folkers 1985 | Not a randomised controlled trial. Cardiac response to Coenzme Q in a group of patients with neuromuscular disease including four patients with unspecified CMT. Design remains unclear. |
| Folkers 1995 | Not a randomised controlled trial. Double‐blinded study assessing the effect of 100mg coenzyme Q and physical performance per day on cardiac function. Three unspecified CMT patients included. |
| Jaradeh 1992 | Not a randomised controlled trial. |
| Lindeman 1994b | Not a randomised controlled trial. |
| Lindeman 1998 | No intervention. |
| Lindeman 1999b | No intervention. |
| Lissett 1998 | Case description, growth hormone replacement |
| Lobzin 1986 | Not a randomised controlled trial. |
| Lobzin 1987 | Not a randomised controlled trial. Open comparison of 118 CMT patients either receiving electrophoresis and benzohexonium versus treatment with electrophoresis and benzohexonium without different changing electrophoresis protocol. |
| Lobzin 1988 | Not a randomised controlled trial. |
| Lobzin 1990 | Not a randomised controlled trial. A comparative evaluation of the efficancy of physical treatment methods in unspecified CMT. |
| Lobzin 1992 | Not a randomised controlled trial. Autonomic vascular changes in a huge group of neuropathies including 315 CMT patients. |
| Mann 1992 | Not a randomised controlled trial, case series, 10 unspecified CMT patients. |
| Miller 1982 | Not a randomised controlled trial, retrospective case series, five feet in three unspecified CMT patients. |
| Oganesyan 1996 | Not a randomised controlled trial. Retrospective analysis of the treatment with a Hinged distraction apparatus in a group of patients including three unspecified CMT patients suffering from pes equinovarus. No subgroup analysis given. |
| Prensky 1984 | Not a randomised controlled trial. High dose of steroid in 10 CMT1 and 2 CMT2 patients. Cross‐over design. Initially blinded but blinding was impossible due to severe side effects. Study had to be stopped due to serious side effects at a dose of 45‐60mg/sqM methylprednisolone |
| Riley 1980 | Not a randomised controlled trial. |
| Russell 1995 | RCT but treatment and observational period was just four days and another four days in a cross over design in 27 CMT1 patients. |
| Saltzman 1999 | Not a randomised controlled trial. Retrospective evaluation of a group of patients suffering from different diseases who had received triple arthrodesis. No CMT specific subgroup analysis. |
| Trumble 1999 | Case series including unspecified CMT patients. |
| Wetmore 1989 | Not a randomised controlled trial. Retospective evaluation of 16 CMT patients after having received triple arthrodeses. |
| Williams 1986 | Not a randomised controlled trial. Linoleic acid, vitamin E and supplementation in 20 CMT 1 patients with matched controls. Nerve conduction velocities after one year Neuropathy disability scale after one year. |
| Wood 1995 | Not a randomised controlled trial. Retrospective analysis of different upper limb surgical treatments in clinical CMT. |
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Information requested |
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Starting date | |
| Contact information | |
| Notes | Personal communication |
| Trial name or title | Single center randomized double blind placebo controlled |
| Methods | |
| Participants | CMT1A patients genetically carrying the duplication on chromosome 17q11.2 between 5‐16 years |
| Interventions | Ascorbic acid for 2 years in a dose of 1500 mg/day versus placebo. |
| Outcomes | Charcot‐Marie‐Tooth neuropathy scale |
| Starting date | September 2007 |
| Contact information | Prof. Dr . G. Kurlemann, Dept. for Pediatric medicine, Neuropediatrics, Albert‐Schweitzer‐Str. 33, 48129 Münster Prof. Dr. P. Young and Dr. G. Kuhlenbäumer Department of Neurology, University of Muenster, Albert‐Schweitzer‐Str‐ 33, 48129 Muenster, Tel: +49‐251‐8348331, Fax: +49‐251‐8348181, young@uni‐muenster.de, gkuhlen@uni‐munester.de |
| Notes | Personal communication |
| Trial name or title | A multicenter, randomized, double‐blind, placebo‐controlled trial of long term ascorbic acid in Charcot‐Marie‐tooth disease type 1A (CMT‐TRIAAL) |
| Methods | |
| Participants | CMT1A patients genetically carrying the duplication on chromosome 17q11.2 between 18‐70 years |
| Interventions | Ascorbic acid for 2 years in a dose of 1500 mg/day versus placebo |
| Outcomes | Charcot‐Marie‐Tooth neuropathy scale , MVIC, 10m walking, nine‐hole‐peg test, ONLS, VAS for pain and fatigue, SF36, skin biopsy (expression of PMP22) |
| Starting date | Autumn 2006 |
| Contact information | Dr. D. Pareyson, Department of Biochemistry and Genetics, C. Besta National Neurological Institute, Via Celoria11, 20133 Milan, Italy Tel: +390223943001 Fax +39022664236 e‐mail: dpareys@istituto‐besta.it |
| Notes | Pareyson 2006 |
| Trial name or title | A Randomized, placebo‐controlled, double masked 120 subject "Futility Design" clinical trial of Ascorbic acid treatment of Charcot‐Marie‐Tooth Disease Type 1A. |
| Methods | |
| Participants | CMT1A patients genetically carrying the duplication on chromosome 17q11.2 between 18‐70 years |
| Interventions | Ascorbic acid (Vitamin C) (4 grams/day). |
| Outcomes | Mean change in the CMT Neuropathy Scale following high dose ascorbic acid ingestion, assessed at baseline and every 6 months throughout the trial. (Time Frame: 25 months per subject from baseline to completion).CMT‐Neuropathy scale as primary outcome. |
| Starting date | April 2007 |
| Contact information | United States, Maryland Johns Hopkins University, Dept of Neurology, Baltimore, Maryland, 21287, United States; Recruiting Lora Clawson 410‐624‐4346 lclawson@jhmi.edu Ahmet Hoke, MD, Principal Investigator United States, Michigan Wayne State University, Dept of Neurology, Detroit, Michigan, 48201, United States; Recruiting Lisa Rowe 313‐577‐1689 lrowe@med.wayne.edu Michael Shy, MD, Principal Investigator United States, New York University of Rochester Medical Center, Dept of Neurology, Rochester, New York, 14642, United States; Recruiting Patty Smith 585‐275‐0581 Patty_Smith@urmc.rochester.edu David Herrmann, MD, Principal Investigator |
| Notes | Listed at ClinicalTrials.gov |
| Trial name or title | Ascorbic Acid Treatment in CMT1A Trial (AATIC) |
| Methods | |
| Participants | CMT1A patients genetically carrying the duplication on chromosome 17q11.2 between 12‐25 years |
| Interventions | Ascorbic acid for 1 year. |
| Outcomes | Change in motor nerve conduction velocity of the median nerve after 1 year primary outcome, Secondary Outcome Measures: * Change in minimal F response latency of the median nerve after 1 year. * Changes in compound muscle action potential amplitude and area after 1 year. * Change in motor unit number estimation of the abductor pollicis brevis muscle after 1 year. * Changes in handgrip strength, strength of armflexors, foot dorsiflexors, knee extensors and hip flexors after 1 year. * Change in overall disability sum score after 1 year. * Change in AMC Linear Disability Scale score after 1 year. * Evaluation of serum ascorbic acid concentrations during 1 year. * Evaluation of side effects during 1 year. |
| Starting date | January 2006 |
| Contact information | Department of Neurology Academic Medical Center University of Amsterdam, Amsterdam, P.O.Box 22660, 1100 DD, Netherlands; Recruiting C. Verhamme, MD +31‐20‐5663856 c.verhamme@amc.uva.nl |
| Notes | Listed at ClinicalTrials.gov |
Ascorbic acid in CMT1A trial (AATIC) Maximum voluntary isometric contraction (MVIC) Academic medical center Linear Disability Scale (AMC Linear Disability Scale) Visual analog scale (VAS) Overall neuropathy limitations scale (ONLS)
Contributions of authors
Peter Young wrote the first and all subsequent drafts. Florian Stögbauer and Peter De Jonghe commented on the first and subsequent drafts. Trude Butterfass‐Bahloul commented on subsequent drafts and supported the systematic search for literature, the review of the literature and systematic biometric analysis. All four authors agreed the final text.
Sources of support
Internal sources
University of Münster, Germany.
University of Antwerp, Belgium.
External sources
European Neuromuscular Centre, Netherlands.
Declarations of interest
None
Edited (no change to conclusions)
References
References to studies included in this review
- Bradley WG, Badger GJ, Tandan R, Fillyaw MJ, Young J, Fries TJ, et al. Double‐blind controlled trials of Cronassial in chronic neuromuscular diseases and ataxia. Neurology 1988;38(11):1731‐1739. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Burns J, Crosbie J, Ouvrier R, Hunt A. Effective orthotic therapy for the painful cavus foot: a randomized controlled trial. Journal of the American Podiatric Medical Association 2006;96(3):205‐211. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chetlin RD, Gutmann L, Tarnopolsky MA, Ullrich IH, Yeater RA. Resistance training exercise and creatine in patients with Charcot‐Marie‐Tooth disease. Muscle & Nerve 2004;30(1):69‐76. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Doherty TJ, Lougheed K, Markez J, Tarnopolsky MA. Creatine monohydrate does not increase strength in patients with hereditary neuropathy. Neurology 2001;57(3):559‐60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Lindeman E, Leffers P, Spaans F, Drukker J, Reulen J, Kerckhoffs M, et al. Strength training in patients with myotonic dystrophy and hereditary motor and sensory neuropathy: a randomized clinical trial. Archives of Physical and Medical Rehabilitation 1995;76(7):612‐20. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Lindeman E, Spaans F, Reulen J, Leffers P, Drukker J. Progressive resistance training in neuromuscular patients. Effects on force and surface EMG. Journal of Electromyography and Kinesiology 1999;9(6):379‐84. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Lindeman E, Drukker J. Specificity of strength training in neuromuscular disorders. Journal of Rehabilitation Sciences 1994;7(Suppl.):13‐15. [Google Scholar]
- Sahenk Z, Nagaraja HN, McCracken BS, King WM, Freimer ML, Cedarbaum JM, et al. NT‐3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology 2005;65(5):681‐9. [DOI] [PubMed] [Google Scholar]
- Smith CA, Chetlin RD, Gutmann L, Yeater RA, Alway SE. Effects of exercise and creatine on myosin heavy chain isoform composition in patients with Charcot‐Marie‐Tooth disease. Muscle & Nerve 2006;34(5):586‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
- Aras O, Karaduman A, Yilmaz O, Basoglu B. The effect of electrical stimulation on muscle strength in neuromuscular diseases. [Turkish]. Fizyoterapi Rehabilitasyon Vol 16(2)()(pp 45‐50), 2005 2005;16(2):45‐50. [MEDLINE: ] [Google Scholar]
- Bean J, Walsh A, Frontera W. Brace modification improves aerobic performance in Charcot‐Marie‐Tooth disease: a single‐subject design. American Journal of Physical Medicine and Rehabilitation 2001;80(8):578‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Brown RE, Zamboni WA, Zook EG, Russell RC. Evaluation and management of upper extremity neuropathies in Charcot‐Marie‐Tooth disease. Journal of Hand Surgery 1992;17(3):523‐30. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chetlin RD, Gutmann L, Tarnopolsky M, Ullrich IH, Yeater RA. Resistance training effectiveness in patients with Charcot‐Marie‐Tooth disease: recommendations for exercise prescription. Archive of Physical Medicine and Rehabilitation 2004;85(8):1217‐23. [DOI] [PubMed] [Google Scholar]
- Congia S, Tronci S, Ledda M, Porcella A, Coppola G. Low doses of TRH in amyotrophic lateral sclerosis and in other neurological diseases. Italian Journal of Neurological Sciences 1991;12(2):193‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Dahl A, Skjeldal OH, Simensen A, Dalen HE, Brathen T, Ahlvin P, et al. [Treatment of patients with neuromuscular disease in a warm climate]. Tidsskr Nor Laegeforen 2004;124(13‐14):1795‐8. [MEDLINE: ] [PubMed] [Google Scholar]
- Matas M, Francis P, Miles JB. Microvascular decompression for trigeminal neuralgia in Charcot‐Marie‐Tooth disease. Journal of Neurosurgery 2000;92(4):715‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Swanson CJ, Low PA, Bartleson JD, Lambert EH. Prednisone‐responsive hereditary motor and sensory neuropathy. Mayo Clinic Proceedings 1982;57(4):239‐46. [PubMed] [Google Scholar]
- Dyck PJ, Swanson CJ, Low PA. Prednisone‐responsive hereditary motor and sensory neuropathy. Neurology 1982;32(4II):710. [PubMed] [Google Scholar]
- Folkers K, Wolaniuk J, Simonsen R, Morishita M, Vadhanavikit S. Biochemical rationale and the cardiac response of patients with muscle disease to therapy with coenzyme Q10. Proceedings of the National Academy of Sciences of the United States of America 1985;82(13):4513‐6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkers K, Simonsen R. Two successful double‐blind trials with coenzyme Q10 (vitamin Q10) on muscular dystrophies and neurogenic atrophies. Biochimica et Biophysica Acta 1995;1271(1):281‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Jaradeh S, Dyck PJ. Hereditary motor and sensory neuropathy with treatable extrapyramidal features. Archives of Neurology 1992;49(2):175‐8. [DOI] [PubMed] [Google Scholar]
- Lindeman E, Leffers P, Reulen J, Spaans F, Drukker J. Reduction of knee torques and leg‐related functional abilities in hereditary motor and sensory neuropathy. Archives of Physical Medicine and Rehabilitation 1994;75(11):1201‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Lindeman E, Leffers P, Reulen J, Spaans F, Drukker J. Quadriceps strength and timed motor performances in myotonic dystrophy, Charcot‐Marie‐Tooth disease, and healthy subjects. Clinical rehabilitation 1998;12(2):127‐35. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Lindeman E, Spaans F, Reulen JP, Leffers P, Drukker J. Surface EMG of proximal leg muscles in neuromuscular patients and in healthy controls. Relations to force and fatigue. Journal of Electromyography and Kinesiology 1999;9(5):299‐307. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Lissett CA, Toogood AA, Didi M, Shalet SM. Growth hormone replacement in an adult with mild growth hormone deficiency and hereditary motor and sensory neuropathy: growth hormone restores independent mobility. Hormone Research 1998;50(4):232‐6. [DOI] [PubMed] [Google Scholar]
- Lobzin VS, Saikova LA, Kosachev VD. Pathogenesis and treatment of neural amyotrophy [Russian]. Zhurnal Nevropatologii i Psikhiatrii Imeni S.S. Korsakova 1986;86(11):1629‐33. [MEDLINE: ] [PubMed] [Google Scholar]
- Lobzin VS, Saikova LA, Kosachev VD, Shiman AG. [Physical therapy of hereditary neuropathies]. Zhurnal Nevropatologii i Psikhiatrii Imeni S.S. Korsakova 1987;87(3):342‐5. [MEDLINE: ] [PubMed] [Google Scholar]
- Lobzin VS, Saikova LA, Shiman AG, Kosachev VD. [Treatment of patients with Charcot‐Marie‐Tooth neural amyotrophy]. Vrach Delo 1988;Dec.(12):73‐4. [MEDLINE: ] [PubMed] [Google Scholar]
- Lobzin VS, Saikova LA, Shiman AG, Kosachev VD. [A comparative evaluation of the efficacy of physical treatment methods in Charcot‐Marie‐Tooth neural amyotrophy]. Vopr Kurortol Fizioter Lech Fiz Kult 1990;Nov‐Dec(6):53‐6. [MEDLINE: ] [PubMed] [Google Scholar]
- Lobzin VS, Zhulev NM, Kosachev VD, Tiukarkina AB, Badzgaradze I, Zavolokov IG. [Autonomic vascular disorders in neuropathies and the methods for their pathogenetic therapy]. Zhurnal Nevropatologii i Psikhiatrii Imeni S.S. Korsakova 1992;92(5‐12):32‐5. [MEDLINE: ] [PubMed] [Google Scholar]
- Mann DC, Hsu JD. Triple arthrodesis in the treatment of fixed cavovarus deformity in adolescent patients with Charcot‐Marie‐Tooth disease. Foot & Ankle 1992;13(1):1‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Miller GM, Hsu JD, Hoffer MM, Rentfro R. Posterior tibial tendon transfer: a review of the literature and analysis of 74 procedures. Journal of Pediatric Orthopedics 1982;2(4):363‐70. [MEDLINE: ] [PubMed] [Google Scholar]
- Oganesyan OV, Istomina IS, Kuzmin VI. Treatment of equinocavovarus deformity in adults with the use of a hinged distraction apparatus. The Journal of Bone and Joint Surgery. American Volume 1996;78(4):546‐56. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Prensky AL, Dodson WE. The steroid treatment of hereditary motor and sensory neuropathy. Neuropediatrics 1984;15(4):203‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Riley WB, Mann RJ, Burkhalter WE. Extensor pollicis longus opponensplasty. Journal of Hand Surgery [Am.] 1980;5(3):217‐20. [DOI] [PubMed] [Google Scholar]
- Russell JW, Windebank AJ, Harper CM. Treatment of stable chronic demyelinating polyneuropathy with 3,4‐diaminopyridine. Mayo Clinic Proceedings 1995;70(6):532‐39. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Saltzman CL, Fehrle MJ, Cooper RR, Spencer EC, Ponseti IV. Triple arthrodesis: twenty‐five and forty‐four‐year average follow‐up of the same patients. The Journal of Bone and Joint Surgery. American Volume 1999;81(10):1391‐1402. [MEDLINE: ] [PubMed] [Google Scholar]
- Trumble SJ, Mayo KA, Mast JW. The periacetabular osteotomy. Minimum 2 year followup in more than 100 hips. Clinical Orthopaedics and Related Research 1999;363:54‐63. [PubMed] [Google Scholar]
- Wetmore RS, Drennan JC. Long‐term results of triple arthrodesis in Charcot‐Marie‐Tooth disease. The Journal of Bone and Joint Surgery. American Volume 1989;71(3):417‐22. [MEDLINE: ] [PubMed] [Google Scholar]
- Williams LL, O'Dougherty MM, Wright FS, Bobulski RJ, Horrocks LA. Dietary essential fatty acids, vitamin E, and Charcot‐Marie‐Tooth disease. Neurology 1986;36(9):1200‐1205. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Wood VE, Huene D, Nguyen J. Treatment of the upper limb in Charcot‐Marie‐Tooth disease. Journal of Hand Surgery (Edinburgh, Lothian) 1995;20(4):511‐18. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
- Information requested. Ongoing study Starting date of trial not provided. Contact author for more information.
- Single center randomized double blind placebo controlled. Ongoing studySeptember 2007.
- A multicenter, randomized, double‐blind, placebo‐controlled trial of long term ascorbic acid in Charcot‐Marie‐tooth disease type 1A (CMT‐TRIAAL). Ongoing studyAutumn 2006. [DOI] [PubMed]
- A Randomized, placebo‐controlled, double masked 120 subject "Futility Design" clinical trial of Ascorbic acid treatment of Charcot‐Marie‐Tooth Disease Type 1A.. Ongoing studyApril 2007.
- Ascorbic Acid Treatment in CMT1A Trial (AATIC). Ongoing studyJanuary 2006.
Additional references
- Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22‐deficient mice. Nature Genetics 1995;11(3):274‐80. [DOI] [PubMed] [Google Scholar]
- Adlkofer K, Frei R, Neuberg DH, Zielasek J, Toyka KV, Suter U. Heterozygous peripheral myelin protein 22‐deficient mice are affected by a progressive demyelinating tomaculous neuropathy. Journal of Neuroscience 1997;17(12):4662‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RT, Aaronson NK, Bullinger M, McBee WL. A review of the progress towards developing health‐related quality‐of‐life instruments for international clinical studies and outcomes research. Pharmacoeconomics 1996;10(4):336‐55. [DOI] [PubMed] [Google Scholar]
- Cornblath DR, Chaudhry V, Carter K, Lee D, Seysedadr M, Miernicki M, et al. Total neuropathy score: validation and reliability study. Neurology 1999;53(8):1660‐4. [DOI] [PubMed] [Google Scholar]
- Shy ME, Lupski JR, Chance PF, Klein CJ, Dyck PJ. Hereditary Motor And Sensory Neuropathy. In: Dyck PJ, Thomas PK editor(s). Peripheral Neuropathy. Vol. 2, Philadelphia: WB Saunders, 2005:1623‐58. [Google Scholar]
- Dyck PJ, Hughes RAC, O'Brien PC. Quantitating overall neuropathic symptoms, impairments, and outcomes. In: Dyck PJ, Thomas PK editor(s). Peripheral Neuropathy. 4th Edition. Philadelphia: WB Saunders, 2005:1031‐52. [Google Scholar]
- Gabreels‐Festen A, Gabreels F. Hereditary demyelinating motor and sensory neuropathy. Brain Pathology 1993;3(2):135‐46. [DOI] [PubMed] [Google Scholar]
- Grandis M, Shy ME. Current therapy for Charcot‐Marie‐Tooth disease. Current Treatment Options in Neurology 2005;7(1):23‐31. [DOI] [PubMed] [Google Scholar]
- Hahn AF, Ainsworth PJ, Bolton CF, Bilbao JM, Vallat JM. Pathological findings in the x‐linked form of Charcot‐Marie‐Tooth disease: a morphometric and ultrastructural analysis. Acta Neuropathologica (Berlin) 2001;101(2):129‐39. [DOI] [PubMed] [Google Scholar]
- Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980;103(2):259‐80. [DOI] [PubMed] [Google Scholar]
- Hayasaka K, Himoro M, Takada G, Takahashi E, Minoshima S, Shimizu N. Structure and localization of the gene encoding human peripheral myelin protein 2 (PMP2). Genomics 1993;18:244‐8. [DOI] [PubMed] [Google Scholar]
- Hayasaka K, Himoro M, Sawaishi Y, Nanao K, Takahashi T, Takada G, et al. De novo mutation of the myelin Po gene in Dejerine‐Sottas disease (hereditary motor and sensory neuropathy type III). Nature Genetics 1993;5:266‐8. [DOI] [PubMed] [Google Scholar]
- Hayasaka K, Ohnishi A, Takada G, Fukushima Y, Murai Y. Mutation of the myelin P0 gene in Charcot‐Marie‐tooth neuropathy type 1. Biochemical and Biophysical Research Communications 1993;194(3):1317‐22. [DOI] [PubMed] [Google Scholar]
- Jenkinson C, Coulter A, Wright L. Short form 36 (SF36) health survey questionnaire: normative data for adults of working age. British Medical Journal 1993;306(6890):1437‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski K, Turansky C, Lewis R, Garbern J, Hinderer S, Kamholz J, et al. Correlation between weakness and axonal loss in patients with CMT1A. Annals of the New York Academy of Science 1999;883:490‐2. [PubMed] [Google Scholar]
- Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, et al. Neurological dysfunction and axonal degeneration in Charcot‐Marie‐Tooth disease type 1A. Brain 2000;123(Pt 7):1516‐27. [DOI] [PubMed] [Google Scholar]
- Kuhlenbaumer G, Young P, Hunermund G, Ringelstein B, Stogbauer F. Clinical features and molecular genetics of hereditary peripheral neuropathies. Journal of Neurology 2002;249(12):1629‐50. [DOI] [PubMed] [Google Scholar]
- Lupski J, Oca‐Luna R, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, et al. DNA duplication associated with Charcot‐Marie‐Tooth disease type 1A. Cell 1991;66(2):219‐32. [DOI] [PubMed] [Google Scholar]
- Lupski J, Wise C, Kuwano A, Pentao L, Parke J, Glaze DG, et al. Gene dosage is a mechanism for Charcot‐Marie‐Tooth disease type 1A. Nature Genetics 1992;1(1):29‐33. [DOI] [PubMed] [Google Scholar]
- Martini R. Animal models for inherited peripheral neuropathies. Journal of Anatomy 1997;191(Pt 3):321‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R. Animal models for inherited peripheral neuropathies: chances to find treatment strategies?. Journal of Neuroscience Research 2000;61(3):244‐50. [DOI] [PubMed] [Google Scholar]
- Martini R. The effect of myelinating Schwann cells on axons. Muscle & Nerve 2001;24(4):456‐66. [DOI] [PubMed] [Google Scholar]
- Maurer M, Kobsar I, Berghoff M, Schmid CD, Carenini S, Martini R. Role of immune cells in animal models for inherited neuropathies: facts and visions. Journal of Anatomy 2002;200(4):405‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkies IS, Schmitz PI, Meche FG, Doorn PA. Psychometric evaluation of a new sensory scale in immune‐mediated polyneuropathies. Inflammatory Neuropathy Cause and Treatment (INCAT) Group. Neurology 2000;54(4):943‐9. [DOI] [PubMed] [Google Scholar]
- Nelis E, Broeckhoven C, Jonghe P, Lofgren A, Vandenberghe A, Latour P, et al. Estimation of the mutation frequencies in Charcot‐Marie‐Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. European Journal of Human Genetics 1996;4(1):25‐33. [DOI] [PubMed] [Google Scholar]
- Nicholson G, Nash J. Intermediate nerve conduction velocities define X‐linked Charcot‐Marie‐Tooth neuropathy families. Neurology 1993;43(12):2558‐64. [DOI] [PubMed] [Google Scholar]
- Pareyson D. Differential diagnosis of Charcot‐Marie‐Tooth disease and related neuropathies. Neurological Science 2004;52(2):72‐82. [DOI] [PubMed] [Google Scholar]
- Pareyson D, Schenone A, Fabrizi GM, Santoro L, Padua L, Quattrone A, et al. A multicenter, randomized, double‐blind, placebo‐controlled trial of long‐term ascorbic acid treatment in Charcot‐Marie‐Tooth disease type 1A (CMT‐TRIAAL): the study protocol [EudraCT no.: 2006‐000032‐27]. Pharmacological Research 2006;54(6):436‐41. [DOI] [PubMed] [Google Scholar]
- Passage E, Norreel JC, Noack‐Fraissignes P, Sanguedolce V, Pizant J, Thirion X, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot‐Marie‐Tooth disease. Nature Medicine 2004;10(4):396‐401. [DOI] [PubMed] [Google Scholar]
- Reilly MM, Jonghe P, Pareyson D. 136th ENMC International Workshop: Charcot‐Marie‐Tooth disease type 1A (CMT1A)8‐10 April 2005, Naarden, The Netherlands. Neuromuscular Disorders 2006;16(6):396‐402. [DOI] [PubMed] [Google Scholar]
- Sereda MW, Meyer zu Horste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot‐Marie‐Tooth disease (CMT‐1A). Nature Medicine 2003;9(12):1533‐7. [DOI] [PubMed] [Google Scholar]
- Shy ME, Blake J, Krajewski K, Fuerst DR, Laura M, Hahn AF, et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 2005;64(7):1209‐14. [DOI] [PubMed] [Google Scholar]
- Street VA, Bennett CL, Goldy JD, Shirk AJ, Kleopa KA, Tempel BL, et al. Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot‐ Marie‐Tooth disease 1C. Neurology 2003;60(1):22‐6. [DOI] [PubMed] [Google Scholar]
- Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nature Reviews Neuroscience 2003;4(9):714‐26. [DOI] [PubMed] [Google Scholar]
- Timmerman V, Nelis E, Hul W, Nieuwenhuijsen B, Chen K, Wang S, et al. The peripheral myelin protein gene PMP‐22 is contained within the Charcot‐Marie‐Tooth disease type 1A duplication. Nature Genetics 1992;1(3):171‐5. [DOI] [PubMed] [Google Scholar]
- Kooi EL, Lindeman E, Riphagen I. Strength training and aerobic exercise training for muscle disease. Cochrane Database of Systematic Reviews 2005;The Cochrane Database of Systematic Reviews 2005(1):CD003907. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke 1988;19(5):604‐7. [DOI] [PubMed] [Google Scholar]
- Verhoeven K, Claeys KG, Zuchner S, Schroder JM, Weis J, Ceuterick C, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot‐Marie‐Tooth type 2. Brain 2006;129(Pt 8):2093‐102. [DOI] [PubMed] [Google Scholar]
- White CM, Pritchard J, Turner‐Stokes L. Exercise for people with peripheral neuropathy. Cochrane Database of Systematic Reviews 2004, Issue 4. [MEDLINE: ; CD003904] [DOI] [PubMed] [Google Scholar]
- Young P, Suter U. Disease mechanisms and potential therapeutic strategies in Charcot‐Marie‐Tooth disease. Brain Research. Brain Research Reviews 2001;36(2‐3):213‐21. [DOI] [PubMed] [Google Scholar]
- Young P, Suter U. The causes of Charcot‐Marie‐Tooth disease. Cellular and Molecular Life Sciences 2003;60(12):2547‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar‐Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nature Genetics 2004;36(5):449‐51. [DOI] [PubMed] [Google Scholar]